
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntratumoral injection of hydrogel-embedded nanoparticles enhances retention in glioblastoma†
Giulia
Brachi
 ab,
Javier
Ruiz-Ramírez
c,
Prashant
Dogra
c,
Zhihui
Wang
c,
Vittorio
Cristini
c,
Gianluca
Ciardelli
a,
Robert C.
Rostomily
d,
Mauro
Ferrari‡
b,
Andrei M.
Mikheev§
d,
Elvin
Blanco§
b and
Clara
Mattu§
*ab
ab,
Javier
Ruiz-Ramírez
c,
Prashant
Dogra
c,
Zhihui
Wang
c,
Vittorio
Cristini
c,
Gianluca
Ciardelli
a,
Robert C.
Rostomily
d,
Mauro
Ferrari‡
b,
Andrei M.
Mikheev§
d,
Elvin
Blanco§
b and
Clara
Mattu§
*ab
aPolitecnico di Torino, DIMEAS, C.so Duca degli Abruzzi 24, 10129 Torino, Italy. E-mail: clara.mattu@polito.it; Tel: +390110906792
bDepartment of Nanomedicine, Houston Methodist Research Institute, 6670 Bertner Ave, Houston, TX 77030, USA
cMathematics in Medicine Program, Houston Methodist Research Institute, 6670 Bertner Ave, Houston, TX 77030, USA
dDepartment of Neurosurgery, Houston Methodist Research Institute, 6670 Bertner Ave, Houston, TX 77030, USA
First published on 25th November 2020
Abstract
Intratumoral drug delivery is a promising approach for the treatment of glioblastoma multiforme (GBM). However, drug washout remains a major challenge in GBM therapy. Our strategy, aimed at reducing drug clearance and enhancing site-specific residence time, involves the local administration of a multi-component system comprised of nanoparticles (NPs) embedded within a thermosensitive hydrogel (HG). Herein, our objective was to examine the distribution of NPs and their cargo following intratumoral administration of this system in GBM. We hypothesized that the HG matrix, which undergoes rapid gelation upon increases in temperature, would contribute towards heightened site-specific retention and permanence of NPs in tumors. BODIPY-containing, infrared dye-labeled polymeric NPs embedded in a thermosensitive HG (HG–NPs) were fabricated and characterized. Retention and distribution dynamics were subsequently examined over time in orthotopic GBM-bearing mice. Results demonstrate that the HG–NPs system significantly improved site-specific, long-term retention of both NPs and BODIPY, with co-localization analyses showing that HG–NPs covered larger areas of the tumor and the peri-tumor region at later time points. Moreover, NPs released from the HG were shown to undergo uptake by surrounding GBM cells. Findings suggest that intratumoral delivery with HG–NPs has immense potential for GBM treatment, as well as other strategies where site-specific, long-term retention of therapeutic agents is warranted.
Introduction
Glioblastoma multiforme (GBM) is the most common primary brain tumor in adults with a median survival of only 12–14 months.1–3 Despite novel insights into the mechanisms underlying tumorigenesis nearly all patients recur and succumb to their disease.4,5 The 5-year survival of GBM patients receiving standard of care is limited to a dismal 5%.6,7 In light of the severe morbidity and mortality associated with GBM, innovative therapeutic interventions are urgently needed to impact patient outcomes.One of the main reasons why systemic chemotherapy remains largely ineffective against GBM is the presence of the blood brain barrier (BBB) that hampers adequate drug distribution to the tumor, resulting in sub-therapeutic concentrations in the brain and severe systemic side effects.8–10 The accumulation profile of temozolomide (TMZ), a drug routinely used for the treatment of GBM, in the brain is limited to ∼20% of the initial dose.8 Local drug delivery has been proposed as an alternative to circumvent the BBB, enhance drug concentrations at the tumor site, and reduce systemic toxicity.11,12 For instance, polymer implants comprised of polifeprosan20 loaded with carmustine (BCNU) (Gliadel®) received clinical approval for the intracranial treatment of resectable GBM.13,14 Direct fluid infusion in the tumor bed via convection-enhanced delivery (CED) has also been explored clinically.15 However, complex implantation procedures, risk of infections, local drug toxicity, and rapid drug clearance from the brain parenchyma have limited the applicability of these strategies.16–20
Intratumoral administration of drug-containing polymer nanoparticles (NPs) is an attractive strategy to deliver drugs to the brain, as NP formulations allow for solvent-free drug administration and protect therapeutic agents from metabolism and degradation.21 Moreover, drug clearance and diffusion-based transport across the tumor interstitium can be modulated by tuning NPs size.22,23 NPs can also be precisely tailored to modulate drug release, resulting in sustained release profiles over time, allowing to increase the drug dose without toxicity.24–28 Injectable hydrogels (HGs) also represent a viable strategy for local drug delivery in resectable and non-resectable brain tumors.29–32 Thermo-sensitive HGs are advantageous compared to other hydrogel systems, as they undergo a sol–gel transition driven by temperature changes, without the need of external cross-linking (e.g. UV-irradiation) or addition of potentially toxic chemicals.33 They can be prepared as a polymer solution at room temperature and injected either into the tumor bulk or into the resection cavity, where the temperature increase triggers solidification into a 3D hydrated matrix.34 Additional advantages include ease of administration during standard surgical procedures, and lower risks of trauma and infection compared to CED.35,36
To fully exploit advantages of NPs for localized treatment, their combination with thermo-responsive HGs (HG–NPs) has been proposed.37,38 By forming a depot at the injection site, HGs may contribute to NP localization over time. In addition, the HG matrix can modulate NPs release kinetics and preserve their surface functionalities from degradation/deactivation.39,40 Despite the potential for combining HG and NPs, the transport kinetics and tumor distribution of HG–NPs after intratumoral administration in GBM remain unexplored. In this work, a multi-component strategy composed of HG-embedded drug-loaded NPs was designed for intratumoral injection in mice bearing a highly infiltrative orthotopic GBM model (Fig. 1).
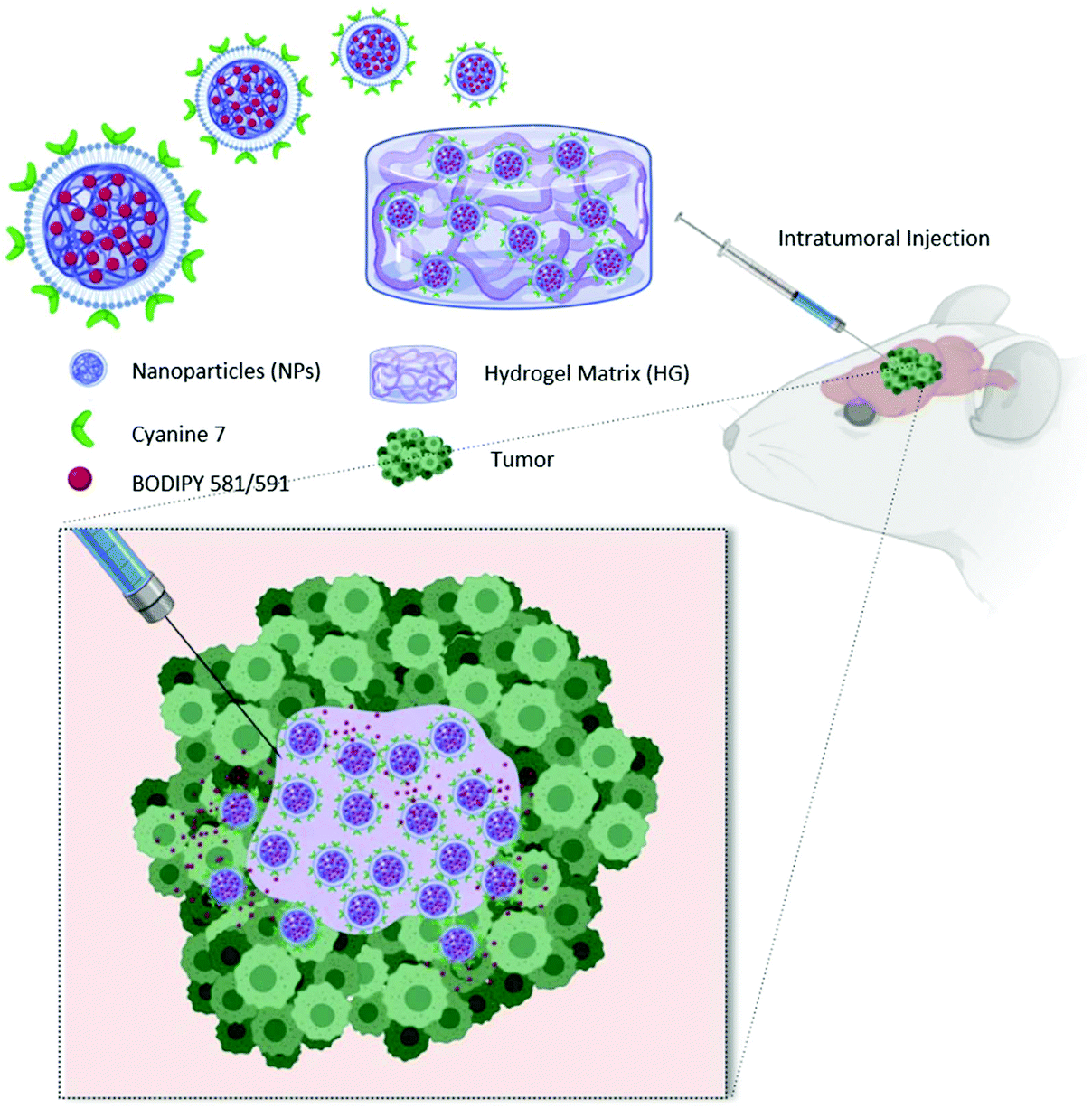 | ||
| Fig. 1 Schematic representation of the HG–NPs system for intratumoral delivery. The HG–NPs solution is injected at room temperature into the tumor. The HG component rapidly solidifies after administration due to the increase in temperature. The result is an HG depot that entraps NPs, enhancing their local retention and mitigating their migration away from the tumor bulk. Image created with BioRender.com. | ||
Our objective was to investigate the distribution dynamics and retention of HG–NPs following intratumoral administration. We exploited the thermosensitive properties of a commercial polymer of well-reported biocompatibility41 (Poloxamer 407) to achieve non-invasive gelation conditions, triggered by body temperature without requiring any additional energy source.34 We hypothesized that the HG matrix would retain NPs near the administration site, reducing their migration away from the tumor bulk, and enhance tumor coverage and NP retention over time. To achieve this, a fluorophore (BODIPY) was encapsulated inside infrared dye (cyanine 7)-labeled polyurethane (PUR) NPs. NPs were embedded in a Poloxamer 407-based thermo-sensitive HG, the system administered intratumorally in mice, and the distribution of NPs and BODIPY examined at different timepoints. Results show that HG–NPs had a significantly longer intratumoral retention compared to free NPs and covered a significantly larger area of the tumor and the peri-tumor region. These findings suggest that the local retention effect of HGs combined with NP-based drug delivery has the potential to extend the therapeutic window of intratumoral-administered drugs, opening avenues for HG–NPs-based local delivery strategies in GBM.
Materials and methods
Materials
Poly(ε-caprolactone)-diol (PCL-diol (2000 g mol−1), n-BOC serinol, dibutyl dilaurate (DBTL), and 1,6-hexamethylene diisocyanate (HDI) were purchased from Sigma Aldrich (St Louis, MO, USA). L-α-Phosphatidylglycerol (Egg-PG), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethyleneglycol)-2000] (DSPE-PEG-NH2) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(cyanine 7) (Cy7-PE) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). BODIPY 581/591-NHS-ester was purchased from Lumiprobe (Hunt Valley, MD, USA). Poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (Poloxamer 407) was purchased from Sigma Aldrich (St Louis, MO, USA). All solvents were of analytical grade.Polyurethane synthesis and characterization
The PCL-based polyurethane used in this study was synthesized following a two-step synthesis procedure.42,43 PCL-diol and HDI were dissolved in anhydrous dichloroethane (DCE) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio) and allowed to react at 85 °C for 150 min in the presence of a catalytic amount of DBTL to obtain a pre-polymer. After addition of n-BOC serinol (1:1 molar ratio to PCL-diol), the reaction continued at RT for 16 h, after which the polymer was purified in diethyl-ether/methanol (95:5) and collected by decantation/precipitation. The molecular weight was analyzed by size exclusion chromatography (SEC, Agilent Technologies 1200 Series, Santa Clara, CA, USA) using a refractive index detector and two Waters Styragel columns (HT2 and HT4) conditioned at 35 °C. Tetrahydrofuran was used as a mobile phase at a flow rate of 0.5 mL min−1 with an injection volume of 20 μL. Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) was performed on a Perkin Elmer Spectrum 100 equipped with diamond crystal (UATR KRS5, Perkin Elmer, Milan). A total of 16 scans with a resolution of 4 cm−1 were acquired.
:2 molar ratio) and allowed to react at 85 °C for 150 min in the presence of a catalytic amount of DBTL to obtain a pre-polymer. After addition of n-BOC serinol (1:1 molar ratio to PCL-diol), the reaction continued at RT for 16 h, after which the polymer was purified in diethyl-ether/methanol (95:5) and collected by decantation/precipitation. The molecular weight was analyzed by size exclusion chromatography (SEC, Agilent Technologies 1200 Series, Santa Clara, CA, USA) using a refractive index detector and two Waters Styragel columns (HT2 and HT4) conditioned at 35 °C. Tetrahydrofuran was used as a mobile phase at a flow rate of 0.5 mL min−1 with an injection volume of 20 μL. Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) was performed on a Perkin Elmer Spectrum 100 equipped with diamond crystal (UATR KRS5, Perkin Elmer, Milan). A total of 16 scans with a resolution of 4 cm−1 were acquired.
Preparation of NPs and HG–NPs
Cy7-labeled, BODIPY-loaded NPs were prepared by a nanoprecipitation/self-assembly method.44,45 BODIPY (25 μg) was dissolved in acetonitrile (1 mL) with the polymer (1 mg) and added drop-wise into 2 mL of water containing 200 μg of Egg-PG, 240 μg of DSPE-PEG-NH2, and 30 μg of Cy7-PE at 60 °C. NPs formed immediately upon precipitation of the polymer in water followed by self-assembly of the mixture of phospholipids on the outer surface. The particle suspension was centrifuged at 3200 rpm and washed three times, using molecular concentrators with a cutoff size of 10 kDa.To prepare HG–NPs, NPs (1 mg) were re-suspended at 4 °C in saline containing different concentrations of Poloxamer 407 (P407) (i.e. 20% w/v, 25% w/v and 30% w/v) and incubated at 37 °C to obtain a 3D hydrogel matrix embedding NPs.
Characterization of NPs and HG–NPs
Size, polydispersity index (PDI), and surface charge (zeta potential) of NPs were analyzed by dynamic light scattering (DLS) (Malvern, Zetasizer Nano S90, Malvern, UK). Stability of NPs was assessed in PBS at 37 °C by daily measurements of size distribution over a period of 7 d. In vitro toxicity of empty NPs was evaluated on GBM8 cells. Briefly, cells (10000 cells per well) were cultured in 96 well plates for 2 days, followed by incubation with empty NPs at different concentrations. Cell viability was assessed at 24 h, 48 h, and 72 h by MTS assay.
NPs morphology was characterized by transmission electron microscopy (TEM) analysis (Cryo-Electron Microscopy Core Facility, Baylor College of Medicine, Houston, TX, USA). For sample preparation, lipids were stained with phosphotungstic acid (1%)45 to enhance contrast and 3 μl of NPs solution were loaded into holey carbon grids (Quantifoil Micro Tools GmbH, Jena, Germany) and imaged with a 200 kV JEOL Model 2200 Electron Microscope (JEOL Ltd, Japan).
BODIPY loading and Cy7 labelling efficiency were quantified by UV spectroscopy (Perkin Elmer, Waltham, MA, USA, Lambda 365,). Briefly, NPs were freeze dried, dissolved in acetonitrile and then analyzed at 580 nm for BODIPY and at 750 nm for Cy7, respectively. Encapsulation efficiency (EE) was determined according to the following equation:
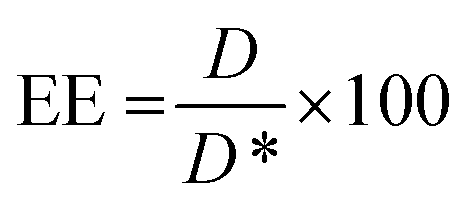 | (1) |
Sol–gel transition of HG–NPs was evaluated by a tube-inverting test. P407 solutions in DI water, with or without NPs, were prepared in triplicate at 4 °C and incubated at 37 °C. At predetermined time points, the tubes were inverted to determine whether the solution could flow under its own weight. Complete gelation was achieved in the absence of flow.
To evaluate the kinetics of BODIPY release, NPs and HG–NPs were incubated in PBS at 37 °C under moderate shaking. At pre-determined time points, the supernatant was collected and analyzed by UV spectroscopy at 580 nm. The supernatant from HG–NPs was also analyzed by DLS.
Cell culture
Cells (human GBM-derived stem cells, GBM-8)46 were cultured in Neurobasal medium supplemented with heparin, B27, N2, penicillin/streptomycin, N-glutamine, epidermal growth factor (EGF, 20 ng ml−1) and fibroblast growth factor (FGF, 20 ng ml−1).6 All cell culture reagents were purchased from Thermofisher (Waltham, MA, USA). Growth factors were purchased from Peprotech (London, UK), cell culture supplements were from Invitrogen (Carlsbad, CA, USA).Orthotopic glioblastoma model
All animal studies were performed according to the Guidelines for Care and Use of Laboratory Animals of the Houston Methodist Research Institute and were approved by the Institutional Animal Care and Use Committee at the Houston Methodist Research Institute.Pre-operative analgesia was administered to mice by subcutaneous injection of lidocaine and buprenorphine. Female nude mice (Envigo) were anesthetized by intraperitoneal (IP) injection of ketamine/xylazine and immobilized in a stereotactic frame (Stoelting).4 The scalp was disinfected and a midline incision was made in the skin. A hole was drilled 1.5 mm anterior and 2 mm lateral to the bregma. GFP-labelled GBM-8 cells (12 × 104 in 3 μL) were injected into the brain using a 25 μL Hamilton syringe equipped with a 26G needle at a depth of 3 mm and flow rate of 1.7 μL min−1 using computer controlled microinjector. Precautions were taken to minimize leakage from the needle tract, including slow administration of the injectate and leaving the needle in place for 1 min before removal. The skull was sealed with bone wax and the incision wound was closed.
Animal injection was performed with the same cell preparation, and injected animals were randomly assigned for subsequent groups (e.g. HG–NPs of free NPs). Based on our previous experience with this tumor model, the percentage of tumor development is 100% and the median survival is 30–32 days (Fig. S1D†). To verify the stages of tumor development, a subset of 2 mice was injected with luciferase-transfected GBM-8 cells. Tumor growth in these mice was monitored by bioluminescence in vivo imaging system (IVIS, PerkinElmer Inc, Waltham, MA), 5 min after intraperitoneal (IP) administration of 20 μL of luciferin solution (Fig. S1A†) and by detection of the GFP signal on the harvested brain (Fig. S1B†). H&E staining was also performed to determine extent of tumor growth (Fig. S1C†).
Intracranial injection of NPs and HG–NPs
At a time point of 18 d post cell injection, mice were prepared for surgery as described in the previous section and the hole re-opened by removing the bone wax. A dose of 2.5 mg of BODIPY-loaded, Cy7-labelled NPs in a volume of 5 μL of PBS (NPs group) or HG solution (HG–NPs group) were injected into the brain using a 25 μL Hamilton syringe equipped with a 26G needle, at a flow rate of 1 μL min−1 as described above. HG–NPs preloaded syringe was kept at 4 °C to prevent HG gelation between injections. The hole was sealed with bone wax and the incision wound closed. BODIPY-loaded, Cy7-labelled NPs (or HG–NPs) were similarly injected in tumor-free mice.Examination of NP retention
NP retention in the brain of tumor-free (n = 3) and tumor-bearing mice (4 mice per group) was evaluated by 3D in vivo imaging system (IVIS, PerkinElmer Inc, Waltham, MA). Mice were anesthetized by isoflurane and the 3D fluorescent imaging tomography (FLIT) modality of IVIS was used to image the brain area. Mice were examined at 1 d, 5 d, and 10 d post-injection. The amount of Cy7 calculated by the software was plotted over time as a percentage of the amount detected at the first timepoint to evaluate the retention of NPs and HG–NPs.Brain tissue processing
At pre-determined time points post injection (1, 5, and 10 d, 4 animals per group), mice were sacrificed by CO2 and brains and major organs immediately harvested. 2D IVIS imaging was performed on the extracted brains and major organs to determine fluorescence from Cy7 in the NPs. Following 2D IVIS imaging, brains were extracted, frozen, and embedded in OCT by immersion in 2,2-dimethylbutane in dry ice. Coronal sections of 20 μm in thickness were obtained with a Leica CM 1800 Microtome and collected on super-frost glass-slides. The slides were then fixed with PFA for 1 min, rinsed thrice with PBS, and nuclei were stained with DAPI. Coverslips were mounted and sealed using Prolong antifade cell mounting medium (Fisher Scientific, Hampton, NH). This process was repeated for all extracted brains (n = 4 brains per group per time point). Coronal sections at 12 different distance points (300 μm step) were collected from anterior (n = 6) and posterior (n = 6) positions from the injection site (0 μm), and then imaged using an EVOS FL imaging system (Thermofisher, Waltham, MA, USA) equipped with fluorescent filters to measure fluorescence from Cy7, BODIPY, GFP and DAPI.Distribution profiles of NPs and BODIPY in the brain
Whole brain images corresponding to entire coronal sections taken at 12 different distance points from the injection site (as detailed in the above section) were analyzed using ImageJ software.47 For each image, the RGB channels were split and the integrated density was calculated for the BODIPY and the Cy7 channels in a selected region of interest, followed by background subtraction. The corrected total cell fluorescence (CTCF) of each section was then determined according to the following equation for BODIPY and Cy7 separately: | (2) |
The overall fluorescence of BODIPY and Cy7 (CTCFtot) in the brain were then calculated by adding up the CTCF values for each coronal section.
To plot the distribution of Cy7 and BODIPY on the sagittal plane the CTCF values were expressed as a percentage of overall brain fluorescence, according to the following:
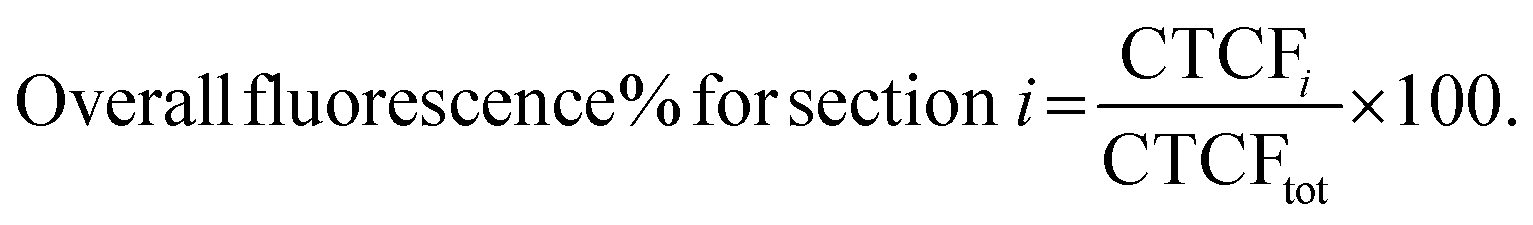 | (3) |
Analysis of tumor co-localization
Co-localization of NPs and BODIPY with tumors was determined using ImageJ and MATLAB48 on regions of the tumor located near the injection site (tumor center) and at the tumor periphery, anterior and posterior to the injection site at 1 d and 10 d post-injection. To achieve this, the RGB channels were split, with Cy7 representing NPs, BODIPY representing the model drug, and GFP or nuclei clusters representing the tumor.49 Contrast was enhanced and images binarized, by means of Otsu's algorithm.50,51 Finally, the arrays of binary pixels representing the NPs and the tumor, as well as the arrays representing BODIPY and the tumor, were multiplied (entrywise product) and the resulting number of non-zero pixels was computed. These quantities indicate the overlap between NPs and model drug, respectively, with the tumor, and were later divided by the total number of nonzero tumor pixels (corresponding to the total area occupied by the tumor) to provide a coverage percentage, according to: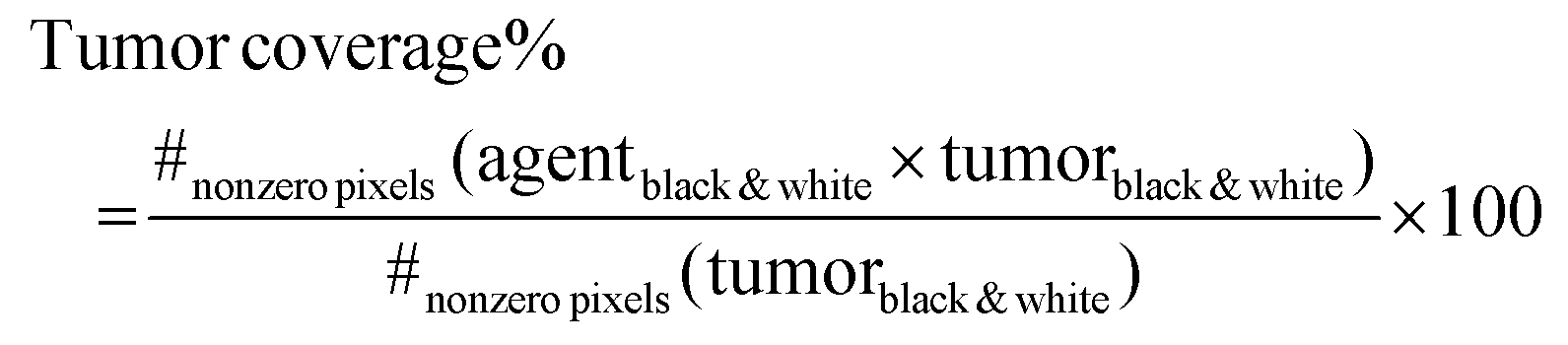 | (4) |
To display tumor coverage on different tumor regions, binary images of Cy7 and BODIPY were merged with the corresponding GFP channel, followed by background subtraction. Images were adapted on representative coronal brain sections.52,53
To gain a better understanding of NPs or HG–NPs trafficking in the intracellular and extracellular space, confocal images were acquired on selected brain slices from tumor-bearing mice at different distances from the injection site at 1 d and 10 d post injection using a Fluoview FV 1000 confocal microscope (Olympus, USA) at a magnification of 40×.
Characterizing NPs and HG–NPs transport in the brain
Several models in the literature have been designed to model the spatiotemporal distribution of NPs across the body.54–56 Based on the nature of the data in this study, we opted for a macroscopic scale continuum modeling approach focused solely on the brain compartment. To quantitatively characterize the effects of the HG on the propagation of NPs after intratumoral administration, the following isotropic diffusion-reaction model was considered:57,58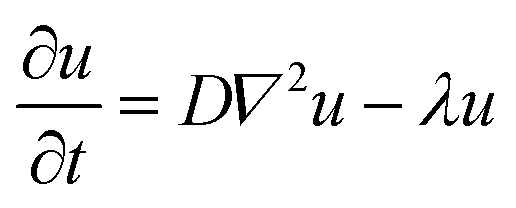 | (5) |
Under the assumption of an isotropic diffusive process (D is considered constant throughout the tissue), we fitted the coefficients D and λ for the NPs and HG–NPs of tumor-bearing mice by solving eqn (5) over the geometry corresponding to the coronal sections located at −2 mm from the injection site. Fitting of the parameters was performed as follows: (1) the Cy7 intensity signal of the coronal sections was mapped to a common (physiologically-accurate) geometry to reduce image distortion and dimension bias; (2) the common geometrical region was partitioned into triangles and a 2D mesh was generated; (3) the available experimental data for the time point t = 1 d was interpolated on the mesh and used as an initial condition for the model; (4) homogeneous (zero) conditions were imposed along the boundary of the mesh; and (5) a least squares approach was used to estimate the parameters of the equation by minimizing the distance between the computed numerical approximations using the finite element method60 and the available experimental data for the time points t = 5 d and t = 10 d.
Statistical analysis
All results are presented as mean ± standard deviation (SD). T-Test analysis with a 95% confidence interval was used for comparisons between the HG–NPs- and NPs groups. For multiple comparisons, one-way ANOVA with Tukey correction was used with a confidence interval of 95%. Statistical analysis was performed with GraphPad Prism software (GraphPad, San Diego, CA).Results
Polymer, NPs and HG–NPs characterization
A polyurethane with the desired structure was successfully obtained, as confirmed by SEC analysis and by ATR-FTIR (reported in Fig. S2†). Signals at 3300 cm−1, 1720 cm−1, 1200 cm−1 were attributed to –NH stretching, urethane stretching, and –C–O–C– stretching of ester groups in the PCL-containing segments. The molecular weight (Mw) of the polymer, measured by SEC, was 46000 Da with a polydispersity (D) of 1.2.
Results for BODIPY loading efficiency, Cy7 labeling efficiency, NPs size, PDI and zeta-potential calculated on three independent NPs batches are summarized in Fig. 2A.
 | ||
| Fig. 2 In vitro characterization of NPs and HG–NPs. (A) Size, PDI, Cy7-labelling efficiency and BODIPY encapsulation efficiency (EE) of NPS. (B) Daily measurements of mean size and PDI of Cy7-labelled NPs loaded with BODIPY. (C) Transmission electron microscopy (TEM) image of hybrid core–shell NPs. (D) Kinetics of gelation of Poloxamer 407 solutions in PBS (37 °C) at different concentrations ranging from 20 to 30 wt% (n = 3). (E) Release kinetics of BODIPY from free NPs and HG–NPs in PBS (37 °C) (n = 4). Insets represent DLS analyses of the supernatant of HG–NPs. | ||
NPs were stable over time, as no significant change in size or PDI was observed after 7 d incubation in PBS (Fig. 2B). NPs did not show toxicity in vitro on GBM-8 cells, regardless of the concentration, up to 1 mg ml−1 (Fig. S3†).
TEM imaging shows that the obtained nanoparticles are spherical and homogeneous in size. Moreover, the presence of a lipid shell surrounding the polymer core can be observed in high-resolution images, as evidenced by negative lipid staining with tungstic acid (Fig. 2C).
Gelation time at 37 °C was tested at different concentrations of Poloxamer 407 (Fig. 2D). Gelation time was concentration-dependent and averaged 8–9 min for 20 wt%, and 3 min for 30 wt%, the latter being the most viscous solution at RT which did not allow for injection through a 26G needle. Thus, an intermediate concentration of 25 wt% (gelation time 4 min) was selected for preparation of HG–NPs. Previous studies on Poloxamer 407 indicate that 20% w/v HG completely dissolves within 5 days, without significant swelling.61 In line with these results, in vitro release of BODIPY from HG–NPs follows zero-order kinetics, characterized by an initial burst of ∼30% within the first 1 d, followed by a more sustained release over 5 d, when the HG matrix was completely dissolved61 (Fig. 2E). DLS analysis of the supernatant also confirmed release of NPs from HG.
In vivo distribution of NPs and HG–NPs in the brain following intracranial administration
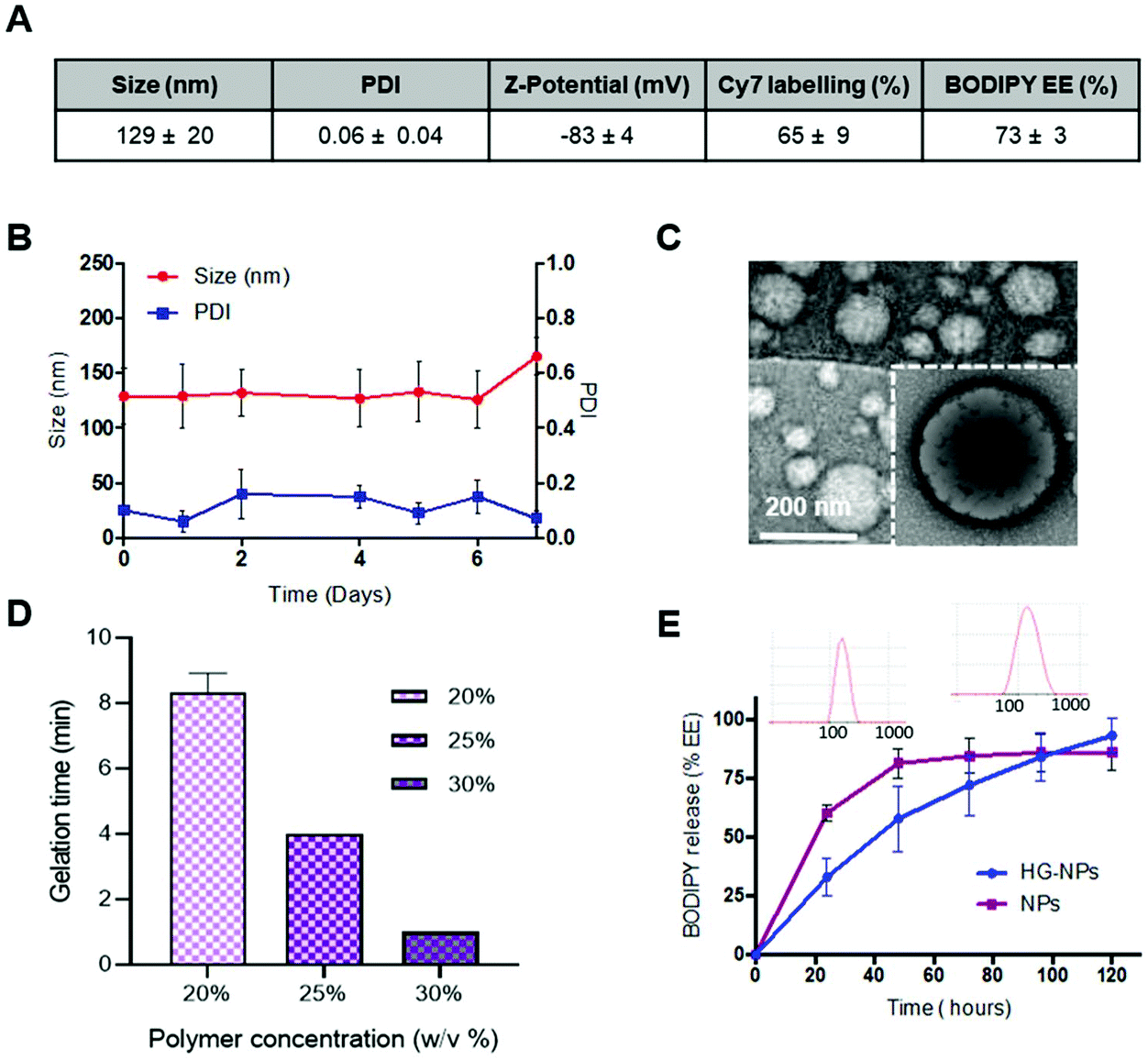
Intracranial injection of NPs and HG–NPs was well tolerated based on animal weight (Fig. S4†) and absence of any neurological signs of toxicity in any of the injected animals. No fluorescence from Cy7 was detected in the harvested organs (Fig. S5†) at any time point.Retention of NPs and HG–NPs was examined over time by 3D fluorescence imaging tomography (FLIT) using IVIS (Fig. 3A).
 | ||
| Fig. 3 Epifluorescence visualization of representative brains from tumor-bearing mice injected with NPs or HG–NPs. (A) Cy7 dye quantification in live mice, expressed as % of dose determined at the first time-point (% ID), following injection of free NPs (dashed line) or HG–NPs (solid line). Results are presented as mean ± SD. (B) 2D IVIS imaging of tumor-bearing mice at timepoints of 1, 5, and 10 d post injection. | ||
Cy7 quantification was ∼2.1-fold higher in HG–NPs compared to free NPs (p = 0.003) at the 10-day time point, corroborating that HG provided a significant improvement in NP retention. Excised brains were then qualitatively examined via 2D IVIS imaging, highlighting NP retention in tumors at each of the timepoints (Fig. 3B). Of note, the epifluorescence signal was stronger in mice injected with HG–NPs, indicating enhanced NP retention near the injection site. In tumor-free mice, no significant difference in retention was observed between free NPs and HG–NPs up to 5 days (Fig. S6A†). Injectate was retained over a period of 10 d, with a decrease in Cy7 signal of approximately 7 ± 0.5% and 27 ± 2% for HG–NPs and NPs, respectively (p = 0.014).
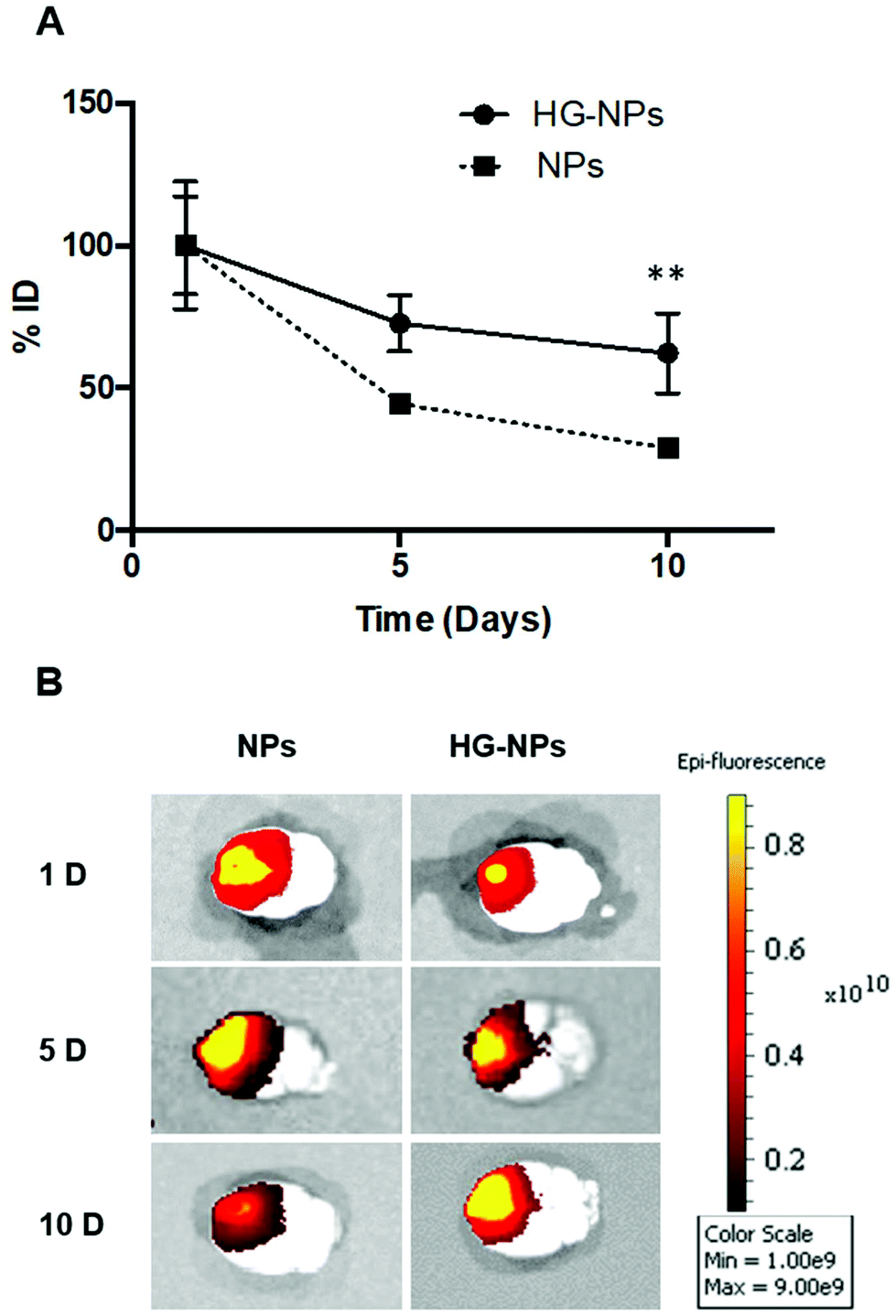
To gain further insights into the distribution of NPs and HG–NPs, confocal images of coronal sections were taken at different distances from the injection site. As shown in Fig. 4A, intratumorally-administered NPs and HG–NPs are initially confined near the injection site. In the case of free NPs, early NP leakage accompanied by backflow towards the injection tract was observed at 1 d, as shown by superficial localization of the fluorescent signal.
 | ||
| Fig. 4 Distribution profiles of Cy7-labeled NPs in the coronal and sagittal plane at 1, 5, and 10 d post injection. (A) Representative confocal images of coronal brain sections at the injection site from tumor-bearing mice injected with NPs and HG–NPs. Nuclei were stained with DAPI (blue); NPs are represented by red fluorescence. (B) Sagittal distribution profiles measured by detecting the overall fluorescence intensity of Cy7 on coronal sections from tumor-bearing mice injected with NPs and HG–NPs. | ||
The analysis of fluorescence distribution along the sagittal direction (Fig. 4B), shows that free NPs distribute well along the sagittal direction and can be detected up to 3 mm posterior to the injection site by day 5 post injection. In the case of HG–NPs, NPs were detected in the sagittal direction up to 4 mm posterior to the injection site, revealing a sustained diffusion of NPs from the HG matrix. These results suggest that the polymer matrix enhanced NP confinement within the injection area near the tumor and delayed, but did not limit, NPs migration in the coronal plane over time. In tumor free mice (Fig. S7†), an enhanced confinement near the injection site was also detected for HG–NPs (Fig. S7A†) up to 5 d post-injection, followed by spreading of the fluorescent signal in the sagittal direction (Fig. S7B†). NPs distributed more rapidly throughout the normal brain as early as 1 d post-injection.
Fig. 5 shows the distribution of the model drug BODIPY in representative coronal sections near the injection site and along the sagittal direction in tumor-bearing mice.
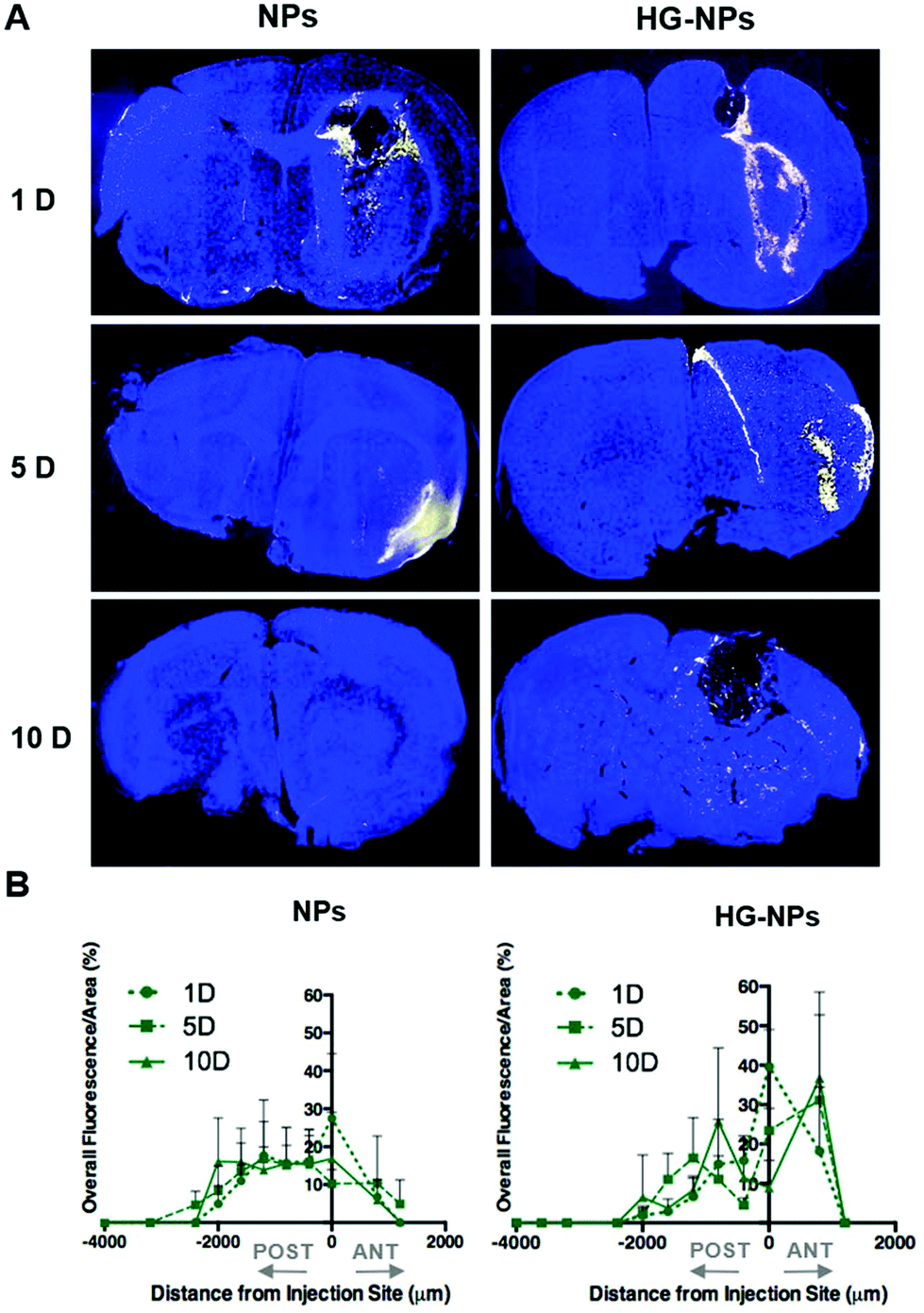 | ||
| Fig. 5 Distribution profiles of BODIPY in the coronal and sagittal plane at 1, 5, and 10 d post injection. (A) Representative confocal images of coronal brain sections at the injection site from tumor-bearing mice injected with NPs and HG–NPs. Nuclei were stained with DAPI (blue); BODIPY is represented by the yellow color. (B) Sagittal distribution profiles measured by detecting the overall fluorescence intensity of BODIPY on coronal sections from tumor-bearing mice injected with NPs and HG–NPs. | ||
At the 24-hour timepoint, the fluorescence signal from BODIPY was co-localized with the NP signal, suggesting that BODIPY was still encapsulated (Fig. 5A). The fluorescent signal of BODIPY was also detected at each timepoint, albeit confirming progressive release of the dye from NPs (Fig. 5B). In tumor-bearing mice, a 10–12% higher BODIPY retention at the injection site was achieved when NPs were embedded within an HG at 24 h, indicating that HGs facilitate confinement of NPs. At later-stage timepoints, results confirm that the presence of the HG matrix did not limit the ability of NPs to diffuse in the sagittal direction, as the signal from BODIPY was detected at comparable distances from the injection site at 5 d and 10 d post injection. Fig. S8† shows the distribution of BODIPY in tumor free mice. BODIPY appeared to be co-localized with NP signal at the early timepoint, after which the signal was mainly detected in the corpus callosum.
Computational analysis of tumor co-localization
A goal for GBM therapeutics is to maximize delivery to match tumor growth patterns. Therefore, we quantified tumor coverage by NPs (Fig. 6A and C) and BODIPY (Fig. 6B and D) in the center of the tumor mass and at the peripheries, anterior and posterior to the injection site.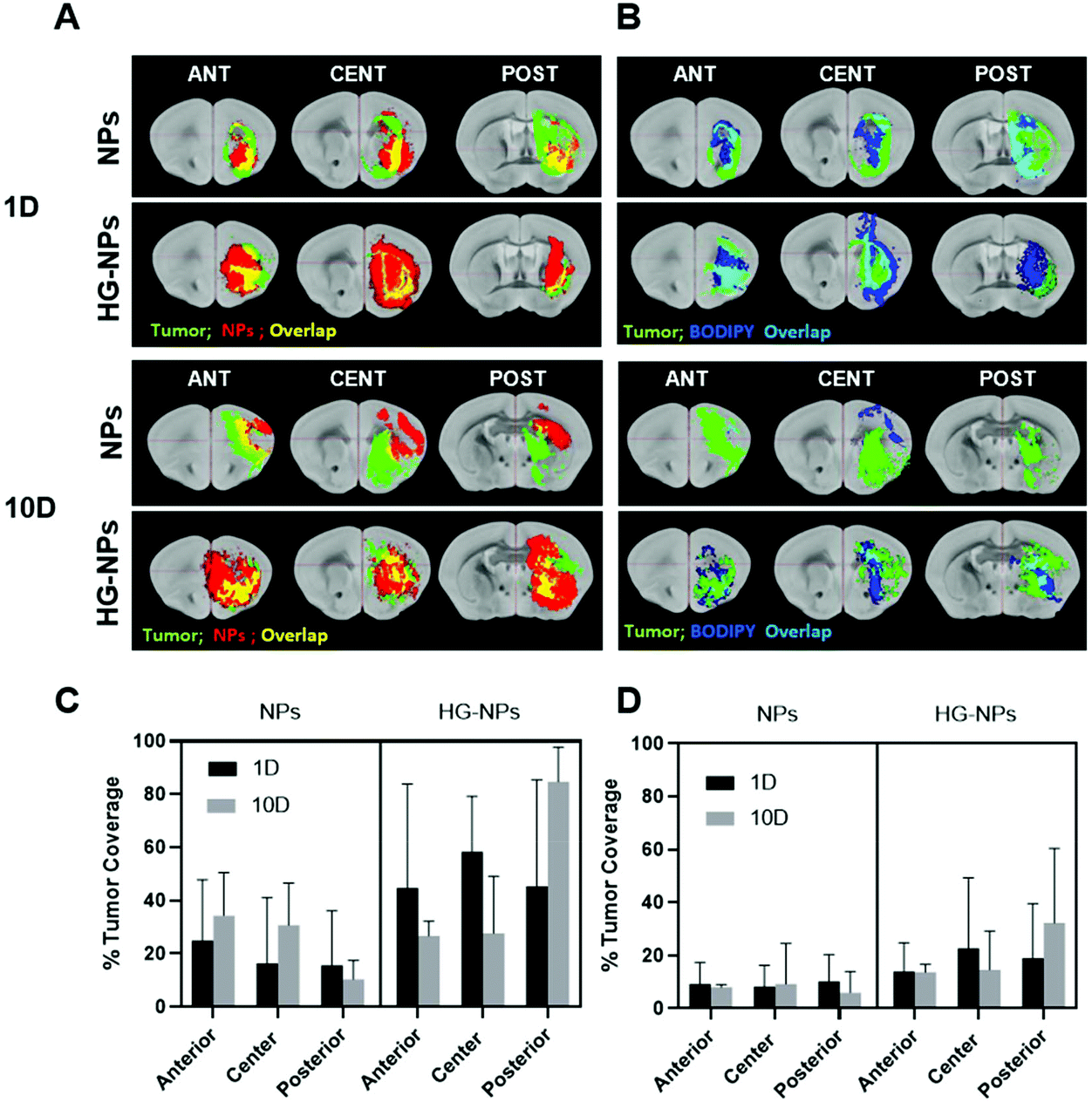 | ||
| Fig. 6 Computational analysis of NP and BODIPY tumor coverage at 1 and 10 d. (A) Representation of tumor coverage by NPs on coronal sections, anterior to the injection site (ant), in the tumor center (cent) and posterior to the injection site (post). Brain section masks (in grey) are from https://tissuestack.org/desktop.html. The tumor reconstruction is shown in green, NPs in red, regions of overlap appear in yellow. (B) Representation of tumor coverage by BODIPY on coronal sections, anterior to the injection site (ant), in the tumor center (cent) and posterior to the injection site (post). The tumor reconstruction is shown in green, BODIPY in blue, regions of overlap appear in cyan. (C) Quantification of the tumor area covered by NPs and (D) quantification of the tumor area covered by BODIPY. Tumor coverage was calculated at 1 d and 10 d post-injection (one-way ANOVA, confidence interval 95%). | ||
Fig. 6A and C indicate that the highest tumor coverage by NPs (58%) was achieved at the injection site (tumor center) for HG–NPs, while free NPs resulted in a lower coverage of nearly 16%. After 10 d, higher tumor coverage was maintained in the HG–NPs group. Coverage of the tumor area in the posterior periphery reached 80%, compared to ∼10% for the NPs group (p = 0.0009). Coverage of the tumor area by BODIPY (Fig. 6B and D) was also higher for HG-NPs compared to free NPs, with BODIPY covering 22% of the tumor area at the injection site 1 d post injection, compared with 8% coverage from free NPs. At the 10 d timepoint, coverage by BODIPY remained higher in the HG-NPs group, reaching ∼35% at the tumor periphery (compared to 10% for free NPs). High magnification confocal images (40×) taken on tumor slide samples confirm that NPs can be found co-localized with the GFP signal associated with tumor cells, as well as in the interstitial space between cells, in both groups (Fig. 7).
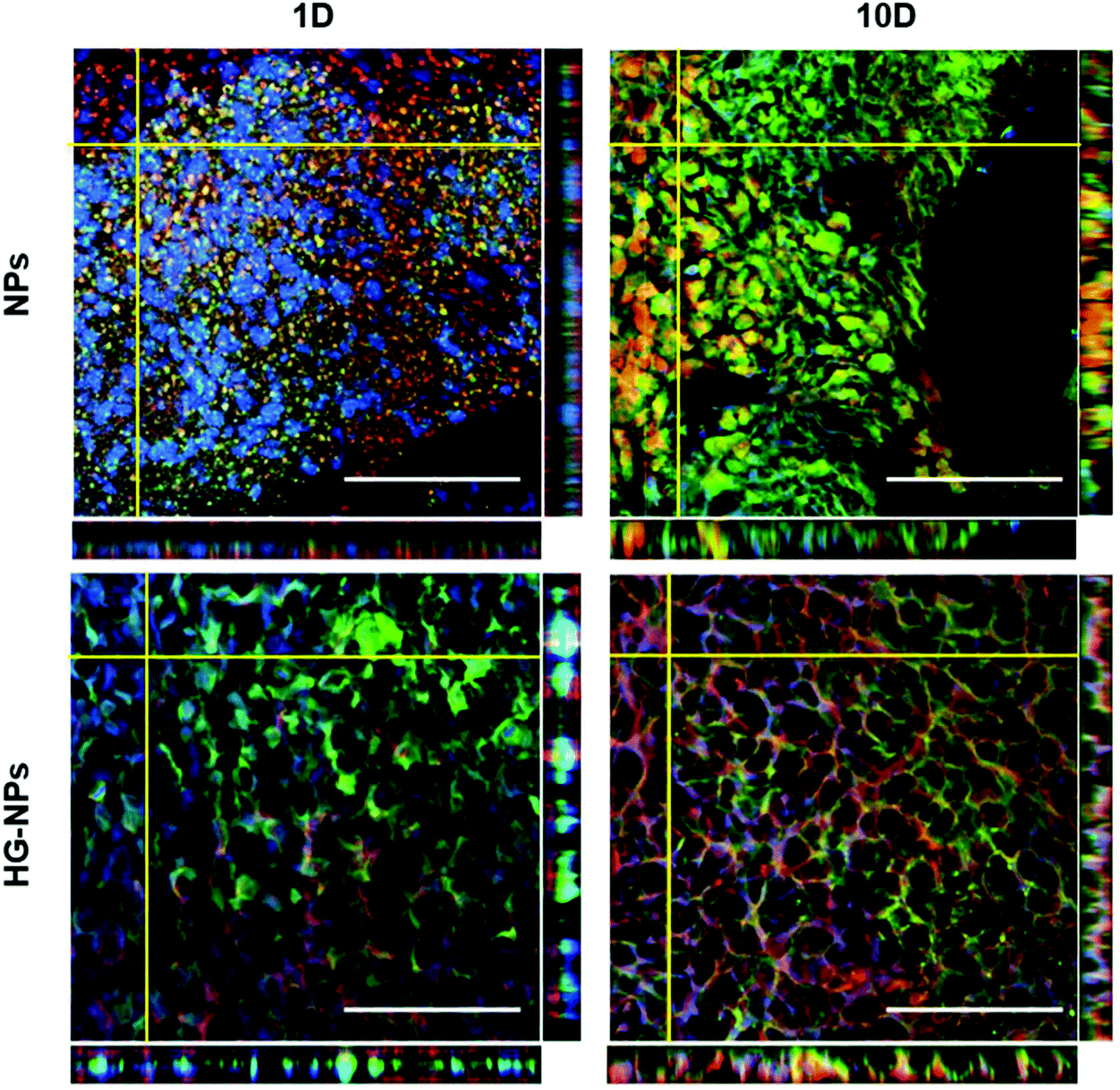 | ||
| Fig. 7 NPs penetration into tumor tissue. 40× confocal images showing internalization of NPs (Cy7, shown in red) by cancer cells (GFP, shown in green). Nuclei were stained with DAPI (blue). Accompanying z-stacks evidence the penetration of NPs in the tissue and the overlap with tumor cells (yellow color). Scale bars represent 100 μm. | ||
Finite element (FEM) modelling was applied to quantify NP diffusion from the injection site over time (Fig. 8).
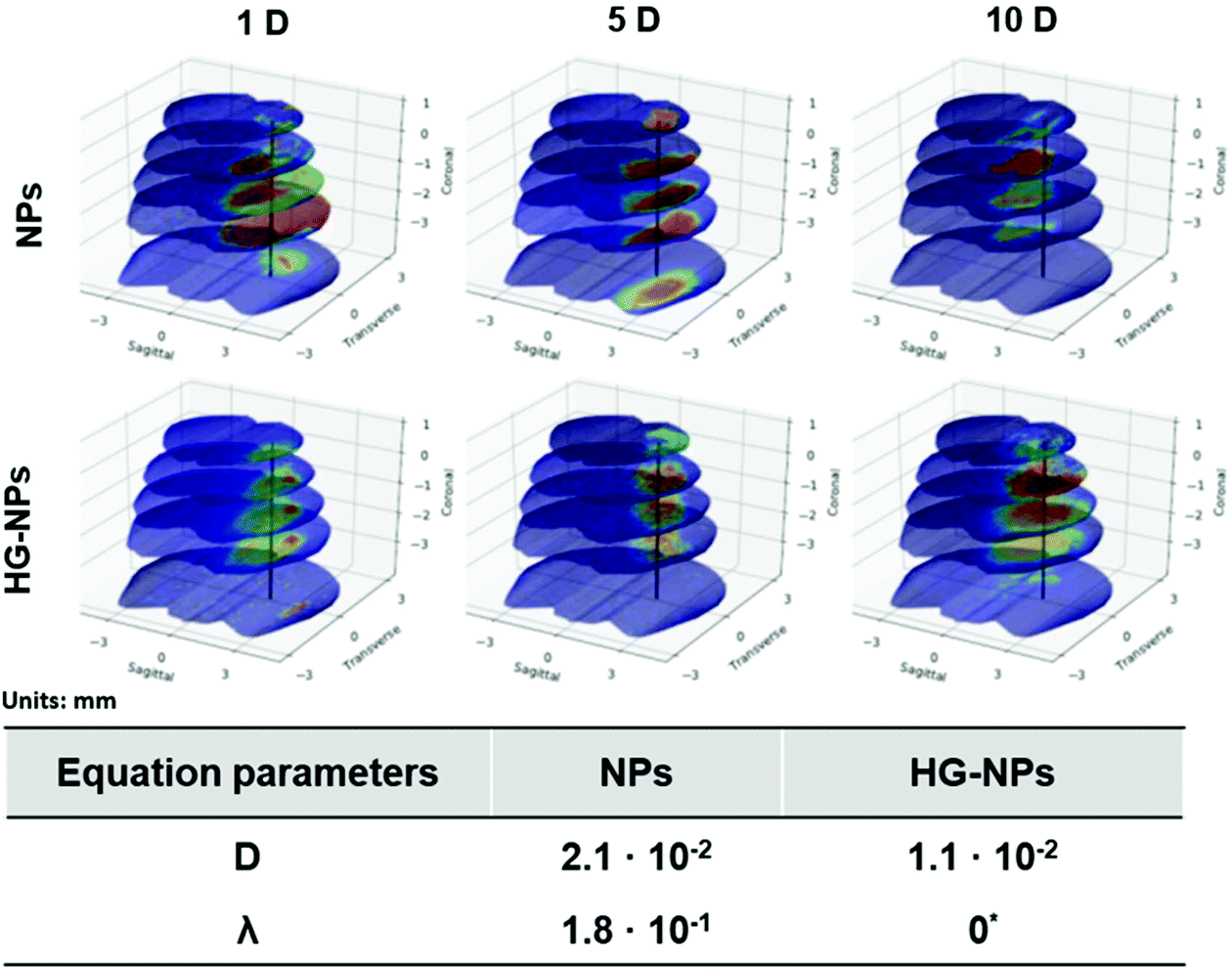 | ||
| Fig. 8 Mathematical modeling of the NPs intensity profile in different areas of the brain of tumor-bearing mice (units are in mm). Estimates for the diffusion (D) and uptake (λ) parameters for the HG–NPs and NPs groups are summarized in the table insert. | ||
The diffusion coefficient for the HG–NPs group is approximately half of that obtained for the free NPs group. This supports the hypothesis that the HG aids in the retention of the NPs to the site of injection. Moreover, we note that the uptake term (λ) for the HG–NPs group is negligible when compared to that corresponding to the free NPs group. Again, this is consistent with the hypothesis that HG enhances localization of NPs to the target site. The reconstruction of the spatiotemporal evolution of NPs and HG–NPs from the injection site is represented in Fig. 8. In accordance with the experimental data, the signal from NPs tends to decrease over time. HG–NPs showed an opposite trend, characterized by an increasing signal intensity over time, which stems from a more controlled NPs release from the HG, resulting in higher retention.
Discussion
Overcoming the restrictions posed by the BBB on systemic drug delivery remains a major challenge in GBM treatment. Therefore, intratumoral drug delivery has emerged as a viable alternative to systemic chemotherapy because of its potential to maximize drug concentration at the site of interest while minimizing systemic side effects.10,29 However, rapid wash-out of drug caused by increased tumor interstitial fluid pressure (IFP) remains a significant obstacle to achieving benefits from intratumoral delivery.62,63 In GBM, increased vascular permeability, together with high resistance to fluid flow in the interstitial space, results in a non-uniform pressure gradient that ultimately leads to rapid drug migration from the tumor towards the surrounding healthy tissue, where the IFP approximates 0 mmHg.64,65 Therefore, this study aimed to test the hypothesis that an intratumoral drug delivery strategy comprised of a combination of thermoresponsive HG and NPs, would reduce drug dispersion from the tumor bulk and result in enhanced delivery, retention and coverage of an intratumorally delivered model drug, BODIPY.In previous studies, examination of drug distribution after CED to GBM showed a consistent pattern of drug migration from the tumor bulk to the surrounding tissues and a considerable reduction in tumor coverage within 1 d after administration.53 Saucier-Sawyer and co-workers observed NP migration towards the peripheral regions of the tumor 1 d after intratumoral administration regardless of tumor stage.53 Similarly, Yang et al. found extensive leakage of liposomal doxorubicin (Doxil®) after intratumoral or peritumoral infusion via CED as early as 1 h after delivery.20 Findings also show more evident drug clearance in the case of intratumoral infusion, while peritumoral infusion resulted in more effective tumor coverage. These observations suggest that intratumoral pressure is principally responsible for drug elimination after CED administration, and that alternative strategies to increase intratumoral confinement of therapeutic agents are warranted.
In this study, epifluorescence examination of brains receiving only NPs demonstrated that the amount of NP wash-out was larger in tumor-bearing mice (Fig. 3) compared to tumor-free controls (Fig. S6†), suggesting that the tumor might contribute to enhanced clearance either by damaging the integrity of the BBB, or by generating high interstitial pressure which facilitates particle removal.10,66,67 Microscopic examination via fluorescence microscopy of tumor-bearing brain sections showed NPs migration towards more superficial areas of the brain (i.e. along the injection pattern), at timepoints as early as 1 d after administration (Fig. 4). This phenomenon was not observed in HG–NPs-injected mice, which showed NPs principally confined to the injection area at the tumor site. Of note, in GBM-bearing mice, significantly higher retention was achieved with HG–NPs at 5 and 10 d post-injection, compared to free NPs (Fig. 4A). Our findings are similar to those of Yang and co-workers wherein NPs were found to undergo migration to the brain periphery after intratumoral injection.20 In our study, embedding NPs in a dense hydrogel matrix seemed to counteract or reduce the effect of tumor IFP, improving the residence time of NPs in the tumor.
Bastiancich and co-workers reported enhanced survival of GBM-bearing mice after local treatment with a gemcitabine-loaded polymer HG.68 Of note, significant survival differences between free drug and HG-embedded drug was achieved mainly with intratumoral administration, confirming the beneficial effect of HGs in retaining treatments in the tumor bulk by reducing the detrimental effect of IFP.
While HGs and NPs have been mainly proposed as strategies to facilitate drug delivery to GBM, they also allow for expansion of the portfolio of drugs that can be applied to GBM.69 Due to low affinity with the BBB, drugs such as paclitaxel (PTX), doxorubicin, camptothecin (CPT), and cisplatin are not clinically applied in GBM,70,71 while the brain accumulation profile of temozolomide (TMZ), routinely used in GBM, is limited to approximately 20% of the systemic dose.8 Consequently, photo-polymerizable HGs have been used to facilitate intracranial delivery of PTX72 and TMZ73 after tumor resection, achieving higher local doses, long-term survival, and reduced adverse effects. Moreover, combining HG with NPs may facilitate combinatorial treatments consisting of water-soluble and non-soluble drugs.74,75
Analysis of NPs and HG–NPs distribution in the sagittal direction corroborated higher confinement of the model drug (BODIPY) at the injection site when formulated in the HG–NPs platform (Fig. 5), suggesting that the hydrogel may also result in a more sustained release of drug from the overall system. Interestingly, the HG delayed, but did not limit NP distribution. Analysis of tumor co-localization confirmed preferential tumor accumulation and retention of BODIPY in the HG–NPs group (Fig. 6). At 1 d post-injection, HG–NPs (Fig. 6C) were found at tumor periphery and in more than 58% of the tumor area near the injection site. In contrast, free NPs were found at anterior tumor border (∼25%), which might be useful for regional targeting of the invasive tumor front in conjunction with HG–NPs that maximize local drug delivery in the tumor core. Concurrent with NPs, BODIPY was localized to a higher extent in the tumor core by HG–NPs, reaching ∼22% co-localization in the tumor center (as compared to an 8% coverage achieved with free NPs). These findings are in agreement with previous reports that show preferential localization of NPs to the peri-tumor area due to NP elimination from the tumor bulk.12,53 Importantly, at the long-term timepoint of 10 d after administration, a more comprehensive coverage of the tumor was achieved with the HG–NPs delivery system, both near the injection site and in the tumor periphery, while tumor coverage by free NPs was limited. At the 10 d timepoint, coverage by BODIPY remained higher in the HG–NPs group, reaching ∼35% at the tumor periphery (compared to 10% for free NPs). These findings mimic those of Shen et al., who recently demonstrated enhanced tumor penetration by NPs when delivered locally with an injectable HG in a flank tumor model of breast cancer.38 Their findings showed that the hydrogel effectively retained NPs, gradually releasing them into the tumor/peri-tumor environment. Using an FEM model to determine the transport characteristic of intratumorally-administered NPs and HG–NPs, we observed a twice higher diffusion coefficient and a non-negligible uptake coefficient for the free NPs, indicating that the HG retained the NPs and reduced their transport. This is compatible with the temporal evolution of NP distribution in tumor-bearing mice, as shown in Fig. 8. Lastly, high-magnification confocal analysis in selected tumor areas showed intratumoral accumulation of NPs in both groups (Fig. 7), suggesting that intact NPs are released from the HG matrix, which subsequently undergo internalization by GBM cells.
Taken together, our findings suggest that embedding NPs in a hydrated HG matrix contributes to their retention and confinement in the tumor area, without limiting their mobility and their penetration into tumor tissue.
Conclusion
Our results suggest that the combination of NPs with HGs has great potential for the treatment of GBM. The enhanced confinement of NPs and BODIPY provided by the HG is expected to be beneficial in GBM to address the remaining cells at the tumor margins without affecting the normal brain, potentially reducing recurrences and toxicity. Moreover, the complexity of GBM could be better addressed with multi-modal drug delivery systems, in which the HG itself could be used as a secondary delivery carrier in combination with NPs or CED to enhance drug exposure of the advancing tumor front. Given that the majority of GBM patients undergo at least partial tumor resection, future work will consist of evaluating transport and treatment efficacy in a resection model.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been supported in part by the National Science Foundation grant DMS-1930583 (ZW, VC), the National Institutes of Health (NIH) grants 1U01CA196403 (ZW, VC), 1U01CA213759 (ZW, VC), 1R01CA226537 (ZW, VC), 1R01CA222007 (ZW, VC), U54CA210181 (ZW, VC), R01NS091251 (RCR), R01CA136808 (RCR), the European Union's Horizon 2020 research and innovation programme, Marie Sklodowska-Curie grant agreement no. 658665 (CM). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.References
- J. R. Schneider, N. V. Patel, K. Kwan and J. A. Boockvar, Neurosurgery, 2018, 83, E200 CrossRef.
- A. Giese, R. Bjerkvig, M. E. Berens and M. Westphal, J. Clin. Oncol., 2003, 21, 1624–1636 CrossRef CAS.
- A. Vehlow and N. Cordes, Biochim. Biophys. Acta, Rev. Cancer, 2013, 1836, 236–244 CrossRef CAS.
- A. M. Mikheev, S. A. Mikheeva, M. Tokita, L. J. Severs and R. C. Rostomily, Oncotarget, 2017, 8, 107716–107729 CrossRef.
- A. Kumar, E. A. Boyle, M. Tokita, A. M. Mikheev, M. C. Sanger, E. Girard, J. R. Silber, L. F. Gonzalez-Cuyar, J. B. Hiatt, A. Adey, C. Lee, J. O. Kitzman, D. E. Born, D. L. Silbergeld, J. M. Olson, R. C. Rostomily and J. Shendure, Genome Biol., 2014, 15, 530 CrossRef.
- R. L. Siegel, K. D. Miller and H. Jemal, CA Cancer J. Clin., 2017, 67, 7–30 CrossRef.
- R. Stupp, S. Taillibert, A. Kanner, W. Read, D. M. Steinberg, B. Lhermitte, S. Toms, A. Idbaih, M. S. Ahluwalia, K. Fink, F. Di Meco, F. Lieberman, J.-J. Zhu, G. Stragliotto, D. D. Tran, S. Brem, A. F. Hottinger, E. D. Kirson, G. Lavy-Shahaf, U. Weinberg, C.-Y. Kim, S.-H. Paek, G. Nicholas, J. Bruna, H. Hirte, M. Weller, Y. Palti, M. E. Hegi and Z. Ram, J. Am. Med. Assoc., 2017, 318, 2306 CrossRef CAS.
- S. Ostermann, Clin. Cancer Res., 2004, 10, 3728–3736 CrossRef CAS.
- M. M. Patel and B. M. Patel, CNS Drugs, 2017, 31, 109–133 CrossRef CAS.
- J. N. Sarkaria, L. S. Hu, I. F. Parney, D. H. Pafundi, D. H. Brinkmann, N. N. Laack, C. Giannini, T. C. Burns, S. H. Kizilbash, J. K. Laramy, K. R. Swanson, T. J. Kaufmann, P. D. Brown, N. Y. R. Agar, E. Galanis, J. C. Buckner and W. F. Elmquist, Neuro-Oncology, 2018, 20, 184–191 CrossRef CAS.
- A. B. Fleming, K. Haverstick and W. M. Saltzman, Bioconjugate Chem., 2004, 15, 1364–1375 CrossRef CAS.
- A. Gaudin, E. Song, A. R. King, J. K. Saucier-Sawyer, R. Bindra, D. Desmaële, P. Couvreur and W. M. Saltzman, Biomaterials, 2016, 105, 136–144 CrossRef CAS.
- A. B. Fleming and W. M. Saltzman, Clin. Pharmacokinet., 2002, 41, 403–419 CrossRef CAS.
- L. S. Ashby, K. A. Smith and B. Stea, World J. Surg. Oncol., 2016, 14, 225 CrossRef.
- M. Vogelbaum and A. Healy, Surg. Neurol. Int., 2015, 6, 59 CrossRef.
- A. Bregy, A. H. Shah, M. V. Diaz, H. E. Pierce, P. L. Ames, D. Diaz and R. J. Komotar, Expert Rev. Anticancer Ther., 2013, 13, 1453–1461 CrossRef CAS.
- P. Szyszka, J. S. Stierle, S. Biergans and C. G. Galizia, PLoS One, 2012, 7, e36096 CrossRef CAS.
- A. I. Mehta, A. Linninger, M. S. Lesniak and H. H. Engelhard, J. Neurooncol., 2015, 125, 1–7 CrossRef CAS.
- M. Yoshida, S. Yamaguchi, Y. Ishi, S. Endo, H. Motegi, H. Kobayashi, K. Asaoka, Y. Kamoshima, S. Terasaka and K. Houkin, No Shinkei Geka, 2015, 43, 603–610 Search PubMed.
- X. Yang, R. Saito, T. Nakamura, R. Zhang, Y. Sonoda, T. Kumabe, J. Forsayeth, K. Bankiewicz and T. Tominaga, Drug Delivery, 2016, 23, 771–776 CrossRef CAS.
- A. Schnyder and J. Huwyler, NeuroRX, 2005, 2, 99–107 CrossRef.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–515 CrossRef CAS.
- Z. Shen, T. Liu, Z. Yang, Z. Zhou, W. Tang, W. Fan, Y. Liu, J. Mu, L. Li, V. I. Bregadze, S. K. Mandal, A. A. Druzina, Z. Wei, X. Qiu, A. Wu and X. Chen, Biomaterials, 2020, 235, 119783 CrossRef CAS.
- V. Segura-Ibarra, F. E. Cara, S. Wu, D. A. Iruegas-Nunez, S. Wang, M. Ferrari, A. Ziemys, M. Valderrabano and E. Blanco, J. Controlled Release, 2017, 262, 18–27 CrossRef CAS.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS.
- O. F. Turkoglu, H. Eroglu, O. Gurcan, E. Bodur, M. F. Sargon, L. Öner and E. Beskonakli, Br. J. Neurosurg., 2010, 24, 578–583 CrossRef.
- Y. Wang, Z. Jiang, B. Yuan, Y. Tian, L. Xiang, Y. Li, Y. Yang, J. Li and A. Wu, Biomater. Sci., 2019, 7, 4748–4757 RSC.
- Z. Shen, T. Liu, Y. Li, J. Lau, Z. Yang, W. Fan, Z. Zhou, C. Shi, C. Ke, V. I. Bregadze, S. K. Mandal, Y. Liu, Z. Li, T. Xue, G. Zhu, J. Munasinghe, G. Niu, A. Wu and X. Chen, ACS Nano, 2018, 12, 11355–11365 CrossRef CAS.
- C. Bastiancich, P. Danhier, V. Préat and F. Danhier, J. Controlled Release, 2016, 243, 29–42 CrossRef CAS.
- S. H. Ranganath, I. Kee, W. B. Krantz, P. K.-H. Chow and C.-H. Wang, Pharm. Res., 2009, 26, 2101–2114 CrossRef CAS.
- C. Gong, T. Qi, X. Wei, Y. Qu, Q. Wu, F. Luo and Z. Qian, Curr. Med. Chem., 2012, 20, 79–94 CrossRef.
- P. Menei, M.-C. Venier, E. Gamelin, J.-P. Saint-André, G. Hayek, E. Jadaud, D. Fournier, P. Mercier, G. Guy and J.-P. Benoit, Cancer, 1999, 86, 325–330 CrossRef CAS.
- M. Boffito, E. Gioffredi, V. Chiono, S. Calzone, E. Ranzato, S. Martinotti and G. Ciardelli, Polym. Int., 2016, 65, 756–769 CrossRef CAS.
- Y. Jung, W. Park, H. Park, D.-K. Lee and K. Na, Carbohydr. Polym., 2017, 156, 403–408 CrossRef CAS.
- S. Kim, S. K. Nishimoto, J. D. Bumgardner, W. O. Haggard, M. W. Gaber and Y. Yang, Biomaterials, 2010, 31, 4157–4166 CrossRef CAS.
- B. Tyler, K. D. Fowers, K. W. Li, V. R. Recinos, J. M. Caplan, A. Hdeib, R. Grossman, L. Basaldella, K. Bekelis, G. Pradilla, F. Legnani and H. Brem, J. Neurosurg., 2010, 113, 210–217 CAS.
- Y. Xu, M. Shen, Y. Sun, P. Gao and Y. Duan, J. Nanosci. Nanotechnol., 2016, 16, 12288–12298 CrossRef CAS.
- H. Shen, Q. Gao, Q. Ye, S. Yang, Y. Wu, Q. Huang, X. Wang and Z. Sun, Int. J. Nanomed., 2018, 13, 7409–7426 CrossRef CAS.
- W. Gao, Y. Zhang, Q. Zhang and L. Zhang, Ann. Biomed. Eng., 2016, 44, 2049–2061 CrossRef.
- M. D. Baumann, C. E. Kang, C. H. Tator and M. S. Shoichet, Biomaterials, 2010, 31, 7631–7639 CrossRef CAS.
- G. Dumortier, J. L. Grossiord, F. Agnely and J. C. Chaumeil, Pharm. Res., 2006, 23, 2709–2728 CrossRef CAS.
- C. Mattu, M. Boffito, S. Sartori, E. Ranzato, E. Bernardi, M. P. Sassi, A. M. Di Rienzo and G. Ciardelli, J. Nanopart. Res., 2012, 14, 1306 CrossRef.
- C. Mattu, R. M. Pabari, M. Boffito, S. Sartori, G. Ciardelli and Z. Ramtoola, Eur. J. Pharm. Biopharm., 2013, 85, 463–472 CrossRef CAS.
- C. Iodice, A. Cervadoro, A. Palange, J. Key, S. Aryal, M. R. Ramirez, C. Mattu, G. Ciardelli, B. E. O'Neill and P. Decuzzi, Opt. Lasers Eng., 2016, 76, 74–81 CrossRef.
- C. Mattu, G. Brachi, L. Menichetti, A. Flori, P. Armanetti, E. Ranzato, S. Martinotti, S. Nizzero, M. Ferrari and G. Ciardelli, Acta Biomater., 2018, 80, 341–351 CrossRef CAS.
- H. Wakimoto, S. Kesari, C. J. Farrell, W. T. Curry, C. Zaupa, M. Aghi, T. Kuroda, A. Stemmer-Rachamimov, K. Shah, T.-C. Liu, D. S. Jeyaretna, J. Debasitis, J. Pruszak, R. L. Martuza and S. D. Rabkin, Cancer Res., 2009, 69, 3472–3481 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- A. Barriga-Rivera and G. J. Suaning, in 2011 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, IEEE, 2011, pp. 4860–4863.
- M. Abdolhoseini, M. G. Kluge, F. R. Walker and S. J. Johnson, Sci. Rep., 2019, 9, 4551 CrossRef.
- N. Otsu, IEEE Trans. Syst. Man Cybern., 1979, 9, 62–66 Search PubMed.
- J. Fan and F. Zhao, Acta Electron. Sin, 2007, 35, 1398–1402 Search PubMed.
- Y.-E. Seo, H.-W. Suh, R. Bahal, A. Josowitz, J. Zhang, E. Song, J. Cui, S. Noorbakhsh, C. Jackson, T. Bu, A. Piotrowski-Daspit, R. Bindra and W. M. Saltzman, Biomaterials, 2019, 201, 87–98 CrossRef CAS.
- J. K. Saucier-Sawyer, Y.-E. Seo, A. Gaudin, E. Quijano, E. Song, A. J. Sawyer, Y. Deng, A. Huttner and W. M. Saltzman, J. Controlled Release, 2016, 232, 103–112 CrossRef CAS.
- P. Dogra, J. D. Butner, Y. Chuang, S. Caserta, S. Goel, C. J. Brinker, V. Cristini and Z. Wang, Biomed. Microdevices, 2019, 21, 40 CrossRef.
- P. Dogra, N. L. Adolphi, Z. Wang, Y.-S. Lin, K. S. Butler, P. N. Durfee, J. G. Croissant, A. Noureddine, E. N. Coker, E. L. Bearer, V. Cristini and C. J. Brinker, Nat. Commun., 2018, 9, 4551 CrossRef.
- Y. Liu, S. Shah and J. Tan, Rev. Nanosci. Nanotechnol., 2012, 1, 66–83 CrossRef CAS.
- V. Cristini, E. J. Koay and Z. Wang, An Introduction to Physical Oncology, Chapman and Hall/CRC, Boca Raton, FL, CRC Press, [2017] | Series: Chapman & Hall/CRC mathematical and computational biology, 2017 Search PubMed.
- J. Sneyd, B. T. Wetton, A. C. Charles and M. J. Sanderson, Am. J. Physiol.: Cell Physiol., 1995, 268, C1537–C1545 CrossRef CAS.
- R. B. Bird, W. E. Stewart and E. N. Lightfoot, AIChE J., 1961, 7, 5J–6J Search PubMed.
- S. Brenner and R. Scott, The mathematical theory of finite element methods, Springer-Verlag, New York, 2008, vol. 15 Search PubMed.
- M. Boffito, E. Gioffredi, V. Chiono, S. Calzone, E. Ranzato, S. Martinotti and G. Ciardelli, Polym. Int., 2016, 65, 756–769 CrossRef CAS.
- Y. Esquenazi, V. P. Lo and K. Lee, J. Intensive Care Med., 2017, 32, 15–24 CrossRef.
- Y. Boucher, H. Salehi, B. Witwer, G. Harsh and R. Jain, Br. J. Cancer, 1997, 75, 829–836 CrossRef CAS.
- G. A. Rosenberg, W. T. Kyner and E. Estrada, Am. J. Physiol., 1980, 238, F42–F49 CAS.
- Y. Navalitloha, Neuro-Oncology, 2006, 8, 227–233 CrossRef.
- M. R. Sage and A. J. Wilson, Am. J. Neuroradiol., 1994, 15, 601–622 CAS.
- D. F. Quail and J. A. Joyce, Cancer Cell, 2017, 31, 326–341 CrossRef CAS.
- C. Bastiancich, J. Bianco, K. Vanvarenberg, B. Ucakar, N. Joudiou, B. Gallez, G. Bastiat, F. Lagarce, V. Préat and F. Danhier, J. Controlled Release, 2017, 264, 45–54 CrossRef CAS.
- T. Ozeki, D. Kaneko, K. Hashizawa, Y. Imai, T. Tagami and H. Okada, Int. J. Pharm., 2012, 427, 299–304 CrossRef CAS.
- L. Nam, C. Coll, L. Erthal, C. de la Torre, D. Serrano, R. Martínez-Máñez, M. Santos-Martínez and E. Ruiz-Hernández, Materials, 2018, 11, 779 CrossRef.
- J. Zhou, T. R. Patel, R. W. Sirianni, G. Strohbehn, M.-Q. Zheng, N. Duong, T. Schafbauer, A. J. Huttner, Y. Huang, R. E. Carson, Y. Zhang, D. J. Sullivan, J. M. Piepmeier and W. M. Saltzman, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11751–11756 CrossRef CAS.
- M. Zhao, F. Danhier, C. Bastiancich, N. Joudiou, L. P. Ganipineni, N. Tsakiris, B. Gallez, A. des Rieux, A. Jankovski, J. Bianco and V. Préat, Int. J. Pharm., 2018, 548, 522–529 CrossRef CAS.
- T. Fourniols, L. D. Randolph, A. Staub, K. Vanvarenberg, J. G. Leprince, V. Préat, A. des Rieux and F. Danhier, J. Controlled Release, 2015, 210, 95–104 CrossRef CAS.
- W. Wang, L. Deng, S. Xu, X. Zhao, N. Lv, G. Zhang, N. Gu, R. Hu, J. Zhang, J. Liu and A. Dong, J. Mater. Chem. B, 2013, 1, 552–563 RSC.
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Nat. Commun., 2015, 6, 6295 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr05053a |
| ‡ Current affiliation: University of Washington, School of Pharmacy, Seattle WA; DXT Pharmaceuticals, San Mateo, CA. |
| § Senior authors. |
| This journal is © The Royal Society of Chemistry 2020 |