
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNucleic acid peptide nanogels for the treatment of bacterial keratitis†
Sybil
Obuobi‡
 a,
Venkatesh
Mayandi
b,
Nurul Azlyn Mohd
Nor
a,
Benedict Jiasheng
Lee
a,
Rajamani
Lakshminarayanan
ab and
Pui Lai Rachel
Ee
*ac
a,
Venkatesh
Mayandi
b,
Nurul Azlyn Mohd
Nor
a,
Benedict Jiasheng
Lee
a,
Rajamani
Lakshminarayanan
ab and
Pui Lai Rachel
Ee
*ac
aDepartment of Pharmacy, National University of Singapore, 18 Science Drive 4, Singapore 117543. E-mail: phaeplr@nus.edu.sg; Fax: +65 6779 1554; Tel: +65 6516 2653
bSingapore Eye Research Institute, The Academia, Discovery Tower Level 6, 20 College Road, Singapore 169857
cNUS Graduate School for Integrative Sciences and Engineering, 21 Lower Kent Ridge Road, Singapore 119077
First published on 14th August 2020
Abstract
Cage-shaped nucleic acid nanocarriers are promising molecular scaffolds for the organization of polypeptides. However, there is an unmet need for facile loading strategies that truly emulate nature's host–guest systems to drive encapsulation of antimicrobial peptides (AMPs) without loss of biological activity. Herein, we develop DNA nanogels with rapid in situ loading of L12 peptide during the thermal annealing process. By leveraging the binding affinity of L12 to the polyanionic core, we successfully confine the AMPs within the DNA nanogel. We report that the thermostability of L12 in parallel with the high encapsulation efficiency, low toxicity and sustained drug release of the pre-loaded L12 nanogels can be translated into significant antimicrobial activity. Using an S. aureus model of infectious bacterial keratitis, we observe fast resolution of clinical symptoms and significant reduction of bacterial bioburden. Collectively, this study paves the way for the development of DNA nanocarriers for caging AMPs with immense significance to address the rise of resistance.
1. Introduction
To emulate nature's precise spatial co-location of bioactive molecules, scientists are adapting the elegance of rational design and self-assembly to create addressable, nanoscale biologically inspired containers.1–4 These nanocarriers are attractive candidates for drug delivery applications given their large loading capabilities, excellent in vivo targeting and enhanced stability features.5–7 Notably, the non-toxicity, size homogeneity and biodegradability of biomimetic nanocontainers favor their use over synthetic nanoscale objects.8–10 To date, a vast majority of self-assembled biological containers reported hitherto are fabricated by coupling highly ordered protein architectures, which may comprise thousands of copies of different proteins and are challenged by chemical/thermal instability or pose immunogenicity issues.11–15 Moreover, there is an unmet need to develop biomimetic cages where spatial control over drug entrapment can easily be achieved.As a burgeoning field in nanomedicine, advances in nucleic acid nanotechnology provide an unparalleled rich toolbox for the bottom-up construction of deoxyribonucleic acid (DNA) nanostructures of variable sizes, mechanical rigidity and enveloping capability.16–19 DNA nanostructures can be reproducibly and rapidly prepared in high yield using minimal sequence composition, and their negative surface potential confers significantly greater stability than their component oligonucleotides.20–24 Owing to their unprecedented control over size, shape and presentation of targeting ligands, cage-shaped DNA polyhedral structures have emerged as promising molecular scaffolds for the organization of biomolecules.25,26 To this end, a large number of studies exploit cargo entrapment via chemical or supramolecular interactions for molecular payloads such as proteins27–29 and anticancer therapies.30,31 Chemical crosslinking strategies are challenged by the formation of heterogeneous product mixtures and can introduce permanent cargo modifications which can interfere with activity.32 On the other hand, while non-covalent strategies have demonstrated enhanced efficacy, many rely on post-loading strategies which may be challenged by long incubation times.33 Recently, de Vries and colleagues reported on novel DNA amphiphiles wherein the corona was extended with DNA aptamer binding kanamycin B or ribonucleic acid (RNA) aptamer binding neomycin B for antimicrobial drug delivery.34 While fabrication was achieved rapidly, drug loading requires specific aptamer binding sequences and the externally bound post-loaded antibiotics may compromise the surface potential due to the presence of positively charged amino acid groups, induce mammalian cell toxicity or even distort the structural features of the nanocarrier. Therefore, non-covalent universal strategies that rapidly immobilize and retain antimicrobials within DNA nanocarriers in vitro without loss of biological activity, structural integrity or compatibility are highly desirable for in vivo applications against infectious diseases.
In this context, specific binding sites within pristine DNA nanocarriers comprise of abundant negatively charged phosphate pools.33 Therefore, taking full advantage of the polyanionic backbone of DNA, cationic antimicrobial cargos can easily be encapsulated within DNA nanocontainers via electrostatic interactions. Herein, a facile strategy that fabricates antimicrobial peptide (AMP) encapsulated DNA nanogels wherein the thermal annealing process proceeds in the presence of the peptides without loss in biological activity is reported. We leverage electrostatic interactions between the DNA monomers and the charged residues of the AMPs to spontaneously encapsulate the peptide during nanogel self-assembly. Fabrication of the DNA nanogels was achieved under physiological conditions and rapid immobilization of the AMP was visibly conserved within the caged structures. Converse to other drug loading strategies with long incubation times, we demonstrate rapid encapsulation, high encapsulation efficiency, peptide-dependent modulation of nanogel physicochemical features alongside improved cell viability. Furthermore, with the peptide loaded DNA nanogels, in vitro time-dependent antimicrobial potency against Staphylococcus aureus infections was observed. Finally, we demonstrate that the efficient caging of the AMP can be translated into significant in vivo antimicrobial efficacy against S. aureus keratitis in mice (Scheme 1).
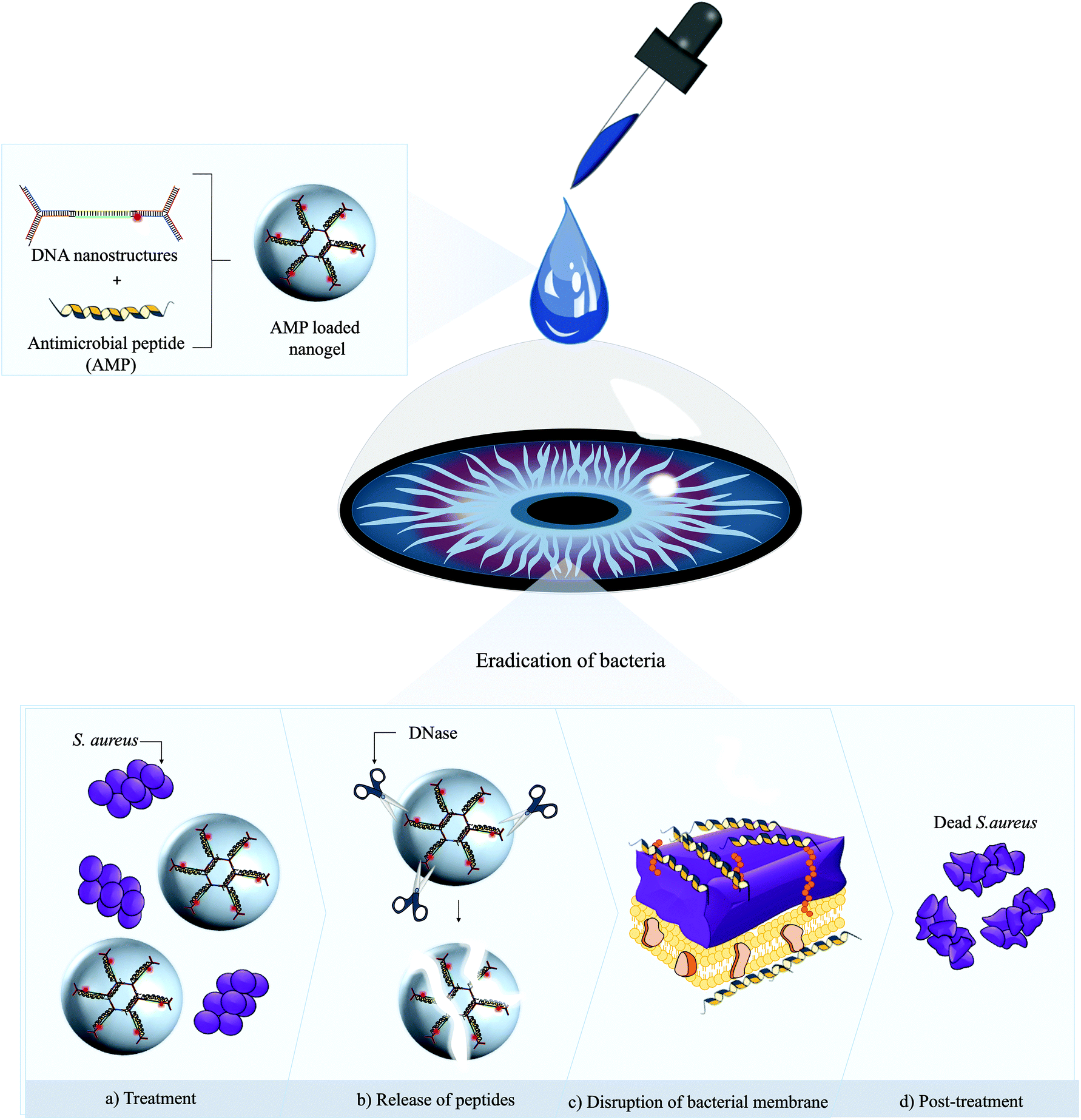 | ||
| Scheme 1 Utility of AMP loaded DNA nanogels for treatment of topical bacterial keratitis. We leveraged electrostatic interactions between self-assembled DNA nanostructures and cationic AMP to spontaneously cage the α-helical L12 AMP (a–d). Following ophthalmic application in vivo, inherent DNase promotes the degradation of the nanogel to release of the encapsulated AMPs, thereby driving rapid and potent bactericidal activity. | ||
2. Materials and methods
2.1. Materials
Synthetic oligonucleotides (see ESI Table 1† for sequences) and L12 antimicrobial peptides (non-FITC and FITC-labeled: >95%) were obtained from Integrated DNA Technologies, Inc (CA, USA) and GL Biochem Ltd (Shanghai, China) respectively. Staphylococcus aureus (ATCC 29737) and Pseudomonas aeruginosa (ATCC 25922) were obtained from American Type Culture Collection (DC, USA). Human dermal fibroblast (HDF) cells were purchased from Lonza (Basel, Switzerland) and human dermal keratinocytes (HaCaT) were a kind gift from Dr Lifeng Kang (Sydney, Australia). Tris base and ethylenediaminetetraacetic acid (EDTA) pH 8.0 were purchased from Axil Scientific (Singapore). Sodium chloride was purchased from Uni-Chem Chemical Reagents (Serbia, EU). Sodium bicarbonate, sodium hydroxide, boric acid and hydrochloric acid, cation-adjusted Mueller Hinton Broth (MHB) and Dulbecco's Modified Eagle's Medium (DMEM) were purchased from Sigma-Aldrich (MO, USA). Fetal bovine serum (FBS) and penicillin–streptomycin were purchased from HyClone Laboratories (UT, USA). 0.5% trypsin–EDTA was purchased from Life Technologies (CA, USA). Triton X-100 and phosphate-buffered saline (PBS) solution were purchased from Bio-Rad Laboratories (CA, USA). MTS colorimetric cell proliferation CellTiter 96® AQueous One Solution Reagent was purchased from Promega (WI, USA). All other analytical grade chemicals were obtained from commercial sources and used as purchased.2.2. Preparation of the monomers
To prepare the Y-shaped monomer A (YMA), Y-shaped monomer B (YMB) and L-shaped linking unit (DL), stoichiometric quantities of the oligonucleotides (Table S1†) were added into either sodium borate (SB, 10 mM NaOH and 36 mM boric acid pH 8.8), Tris/borate/EDTA (TBE, 0.89 mM Tris base, 0.89 mM boric acid, 0.02 M EDTA pH 8.5) or Tris buffered saline (TBS, 20 mM Tris-HCl and 100 mM NaCl pH 7.5) buffer solutions and made up to a final volume of 100 μL with grade 1 water. YMA and DL solutions were heated at 95 °C for 5 min, 60 °C for 30 min, 50 °C for 30 min, 37 °C for 30 min, 25 °C for 30 min and cooled for 2 h at 4 °C. YMB was hybridized at 95 °C for 5 min, 51 °C for 30 min, 50 °C for 30 min, 37 °C for 30 min, 25 °C for 30 min and cooled for 2 h at 4 °C. The annealing process was performed in a DNA Engine Tetrad® Thermal Cycler (BioRad, CA, USA).2.3. Preparation of blank and L12 loaded DNA nanogels
Equal volumes of 4 μM YMA, 1 μM YMB and 6.5 μM DL were mixed together and subjected to hybridization (95 °C for 5 min, 25 °C for 30 min and cooled for 3.5 h at 4 °C) to form the blank DNA nanogels. To prepare the L12 loaded nanogels via the post-loading method, 30 μL of blank DNA nanogels was added into 30 μL of L12 solution and gently mixed to yield L12 nanogel formulations with final peptide concentration of 5.8 μM, 11.5 μM, 23 μM or 46 μM. Conversely, nanogels prepared via the pre-loading method were formed by adding 10 μL YMA, 10 μL YMB and 10 μL DL into 30 μL of L12. The mixtures were annealed (95 °C for 5 min, 25 °C for 30 min and cooled for 3.5 h at 4 °C) to form the pre-loaded L12 nanogels with final peptide concentrations of 5.8 μM, 11.5 μM, 23 μM or 46 μM.2.4. Characterization of the blank and L12 loaded DNA nanogels
Gel electrophoretic measurements were conducted at 100 V, 25 °C for 45 min using 10 μM of each sample and with 3% w/v agarose gels in 1× TBE buffer solutions. For the run, YMA, YMB, DL monomers and the blank DNA nanogels were made up to volume in 5% v/v glycerol solutions. Agarose gels were stained with SYBR safe dye (Invitrogen) and scanned using the Biorad Gel Doc XR system for visualization.The hydrodynamic diameter and zeta potential of the DNA nanogels was carried out using a Litesizer™ 500 (Anton Paar, Austria). The hydrodynamic sizes of the DNA nanogels and L12 nanogel samples was determined on a Kalliope™ (Anton Paar, Austria) software using a quartz low volume cuvette (105-251-85-40) (Hellma Analytics, Germany) operating at a 90 °C angle (side scatter). Surface potential measurements were also carried out using omega zeta potential cuvettes (Anton Paar, Austria). The effect of temperature (4 °C and 25 °C), pH (7.5, 8.5 and 8.8) and ionic strength (100 mM and 200 mM NaCl) was examined using DLS measurements. All reported data were expressed as mean ± standard deviation (n = 3).
The external morphologies of the freeze-dried DNA nanogels was imaged using the JEOL JEM 2010F HRTEM (JEOL Ltd, Tokyo, Japan) for cryo-EM examination. Briefly, DNA nanogels were concentrated by centrifugation and thereafter flash-frozen in liquid nitrogen prior to examination with the electron microscope. High resolution TEM imaging was also performed using JEOL TEM 2200FS operated at 200 kV (JEOL Ltd, Tokyo, Japan). Air-dried nanogel samples on Tedpella carbon coated copper grids (200 mesh) were stained with Uranyless EM stain (Electron Microscopy Sciences, USA) solutions for 3 h at RT, washed with grade 1 water and air-dried overnight prior to examination with the electron microscope.
To examine the L12 nanogels using confocal microscopy, DNA nanogels were labelled with Alexa-594 and the L12 peptide was labelled with FITC. L12 loaded nanogels (20 μL) were added onto polylysine slides and secured in position using cover slips and nail varnish. Conventional confocal microscopy was performed using the Olympus FluoView™ instrument (FV1000, Tokyo, Japan) equipped with a solid laser diode 594 nm and 490 nm. SIM imaging was performed with a DeltaVision OMX Imaging System (GE Healthcare).
2.5. Encapsulation efficiency
To quantify the encapsulation efficiency of the formulations, pre-loaded L12 nanogels containing 92 μM of L12 were prepared. The nanogels were centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min and the supernatant was subjected to fluorescence quantification. Using the indirect method, encapsulation efficiency (EE%) of L12 was calculated using the following equation:
000 rpm for 30 min and the supernatant was subjected to fluorescence quantification. Using the indirect method, encapsulation efficiency (EE%) of L12 was calculated using the following equation: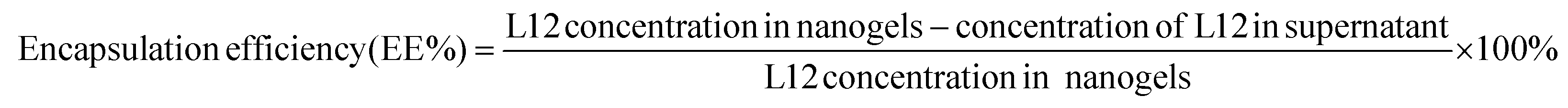
2.6. In vitro release of L12
In vitro release of FITC labelled L12 from the pre-loaded nanogels was examined in TBS buffer, pH 7.5 by monitoring changes in fluorescence intensity. The excitation and emission wavelengths were set at 490 nm and 535 nm respectively. L12 loaded nanogels containing 46 μM (n = 3) of L12 were dispersed in TBS buffer, pH 7.5 at 37 °C with mild shaking. At pre-determined time points, samples were collected from the solutions and centrifuged (13000 rpm, 15 minutes). The supernatant containing released L12 was collected and analyzed. The released concentrations of L12 was determined using a calibration curved obtained from standard solutions in TBS.
2.7. Stability of L12 nanogels
Changes in hydrodynamic diameter of the L12 nanogels was examined over 7 days. Pre-loaded L12 nanogels (n = 3) containing 20 μM L12 were prepared in TBS and stored at 4 °C, 25 °C and 37 °C. Size measurements was monitored over 7 days for each group and reported as the average hydrodynamic diameters ± standard deviation. All readings were performed in triplicates.2.8. In vitro biocompatibility of the nanogels
Biocompatibility of the blank nanogels was evaluated against HDF and HaCaT cells. HDF and HaCaT cells were seeded into 96-well plate at a density of 3000 cells or 4000 per well respectively. After 24 h, the culture medium was aspirated and replaced with 50 μL DMEM added to 50 μL of the blank nanogels prepared using monomer ratios (μM) of 4/1/6.5, 8/2/13 or 16/4/26 (YMA/YMB/DL) in 1× TBS buffers. 1× TBS solutions served as controls. The cells were incubated for 72 h and cell viability determined using MTS solutions as described.The toxicity of the pre-loaded L12-loaded nanogels was evaluated against HDF and HaCaT cells using the MTS colorimetric cell proliferation assay. HDF and HaCaT cells were seeded into 96-well plate at a density of 3000 or 6000 cells per well respectively. The cells were cultivated in 200 μL cell culture media (DMEM – high glucose (D1152-10X1L) supplemented with 10% FBS and 1% penicillin–streptomycin solution) at 37 °C and 5% CO2 in the incubator. After 24 h, the cell culture media was aspirated and replaced with fresh media (50 μL) together with 50 μL L12 nanogels (L12 final concentration range: 11.5 μM, 23 μM and 46 μM). Peptide treated cells served as controls. After 24 h, 20 μL of CellTiter 96® AQueous One solution was added into each well and incubated for 2 h. 100 μL of the media was added into 96 well plates in triplicates and the absorbance was read (490 nm) using HIDEX microplate reader (HIDEX, Turku, Finland).
2.9. In vitro antibacterial activity
MIC of L12 was tested against antibiotic susceptible and resistant pathogens i.e. Staphylococcus aureus (S. aureus; ATCC 29737), methicillin resistant Staphylococcus aureus (MRSA) and Pseudomonas aeruginosa (P. aeruginosa; ATCC 9027). Bacteria strains were cultured on TSB agar plates overnight and single colonies were inoculated into cation-adjusted Mueller Hinton Broth until the cultures reached mid-log phase. Optical density at 600 nm (OD600) of the cultures was measured, adjusted to 0.07 (108 CFU ml−1) and further diluted to a final concentration of 105 CFU ml−1. L12 test solutions subjected to nanogel hybridizing conditions (95 °C for 5 min, 25 °C for 30 min and 4 °C for 3.5 h) were added to the inoculum in 2-fold serial dilutions to give peptide concentration ranging between 2.5–32 μM. Non-thermocycled peptide solutions and pre-loaded L12 nanogels were added to the bacteria inoculum in a similar fashion. Growth media containing only bacteria and the blank nanogels served as negative controls. The plates were incubated at 37 °C with constant agitation at 100 rpm. OD600 was used as an indicator for growth and measured after 18 h using the HIDEX microplate reader (HIDEX, Turku, Finland). Minimum inhibitory concentration (MIC90) was defined as the lowest concentration of antimicrobial peptide which resulted in 90% growth inhibition as compared to the untreated control. All MIC readings were performed in triplicates.In vitro killing kinetics of the pre-loaded L12 nanogel formulations was performed against S. aureus and P. aeruginosa. Briefly, tubes containing 500 μL of MHB with L12 nanogels containing 0.5×, 1×, 2× and 4× MIC L12 was inoculated with S. aureus and P. aeruginosa at an inoculum density of 105–106 CFU mL−1 and incubated at 37 °C for 24 h under continuous agitation (100 rpm). At pre-determined time points, aliquots were removed and serially diluted in MHB. The diluted samples were then plated in triplicates on MHB agar plates and further incubated at 37 °C for 24 h. The total bacterial counts was determined by manual counting and results expressed as the CFU mL−1 ± standard deviation.
2.10. Corneal wound infection model
Pathogen free 6–8 weeks old female mice (wild type C57BL/6) with average body weight of 19 g were used to study the in vivo efficacy of the L12 nanogels. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the Singapore Eye Research Institute and approved by the Animal Ethics Committee known as the Sing-Health Institutional Animal Care and Use Committee (IACUC) (protocol no. 2016/SHS/1204). Additionally, all the animals were handled in accordance with the guidelines of Association for Research in Vision and Ophthalmology. Prior to wounding and infection, all mice eyes were examined by slit-lamp photography and AS-OCT to ensure that no corneal aberrations such as vascularization or other ocular defects were present. Mice were anesthetized by intraperitoneal injection of xylazine (10 mg kg−1, Troy Laboratories, Smithfield, Australia) and ketamine (80 mg kg−1, Parnell Laboratories, Australia) under a dissecting microscope (Zeiss, Stemi-2000 C). The mice corneal epithelium was then scratched and removed using a sterile Beaver6400 Mini-Blade to create a superficial wound without damaging the stroma. A drop of 1–5% lignocaine hydrochloride was topically applied as an anesthesia agent and cornea infection was induced immediately by applying 15 μL of bacteria suspension (S. aureus, ATCC 29213) containing 1–5 × 106 CFU mL−1 on the corneal surface. Each group was assigned 5–6 mice and after 6 h post-infection, mice were treated with 15 μL pre-loaded L12 nanogel (0.15% or 0.3% L12) test compounds which was topically applied every one and half hours for 3 days. Zymar eye drops containing 0.3% gatifloxacin served as a positive control for the S. aureus keratitis and the blank nanogels served as negative control treatments. The eyes were examined daily using slit-lamp and AS-OCT and the animal were sacrificed on day 3 for bacteria quantification analysis. Changes in corneal thickness (CT) was measured pre-inoculation and post-inoculation over the treatment period.2.11. Quantification of viable bacteria from infected mice cornea
Three days after treatment with the blank nanogels, pre-loaded L12 nanogel and gatifloxacin, the infected eyes were enucleated and used for bacterial quantification. Briefly, the mice corneas were dissected and homogenized in sterile PBS using sterile plastic pestles (Z359971, Sigma) followed by fine homogenization with sterile glass beads (2 mm). Homogenized mixtures were vortexed and 10-fold serial dilutions were prepared using sterile saline and inoculated onto Tryptic Soy Agar (TSA) plates. The plates were incubated at 37 °C for 48 h and the number of colonies were counted and expressed as the log10 number of CFU/cornea.2.12. Statistical evaluation
All statistical analyses were performed with software Graphpad Prism (Version 7.0, GraphPad Software Inc., CA, USA). Between two groups with statistical difference, the student's t-test was used. To compare experimental results from three or more groups, one-way Anova or two-way Anova with Turkey's post hoc analysis was performed. Results were represented as mean ± standard deviation and p values of <0.05 were considered significant.3. Results and discussion
3.1. Synthesis and characterization of the blank DNA nanogels
In this work, we fabricated a DNA nanogel platform (Fig. 1a) which features three monomers (Y-shaped monomer A: YMA, Y-shaped monomer B: YMB and L-shaped linking unit: DL) that are terminated by complementary ‘sticky end’ units (Table S1†) as previously reported.35 The terminating unit YMB, which comprises of one sticky end unit allows the nanogel formation process to be controlled and its sequences can easily be modified to incorporate fluorophores or bacteria specific aptamer sequences. Moreover, inherent enzymes (e.g. DNases) in the ocular fluid and infection milieu (e.g. S. aureus) can facilitate sustained release of encapsulated therapeutics.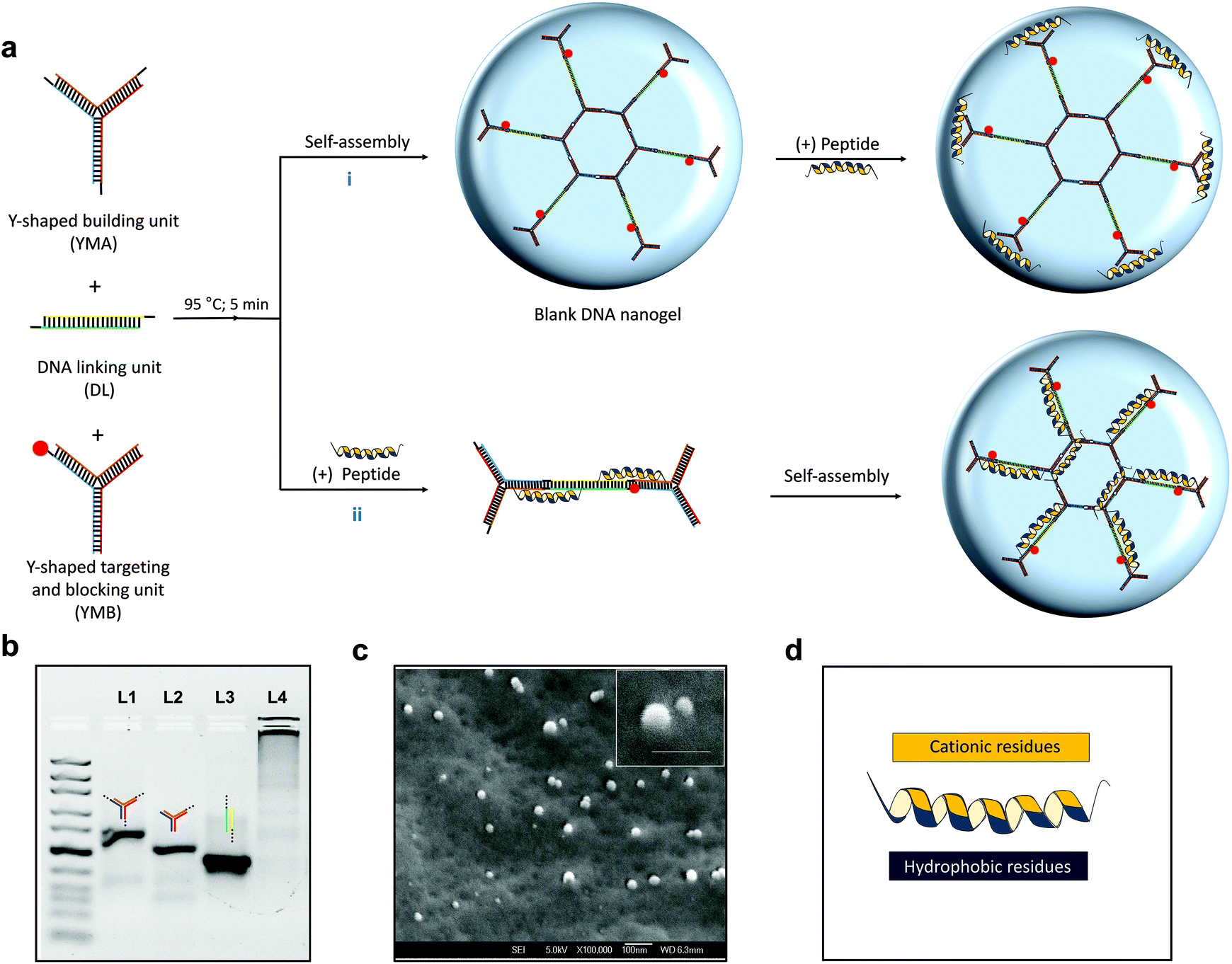 | ||
| Fig. 1 Schematic illustration of L12 nanogels and L12 peptide, hybridization and morphology of blank nanogels. a. Synthesis of L12 nanogels via (i) post-loading and (ii) pre-loading strategies. b. Agarose gel electrophoresis to verify self-assembly of DNA nanostructures (lane 1 = YMA; lane 2 = YMB; lane 3 = DL) and blank nanogels (lane 4). c. Cryo-electron microscopy image of the blank DNA nanogels (scale bar: 100 nm). d. Schematic illustration of L12 antimicrobial peptide (sequence: LKKLLKKLLKKL). | ||
While fabrication of rigid DNA nanostructures (e.g. tetrahedrons) and nucleic acid hybrids (e.g. micelles using amphiphilic DNA) has been demonstrated to enhance nuclease stability, the reported nuclease responsiveness of other pristine nucleic acid nanostructures such as nanogels can be exploited to enhance in vivo responsiveness.36,37 Fabrication of the blank nanogels is facile and controllable. First, the DNA nanostructures are individually hybridized via thermal annealing and then mixed together to self-assemble. Although previous studies have shown that the size of the nanogels can be controlled by varying the concentration of the monomers, exploiting this platform for drug loading has not been performed. For drug entrapment, a post-loading (Fig. 1a [i]) and pre-loading method (Fig. 1a [ii]) was adopted using our previously published L12 peptide44 (Fig. 1d) to fabricate the antimicrobial nanogel system. Using agarose gel electrophoresis, a distinct band was observed for lanes 1, 2 and 3 (Fig. 1b) demonstrating that stable assembled nanostructures were formed. After mixing the three nanostructures, the bands were replaced by an intense band in the sample loading well (Fig. 1b; lane 4) which corresponds with an increase in molecular weight and confirms the formation of a new product of larger size. The product was characterized using cryo-scanning electron microscopy to visualize its morphology and confirm the formation of the nanogels. As shown in Fig. 1c, we observed a smooth surface and spherical appearance of the products which provides clear evidence of the formation of the nanogels.
The impact of buffer identity and composition on the physicochemical features of the nanogels was investigated using dynamic light scattering (DLS) and zeta potential experiments. Significant variation in hydrodynamic diameter and zeta potential was observed for nanogels prepared in the three different buffer systems. For instance, DLS revealed larger hydrodynamic diameters (493.4 ± 10.2 nm) and a highly negative surface potential (−59.2 ± 1.8 mV) for nanogels prepared in sodium borate (SB) buffer (Fig. 2a & b). Conversely, we observed significantly smaller hydrodynamic diameters of 290 ± 68.5 nm (p < 0.01) and 108.8 ± 5.3 nm (p < 0.001) for nanogels prepared in Tris/borate/EDTA (TBE) and Tris buffered saline (TBS) buffers respectively. This reduction in size corresponded with the observed changes in surface potential to −35.1 ± 0.7 mV (TBE buffer; p < 0.001) and −29.9 ± 3.3 mV (TBS buffer; p < 0.001).
 | ||
| Fig. 2 Characterization of blank DNA nanogels. a. Effect of buffer composition on hydrodynamic diameter. b. Effect of buffer composition on zeta potential. c. Effect of pH on hydrodynamic diameter. d. Effect of ionic strength on hydrodynamic diameter. e. Percentage change in hydrodynamic diameter at 4 °C and 25 °C storage temperatures over 24 h. f. Transmission electron micrograph (TEM) images of optimized DNA nanogels in TBS buffer (scale bar: 200 nm). Average diameters of the nanogels was 65.00 ± 6.0 nm as measured with image J (*, p < 0.05; **, p < 0.01; ***, p < 0.001; **** p < 0.0001). | ||
To identify the properties of the buffer systems that control the hydrodynamic diameter and net charge of the DNA nanogels, DLS measurements were performed on nanogels prepared in TBS buffers of varying pH and ionic strength. As shown in Fig. 2c, changing the pH of the buffer from 7.5 to 8.8 steadily increased the hydrodynamic diameter of the nanogels. The average hydrodynamic diameter of the nanogels was lowest at pH 7.5 (86.98 ± 20.78 nm). We observed an increase in the measured size to 177.4 ± 29.6 nm at pH 8.5 and 305.9 ± 19.8 nm at pH 8.8. The considerable size increase at higher pH is likely attributed to the increased ionic strength of the buffer which contributes to the growth of the nanogel.
To confirm this, the effect of ionic strength on the hydrodynamic diameter was determined under different sodium chloride concentrations. Increasing the salt concentration of the external buffer solution from 100 mM to 200 mM resulted in a significant increase in size from 64.7 ± 5.0 nm (100 mM) to 136.5 ± 10.0 nm (200 mM) as shown in Fig. 2d. This can be attributed to nanogel instability or charge screening due to increasing counterions that mediates an effective electrostatic attraction between the DNA strands or nanogels.38–40 Subsequently, temperature effects were determined by measuring percentage changes in hydrodynamic diameter of the blank nanogels at storage temperatures of 4 °C and 25 °C over 24 h. We found that at low temperatures (4 °C), the hydrodynamic diameter of the blank nanogels significantly reduced to 53.8 ± 10.6% of its initial value (Fig. 2e) and remained stable for 5 days (Fig. S1†). On the other hand, increasing the temperature of the buffer solution to 25 °C resulted in a significant percentage increase in the hydrodynamic diameter (153.8 ± 37.3%). These results demonstrate that the crosslinked blank DNA nanogels display reversible volume transition in response to pH, ionic strength and temperature. These features justify their characterization as nanogels and are central for developing nanocarriers with controlled loading and release characteristics.41 Accordingly, nanogels prepared in TBS buffer at pH 7.5 and salt concentration of 100 mM were used for all subsequent experiments as the optimal conditions. TEM micrograph images (Fig. 2f) of the blank nanogels prepared in TBS buffers displayed average diameters of 65.00 ± 6.0 nm which correlated well with DLS measurements (64.7 ± 5.0 nm) and the 3D spherical morphologies of the nanogels were also apparent. The tunability of the hydrodynamic diameter of the nanogels is highly advantageous for extending applications in systemic, intracellular and topical formulations.
3.2. Synthesis and characterization of the L12 loaded DNA nanogels
The growing challenge of drug resistance and tolerance of quiescent bacteria to conventional antibiotics has prompted the development of novel antimicrobial peptides as promising new solutions.42 Antimicrobial nanogel formulations have been fabricated via physical loading and reported to reduce drug toxicity, improve drug stability and enhance therapeutic activity.43 We selected L12 (Table S2†), a novel α-helical peptide in this study given its high selectivity, superior potency against drug-susceptible and multi-drug resistant Staphylococcus aureus, anti-biofilm and anti-endotoxin activity.44 To encourage high local concentrations for fast and profound antimicrobial effects, our approach seeks to non-covalently encapsulate L12 within the DNA nanogels for sustained delivery. To achieve this, we adopted two strategies for drug loading as described in Fig. 1a. Converse to the post-loading method which bears similarity to other reports in literature, the pre-loading strategy was introduced as a novel one-step peptide encapsulation process and hypothesized to produce size-tunable monodispersed nanogels without affecting the surface potential. Moreover, we envisioned that strong electrostatic binding to the DNA nanocarrier and conformational changes of the α-helical peptide, L12 would drive high loading and efficient caging (Fig. S2†).Using the post-loading strategy (Fig. 3a) wherein increasing peptide concentrations were added to the pre-formed nanogels, no significant difference in hydrodynamic diameter was observed at 5.8 μM (97.2 ± 12.5 nm) and 11.6 μM (74.3 ± 9.8 nm) of L12 in comparison to the blank nanogels (81.1 ± 14.1 nm).
 | ||
| Fig. 3 Characterization of L12 nanogels. a. Effect of post-loading L12 peptide on hydrodynamic diameter of DNA nanogels. b. Effect of pre-loaded L12 peptide on hydrodynamic diameter of DNA nanogels. c. Size distribution of pre-loaded L12 nanogels. d. TEM images of L12 nanogels. Scale bar is 200 nm. e. Effect of pre-loading and post-loading L12 peptide on zeta potential (**** p < 0.0001). f. Effect of increasing monomer ratio and peptide concentration on hydrodynamic diameter (*, p < 0.05; **, p < 0.01; **** p < 0.0001) of the pre-loaded L12 nanogels. g. Effect of temperature changes on hydrodynamic diameter of pre-loaded L12 nanogels for 7 days. | ||
The slight reduction in hydrodynamic diameter at 11.6 μM of L12 can be attributable to DNA condensation as the peptide binds to the surface of the nanogel. A drastic increase in size to 144.7 ± 6.1 nm and 342.3 ± 26.6 nm was observed at 23 μM and 46 μM respectively. Conversely, nanogels encapsulating L12 via the pre-loading strategy showed no significant difference in hydrodynamic diameter up to 23 μM (113.2 ± 11.7 nm) in comparison to blank nanocarrier (96.3 ± 7.1 nm). However, further increment of the peptide concentration to 32 μM (160.6 ± 9.3 nm) and 46 μM (379.9 ± 2.8 nm) resulted in a significant increase in hydrodynamic diameter as shown in Fig. 3b. Remarkably using the pre-loading strategy for nanogel fabrication, we observed that the concentration of L12 was inversely proportional to polydispersity and the formulations displayed highly monomodal size distribution at 46 μM of L12 (Fig. 3b & c) compared to the blank nanogels or the post-loaded formulations (Fig. S3−S5†). These results clearly demonstrate that the pre-loading strategy enabled L12 encapsulated nanogels to be prepared with highly controllable and homogenous size distribution. This observation is credited to the highly cationic and hydrophobic nature of L12 which acts as a driving force for secure encapsulation within the nanogel core. Subsequently, TEM images confirmed the spherical morphology of the peptide loaded nanogel formulations as shown in Fig. 3d.
Next, we investigated the surface potential of the pre-loaded and post-loaded L12 nanogel formulations. As shown in Fig. 3e, post-loading L12 resulted in a steady increase in zeta potential with a drastic change from the highly anionic surface charge of −36.9 ± 1.5 mV for the blank nanogel to +3.9 ± 0.1 mV and +5.6 ± 0.1 mV at L12 concentrations of 23 μM and 46 μM respectively. This observation is attributable to complete neutralization of the negative charge on the surface of the DNA nanogels as a result of electrostatic interaction and binding of L12 and corresponds with DLS measurements. Conversely, we observed no significant difference in zeta potential using the pre-loading method at 23 μM (−30.0 ± 0.8 mV) and 46 μM (−34.4 ± 0.9 mV) L12. These results clearly demonstrate that pre-loading L12 does not influence the surface potential of the DNA nanogels and serve as a prime indication of peptide caging in contrast to the post-loading strategy wherein surface bound peptides account for the highly positive zeta potential.
Having demonstrated the peptide-dependent variation in nanogel size, we sought to illustrate flexibility of drug loading by increasing monomer ratio of the nanostructures to drive higher loading capacity (Fig. 3f). Doubling the monomer ratio of YMA:YMB:DL from 4:1:6.5 to 8:2:13, resulted in a significant reduction (p < 0.1) in hydrodynamic diameter from 1182.8 ± 156.9 nm to 584.9 ± 85.8 nm for pre-loaded nanogels containing 92 μM of L12. Similarly, while a significant increase in hydrodynamic diameter at 4:1:6.5 monomer ratio (3337.3 ± 669.8 nm) was seen at 184 μM L12, further increment of the monomer ratio to 8:2:13 and 16:4:26 subsequently resulted in drastic reduction to 2436.4 ± 774.3 nm and 789.8 ± 116.6 nm respectively. This may be attributable to the increase in polyanionic DNA monomers for L12 binding during nanogel formation. We confirmed this by showing that by increasing the monomer ratio of the nanostructures, a significant increase in the number (kcount per s) of blank nanogels can be quantified (Fig. S6†). Taken together, these results demonstrate that, the loading capacity of the DNA nanogels can be expanded by tuning the monomer ratio of the nanostructures.
The thermal stability of the pre-loaded L12 nanogel formulations was examined over 7 days by monitoring the changes in hydrodynamic diameter below dehybridization conditions. We prepared pre-loaded L12 nanogels loaded with 20 μM L12 in TBS buffer and stored the samples at 4 °C, 25 °C and 37 °C. As shown in Fig. 3g, we observed no apparent change in the hydrodynamic diameter when the L12 nanogels were stored at 4 °C for 7-days. At 25 °C, no difference in hydrodynamic diameter was shown after 24 h. However, on longer storage, a large increase in the average hydrodynamic diameter of the nanogels by 62.2 ± 26.7% (day 3) and 148.1 ± 73.8% (day 7) was observed. It was found that, heating the L12 nanogels to 37 °C under mild agitation resulted in rapid increase in the average hydrodynamic diameter by 87.1 ± 27.7% (day 1), 189.9 ± 86.2% (day 3) and 241.4 ± 32.4% (day 7). The observed dramatic increase in hydrodynamic diameter may be attributable to the temperature responsiveness of the peptide loaded DNA nanogels and highlight their volume phase transition at physiological conditions which is hypothesized to drive peptide release. Moreover, these results also clearly demonstrate that the peptide loaded nanogels do not aggregate during storage at 4 °C and remained stable for at least 7-days.
Visualized TEM images of the formulations could not distinguish between the encapsulated peptide and the DNA nanogels while using negative stain. Thus, to detect the encapsulated peptide within the formulations, we performed confocal microscopy using alexa-594 labelled DNA nanogels containing the FITC labelled L12. As shown in Fig. 4a, spherical swollen networks were observed in pre-loaded L12 nanogel formulations that were homogenously dispersed in the aqueous buffer solution. Analysis of the superimposed images revealed a yellow core surrounded by a red corona which clearly confirms FITC-labelled peptide caging within the nanogels (Fig. 4a). The post-loaded L12 nanogels however demonstrated attachment of L12 to the nanogel surface which was visualized as yellow cores surrounded by green coronas and is in good agreement with surface potential measurements (Fig. 4b). To improve the resolution of the nanogels and to visualize the entrapped L12 peptide within, super resolution structured illumination (SIM) confocal microscopy was used and showed that the DNA nanogels displayed a well-organized, highly porous internal structure when loaded with the unlabeled and labelled L12 peptide (Fig. 4c & d). Within the matrix of the L12 nanogels, we observed that the FITC labelled L12 peptide was strictly bound to the self-assembled DNA nanostructures, assumed its architecture and was evenly distributed within the Alexa-594 labeled DNA nanogels as shown in Fig. 4d. Converse to the post-loaded formulation, the pre-loaded L12 nanogels displayed homogeneity, stability of the surface potential with increasing concentration of L12 and capacity to entrap the peptide fully into its core. Thus, we therefore chose the pre-loaded formulation for further in vitro and in vivo evaluations.
 | ||
| Fig. 4 Visualization of the preloaded L12 nanogels. DNA nanogels are labelled with Alexa-594 and L12 peptide is labelled with FITC. a. Conventional confocal microscopy of pre-loaded L12 nanogels. b. Conventional confocal microscopy of post-loaded L12 nanogels. c. Structured illumination microscopy (SIM) images of DNA nanogels containing unlabeled (UL) antimicrobial L12 peptide. DNA nanogels are labelled with Alexa-594 and L12 peptide is unlabeled. d. SIM confocal image of the L12 nanogels. Individual frames of Z-stacks (step size: 0.125 μm) are shown in the right panel to demonstrate distribution of L12 within the nanogels. DNA nanogels are labelled with Alexa-594 and L12 peptide is labelled with FITC. | ||
Finally, quantification of the encapsulation efficiency of the pre-loaded L12 nanogels revealed high encapsulation efficiencies of 92.1 ± 0.4% as determined by fluorescence spectroscopy. These results are comparable to the reported encapsulation efficiency of other antimicrobial peptides in natural and synthetic nanocarriers.45–47 Taken together, the observed homogenous distribution and strict association of the peptide within the pre-loaded nanogel matrix in parallel with the high encapsulation efficiency corroborates our hypothesis that the encapsulation of L12 is facilitated by non-covalent electrostatic interactions. While there is concern that there is limited room to increasing the loading of cationic cargos without distorting the nanostructures, we have demonstrated in this present work that by simply optimizing the monomer concentration, we can potentially overcome this issue (Fig. 3f). Indeed, the cost of DNA in comparison to other biopolymers e.g. natural polymers is one of the barriers to clinical translation. Nevertheless, compared to bulk gel systems (e.g. hydrogel), the lower amount (%w/w) of DNA required for fabricating nanogels may translate to overall lower cost. Of interest, a recent report indicates that an estimated cost of less than €10 per dose of DNA nanostructured formulations is perceivable without incorporating costs associated with sterilization, purity and batch-to-batch consistency.48 Ultimately, large scale synthesis (multi kg range) is expected to reduce overall cost of production, drive fairly moderate competitive market prices that are comparable to other approved formulations (e.g. monoclonal antibodies). This would render the development of nucleic acid systems promising for future therapeutics.48
3.3. Antibacterial activity, in vitro drug release and cellular toxicity of DNA nanogels
We have previously reported L12 as a highly potent synthetic peptide against Gram positive and Gram negative pathogens.44 Having demonstrated high encapsulation efficiency and homogenous distribution of L12 within the DNA nanogels, time-kill kinetic studies were performed to monitor the time-course of the antimicrobial effect of the L12 nanogels against S. aureus. Rapid release of L12 is essential to ensure that lethal antibacterial concentrations are available to avert undesirable complications attributable to the circulation of bacterial endotoxins or exotoxins.49,50 The pre-loaded L12 nanogels displayed rapid onset of action, reducing bacterial inoculum by 89.2% (0.96 log reduction) at 4× MIC within 2 h of treatment (Fig. 5a). After 4 h, 2.27 log, 2.65 log and 2.87 log reduction of S. aureus colonies was attained for cells treated with 1×, 2× and 4× MIC of the L12 nanogel formulations. A strong and prolonged antibacterial activity was observed over 24 h for cells treated with 2× (6.7 log reduction) and 4× (6.7 log reduction) MIC concentrations of the L12 nanogels.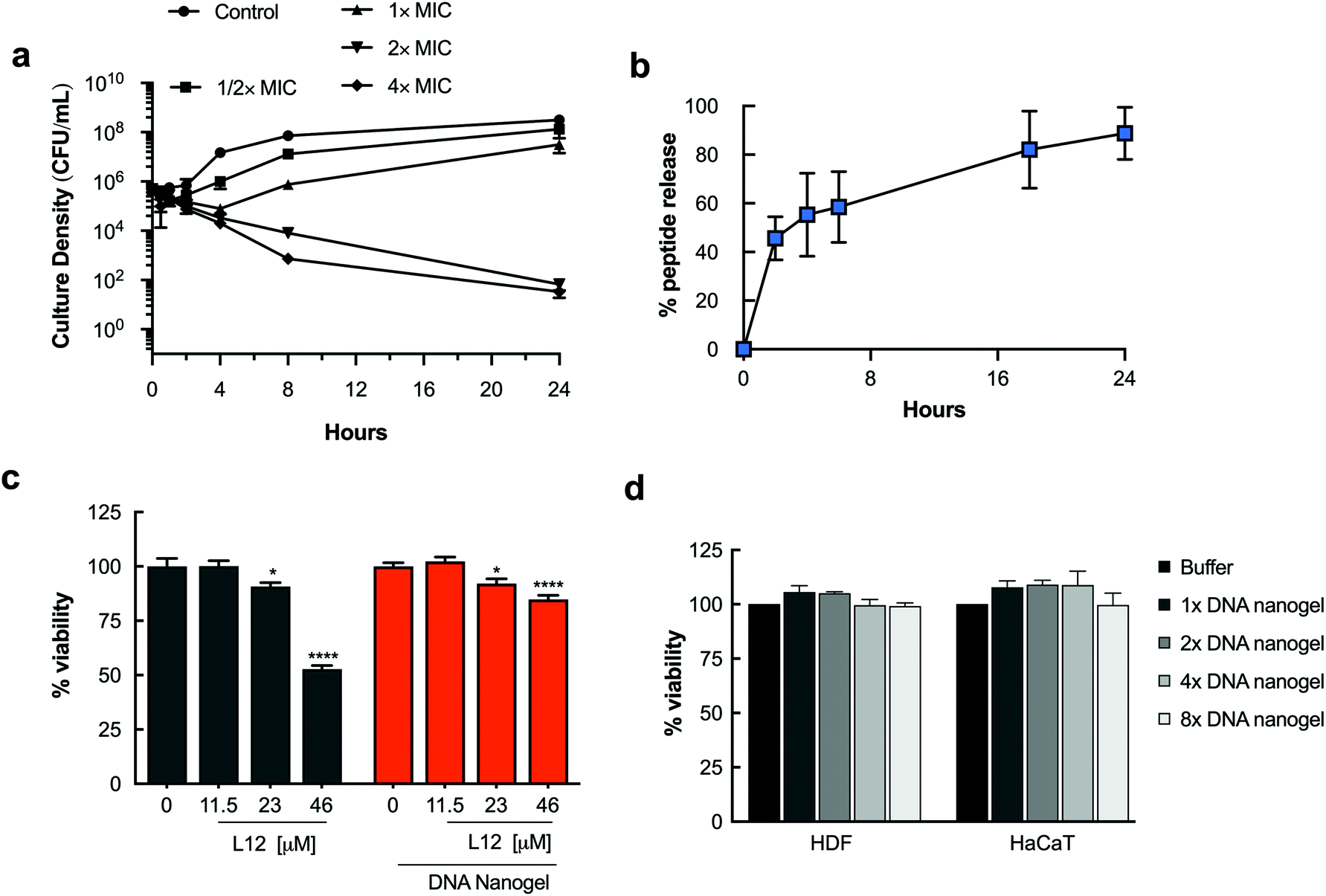 | ||
| Fig. 5 In vitro antimicrobial activity, drug release and cytotoxicity evaluations. a. Time kill kinetics of pre-loaded L12 nanogels against Staphylococcus aureus (106 CFU mL−1). b. In vitro release of pre-loaded L12 nanogels. c. Toxicity of pre-loaded L12 nanogels against HDF cells over 24 h (*, p < 0.05; ****, p < 0.0001). d. Toxicity of blank nanogels against HDF and HaCaT cells over 72 h treated with YMA/YMB/DL monomer ratios of 1× (4/1/6.5), 2× (8/2/13), 4× (16/4/26) and 8× (32/8/52) to prepare the DNA nanogel. | ||
Notably, the empty nanogel control group had no antibacterial effect on S. aureus in vitro leading to the conclusion that L12 is rapidly released in vitro to exert an antibacterial effect despite being securely loaded and bound to the nanogel core. Similar efficacy was observed against P. aeruginosa (Fig. S10†). Thereafter, in vitro release of L12 from the pre-loaded DNA nanogels incubated at 37 °C was monitored over 24 h by centrifuging aliquots of the formulations at pre-determined intervals and measuring the concentration of FITC-labelled L12 in the supernatant. As shown in Fig. 5b, 45.59% of L12 was released within 2 h which is in good agreement with the rapid onset of antibacterial activity. By 24 h, 88.13% of L12 was released from the L12 nanogels. The initial rapid release of L12 from the nanogel system is likely due to simple diffusion of the cationic cargo from the nanostructures.51,52 Conversely, the sustained release profile between 2 h and 24 h can potentially be attributed to swelling of the nanogels under elevated temperatures as shown in Fig. 3f. The release profile of the L12 nanogels correlated well with the time-kill kinetics and the observed antimicrobial efficacy in vitro.
Nanocarriers that prolong the release of encapsulated drugs have the advantage of maintaining peptide concentrations within the therapeutic window over an extended period, minimizing underexposure and reducing drug toxicity. Thus, we quantified the ability of the pre-loaded L12 nanogels to improve toxicity of L12 over 24 h. Against HDF cells, we observed a 32 ± 1.2% increase in the viability (p < 0.01) of cells treated with L12 nanogels at 46 μM compared to the peptide controls (Fig. 5c). A 20.60 ± 4.4% and 22.3 ± 5.7% increase in cell viability (p < 0.01) was observed for HaCaT cells treated with 23 μM and 46 μM of L12 nanogels respectively compared to the peptide controls (Fig. S11†). These results are caused by the sustained release of L12 from the nanogels and not associated with the toxicity of the DNA nanogel itself. To demonstrate the biocompatibility of the blank DNA nanogels, cell proliferation studies were performed. HDF and HaCaT cells were treated with DNA nanogels prepared using four different monomer ratios up to 8-fold increase. As shown in Fig. 5d, blank DNA nanogels and their degradation products did not induce cell death after 72 h and were biocompatible with mammalian cells. Overall, these results underscore the advantage of using DNA-based materials as nanocarriers for drug delivery and clearly highlight the ability of the DNA nanogels to reduce the toxicity of encapsulated peptides.
3.4. In vivo testing of the L12 nanogels in a corneal infection wound model
Corneal infections caused by S. aureus are the leading cause of blinding microbial keratitis.53 Recent documented cases of keratitis caused by MRSA have shown resistance against first line antimicrobials such as fluoroquinolones, highlighting the need for new therapies.54,55 Moreover, the promising in vitro activity of the formulation warrants further in vivo studies. Thus, using a mouse model of bacterial keratitis, we sought to test whether the nanogels could be used as antimicrobial delivery systems by assessing the efficacy of the L12 nanogels in vivo. Superficial wounds created by carefully removing the corneal epithelium were inoculated with S. aureus to create the corneal infection wound model. Following clinical diagnosis of ocular infection, initial treatment usually involves intensive topical antibiotics,56 thus we compared intensive ophthalmic application of a clinically approved formulation (0.3% gatifloxacin) with our pre-loaded L12 nanogels and monitored the progression of the infection using Slit-lamp (SL) biomicroscopy and anterior-segment optical coherence tomography (AS-OCT). The mice were treated with gatifloxacin (0.3% w/v), L12 nanogels (0.15% w/v) or L12 nanogels (0.3% w/v) for 48 h (Fig. 6a). After 48 h, mice were sacrificed and the infected eyes were enucleated for bacterial quantification.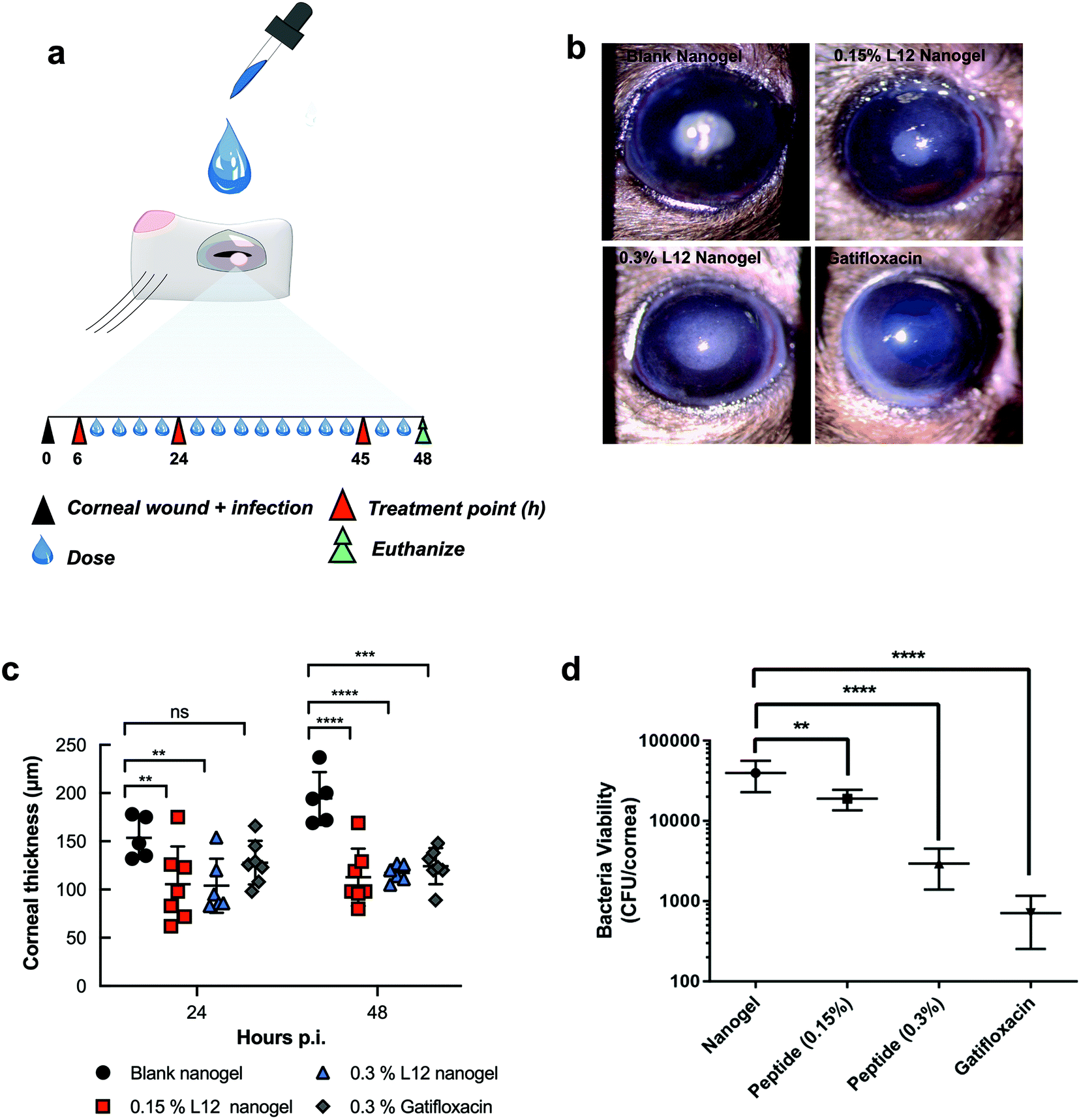 | ||
| Fig. 6 Analysis of in vivo antimicrobial efficacy in murine corneal wounds infected with S. aureus. a. Schematic representation of experimental design for the bacterial keratitis wound model of S. aureus and timescale of topical treatment. A total of 14 doses of pre-loaded L12 nanogels and gatifloxacin was administered over 48 h and the bacterial bioburden from the infected eyes post-treatment was compared to the blank nanogel control (n = 5–7 infected eyes from 5–7 mice per group was used). b. Slit lamp images of the murine corneal wounds after treatment with the blank nanogels, 0.15% pre-loaded L12 nanogels, 0.3% pre-loaded L12 nanogels and 0.3% gatifloxacin formulations. c. Time dependent changes in corneal thickness measurement at 24 h and 48 h post-inoculation by AS-OCT for mice treated with the blank nanogels, 0.15% pre-loaded L12 nanogels, 0.3% pre-loaded L12 nanogels and 0.3% gatifloxacin formulations. Results are reported as the means ± standard deviations for 5–7 corneas per group. d. Quantitative S. aureus burden in infected corneas following treatment with blank nanogel, pre-loaded L12 nanogels (0.15% and 0.3% L12) and 0.3% gatifloxacin (Zymar) at 48 h. Results are reported as the means ± standard deviations for 5–7 corneas per group (ns, no statistical significance; *, p < 0.05; **p < 0.01; ***, p < 0.001; **** p < 0.0001). | ||
Slit lamp (SL) examination of S. aureus infected cornea treated with various groups displayed different degrees of corneal opacity (Fig. 6b).57 SL images of infected cornea treated with nanogel indicated dense opacities on corneal surface over pupil. 0.3% L12 nanogel treated cornea, however, contained readily detectable opacity that was lower than 0.15% L12 nanogel. Infected cornea treated with gatifloxacin appeared clear and normal and superior clinical presentation among all the groups, confirming potency of the antibiotics in resolving S. aureus infections. Consistent with the SL examination, AS-OCT images indicated the presence of hyper-reflective materials and edematous cornea in the nanogel treated groups.
The baseline corneal thickness of mice eye was ranged from 80–98 μm (mean thickness 92.0 ± 5.8 μm). Within 24 h of S. aureus wound infection, the mean corneal thickness in the control group increased to 153.6 ± 21.8 μm (Fig. 6c), suggesting exaggerated inflammatory response. Conversely, a significant reduction in the mean corneal thickness was observed for the 0.15% pre-loaded L12 nanogel (105.6 ± 39.0 μm) and 0.3% pre-loaded L12 nanogel (104.0 ± 28.0 μm) treated mice over 24 h. However, no significant difference in mean corneal thickness was observed for the 0.3% gatifloxacin (127.9 ± 22.6 μm) infected eyes. The significant decrease in corneal thickness for the L12 nanogel treated group at 24 h highlight fast resolution of infection-induced inflammation which is potentially attributable to the anti-inflammatory action of the L12 peptide and the DNA nanostructures as previously reported.26,44,58 At 48 h post-inoculation the mean corneal thickness continued to increase for the control group (194.4 ± 27.4 μm) which indicates increased severity of the infection.59 An even more intense reduction in the average corneal thickness was seen in the 0.15% pre-loaded L12 nanogel (112.7 ± 29.6 μm), 0.3% pre-loaded L12 nanogel (117.2 ± 8.7 μm) and 0.3% gatifloxacin (124.3 ± 18.7 μm) treated groups after 48 h.
A severe clinical course of ocular infection in the control group was observed along with increased bacterial bioburden in infected mice treated with the blank nanogels. Microbiological determination of bacterial viability indicated a significant reduction in bacterial bioburden for mice treated with 0.15% pre-loaded L12 nanogel (p < 0.01) (Fig. 6d) at 48 h. However, a more substantial decrease in the number of viable bacterial cells was recovered from corneal wounds treated with 0.3% pre-loaded L12 nanogels (p < 0.0001) and 0.3% gatifloxacin (p < 0.0001). The slightly better efficacy of gatifloxacin than 0.3% pre-loaded L12 nanogels could be attributed to the higher molar concentrations of the fluoroquinolone drug. Alongside the in vitro antimicrobial activity of the formulation, these results establish the excellent antimicrobial properties of pre-loaded L12 nanogels against S. aureus keratitis. Notably, the topical application of the formulations over 48 h demonstrated excellent ocular tolerance and no significant changes in mice weight or behavior was observed. These results corroborate the in vitro biocompatibility of the nanogels and are in good agreement with other reports on DNA nanocarriers.
4. Conclusion
Our work highlights a facile strategy to cage antimicrobial peptides within DNA nanocarriers in a tunable manner without loss of biological activity or structural integrity for local drug delivery applications. We demonstrate that the sustained release of encapsulated peptides can be translated into significant in vitro and in vivo antimicrobial efficacy against S. aureus. The progressive decrease in corneal thickness and quantitative analysis of bacterial viability demonstrate that the topical application of L12 nanogels can reduce the clinical symptoms and bacterial bioburden in S. aureus models of infectious bacterial keratitis. Considering that S. aureus remains the leading cause of blinding bacterial keratitis, the observed fast resolution of infection-induced inflammation and comparable efficacy of topical 0.3% L12 nanogels to gatifloxacin is clinically advantageous. Moreover, these nanocarriers are biocompatible against mammalian cells, significantly improve toxicity of encapsulated peptides and possessed excellent ocular tolerance in vivo. For future applications, targeted delivery against pathogenic bacteria and transport across ocular barriers should be explored.Author contributions
S.O. and P.L.R.E. conceived the project; S.O., V.M., R.L. and P.L.R.E. designed the experiments; S.O., V.M., N.A.M.N. and B.L.J. performed experiments and collated the experimental results; S.O. wrote the manuscript with editing and suggestions from V.M., N.A.M.N., R.L. and P.L.R.E. Schematic diagrams were prepared by N.A.M.N and S.O. All authors approved the final version of the manuscript.Conflicts of interest
The authors declare that no competing interest exists.Acknowledgements
The authors would like to acknowledge research funding and facilities provided by the National University of Singapore and Ministry of Education Academic Research Fund (R148000240114) awarded to P. L. R. Ee and the Singapore International Graduate Award (SINGA) to S. O. R. L. would like to thank funding support from the Singapore Ministry of Health's National Medical Research Council under its Centre Grant Programme – Optimization of Core Platform Technologies for Ocular Research (INCEPTOR)-NMRC/CG/M010/2017_SERI and the SingHealth Foundation (SHF/FG663P/2017). We would like to thank the A*STAR Microscopy Platform for technical guidance in the SIM studies. Art work acknowledgement to N. A. M. N and S. O.References
- R. M. Putri, J. J. Cornelissen and M. S. Koay, Self-assembled cage-like protein structures, ChemPhysChem, 2005, 16, 911–918 CrossRef PubMed.
- N. P. King and Y. T. Lai, Practical approaches to designing novel protein assemblies, Curr. Opin. Struct. Biol., 2013, 23, 632–638 CrossRef CAS PubMed.
- S. Hurtley, Spatial cell biology. Location, location, location. Introduction, Science, 2009, 326, 1205 CrossRef CAS PubMed.
- A. H. Chen and P. A. Silver, Designing biological compartmentalization, Trends Cell Biol., 2012, 22, 662–670 CrossRef CAS PubMed.
- S. Deshayes and R. Gref, Synthetic and bioinspired cage nanoparticles for drug delivery, Nanomedicine, 2014, 9, 1545–1564 CrossRef CAS PubMed.
- M. Uchida, et al., Biological containers: Protein cages as multifunctional nanoplatforms, Adv. Mater., 2007, 19, 1025–1042 CrossRef CAS.
- J. G. Heddle, S. Chakraborti and K. Iwasaki, Natural and artificial protein cages: design, structure and therapeutic applications, Curr. Opin. Struct. Biol., 2017, 43, 148–155 CrossRef CAS PubMed.
- M. Rother, M. G. Nussbaumer, K. Renggli and N. Bruns, Protein cages and synthetic polymers: a fruitful symbiosis for drug delivery applications, bionanotechnology and materials science, Chem. Soc. Rev., 2016, 45, 6213–6249 RSC.
- D. Jiang, C. G. England and W. Cai, DNA nanomaterials for preclinical imaging and drug delivery, J. Controlled Release, 2016, 239, 27–38 CrossRef CAS PubMed.
- Y. Jin and Z. Li, et al., Biodegradable, multifunctional DNAzyme nanoflowers for enhanced cancer therapy, NPG Asia Mater., 2017, 9, 1–10 Search PubMed.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Protein Assembly: Versatile Approaches to Construct Highly Ordered Nanostructures, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- J. F. Ross, et al., Decorating Self-Assembled Peptide Cages with Proteins, ACS Nano, 2017, 11, 7901–7914 CrossRef CAS PubMed.
- W. M. Aumiller, M. Uchida and T. Douglas, Protein cage assembly across multiple length scales, Chem. Soc. Rev., 2018, 47, 3433–3469 RSC.
- E. Sasaki, et al., Structure and assembly of scalable porous protein cages, Nat. Commun., 2017, 8, 14663 CrossRef PubMed.
- C. R. Kaiser, et al., Biodistribution studies of protein cage nanoparticles demonstrate broad tissue distribution and rapid clearance in vivo, Int. J. Nanomed., 2007, 2, 715–733 CAS.
- R. Chhabra, J. Sharma, Y. Liu, S. Rinker and H. Yan, DNA self-assembly for nanomedicine, Adv. Drug Delivery Rev., 2010, 62, 617–625 CrossRef CAS PubMed.
- C. Angell, S. Xie, L. Zhang and Y. Chen, DNA Nanotechnology for Precise Control over Drug Delivery and Gene Therapy, Small, 2016, 12, 1117–1132 CrossRef CAS PubMed.
- Y. Zhang, H. F. Chan and K. W. Leong, Advanced materials and processing for drug delivery: the past and the future, Adv. Drug Delivery Rev., 2013, 65, 104–120 CrossRef CAS PubMed.
- S. H. Um, J. B. Lee, N. Park, S. Y. Kwon, C. C. Umbach and D. Luo, Enzyme-catalused assembly of DNA hydrogel, Nat. Mater., 2006, 5, 104–120 CrossRef PubMed.
- G. D. Hamblin, K. M. Carneiro, J. F. Fakhoury, K. E. Bujold and H. F. Sleiman, Rolling circle amplification-templated DNA nanotubes show increased stability and cell penetration ability, J. Am. Chem. Soc., 2012, 134, 2888–2891 CrossRef CAS PubMed.
- J. W. M. Conway, C. K. McLaughlin, K. J. Castor and H. Sleiman, DNA nanostructure serum stability: Greater than the sum of its parts, Chem. Commun., 2013, 49, 1172–1174 RSC.
- J. W. Keum and H. Bermudez, Enhanced resistance of DNA nanostructures to enzymatic digestion, Chem. Commun., 2009, 7036–7038 RSC.
- R. P. Goodman, R. M. Berry and A. J. Turberfield, The single-step synthesis of a DNA tetrahedron, Chem. Commun., 2004, 12, 1372–1373 RSC.
- R. P. Goodman, et al., Rapid chiral assembly of rigid DNA building blocks for molecular nanofabrication, Science, 2005, 310, 1661–1665 CrossRef CAS PubMed.
- A. S. Walsh, H. Yin, C. M. Erben, M. J. Wood and A. J. Turberfield, DNA cage delivery to mammalian cells, ACS Nano, 2011, 5, 5427–5432 CrossRef CAS PubMed.
- S. Obuobi, H. K. Tay, N. D. T. Tram, V. Selvarajan, J. S. K. Khara, Y. Wang and P. L. R. Ee, Facile and Efficient encapsulation of antimicrobial peptides via crosslinked DNA nanostructures and their application in wound therapy, J. Controlled Release, 2019, 313, 120–130 CrossRef CAS PubMed.
- C. M. Erben, R. P. Goodman and A. J. Turberfield, Single-molecule protein encapsulation in a rigid DNA cage, Angew. Chem., Int. Ed., 2006, 45, 7414–7417 CrossRef CAS PubMed.
- R. Crawford, et al., Non-covalent single transcription factor encapsulation inside a DNA cage, Angew. Chem., Int. Ed., 2013, 52, 2284–2288 CrossRef CAS PubMed.
- Z. Zhao, et al., Nanocaged enzymes with enhanced catalytic activity and increased stability against protease digestion, Nat. Commun., 2016, 7, 10619 CrossRef CAS PubMed.
- V. Kumar, et al., DNA Nanotechnology for Cancer Therapy, Theranostics, 2016, 6, 710–725 CrossRef CAS PubMed.
- H. V. P. Thelu, et al., Size controllable DNA nanogels from the self-assembly of DNA nanostructures through multivalent host-guest interactions, Nanoscale, 2018, 10, 222–230 RSC.
- A. Sprengel, et al., Tailored protein encapsulation into a DNA host using geometrically organized supramolecular interactions, Nat. Commun., 2017, 8, 14472 CrossRef CAS PubMed.
- E. Kim, et al., One-Pot Synthesis of Multiple Protein-Encapsulated DNA Flowers and Their Application in Intracellular Protein Delivery, Adv. Mater., 2017, 29, 1701086 CrossRef PubMed.
- J. Willem de Vries, et al., DNA nanoparticles for ophthalmic drug delivery, Biomaterials, 2018, 157, 98–106 CrossRef CAS PubMed.
- J. Li, et al., Self-assembly of DNA nanohydrogels with controllable size and stimuli-responsive property for targeted gene regulation therapy, J. Am. Chem. Soc., 2015, 137, 1412–1415 CrossRef CAS PubMed.
- A. M. Rush, M. P. Thompson, E. T. Tatro and N. C. Gianneschi, Nuclease-resistant DNA via high-density packing in polymeric micellar nanoparticle coronas, ACS Nano, 2013, 7, 1379–1387 CrossRef CAS PubMed.
- Y. Ma, H. Liu, Q. Mou, D. Yan, X. Zhu and C. Zhang, Floxuridine-containing nucleic acid nanogels for anticancer drug delivery, Nanoscale, 2018, 10, 8367–8371 RSC.
- Y. Burak, G. Ariel and D. Andelman, Onset of DNA aggregation in presence of monovalent and multivalent counterions, Biophys. J., 2003, 85, 2100–2110 CrossRef CAS PubMed.
- G. C. L. Wong and L. Pollack, Electrostatics of Strongly Charged Biological Polymers: Ion-Mediated Interactions and Self-Organization in Nucleic Acids and Proteins, Annu. Rev. Phys. Chem., 2010, 61, 171–189 CrossRef CAS PubMed.
- S. Kewalramani, G. I. Guerrero-Garcia, L. M. Moreau, J. W. Zwanikken, C. A. Mirkin, M. O. de la Cruz and M. J. Bedzyk, Electrolyte-Mediated Assembly of Charged Nanoparticles, ACS Cent. Sci., 2016, 2, 219–224 CrossRef CAS PubMed.
- S. Bontha, A. V. Kabanov and T. K. Bronich, Polymer micelles with cross-linked ionic cores for delivery of anticancer drugs, J. Controlled Release, 2006, 114, 163–174 CrossRef CAS PubMed.
- A. G. McArthur, et al., The comprehensive antibiotic resistance database, Antimicrob. Agents Chemother., 2013, 57, 3348–3357 CrossRef CAS PubMed.
- J. P. Silva, Delivery of LLKKK18 loaded into self-assembling hyaluronic acid nanogel for tuberculosis treatment, J. Controlled Release, 2016, 235, 112–124 CrossRef CAS PubMed.
- J. S. Khara, et al., Disruption of drug-resistant biofilms using de novo designed short alpha-helical antimicrobial peptides with idealized facial amphiphilicity, Acta Biomater., 2017, 57, 103–114 CrossRef CAS PubMed.
- M. Fumakia and E. A. Ho, Nanoparticles Encapsulated with LL37 and Serpin A1 Promotes Wound Healing and Synergistically Enhances Antibacterial Activity, Mol. Pharm., 2016, 13, 2318–2331 CrossRef CAS PubMed.
- A. M. Piras, Chitosan nanoparticles loaded with the antimicrobial peptide temporin B exert a long-term antibacterial activity in vitro against clinical isolates of Staphylococcus epidermidis, Front. Microbiol., 2015, 6, 372 Search PubMed.
- K. K. Chereddy, et al., PLGA nanoparticles loaded with host defense peptide LL37 promote wound healing, J. Controlled Release, 2014, 194, 138–147 CrossRef CAS PubMed.
- A. Keller and V. Linko, Challenges and Perspectives of DNA Nanostructures in Biomedicine, Angew. Chem., Int. Ed., 2020, 59, 2–18 CrossRef.
- G. A. Pankey and L. D. Sabath, Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections, Clin. Infect. Dis., 2004, 38, 864–870 CrossRef CAS PubMed.
- A. Ebbensgaard, et al., Comparative Evaluation of the Antimicrobial Activity of Different Antimicrobial Peptides against a Range of Pathogenic Bacteria, PLoS One, 2015, 10, e0144611 CrossRef PubMed.
- G. Zhu, et al., Self-assembled, aptamer-tethered DNA nanotrains for targeted transport of molecular drugs in cancer theranostics, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7998–8003 CrossRef CAS PubMed.
- D. Mathur and I. L. Medintz, The Growing Development of DNA Nanostructures for Potential Healthcare-Related Applications, Adv. Healthcare Mater., 2019, 8, e1801546 CrossRef PubMed.
- R. J. O'Callaghan, The Pathogenesis of Staphylococcus aureus Eye Infections, Pathogens, 2018, 7, 1–22 Search PubMed.
- A. Lichtinger, et al., Shifting trends in bacterial keratitis in Toronto: an 11-year review, Ophthalmology, 2012, 119, 1785–1790 CrossRef PubMed.
- N. Ni, et al., Seasonal, geographic, and antimicrobial resistance patterns in microbial keratitis: 4-year experience in eastern Pennsylvania, Cornea, Cornea, 2015, 34, 294–302 Search PubMed.
- X. W. Tan, et al., Effectiveness of antimicrobial peptide immobilization for preventing perioperative cornea implant-associated bacterial infection, Antimicrob. Agents Chemother., 2014, 58, 5229–5238 CrossRef PubMed.
- L. D. Hazlett, M. M. Moon, M. Strejc and R. S. Berk, Evidence for N-acetylmannosamine as an ocular receptor for P. aeruginosa adherence to scarified cornea, Invest. Ophthalmol. Visual Sci., 1987, 28, 1978–1985 CAS.
- Q. Zhang, et al., Anti-inflammatory and Antioxidative Effects of Tetrahedral DNA Nanostructures via the Modulation of Macrophage Responses, ACS Appl. Mater. Interfaces, 2018, 10, 3421–3430 CrossRef CAS PubMed.
- M. Venkatesh, et al., Antimicrobial Activity and Cell Selectivity of Synthetic and Biosynthetic Cationic Polymers, Antimicrob. Agents Chemother., 2017, 61, e00469–e00417 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr03095c |
| ‡ Present address: Drug Transport and Delivery Research Group, Department of Pharmacy, Faculty of Health Sciences, UiT-The Arctic University of Norway, N-9037, Tromsø, Norway. |
| This journal is © The Royal Society of Chemistry 2020 |