
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalently functionalized layered MoS2 supported Pd nanoparticles as highly active oxygen reduction electrocatalysts†
Dimitrios K.
Perivoliotis
 *a,
Yuta
Sato
b,
Kazu
Suenaga
b and
Nikos
Tagmatarchis
*a
*a,
Yuta
Sato
b,
Kazu
Suenaga
b and
Nikos
Tagmatarchis
*a
aTheoretical and Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vassileos Constantinou Avenue, 11635 Athens, Greece. E-mail: tagmatar@eie.gr; dimperiv@eie.gr
bNanomaterials Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Central 5, 1-1-1 Higashi, Tsukuba 305-8565, Japan
First published on 6th August 2020
Abstract
Molybdenum disulfide nanosheets covalently modified with a 1,2-dithiolane derivative were used as a novel substrate for the immobilization of Pd nanoparticles (PdNPs) towards the development of a highly efficient hybrid electrocatalyst, namely PdNPs/f-MoS2, for the oxygen reduction in an alkaline medium. The newly prepared hybrid material was thoroughly characterized through complementary techniques such as Raman and IR spectroscopy, TGA, HRTEM, STEM/EELS, and EDS. The PdNPs/f-MoS2 nanohybrid exhibited excellent performance towards oxygen electroreduction with a positive onset potential of +0.066 V and a half-wave potential of −0.116 V vs. Hg/HgO, along with a high current response, which are superior to those of its graphene counterpart and comparable to those of the benchmark Pd/C product. Moreover, PdNPs/f-MoS2 was proved to be remarkably stable as chronoamperometric assays showed minimum activity loss among the tested materials, clearly outperforming the commercial catalyst. The excellent performance of PdNPs/f-MoS2 is attributable to (i) the high affinity of the catalytic PdNPs with the f-MoS2 substrate, (ii) the absence of any capping agent for the stabilization of PdNPs onto f-MoS2, and more importantly (iii) the preservation of the integrity of the MoS2 basal plane during the functionalization process. Lastly, the oxygen reduction on PdNPs/f-MoS2 proceeded through the energy efficient four-electron pathway, showing great potential for the use of layered transition metal dichalcogenides in energy conversion applications, comprising fuel cells.
Introduction
Tailoring electrocatalysts to accelerate the sluggish kinetics of the oxygen reduction reaction (ORR) still remains a key challenge towards the commercialization of the fuel cell technology.1 In this regard, supported catalysts for the ORR have drawn much attention, since they offer not only enhanced electrocatalytic activities, but also more importantly improved durability. Notably, the efficient interactions between a nanoparticle catalyst and a support could modify the electronic structure of the catalyst, facilitating the oxygen adsorption and dissolution, while, at the same time, prevent nanoparticles from aggregation, contributing to long term stability.2Graphene, a two-dimensional (2D) sp2 hybridized carbon allotrope, has been widely used as a supporting material, owing to its high surface area and electrical conductivity as well as good chemical stability.3 Beyond graphene, layered transition metal dichalcogenides, abbreviated as TMDs, constitute another class of 2D materials that combine the exceptional properties of graphene with remarkable intrinsic electrocatalytic activities toward key electrochemical reactions, originating from a large number of exposed prismatic edges.4,5 Actually, TMDs are atomically thick nanocrystals with MX2 stoichiometry, where M refers to a transition metal atom (e.g. Mo, W, Ta) and X to a chalcogen atom (S, Se, Te).6 Among various TMDs, molybdenum disulfide (MoS2) nanosheets have received special scientific attention as they are readily accessible and chemically stable.7 Liquid phase exfoliation from the bulk material is the most effective way to obtain high quality layered MoS2 on a large scale.8–11 Similar to other TMDs, MoS2 is characterized by a thickness dependent semiconducting behavior and an indirect-to-direct bandgap transition occurs when its thickness decreases to a single layer.12 Moreover, the MoS2 monolayer presents a much lower bandgap (∼1.2 eV) than typical semiconducting materials (>3 eV),13 while its electronic structure is closely related to the coordination of the Mo atoms. All in all, these fascinating properties give layered MoS2 a clear advantage over graphene, making it an attractive component for potential use in energy conversion (electrocatalysis14 and (photo)-electrocatalysis15) and storage16–19 applications.
Similar to graphene, in order to fully exploit the intriguing properties of layered TMDs, their chemical functionalization is an absolute necessity.20 Needless to mention, the introduction of functional groups on their surface could not only solve the problem of their limited solubility in common solvents and restacking of nanosheets, but also allows the design of entirely new hybrid materials. Markedly, the covalent attachment of the appropriate organic moieties on MoS2 sheets’ surface makes them ideal platforms for the immobilization of various catalytic nanoparticles by effectively tuning the interfacial interactions and thereby leading to the formation of hybrids with increased electrocatalytic activity and excellent durability. To date, diverse methodologies have been developed towards the covalent functionalization of layered MoS2,21 including the reaction with electrophiles e.g. organoiodides and aryldiazonium salts22 and organic thiols.23 In the latter case, a Mo–S bond is formed between an unsaturated molybdenum atom in MoS2 and a sulfur atom in the organic functional group. Interestingly, unsaturated Mo atoms are dominant at the edges of the MoS2 nanosheets, where structural defects (S vacancies) have been naturally introduced during the exfoliation process.8 Therefore, the MoS2 basal plane remains intact and hence its exceptional properties, which are essential when considering the design of high performance electrocatalysts, are preserved. However, a major drawback of this approach is related to the possible conversion of thiols into the corresponding disulfides – a reaction that is activated in the presence of MoS2. Based on this, an alternative route employing 1,2-dithiolanes as robust thiol groups to react with exfoliated MoS2 sheets has been recently developed.24
Despite the huge potential of MoS2 as a catalyst substrate, only a few reports have been published employing MoS2 sheets as a supporting material for metal-based catalytic nanoparticles towards the ORR.25–27 Actually, the main bottlenecks that keep MoS2 behind the scenes are related to their low conductivity and the poor dispersity and the homogeneity of the resulting hybrids.14 Therefore, developing efficient synergistic interactions between MoS2 sheets and noble metal nanoparticles is essential to overcome these limitations.28 To this end, diverse methodologies25,26e.g. epitaxial growth, physical mixing, and sonochemical approaches, have been involved for the preparation of such hybrid materials aiming at the optimization of the ORR performance of the supported catalysts. Despite the advancements that have been made in the field, the ORR performance of the reported hybrids still needs improvement to compete with the benchmark catalysts. In the same context, the surface functionalization of MoS2 sheets could tune the intra-hybrid interactions, and hence significantly boost the performance of the hybrid electrocatalysts. However, to the best of our knowledge, the use of modified layered MoS2 as an ORR catalyst substrate has not been reported yet.
The present work goes beyond the current state-of-the-art by employing covalent functionalized MoS2 nanosheets with a 1,2-dithiolane derivative (f-MoS2), featuring free amino groups, as a substrate for immobilization of Pd nanoparticles (PdNPs). The obtained hybrid, abbreviated as PdNPs/f-MoS2, was examined as the electrocatalyst for the ORR under alkaline conditions and its performance was further compared with that of its counterpart based on functionalized graphene (f-G), abbreviated as PdNPs/f-G. The results clearly demonstrated an overall improved performance of PdNPs/f-MoS2 over that of PdNPs/f-G, highlighting the benefits of using the TMD-based substrate. The exceptional ORR activity and stability of PdNPs/f-MoS2 was ascribed to (i) the high affinity of PdNPs with the f-MoS2 substrate, (ii) the absence of any capping agent for the stabilization of PdNPs onto f-MoS2 and more importantly (iii) the preservation of the integrity of the MoS2 basal plane during the functionalization process. Markedly, the latter explains to a large extent why PdNPs/f-MoS2 outperforms the graphene-based PdNPs/f-G electrocatalyst as in this case the covalent functionalization induces damage to the graphene lattice leading to the deterioration of its inherent properties. Generally speaking, the latter is the main obstacle in using covalently functionalized graphene as a component in electrocatalytic applications and thus the current research on graphene-based electrocatalysts has been focused on non-covalently modified as well as doped graphene.3,29–31 This work proposes the use of covalently edge-functionalized MoS2 nanosheets as platforms for the fabrication of the next generation of hybrid electrocatalysts, paving the way for their further use in real energy conversion applications.
Results and discussion
Preparation
The preparation of PdNPs/f-MoS2 and PdNPs/f-G hybrids is illustrated in Scheme 1. First, MoS2 and graphene sheets were covalently functionalized with organic moieties bearing free amino groups to enable the attachment of PdNPs on their surface. As for MoS2, exfoliated MoS2 nanosheets (exf-MoS2) were obtained upon chlorosulfonic acid treatment of the bulk material8 and modified with the 1,2-dithiolane N-tert-butoxycarbonyl (BOC) derivative. Notably, the latter reaction is based on the high binding affinity of the 1,2-dithiolane component for the Mo atoms that are located at the edges of MoS2, where sulfur vacancy sites were introduced during the exfoliation process.24 On the other hand, graphite was oxidized to graphene oxide (GO) and the GO functionalization was realized upon (i) activation of the carboxylic acid groups by acyl chloride followed by the condensation reaction with BOC-2,2′-(ethylenedioxy)bis-ethylamine32 and (ii) the ring-opening reaction of the epoxy groups present on the graphene lattice by the nucleophilic attack of the BOC-2,2′-(ethylenedioxy)bis-ethylamine, yielding covalently functionalized graphene sheets at both edges and the basal plane simultaneously.33 Importantly, in both MoS2 and graphene modified nanosheets, the attached amine species were masked with the BOC-protecting group, which was effectively cleaved after acidic treatment, yielding amino functionalized MoS2 and graphene materials, respectively (Scheme 1). Afterwards, a modified polyol method was employed for the in situ synthesis and immobilization of PdNPs onto f-MoS2 and f-G substrates towards the realization of PdNPs/f-MoS2 and PdNPs/f-G hybrids. At this point, it is worth noting that the free amino groups serve as anchoring sites for metal-ion nucleation and subsequent nanoparticle growth, stabilizing effectively the in situ formed nanoparticles without using any additional capping agent, which significantly impair their electrocatalytic activity.34,35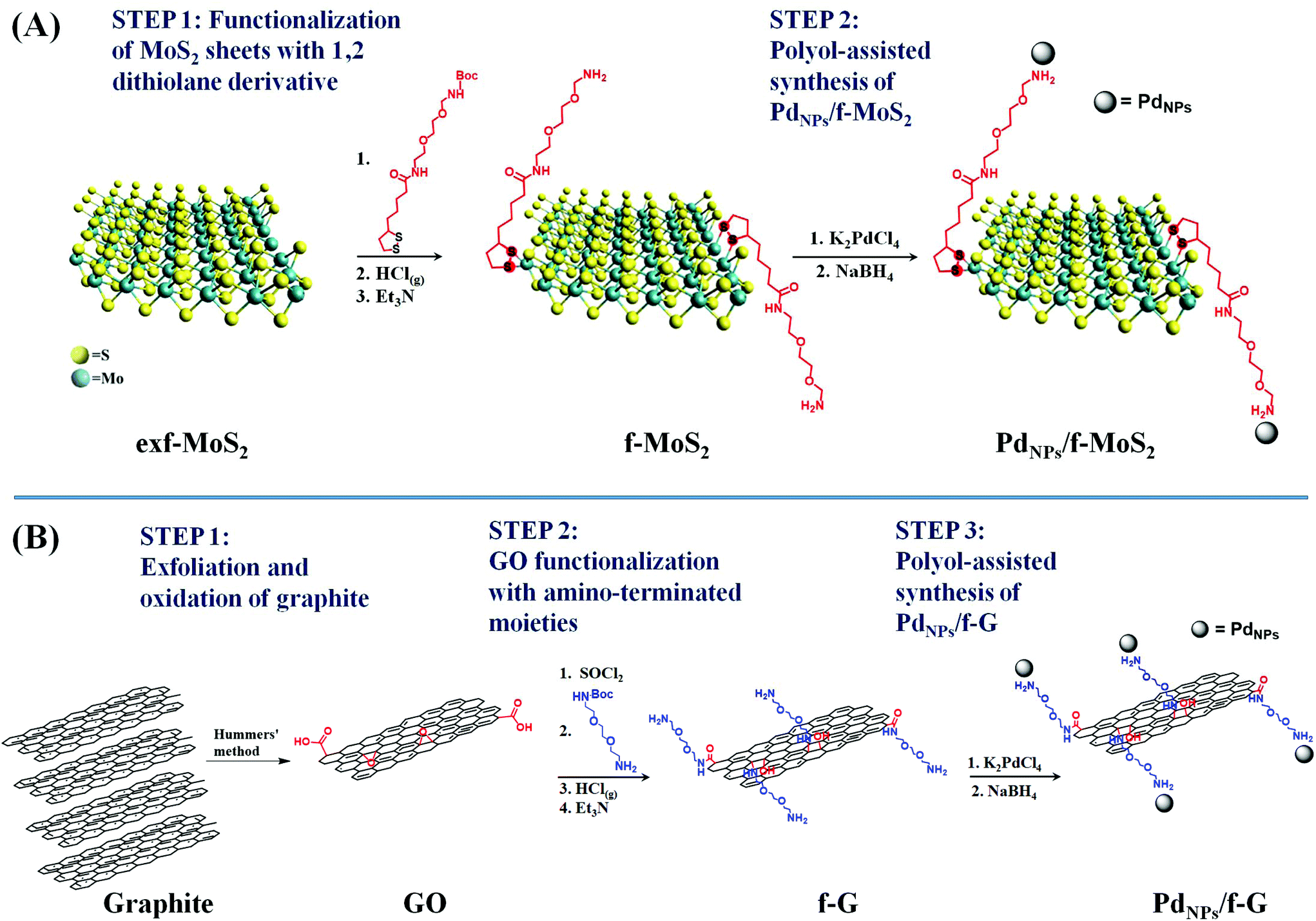 | ||
| Scheme 1 Illustrative scheme for the preparation protocol of (A) PdNPs/f-MoS2 and (B) PdNPs/f-G hybrids. | ||
Characterization
Direct proof for the formation of f-MoS2 was given by complementary characterization tools. In this context, the ATR-IR spectrum of BOC-modified MoS2 is governed by bands in the 2840–2960 cm−1 range owing to the alkyl chain C–H vibration modes, while two discrete bands at 1645 and 1707 cm−1 attributed to carbonyl amide and BOC units, respectively, are also present (Fig. 1, panel A). Interestingly, the band of the latter is absent in the ATR-IR spectrum of f-MoS2, indicating the successful BOC deprotection process. Moreover, a band at 3225 cm−1 owing to the N–H stretching can be observed in the ATR-IR spectrum of f-MoS2, further proving the presence of the free amine group on the MoS2 surface. Next, Raman analysis of f-MoS2 and exf-MoS2 sheets further confirmed the accomplishment of MoS2 covalent functionalization (Fig. 1, panel B). Upon excitation at 633 nm, which produces resonance Raman enhancement of the first and second order vibrational modes, both exf-MoS2 and f-MoS2 Raman spectra clearly revealed four dominant Raman bands, namely A1g-LA(M), E12g, A1g and 2LA(M) at 177, 378, 405 and 450 cm−1, respectively. The E12g and A1g modes derive from the in-plane and out-of-plane vibrations within the S–Mo–S layer, while the 2LA(M) vibration mode is closely related to the S vacancies.36,37 Therefore, the intensity ratio of 2LA(M) to A1g is commonly used as an indicator to quantify the functionalization degree in layered MoS2 materials.38–40 Here, the 2LA(M)/A1g value was found to be decreased for f-MoS2, implying the elimination of S vacancies due to the functionalization process. The latter observation was further confirmed through complementary Raman mapping assays, revealing an average intensity ratio 2LA(M)/A1g of 0.75 for f-MoS2versus 1.35 for the exfoliated MoS2 sheets, as presented in panels C and D of Fig. 1. Please note that we have performed various mappings in both exf-MoS2 and f-MoS2 materials (5 mappings with 121 acquisition points each) and we here present a representative one close to the total average. It is also worth mentioning that no significant changes in the f-MoS2 Raman spectrum are observed after the immobilization of PdNPs, suggesting that no structural alterations occurred during the synthesis of the hybrid material (Fig. S1†). Lastly, the functionalization degree was quantified by means of thermogravimetric analysis (TGA). Given that exf-MoS2 sheets are thermally stable up to 900 °C, the weight loss (around 6%) observed in the temperature range 200–500 °C for f-MoS2 (Fig. 1, panel E) is ascribed to the thermal decomposition of the covalently grafted organic functionalities onto f-MoS2. Based on these data, the loading of the organic material was calculated to be 186 μmol g−1.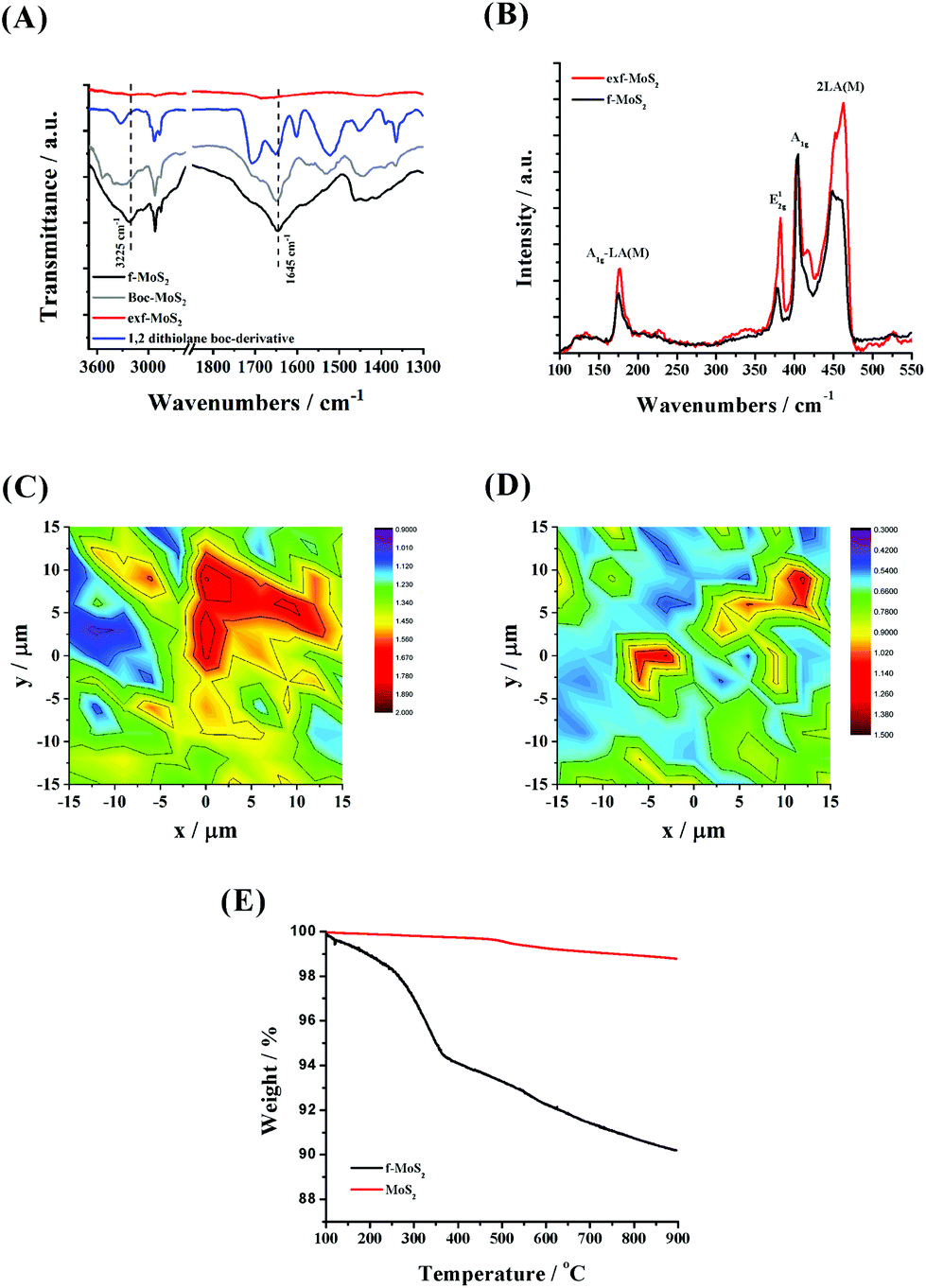 | ||
| Fig. 1 (A) ATR-IR spectra for amino functionalized MoS2 (black), BOC modified MoS2 (gray) and 1,2-dithiolane derivatives (blue). (B) Raman spectra for amino functionalized MoS2 (black) and exfoliated MoS2 (red) upon excitation at 633 nm. (C and D) Raman mapping upon excitation at 633 nm of the 2LA(M)/A1g intensity ratio of a 30 × 30 mm2 area (121 acquisition points in total) for exf-MoS2 and f-MoS2, respectively. (E) Thermographs for amino functionalized MoS2 (black) and exfoliated MoS2 (red). | ||
Concerning f-G, ATR-IR assays provided spectroscopic proof for the success of the functionalization (Fig. S2,† panel A). In short, although the IR spectrum of GO is governed by bands centered at 1720 cm−1 (carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), the BOC-functionalized graphene material manifests a different and broad band at 1708 cm−1 due to the BOC protecting groups and the carbonyl amide formed. Moreover, strong vibration bands are recorded in the region 2840–2960 cm−1 (C–H stretching and bending) and at 3225 cm−1 (N–H stretching), further proving the attachment of the organic moiety onto graphene nanosheets. Notably, in the IR spectrum of f-G, in which free amine functionalities are present, the band related to the BOC unit was disappeared, while a pronounced peak at 1655 cm−1 derived from the carbonyl amide is present. The epoxy group ring-opening reaction is also confirmed via ATR-IR measurements, as the bands assigned to the C–O–C vibrations (at 1200–1250 cm−1) that are observed in the GO spectrum are absent in both BOC-G and f-G materials. Raman spectroscopy was employed to estimate the disorder degree in f-G (Fig. S2,† panels B–D) through the evaluation of the relative intensity of the defect activated D-band at around 1350 cm−1.41 Briefly, the D/G intensity ratio for f-G was found to be close to 1.0, witnessing its defective nature. Notably, the presence of sp3 hybridized carbon atoms is mainly related to the introduction of the oxygen containing groups (the D/G intensity ratio for GO is around to 0.90) which are essential for further graphene functionalization. It was also demonstrated (Fig. S3†) that the in situ synthesis and immobilization of PdNPs were not affected by the graphene structure. Lastly, the loading of the organic functionalities was determined through TGA (Fig. S2,† panel E) and it was found to be 840 μmol g−1, being considerably higher than that registered for the f-MoS2 material; in contrast to MoS2, the GO functionalization takes place at both edges and the basal plane.
O), the BOC-functionalized graphene material manifests a different and broad band at 1708 cm−1 due to the BOC protecting groups and the carbonyl amide formed. Moreover, strong vibration bands are recorded in the region 2840–2960 cm−1 (C–H stretching and bending) and at 3225 cm−1 (N–H stretching), further proving the attachment of the organic moiety onto graphene nanosheets. Notably, in the IR spectrum of f-G, in which free amine functionalities are present, the band related to the BOC unit was disappeared, while a pronounced peak at 1655 cm−1 derived from the carbonyl amide is present. The epoxy group ring-opening reaction is also confirmed via ATR-IR measurements, as the bands assigned to the C–O–C vibrations (at 1200–1250 cm−1) that are observed in the GO spectrum are absent in both BOC-G and f-G materials. Raman spectroscopy was employed to estimate the disorder degree in f-G (Fig. S2,† panels B–D) through the evaluation of the relative intensity of the defect activated D-band at around 1350 cm−1.41 Briefly, the D/G intensity ratio for f-G was found to be close to 1.0, witnessing its defective nature. Notably, the presence of sp3 hybridized carbon atoms is mainly related to the introduction of the oxygen containing groups (the D/G intensity ratio for GO is around to 0.90) which are essential for further graphene functionalization. It was also demonstrated (Fig. S3†) that the in situ synthesis and immobilization of PdNPs were not affected by the graphene structure. Lastly, the loading of the organic functionalities was determined through TGA (Fig. S2,† panel E) and it was found to be 840 μmol g−1, being considerably higher than that registered for the f-MoS2 material; in contrast to MoS2, the GO functionalization takes place at both edges and the basal plane.
The surface morphology of PdNPs/f-MoS2 and PdNPs/f-G hybrids was thoroughly investigated via high-resolution and annular dark-field scanning transmission electron microscopy (HRTEM and ADF-STEM, respectively), whereas their elemental composition was probed by energy dispersive spectroscopy (EDS) and electron energy loss spectroscopy (EELS) chemical mapping. The HRTEM images of PdNPs/f-MoS2 and PdNPs/f-G ensembles (Fig. 2, panels A and D) revealed the uniform distribution of PdNPs onto f-MoS2 and f-G, respectively, as only a few agglomerations are observed. Actually, the free amino groups possess high affinity with PdNPs, stabilizing them effectively on f-MoS2 and f-G substrates.34 Interestingly, focusing on the PdNPs/f-MoS2 HRTEM image, the presence of a few overlapping MoS2 monolayers is observed, while PdNPs is mainly located at the edges, in contrast to the case of PdNPs/f-G hybrids in which PdNPs can be found on the entire f-G surface. This finding is in good agreement with the hypothesis that the covalent modification of MoS2 sheets with 1,2-dithiolane derivatives takes place only at their edges. Next, higher magnification ADF-STEM images revealed that the average size of PdNPS is 2–3 nm for both hybrid materials (Fig. 2, panels B and E), while at the same time, EDS and EELS elemental analysis (Fig. 2, panels C and F) confirmed the existence of Mo, S and Pd for PdNPs/f-MoS2 as well as the presence of C and Pd for the PdNPs/f-G hybrid.
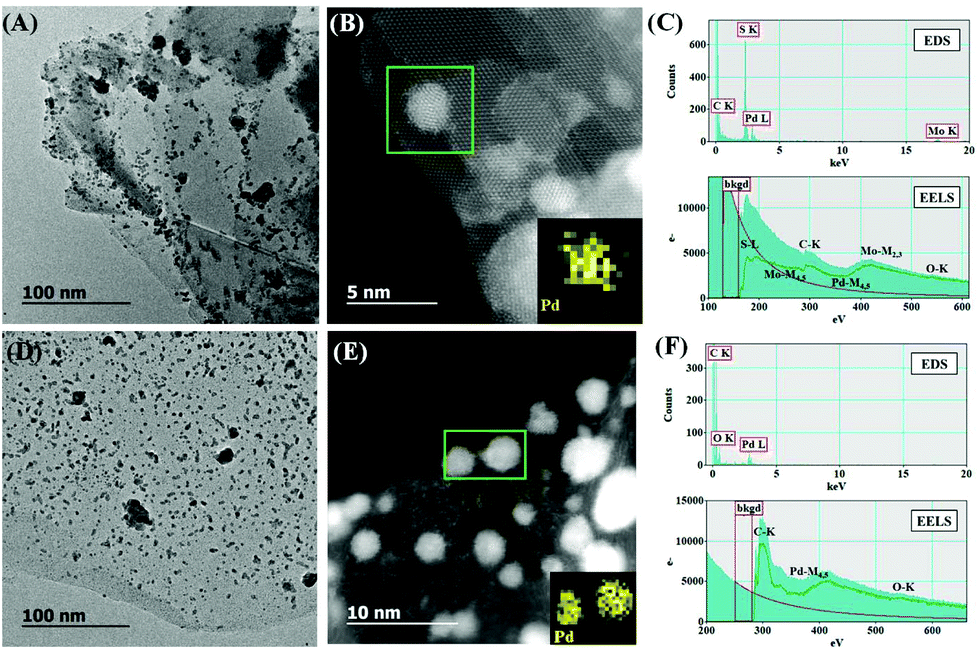 | ||
| Fig. 2 (A–C) Representative HRTEM and ADF-STEM image along with EELS chemical mapping for Pd and EDS and EELS spectra for PdNPs/f-MoS2. (D–F) Representative HRTEM and ADF-STEM image along with EELS chemical mapping for Pd and EDS and EELS spectra for PdNPs/f-G. | ||
Electrocatalytic performance toward the ORR
To explore the ORR performance of PdNPs/f-MoS2 and PdNPs/f-G electrocatalysts, linear sweep voltammetry (LSV) measurements on the rotating ring-disk electrode (RRDE) were carried out in O2-saturated 0.1 M KOH electrolyte at a rotation rate of 1600 rpm. As a control, the performance towards the ORR of the benchmark Pd/C catalyst was also recorded. As can be seen in Fig. 3, the PdNPs/f-MoS2 hybrid presented by far improved the ORR activity, clearly outperforming its counterpart based on graphene. Indeed, the ORR onset and half-wave potential values of PdNPs/f-MoS2 were determined to be +66 and −116 mV (vs. Hg/HgO), respectively, being more positive by 50 mV when compared with those recorded for the PdNPs/f-G hybrid. In addition, the diffusion-limited current density (jd) for PdNPs/f-MoS2 reached 5.70 mA cm−2, whereas its kinetic current density (jk) was found to be 2.65 mA cm−2 at −60 mV (vs. Hg/HgO). Notably, these values are 46% and 4.5 times higher over those of PdNPs/f-G. Next, the ORR activity of PdNPs/f-MoS2 was further compared with that of the state-of-the-art catalyst, showing analogous or enhanced performance, especially in terms of onset potential and kinetic current density. Needless to mention, PdNPs/f-MoS2 manifested a more positive onset potential by 53 mV along with a 32.5% greater kinetic current density value at −60 mV (vs. Hg/HgO). Overall, the ORR performance of the tested catalysts was in the order of PdNPs/f-MoS2 > Pd/C > PdNPs/f-G. Interestingly, a consistent result was also obtained through durability experiments, in which the chronoamperometric assays of PdNPs/f-MoS2, PdNPs/f-G and Pd/C were performed at a constant applied potential of −0.45 V (vs. Hg/HgO) for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s and a rotation speed of 1600 rpm. As shown in panel B of Fig. 3, the current response of PdNPs/f-MoS2 retained 84% of its initial value, whereas an activity loss of 31.8 and 29.7 was recorded for PdNPs/f-G and Pd/C, respectively. Next, the LSV profiles for all the tested materials after 10000 s of chronoamperometric assays were recorded in O2-saturated 0.1 M KOH electrolyte at a rotation rate of 1600 rpm (panel A of Fig. 3). The results clearly demonstrate that the PdNPs/f-MoS2 electrocatalyst is remarkably stable, being in total agreement with the chronoamperometric experiments. Needless to say, after 10000 s, PdNPs/f-MoS2 presented less than 10% decrease in its initial diffusion-limited and kinetic current density values, while its half-wave potential value remained almost unchanged. On the other hand, the PdNPs/f-G material experienced a significant loss of its initial performance (23 and 40% decrease in diffusion-limited and kinetic current density values, respectively, as well as a 26 mV negative shift in the half-wave potential value). Last but not least, after 10000 s, the MoS2 based electrocatalyst continues to outperform the commercial Pd/C catalyst by manifesting almost 1.5 and 3.5 times greater diffusion-limited and kinetic current density values, respectively. Interestingly, the observed activity loss after the chronoamperometric assays is mainly attributable to the reduction of the electrochemical active surface area (EASA) of each hybrid electrocatalyst owing to the possible agglomeration of the metal nanoparticles during the chronoamperometric experiment. Given the fact that the EASA is closely related to capacitance,42–44 the capacitance values for PdNPs/f-MoS2 and PdNPs/f-G hybrids as well as the Pd/C were calculated by integrating the graph area derived from cyclic voltammographs obtained in the “double layer region”, according to eqn (6), before and after the chronoamperometric assays.42 Particularly, it was found that the capacitance value of PdNPs/f-MoS2 decreased only by 13.8%, while the same value for PdNPs/f-G and Pd/C was 43 and 31.5%, respectively (Fig. S4†). Based on these data, the superior stability of PdNPs/f-MoS2 is well justified, as it presents the minimum EASA loss, implying the minimum agglomeration.
000 s and a rotation speed of 1600 rpm. As shown in panel B of Fig. 3, the current response of PdNPs/f-MoS2 retained 84% of its initial value, whereas an activity loss of 31.8 and 29.7 was recorded for PdNPs/f-G and Pd/C, respectively. Next, the LSV profiles for all the tested materials after 10000 s of chronoamperometric assays were recorded in O2-saturated 0.1 M KOH electrolyte at a rotation rate of 1600 rpm (panel A of Fig. 3). The results clearly demonstrate that the PdNPs/f-MoS2 electrocatalyst is remarkably stable, being in total agreement with the chronoamperometric experiments. Needless to say, after 10000 s, PdNPs/f-MoS2 presented less than 10% decrease in its initial diffusion-limited and kinetic current density values, while its half-wave potential value remained almost unchanged. On the other hand, the PdNPs/f-G material experienced a significant loss of its initial performance (23 and 40% decrease in diffusion-limited and kinetic current density values, respectively, as well as a 26 mV negative shift in the half-wave potential value). Last but not least, after 10000 s, the MoS2 based electrocatalyst continues to outperform the commercial Pd/C catalyst by manifesting almost 1.5 and 3.5 times greater diffusion-limited and kinetic current density values, respectively. Interestingly, the observed activity loss after the chronoamperometric assays is mainly attributable to the reduction of the electrochemical active surface area (EASA) of each hybrid electrocatalyst owing to the possible agglomeration of the metal nanoparticles during the chronoamperometric experiment. Given the fact that the EASA is closely related to capacitance,42–44 the capacitance values for PdNPs/f-MoS2 and PdNPs/f-G hybrids as well as the Pd/C were calculated by integrating the graph area derived from cyclic voltammographs obtained in the “double layer region”, according to eqn (6), before and after the chronoamperometric assays.42 Particularly, it was found that the capacitance value of PdNPs/f-MoS2 decreased only by 13.8%, while the same value for PdNPs/f-G and Pd/C was 43 and 31.5%, respectively (Fig. S4†). Based on these data, the superior stability of PdNPs/f-MoS2 is well justified, as it presents the minimum EASA loss, implying the minimum agglomeration.
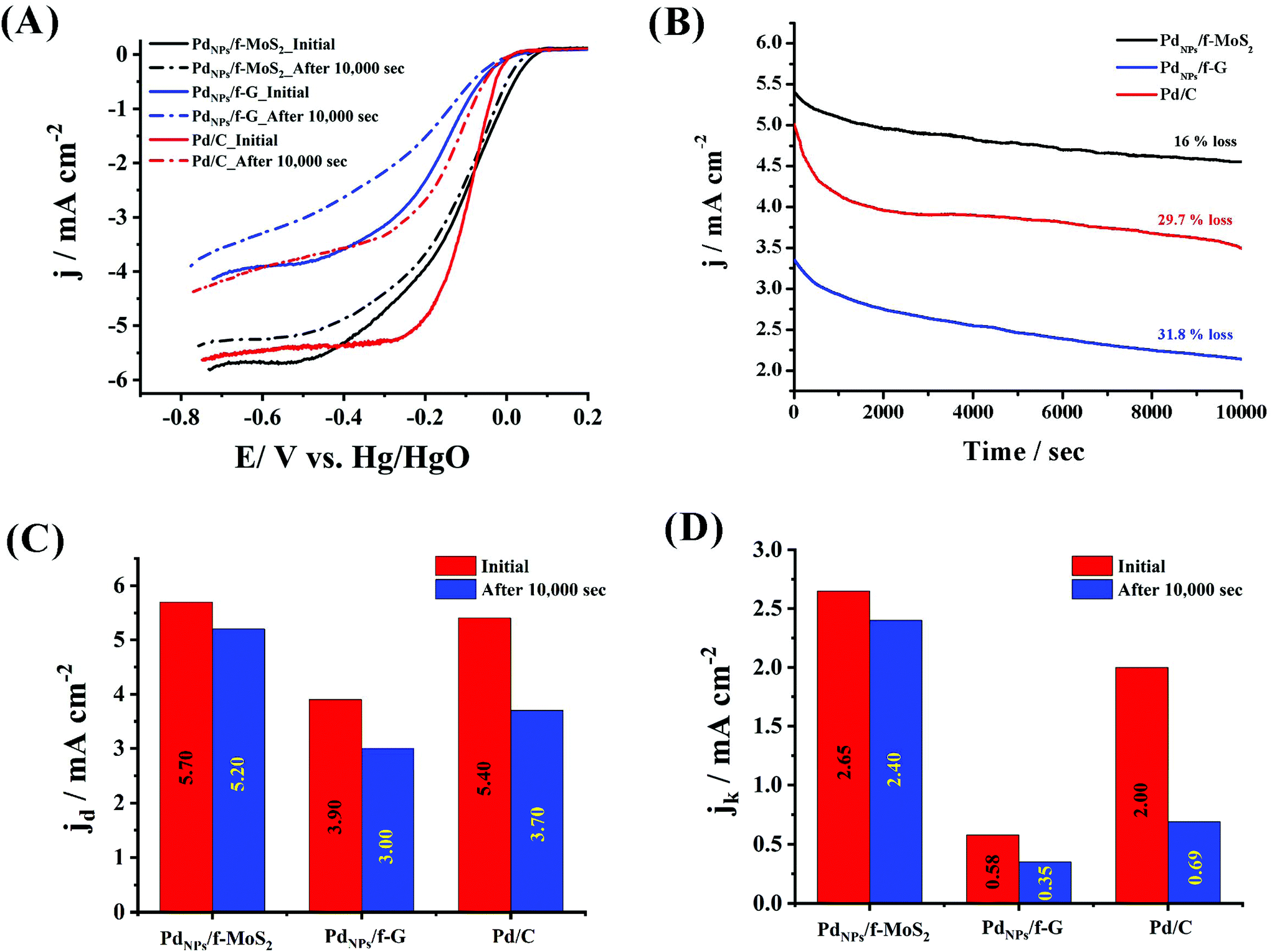 | ||
| Fig. 3 (A) LSV ORR polarization curves obtained at a rotation speed of 1600 rpm for PdNPs/f-MoS2 and PdNPs/f-G before and after 10000 s of chronoamperometric assays as compared to that due to Pd/C; (B) ORR chronoamperometric response for all materials at −0.45 V (vs. Hg/HgO) for 10000 s and (C and D) corresponding diffusion limited and kinetic current density values. All measurements were conducted in O2-saturated 0.1 M KOH electrolyte, and the corresponding electrocatalyst LSVs were recorded at a scan rate of 5 mV s−1. In all graphs, the current densities are normalized to the geometric electrode area. | ||
To elucidate the reaction kinetics, hydrodynamic voltammetry experiments were conducted by altering the electrode's rotation speed. The related ORR polarization curves for PdNPs/f-MoS2 and PdNPs/f-G obtained at different rotation rates between 400 and 3600 rpm are presented in panel A of Fig. 4 and panel A of Fig. S5,† respectively, showing the typical increase in current with the increase of the rotation speed due to the shortening of the diffusion layer at high speeds. Next, the corresponding Koutecký–Levich (K–L) plots (Fig. 4, panel B and Fig. S5,† panel B) were constructed at different potentials based on the inverse of the current density (j−1) against the inverse of the square root of the rotation rate (ω−1/2). As can be seen, these plots presented good linearity, implying first order reaction kinetics towards the dissolved O2 concentration. Based on these analyses, the electron transfer number per oxygen molecule (n) was estimated and it was found to be 3.6–4.0 and 3.4–3.6 for PdNPs/f-MoS2 and PdNPs/f-G, respectively. Such an n value clearly suggests a four electron selectivity for the MoS2 based ensemble, in which oxygen reduced directly into water. Lastly, for comparison reasons, the electrocatalytic properties of the f-MoS2 substrate have also been investigated (Fig. S6†). Briefly, f-MoS2 was found to present a substantial intrinsic oxygen reduction activity, mainly proceeding through the two-electron pathway (n = 2.1–2.3).
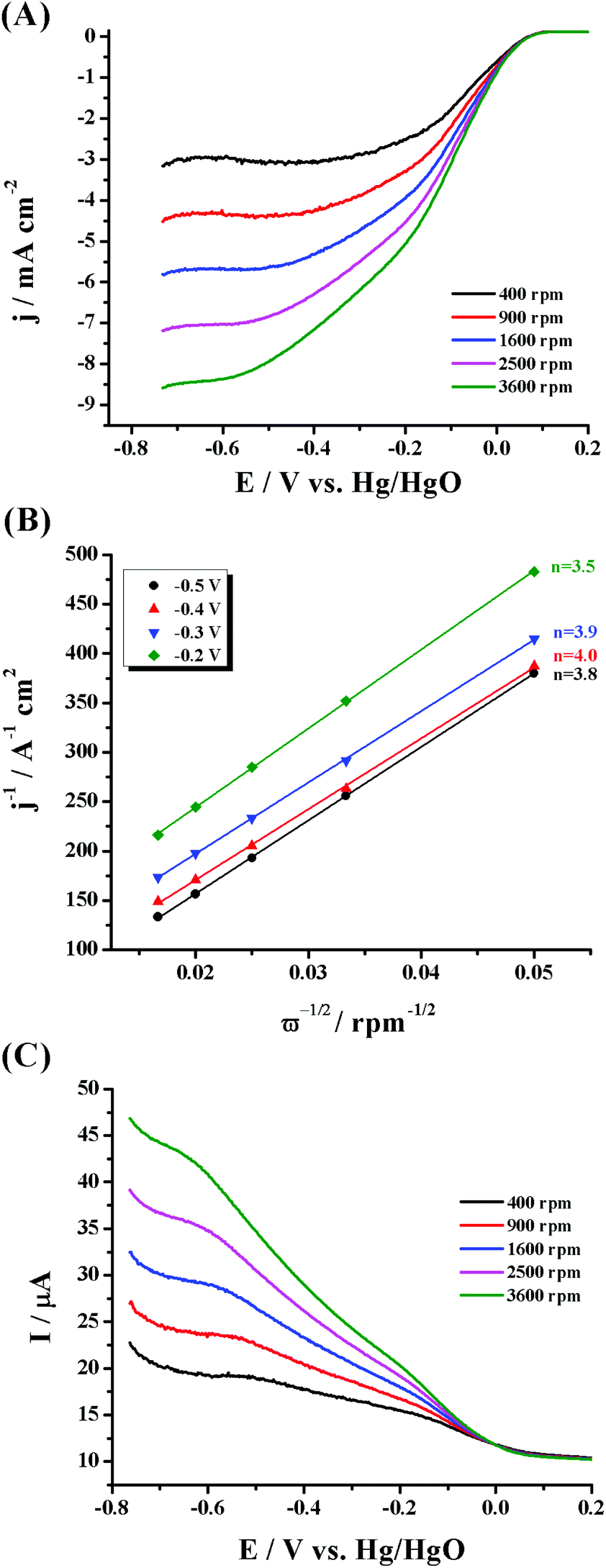 | ||
| Fig. 4 (A) ORR polarization curves at different rotation rates (400–3600 rpm) for PdNPs/f-MoS2, (B) the corresponding K–L plots, and (C) ring response. All measurements were conducted in O2-saturated aqueous 0.1 M KOH electrolyte, and the corresponding LSV polarization curves were recorded at a scan rate of 5 mV s−1. The ring potential was fixed at 1.0 V vs. Hg/HgO. The current densities in (A) are normalized to the geometric electrode area. | ||
The RRDE approach was applied to further scrutinize the ORR kinetics of PdNPs/f-MoS2 and PdNPs/f-G hybrids (Fig. 4, panel C and Fig. S5,† panel C, respectively). Markedly, the recorded ring current corresponds to the amount of the hydrogen peroxide intermediate produced at the disk electrode during the ORR, providing us not only a more accurate way to calculate the n value, but also a reliable method to estimate the percentage (%) of the produced H2O2.45 In this regard, the electron transfer number and the H2O2 yield for PdNPs/f-MoS2 in the potential range from 0.2 to 0.5 V (vs. Hg/HgO) were found to be 3.6 and 20%, respectively, suggesting that the oxygen reduction was mostly proceeded through the four electron transfer route and the main product was water. In general, the four-electron pathway is considered as more efficient and it is highly preferred in energy conversion applications. On the other hand, in the case of PdNPs/f-G, the two-electron and four-electron pathways coexist as n and % H2O2 were calculated to be 3.1 and 47%, respectively. Markedly, these results are consistent with those extracted through the K–L method.
Next, to gain insight into the rate-determining step involved in the oxygen electroreduction, the mass transfer corrected Tafel plots for all tested catalysts were constructed. As shown in Fig. 5, in the low current density region (region I), the PdNPs/f-MoS2 and PdNPs/f-G hybrids exhibited a slope of −40 and −70 mV dec−1, respectively, which are roughly close to that of Pd/C (−46 mV dec−1). Actually, a Tafel slope near −60 mV dec−1 in this region indicates that the reaction rate is governed by the surface oxide coverage (Temkin adsorption mechanism).46 In the high current density region (region II), a Tafel slope value of −180 mV dec−1 was recorded for both PdNPs/f-MoS2 and PdNPs/f-G, whereas the corresponding value for the commercial catalyst was found to be −112 mV dec−1. The higher Tafel value in region II is ascribed to a switch in the oxygen reduction mechanism since at these potentials the significant oxide coverage ceases to exist. In more depth, a slope close to −120 mV dec−1 indicates that the ORR is limited by the transfer of the first electron to the oxygen molecule (Langmuir adsorption), while for a value around −200 mV dec−1, the adsorption of molecular oxygen has been proposed as the rate-determining step.47,48 Therefore, the abovementioned results suggest a different rate-determining step in region II for the hybrid materials and the benchmark Pd/C catalyst.
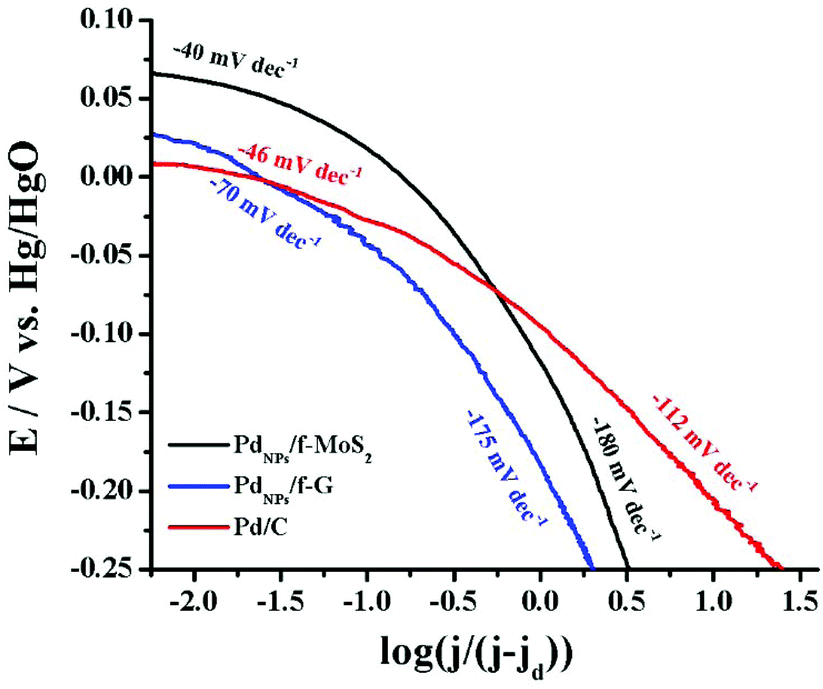 | ||
| Fig. 5 Tafel plot for the PdNPs/f-MoS2 and PdNPs/f-G hybrid electrocatalyst as compared to those due to Pd/C. Data derived from Fig. 3A. | ||
Table 1 summarizes the electrochemical performance towards the ORR for all tested materials. Overall, it can be deduced that PdNPs immobilized onto MoS2 sheets covalently functionalized with free amino groups (PdNPs/f-MoS2) manifested improved electrocatalytic activity and stability as compared with those supported on graphene layers bearing analogous functionalities (PdNPs/f-G). Therefore, these results indicate that TMD nanosheets can be used as an alternative to graphene substrates for the development of novel and highly active hybrid electrocatalysts. Interestingly, the superior performance of the MoS2 based hybrid over PdNPs/f-G is attributed to the following reasons. First, the covalent functionalization of graphene induces interruption of its continuous π-electron network through the introduction of sp3 defect sites, thereby resulting in the degradation of its extraordinary electronic properties.49 On the other hand, in the case of MoS2, as mentioned before, the covalent attachment of the organic moieties takes place at the edges of the MoS2 layers, keeping the basal plane intact and thus preserving its intrinsic properties.24,50 At the same time, the high affinity of PdNPs with the free amino groups implies enhanced electronic interactions between them and the f-MoS2 substrate, which are essential for the good ORR performance and long-term stability of the resulting hybrids. Here, it is worth noting that the high concentration of d electrons on the MoS2 surface provides an additional mechanism for the nanoparticle stabilization, contributing to the remarkable durability of the PdNPs/MoS2 catalyst.51 Lastly, the surfactant-free synthesis of the hybrid material further improves its ORR activity by providing more active sites on the nanoparticle surface.35,52
| Catalyst | Onset potential (mV vs. Hg/HgO) | Half-wave potential (mV vs. Hg/HgO) | Diffusion-limited current densitya (mA cm−2) | Kinetic current densityb (mA cm−2) | Tafel slopes | n K–L | n RRDE | H2O2 yield (%) |
|---|---|---|---|---|---|---|---|---|
|
a At 1600 rpm rotation rate (at ∼0.50 V vs. Hg/HgO).
b Calculated at −60 mV vs. Hg/HgO using the K–L equation.
c After 10000 s of the chronoamperometric experiment.
|
||||||||
| PdNPs/f-MoS2 | +66 | −116 | 5.70 | 2.65 | −40/−180 | 3.6–4.0 | 3.6 | 20 |
| PdNPs/f-MoS2c | +52 | −120 | 5.20 | 2.40 | — | — | — | — |
| PdNPs/f-G | +15 | −168 | 3.90 | 0.58 | −70/−175 | 3.4–3.6 | 3.1 | 47 |
| PdNPs/f-Gc | 0 | −194 | 3.00 | 0.35 | — | — | — | — |
| f-MoS2 | −236 | −360 | 1.47 | 0.00 | — | 2.1–2.3 | — | — |
| Pd/C | +13 | −93 | 5.40 | 2.00 | −46/−112 | 3.8–4.0 | 3.8 | <10 |
| Pd/Cc | −11 | −135 | 3.70 | 0.69 | — | — | — | — |
An overview of the recently developed hybrid ORR electrocatalysts based on PdNPs and MoS2 nanosheets is provided in Table S1.† For instance, PdNPs was formed and in situ immobilized on exfoliated MoS2via a sonochemical method.26 The obtained Pd/MoS2 hybrid exhibited an onset potential of −100 mV vs. SCE along with a diffusion-limited current density of 5.20 mA cm−2 in alkaline medium. Furthermore, the hybrid electrocatalyst outperformed the commercial one in terms of long-term stability. In a more recent work, a hybrid material based on palladium sulfide (PdS) nanoparticles supported on the MoS2/N-doped graphene heterostructure (PdS–MoS2/N-G) has been developed through a two-step thermolysis process while its ORR performance was examined under alkaline conditions.27 Markedly, the hybrid catalyst demonstrated more positive onset and half-wave potential values (−146 and −214 mV vs. SCE, respectively) as well as a greater diffusion limited current density (4.1 mA cm−2) than those of MoS2/N-G and PdS/N-G references, despite failing to surpass the commercial Pt/C catalyst. Based on these data, the current PdNPs/f-MoS2 hybrid is ranked among the top TMD-supported ORR electrocatalysts reported in the literature.
Experimental section
Materials
All chemicals and solvents were purchased from Aldrich and used without further purification unless otherwise stated.Preparation of functionalized MoS2 (f-MoS2)
First, a chlorosulfonic acid assisted method was applied for MoS2 exfoliation from the bulk in the form of semiconducting nanosheets.8 Next, the covalent functionalization of MoS2 sheets was achieved by employing 1,2-dithiolane units bearing an ethylene glycol alkyl chain terminated to a butoxycarbonyl (BOC)-protected amine.24 In short, exfoliated MoS2 (25 mg) and a 1,2-dithiolane derivative (30 mg) were dissolved in N,N-dimethylformamide (15 mL) and the reaction mixture was heated at 70 °C under stirring for 3 days. After this period, the mixture was filtered through a PTFE membrane filter (0.2 μm pore size) and the solid residue was extensively washed with dichloromethane. To cleave the BOC protecting group, the BOC modified MoS2 nanosheets (20 mg) were redispersed in dichloromethane and treated with gaseous HCl. Finally, the reaction mixture was left under stirring overnight, filtered through a PTFE membrane (0.2 μm pore size), washed with dichloromethane and neutralized with triethylamine to yield the amino functionalized MoS2 sheets (f-MoS2).Preparation of functionalized graphene (f-G)
Graphene oxide (GO) was prepared by following a modified Hummers’ method.53 Briefly, graphite powder was mixed with concentrated sulfuric acid, sodium nitrate and potassium permanganate to obtain a brownish slurry and the mixture was heated at 50 °C for 24 h. The suspension was diluted with water and hydrogen peroxide was added to get a higher oxidation degree. GO was finally obtained after washing with distilled water when the pH value became neutral. To prepare amino functionalized graphene (f-G), first, GO (20 mg) was activated by treatment with thionyl chloride (7 mL) at 66 °C overnight under an inert atmosphere. Next, the excess thionyl chloride was evaporated under reduced pressure and the remaining solid was carefully washed with dry tetrahydrofuran under nitrogen. The as-produced acyl-chlorinated graphene was dispersed in dry tetrahydrofuran (10 mL) followed by the addition of N-tert-butoxycarbonyl-2,2′-(ethylenedioxy)bis-ethylamine (300 mg) dissolved in dry tetrahydrofuran (5 mL). The reaction mixture was refluxed for 3 days. The resulting suspension was filtered over a PTFE membrane filter (0.2 μm pore size) and the filtrate was extensively washed with dichloromethane to remove organic residues before being dried under vacuum. Afterwards, the material was dispersed in dry dichloromethane and treated with gaseous HCl overnight to cleave the N-tert-butoxycarbonyl moieties. Finally, the reaction mixture was filtered through a PTFE membrane (0.2 μm pore size), washed with dichloromethane and neutralized with triethylamine to yield the amino functionalized graphene sheets (f-G).Preparation of PdNPs/f-MoS2 and PdNPs/f-G hybrids
For a typical synthesis of PdNPs/f-MoS2, f-MoS2 (8 mg) was ultrasonically dispersed in ethylene glycol (20 mL) to form a black homogeneous dispersion. Subsequently, potassium tetrachloropallade (6 mg) dissolved in ethylene glycol (5 mL) was mixed with the as-prepared polyol solution, degassed, and stirred for 30 min. Then, sodium borohydride (3.5 mg) dissolved in ethylene glycol (3 mL) was added gradually, and the reaction mixture was stirred under nitrogen for 60 min. The resulting suspension was washed with ethanol and vacuum dried. The same procedure was followed to prepare the PdNPs/f-G hybrid. In both PdNPs/f-MoS2 and PdNPs/f-G hybrids, the designed PdNPs loading was 20 wt%.Physical characterization
Scanning transmission electron microscopy (STEM) combined with EDS and electron energy-loss spectroscopy (EELS) were performed using a JEOL JEM-2100F microscope equipped with a couple of JEOL Centurio EDS detectors and a Gatan Quantum electron spectrometer at an electron accelerating voltage of 60 kV. An EELS chemical map of Pd was obtained by measuring its M4,5 edge signals at each point of a scanned area. Infrared (IR) spectra were acquired on a Fourier transform IR spectrometer (Equinox 55 from Bruker Optics) equipped with a single reflection diamond ATR accessory (DuraSamp1IR II by SensIR Technologies). Raman measurements were performed with a Renishaw confocal spectrometer at 633 nm. Thermogravimetric analysis (TGA) was carried out using a TGA Q500 V20.2 Build 27 instrument by TA under a nitrogen inert atmosphere.Catalyst preparation and deposition onto the working electrode
To prepare the catalyst ink, 4.0 mg of the hybrid catalytic powder were dispersed in a mixture of solvents (1 mL) containing water, isopropanol, and 5% Nafion (v/v/v = 4:1:0.02) and sonicated for 15 min. The working electrode was first cleaned through polishing using 6, 3 and 1 mm diamond pastes, rinsed with deionized water, and sonicated in double-distilled water. Then, 8.5 μL aliquots of the catalyst ink were cast on the electrode surface and dried at room temperature.
Electrochemical testing and calculations
All the electrochemical measurements were carried out using an Autolab PGSTAT128 N potentiostat/galvanostat equipped with a dual mode bipotentiostat (BA module). The working electrode (Autolab RRDE electrode tips) consisted of a Teflon-embedded glassy carbon (GC) disk/Pt ring rotating assembly (5 mm electrode disk diameter, 375 μm electrode gap, collection efficiency N = 0.249). As the counter electrode, a platinum wire was used and as the reference electrode an Hg/HgO (0.1 M KOH) electrode was placed into the Luggin capillary. The ORR measurements were realized at room temperature in O2-saturated aqueous 0.1 M KOH electrolyte. Linear sweep voltammetry (LSV) measurements on the disk electrode were conducted from −0.8 to +0.2 V vs. Hg/HgO at different rotation rates with a scan rate of 5 mV s−1. At the ring, the potential was fixed at +1.0 V vs. Hg/HgO and the current response was recorded. Chronoamperometric measurements for all materials were probed at −0.40 V vs. Hg/HgO at a rotation rate of 1600 rpm for 10000 s.
The kinetic current densities (jk) were calculated using the Koutecký–Levich (K–L) equation:
| 1/j = 1/jd + 1/jk | (1) |
| 1/j = 1/jd + 1/jk = 1/Bω1/2 + 1/jk | (2) |
| B = 0.20nFC0D2/30ν1/6. | (3) |
Here, n is the electron transfer number, F is the Faraday constant (F = 96485 C mol−1), D0 is the diffusion coefficient of O2 (D0 = 1.9 × 10−5 cm2 s−1), ν is the kinematic viscosity of the solution (ν = 0.01 cm2 s−1), and C0 is the concentration of dissolved O2 in the solution (C0 = 1.2 × 10−6 mol cm−3). The constant of 0.2 is adopted when the rotation speed is expressed in revolutions per minute (rpm). Tafel plots (potential vs. log[j/(jd − j)]) were calculated in the mixed kinetic–diffusion region at a single electrode rotation rate (ω = 1600 rpm).
Based on the RRDE data, the n value and the percentage (%) of the produced H2O2 can be determined using the following equations:
| n = 4IDisk/(IDisk + IRing/N) | (4) |
| %H2O2 = (200 IRing/N)/(IDisk + IRing/N) | (5) |
The capacitance values were calculated from the CV curves obtained in N2-saturated aqueous 0.1 M KOH according to eqn (6):
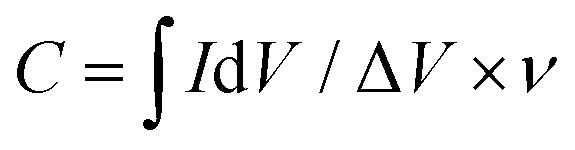 | (6) |
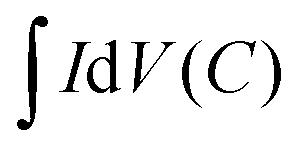 is the integrated area of the CV curve; ΔV (V) is the potential window and v is the scan rate (V s−1).
is the integrated area of the CV curve; ΔV (V) is the potential window and v is the scan rate (V s−1).
Conclusions
In summary, the fabrication of a novel hybrid ORR electrocatalyst based on covalently functionalized MoS2 nanosheets bearing free amino moieties and Pd nanoparticles, PdNPs/f-MoS2, through a simple procedure was accomplished. For comparison, an analogous catalyst based on modified graphene, PdNPs/f-G, was also obtained. Complementary spectroscopic techniques and thermogravimetric analysis confirmed and quantified the covalent attachment of the organic functionalities onto MoS2 and graphene. TEM imaging along with EDS/EELS elemental analysis revealed the immobilization of 2–3 nm PdNPs onto both 2D substrates, while the presence of agglomeration was not observed. The preservation of the MoS2 intrinsic properties during the functionalization process allowed us, in contrast to graphene, to fully exploit the benefits of the covalent modification, leading to improved electrocatalytic activities and stability toward the ORR in an alkaline medium for the PdNPs/f-MoS2 hybrid, outperforming not only its graphene based counterpart, but also the benchmark Pd/C catalyst. In more detail, the onset potential value for PdNPs/f-MoS2 was found to be approximately 50 mV more positive than that of PdNPs/f-G and Pd/C, whereas, with a kinetic current density value of 2.65 mA cm−2, the MoS2 based electrocatalyst possesses a 4.5-fold and by 46% greater activity over that registered for the graphene based catalyst and the commercial catalyst, respectively. Furthermore, PdNPs/f-MoS2 proved to be remarkably stable, exhibiting the lowest activity loss (∼16%) after 10000 s, among the tested electrocatalysts. A detailed analysis of the reaction kinetics suggested that the reduction of oxygen on the PdNPs/f-MoS2 hybrid abided mainly by the direct four-electron route, with a low H2O2 yield. All in all, these findings pave the way for future designs of high-performance TMD-based ORR electrocatalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is co-financed by Greece and the European Union (European Social Fund- ESF) through the Operational Programme “Human Resources Development, Education and Lifelong Learning” in the context of the project “Reinforcement of Postdoctoral Researchers - 2nd Cycle” (MIS-5033021), implemented by the State Scholarships Foundation (IKY). Y. S. and K. S. acknowledge JSPS KAKENHI (Grant No. JP19K05223 and JP16H06333, respectively) for financial support.Notes and references
- X. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 4, 45–68 CrossRef CAS.
- H. Li, C. Chen, D. Yan, Y. Wang, R. Chen, Y. Zou and S. Wang, J. Mater. Chem. A, 2019, 7, 23432–23450 RSC.
- D. K. Perivoliotis and N. Tagmatarchis, Carbon, 2017, 118, 493–510 CrossRef CAS.
- S. Jayabal, G. Saranya, J. Wu, Y. Liu, D. Geng and X. Meng, J. Mater. Chem. A, 2017, 5, 24540–24563 RSC.
- O. My Na, N. T. Xuan Huynh, P. T. Thi, V. Chihaia and D. Ngoc Son, RSC Adv., 2020, 10, 8460–8469 RSC.
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419–425 CrossRef CAS PubMed.
- X. Li and H. Zhu, J. Materiomics, 2015, 1, 33–44 CrossRef.
- G. Pagona, C. Bittencourt, R. Arenal and N. Tagmatarchis, Chem. Commun., 2015, 51, 12950–12953 RSC.
- X. Fan, P. Xu, Y. C. Li, D. Zhou, Y. Sun, M. A. T. Nguyen, M. Terrones and T. E. Mallouk, J. Am. Chem. Soc., 2016, 138, 5143–5149 CrossRef CAS PubMed.
- Z. Lei, J. Zhan, L. Tang, Y. Zhang and Y. Wang, Adv. Energy Mater., 2018, 8, 1703482 CrossRef.
- S. Shi, Z. Sun and Y. H. Hu, J. Mater. Chem. A, 2018, 6, 23932–23977 RSC.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed.
- G. Eda, H. Yamaguchi, D. Voiry, T. Fujita, M. Chen and M. Chhowalla, Nano Lett., 2011, 11, 5111–5116 CrossRef CAS PubMed.
- C. Zhu, D. Gao, J. Ding, D. Chao and J. Wang, Chem. Soc. Rev., 2018, 47, 4332–4356 RSC.
- Z. Li, X. Meng and Z. Zhang, J. Photochem. Photobiol., C, 2018, 35, 39–55 CrossRef CAS.
- K. Wang, L. Li, Y. Liu, C. Zhang and T. Liu, Adv. Mater. Interfaces, 2016, 3, 1600665 CrossRef.
- Y. Wang, K. Wang, C. Zhang, J. Zhu, J. Xu and T. Liu, Small, 2019, 15, 1903816 CrossRef CAS PubMed.
- K. Wang, J. Yang, J. Zhu, L. Li, Y. Liu, C. Zhang and T. Liu, J. Mater. Chem. A, 2017, 5, 11236–11245 RSC.
- J. Yang, J. Zhu, J. Xu, C. Zhang and T. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 44550–44559 CrossRef CAS PubMed.
- X. Chen and A. R. McDonald, Adv. Mater., 2016, 28, 5738–5746 CrossRef CAS PubMed.
- A. Stergiou and N. Tagmatarchis, Chem. – Eur. J., 2018, 24, 18246–18257 CrossRef CAS PubMed.
- P. Vishnoi, A. Sampath, U. V. Waghmare and C. N. R. Rao, Chem. – Eur. J., 2017, 23, 886–895 CrossRef CAS PubMed.
- X. Chen, N. C. Berner, C. Backes, G. S. Duesberg and A. R. McDonald, Angew. Chem., Int. Ed., 2016, 55, 5803–5808 CrossRef CAS PubMed.
- R. Canton-Vitoria, Y. Sayed-Ahmad-Baraza, M. Pelaez-Fernandez, R. Arenal, C. Bittencourt, C. P. Ewels and N. Tagmatarchis, npj 2D Mater. Appl., 2017, 1, 13 CrossRef.
- E. Lee and Y.-U. Kwon, RSC Adv., 2016, 6, 47468–47473 RSC.
- L.-X. Zuo, L.-P. Jiang and J.-J. Zhu, Ultrason. Sonochem., 2017, 35, 681–688 CrossRef CAS PubMed.
- L. G. Bach, M. L. N. Thi, Q. B. Bui and H. T. Nhac-Vu, Synth. Met., 2019, 254, 172–179 CrossRef CAS.
- S. Su, W. Cao, C. Zhang, X. Han, H. Yu, D. Zhu, J. Chao, C. Fan and L. Wang, RSC Adv., 2016, 6, 76614–76620 RSC.
- M. Liu, R. Zhang and W. Chen, Chem. Rev., 2014, 114, 5117–5160 CrossRef CAS.
- D. Perivoliotis, Y. Sato, K. Suenaga and N. Tagmatarchis, Chem. – Eur. J., 2019, 25, 11105–11113 CrossRef CAS.
- D. K. Perivoliotis, Y. Sato, K. Suenaga and N. Tagmatarchis, ACS Appl. Energy Mater., 2018, 1, 3869–3880 CrossRef CAS.
- M. A. Koklioti, T. Skaltsas, Y. Sato, K. Suenaga, A. Stergiou and N. Tagmatarchis, Nanoscale, 2017, 9, 9685–9692 RSC.
- S. Park, D. A. Dikin, S. T. Nguyen and R. S. Ruoff, J. Phys. Chem. C, 2009, 113, 15801–15804 CrossRef CAS.
- J. Liu, J. Hao, C. Hu, B. He, J. Xi, J. Xiao, S. Wang and Z. Bai, J. Phys. Chem. C, 2018, 122, 2696–2703 CrossRef CAS.
- D. Alba-Molina, A. R. Puente Santiago, J. J. Giner-Casares, E. Rodríguez-Castellón, M. T. Martín-Romero, L. Camacho, R. Luque and M. Cano, J. Mater. Chem. A, 2019, 7, 20425–20434 RSC.
- H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin, A. Olivier and D. Baillargeat, Adv. Funct. Mater., 2012, 22, 1385–1390 CrossRef CAS.
- S. Bae, N. Sugiyama, T. Matsuo, H. Raebiger, K.-I. Shudo and K. Ohno, Phys. Rev. Appl., 2017, 7, 024001 CrossRef.
- K. C. Knirsch, N. C. Berner, H. C. Nerl, C. S. Cucinotta, Z. Gholamvand, N. McEvoy, Z. Wang, I. Abramovic, P. Vecera, M. Halik, S. Sanvito, G. S. Duesberg, V. Nicolosi, F. Hauke, A. Hirsch, J. N. Coleman and C. Backes, ACS Nano, 2015, 9, 6018–6030 CrossRef CAS PubMed.
- X. Chen, P. Denninger, T. Stimpel-Lindner, E. Spiecker, G. S. Duesberg, C. Backes, K. C. Knirsch and A. Hirsch, Chem. – Eur. J., 2020, 26, 6535–6544 CrossRef CAS PubMed.
- R. Canton-Vitoria, H. B. Gobeze, V. M. Blas-Ferrando, J. Ortiz, Y. Jang, F. Fernández-Lázaro, Á. Sastre-Santos, Y. Nakanishi, H. Shinohara, F. D'Souza and N. Tagmatarchis, Angew. Chem., Int. Ed., 2019, 58, 5712–5717 CrossRef CAS PubMed.
- J.-B. Wu, M.-L. Lin, X. Cong, H.-N. Liu and P.-H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC.
- M. Lukaszewski, M. Soszko and A. Czerwiński, Int. J. Electrochem. Sci., 2016, 11, 4442–4469 CrossRef CAS.
- J. Benson, Q. Xu, P. Wang, Y. Shen, L. Sun, T. Wang, M. Li and P. Papakonstantinou, ACS Appl. Mater. Interfaces, 2014, 6, 19726–19736 CrossRef CAS PubMed.
- A. Stergiou, D. K. Perivoliotis and N. Tagmatarchis, Nanoscale, 2019, 11, 7335–7346 RSC.
- R. Zhou, Y. Zheng, M. Jaroniec and S.-Z. Qiao, ACS Catal., 2016, 6, 4720–4728 CrossRef CAS.
- N. P. Subramanian, T. Greszler, J. Zhang, W. Gu and R. R. Makharia, ECS Trans., 2011, 41, 985–1007 CrossRef CAS.
- Q. He, Q. Li, S. Khene, X. Ren, F. E. López-Suárez, D. Lozano-Castelló, A. Bueno-López and G. Wu, J. Phys. Chem. C, 2013, 117, 8697–8707 CrossRef CAS.
- C. Lee, S. Ozden, C. S. Tewari, O.-K. Park, R. Vajtai, K. Chatterjee and P. M. Ajayan, ChemSusChem, 2018, 11, 2960–2966 CrossRef CAS PubMed.
- Z. Zhang, H. Huang, X. Yang and L. Zang, J. Phys. Chem. Lett., 2011, 2, 2897–2905 CrossRef CAS.
- L. Guardia, J. I. Paredes, J. M. Munuera, S. Villar-Rodil, M. Ayán-Varela, A. Martínez-Alonso and J. M. D. Tascón, ACS Appl. Mater. Interfaces, 2014, 6, 21702–21710 CrossRef CAS PubMed.
- Y. Wang, B. Wang, R. Huang, B. Gao, F. Kong and Q. Zhang, Phys. E, 2014, 63, 276–282 CrossRef CAS.
- X. Zhang, J. Zhu, C. S. Tiwary, Z. Ma, H. Huang, J. Zhang, Z. Lu, W. Huang and Y. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 10858–10865 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization & electrochemical data. See DOI: 10.1039/d0nr04446f |
| This journal is © The Royal Society of Chemistry 2020 |