
Design, synthesis, and evaluation of azo D–π-A dyes as photothermal agents†
Shinichiro
Fuse
 *a,
Tsukasa
Oishi
ab,
Keisuke
Matsumura
a,
Yoshihiro
Hayashi
c,
Susumu
Kawauchi
c and
Hiroyuki
Nakamura
*a
*a,
Tsukasa
Oishi
ab,
Keisuke
Matsumura
a,
Yoshihiro
Hayashi
c,
Susumu
Kawauchi
c and
Hiroyuki
Nakamura
*a
aLaboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, Yokohama, 226-8503, Japan. E-mail: sfuse@res.titech.ac.jp; hiro@res.titech.ac.jp
bSchool of Life Science and Technology, Tokyo Institute of Technology, Yokohama, 226-8501, Japan
cSchool of Materials and Chemical Technology, Tokyo Institute of Technology, Tokyo, 152-8552, Japan
First published on 4th December 2019
Abstract
Thirteen readily accessible azo D–π-A dyes, intended for use as photothermal agents, were synthesized using only a few steps. Absorption wavelengths were readily tuned by changing the building blocks, and 6 of these dyes exhibited NIR absorption that would be useful for biomedical applications. Unexpected suppression of an N–C single bond rotation that neighbors the azo bond was observed in the case of 5 dyes. Photothermal conversion efficiency measurements revealed a significant effect of the D moiety in these synthesized azo D–π-A dyes, but neither the π moiety nor the A moiety showed an obvious influence. The obtained results offer valuable information for the design of high-performance azo D–π-A dyes that have utility as photothermal agents.
Introduction
Photothermal agents/materials that convert light energy into thermal energy have garnered much attention in recent years based on their utility in photothermal therapy (PTT)1–4 and photoacoustic imaging (PAI).5–8 In addition, they are also useful for the efficient release of drugs from carriers,9–12 as well as in tissue repair,13 SPR-mediated photothermal chemistry,14 and enzymatic reactions.15 Inorganic, gold and carbon materials are known to be highly efficient and stable photothermal materials. For biomedical applications, however, concerns that include slow clearance and toxicity in vivo persist.16,17Indocyanine green (ICG), an organic small molecule, is the only agent that is approved for clinical use, but it suffers from instability against oxygen and photoirradiation and low photothermal conversion efficiency due to its fluorescence properties (Fig. 1).18 In order to overcome the drawbacks of ICG, many structurally rigid π-conjugated molecules originally used as organic electronic materials have been reported.8 Although their stability is generally high and controlling their photoabsorption wavelengths is easy, they tend to have undesired fluorescence properties. Therefore, aggregation of the molecules is usually required to suppress the fluorescence. The development of synthetic, readily accessible, and stable organic small molecules that allow easy absorption wavelength tuning and exhibit high photothermal conversion efficiency remains important. However, lack of information concerning the structure–photothermal conversion relationship has hampered the design of high-performance photothermal agents.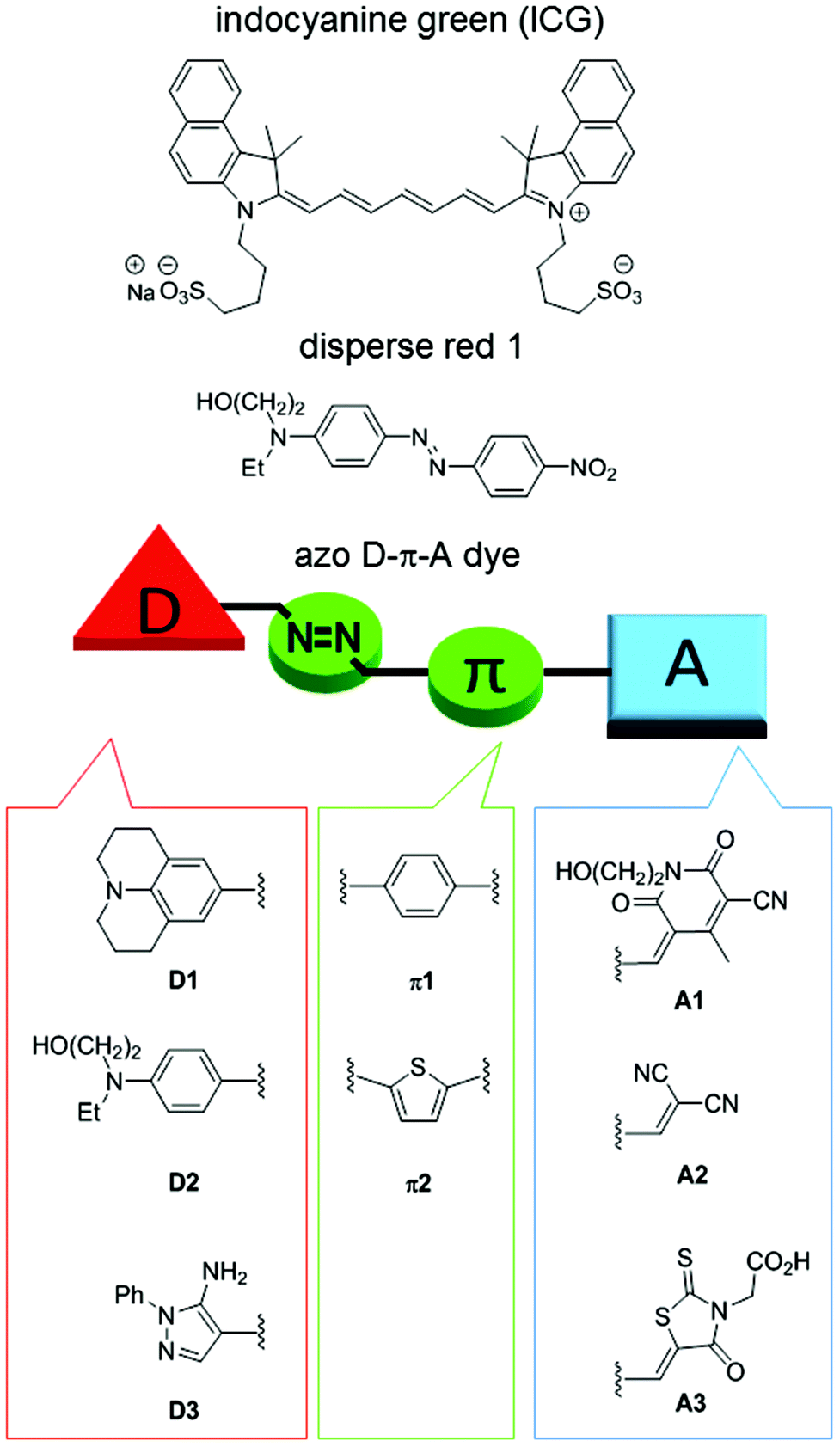 | ||
| Fig. 1 Chemical structures of indocyanine green (ICG), disperse red 1, and azo D–π-A dyes. | ||
We have designed functional D–π-A dyes and developed efficient synthetic approaches for them while constructing a library to help elucidate the structure–function relationship.19–29 As far as we could ascertain, disperse red 1 is the only D–π-A azo dye credited with exerting a photoacoustic effect (Fig. 1),30 but neither the photothermal conversion efficiency nor the structure–photothermal conversion relationship has been reported. In the present study, we designed and synthesized 13 azo D–π-A dyes that combine an electron-donating moiety (D), a π-conjugated bridge that consists of an azo bond (π), and an electron-accepting moiety (A), as shown in Fig. 1. NMR analysis of these synthesized dyes revealed the unexpected slow rotation of a single bond neighboring the azo bond. Evaluation of the photothermal conversion efficiency revealed how each building block contributes to photothermal conversion.
Results and discussion
We have designed azo D–π-A dyes for use as photothermal agents. These dyes can be synthesized using only a small number of steps that involved using azo coupling reactions.31 The absorption wavelengths of these dyes can be readily tuned by changing the building blocks. The azo bond is highly flexible when in an excited state,32 which results in an efficient photothermal conversion that is usually more stable in comparison with disubstituted alkenes. In addition, D–π-A dyes undergo intramolecular charge transfer via photoirradiation wherein a polarity change before and after the photoexcitation causes a rearrangement of the solvent molecules. Therefore, we anticipated the consumption of excited energy by the D–π-A dyes via a nonradiative process that could enhance photoconversion efficiency.33,34Building blocks with different electron-donating or -accepting abilities were used for the synthesis of these azo D–π-A dyes (Fig. 1). Electron-donating ability was decreased on the order of D1 > D2 > D3. Julolidine, D1, demonstrated very strong electron-donating ability. D2 has the donor structure of disperse red 1. Reportedly, a pyrazole ring exhibits a large Stokes shift due to intense motion via kite and butterfly mechanisms while in an excited state.35 Therefore, we anticipated that D3 would cause nonradiative deactivation that would enhance photothermal conversion. Typical π-conjugated benzene and thiophene bridges were selected as π1 and π2. Electron-accepting ability was decreased on the order of A1 > A2 > A3. Cyanopyridone acceptor, A1, demonstrated very strong electron-accepting ability.
Steps for the synthesis of 6 azo D–π-A dyes containing π1 are shown in Scheme 1. In our preliminary study, 4-nitrobenzaldehyde (π1) was in situ reduced to the corresponding 4-aminobenzaldehyde in the presence of SnCl2 and HCl.36 Unstable 4-aminobenzaldehyde was directly converted into diazonium salt without purification, and it was coupled with highly nucleophilic D1. This approach, however, resulted in D1π1 with low purity. Therefore, the polymer form of 4-aminobenzaldehyde π1 was used instead of 4-aminobenzaldehyde.37 Reversible depolymerization occurred under acidic conditions and the in situ-generated 4-aminobenzaldehyde monomer was irreversibly converted to the corresponding diazonium salt to couple with D1. The desired D1π1 was obtained in better purity. This approach also afforded the desired products D2π1 and D3π1. These aldehydes were somewhat unstable and not suitable for storage, and, therefore, they were used for the subsequent Knoevenagel condensation following rough purification by silica-gel column chromatography. D1π1–D3π1 with A2 and A3 afforded the desired 6 azo D–π-A dyes in acceptable to good yields. Attempts to obtain coupling products from D1π1–D3π1 with A1 were in vain, however, probably due to high levels of steric repulsion between the hydrogen atom on the benzene ring of π1 and the methyl and hydroxy substituents on the pyridone ring of A1.
 | ||
| Scheme 1 Synthesis of 6 azo D–π-A dyes containing π1. | ||
Synthesis of 6 azo D–π-A dyes containing π2 is shown in Scheme 2. 5-Nitrothiophene-2-carbaldehyde (π2) was in situ reduced to the corresponding 5-aminothiophene-2-carbaldehyde in the presence of SnCl2 and HCl. The aminothiophene was converted to diazonium salt, and it was coupled with D1 or D2 to obtain D1π2 and D2π2. In contrast to the coupling of D3 with π1, the coupling of D3 with π2 did not proceed probably due to the lower electrophilicity of the diazonium salt delivered from an electron-rich thiophene (π2). The subsequent Knoevenagel condensation of two aldehydes, D1π2 and D2π2, with A1–A3 afforded the desired 6 azo D–π-A dyes. The coupling of less sterically hindered thiophene-containing D1π2 and D2π2 with A1 proceeded smoothly. These results corroborate the previous speculation as to why the coupling between more sterically hindered benzene-containing aldehydes D1π1–D3π1 with A1 did not afford the desired products.
 | ||
| Scheme 2 Synthesis of 6 azo D–π-A dyes containing π2. | ||
The nitro group is a well-known fluorescence-quenching functional group, and, therefore, a nitro-containing D1π1A4 dye was additionally synthesized as shown in Scheme 3. The D1π1A4 dye was obtained as mixtures of E/Z isomers (E/Z = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3). Reportedly, when an electron-donating structure is conjugated to a nitro acrylic acid ester moiety, E/Z isomerization of the alkene readily occurs. Our observation is consistent with this report.38
:1.3). Reportedly, when an electron-donating structure is conjugated to a nitro acrylic acid ester moiety, E/Z isomerization of the alkene readily occurs. Our observation is consistent with this report.38
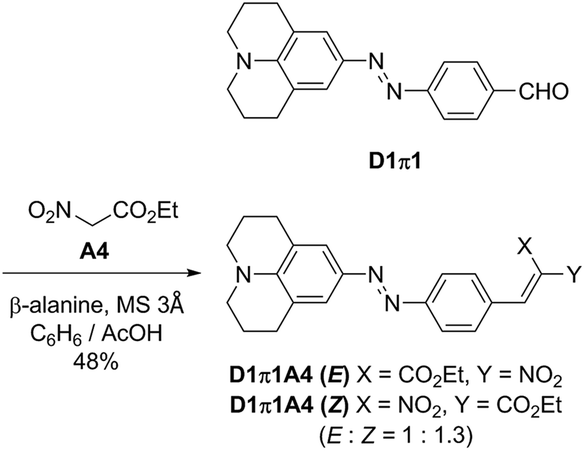 | ||
| Scheme 3 Synthesis of the nitro-containing D1π1A4 dye. | ||
Interestingly, in the 13C NMR measurement of the synthesized azo dyes at 27 °C, the signals of C2 and C3 in D1π2A1–D1π2A3 and D2π2A1–D2π2A3 were significantly broadened (Fig. 2, group A). The 13C NMR signals in D1π2A1–D1π2A3 containing highly electron-donating D1 seemed to be more broadened than those in D2π2A1–D2π2A3, which contained the lesser electron-donating D2. Moreover, the 13C NMR signals of C2 and C3 in D3π1A2 and D3π1A3 dyes were not observed (Fig. 2, group B).
 | ||
| Fig. 2 Structure of the synthesized D1π2A1–D1π2A3 and D2π2A1–D2π2A3 dyes (group A, this study), D3π1A2 and D3π1A3 dyes (group B, this study), and azobenzenes I and II (reported study) showed broadened or undetectable 13C NMR signals. EDG: electron-donating group and EWG: electron-withdrawing group. | ||
Stepanov and coworkers reported the low-temperature 13C NMR analysis of substituted azobenzenes I and they observed broadened signals at −100 °C when dyes I contained a benzene ring with EDG (Fig. 2, reported study).39 They explained that the suppressed rotation of an N–C1 single bond caused the broadening of signals. Enhancement of the electron-accepting ability of another benzene ring further slows down the rotation of the N–C1 single bond. They concluded that an increase in the double bond characteristics of the N–C1 bond by EDG and EWG slowed down the rotation of the N–C1 bond. Savarino and coworkers reported the suppressed rotation of the N–C1 single bond of II when EWG was attached to the benzene ring (Fig. 2, reported study).40 They reported that an enhancement of the electron-withdrawing ability of EWG increases the acidity of N–H protons, which creates a stronger hydrogen bond with a nitrogen atom in an azo group. This slowed down the rotation of the N–C1 single bond.
We assumed that the combination of EDG, EWG, and the thiophene bridge π2 (group A) that did not twist the molecule assisted electron-delocalization around the D–π moiety. This combination enhanced the double bond characteristics of the N–C1 bond and led to a significant broadening of the 13C NMR signals of C2 and C3 even at room temperature. On the other hand, the unobserved 13C NMR signals of C2 and C3 in D3π1A2 and D3π1A3 dyes (group B) arose from the hydrogen bond formation that was similar to the case of azo dyes II. As far as we could ascertain, this is the first report on the significant suppression of a single bond rotation in azo D–π-A dyes even at room temperature.
In order to further confirm our speculation, DFT calculations were performed to estimate the rotational barrier of the N–C1 single bond (Fig. 3). The calculation results indicated that the combination of electron-donating D1 or D2, thiophene bridge π2, and the acceptor A2 resulted in a rotational barrier (11.1, 10.4 kcal mol−1, respectively, Fig. 3a and b) that was higher than that of the combination of D1, benzene bridge π1, and A2 (10.1 kcal mol−1, Fig. 3c). The calculation results indicated that D1π2A2, which contained the highly electron-donating D1, had a higher rotational barrier (11.1 kcal mol−1) than D2π2A2 (10.4 kcal mol−1), which contained the fewer electron-donating D2. In addition, the D3π1A2 dye showed a very high rotational barrier (14.5 kcal mol−1, Fig. 3d). All the calculation results agreed well with the experimental observations, which corroborated our speculation.
 | ||
| Fig. 3 DFT calculations used to estimate the rotational barrier of N–C1 single bond dyes: (a) D1π2A2, (b) D2π2A2, (c) D1π1A2, and (d) D3π1A2. | ||
The absorption spectra of the synthesized dyes were measured (Table 1 and Fig. 4).41 As expected, the absorption wavelengths of the dyes that contained the higher electron-donating form of the D moiety became longer (D3 < D2 < D1) and those of the dyes that contained the higher electron-accepting form of the A moiety also became longer (A4 < A3 < A2 < A1). In addition, the absorption wavelengths of the dyes containing π2 (a thiophene ring) was longer than those of the dyes containing π1 (a benzene ring). The D1π2A1, D1π2A2, D1π2A3, D2π2A1, D2π2A2, and D2π2A3 dyes show near infrared (NIR) absorption that is preferable for biomedical applications.
 | ||
| Fig. 4 Absorption spectra of the synthesized dyes containing π1 in DMSO (a) and the synthesized dyes containing π2 in DMSO (b). | ||
| Dye |
λ
maxa [nm] |
εb [Lmol−1 cm−1] |
Optical edgec [nm] |
|---|---|---|---|
| a Absorption maxima in DMSO. b Molar absorption coefficients in DMSO. c The edge of the absorption spectra was defined by the wavelength for which the absorbance revealed 1/10 of peak top. | |||
| D1π1A2 | 572 | 33200 |
678 |
| D1π1A3 | 550 | 37800 |
643 |
| D1π1A4 | 494 | 26700 |
590 |
| D1π2A1 | 750 | 35500 |
878 |
| D1π2A2 | 674 | 55200 |
790 |
| D1π2A3 | 642 | 64600 |
752 |
| D2π1A2 | 534 | 29900 |
631 |
| D2π1A3 | 517 | 22500 |
604 |
| D2π2A1 | 669 | 31500 |
799 |
| D2π2A2 | 622 | 45200 |
735 |
| D2π2A3 | 604 | 47400 |
703 |
| D3π1A2 | 476 | 29600 |
564 |
| D3π1A3 | 473 | 29000 |
548 |
The photothermal conversion efficiency of the synthesized dyes was evaluated by simple instruments that included a light source, a quartz-derived cell, and a thermocouple (for details, see the ESI†). In order to completely dissolve the synthesized dyes, a DMSO solution (80 mM) was used for the measurement. The photothermal curve and an iterative photoirradiation cooling curve of the D2π2A1 dye are shown in Fig. 5 (for the data of other dyes, see the ESI†). The temperature of the DMSO solution of the D2π2A1 dye was elevated to 27.6 °C from 24.5 °C with photoirradiation. On the other hand, the temperature of the DMSO blank sample was only elevated to ≤0.2 °C. The temperature increase under photoirradiation was not changed in 5 cycles of photoirradiation and cooling, and this indicated that the D2π2A1 dye was stable against photoirradiation and oxygen. The other dyes also showed good stability. The calculated photothermal conversion efficiency42 is listed in Table 2. The donor structure had a significant effect on the photothermal conversion efficiency. The dyes with a D1 donor (entries 1–6) exhibited a decreased photothermal conversion efficiency compared with those of the dyes with D2 or D3 donors (entries 7–13). This is probably due to the rigid structure of a D1 donor that helps avoid the nonradiative process. On the other hand, the π-bridge and A moieties had no obvious influence on the photothermal efficiency with the exception of the nitro-group containing A4 (entry 3). The D1π1A4 dye exhibited the highest level of photothermal conversion efficiency regardless of the existence of a rigid D1 donor. The D2π2A1 dye that was absorbed in the NIR region exhibited the highest photothermal conversion efficiency (entry 9). ICG (80 mM in DMSO) exhibited a photothermal conversion efficiency of 28%, as shown by our measurements. These results clearly indicate the high level of photothermal converting ability achieved by our designed azo D–π-A dyes.
 | ||
| Fig. 5 Photothermal curve (a) and iterative photoirradiation cooling curve of the D2π2A1 dye (b). | ||
| Entry | Dye | Photothermal conversion efficiency |
|---|---|---|
| 1 | D1π1A2 | 29% |
| 2 | D1π1A3 | 31% |
| 3 | D1π1A4 | 53% |
| 4 | D1π2A1 | 34% |
| 5 | D1π2A2 | 31% |
| 6 | D1π2A3 | 32% |
| 7 | D2π1A2 | 60% |
| 8 | D2π1A3 | 41% |
| 9 | D2π2A1 | 66% |
| 10 | D2π2A2 | 43% |
| 11 | D2π2A3 | 62% |
| 12 | D3π1A2 | 40% |
| 13 | D3π1A3 | 59% |
Conclusions
In conclusion, synthetic, readily accessible azo D–π-A dyes were synthesized for use as photothermal agents. These 13 dyes were synthesized using only a small number of synthetic steps and building blocks with different electron-donating or electron-accepting abilities. The absorption wavelength was readily tuned by changing the building blocks. Six dyes showed NIR absorption that will be useful for biomedical applications: D1π2A1, D1π2A2, D1π2A3, D2π2A1, D2π2A2, and D2π2A3. The unexpected suppression of an N–C single bond rotation neighboring the azo bond was observed in the case of 8 dyes: D1π2A1, D1π2A2, D1π2A3, D2π2A1, D2π2A2, D2π2A3, D3π1A2, and D3π1A3. This is the first report on the significant suppression of a single bond rotation in azo D–π-A dyes even at room temperature. Photothermal conversion efficiency measurements revealed a significant effect of the D moiety in our synthesized azo D–π-A dyes, but on the other hand, neither the π moiety nor the A moiety showed an obvious influence, with the exception of A4. The nitro group containing acceptor A4 showed a high level of photothermal conversion ability. These results suggest that the combination of a D moiety that exhibits high photothermal conversion ability and the proper π and A moieties that lead to a desired absorption wavelength is a good approach for synthesizing high performance azo D–π-A dyes with the desired properties.Experimental
General chemistry
The NMR spectra were recorded using either a Bruker Biospin Avance II 400 (400 MHz for 1H, 100 MHz for 13C) or a Bruker Biospin Avance III HD500 (500 MHz for 1H, 125 MHz for 13C) in the indicated solvent. Chemical shifts are reported in units of parts per million (ppm) relative to tetramethylsilane (0.00 ppm) or CDCl3 (7.26 ppm) or DMSO-d6 (2.50 ppm) for 1H NMR and CDCl3 (77.2 ppm) or DMSO-d6 (39.5 ppm) for 13C NMR. Multiplicities are reported using the following abbreviations: s, singlet; d, doublet; dd, doublet of doublets; t, triplet; m, multiplet; and coupling constants J are reported in hertz (Hz). IR spectra were recorded on a JASCO Corporation FT/IR-4100 FT-IR spectrometer. An ATR PRO ONE was attached to the FT/IR-4100 to measure solid IR spectra via single reflection attenuated total reflection. Only the strongest and/or structurally important peaks were reported and the IR data are given in cm−1. The absorption spectra were recorded using a JASCO V-670. All reactions were monitored by thin layer chromatography carried out on 0.2 mm E. Merck silica gel plates (60 F254) with UV light and visualized using p-anisaldehyde solution and 10% ethanolic phosphomolybdic acid. Flash column chromatography was performed on silica gel (Fuji Silysia, CHROMATOREX PSQ 60B, 50–200 μm).General procedure for azo coupling from the polymer form of 4-aminobenzaldehyde (p1)
The polymer form of 4-aminobenzaldehyde (π1) (1.0 equiv.) was stirred in 1 M HCl (3.2 mL per 1 mmol of D block) for 1 h under argon at room temperature. The resulting solution was cooled to 0 °C and a solution of sodium nitrite (1.5 equiv.) in water (0.54 mL per 1 mmol of D block) was added dropwise cautiously and stirred for 2 h at 0 °C. The resulting diazonium salt solution was added dropwise to the D block (1.0 equiv.) and AcONa (400 mg per 1 mmol of D block) solution dissolved in ethanol (3 mL per 1 mmol of D block) and water (3 mL per 1 mmol of D block) at 0 °C. The resulting solution was stirred for 2 h at 0 °C and 2 h at room temperature. The mixture was washed with brine and extracted with CH2Cl2 3 times. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was roughly purified by silica gel column chromatography with CH2Cl2. The obtained aldehydes D1π1–D3π1 were used for the next Knoevenagel condensation without further purification.General procedure for azo coupling from 5-nitrothiophene-2-carbaldehyde (π2)
A mixture of 5-nitrothiophene-2-carbaldehyde (π2) (1.0 equiv.) and stannous chloride (3 equiv.) in conc. HCl (3.3 mL per 1 mmol of D block) and THF (3.3 mL per 1 mmol of D block) was stirred at 60 °C for 1 h and then gradually cooled to room temperature. The resulting mixture was cooled to 0 °C and a solution of sodium nitrite (1.0 equiv.) in water (1.3 mL per 1 mmol of D block) was added dropwise cautiously. The resulting solution was stirred at 0 °C for 2 h to form the diazonium salt, which was used directly in the next step. The resulting diazonium salt solution was added dropwise to the D block (1.0 equiv.) and AcONa (2.5 g per 1 mmol of D block) solution dissolved in ethanol (7.5 mL per 1 mmol of D block) and water (7.5 mL per 1 mmol of D block) at 0 °C. The resulting solution was stirred at 0 °C for 12 h and at room temperature for 8 h. The mixture was washed with brine and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was roughly purified by silica gel column chromatography with CH2Cl2. The obtained aldehydes D1π2 and D2π2 were used for the next Knoevenagel condensation without further purification.General procedure for Knoevenagel condensation
To a solution of semi-pure aldehyde (ca. 1.0 equiv.) and A block (1.5 equiv.) in MeCN (15.0 mL per 1 mmol of aldehyde), piperidine (0.1 equiv.) was added and stirred for 1–6 h. To the reaction mixture was added saturated aq. NH4Cl and the aqueous layer was extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography and/or recrystallization.2-(4-((2,3,6,7-Tetrahydro-1H,5H-pyrido[3,2,1-ij]quinolin-9-yl)diazenyl)benzylidene)malononitrile (D1π1A2)
The residue was purified by silica gel column chromatography with CH2Cl2. Green solid; 57.3 mg, 0.162 mmol, 2 steps 41% yield; 1H NMR (500 MHz, CDCl3): δ 7.98 (d, J = 8.6 Hz, 2H), 7.86 (d, J = 8.6 Hz, 2H), 7.70 (s, 1H), 7.49 (s, 2H), 3.34 (t, J = 5.8 Hz, 4H), 2.82 (t, J = 6.3 Hz, 4H), 2.00 (m, 4H); 13C NMR (125 MHz, CDCl3): δ 158.9, 157.3, 147.4, 143.3, 132.3, 130.5, 124.1, 123.0, 121.3, 114.4, 113.3, 80.8, 50.4, 27.9, 21.5; FT-IR (neat) 2924, 2847, 2219, 1595, 1577, 1556, 1520, 1297, 1266, 1207, 1104 cm−1; HRMS (ESI) calcd for C22H19N5Na [M + Na+] 376.1533, found 376.1535.2-(4-Oxo-5-((Z)-4-((E)-(2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinolin-9-yl)diazenyl)benzylidene)-2-thioxothiazolidin-3-yl)acetic acid (D1π1A3)
The residue was purified by silica gel column chromatography with CH2Cl2 and methanol (9:1) and recrystallized from ethanol. Brownish solid; 65.1 mg, 0.136 mmol, 2 steps 34% yield; 1H NMR (500 MHz, DMSO-d6): δ 7.93 (s, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.79 (d, J = 8.0 Hz, 2H), 7.41 (s, 2H), 4.75 (s, 2H), 3.35 (m, 4H), 2.76 (m, 4H), 1.90 (m, 4H); 13C NMR (125 MHz, DMSO-d6): δ 193.1, 167.4, 166.4, 153.8, 146.7, 142.0, 133.4, 132.8, 132.2, 123.2, 122.5, 122.0, 121.5, 49.5, 45.1, 27.2, 20.9; FT-IR (neat) 1714, 1602, 1581, 1299, 1266, 1184, 1116, 1094, 1045 cm−1; HRMS (ESI) calcd for C24H21N4O3S2 [M − H−] 477.1050, found 477.1044.
2-(5-((Z)-4-((E)-(4-(Ethyl(2-hydroxyethyl)amino)phenyl)diazenyl)benzylidene)malononitrile (D2π1A2)
The residue was purified by silica gel column chromatography with CH2Cl2 and recrystallization from AcOEt and hexane. Red solid; 59.4 mg, 0.172 mmol, 2 steps 71% yield; 1H NMR (500 MHz, CDCl3): δ 8.02 (d, J = 8.0 Hz, 2H), 7.93 (d, J = 8.0 Hz, 2H), 7.90 (d, J = 8.6 Hz, 2H), 7.76 (s, 1H), 6.82 (d, J = 8.6 Hz, 2H), 3.90 (m, 2H), 3.64–3.56 (m, 4H), 1.26 (t, J = 6.3, 3H); 13C NMR (125 MHz, CDCl3): δ 159.0, 156.8, 151.9, 144.1, 132.2, 131.1, 126.5, 123.3, 114.2, 113.1, 111.8, 81.8, 60.5, 52.5, 46.2, 12.3; FT-IR (neat) 2924, 2847, 2219, 1595, 1577, 1556, 1520, 1297, 1266, 1207, 1104, 832 cm−1; HRMS (ESI) calcd for C20H19N5ONa [M + Na+] 368.1482, found 368.1484.2-(5-((Z)-4-((E)-(4-(Ethyl(2-hydroxyethyl)amino)phenyl)diazenyl)benzylidene)-2,4-dioxothiazolidin-3-yl)acetic acid
The residue was filtered and washed with ethanol and recrystallized from ethanol. Brown solid; 59.0 mg, 0.130 mmol, 2 steps 53% yield; 1H NMR (500 MHz, DMSO-d6): δ 7.89 (d, J = 7.6 Hz, 3H), 7.80 (d, J = 8.6 Hz, 4H), 6.85 (d, J = 8.8 Hz, 2H), 4.61 (s, 2H) 3.61 (m, 2H), 3.53–3.49 (m, 4H), 1.15 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 193.0, 167.1, 166.6, 153.4, 151.4, 142.6, 133.4, 132.6, 132.1, 125.7, 122.7, 122.3, 111.4, 58.4, 52.2, 46.3, 45.3, 12.1; FT-IR (neat) 1747, 1712, 1601, 1576, 1526, 1508, 1260, 1167, 773 cm−1; HRMS (ESI) calcd for C22H21N4O4S2 [M − H−] 469.0999, found 469.0990.(E)-2-(4-((5-Amino-1-phenyl-1H-pyrazol-4-yl)diazenyl)benzylidene)malononitrile (D3π1A2)
The residue was purified by silica gel column chromatography by CH2Cl2. Purple solid; 55.0 mg, 0.162 mmol, 2 steps 66% yield; 1H NMR (500 MHz, DMSO-d6): δ 8.50 (s, 1H), 8.10 (brs, 1H), 8.06 (d, J = 8.2 Hz, 2H), 7.94 (d, J = 8.2 Hz, 2H), 7.64 (brs, 2H), 7.63–7.54 (m, 4H), 7.46 (m, 1H); 13C NMR (125 MHz, DMSO-d6): δ 160.3, 156.7, 137.5, 132.2, 130.4, 129.7, 127.9, 126.1, 123.8, 121.8, 114.7, 113.8, 79.6 (Two signals of the pyrazole ring were not observed due to significant broadening.); FT-IR (neat) 2222, 1556, 1516, 1491, 1341, 1308, 1146, 912, 838, 761, 693, 607 cm−1; HRMS (ESI) calcd for C19H13N7Na [M + Na+] 362.1125, found 362.1125.2-(5-((Z)-4-((E)-(5-Amino-1-phenyl-1H-pyrazol-4-yl)diazenyl)benzylidene)-4-oxo-2-thioxothiazolidin-3-yl)acetic acid (D3π1A3)
To a solution of aldehyde D3π1 (1.0 equiv.) and rhodanine A3 (1.5 equiv.) in MeCN (30.0 mL per 1 mmol of aldehyde) was added piperidine (30 μL per 1 mmol of aldehyde) and stirred for 15 h. The precipitate was filtered and washed with MeCN. The residue was purified by recrystallization from ethanol. Purple solid; 59.5 mg, 0.128 mmol, 2 steps 52% yield; 1H NMR (500 MHz, DMSO-d6): δ 8.10 (brs, 1H) 7.92 (d, J = 8.6 Hz, 2H), 7.92 (s, 1H), 7.77 (d, J = 8.6 Hz, 2H), 7.63–7.56 (m, 4H), 7.52 (brs, 2H), 7.46 (m, 1H), 4.73 (s, 2H); 13C NMR (125 MHz, DMSO-d6): δ 193.1, 167.4, 166.5, 154.3, 137.6, 133.4, 132.2, 132.2, 129.6, 127.8, 125.6, 123.7, 122.1, 121.2, 45.3 (Two signals of the pyrazole ring were not observed due to significant broadening.); FT-IR (neat) 1704, 1582, 1520, 1355, 1317, 1291, 1187, 768 cm−1; HRMS (ESI) calcd for C21H17N6O3S2 [M + H+] 465.0798, found 465.0799.(E)-1-(2-Hydroxyethyl)-4-methyl-2,6-dioxo-5-((5-((E)-(2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinolin-9-yl)diazenyl)thiophen-2-yl)methylene)-1,2,5,6-tetrahydropyridine-3-carbonitrile (D1π2A1)
A mixture of aldehyde D1π2 (0.10 mmol, 31.0 mg, 1.0 equiv.) and A1 (0.15 mmol, 29.1 mg, 1.5 equiv.) in ethanol (3.0 mL) was stirred for 24 h. The precipitate was filtered and washed with ethanol. Green solid; 71.2 mg, 0.146 mmol, 3 steps 28% yield; 1H NMR (400 MHz, DMSO-d6): δ 8.15 (d, J = 4.4 Hz, 1H), 8.13 (s, 1H), 7.55 (d, J = 4.4 Hz, 1H), 7.41 (s, 2H), 3.94 (t, J = 6.4 Hz, 2H), 3.51 (t, J = 6.4 Hz, 2H), 3.45 (t, J = 6.4 Hz, 2H), 3.41 (m, 4H), 2.75 (brs, 3H), 2.56 (s, 3H), 1.91 (m, 4H); 13C NMR (100 MHz, DMSO-d6): δ 173.2, 162.4, 160.6, 160.0, 148.8, 148.6, 145.9, 142.8, 134.4, 126.1, 124.6 (broad signal), 122.3, 115.8, 115.4, 100.5, 57.5, 50.0, 41.5, 26.8, 20.5, 18.9; FT-IR (neat) 3446, 2926, 2818, 2224, 1636, 1599, 1519, 1175, 1108, 986 cm−1; HRMS (ESI) calcd for C26H25N5O3S1Na [M + Na+] 510.1570, found 510.1570.(E)-2-((5-((2,3,6,7-Tetrahydro-1H,5H-pyrido[3,2,1-ij]quinolin-9-yl)diazenyl)thiophen-2-yl)methylene)malononitrile (D1π2A2)
The residue was purified by silica gel column chromatography with CH2Cl2 and methanol (9:1). Green solid; 68.3 mg, 0.190 mmol, 3 steps 36% yield; 1H NMR (500 MHz, CDCl3): δ 7.65 (d, J = 4.4 Hz, 2H), 7.62 (s, 1H), 7.44 (s, 2H), 7.41 (d, J = 4.4 Hz, 1H), 3.37 (t, J = 5.8 Hz, 4H), 2.79 (t, J = 6.2 Hz, 4H), 1.98 (m, 4H); 13C NMR (125 MHz, CDCl3): δ 171.2, 150.8, 148.4, 142.9, 139.7, 132.6, 125.5, 125.2 (broad signal), 121.9, 115.1, 113.9, 75.0, 50.7, 27.7, 21.3; FT-IR (neat) 2214, 1602, 1558, 1520, 1226, 1177, 1119, 1037, 986, 772 cm−1; HRMS (ESI) calcd for C20H17N5SNa [M + Na+] 382.1097, found 382.1094.
(E)-2-(4-((4-(Ethyl(2–2-((Z)-4-oxo-5-((5-((E)-(2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinolin-9-yl)diazenyl)thiophen-2-yl)methylene)-2-thioxothiazolidin-3-yl)acetic acid (D1π2A3)
The residue was purified by silica gel column chromatography with CH2Cl2 and methanol (9:1) and recrystallized from ethanol. Green solid; 85.3 mg, 0.176 mmol, 3 steps 33% yield; 1H NMR (400 MHz, DMSO-d6): δ 8.08 (s, 1H), 7.85 (d, J = 4.4 Hz, 1H), 7.59 (d, J = 4.4 Hz, 1H), 7.38 (s, 2H), 4.71 (s, 2H), 3.51 (t, J = 5.4 Hz, 4H), 2.75 (t, 5.6 Hz, 4H), 1.89 (m, 4H); 13C NMR (125 MHz, DMSO-d6): δ 191.6, 167.8, 167.3, 166.0, 147.4, 141.4, 138.0, 134.8, 127.5, 126.9, 124.0 (broad signal), 121.6, 118.7, 49.8, 45.3, 27.0, 20.7; FT-IR (neat) 1714, 1602, 1581, 1299, 1266, 1184, 1116, 1094, 1045 cm−1; HRMS (ESI) calcd for C22H19N4O3S3 [M − H−] 483.0614, found 483.0614.
(E)-5-((5-((E)-(4-(Ethyl(2-hydroxyethyl)amino)phenyl)diazenyl)thiophen-2-yl)methylene)-1-(2-hydroxyethyl)-4-methyl-2,6-dioxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (D2π2A1)
A mixture of aldehyde D2π2 (1.0 equiv.) and A1 (1.5 equiv.) in ethanol (30.0 mL per 1 mmol of aldehyde) was stirred for 24 h. The precipitate was filtered and washed with ethanol. Green solid; 76.7 mg, 0.160 mmol, 3 steps 28% yield; 1H NMR (400 MHz, DMSO-d6): δ 8.25 (s, 1H), 8.23 (d, J = 4.8 Hz, 1H), 7.80 (d, J = 9.2 Hz, 2H), 7.74 (d, J = 4.8 Hz, 1H), 6.91 (d, J = 9.2 Hz, 2H), 4.89 (brs, 1H), 4.81 (brs, 1H), 3.96 (t, J = 6.4 Hz, 2H), 3.62–3.50 (m, 8H), 2.61 (s, 3H), 1.18 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 171.5, 162.6, 160.5, 160.3, 152.5, 148.2, 146.4, 143.1, 135.3, 127.6, 126.9 (broad signal), 116.8, 115.6, 112.4, 101.8, 58.5, 57.5, 52.4, 45.6, 41.6, 19.0, 12.2; FT-IR (neat) 3292, 2213, 1625, 1598, 1525, 1246, 1127, 1040, 778 cm−1; HRMS (ESI) calcd for C24H25N5O4SNa [M + Na+] 502.1519, found 502.1519.(E)-2-((5-((4-(Ethyl(2-hydroxyethyl)amino)phenyl)diazenyl)thiophen-2-yl)methylene)malononitrile (D2π2A2)
The residue was purified by silica gel column chromatography with CH2Cl2 and methanol (9:1). Green solid; 66.8 mg, 0.190 mmol, 3 steps 33% yield; 1H NMR (500 MHz, CDCl3): δ 7.85 (d, J = 9.2 Hz, 2H), 7.71 (d, J = 4.3 Hz, 1H), 7.70 (s, 1H), 7.56 (d, J = 4.3 Hz, 1H), 6.80 (d, J = 9.2 Hz, 2H), 3.91 (t, J = 5.5 Hz, 2H), 3.66–3.59 (m, 4H), 1.27 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 170.0, 152.4, 151.3, 143.6, 139.4, 133.6, 127.4 (broad signal), 127.1, 114.7, 113.5, 112.1, 60.5, 52.6, 46.4, 12.4; FT-IR (neat) 2214, 1602, 1558, 1520, 1226, 1177, 1119, 1037, 986, 772 cm−1; HRMS (ESI) calcd for C18H17N5OSNa [M + Na+] 374.1046, found 374.1046.
2-((Z)-5-((5-((E)-(4-(Ethyl(2-hydroxyethyl)amino)phenyl)diazenyl)thiophen-2-yl)methylene)-4-oxo-2-thioxothiazolidin-3-yl)acetic acid (D2π2A3)
The residue was purified by silica gel column chromatography with CH2Cl2 and methanol (9:1) and recrystallized from ethanol. Green solid; 68.6 mg, 0.144 mmol, 3 steps 25% yield; 1H NMR (500 MHz, DMSO-d6): δ 8.01 (s, 1H), 7.82 (brs, 1H), 7.74 (d, J = 8.7 Hz, 2H), 7.71 (brs, 1H), 6.86 (d, J = 8.7 Hz, 2H), 4.89 (brs, 1H), 4.40 (s, 2H), 3.70–3.45 (m, 6H), 1.15 (t, J = 6.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ 191.5, 167.1, 166.5, 166.3, 151.6, 141.9, 136.9, 136.1, 128.3, 126.2 (broad signal), 125.9, 120.9, 111.9, 58.4, 52.3, 48.0, 45.4, 12.1; FT-IR (neat) 1723, 1313, 1218, 1188, 1036, 636 cm−1; HRMS (ESI) calcd for C20H19N4O4S3 [M − H−] 475.0563, found 475.0569.
Ethyl 2-nitro-3-(4-((E)-(2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinolin-9-yl)diazenyl)phenyl)acrylate (D1π1A4)
Ethyl nitroacetate (A4) (1.0 equiv.) and aldehyde D1π1 (1.0 equiv.) and a catalytic amount of β-alanine (0.2 eq.) were refluxed in acetic acid (4.0 mL per 1 mmol of aldehyde) and benzene (30.0 mL per 1 mmol of aldehyde) with MS3 Å (1.0 g per 1 mmol of aldehyde) for 10 h. After cooling, MS3 Å was filtered out and the mixture was washed with saturated aq. NaHCO3 and extracted with CH2Cl2 3 times. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography with CH2Cl2. Purple solid; 40.4 mg, 0.0960 mmol, 48% yield (mixture of two isomers); 1H NMR (400 MHz, CDCl3) Z (major) isomer: δ 7.80 (d, J = 8.4 Hz, 2H), 7.54 (s, 1H), 7.50 (d, J = 8.4 Hz, 2H), 7.47 (s, 2H), 4.39 (q, J = 7.2 Hz, 2H), 3.32 (m, 4H), 2.82 (m, 4H), 1.99 (m, 4H), 1.37 (t, J = 7.3 Hz, 3H); E (minor) isomer: δ 8.11 (s, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 8.4 Hz, 2H), 7.48 (s, 2H), 4.47 (q, J = 7.2 Hz, 2H), 3.32 (m, 4H), 2.82 (m, 4H), 1.99 (m, 4H), 1.37 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) Z (major) isomer: δ 159.4, 155.6, 146.6, 142.9, 141.3, 132.5, 131.0, 128.5, 123.5, 122.8, 121.0, 63.0, 50.2, 27.8, 21.5, 14.1; E (minor) isomer: δ 161.5, 155.8, 146.8, 142.9, 139.4, 136.3, 131.7, 128.5, 123.6, 122.7, 121.0, 63.2, 50.2, 27.8, 21.4, 13.8; FT-IR (neat) 2931, 2847, 2359, 1601, 1307, 1272, 1122, 771 cm−1; HRMS (ESI) calcd for C23H24N4O4Na [M + Na+] 443.1690, found 443.1692.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr Yoshihisa Sei (Technical Department, Tokyo Institute of Technology) for his technical assistance with NMR measurements. The numerical calculations were carried out on the TSUBAME3.0 supercomputer at the Tokyo Institute of Technology, Tokyo, Japan, and on the supercomputer at the Research Center for Computational Science, Okazaki, Japan. This computational work was supported by a Grant-in-Aid for Young Scientists (B) (JSPS KAKENHI Grant Number JP17K17720 to Y. H.), a Grant-in-Aid for Specially Promoted Research (JSPS KAKENHI Grant Number JP17H06092 to S. K.), and a JST CREST (Grant Number JPMJCR1522 to S. K.).Notes and references
- Q. Ban, T. Bai, X. Duan and J. Kong, Biomater. Sci., 2017, 5, 190–210 RSC.
- J.-J. Hu, Y.-J. Cheng and X.-Z. Zhang, Nanoscale, 2018, 10, 22657–22672 RSC.
- H. S. Jung, P. Verwilst, A. Sharma, J. Shin, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2018, 47, 2280–2297 RSC.
- L. Zou, H. Wang, B. He, L. Zeng, T. Tan, H. Cao, X. He, Z. Zhang, S. Guo and Y. Li, Theranostics, 2016, 6, 762–772 CrossRef CAS PubMed.
- Y. Cai, Z. Wei, C. Song, C. Tang, W. Han and X. Dong, Chem. Soc. Rev., 2019, 48, 22–37 RSC.
- K. Huang, Y. Zhang, J. Lin and P. Huang, Biomater. Sci., 2019, 7, 472–479 RSC.
- Y. Jiang and K. Pu, Adv. Biosyst., 2018, 2, 1700262 CrossRef.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC.
- X. Y. Zhang, X. Nan, W. Shi, Y. N. Sun, H. L. Su, Y. He, X. Liu, Z. Zhang and D. T. Ge, Nanotechnology, 2017, 28, 12 Search PubMed.
- J. J. Shi, L. Wang, J. Zhang, R. Ma, J. Gao, Y. Liu, C. F. Zhang and Z. Z. Zhang, Biomaterials, 2014, 35, 5847–5861 CrossRef CAS PubMed.
- H. Liu, K. Wang, C. Yang, S. Huang and M. Wang, Colloids Surf., B, 2017, 157, 398–406 CrossRef CAS PubMed.
- D. Pemmaraju, T. Appidi, G. Minhas, S. P. Singh, N. Khan, M. Pal, R. Srivastava and A. K. Rengan, Int. J. Biol. Macromol., 2018, 110, 383–391 CrossRef CAS PubMed.
- S. J. Frost, D. Mawad, J. Hook and A. Lauto, Adv. Healthcare Mater., 2016, 5, 401–414 CrossRef CAS PubMed.
- J. Qiu and W. D. Wei, J. Phys. Chem. C, 2014, 118, 20735–20749 CrossRef CAS.
- Y. Cao, Z. Wang, S. Liao, J. Wang and Y. Wang, Chem. – Eur. J., 2016, 22, 1152–1158 CrossRef CAS PubMed.
- T. P. Dasari Shareena, D. McShan, A. K. Dasmahapatra and P. B. Tchounwou, Nano-Micro Lett., 2018, 10, 53 CrossRef PubMed.
- S. Sharifi, S. Behzadi, S. Laurent, M. Laird Forrest, P. Stroeve and M. Mahmoudi, Chem. Soc. Rev., 2012, 41, 2323–2343 RSC.
- H.-J. Yoon, H.-S. Lee, J.-Y. Lim and J.-H. Park, ACS Appl. Mater. Interfaces, 2017, 9, 5683–5691 CrossRef CAS PubMed.
- S. Fuse, Y. Asai, S. Sugiyama, K. Matsumura, M. M. Maitani, Y. Wada, Y. Ogomi, S. Hayase, T. Kaiho and T. Takahashi, Tetrahedron, 2014, 70, 8690–8695 CrossRef CAS.
- S. Fuse, K. Matsumura, Y. Fujita, H. Sugimoto and T. Takahashi, Eur. J. Med. Chem., 2014, 85, 228–234 CrossRef CAS PubMed.
- S. Fuse, K. Matsumura, A. Wakamiya, H. Masui, H. Tanaka, S. Yoshikawa and T. Takahashi, ACS Comb. Sci., 2014, 16, 494–499 CrossRef CAS PubMed.
- S. Fuse, S. Sugiyama, M. M. Maitani, Y. Wada, Y. Ogomi, S. Hayase, R. Katoh, T. Kaiho and T. Takahashi, Chem. – Eur. J., 2014, 20, 10685–10694 CrossRef CAS PubMed.
- S. Fuse, R. Takahashi, M. M. Maitani, Y. Wada, T. Kaiho, H. Tanaka and T. Takahashi, Eur. J. Org. Chem., 2016, 508–517 CrossRef CAS.
- S. Fuse, H. Yoshida and T. Takahashi, Tetrahedron Lett., 2012, 53, 3288–3291 CrossRef CAS.
- S. Irie, S. Fuse, M. M. Maitani, Y. Wada, Y. Ogomi, S. Hayase, T. Kaiho, H. Masui, H. Tanaka and T. Takahashi, Chem. – Eur. J., 2016, 22, 2507–2514 CrossRef CAS PubMed.
- K. Matsumura, S. Yoshizaki, M. M. Maitani, Y. Wada, Y. Ogomi, S. Hayase, T. Kaiho, S. Fuse, H. Tanaka and T. Takahashi, Chem. – Eur. J., 2015, 21, 9742–9747 CrossRef CAS PubMed.
- S. Fuse, M. Takizawa, K. Matsumura, S. Sato, S. Okazaki and H. Nakamura, Eur. J. Org. Chem., 2017, 5170–5177 CrossRef CAS.
- S. Fuse, K. Matsumura, M. Takizawa, S. Sato and H. Nakamura, Bioorg. Med. Chem. Lett., 2018, 28, 3099–3104 CrossRef CAS PubMed.
- S. Fuse, M. Takizawa, S. Sato, S. Okazaki and H. Nakamura, Bioorg. Med. Chem., 2019, 27, 315–321 CrossRef CAS PubMed.
- S. Salmani, M. H. Majles Ara, M. S. Zakerhamidi and E. Safari, Dyes Pigm., 2016, 125, 132–135 CrossRef CAS.
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 RSC.
- C. R. Crecca and A. E. Roitberg, J. Phys. Chem. A, 2006, 110, 8188–8203 CrossRef CAS PubMed.
- A. O. Doroshenko, Theor. Exp. Chem., 2002, 38, 135–155 CrossRef CAS.
- X. Peng, F. Song, E. Lu, Y. Wang, W. Zhou, J. Fan and Y. Gao, J. Am. Chem. Soc., 2005, 127, 4170–4171 CrossRef CAS PubMed.
- F. Vollmer, W. Rettig and E. Birckner, J. Fluoresc., 1994, 4, 65–69 CrossRef CAS PubMed.
- J. Zhou, X. Li and M. Wang, Mater. Lett., 2016, 172, 15–18 CrossRef CAS.
- L.-L. Lai, C.-H. Ho, Y.-J. Lin, E. Wang, Y.-H. Liu, Y. Wang, Y.-C. Lin and K.-L. Cheng, Helv. Chim. Acta, 2002, 85, 108–114 CrossRef CAS.
- R. I. Baichurin, L. V. Baichurina, N. I. Aboskalova and V. M. Berestovitskaya, Russ. J. Gen. Chem., 2013, 83, 1764–1770 CrossRef CAS.
- V. V. Negrebetskii, A. I. Bokanov and V. I. Stepanov, Zh. Org. Khim., 1980, 16, 1708–1712 CAS.
- P. Savarino, G. Viscardi, E. Barni, R. Carpignano and L. A. Fedorov, Dyes Pigm., 1990, 13, 71–80 CrossRef CAS.
- We measured the fluorescence of several azo DπA dyes; however, the fluorescence quantum yield could not be determined because the fluorescence intensity was too weak.
- D. K. Roper, W. Ahn and M. Hoepfner, J. Phys. Chem. C, 2007, 111, 3636–3641 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob02066g |
| This journal is © The Royal Society of Chemistry 2020 |