
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast increase of nanofluidic slip in supercooled water: the key role of dynamics†
Cecilia
Herrero
 a,
Gabriele
Tocci
b,
Samy
Merabia
a and
Laurent
Joly
*ac
a,
Gabriele
Tocci
b,
Samy
Merabia
a and
Laurent
Joly
*ac
aUniv Lyon, Univ Claude Bernard Lyon 1, CNRS, Institut Lumière Matière, F-69622, Villeurbanne, France. E-mail: laurent.joly@univ-lyon1.fr
bDepartment of Chemistry, Universität Zürich, 8057 Zürich, Switzerland
cInstitut Universitaire de France (IUF), 1 rue Descartes, 75005 Paris, France
First published on 29th September 2020
Abstract
Nanofluidics is an emerging field offering innovative solutions for energy harvesting and desalination. The efficiency of these applications depends strongly on liquid–solid slip, arising from a favorable ratio between viscosity and interfacial friction. Using molecular dynamics simulations, we show that wall slip increases strongly when water is cooled below its melting point. For water on graphene, the slip length is multiplied by up to a factor of five and reaches 230 nm at the lowest simulated temperature, T ∼ 225 K; experiments in nanopores can reach much lower temperatures and could reveal even more drastic changes. The predicted fast increase in water slip can also be detected at supercoolings reached experimentally in bulk water, as well as in droplets flowing on anti-icing surfaces. We explain the anomalous slip behavior in the supercooled regime by a decoupling between viscosity and bulk density relaxation dynamics, and we rationalize the wall-type dependence of the enhancement in terms of interfacial density relaxation dynamics. While providing fundamental insights on the molecular mechanisms of hydrodynamic transport in both interfacial and bulk water in the supercooled regime, this study is relevant to the design of anti-icing surfaces, could help explain the subtle phase and dynamical behaviors of supercooled confined water, and paves the way to explore new behaviors in supercooled nanofluidic systems.
1 Introduction
Nanofluidics, i.e. the study of fluidic transport at nanometer scales, has emerged as a new and interesting field in the past few decades due to novel behaviors associated to this length scale1–3 – e.g. dielectric anomalies of confined water4 or intriguing ionic transport,5–7 with promising applications related to new 2D materials such as the development of sustainable energies.8–10 As the channel size decreases, interfacial properties have an increasingly important role. An interfacial characteristic of special concern at the nanoscale is the existence of a velocity jump Δv (‘slippage’) at the liquid–solid interface.11–13 The simplest approach to describe slip, initially proposed by Navier,14 is to consider that the viscous shear stress τ in the liquid at the wall is proportional to the velocity jump, τ = λΔv, where λ is the liquid–solid friction coefficient.Because nanofluidic slip is key to improving the performance of nanofluidic systems,15–21 an intensive experimental effort has been undertaken during the recent years to characterize the ultra-low liquid–solid friction of new 2D materials and their derivative.22–25 On the modeling side, several efforts have been pursued in order to understand the molecular mechanisms that control friction, with special interest on the discussion of the relation between the friction coefficient and the time autocorrelation of the force exerted by the liquid on the wall.26–33 Further work has been performed to study the impact on friction of different wall features such as wettability,34,35 roughness,36 crystallographic orientation,37 electronic structure,38–40 or electrostatic interactions.41 Yet a large number of questions with regard to the interface properties, such as its viscoelastic or purely viscous nature42–44 or the possible link with its interfacial thermal transport equivalents via wall's wetting properties,45–47 remain open nowadays, limiting the perspectives for a rational search of optimal interfaces.
Among all fluids, the study of water has always been of special concern for scientists from a broad variety of research fields.48–50 Its interest not only lies on its ubiquitous nature but also on its many thermodynamic and dynamic anomalies like, among others, the non-monotonous temperature dependence of its isothermal compressibility and density.51,52 These anomalies are enhanced when water is driven to its supercooled regime (i.e. the range of temperatures below the freezing point where water keeps its liquid state), making this regime ideal to test and refine our current understanding of water. In particular, the temperature dependence of the bulk transport properties of supercooled water has been explored both numerically and experimentally over the last decade,53,54 considering especially the connection between viscosity and structural relaxation times.55–59
Confined water has also been explored from an experimental and theoretical point of view, with an special interest in the novel 2D materials such as graphene.60–67
In particular, the temperature evolution of supercooled water under confinement has been the subject of intensive experimental research.56,57,68–71 Broadband dielectric spectroscopy, nuclear magnetic resonance, as well as neutron scattering experiments have successfully probed water confined in pores with sub-nm radii at temperatures as low as about 130 K, in order to connect the dynamical behavior of supercooled confined water to that of bulk water in the so-called no-man's land (150 K to 230 K).70 At temperatures above the no-man's land, marked differences have been found in the time relaxation of supercooled water under confinement compared to bulk water, suggesting that the interfacial water dynamics, and thus water friction, may play an important role. However, the temperature evolution of water friction in the liquid and supercooled regime remains unclear nowadays. Besides achieving a better understanding of interfacial and nanoconfined water dynamics and phase behavior under supercooling, such a knowledge would be instrumental e.g. for the development of innovative nanofluidic systems working in the supercooled regime, and would provide fundamental insight on recent experimental work on anti-icing surfaces.72–74
In that context, we report a study in which the temperature dependence of water viscosity and wall slip are examined in detail, in connection with its bulk and interfacial dynamics in the supercooled regime. To this end we perform extensive molecular dynamics (MD) simulations of a slab of water confined between graphene and generic Lennard-Jones surfaces. In order to assess the role of supercooling, we compare water, which is in its supercooled state for the lower temperatures, and methanol, which remains liquid for the whole range of temperatures considered in our study. We find that whilst the liquid–solid friction coefficient and the viscosity follow the same fundamental laws and are almost proportional to each other in the liquid state, their behavior strikingly differ for water in the supercooled regime As a result, the slip length – defined as the ratio between the viscosity and the friction coefficient – increases fast for water as soon as it goes below its melting point; on graphene, we report a twofold enhancement at ∼240 K, and up to a fivefold enhancement at 225 K, reaching ∼230 nm. Although the presence of impurities may enhance ice nucleation in supercooled water, a number of experimental works have consistently shown that it is possible to cool down water to the range of temperatures explored in this study and even below.53,56,57,68–71,73 Our analysis reveals that the dynamics of interfacial water, specifically the time relaxation of the interfacial density fluctuations, is the most important factor governing the temperature behavior of liquid–solid friction and slip. This fundamental mechanistic insight sheds new light on the general molecular mechanisms underlying water slip.
2 MD simulations
All the simulations were carried out with the LAMMPS package.75 The confined system consisted in a fluid – TIP4P/2005 water76 or methanol (MeOH)77,78 – between two parallel walls – graphene, or a generic hydrophobic wall made of Lennard-Jones (LJ) particles – with periodic boundary conditions applied in the directions parallel to the walls (Fig. 1), see details in the ESI.† The surfaces were characterized by contact angles, at 300 K, of θ ∼ 134° for water-LJ walls, θ ∼ 80° for water–graphene, θ ∼ 100° for MeOH-LJ walls and θ ∼ 0° for MeOH–graphene; these contact angles were obtained through sessile nanodroplet simulations, following a procedure described in ref. 79.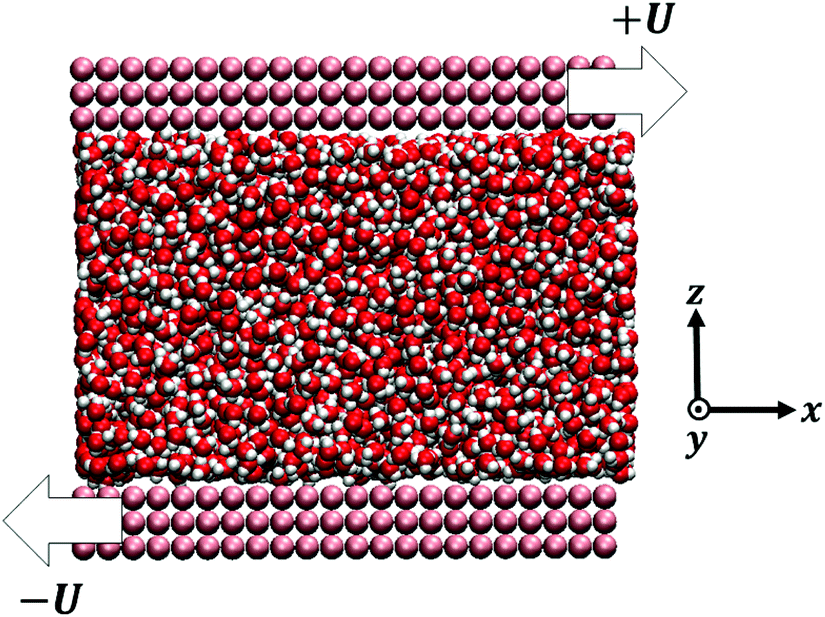 | ||
| Fig. 1 Modelled system constituted by a confined fluid between two planar solid walls. The snapshot corresponds to TIP4P/2005 water enclosed by LJ walls. The arrows indicate the shear velocity U directions by which the system is driven out of equilibrium for the shear flow measurements. | ||
The wall dimensions were Lx = Ly = 58.92 Å for the LJ wall, and Lx = 56.57 Å, Ly = 58.92 Å for graphene. The pressure was set to 1 atm by using the top wall as a piston during a preliminary run and measuring its average equilibrium position; the top wall was then fixed at such equilibrium position during the production run; the corresponding height was H ∼ 40 Å for water and H ∼ 90 Å for MeOH, allowing for a large region of bulk liquid between the walls for both liquids. Equivalent results were obtained when letting the top wall fluctuate around its equilibrium position during the production run, see the ESI.† The temperature T was varied between 225 and 360 K, by applying a Nosé–Hoover thermostat to the liquid (only along the directions perpendicular to the flow for non-equilibrium simulations). Equivalent results were obtained for different damping times, and with a Berendsen thermostat.
To measure the hydrodynamic transport coefficients we performed non-equilibrium molecular dynamics (NEMD) simulations, applying a constant shear velocity U to the walls in opposite x directions for each wall (see Fig. 1), producing a linear velocity profile far from the wall. The friction coefficient was measured from the ratio between the shear stress τ and the velocity jump at the interface Δv – defined at the effective wall position zs:80λ = τ/Δv, and the viscosity was measured from the ratio between the shear stress and the bulk shear rate, η = τ/(∂zvx), see the ESI† for details.
Both interfacial and bulk equations can be combined in the so-called partial slip boundary condition,13
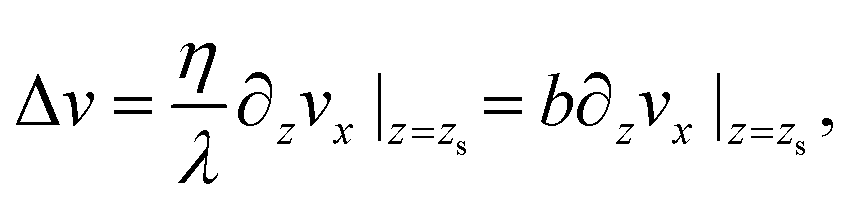 | (1) |
3 Results and discussion
We first computed the shear viscosity η from NEMD with LJ walls to test the applicability of different temperature dependence laws. For water in particular, the temperature dependence of the viscosity deviates from Arrhenius behavior.51 We tested three common alternative laws, the Vogel–Tammann–Fulcher (VTF) law,81–83 the Speedy–Angell (SA) law,84 and the Bässler (B) law,85 respectively: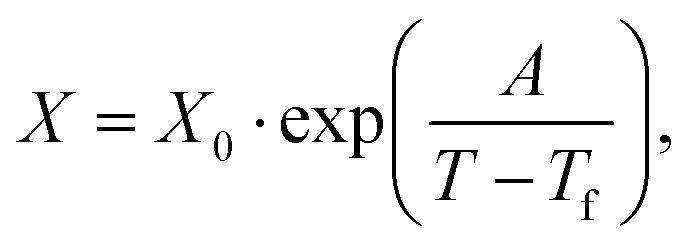 | (2a) |
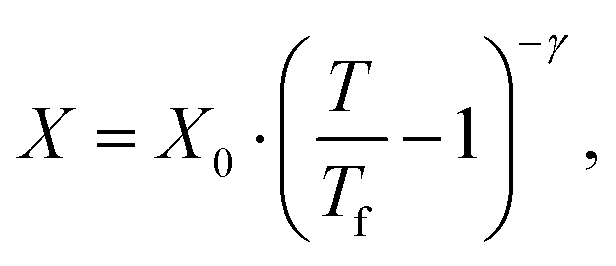 | (2b) |
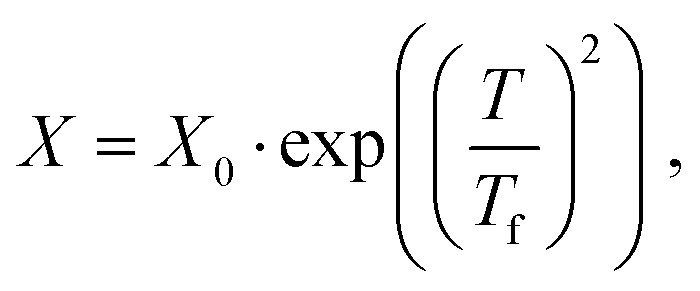 | (2c) |
We then proceeded to explore temperature effects on friction. For each fluid, when varying the wall type, we already saw a difference at a given temperature in the absolute value of λ, being more than one order of magnitude smaller for graphene than for LJ walls (see the ESI†). This effect has already been measured and discussed in previous work38,60,89,90 and it is due to the extreme smoothness of graphene. Additionally, in Fig. 2 one can see that the temperature dependence changes with the fluid, but for a given fluid, depends weakly on the wall type. Interestingly, the temperature dependence of η and λ can be fitted by the same laws (VTF for TIP4P/2005 and Arrhenius for MeOH, corresponding to continuous lines in Fig. 2), although with different parameters.
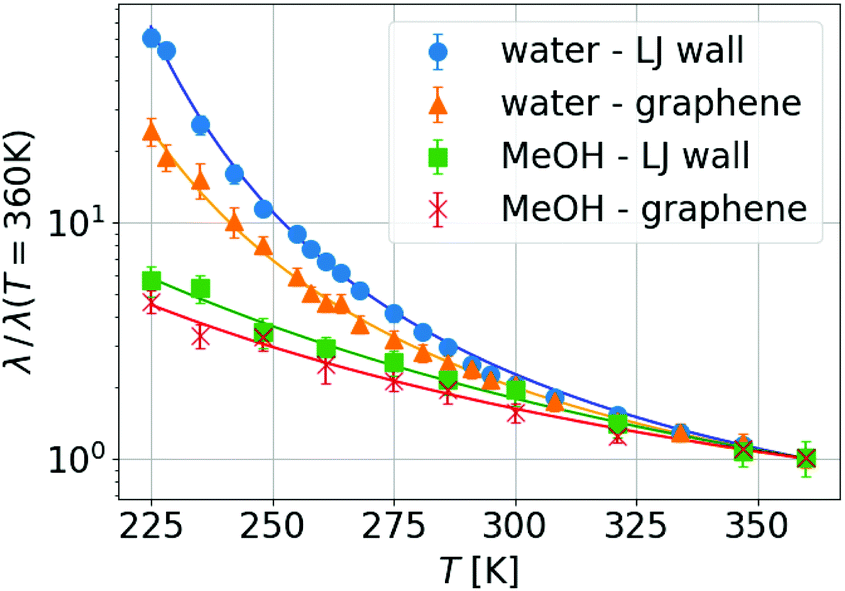 | ||
| Fig. 2 Friction's temperature dependence results normalized by the value at 360 K for each fluid and wall, in order to highlight the similar temperature evolution for a given liquid regardless of the wall type. Blue dots correspond to water with LJ walls, orange triangles to water with graphene walls, green squares to MeOH with LJ walls and red crosses to MeOH with graphene walls. Continuous lines are the respective VTF (for water) and Arrhenius (for MeOH) fits. | ||
We can go further in exploring the relation between η and λ by plotting the slip length b given by the ratio between both transport coefficients, see eqn (1). In Fig. 3 one can see that for a wide range of high temperatures, where the systems are in the stable liquid state, η and λ vary together with T, so their ratio (or equivalently the slip length) is roughly constant. Specifically, for MeOH (which remains a simple liquid for the whole range of simulated temperatures, including those slightly higher than its boiling point at around 338 K), b increases slowly and regularly when T decreases; this indicates a slightly weaker temperature dependence of friction as compared to viscosity. In contrast, for water, b starts to increase very fast when the temperature decreases below the melting point, indicating a much weaker temperature dependence of friction as compared to viscosity, only in the supercooled regime. When comparing these results with those for liquid MeOH one can conclude that this large slip increase is mostly related to the supercooling of water.
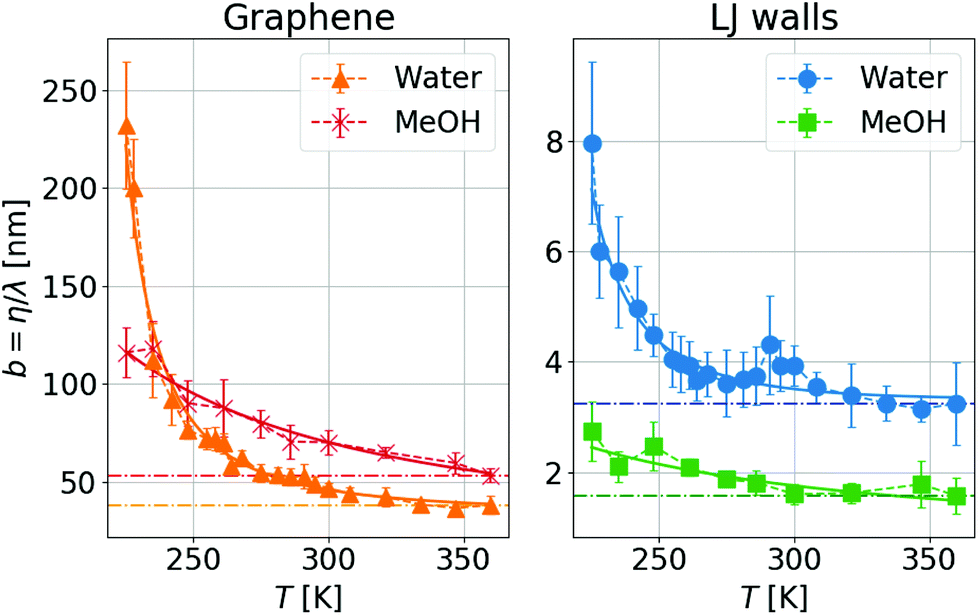 | ||
| Fig. 3 Temperature dependence of the slip length, b = η/λ, with the same symbols as Fig. 2. Dash-dotted lines are guide-to-the-eye for a constant b value. One can see a small temperature variation for the highest temperatures (indicating that η and λ evolve in similar ways), while the slip length increases significantly when decreasing the temperature for the lowest Ts, in the supercooled regime. | ||
The biggest temperature effect on b is observed for water and graphene walls, where it grows by a factor of 5 from the highest to the lowest simulated temperature (225 K), reaching a maximum value of ∼230 nm. Although experiments of interfacial slip in supercooled water have not yet appeared, we envision that experimental verification of our results is within reach of capillary flow measurements of water confined between graphene/silica nanochannels,25 considering that nuclear magnetic resonance and neutron scattering experiments of water confined in graphite oxide and silica nanopores have explored water dynamics down to 130 K and 220 K, respectively.56,68,69 Additionally, recent microscopy studies have investigated the dynamical behavior of supercooled water down to 230 K using polystyrene spheres suspended in water53 and have studied the anti-icing behavior of water droplets sliding on of nanopatterned surfaces around 258 K.73 As we have seen in our simulations, at temperatures between 225 and 270 K, water slippage is largely affected by the wall type, a result consistent with the experimental observation that the water relaxation time in nanopores is strongly dependent on the nature of the solid surface.68,91 Instead, experiments in no-man's land report a universal dynamical behavior in confined water.70 Thus, future measurements of water slippage in different nanopores at lower temperatures than those explored here might elucidate whether or not slippage depends on the wall type below 225 K.
Two main questions remain then to be understood. First, what is the main physical parameter that controls the temperature evolution of the friction coefficient observed in Fig. 2? Second, why bulk and interface have a similar temperature dependence at high temperatures and why they don't at the lower ones (Fig. 3)? From now on we will focus on water to address these questions and explore in particular the effect of supercooling.
In order to better understand the molecular mechanisms that control friction temperature dependence, λ can be related to the autocorrelation of the equilibrium force at the interface through a Green–Kubo formula:26,92
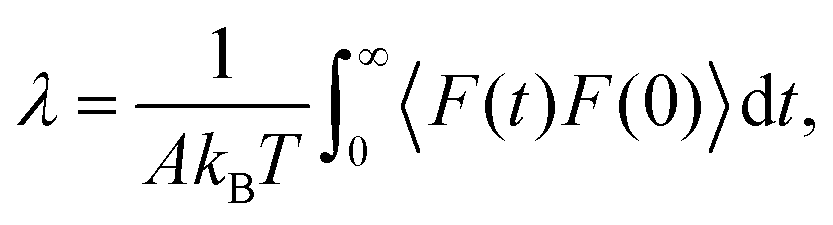 | (3) |
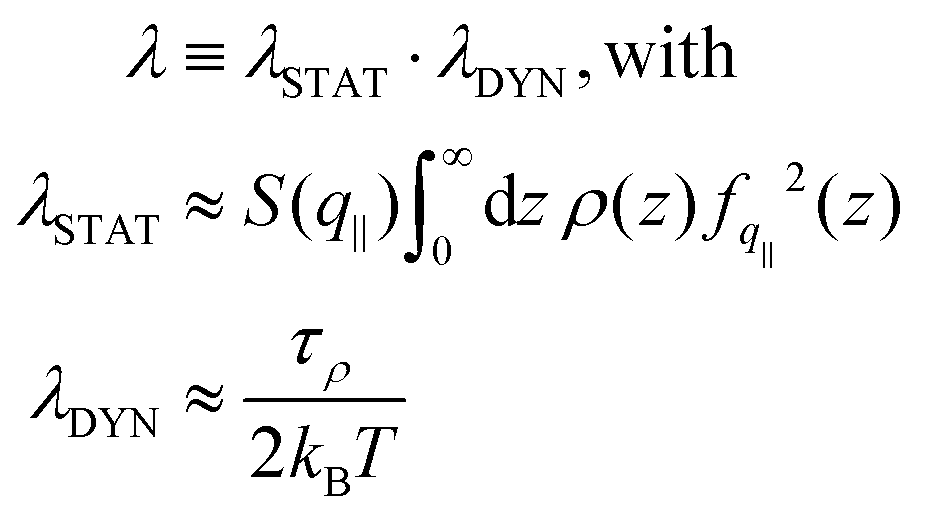 | (4) |
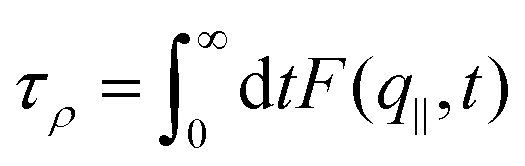 . The contact layer was defined as the liquid region between the wall and the first non-zero minimum of the liquid's density profile. Note that we included the 1/(kBT) term of the Green–Kubo integral in the dynamical part; we will come back to that choice later. Regarding the static terms in eqn (4), we found that S(q‖) remained almost constant with temperature for both graphene and LJ walls (see the ESI†). Accordingly, the main static contribution to friction T dependence comes from the integral in eqn (4). We used for fq‖(z) the analytical expression derived in ref. 93 for LJ walls and the measurements in ref. 60 for graphene (as detailed in the ESI†). For both surfaces, the integral remains constant at low temperatures, and then increases by at most a factor of 2 at higher temperatures. This tendency can be explained by the spreading of the atoms in the contact layer toward the wall due to larger thermal fluctuations; indeed, fq‖(z) decreases very fast with z, so that the integral of the static part is dominated by a small fraction of the atoms in the contact layer that are the closest to the wall, see the ESI.†
. The contact layer was defined as the liquid region between the wall and the first non-zero minimum of the liquid's density profile. Note that we included the 1/(kBT) term of the Green–Kubo integral in the dynamical part; we will come back to that choice later. Regarding the static terms in eqn (4), we found that S(q‖) remained almost constant with temperature for both graphene and LJ walls (see the ESI†). Accordingly, the main static contribution to friction T dependence comes from the integral in eqn (4). We used for fq‖(z) the analytical expression derived in ref. 93 for LJ walls and the measurements in ref. 60 for graphene (as detailed in the ESI†). For both surfaces, the integral remains constant at low temperatures, and then increases by at most a factor of 2 at higher temperatures. This tendency can be explained by the spreading of the atoms in the contact layer toward the wall due to larger thermal fluctuations; indeed, fq‖(z) decreases very fast with z, so that the integral of the static part is dominated by a small fraction of the atoms in the contact layer that are the closest to the wall, see the ESI.†
Overall, the temperature behavior of λSTAT is too weak to explain the exponential decrease of friction for increasing temperature. It is only left to check the dynamical contribution from eqn (4), enclosed in τρ. To measure this parameter we fitted the intermediate scattering function following ref. 94:
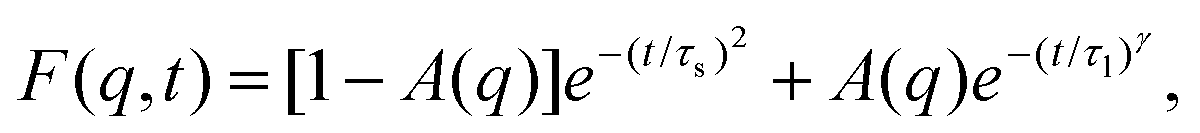 | (5) |
To then understand the temperature dependence of the slip length b = η/λ, we will decompose the viscosity into a static and a dynamical part in the same manner as for the friction coefficient: η = ηSTAT × ηDYN, with ηDTN = τbulkρ/(2kBT) – in analogy with the definition of λDYN, and with ηSTAT = η/ηDYN. The slip length can then be decomposed as follows:
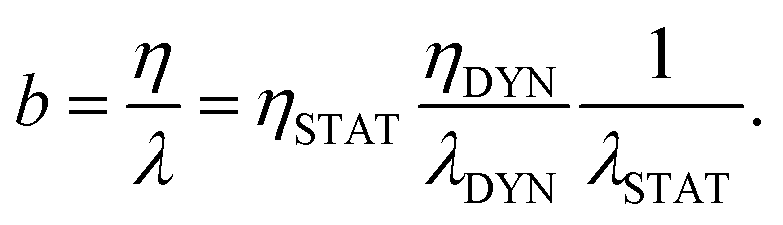 | (6) |
Fig. 4 illustrates the temperature evolution of the three contributions to λ for water on LJ walls and graphene. In this figure, the lines are obtained from the ratios between VTF fits of the simulation results for η, λ, τρ and τBρ: specifically, ηSTAT ∝ Tη/τBρ, ηDYN/λDYN = τBρ/τρ, and λSTAT ∝ Tλ/τρ. One can observe in Fig. 4 that 1/λSTAT increases when T decreases for both interfaces. The temperature variation of 1/λSTAT is slightly larger for the LJ walls (a factor of ∼2) than for graphene (a factor of ∼1.5). As mentioned above, 1/λSTAT is controlled by S(q‖), which remains almost constant with temperature, and by the integral 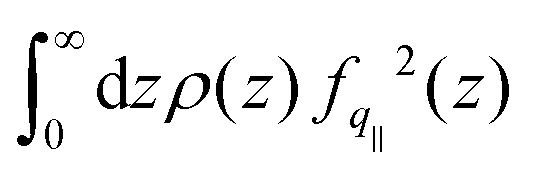 , which increases when the atoms of the contact layer spread toward the wall under larger thermal fluctuations. Therefore, the stronger temperature variation of λSTAT for the LJ walls can be related to the larger extension of the density profiles toward the wall at high temperatures, see the ESI† for detail. In bulk, ηSTAT remains constant at high T, but it increases significantly when water enters its supercooled regime, for T < 273 K, providing a large contribution – independent of the wall type – to the significant increase of b in the same T region. As a side note, following our choice to include 1/(kBT) in ηDYN, the fact that ηSTAT is constant in the liquid state corresponds to η ∝ τBρ/T; we suggest this correlation could replace more traditional ones used when studying supercooled liquids, η ∝ τα or η ∝ Tτα.55,57–59,95,96
, which increases when the atoms of the contact layer spread toward the wall under larger thermal fluctuations. Therefore, the stronger temperature variation of λSTAT for the LJ walls can be related to the larger extension of the density profiles toward the wall at high temperatures, see the ESI† for detail. In bulk, ηSTAT remains constant at high T, but it increases significantly when water enters its supercooled regime, for T < 273 K, providing a large contribution – independent of the wall type – to the significant increase of b in the same T region. As a side note, following our choice to include 1/(kBT) in ηDYN, the fact that ηSTAT is constant in the liquid state corresponds to η ∝ τBρ/T; we suggest this correlation could replace more traditional ones used when studying supercooled liquids, η ∝ τα or η ∝ Tτα.55,57–59,95,96
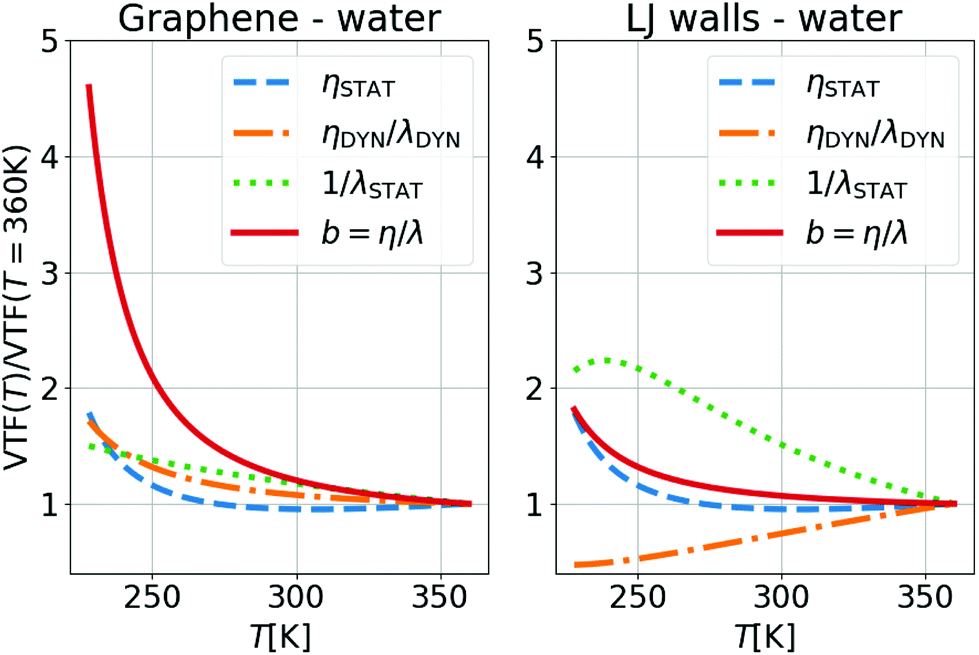 | ||
| Fig. 4 Temperature evolution of the static and dynamical contributions to the slip length b = η/λ of water on graphene and LJ walls, normalized by the values at 360 K. The lines were obtained from VTF fits of the simulation results, see text for details. | ||
Finally, to understand the relative increase of b by ∼2 times for the LJ wall and by ∼5 times for graphene, we looked at the dynamic ratio ηDYN/λDYN. In Fig. 4(right), one can see that for LJ walls the interface relaxation time increases more (i.e. slower dynamics) when decreasing T than the bulk one, compensating the static contribution and resulting in a smaller b variation. In contrast, for graphene, due to the surface smoothness, there is no contribution from the wall to the slowing down of the interface dynamics with T when compared to the bulk dynamics. Therefore, as for the temperature dependence of λ, we conclude that also with regard to b it is not the different interfacial structures which contribute to its T evolution but the different dynamics.
Before concluding, we would like to comment on a prediction for the temperature dependence of b introduced by Bocquet and Barrat,13 who wrote that b should be proportional to (kBT)2/λSTAT, in contrast with our results. This formula can be derived from eqn (4) by relating the density relaxation time τρ to the collective diffusion coefficient Dq‖: τρ = 1/(q‖2Dq‖), and by identifying Dq‖ with the self-diffusion coefficient D0, itself related to the viscosity through the Stokes–Einstein relation: D0 ∝ kBT/η. However, while we found that indeed Dq‖ ≃ D0 at room temperature, their temperature evolution is quite different, especially in the supercooled regime (see the ESI†). Indeed, both diffusion coefficients arise from processes that happen at different scales and their relation is non-trivial: while Dq‖ is related to collective diffusion in the sense that it comes from the density Fourier transform integration to all atoms positions, D0 is referred to the diffusion of one molecule of σl size.
4 Conclusions
In this work we investigated the temperature evolution of bulk and interfacial hydrodynamic transport coefficients for water and MeOH confined between LJ walls and graphene. For a given liquid, the temperature evolution of viscosity and friction were described by the same laws, although with different parameters. The temperature evolution of interfacial friction was weakly affected by the wall type, but changed significantly with the liquid type. We then compared the temperature evolution of viscosity η and friction coefficient λ by considering their ratio, defined as the slip length b = η/λ. We observed, from higher to lower T, that both transport coefficients evolved similarly in the high temperature region where the liquid is stable, but that for water, viscosity increased faster than friction in the supercooled regime, implying a fast growing slip length. The largest temperature effect on b was observed for water and graphene walls, where it grew by a factor of 5 from the highest to the lowest simulated temperature (225 K), reaching a maximum value of ∼230 nm.In order to understand the molecular mechanisms that control friction, we decomposed the friction coefficient λ into the product of a static contribution λSTAT and a dynamical one λDYN, in the form of an interface density relaxation time τρ. We observed a small variation of the static part with T, but the main contribution to the temperature dependence of friction came from the dynamical term. Finally, in order to explain the temperature dependence of the slip length b = η/λ, we also decomposed the viscosity η into a static term ηSTAT and a dynamical term ηDYN, controlled by the bulk density relaxation timeτBρ. The slip length could then be decomposed into three contributions: first, the interfacial static contribution 1/λSTAT; second, the bulk static contribution, ηSTAT; and third, the relation between the bulk and interfacial dynamical terms ηDYN/λDYN = τBρ/τρ. We observed that the viscosity static part, while it remained constant at high temperature, increased significantly in the supercooled regime, representing a major contribution – independent of the wall type – to the slip length temperature evolution. We could finally relate the different slip length temperature dependence on LJ walls and graphene to the difference in interfacial dynamics on these two surfaces.
We suggest that the promising predictions presented here could help explain the subtle phase and dynamical behaviors of supercooled confined water,56,68–70,73 and are within reach of experimental verification, with the recent accurate characterization of liquid–solid slip on new 2D materials and their derivative,22–25 and investigation of supercooled water dynamics down to very small temperatures, e.g. ∼230 K in bulk53 and ∼130 K in confinement.98 Moreover, beyond liquid–solid slip, many other new behaviors could arise in the promising field of supercooled nanofluidics. In particular, due to the high slip values obtained at room temperature for CNT,22 comparable to the ones of the present study for graphene at the lower temperatures, we find as an interesting perspective the study of the curvature effect in combination to the supercooling of the liquid. Additionally, nanopatterned superhydrophobic surfaces have shown increased slip compared to flat surfaces.99 Exploring the effect of supercooling on slip at superhydrophobic surfaces therefore is another relevant venue.
Not only superhydrophobic surfaces are associated with large slippage but also with iceophobicity.74 Thus, examining the coupling between ice nucleation and slippage at such interfaces may be a further direction to embark on, which could be relevant for the development of anti-freezing coatings. Indeed, one of the proposed mechanisms for hindering ice formation of water droplets on superhydrophobic surfaces is to ensure a minimal contact time between the bouncing droplet and the surface. It will be interesting to see whether the increase in water viscosity at low temperature would promote ice nucleation due to increased contact time of a bouncing droplet,100 or instead whether the increase in water slip – as observed here – would actually decrease the contact time,101 thus hindering ice nucleation. An additional mechanism at play on anti-icing coatings is that of retardation of ice nucleation due to the presence of air pockets between the nanoscale patterns which act as insulating layers,74 while the main reason for the increase in slip length observed in nanopatterned surfaces at room temperature is due to the absence of friction at the vapor–liquid interface.99 Therefore, it would be interesting to understand whether the friction reduction at nanopatterned surfaces also slows ice nucleation at such interfaces. Further, in this work we found that the interfacial water dynamics is key to the increase in water slip under supercooled conditions. Because the microscopic dynamics of bulk water has been reported to influence homogeneous ice nucleation,102 it is possible that the interfacial dynamics may instead have an impact on heterogeneous ice nucleation.
Overall we hope the findings obtained here by investigating water friction as a function of temperature down to the supercooled regime will help understanding generally the molecular mechanisms underlying both interfacial and bulk hydrodynamic transport in this fascinating liquid and motivate experimentalists to find protocols to measure water slippage under supercooling.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Li Fu, Simon Gelin, Yasutaka Yamaguchi, Takeshi Omori, Emmanuel Guillaud, Bruno Issenmann for fruitful discussions. We are also grateful for HPC resources from GENCI/TGCC (grants A0050810637 and A0070810637), and from the PSMN mesocenter in Lyon. This work is supported by the ANR, Project ANR-16-CE06-0004-01 NECtAR. LJ is supported by the Institut Universitaire de France. GT is supported by the Swiss National Science Foundation through the project PZ00P2_179964.References
- K. Koga, G. T. Gao, H. Tanaka and X. C. Zeng, Nature, 2001, 412, 802–805 CrossRef CAS.
- R. B. Schoch, J. Han and P. Renaud, Rev. Mod. Phys., 2008, 80, 839 CrossRef CAS.
- L. Bocquet and E. Charlaix, Chem. Soc. Rev., 2010, 39, 1073–1095 RSC.
- L. Fumagalli, A. Esfandiar, R. Fabregas, S. Hu, P. Ares, A. Janardanan, Q. Yang, B. Radha, T. Taniguchi, K. Watanabe, G. Gomila, K. S. Novoselov and A. K. Geim, Science, 2018, 360, 1339–1342 CrossRef CAS.
- J. Feng, K. Liu, M. Graf, D. Dumcenco, A. Kis, M. Di Ventra and A. Radenovic, Nat. Mater., 2016, 15, 850–855 CrossRef CAS.
- N. Kavokine, S. Marbach, A. Siria and L. Bocquet, Nat. Nanotechnol., 2019, 14, 573–578 CrossRef CAS.
- T. Mouterde, A. Keerthi, A. R. Poggioli, S. A. Dar, A. Siria, A. K. Geim, L. Bocquet and B. Radha, Nature, 2019, 567, 87–90 CrossRef CAS.
- A. Siria, M.-L. Bocquet and L. Bocquet, Nat. Rev. Chem., 2017, 1, 0091 CrossRef CAS.
- S. Marbach and L. Bocquet, Chem. Soc. Rev., 2019, 48, 3102–3144 RSC.
- W. Xu, H. Zheng, Y. Liu, X. Zhou, C. Zhang, Y. Song, X. Deng, M. Leung, Z. Yang, R. X. Xu, Z. L. Wang, X. C. Zeng and Z. Wang, Nature, 2020, 578, 392–396 CrossRef CAS.
- E. Lauga, M. P. Brenner and H. A. Stone, 2005, arXiv:preprint cond-mat/0501557.
- C. Neto, D. R. Evans, E. Bonaccurso, H.-J. Butt and V. S. Craig, Rep. Prog. Phys., 2005, 68, 2859 CrossRef CAS.
- L. Bocquet and J.-L. Barrat, Soft Matter, 2007, 3, 685–693 RSC.
- C. Navier, Mem. Acad. Sci. Inst. Fr., 1823, 6, 389 Search PubMed.
- Y. Ren and D. Stein, Nanotechnology, 2008, 19, 195707 CrossRef.
- S. R. Maduar, A. V. Belyaev, V. Lobaskin and O. I. Vinogradova, Phys. Rev. Lett., 2015, 114, 118301 CrossRef CAS.
- T. Q. Vo, M. Barisik and B. Kim, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 92, 053009 CrossRef.
- T. Q. Vo and B. Kim, Sci. Rep., 2016, 6, 33881 CrossRef CAS.
- L. Fu, S. Merabia and L. Joly, Phys. Rev. Lett., 2017, 119, 214501 CrossRef.
- E. F. Silkina, E. S. Asmolov and O. I. Vinogradova, Phys. Chem. Chem. Phys., 2019, 21, 23036–23043 RSC.
- B. L. Werkhoven and R. van Roij, Soft Matter, 2020, 16, 1527–1537 RSC.
- E. Secchi, S. Marbach, A. Niguès, D. Stein, A. Siria and L. Bocquet, Nature, 2016, 537, 210–213 CrossRef CAS.
- Q. Yang, Y. Su, C. Chi, C. T. Cherian, K. Huang, V. G. Kravets, F. C. Wang, J. C. Zhang, A. Pratt, A. N. Grigorenko, F. Guinea, A. K. Geim and R. R. Nair, Nat. Mater., 2017, 16, 1198–1202 CrossRef CAS.
- R. H. Tunuguntla, R. Y. Henley, Y.-C. Yao, T. A. Pham, M. Wanunu and A. Noy, Science, 2017, 357, 792–796 CrossRef CAS.
- Q. Xie, M. A. Alibakhshi, S. Jiao, Z. Xu, M. Hempel, J. Kong, H. G. Park and C. Duan, Nat. Nanotechnol., 2018, 13, 238–245 CrossRef CAS.
- L. Bocquet and J.-L. Barrat, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1994, 49, 3079 CrossRef CAS.
- J. Petravic and P. Harrowell, J. Chem. Phys., 2007, 127, 174706 CrossRef.
- J. S. Hansen, B. Todd and P. J. Daivis, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 016313 CrossRef CAS.
- K. Huang and I. Szlufarska, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 89, 032119 CrossRef.
- H. Oga, Y. Yamaguchi, T. Omori, S. Merabia and L. Joly, J. Chem. Phys., 2019, 151, 054502 CrossRef.
- P. Español, J. de la Torre and D. Duque-Zumajo, Phys. Rev. E, 2019, 99, 022126 CrossRef.
- H. Nakano and S.-i. Sasa, Phys. Rev. E, 2020, 101, 033109 CrossRef CAS.
- A. V. Straube, B. G. Kowalik, R. R. Netz and F. Höfling, Commun. Phys., 2020, 3, 126 CrossRef.
- J.-L. Barrat and L. Bocquet, Faraday Discuss., 1999, 112, 119–128 RSC.
- C. Sendner, D. Horinek, L. Bocquet and R. R. Netz, Langmuir, 2009, 25, 10768–10781 CrossRef CAS.
- X. Gu and M. Chen, Appl. Phys. Lett., 2011, 99, 063101 CrossRef.
- E. Wagemann, E. Oyarzua, J. H. Walther and H. A. Zambrano, Phys. Chem. Chem. Phys., 2017, 19, 8646–8652 RSC.
- G. Tocci, L. Joly and A. Michaelides, Nano Lett., 2014, 14, 6872–6877 CrossRef CAS.
- G. Tocci, M. Bilichenko, L. Joly and M. Iannuzzi, Nanoscale, 2020, 12, 10994–11000 RSC.
- Y. Xie, L. Fu, T. Niehaus and L. Joly, Phys. Rev. Lett., 2020, 125, 014501 CrossRef CAS.
- A. Govind Rajan, M. S. Strano and D. Blankschtein, Nano Lett., 2019, 19, 1539–1551 CrossRef CAS.
- B. Cross, C. Barraud, C. Picard, L. Léger, F. Restagno and É. Charlaix, Phys. Rev. Fluids, 2018, 3, 062001 CrossRef.
- T. Omori, N. Inoue, L. Joly, S. Merabia and Y. Yamaguchi, Phys. Rev. Fluids, 2019, 4, 114201 CrossRef.
- M. Grzelka, I. Antoniuk, E. Drockenmuller, A. Chennevière, L. Léger and F. Restagno, ACS Macro Lett., 2020, 924–928 CrossRef CAS.
- J.-L. Barrat and F. Chiaruttini, Mol. Phys., 2003, 101, 1605–1610 CrossRef CAS.
- M. E. Caplan, A. Giri and P. E. Hopkins, J. Chem. Phys., 2014, 140, 154701 CrossRef.
- A. Giri and P. E. Hopkins, Appl. Phys. Lett., 2014, 105, 033106 CrossRef.
- G. W. Robinson, Water in biology, chemistry, and physics: experimental overviews and computational methodologies, World Scientific, 1996, vol. 9 Search PubMed.
- F. Franks, Water: a matrix of life, Royal Society of Chemistry, 2007 Search PubMed.
- P. Ball, Chem. Rev., 2008, 108, 74–108 CrossRef CAS.
- P. G. Debenedetti, J. Phys.: Condens. Matter, 2003, 15, R1669 CrossRef CAS.
- P. Gallo, K. Amann-Winkel, C. A. Angell, M. A. Anisimov, F. Caupin, C. Chakravarty, E. Lascaris, T. Loerting, A. Z. Panagiotopoulos and J. Russo, et al. , Chem. Rev., 2016, 116, 7463–7500 CrossRef CAS.
- A. Dehaoui, B. Issenmann and F. Caupin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12020–12025 CrossRef CAS.
- P. Montero de Hijes, E. Sanz, L. Joly, C. Valeriani and F. Caupin, J. Chem. Phys., 2018, 149, 094503 CrossRef.
- R. Yamamoto and A. Onuki, Phys. Rev. Lett., 1998, 81, 4915 CrossRef CAS.
- S.-H. Chen, F. Mallamace, C.-Y. Mou, M. Broccio, C. Corsaro, A. Faraone and L. Liu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12974–12978 CrossRef CAS.
- L. Xu, F. Mallamace, Z. Yan, F. W. Starr, S. V. Buldyrev and H. E. Stanley, Nat. Phys., 2009, 5, 565–569 Search PubMed.
- Z. Shi, P. G. Debenedetti and F. H. Stillinger, J. Chem. Phys., 2013, 138, 12A526 CrossRef.
- T. Kawasaki and K. Kim, Sci. Adv., 2017, 3, e1700399 CrossRef.
- K. Falk, F. Sedlmeier, L. Joly, R. R. Netz and L. Bocquet, Nano Lett., 2010, 10, 4067–4073 CrossRef CAS.
- W. Chen, A. S. Foster, M. J. Alava and L. Laurson, Phys. Rev. Lett., 2015, 114, 095502 CrossRef.
- M. H. Köhler, J. R. Bordin, L. B. da Silva and M. C. Barbosa, Phys. Chem. Chem. Phys., 2017, 19, 12921–12927 RSC.
- J. Martí, C. Calero and G. Franzese, Entropy, 2017, 19, 135 CrossRef.
- A. T. Celebi, C. T. Nguyen, R. Hartkamp and A. Beskok, J. Chem. Phys., 2019, 151, 174705 CrossRef.
- X. Cai, W. J. Xie, Y. Yang, Z. Long, J. Zhang, Z. Qiao, L. Yang and Y. Q. Gao, J. Chem. Phys., 2019, 150, 124703 CrossRef.
- A. Zaragoza, M. A. González, L. Joly, I. López-Montero, M. Canales, A. Benavides and C. Valeriani, Phys. Chem. Chem. Phys., 2019, 21, 13653–13667 RSC.
- C. Bakli and S. Chakraborty, Nanoscale, 2019, 11, 11254–11261 RSC.
- A. Buchsteiner, A. Lerf and J. Pieper, J. Phys. Chem. B, 2006, 110, 22328–22338 CrossRef CAS.
- S. Cerveny, F. Barroso-Bujans, A. Alegria and J. Colmenero, J. Phys. Chem. C, 2010, 114, 2604–2612 CrossRef CAS.
- S. Cerveny, F. Mallamace, J. Swenson, M. Vogel and L. Xu, Chem. Rev., 2016, 116, 7608–7625 CrossRef CAS.
- T. Kaneko, J. Bai, T. Akimoto, J. S. Francisco, K. Yasuoka and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4839–4844 CrossRef CAS.
- L. Mishchenko, B. Hatton, V. Bahadur, J. A. Taylor, T. Krupenkin and J. Aizenberg, ACS Nano, 2010, 4, 7699–7707 CrossRef CAS.
- S. Jung, M. K. Tiwari, N. V. Doan and D. Poulikakos, Nat. Commun., 2012, 3, 615 CrossRef.
- M. J. Kreder, J. Alvarenga, P. Kim and J. Aizenberg, Nat. Rev. Mater., 2016, 1, 15003 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- J. L. Abascal and C. Vega, J. Chem. Phys., 2005, 123, 234505 CrossRef CAS.
- T. Schnabel, A. Srivastava, J. Vrabec and H. Hasse, J. Phys. Chem. B, 2007, 111, 9871–9878 CrossRef CAS.
- G. Guevara-Carrion, J. Vrabec and H. Hasse, J. Chem. Phys., 2011, 134, 074508 CrossRef.
- L. Fu, L. Joly and S. Merabia, Phys. Rev. Lett., 2019, 123, 138001 CrossRef CAS.
- C. Herrero, T. Omori, Y. Yamaguchi and L. Joly, J. Chem. Phys., 2019, 151, 041103 CrossRef.
- H. Vogel, Phys. Z, 1921, 22, 645–646 CAS.
- G. Tammann and W. Hesse, Z. Anorg. Allg. Chem., 1926, 156, 245–257 CrossRef CAS.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339–355 CrossRef CAS.
- R. Speedy and C. Angell, J. Chem. Phys., 1976, 65, 851–858 CrossRef CAS.
- H. Bässler, Phys. Rev. Lett., 1987, 58, 767 CrossRef.
- J. Hallett, Proc. Phys. Soc., 1963, 82, 1046 CrossRef CAS.
- A. P. Markesteijn, R. Hartkamp, S. Luding and J. Westerweel, J. Chem. Phys., 2012, 136, 134104 CrossRef CAS.
- E. Guillaud, S. Merabia, D. de Ligny and L. Joly, Phys. Chem. Chem. Phys., 2017, 19, 2124–2130 RSC.
- S. K. Kannam, B. Todd, J. S. Hansen and P. J. Daivis, J. Chem. Phys., 2013, 138, 094701 CrossRef.
- A. Sam, R. Hartkamp, S. Kumar Kannam, J. S. Babu, S. P. Sathian, P. J. Daivis and B. D. Todd, Mol. Simul., 2020, 1–20 CrossRef.
- L. Liu, S.-H. Chen, A. Faraone, C.-W. Yen and C.-Y. Mou, Phys. Rev. Lett., 2005, 95, 117802 CrossRef.
- L. Bocquet and J.-L. Barrat, J. Chem. Phys., 2013, 139, 044704 CrossRef.
- W. A. Steele, Surf. Sci., 1973, 36, 317–352 CrossRef CAS.
- P. Gallo, F. Sciortino, P. Tartaglia and S.-H. Chen, Phys. Rev. Lett., 1996, 76, 2730–2733 CrossRef CAS.
- P. Kumar, S. V. Buldyrev, S. R. Becker, P. H. Poole, F. W. Starr and H. E. Stanley, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9575–9579 CrossRef CAS.
- D. Jeong, M. Y. Choi, H. J. Kim and Y. Jung, Phys. Chem. Chem. Phys., 2010, 12, 2001 RSC.
- A. Ikeda and K. Miyazaki, Phys. Rev. Lett., 2011, 106, 015701 CrossRef.
- J. Sjöström, J. Mattsson, R. Bergman, E. Johansson, K. Josefsson, D. Svantesson and J. Swenson, Phys. Chem. Chem. Phys., 2010, 12, 10452–10456 RSC.
- C. Cottin-Bizonne, J.-L. Barrat, L. Bocquet and E. Charlaix, Nat. Mater., 2003, 2, 237–240 CrossRef CAS.
- T. Maitra, C. Antonini, M. K. Tiwari, A. Mularczyk, Z. Imeri, P. Schoch and D. Poulikakos, Langmuir, 2014, 30, 10855–10861 CrossRef CAS.
- Y. Lu, S. Sathasivam, J. Song, C. R. Crick, C. J. Carmalt and I. P. Parkin, Science, 2015, 347, 1132–1135 CrossRef CAS.
- M. Fitzner, G. C. Sosso, S. J. Cox and A. Michaelides, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2009–2014 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Simulation details; fitting of viscosity and friction by phenomenological laws; discussion of eqn (4); temperature dependence of structure factor; calculation of the static contribution to friction; details on the characteristic times, and on the self and collective diffusion coefficients. See DOI: 10.1039/D0NR06399A |
| This journal is © The Royal Society of Chemistry 2020 |