
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring molecular interactions between microporous polymers in high performance mixed matrix membranes for gas separations†
Cher Hon
Lau
 *a,
Kristina
Konstas
b,
Cara M.
Doherty
b,
Stefan J. D.
Smith
b,
Rujing
Hou
bc,
Huanting
Wang
c,
Mariolino
Carta
d,
Heewook
Yoon
e,
Jaesung
Park
e,
Benny D.
Freeman
e,
Richard
Malpass-Evans
f,
Elsa
Lasseuguette
a,
Maria-Chiara
Ferrari
a,
Neil B.
McKeown
*f and
Matthew R.
Hill
*bc
*a,
Kristina
Konstas
b,
Cara M.
Doherty
b,
Stefan J. D.
Smith
b,
Rujing
Hou
bc,
Huanting
Wang
c,
Mariolino
Carta
d,
Heewook
Yoon
e,
Jaesung
Park
e,
Benny D.
Freeman
e,
Richard
Malpass-Evans
f,
Elsa
Lasseuguette
a,
Maria-Chiara
Ferrari
a,
Neil B.
McKeown
*f and
Matthew R.
Hill
*bc
aSchool of Engineering, University of Edinburgh, Robert Stevenson Road, Edinburgh, EH9 3FB, UK. E-mail: cherhon.lau@ed.ac.uk
bCSIRO, Bag 10, Clayton South, VIC 3169, Australia. E-mail: matthew.hill@csiro.au
cDepartment of Chemical Engineering, Monash University, Clayton, VIC 3169, Australia
dDepartment of Chemistry, College of Science, Grove Building, Singleton Park, Swansea University, Swansea, SA2 8PP, UK
eDepartment of Chemical Engineering, University of Texas, Austin, TX78758, USA
fEastChem, School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: neil.mckeown@ed.ac.uk
First published on 7th August 2020
Abstract
Membranes are crucial to lowering the huge energy costs of chemical separations. Whilst some promising polymers demonstrate excellent transport properties, problems of plasticisation and physical aging due to mobile polymer chains, amongst others, prevent their exploitation in membranes for industrial separations. Here we reveal that molecular interactions between a polymer of intrinsic microporosity (PIM) matrix and a porous aromatic framework additive (PAF-1) can simultaneously address plasticisation and physical aging whilst also increasing gas transport selectivity. Extensive spectroscopic characterisation and control experiments involving two near-identical PIMs, one with methyl groups (PIM-EA(Me2)-TB) and one without (PIM-EA(H2)-TB), directly confirm the key molecular interaction as the adsoprtion of methyl groups from the PIM matrix into the nanopores of the PAF. This interaction reduced physical aging by 50%, suppressed polymer chain mobilities at high pressure and increased H2 selectivity over larger gases such as CH4 and N2.
Introduction
Chemical separations are a major consumer of energy on a global scale, with estimates that they demand as much as 15% of global energy production.1 As membranes operate in a continuous fashion at ambient conditions, energy requirements could be lowered by up to 90%.2,3 However, for this potential to be realized, significant challenges must be overcome. Membrane materials must exhibit remarkable inherent transport behavior and maintain performance under operating conditions. Polymers are attractive candidate membrane materials due to their useful transport behavior coupled with ready processibility. In fact, polymer membranes have been actively deployed for gas4 and liquid5 separations for over 40 years, while hundreds of new polymers with superior transport behavior have been reported.Despite advances in polymer science, challenges beyond their inherent transport behavior have limited the implementation of new polymers into commercial modules. For example, regardless of film thickness, polymers with intrinsic microporosity (PIMs) are susceptible to CO2-induced plasticisation and physical aging6–8 and possess insufficient gas selectivity. Plasticisation arises from sorbed molecules mobilising polymer chains, reducing gas selectivities.9 Due to dual mode sorption, it can also cause gas permeability to reduce before increasing at higher pressures.10 Meanwhile, physical aging occurs naturally as polymer chains converge, reducing gas permeabilities.11
A strategy to reduce these effects is to rigidify polymer chains. This has been achieved with mixed matrix membranes (MMMs) comprising microporous additives such as metal organic frameworks,12 porous aromatic frameworks (PAFs),13 covalent organic frameworks,14 metal organic polyhedra,15 and hypercrosslinked polymers (HCPs).16,17 Amongst these additives, PAFs can best control physical aging of PIMs where PIM/PAF composites only lose 20% of their CO2 permeabilities.18 Meanwhile PIM composites comprising other microporous additives (MOFs,19 HCPs,17 fumed silica19) lose up to 80% of their CO2 permeabilities. To date, it has been demonstrated that such approaches can only address a single limitation of polymer membrane.13,15,16,19,20 Moreover, this approach may reduce gas selectivities when incompatible additives are used.
Here we show that plasticisation, physical aging and poor gas selectivity of polymer membranes can be simultaneously addressed in a single MMM by tailoring interactions between additives and specific sites of the polymer matrix. The polymer matrices deployed in this work were PIMs that contained Tröger base (TB) and ethanoanthracene (EA), with (PIM-EA(Me2)-TB)22 and without (PIM-EA(H2)-TB)23 methyl pendent groups at its two bridgehead positions (Fig. 1). Due to the better capability to control physical aging of PIMs,18 PAF-1, a microporous network polymer comprised of tetrahedral carbon atoms linked by biphenyl groups with 2 pore sizes centred at 0.2 and 1.3 nm (ref. 13 and 21) was deployed as an additive. The average diameter of PAF-1 nanoparticles deployed here in this work were 200 nm (Fig. S1–4†). The non-methylated PIM-EA(H2)-TB served as a control to demonstrate the impact of a trivial chemical structural difference in the polymer matrix on additive interactions and compatibility. This led to contrasting PIM chain mobility, aging mechanisms and gas transport behaviour at various operating conditions.
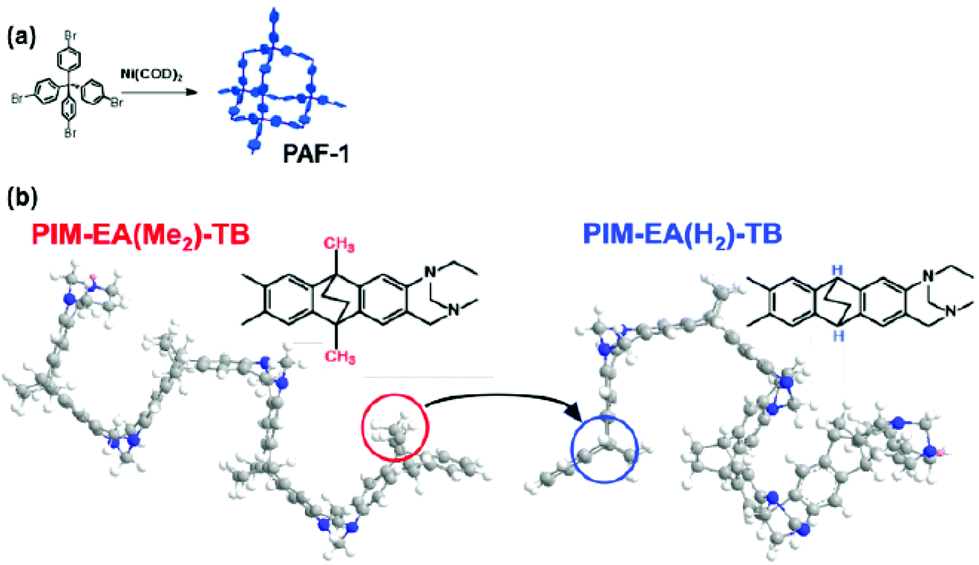 | ||
| Fig. 1 (a) Chemical structure of PAF-1, a highly microporous polymer first reported in 2009.21 (b) The methyl groups in PIM-EA(Me2)-TB22 are absent in the new polymer, PIM-EA(H2)-TB, which results in reduction of inter-chain distances (d-space), alteration of preferred interaction sites on PIM chains with PAF-1, and impacting chain mobility during physical aging and plasticisation. | ||
Results and discussion
Impact of methyl groups on interactions with PAF-1 and physical aging
T 1 relaxation times of carbon atoms in polytrimethylsilylpropyne24 and PIM-125 obtained from solid state 13C NMR could be used to describe polymer chain mobility, where relative decreases in T1 relaxation times (w.r.t those of as-cast polymers) infer a greater mobility of carbon atoms, while increments in T1 relaxation times indicate less mobile carbon atoms. Hence, the mobility of carbon atoms in functional groups located on the bulky side chains or flexible points on these polymer chains are excellent indicators of plasticisation and physical aging.25 Based on these observations, here we hypothesise that the key drivers of physical aging in PIMs studied here were; namely, ethylene bridges, TB methylene bridge, methyl group (PIM-EA(Me2)-TB only) and the methine group (PIM-EA(H2)-TB only). This hypothesis was validated here with solid state 13C NMR.The carbon atoms in methyl groups, ethylene bridge, and TB methylene bridge of PIM-EA(Me2)-TB became more mobile as the polymer chains converged during physical aging (Fig. 2). The degree of freedom in these functional groups was as follows: methyl (C9) > TB methylene bridge (C1) > ethylene bridge (C7, C8); indicating that PIM-EA(Me2)-TB chain convergence was primarily driven by the EA methyl groups. PAF-1 preferred to interact with methylated bulky side-chains of polyacetylenes26 and PIM-1.18 However, here, we observed that PAF-1 only appeared to immobilize the EA ethylene and TB methylene bridges in PIM-EA(Me2)-TB, but not the EA methyl groups. This was because the presence of other CH-based functional groups altered the preferred interaction sites between PAF-1 and the polymer matrix. Facing competition from both ethylene and methylene bridges where PAF-1 preferred to interact with and rigidify the EA ethylene bridge, interactions between EA methyl groups and PAF-1 were minimized.
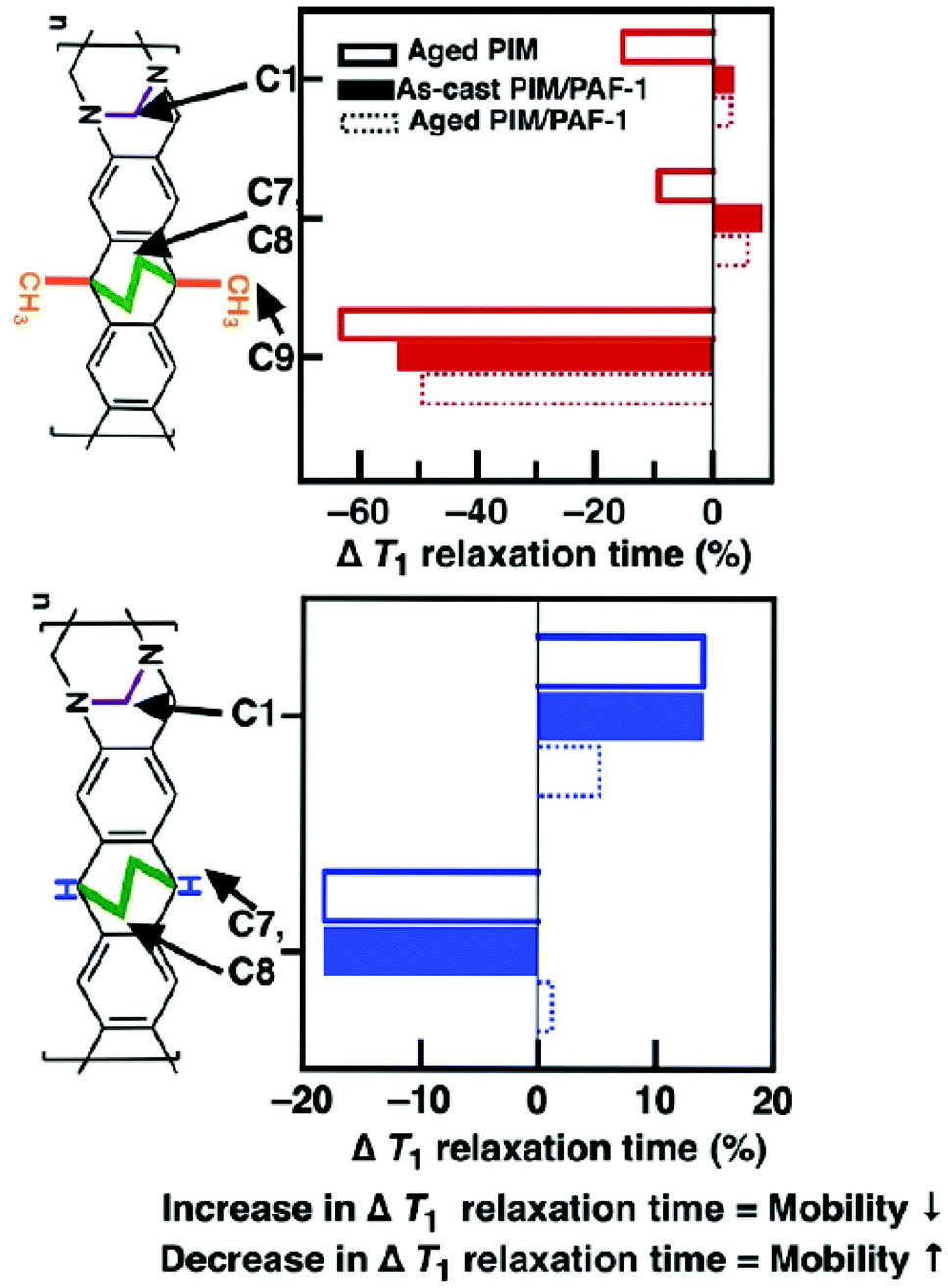 | ||
| Fig. 2 T 1 relaxation times of carbon atoms obtained from 13C solid state NMR spectroscopy correlate to molecular mobility. Relative T1 decrease (w.r.t. as-cast polymer – black line at 0%) correlates to more mobile atoms, and vice versa.24 PAF-1 rigidifies the EA ethylene and TB methylene bridges in PIM-EA(Me2)-TB, while having minimal impact on these bridges in PIM-EA(H2)-TB. T1 values of main chain carbon atoms are shown in Fig. S19, ESI.† | ||
The immobilization of both EA ethylene and TB methylene bridges was achieved through non-bonding physical interactions for example, weak aliphatic–aromatic CH/π interactions, as the NMR spectra of both the neat polymer and nanocomposite film contained similar peak positions (Fig. S20†).
Such interactions were most likely to happen between the ethylene and methylene bridges and the aromatic rings of PAF-1, similar to the stabilization of biomolecules27,28 and organic crystals29via London dispersion forces governing CH/π interactions between aromatic and aliphatic structures.30 The absence of pendant methyl groups in PIM-EA(H2)-TB changed the way these polymer chains aggregated and interacted with PAF-1. As PIM-EA(H2)-TB aged, both the ethylene bridge and methine group on the EA(H2) unit became more mobile, while the TB methylene bridge lost mobility.
Clearly, PIM-EA(H2)-TB chain convergence during aging was driven primarily by the EA(H2) unit; leading to smaller and less free volume (Tables S2 and 3†) that restricted the mobility of TB methylene bridges. Meanwhile, although PAF-1 preferred to interact with both the methine group and ethylene bridges of PIM-EA(H2)-TB, the mobility of these functional groups was retained. Conversely, the mobility of TB methylene bridges in PIM-EA(H2)-TB/PAF-1 nanocomposites was enhanced, most likely due to minimal interactions between this functional group and PAF-1. The negligible influence of PAF-1 on the mobility of a CH-based functional group, possibly due to the inability of PAF-1 to break up the inter-PIM chain interactions in PIM-EA(H2)-TB. This was validated here by characterising the behaviour of PIMs studied here and their interactions with PAF-1 in solution phase. The viscosity of solutions containing 2 wt% PIM-EA(H2)-TB in chloroform was 100% higher than that of PIM-EA(Me2)-TB (Fig. 3). This observed effect is not an artefact of polymer molecular weight where high molecular weight PIM typically increase the viscosity of solutions.31 The molecular weight of PIM-EA(Me2)-TB was 155![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 ∼ 3 times larger than that of PIM-EA(H2)-TB.
800 ∼ 3 times larger than that of PIM-EA(H2)-TB.
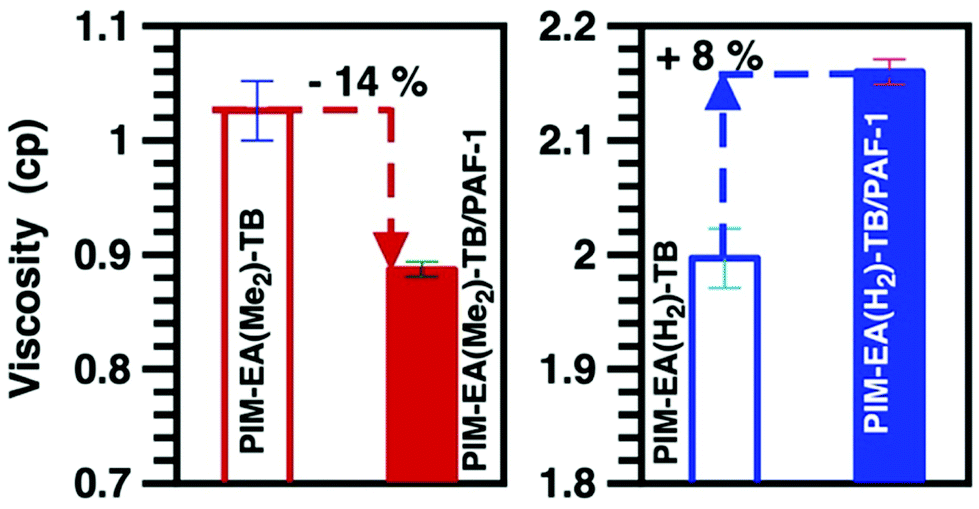 | ||
Fig. 3 Viscosity measurements of solutions containing 2 wt% PIM-EA(Me2)-TB ( ), PIM-EA(Me2)-TB/PAF-1 ( ), PIM-EA(Me2)-TB/PAF-1 ( ), PIM-EA(H2)-TB ( ), PIM-EA(H2)-TB ( ), and PIM-EA(H2)-TB/PAF-1 ( ), and PIM-EA(H2)-TB/PAF-1 ( ). The concentration of PAF-1 in these solutions is 10 wt% w.r.t. to PIM weight. ). The concentration of PAF-1 in these solutions is 10 wt% w.r.t. to PIM weight. | ||
Here the lack of methyl groups promoted inter-chain interactions;32 leading to greater PIM-EA(H2)-TB chain agglomeration. In contrast, for PIM-EA(Me2)-TB, the pendant methyl groups reduced polymer chain interactions thus lowering viscosity. The viscosity of solution mixtures provides insight into the nature of polymer-PAF interactions.18 Typical of systems with non-compatible components, 10 wt% PAF-1 (w.r.t. polymer concentration) increased the solution viscosity of PIM-EA(H2)-TB by 8%. In contrast, PAF-1 reduced the viscosity of PIM-EA(Me2)-TB solutions. This could be ascribed to better interactions between the PAF additive and the PIM chains, possibly due to adsorption of polymer chains on the additive, the threading of polymer chains into additive pores and enhanced porosity within the mixture.18,33 The degree of interaction between PAF-1 and PIMs also impacted on the physicochemical structure of resultant films.
From SAXS/WAXS spectra of neat PIM films, the inter-chain distance between PIM chains was smaller by 30% for PIM-EA(H2)-TB (d-space = 7.7 Å, Fig. 4a) relative to PIM-EA(Me2)-TB (d-space = 11 Å, Fig. 4b). Typical of non-compatible systems,34 PAF-1 enhanced d-spacing in PIM-EA(H2)-TB by 200% (from 7.7 Å to 23.6 Å) (Fig. 4c). This d-space was reduced by 40% to 14.8 Å upon aging. Meanwhile, for the PIM-EA(Me2)-TB/PAF-1 composite, the polymer chains were only propped further apart by 60% (Fig. 4d) relative to the neat film and the largest pore size distributions in PIM-EA(Me2)-TB/PAF-1 were only reduced by 4% after aging. It appeared that PAF-1 caused PIM-EA(H2)-TB to age faster while reducing the physical aging rate of PIM-EA(Me2)-TB. This was also validated with single gas permeation results collected from samples aged naturally (stored in ambient conditions) over 730 days (Fig. S22, ESI†). This significant structural change was reflected in data from positron annihilation lifetime spectroscopy (PALS) (Tables S2 and 3, ESI†). Although the PALS average pore sizes remained relatively stable from aging, there was an overall drop in fractional free volume as the number of large free volume elements decreased while increasing the number of smaller free volume elements. Without PAF-1, there was an overall decrease in size and number of free volume elements with aging. To untangle the effects of physical aging rates due to film thicknesses, we measured the gas permeability loss rates of 15 μm thin and 139 μm thick PIM-EA(Me2)-TB/PAF 1 films (Fig S22, ESI†). The loss in H2 permeabilities across these films of different thicknesses is identical (10–20%), indicating that physical aging occurs at the same rate in PIM-EA(Me2)-TB/PAF-1, regardless of film thickness.
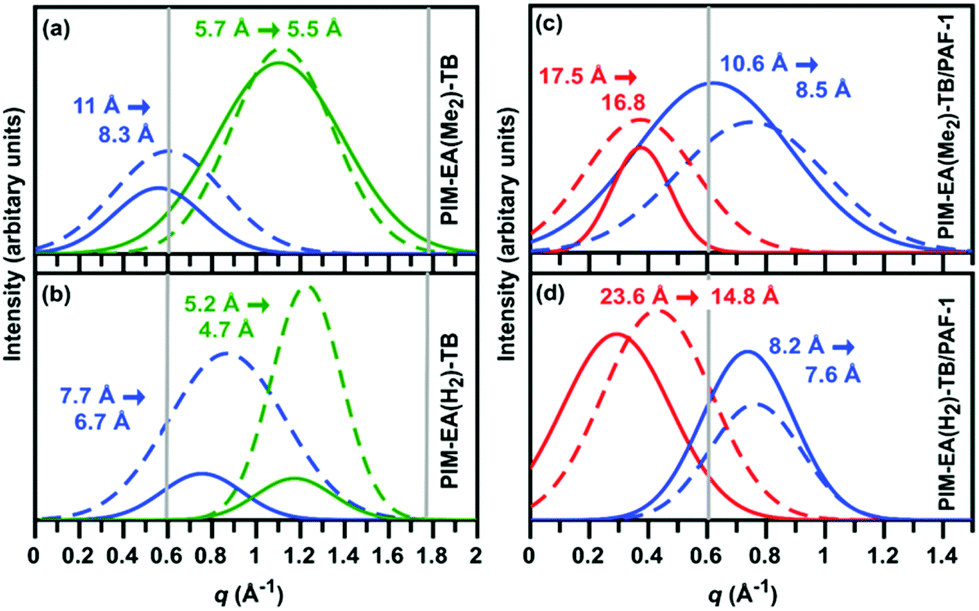 | ||
| Fig. 4 SAXS/WAXS raw data was fitted with a sum of Gaussian plots using MagicPlot to obtain various pore size distributions (as-cast samples – solid lines, aged samples (90 days) – dashed lines): (a) neat film of PIM-EA(Me2)-TB; (b) neat film of PIM-EA(H2)-TB; (c) film of PIM-EA(Me2)-TB/PAF-1 and (d) film of PIM-EA(H2)-TB/PAF-1. | ||
Impact of methyl groups on interactions with PAF-1 and plasticisation
Gas-induced plasticisation (gas pressure increase, gas permeability increase) and dual mode sorption (gas pressure increase, gas permeability decrease) in polymers are best illustrated with a highly sorbing molecule like CO2.8,35 Here, we used both pure and mixed (50:50, stage cut 0.1%) CO2 and CH4 gases over pressures or partial pressures of 2 to 20 atm (Fig. 5). Typical of dual mode sorption36 and widely observed in polymer membranes that were not plasticised significantly by the permeating gas,37 the CO2 permeabilities of PIM-EA(Me2)-TB/PAF-1 and PIM-EA(H2)-TB/PAF-1 were reduced by 2% and 20%, respectively (Fig. 5a and b) as single gas CO2 pressure increased from 2 to 20 bar. Likewise, the mixed gas CO2 permeabilities of both PIM-EA(Me2)-TB/PAF-1 and PIM-EA(H2)-TB/PAF-1 were also reduced by similar amounts at higher CO2 partial pressures. The moderation in CO2-induced swelling for PIM-EA(Me2)-TB was consistent with the greater interaction of this polymer host with the PAF-1 additive where rigidified PIM EA(Me2)-TB chains were less likely to swell. Meanwhile, higher CH4 pressures did not result in any significant changes in single gas CH4 permeabilities (Table S9, ESI†). The main difference here was in the 63 and 39% increase in mixed gas CH4 permeabilites of PIM-EA(Me2)-TB/PAF-1 and PIM-EA(H2)-TB/PAF-1, respectively. The enhancements in mixed gas CH4 permeabilities underpinned an unexpected loss in mixed gas CO2/CH4 selectivities, possibly due to a combination of dual mode sorption effect reducing mixed gas CO2 permeability and moderated swelling of the PIM matrices at high CO2 pressures that enhanced the permeation of larger gases such as CH4 (Fig. 5c–e). Similar to physical aging, where permeation of small gas molecules in PIM-EA(Me2)-TB/PAF-1 nanocomposites were less affected (Fig. S23, ESI†), the impact of CO2 induced swelling and plasticisation favoured the mixed gas permeation of smaller gases (CO2 kinetic diameter 3.3 Å) over larger gases (CH4 kinetic diameter 3.8 Å) as a function of pressure.
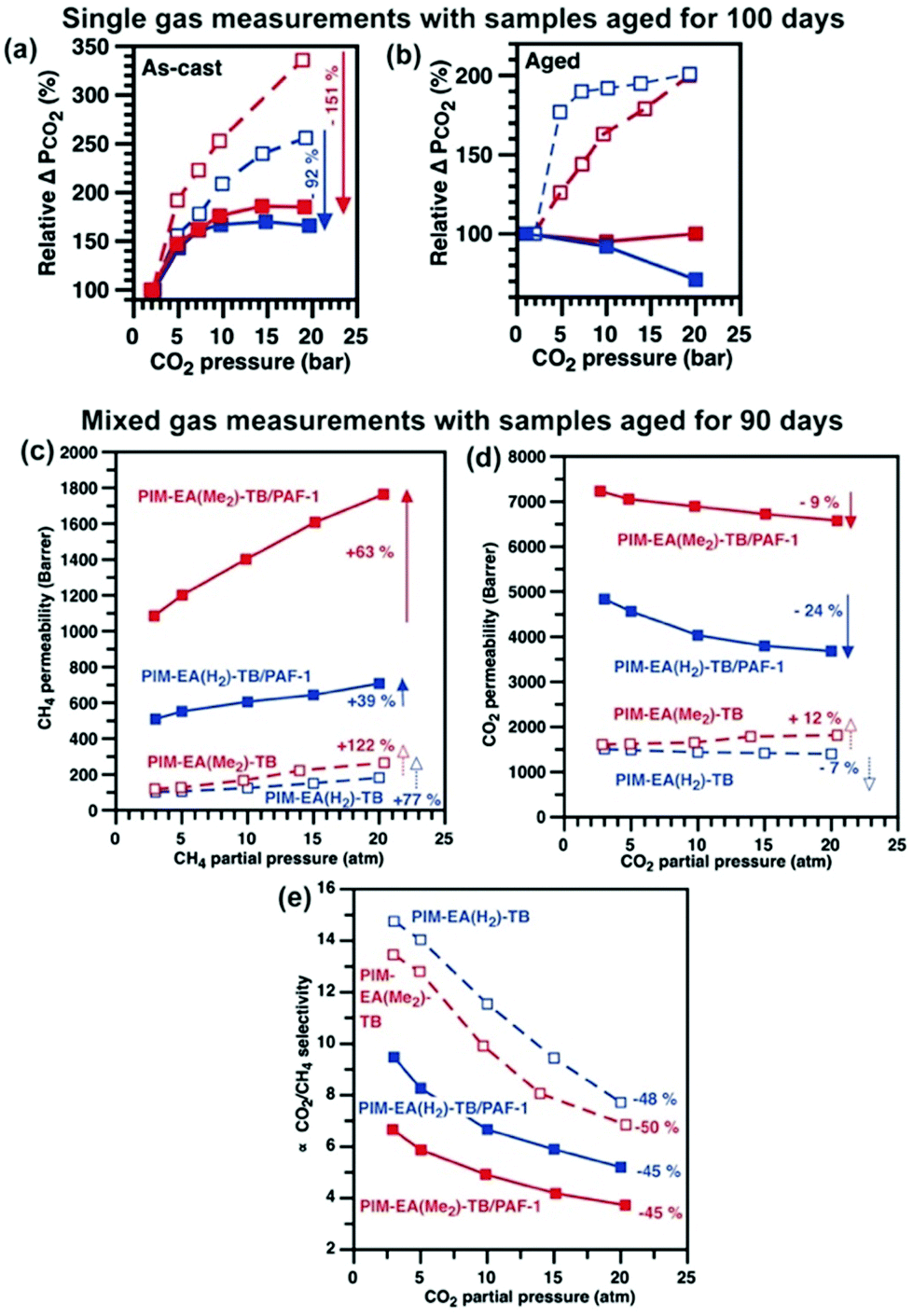 | ||
Fig. 5 The effects of increasing CO2 pressure on CO2 permeabilities of (a) as-cast and (b) aged (100 days) PIM-EA(Me2)-TB ( ), PIM-EA(Me2)-TB/PAF-1 ( ), PIM-EA(Me2)-TB/PAF-1 ( ), PIM-EA(H2)-TB ( ), PIM-EA(H2)-TB ( ), and PIM-EA(H2)-TB/PAF-1 ( ), and PIM-EA(H2)-TB/PAF-1 ( ) membranes are investigated here with CO2. The effects of CO2 plasticization on mixed (c) CH4, (d) CO2 gas permeabilities and (e) CO2/CH4 mixed gas selectivity were characterized using samples that have aged for 90 days, and over a range of CO2 partial pressures at 35 °C with gas mixtures containing 50:50 mol% of CO2:CH4 and a stage cut <0.1. Mixed gas data for plasticization were collected from the Freeman labs at UT, Austin. Lines were drawn to guide the eye, and permeability values are ±5% within standard deviation. ) membranes are investigated here with CO2. The effects of CO2 plasticization on mixed (c) CH4, (d) CO2 gas permeabilities and (e) CO2/CH4 mixed gas selectivity were characterized using samples that have aged for 90 days, and over a range of CO2 partial pressures at 35 °C with gas mixtures containing 50:50 mol% of CO2:CH4 and a stage cut <0.1. Mixed gas data for plasticization were collected from the Freeman labs at UT, Austin. Lines were drawn to guide the eye, and permeability values are ±5% within standard deviation. | ||
Impact of methyl groups on interactions with PAF-1 on gas separations
10 wt% PAF-1 enhanced all gas permeabilities of PIM-EA(H2)-TB by at least 110%, but majorly reduced gas selectivities (Fig. S21†). This was typical of nanocomposites containing components with low compatibility,38 where the bulk polymer phase was not in intimate contact with the dispersed phase.39 As PAF-1 increased the d-space in PIM-EA(H2)-TB by 200%, the Knudsen diffusion of H2, N2, and CH4 were facilitated; leading to drastic gas permeability enhancements. Rapid physical aging of the PIM-EA(H2)-TB/PAF-1 nanocomposites was apparent, consistent with the reduction in d-spacing indicated by SAXS/WAXS.In contrast, the strong interaction between PAF-1 and PIM-EA(Me2)-TB delivered a remarkable increase in both permeability and selectivity, surpassing the 2015 upper bounds for light gas separations including H2/N2, H2/CH4, and O2/N2 reported by Pinnau et al.44 (Fig. 6a–c). 10 wt% PAF-1 enhanced the H2 permeability of PIM-EA(Me2)-TB by 71% while reducing N2 and CH4 permeabilities by 5 and 11%, respectively (Fig. S21†). This contrasting effect of PAF-1 on gas permeation in PIMs with similar chemical structures was attributed to the difference in gas diffusion modes. The 60% increase in PIM-EA(Me2)-TB d-spacing due to blending with PAF-1 was sufficient to support a Knudsen diffusion mechanism for H2 but not for larger gases like N2 and CH4.45 This enhanced H2 separation from CH4 and N2, leading to H2/CH4 separation performances. The compatibility of PIM-EA(Me2)-TB/PAF-1 resulted in selective aging that benefitted the transport of He and H2 gas molecules, similar to other PIM/PAF membranes, where some physical aging drivers remain untethered to PAF-1.18 The excellent H2 permeabilities and H2/CH4 selectivities of aged PIM-EA(Me2)-TB/PAF-1 at realistic operating conditions (Table S10, Fig. S26, ESI†) are ideal for exploiting these mixed matrix membranes for recovering H2 from the off-gas of hydrocracker refineries in high temperature and pressure conditions.46
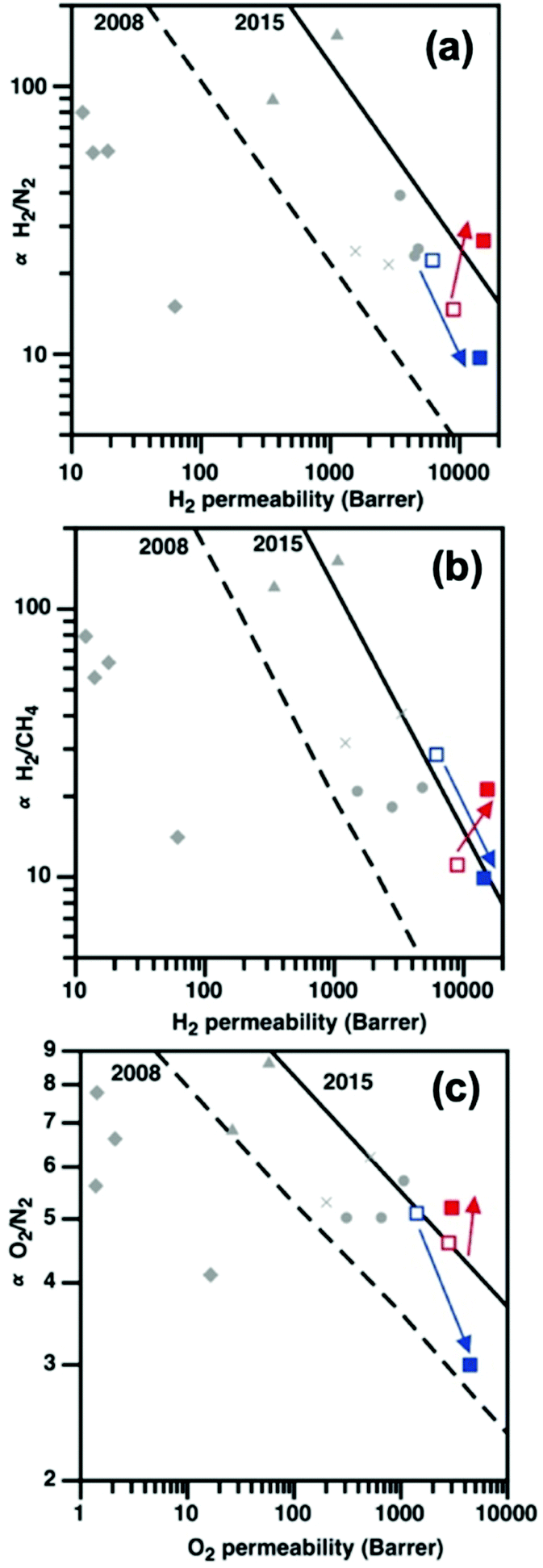 | ||
Fig. 6 The initial impact of PAF-1 incorporation on (a) H2/N2, (b) H2/CH4, and (c) O2/N2 separation for as-cast PIM-EA(Me2)-TB ( ), PIM-EA(Me2)-TB/PAF-1 ( ), PIM-EA(Me2)-TB/PAF-1 ( ), PIM-EA(H2)-TB ( ), PIM-EA(H2)-TB ( ), and PIM-EA(H2)-TB/PAF-1 ( ), and PIM-EA(H2)-TB/PAF-1 ( ) films. Also shown for comparison are data for current state-of-the-art polymers such as triptycene PIMs ( ) films. Also shown for comparison are data for current state-of-the-art polymers such as triptycene PIMs ( ),40 triptycene polyimides ( ),40 triptycene polyimides ( ),41 commercial polymers ( ),41 commercial polymers ( ),42 and other PIMs ( ),42 and other PIMs ( ).43 ).43 | ||
Conclusions
In summary, through a very simple change in the structure of a PIM, we elucidated the mechanism leading to controlling both plasticisation and physical aging in ultrapermeable nanocomposites. The absence of pendant methyl groups resulted in PIM chains that aggregated and prevented fine intercalation with PAF-1 nanoparticles; minimizing the benefits of PAF-1 towards enhancing membrane separation and the effects of aging and rigid polymer chains. Hence, polymer additive compatibility could be manipulated to direct specific molecular-scale interactions between the bulk matrix and the additive at precise locations of the polymer chains; allowing facile design of mixed matrix membranes that age differently. In addition, these ultrapermeable composites demonstrated some form of chain rigidification, even at high pressures and temperatures. PIM/PAF composites have potential for membrane gas separations involving important gases for the global economy, in addition to the described performance of separating H2 from natural gas. These include He mining from natural gases, O2 or N2 enrichment of air and H2 separation from ammonia purge gases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Adrian Hawley at the Australian Synchrotron for setting up the SAXS/WAXS beamline, and Dr Roger Mulder at CSIRO for performing solid state NMR experiments. CHL acknowledges funding support from the University of Edinburgh Chancellor's Fellowship.References
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed
.
- D.-Y. Koh, B. A. McCool, H. W. Deckman and R. P. Lively, Science, 2016, 353, 804–807 CrossRef CAS PubMed
- D. F. Sanders, Z. P. Smith, R. Guo, L. M. Robeson, J. E. McGrath, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 4729–4761 CrossRef CAS
- R. W. Baker and B. T. Low, Macromolecules, 2014, 47, 6999–7013 CrossRef CAS
- P. Marchetti, M. F. Jimenez Solomon, G. Szekely and A. G. Livingston, Chem. Rev., 2014, 114, 10735–10806 CrossRef CAS PubMed
- H. Wang, T. S. Chung and D. R. Paul, J. Membr. Sci., 2014, 458, 27–35 CrossRef CAS
- S. Kim and Y. M. Lee, Prog. Polym. Sci., 2015, 43, 1–32 CrossRef CAS
- R. Swaidan, B. Ghanem, E. Litwiller and I. Pinnau, Macromolecules, 2015, 48, 6553–6561 CrossRef CAS
- N. R. Horn and D. R. Paul, Macromolecules, 2012, 45, 2820–2834 CrossRef CAS
- M. Wessling, S. Schoeman, T. van der Boomgaard and C. A. Smolders, Gas Sep. Purif., 1991, 5, 222–228 CrossRef CAS
- B. W. Rowe, B. D. Freeman and D. R. Paul, Polymer, 2009, 50, 5565–5575 CrossRef CAS
- S. Japip, K.-S. Liao and T.-S. Chung, Adv. Mater., 2017, 29, 1603833 CrossRef PubMed
- C. H. Lau, P. T. Nguyen, M. R. Hill, A. W. Thornton, K. Konstas, C. M. Doherty, R. J. Mulder, L. Bourgeois, A. C. Y. Liu, D. J. Sprouster, J. P. Sullivan, T. J. Bastow, A. J. Hill, D. L. Gin and R. D. Noble, Angew. Chem., Int. Ed., 2014, 53, 5322–5326 CrossRef CAS PubMed
- Z. Kang, Y. Peng, Y. Qian, D. Yuan, M. A. Addicoat, T. Heine, Z. Hu, L. Tee, Z. Guo and D. Zhao, Chem. Mater., 2016, 28, 1277–1285 CrossRef CAS
- M. Kitchin, J. Teo, K. Konstas, C. H. Lau, C. J. Sumby, A. W. Thornton, C. J. Doonan and M. R. Hill, J. Mater. Chem. A, 2015, 3, 15241–15247 RSC
- C. H. Lau, X. Mulet, K. Konstas, C. M. Doherty, M.-A. Sani, F. Separovic, M. R. Hill and C. D. Wood, Angew. Chem., Int. Ed., 2016, 55, 1998–2001 CrossRef CAS PubMed
- T. Mitra, R. S. Bhavsar, D. J. Adams, P. M. Budd and A. I. Cooper, Chem. Commun., 2016, 52, 5581–5584 RSC
- C. H. Lau, K. Konstas, A. W. Thornton, A. C. Y. Liu, S. Mudie, D. F. Kennedy, S. C. Howard, A. J. Hill and M. R. Hill, Angew. Chem., Int. Ed., 2015, 54, 2669–2673 CrossRef CAS PubMed
- S. J. D. Smith, K. Konstas, C. H. Lau, Y. M. Gozukara, C. D. Easton, R. J. Mulder, B. P. Ladewig and M. R. Hill, Cryst. Growth Des., 2017, 17, 4384–4392 CrossRef CAS
- J. E. Bachman, Z. P. Smith, T. Li, T. Xu and J. R. Long, Nat. Mater., 2016, 15, 845–849 CrossRef CAS PubMed
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., 2009, 121, 9621–9624 CrossRef
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed
- P. Bernardo, V. Scorzafave, G. Clarizia, E. Tocci, J. C. Jansen, A. Borgogno, R. Malpass-Evans, N. B. McKeown, M. Carta and F. Tasselli, J. Membr. Sci., 2019, 569, 17–31 CrossRef CAS
- A. J. Hill, S. J. Pas, T. J. Bastow, M. I. Burgar, K. Nagai, L. G. Toy and B. D. Freeman, J. Membr. Sci., 2004, 243, 37–44 CrossRef CAS
- C. L. Staiger, S. J. Pas, A. J. Hill and C. J. Cornelius, Chem. Mater., 2008, 20, 2606–2608 CrossRef CAS
- C. H. Lau, K. Konstas, C. M. Doherty, S. Kanehashi, B. Ozcelik, S. E. Kentish, A. J. Hill and M. R. Hill, Chem. Mater., 2015, 27, 4756–4762 CrossRef CAS
- M. T. Pastor, A. Giménez-Giner and E. Pérez-Payá, ChemBioChem, 2005, 6, 1753–1756 CrossRef CAS PubMed
- R. E. Koeppe, J. Hatchett, A. R. Jude, L. L. Providence, O. S. Andersen and D. V. Greathouse, Biochemistry, 2000, 39, 2235–2242 CrossRef CAS PubMed
- Y. Umezawa, S. Tsuboyama, H. Takahashi, J. Uzawa and M. Nishio, Tetrahedron, 1999, 55, 10047–10056 CrossRef CAS
- S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2000, 122, 3746–3753 CrossRef CAS
- A. B. Foster, M. Tamaddondar, J. M. Luque-Alled, W. J. Harrison, Z. Li, P. Gorgojo and P. M. Budd, Macromolecules, 2020, 53, 569–583 CrossRef CAS
- C. R. Mason, L. Maynard-Atem, K. W. J. Heard, B. Satilmis, P. M. Budd, K. Friess, M. Lanč, P. Bernardo, G. Clarizia and J. C. Jansen, Macromolecules, 2014, 47, 1021–1029 CrossRef CAS PubMed
- Z.-X. Wang, C.-H. Lau, N.-Q. Zhang, Y.-P. Bai and L. Shao, J. Mater. Chem. A, 2015, 3, 2650–2657 RSC
- R. J. Hill, Phys. Rev. Lett., 2006, 96, 216001 CrossRef PubMed
- R. R. Tiwari, J. Jin, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2017, 537, 362–371 CrossRef CAS
- O. Vopička, M. G. De Angelis, N. Du, N. Li, M. D. Guiver and G. C. Sarti, J. Membr. Sci., 2014, 459, 264–276 CrossRef
-
D. R. Paul, in Encyclopedia of Membranes, ed. E. Drioli and L. Giorno, Springer Berlin Heidelberg, Berlin, Heidelberg, 2016, pp. 1–2, DOI:10.1007/978-3-642-40872-4_662-3
- Y. Xiao, B. T. Low, S. S. Hosseini, T. S. Chung and D. R. Paul, Prog. Polym. Sci., 2009, 34, 561–580 CrossRef CAS
- T.-S. Chung, L. Y. Jiang, Y. Li and S. Kulprathipanja, Prog. Polym. Sci., 2007, 32, 483–507 CrossRef CAS
- M. Carta, M. Croad, R. Malpass-Evans, J. C. Jansen, P. Bernardo, G. Clarizia, K. Friess, M. Lanč and N. B. McKeown, Adv. Mater., 2014, 26, 3526–3531 CrossRef CAS PubMed
- B. S. Ghanem, R. Swaidan, E. Litwiller and I. Pinnau, Adv. Mater., 2014, 26, 3688–3692 CrossRef CAS PubMed
- L. M. Robeson, W. F. Burgoyne, M. Langsam, A. C. Savoca and C. F. Tien, Polymer, 1994, 35, 4970–4978 CrossRef CAS
- P. M. Budd, K. J. Msayib, C. E. Tattershall, B. S. Ghanem, K. J. Reynolds, N. B. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263–269 CrossRef CAS
- R. Swaidan, B. Ghanem and I. Pinnau, ACS Macro Lett., 2015, 4, 947–951 CrossRef CAS
- A. W. Thornton, T. Hilder, A. J. Hill and J. M. Hill, J. Membr. Sci., 2009, 336, 101–108 CrossRef CAS
- R. W. Baker, Ind. Eng. Chem. Res., 2002, 41, 1393–1411 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr04801a |
| This journal is © The Royal Society of Chemistry 2020 |