
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA CO2-gated anodic aluminum oxide based nanocomposite membrane for de-emulsification†
Xia
Huang
ab,
Hatice
Mutlu
 b and
Patrick
Theato
*ab
b and
Patrick
Theato
*ab
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstr.18, D-76131, Karlsruhe, Germany. E-mail: patrick.theato@kit.edu
bSoft Matter Synthesis Laboratory, Institute for Biological Interfaces III (IBG 3), Karlsruhe Institute of Technology (KIT), Herrmann-von-Helmholtz-Platz 1, D-76344, Eggenstein-Leopoldshafen, Germany
First published on 28th September 2020
Abstract
A carbon-dioxide-responsive organic–inorganic nanocomposite membrane based on a through-hole anodic aluminum oxide (AAO) template was constructed. The composite was prepared via a surface-initiated reversible addition–fragmentation chain-transfer (SI-RAFT) polymerization strategy to achieve the grafting of poly(methyl methacrylate-co-2-(diethylamino)ethyl methacrylate) brushes on the AAO membrane. The grafted polymer chain length could be controlled based on the feed ratio between the free chain transfer agent (CTA) and reactive monomer, e.g., methyl methacrylate and 2-(diethylamino)ethyl methacrylate, resulting in a membrane that features adjustable water permeability. Importantly, the membrane pore size and surface wettability could be switched from hydrophobic to hydrophilic upon the introduction of carbon dioxide and nitrogen gases. This allowed for the nanocomposite membrane to be utilized for controlled water flux and oil/water emulsion separation. The simple fabrication methodology as well as sustainable gaseous stimulus will be useful for the construction of future smart membranes.
Introduction
Biological cell membranes exist everywhere in living systems, in order to separate the intracellular environment from the extracellular environment. The cell membrane self-controls, in a precisely selective manner, the permeability of substances in and out of cells and organelles.1 Inspired by biological cell membranes, artificial membrane chromatography, which is mainly based on a thin layer of a well-organized porous stationary phase, has been extensively explored for controlling, on demand, the permeability through a membrane.2,3 Artificial smart gating membranes can be fabricated by chemically or physically by incorporating stimuli-responsive materials into porous membranes as functional gates.4,5 Accordingly, in response to diverse stimuli, such as temperature, pH, light, magnetic field or redox agents, the pore sizes and/or the surface properties of the membranes are adjusted by a responsive material, which in turn is manipulating the permeability and selectivity of the membrane. Indeed, mimetic-membranes have been widely used for water purification,6 bioprocessing,7,8 energy devices,9 molecule sieving,10 and so on. However, materials with sufficient uniformity, controlled pore diameter and length as well as a particular surface chemistry are required for artificial membrane chromatography.11 Generally, anodic aluminum oxide (AAO) membranes stand out from other artificial membranes, because it can be easily fabricated via a two-step anodization method resulting in monodispersed, geometrically regular and hexagonally packed porous structures.12 In addition, the pore size and length could be further customized. The AAO pore walls which are rich in hydroxyl groups are accessible for further functionalization via a general modification method.13 Such porous membranes feature a high surface area, exceptional thermo-, chemical- and mechanical-resistance as well as excellent biocompatibility.14 Therefore, the AAO membrane has been exclusively used as a filtration membrane in diverse fields of molecule separation such as amino acids,15 nucleic acids,16 proteins,17 and others,18 or water purification of various contaminated sources, e.g., dyes,19 oil fouling,20 heavy metal ions,21 or desalination,22 and so on. For example, Song et al. prepared a polyrhodanine grafted AAO membrane via vapor deposition polymerization and the polyrhodanine-AAO composites obtained were utilized for the selective removal of Hg(II), Ag(I) and Pb(II) ions from contaminated water.23 In another example, Li et al. prepared a thermo-responsive gating membrane by grafting poly(N-isopropylacrylamide) (PNIPAm) on an AAO membrane via surface-initiated atom transfer radical polymerization (SI-ATRP). The PNIPAm-AAO membrane obtained exhibited reversible thermo-responsive permeation.24 In addition, Tufani and Ince anchored pH sensitive poly(methylacrylic acid-co-ethylene glycol dimethacrylate) brushes onto an AAO template and the filtering performance of the membrane obtained could be tuned by changes in pH.25Although plenty of stimuli responsive polymers have been applied in the preparation of AAO membranes with smart gating, there are still certain drawbacks that hinder their broader applicability. Despite the advantages of pH-responsiveness, for example, its reversibility requires repeated addition of acids and bases, which causes salt accumulation, and thus contaminates the system.26,27 Hence, in order to eliminate the formation of by-products during a repeated stimulation process, it is essential that the use of “sustainable triggers” is explored.28 Indeed, the result of a literature survey reveals the recent trend in the development of membrane technologies, which are controlled by the manipulation of surface wettability in the presence of gases, such as carbon dioxide (CO2).29,30 Undoubtedly, CO2, as a nontoxic, inexpensive, benign and abundant gas, has emerged as the most studied gas trigger during the past two decades.31 Indeed, CO2 can react selectively with amine and amidine groups, and thus can be used to alter the hydrophilicity and polarity of diverse systems. The construction of CO2 gated membranes has attracted great interest and research.32,33 Recently, Li et al. have prepared a CO2-responsive cellulose nanofiber aerogel which can be used for switchable oil–water separation.34 Nevertheless, until now, studies focused on the fabrication of smart AAO membranes utilizing this sustainable gas trigger are unreported. Indeed, more effort is essential to develop efficient methods for the preparation of AAO membranes decorated with CO2 stimuli-responsive polymers, particularly, for specific targeted applications, such as emulsion separation.
Therefore, the aim of this research is to develop a “sustainable gas” (i.e., CO2) gated AAO membrane (Fig. 1) for controlled permeation. Accordingly, a simple method to fabricate a smart AAO membrane decorated with CO2 stimuli-responsive polymer is reported. By adopting the surface-initiated reversible addition–fragmentation chain-transfer (SI-RAFT) polymerization strategy, poly(methyl methacrylate-co-2-(diethylamino)ethyl methacrylate) (poly(MMA-co-DEAEMA); PMD) brushes are “grafted” from the AAO membrane. The synthesized composite membrane AAO-g-PMD features a uniform porous structure and a thin stimuli-responsive copolymer-brush layer. The grafted PDEAEMA chain endows the system with a gas switchable hydrophobic–hydrophilic surface, and in addition, combined with the volume variation of the polymer chains under gaseous stimulation, the functionalized membrane has beneficial switchable permeation properties and is suitable for the separation of oil/water emulsions. It is postulated that this smart membrane could be utilized for gaseous controlled water fluxing and emulsion separation.
 | ||
| Fig. 1 A schematic illustration of the preparation of the poly(methyl methacrylate-co-2-(diethylamino)ethyl methacrylate) grafted AAO membrane (AAO-g-PMD): (I) the pristine AAO membrane; (II) the AAO-silica membrane (AAO-g-SiO2); (III) a chain transfer agent immobilized onto the silanized AAO membrane (AAO-g-CTA); and (IV) AAO-g-PMD. | ||
Experimental
Materials
The 2-(N,N-diethylamino)ethyl methacrylate (DEAEMA, 99%, Sigma-Aldrich) and methyl methacrylate (MMA, 99%, Sigma-Aldrich) were purified by passing them through a neutral aluminum column; and 2,2′-azobis(isobutyronitrile) (AIBN, 98%, Sigma-Aldrich) was recrystallized from methanol. Aluminum discs (99.999%, Goodfellow), perchloric acid (HClO4, 70%, Carl Roth), ethanol (EtOH, 96%, Carl Roth), ortho-phosphoric acid (H3PO4, 85%, Carl Roth), chromium(IV) oxide (CrO3, ≥98%, Sigma-Aldrich), hydrogen peroxide (H2O2, 35%, Carl Roth), buffer solution pH 5 ± 0.02 (PBS, Carl Roth), sodium dodecyl sulfate (SDS, ≥99%, Fischer), anhydrous 1,4-dioxane (99.8%, Acros), toluene (≥99.5%, Roth), anhydrous toluene (99.8%, Acros), silica nanoparticles (SiO2 NP, 10–20 nm, Sigma-Aldrich), tetraethyl orthosilicate (TEOS, 98%, Sigma-Aldrich), hydrochloric acid (HCl, 37%, Carl Roth), S-(thiobenzoyl)thioglycolic acid (99%, Sigma-Aldrich), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, >98%, ABCR), N-hydroxysuccinimide (NHS, 98%, ABCR), anhydrous dichloromethane (DCM, 99.8%, Acros), (3-aminopropyl)triethoxysilane (APTES, ≥98%, ABCR) and other chemicals were used as received.Preparation of a through-hole anodic aluminum oxide template
The AAO membranes were prepared using a two-step anodization method. Pure aluminum (99.999%) discs were mounted in a tetrafluoroethylene case and exposed to electro-polishing at 21 V, 5 °C for 6 min in an electrolyte bath mixed with EtOH and HClO4 (v/v, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to form a flat surface. Then the first anodization process was carried out as follows. Upon cooling the electropolished aluminum disks were treated at 3–5 °C at a voltage of 175 V for 3 h, followed by an increase of the voltage to 195 V for 20 h, and in this process, 1 wt% H3PO4 was used as the electrolyte solution. After the first anodization treatment, an irregular layer of aluminum oxide pores was formed and this layer was removed by an etching step using a mixed aqueous solution of chromic acid (1.8 wt%) and H3PO4 (6.0 wt%) at 45 °C for 48 h. Afterwards, the chamber containing the aluminum disks was washed thoroughly with deionized water. The second anodization step then proceeded at a voltage of 195 V for 20 h in 1 wt% H3PO4 at 3–5 °C. The AAO templates obtained were washed and treated ultrasonically with acetone and then dried. Next, the AAO membranes were mounted with their backside pointing upwards into the chamber, and the bare backside of the aluminum substrate of the AAO membrane was removed via etching with CuCl2/HCl solution. The exposed alumina barrier layer was removed with 10 wt% H3PO4 in a 45 °C oven for 3 h. Ultimately, the through-hole isotropic AAO membranes were obtained after rinsing with DI water and drying in an oven at 45 °C.
:1) to form a flat surface. Then the first anodization process was carried out as follows. Upon cooling the electropolished aluminum disks were treated at 3–5 °C at a voltage of 175 V for 3 h, followed by an increase of the voltage to 195 V for 20 h, and in this process, 1 wt% H3PO4 was used as the electrolyte solution. After the first anodization treatment, an irregular layer of aluminum oxide pores was formed and this layer was removed by an etching step using a mixed aqueous solution of chromic acid (1.8 wt%) and H3PO4 (6.0 wt%) at 45 °C for 48 h. Afterwards, the chamber containing the aluminum disks was washed thoroughly with deionized water. The second anodization step then proceeded at a voltage of 195 V for 20 h in 1 wt% H3PO4 at 3–5 °C. The AAO templates obtained were washed and treated ultrasonically with acetone and then dried. Next, the AAO membranes were mounted with their backside pointing upwards into the chamber, and the bare backside of the aluminum substrate of the AAO membrane was removed via etching with CuCl2/HCl solution. The exposed alumina barrier layer was removed with 10 wt% H3PO4 in a 45 °C oven for 3 h. Ultimately, the through-hole isotropic AAO membranes were obtained after rinsing with DI water and drying in an oven at 45 °C.
Synthesis of the silanized chain transfer agent 2-oxo-2-((3-(triethoxysilyl)propyl)amino)ethyl benzodithioate (SiO-CTA)
The SiO-CTA was synthesized as follows: S-(thiobenzoyl)thioglycolic acid (2 g, 9.4 mmol), EDC hydrochloride (2 g, 10.32 mmol), and NHS (1.2 g, 10.32 mmol) were dissolved in 70 mL of anhydrous DCM, and then cooled in an ice bath. Subsequently, APTES (2.08 g, 9.4 mmol) in 10 mL of cold anhydrous DCM was added dropwise into the mixture. The reaction was then continuously stirred at ambient temperature for 5 h. Next, the mixture was washed three times with brine and then dried over anhydrous Na2SO4. A red oily product was obtained after rotary evaporation of the solvent, with a yield of 88.7%. The SiO-CTA was stored at −20 °C before use. The 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.02 (dd, H, –CH– from aryl), 7.55 (t, 2H, –CH– from aryl), 7.39 (t, 2H, –CH– from aryl), 6.45 (t, H, –NH–), 4.15 (s, 2H, –SCH2C–), 3.76 (q, 6H, –OCH2CH3), 3.24 (m, 2H, –NHCH2CH2–), 1.57 (m, 2H, –NHCH2CH2–), 1.19 (t, 9H, –OCH2CH3), 0.55 (t, 2H, –CH2CH2Si–).Immobilization of SiO-CTA onto the AAO membrane (AAO-g-CTA)
The pristine AAO membrane was immersed in concentrated H2O2 for 15 min at room temperature and then exposed to an oxygen plasma treatment for 10 min. While this was occurring, the silica solution consisting of 100 mg of SiO2 NPs, 10 mL of EtOH, 200 μL of 0.1 M HCl and 1 mL of TEOS was prepared, and mixed ultrasonically to form a homogenous solution. Subsequently, the silica solution was spin coated onto the AAO membrane at a speed of 1500 rpm for 30 s. Afterwards the AAO membrane was cured in oven at 120 °C for 2 h. This pre-treated AAO membrane was then immersed in a solution containing 100 mg of SiO-CTA in 10 mL of dry toluene in a round bottomed flask sealed with a rubber septum in water bath shaker at 40 °C overnight. Next, the membrane was rinsed with DCM and cured in an oven at 120 °C for 2 h. The membranes obtained were then washed and dried under reduced pressure before any use.SI-RAFT polymerization of poly(methyl methacrylate-co-2-(diethylamino)ethyl methacrylate) on the AAO membrane (AAO-g-PMD)
The preparation of AAO-g-PMD was achieved by SI-RAFT polymerization of MMA and DEAEMA using the SiO-CTA anchored AAO membrane according to the following procedure. AAO-g-CTA, MMA, DEAEMA, AIBN, free CTA and 10 mL anhydrous 1,4-dioxane were added into a Schlenk flask in varying ratio as summarized in Table 1, and then the mixture was degassed by three freeze–pump–thaw cycles. Simultaneously, the AAO membrane was degassed with N2 in another Schlenk flask. Afterwards, the monomer mixture was transferred into the AAO containing flask and then immersed in a preheated shaking bath at 70 °C for 4 h. After polymerization, the AAO membrane was taken out and washed sequentially with DMF, DCM and acetone before drying at ambient environment. 1H-NMR (400 MHz, CDCl3) δ (ppm) 8.05 (d, 1H, –CH– from aryl), 7.55–7.44 (m, 1H, –CH– from aryl), 7.36 (t, 1H, –CH– from aryl), 4.17 (s, 2H, –CH2–COOH), 4.02 (s, 19H, –O–CH2–CH2–), 3.59 (s, 3H, –O–CH3), 2.71 (t, 2H, –O–CH2–CH2–), 2.59 (d, 2H, –N–CH2–CH3), 2.10–1.68 (m, 2H, –CH2– from the polymer backbone), 1.16–0.95 (m, 3H, –CH2–CH3 from the polymer backbone), 0.87 (d, 3H, –N–CH2–CH3).| MMA | DEAEMA | AIBN | CTA | 1,4-Dioxane | |
|---|---|---|---|---|---|
| PMD-1 | 600 mg, 6 mmol | 740 mg, 4 mmol | 3.28 mg, 0.02 mmol | 42.4 mg, 0.2 mmol | 10 mL |
| PMD-2 | 600 mg, 6 mmol | 740 mg, 4 mmol | 1.64 mg, 0.01 mmol | 21.2 mg, 0.1 mmol | 10 mL |
| PMD-3 | 600 mg, 6 mmol | 740 mg, 4 mmol | 0.328 mg, 0.002 mmol | 4.24 mg, 0.02 mmol | 10 mL |
Flux experiment
The flux test was carried out using a commercially available filtration funnel (Fig. S3, ESI†). For this, the AAO membrane was placed on fritted sand core support and tightly sealed with an O-ring to prevent any leakage. Permeation experiments were performed at a pressure of 0.11 MPa at room temperature and all the eluate was collected in a conical flask. For controlled water flux testing, the eluate was weighed five times every 5 min at each set of conditions. The transition was achieved by alternatively purging with CO2 or N2 for 5 min in an ambient environment.Emulsion preparation
The typical recipe of the oil-in-water emulsion was as follows: toluene and PBS solution (pH 5) mixed in 1/100 (v/v) with the addition of 0.1 mg mL−1 SDS. The emulsion was mixed ultrasonically for 15 min to form a homogenous emulsion solution before use. For the water-in-oil emulsion, the volume ratio of distilled water and toluene was 1/100 (v/v). The solution was mixed ultrasonically for 15 min.Characterization
The 1H-NMR spectra were recorded on a Bruker 400 NMR in CDCl3, and all chemical shifts are reported in ppm (δ) and calibrated on characteristic solvent signals as internal standards. All data were reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet). The Fourier-transform infrared (FT-IR) spectra were measured using an attenuated total reflectance infrared spectroscopy (ATR-IR, ThermoFisher Smart iTR) unit on a Bruker VERTEX 80 V FT-IR spectrometer with a range of 500 to 4000 cm−1 at ambient temperature. The scanning electron microscope (SEM) images were taken with a Zeiss LEO 1530 microscope operating at 5 kV. Samples were sputtered with gold prior to measurement. The water and underwater oil contact angles were measured using a Krüss DSA25S drop shape analyzer. Gel permeation chromatography (GPC) measurements were performed using a Polymer Laboratories (Varian) PL-GPC 50 Plus integrated system equipped with two PLgel 5 m MIXED columns (300 × 7.5 mm), and related to poly(methyl methacrylate) standards. All the measurements were carried with DMAc as eluent at 50 °C with a flow rate of 1 mL min−1. Dynamic light scattering measurements of the emulsions were recorded with a Malvern Zetasizer Nano ZS equipped with a 632 nm He–Ne laser at 25 °C. Optical images of the emulsions and eluates were recorded using a Keyence BIOREVO fluorescence microscope.Results and discussion
For the creation of the gas sensitive polymer membranes, initially, the pristine AAO on an aluminum membrane was obtained using a two-step anodization method using aqueous H3PO4 electrolyte.34 Next, the backside of the aluminum substrate and the corresponding alumina barrier layer of pristine AAO membrane were partially removed. Ultimately, through-hole isotropic AAO membranes on a support of circumferentially connected aluminum and alumina substrate were obtained. The optical image, and the SEM image as well as a schematic illustration of the AAO membrane obtained are shown in Fig. 2.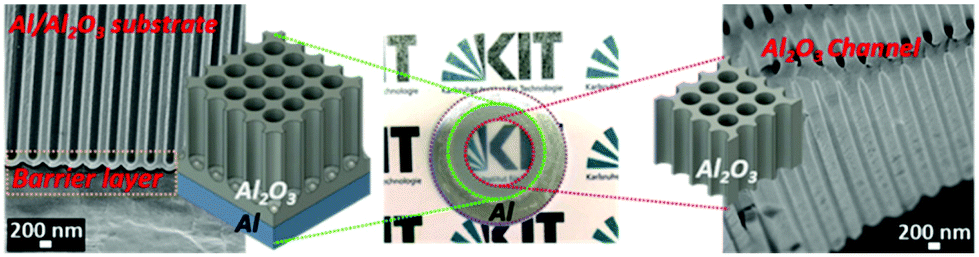 | ||
| Fig. 2 An optical image, SEM image, and corresponding schematic illustration of the through-hole AAO membrane. | ||
As reported in the literature, a pure free standing through-hole AAO membrane only consisted of alumina, which had a low mechanical strength. It was brittle and fragile, hence the treatment during the modification process was hard to handle without the assistance of a supporting layer.35,36 In order to solve the problem and increase the mechanical property of the through-hole AAO membrane, the backside was fixed in a trepanning position and only the inner circle of AAO membrane was opened as shown by a red dotted-line in Fig. 2. A bigger concentric circle was left that retained the barrier alumina layer and the aluminum underneath (green dotted line). The AAO membrane obtained was composed of through-hole channels that were responsible for fluxing and a build-in supporting layer that guaranteed the manoeuvrability during post-processing. The SEM images that depict the morphology of both channel and substrate structures are shown in Fig. 2, indicating the successful fabrication of the membrane.
Once the desired through-hole AAO membrane was obtained, essential post-processing procedures for the construction of a functional membrane were carried out as shown in Fig. 1. The AAO membranes were decorated by a surface enriched hydroxyl group, however, in order to increase the amount of active groups on the membrane surface for stabilizing further modifications, hydrolysis via H2O2 and oxygen plasma treatment were carried out.37 Next, a thin layer of SiO2 NPs (diameter 10–20 nm) was deposited and anchored onto the membrane surface via sol–gel impregnation to enhance the surface roughness (Fig. 1, II). Subsequently, after immobilization of the pre-prepared surface CTA (SiO-CTA), SI-RAFT polymerization was carried on the AAO-grafted-CTA which resulted in grafting of poly(MMA-co-DEAEMA) brushes on AAO, i.e., AAO-g-PMD (Fig. 1, III and IV). The brushes were grown on the AAO surface in anhydrous 1,4-dioxane in the presence of 2,2′-azobis(isobutyronitrile) as initiator at 70 °C for 4 h. Indeed, this SI-RAFT process was versatile and facilitated the attachment of a wide range of chemical functionalities to the AAO surface, which would allow simple tailoring of the AAO properties by the incorporation of specific functional groups.
The surface morphology of pristine AAO and AAO-g-PMD were characterized by SEM, and the results are shown in Fig. 3. From the SEM image of the pristine AAO membrane (Fig. 3A and B) hexagonally arranged pores with a narrow size distribution were observed over the aluminum sheets and the surface was found to be smooth with an average pore size of ∼261 nm. After deposition of the SiO2 NPs and grafting with the polymer brushes, the membrane pore size decreased to ∼213 nm. In addition, the surface roughness was found to increase, as can be seen in Fig. 3C and D, and the aggregated SiO2 NPs were evenly distributed on the membrane surface.
 | ||
| Fig. 3 Comparative top-view SEM images of (A and B) pristine AAO, and (C and D) AAO-g-PMD. | ||
In order to monitor the surface component variation of the AAO membrane during each sequential synthetic step, FT-IR measurements were performed. As shown in Fig. 4, by comparing spectra (a) and (b), it was seen that a new band appeared at 1090 cm−1 that was ascribed to the Si–O–Si asymmetric stretching vibration of silica after deposition of SiO2 NPs onto the pristine AAO.38 Whereas, upon tethering the surface CTA onto the silanized AAO membrane, bands that could be ascribed to the symmetric C–N stretching vibration, N–H deformation and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, were observed at 1445 cm−1, 1533 cm−1 and 1647 cm−1, respectively. Therefore, the successful immobilization of the surface CTA was confirmed.39 In a similar manner, the results of the FT-IR analysis confirmed the SI-RAFT polymerization; and a peak ascribed to the carbonyl vibration of PDEAEMA was observed at 1730 cm−1.40 In addition, a strong absorption band was detected in the range of 2821 to 3005 cm−1, that was attributed to the stretching vibrations of –CH2 and –CH3 functional groups from the polymer backbone.41
O stretching, were observed at 1445 cm−1, 1533 cm−1 and 1647 cm−1, respectively. Therefore, the successful immobilization of the surface CTA was confirmed.39 In a similar manner, the results of the FT-IR analysis confirmed the SI-RAFT polymerization; and a peak ascribed to the carbonyl vibration of PDEAEMA was observed at 1730 cm−1.40 In addition, a strong absorption band was detected in the range of 2821 to 3005 cm−1, that was attributed to the stretching vibrations of –CH2 and –CH3 functional groups from the polymer backbone.41
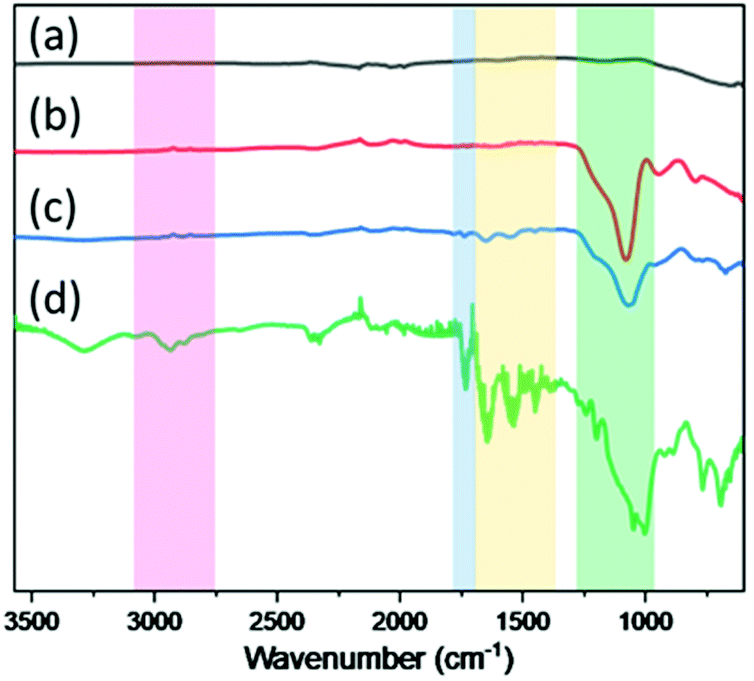 | ||
| Fig. 4 The FT-IR spectra of (a) the pristine AAO membrane, (b) AAO-g-SiO2, (c) AAO-g-CTA, and (d) AAO-g-PMD. | ||
Static water contact angle (WCA) measurements also confirmed the successful AAO functionalization. Therefore, changes in the wettability of the membrane at each modification process were tracked, and the optical images obtained are shown in Fig. 5. The surface of the AAO membranes converted from hydrophilic (WCA: ∼30 °C) to hydrophobic (WCA: ∼100°) after deposition with SiO2 NPs. Subsequently, the hydrophobicity of the AAO membrane surface was further increased (WCA: ∼130°) when the surface CTA was immobilized, and this was particularly attributed to the hydrocarbon chain and siloxane head group of the as-prepared surface CTA. Upon SI-RAFT polymerization, grafted polymer chains introduced additional hydrophobicity to the modified AAO (WCA: ∼141°), which was revealed by the enhanced surface roughness after the polymerization.
 | ||
| Fig. 5 Water droplets on different surfaces: (A) pristine AAO, (B) AAO-g-SiO2, (C) AAO-g-CTA, and (D) AAO-g-PMD. | ||
Furthermore, to shed light on conformation change of PMD chains grafted onto AAO template, static WCA measurements were carried out via treating AAO-g-PMD membrane with CO2 and N2. As illustrated in Fig. 6A, upon exposure to a CO2-purged aqueous solution for 5 min, the WCA showed a drastic decrease from 140.38 ± 0.88° to 12.80 ± 3.77°, which was indicative of polymer brush hydration.42,43 Whereas, after passing N2 gas through the membrane for 10 min, the initial WCA was recovered, which suggested that the deprotonation of PDEAEMA resulted in loss of water from the polymer brushes and the formation of a collapsed state. The experiments were repeated 10 times (Fig. 6B), showing the reversibility of the WCA by alternating bubbling with CO2 and N2. The responsive mechanism (Fig. 6A) was typically a variation of pH control and the result of the reaction of CO2 with a functional group. In the initial state, the AAO-g-PMD membrane surface was hydrophobic, because the polymeric tertiary amines were hydrophobic in their neutral form. By exposure to CO2, the tertiary amine groups in PDEAEMA reacted with CO2 in an aqueous medium, forming charged ammonium bicarbonate salts.44,45 The latter caused the PDEAEMA brushes to swell in water, thus adopting a chain-extended state. Subsequently, passing N2 through the solution expelled CO2 and the PDEAEMA brushes returned back to the collapsed hydrophobic state. As mentioned, previously, this CO2 switching can be repeated many times without the addition of any chemicals that could remain in the solution. Indeed, the advantage of gas responsive membranes compared to a conventional pH-responsive membrane is that it functions without using strong chemicals (such as acids and bases) to change pH and generally requiring repeated addition of these chemicals into the solution, producing chemical wastes such as excessive salts. This switchable wettability between surface hydrophobicity and hydrophilicity makes the membrane a very promising candidate for use in oil/water separation.
 | ||
| Fig. 6 (A) An optical image and chain transformation illustration, and (B) reversible contact angle (CA) data of AAO-g-PMD triggered by CO2 and N2. | ||
It has been reported that the molecular weight of surface initiated polymers is equal to that of free polymers synthesized in bulk.46 Therefore, grafted polymers are often characterized by polymerizing the same monomers under bulk conditions using free CTA. In order to further investigate the influence of polymer molecular weight on the membrane permeability, a series of RAFT and SI-RAFT polymerizations of MMA and DEAEMA were processed. During the reactions, different amounts of free chain transfer agent (S-(thiobenzoyl)thioglycolic acid) were added into each reaction system to control the degree of polymerization in bulk as well as on the AAO membrane. The free copolymer chains were used as a reference system to estimate the apparent number average molecular weight (Mn) and molecular weight distribution (Đ) of the grafted polymer chains. Detailed feed ratios and the characterization data of the polymers obtained are given in Table 2. As expected, higher feed ratios of CTA resulted in lower number average molecular weights.
| Code | MMA | DEAEMA | CTA | AIBN |
M
n,theoa (g mol−1) |
M
n,H NMRa (g mol−1) |
M
n,GPCb (g mol−1) |
M w/Mnb |
|---|---|---|---|---|---|---|---|---|
| (Ratio between the reagents) | ||||||||
| a Calculated by 1H-NMR in CDCl3. b Obtained from GPC in DMAc. | ||||||||
| AAO-g-PMD-1 | 200 | 300 | 10 | 1 | 7762 | 2320 | 20300 |
1.94 |
| AAO-g-PMD-2 | 400 | 600 | 10 | 1 | 15312 |
6480 | 33300 |
2.04 |
| AAO-g-PMD-3 | 2000 | 3000 | 10 | 1 | 75712 |
17820 |
85000 |
2.27 |
The grafting of polymers with varying molecular weights on porous AAO membranes lead to the variation of membrane permeability. As shown in Fig. 6, water flux measurements indicated that the higher molecular weights of the grafted polymer chains resulted in a relatively low water flux. For example, the AAO-g-PMD-1 membrane with a molecular weight of 2320 g mol−1 exhibited an average water flux of 510 L m−2 h−1, however, when the grafting molecular weight was increased to 17820 g mol−1 (AAO-g-PMD-3), the average water flux decreased to 462 L m−2 h−1.
In addition, it has been mentioned previously that grafted PDEAEMA chains could undergo a transition of polymer conformation between the collapsed and stretched states.45 The volume change of polymer brushes could control the water permeance of the AAO-g-PMD membrane. Fig. 7 shows the change of water flux through AAO-g-PMD-(1, 2, 3, from Table 2) membranes depending on CO2 stimulation. In neutral media, the AAO-g-PMD membrane exhibited a water flux of 462–510 L m−2 h−1. Upon triggering with CO2, the water flux decreased sharply to 187–69 L m−2 h−1. Furthermore, when purging the membrane with N2 to expel the CO2, the PDEAEMA brushes changed from stretched to collapsed, which in turn lead to the increase of permeability for all AAO-g-PMD membranes. As expected, the permeability of the AAO-g-PMD membrane also showed good CO2-reversibility when the aqueous media changed from acidic to neutral, or vice versa. Moreover, it is worth noting that the molecular weight of the grafted polymer chains also effected the stimuli-triggered change of the membrane permeability. As illustrated in Fig. 7, AAO-g-PMD-3 grafted with a higher molecular weight polymer resulted in a broader variation of water fluxing of 392 L m−2 h−1 upon stimulation. However, AAO-g-PMD-1 with shorter polymer chains exhibited a narrower water flux variation of 323 L m−2 h−1.
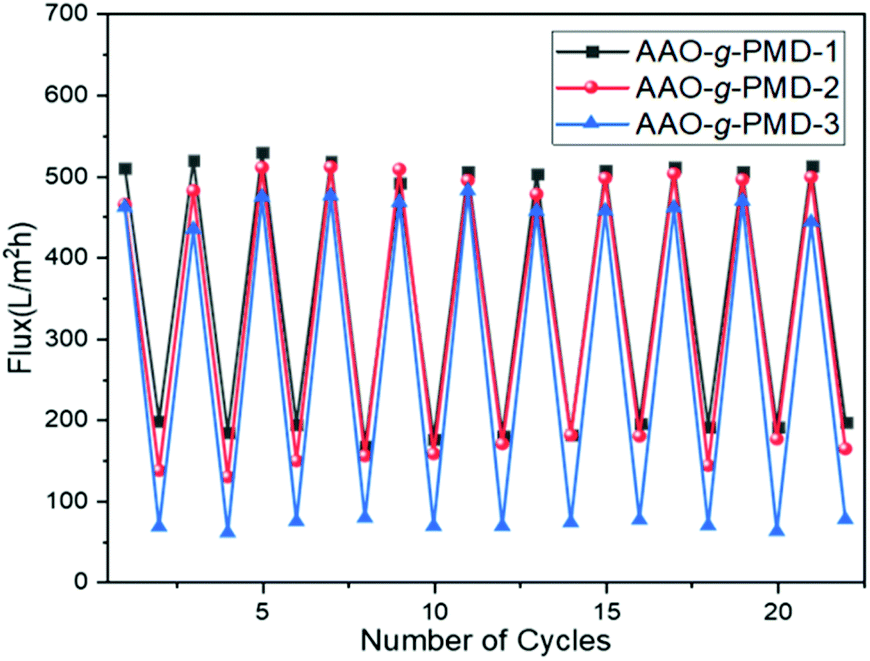 | ||
| Fig. 7 Reversible behaviour of the water flux of AAO-g-PMD 1, 2, and 3 (Table 2), upon triggering with CO2 (bottom) and N2 (top). | ||
As previously mentioned, the gas-controlled reversible change in membrane pore size and wettability are useful for selective separation.47 Therefore, the AAO-g-PMD membranes were tested for oil/water emulsion separation by making a stabilized emulsified toluene-in-water or water-in-toluene mixture, and using them as representative emulsions to permeate the membranes. Under neutral conditions, when filtering a water-in-oil emulsion through a modified AAO membrane, the continuous oil phase had a higher affinity for the hydrophobic surface of the narrow channels of the membrane and preferred to spread over the membrane, while water droplets were repelled from the membrane. As a consequence, the oil phase was selectively filtered through the AAO-g-PMD membrane and water droplets were rejected. Correspondingly, upon CO2 bubbling, the protonation of the PDEAEMA brushes resulted in a hydrophilic membrane surface. In this extended state, the AAO-g-PMD membrane was able to separate the water phase from oil-in-water emulsion. During filtration, a continuous water phase was preferentially allowed to penetrate the membrane, while the oil droplets were retained. Consequently, the AAO-g-PMD membrane could be used for selective separation of oil-in-water and water-in-oil emulsions under CO2 regulation. The filtration setup is shown in Fig. S3 (ESI†) and all the percolations were carried out at a pressure of 0.11 MPa with the eluate being collected for further characterization. Using AAO-g-PMD-1 as an example, the results for emulsion separation are given in Fig. 8 and 9.
 | ||
| Fig. 8 Water-in-oil emulsion separation using the AAO-g-PMD-1 membrane. (A) Optical images and (B) the droplet size distribution from DLS of a water-in-toluene emulsion before and after filtration. (C) The permeation of a water-in-toluene emulsion. | ||
 | ||
| Fig. 9 Oil-in-water separation using AAO-g-PMD-1. (A) Optical images and (B) the droplet size distribution from DLS of a toluene-in-water emulsion before and after filtration. (C) The permeation of the toluene-in-water emulsion. | ||
Then, the emulsion and the eluate remaining after filtration were analysed using dynamic light scattering (DLS) measurements. From the DLS spectra in Fig. 8B, the droplet size distribution of a water-in-toluene emulsion showed a range from 2 μm to 4 μm with a maximum at 3 μm. After filtration through the AAO-g-PMD-1 membrane, the eluate was clear and no obvious water droplets were found under the optical microscope, and the droplet sizes were only in the range of tens of nanometers. Furthermore, in order to verify the stability and recovery capacities of the as-prepared membrane, a continuous emulsion separation test was carried out, which was in agreement with the optical image of the emulsion before and after filtration. From the optical image (Fig. 8A) it could be seen that prior to filtration, the water-in-toluene emulsion revealed a milky but homogenous state with dispersed water droplets in the oil continuous phase. Whereas, after passing through the AAO-g-PMD membrane, the eluate exhibited a translucent appearance. The permeability behaviour of the water-in-toluene emulsion is shown in Fig. 8C. As illustrated, the initial flux of the emulsion is 300 L m−2 h−1, while the flux decreased over time due to the accumulated water droplets spreading on the membrane surface and partially blocking the channel for oil fluxing. After rinsing with ethanol, the permeability recovered to 82%, and kept decreasing upon further filtration of the emulsion. Noticeably, when the membrane was washed again with ethanol, the emulsion flux could again be recovered. The results indicated substantial reversibility of the membrane for water-in-oil separation under neutral working conditions.
Meanwhile, the AAO-g-PMD membranes were able to separate the water phase from the oil-in-water emulsion upon triggering with CO2. The results are shown in Fig. 9. From the recorded optical image, when the milky emulsion passed through the CO2 triggered membrane, a transparent clear eluate was obtained. In similar manner, the DLS measurements showed clearly the absence of large emulsion droplets ranging from 3 μm to 6 μm. After filtration, the residual particles with a size of a few to tens of nanometers were observed that can be ascribed to the dissolved surfactant.48 In addition, the permeability and separation reversibility of the CO2 triggered membrane were investigated by continuous filtration over a toluene-in-water emulsion. The initial fluent was 100 L m−2 h−1, which was apparently lower than the flux of the water-in-toluene emulsion. The results of membrane de-emulsification depended on a combination of chemical composition, a rough nanostructured surface and an inherent porous membrane. Upon CO2 purging, the tertiary amine group in the grafted polymer chains protonated, and transferred the polymer chains from the hydrophobic collapsed state to the extended hydrophilic extended state. In addition, as expected, the permeability of the as-prepared membrane decreased over filtration time due to the blocking of the oil droplet in the membrane pores. Nevertheless, further washing of the membrane with ethanol resulted in 90% recovery of the initial permeability.
As mentioned previously, upon CO2 purging, the grafted polymer chains became hydrophilic upon protonation, due to the conformational transition of the polymer brush from a collapsed state to an extended state. In fact, the intermolecular forces between the hydrophilic polymer chains and the hydrophilic surface of the emulsified oil-in-water droplets acted as the driving forces for de-emulsification. These intermolecular forces, including hydrogen bonding and van der Waals forces, facilitate the breaking of the stable emulsions. The straining of the emulsified droplets during the so-called absorption–extrusion process also increased the destabilization of the emulsion.
Conclusions
In conclusion, poly(methyl methacrylate-co-2-(diethylamino)ethyl methacrylate) brushes have been grafted onto a through-hole AAO membrane via SI-RAFT. The obtained AAO composites featured CO2-stimulus-response behaviour, which influenced wettability and permeability. In addition, the molecular weight of the grafted polymer chains could be controlled through varying the feed ratio between the chain transfer agent and monomers during the RAFT polymerization. A higher molecular weight of the grafted polymer chains resulted in broader flux variation upon external stimulation. Ultimately, the functionalized AAO membrane possessed CO2-switchable wettability and permeability, which were used for oil/water emulsion separation. Thus, this study provides a simple way for fabricating gas-induced stimuli-responsive membranes that could be of enormous scientific and technological interest because the stimulation by CO2 is economically viable. Indeed, these membranes may offer many possibilities for developing new smart materials that, for example, could be applied to industrial sewage treatment, due to their continuous processing ability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Xia Huang gratefully acknowledges the China Scholarship Council (CSC grants 201506240019) for partial financial support of this work.References
- P. Singleton and P. Mazliak, Bacteria in Biology, Biotechnology and Medicine, Wiley Chichester, 1997 Search PubMed.
- T. B. Tennikova, M. Bleha, F. Švec, T. V. Almazova and B. G. Belenkii, High-Performance Membrane Chromatography of Proteins, a Novel Method of Protein Separation, J. Chromatogr. A, 1991, 555(1–2), 97–107 CrossRef CAS.
- R. Ghosh, Protein Separation Using Membrane Chromatography: Opportunities and Challenges, J. Chromatogr. A, 2002, 952(1–2), 13–27 CrossRef CAS.
- Z. Zhang, L. Wen and L. Jiang, Bioinspired Smart Asymmetric Nanochannel Membranes, Chem. Soc. Rev., 2018, 47(2), 322–356 RSC.
- Z. Liu, W. Wang, R. Xie, X.-J. Ju and L.-Y. Chu, Stimuli-Responsive Smart Gating Membranes, Chem. Soc. Rev., 2016, 45(3), 460–475 RSC.
- G. Kang and Y. Cao, Application and Modification of Poly (Vinylidene Fluoride)(PVDF) Membranes–a Review, J. Membr. Sci., 2014, 463, 145–165 CrossRef CAS.
- A. Tufani and G. Ince, Protein Gating by Vapor Deposited Janus Membranes, J. Membr. Sci., 2019, 575(March 2018), 126–134, DOI:10.1016/j.memsci.2019.01.013.
- S. Y. Yang, I. Ryu, H. Y. Kim, J. K. Kim, S. K. Jang and T. P. Russell, Nanoporous Membranes with Ultrahigh Selectivity and Flux for the Filtration of Viruses, Adv. Mater., 2006, 18(6), 709–712, DOI:10.1002/adma.200501500.
- H. Zhao, L. Liu and Y. Lei, A Mini Review: Functional Nanostructuring with Perfectly-Ordered Anodic Aluminum Oxide Template for Energy Conversion and Storage, Front. Chem. Sci. Eng., 2018, 12(3), 481–493, DOI:10.1007/s11705-018-1707-x.
- S. P. Pujari, L. Scheres, A. T. M. Marcelis and H. Zuilhof, Covalent Surface Modification of Oxide Surfaces, Angew. Chem., Int. Ed., 2014, 53(25), 6322–6356, DOI:10.1002/anie.201306709.
- H. U. Osmanbeyoglu, T. B. Hur and H. K. Kim, Thin Alumina Nanoporous Membranes for Similar Size Biomolecule Separation, J. Membr. Sci., 2009, 343(1–2), 1–6 CrossRef CAS.
- A. Rath and P. Theato, Advanced AAO Templating of Nanostructured Stimuli–Responsive Polymers: Hype or Hope?, Adv. Funct. Mater., 2020, 30(2), 1902959, DOI:10.1002/adfm.201902959.
- A. Santos, T. Kumeria and D. Losic, Nanoporous Anodic Aluminum Oxide for Chemical Sensing and Biosensors, TrAC, Trends Anal. Chem., 2013, 44, 25–38, DOI:10.1016/j.trac.2012.11.007.
- A. M. M. Jani, E. J. Anglin, S. J. P. McInnes, D. Losic, J. G. Shapter and N. H. Voelcker, Nanoporous Anodic Aluminium Oxide Membranes with Layered Surface Chemistry, Chem. Commun., 2009, 21, 3062–3064, 10.1039/b901745c.
- S. B. Lee, D. T. Mitchell, L. Trofin, T. K. Nevanen, H. Söderlund and C. R. Martin, Antibody-Based Bio-Nanotube Membranes for Enantiomeric Drug Separations, Science, 2002, 296(5576), 2198–2200, DOI:10.1126/science.1071396.
- S. U. Hong and M. L. Bruening, Separation of Amino Acid Mixtures Using Multilayer Polyelectrolyte Nanofiltration Membranes, J. Membr. Sci., 2006, 280(1–2), 1–5 CAS.
- W. Shi, Y. Shen, D. Ge, M. Xue, H. Cao, S. Huang, J. Wang, G. Zhang and F. Zhang, Functionalized Anodic Aluminum Oxide (AAO) Membranes for Affinity Protein Separation, J. Membr. Sci., 2008, 325(2), 801–808 CrossRef CAS.
- S. Kipke and G. Schmid, Nanoporous Alumina Membranes as Diffusion Controlling Systems, Adv. Funct. Mater., 2004, 14(12), 1184–1188 CrossRef CAS.
- N. T. Phuong, A. Andisetiawan, D. Van Lam, J. H. Kim, D. S. Choi, K. H. Whang, J. Nham, Y. J. Lee, Y. E. Yoo and J. S. Yoon, Nano Sand Filter with Functionalized Nanoparticles Embedded in Anodic Aluminum Oxide Templates, Sci. Rep., 2016, 6(November), 1–8, DOI:10.1038/srep37673.
- Y. Huang, H. Li, L. Wang, Y. Qiao, C. Tang, C. Jung, Y. Yoon, S. Li and M. Yu, Ultrafiltration Membranes with Structure-Optimized Graphene-Oxide Coatings for Antifouling Oil/Water Separation, Adv. Mater. Interfaces, 2015, 2(2), 1400433 CrossRef.
- M. Agheli and A. Habibolahzadeh, Study of Hexavalent Chromium Ion Adsorption on Nano-Porous Anodic Aluminum Oxide, Prot. Met. Phys. Chem. Surf., 2016, 52(6), 972–974 CrossRef CAS.
- M. Fujiwara and T. Imura, Photo Induced Membrane Separation for Water Purification and Desalination Using Azobenzene Modified Anodized Alumina Membranes, ACS Nano, 2015, 9(6), 5705–5712, DOI:10.1021/nn505970n.
- J. Song, H. Oh, H. Kong and J. Jang, Polyrhodanine Modified Anodic Aluminum Oxide Membrane for Heavy Metal Ions Removal, J. Hazard. Mater., 2011, 187(1–3), 311–317, DOI:10.1016/j.jhazmat.2011.01.026.
- P.-F. Li, R. Xie, J.-C. Jiang, T. Meng, M. Yang, X.-J. Ju, L. Yang and L.-Y. Chu, Thermo-Responsive Gating Membranes with Controllable Length and Density of Poly(N-Isopropylacrylamide) Chains Grafted by ATRP Method, J. Membr. Sci., 2009, 337(1–2), 310–317, DOI:10.1016/j.memsci.2009.04.010.
- A. Tufani and G. Ince, Smart Membranes with PH-Responsive Control of Macromolecule Permeability, J. Membr. Sci., 2017, 537(January), 255–262, DOI:10.1016/j.memsci.2017.05.024.
- M. Motornov, R. Sheparovych, E. Katz and S. Minko, Chemical Gating with Nanostructured Responsive Polymer Brushes: Mixed Brush versus Homopolymer Brush, ACS Nano, 2007, 2(1), 41–52 CrossRef.
- L. A. Fielding, S. Edmondson and S. P. Armes, Synthesis of PH-Responsive Tertiary Amine Methacrylate Polymer Brushes and Their Response to Acidic Vapour, J. Mater. Chem., 2011, 21(32), 11773–11780 RSC.
- Q. Yan, R. Zhou, C. Fu, H. Zhang, Y. Yin and J. Yuan, CO2-Responsive Polymeric Vesicles That Breathe, Angew. Chem., Int. Ed., 2011, 50(21), 4923–4927 CrossRef CAS.
- S. Lin and P. Theato, CO2-Responsive Polymers, Macromol. Rapid Commun., 2013, 34(14), 1118–1133 CrossRef CAS.
- H. Liu, S. Lin, Y. Feng and P. Theato, CO 2-Responsive Polymer Materials, Polym. Chem., 2017, 8(1), 12–23 RSC.
- Q. Zhang, L. Lei and S. Zhu, Gas-Responsive Polymers, ACS Macro Lett., 2017, 6(5), 515–522, DOI:10.1021/acsmacrolett.7b00245.
- Z. Guo, Y. Feng, S. He, M. Qu, H. Chen, H. Liu, Y. Wu and Y. Wang, CO2-Responsive “Smart” Single-Walled Carbon Nanotubes, Adv. Mater., 2013, 25(4), 584–590 CrossRef CAS.
- H. Che, M. Huo, L. Peng, T. Fang, N. Liu, L. Feng, Y. Wei and J. Yuan, CO2-Responsive Nanofibrous Membranes with Switchable Oil/Water Wettability, Angew. Chem., Int. Ed., 2015, 54(31), 8934–8938 CrossRef CAS.
- Y. Li, L. Zhu, N. Grishkewich, K. C. Tam, J. Yuan, Z. Mao and X. Sui, CO 2 -Responsive Cellulose Nanofibers Aerogels for Switchable Oil–Water Separation, ACS Appl. Mater. Interfaces, 2019, 11(9), 9367–9373, DOI:10.1021/acsami.8b22159.
- Z. Zhang, T. Shimizu, S. Senz and U. Gösele, Ordered High-Density Si [100] Nanowire Arrays Epitaxially Grown by Bottom Imprint Method, Adv. Mater., 2009, 21(27), 2824–2828 CrossRef CAS.
- G. E. J. Poinern, X. Le, C. Loomes and D. Fawcett, Biocompatibility of Composite Membranes Composed of Anodic Aluminium Oxide (AAO) and Poly (2-Hydroxyethylmethacrylate) for Use as a Cell Culture Substrate, Mater. Lett., 2014, 131, 182–185 CrossRef CAS.
- C. P. Sharma and M. C. Sunny, Albumin Adsorption on to Aluminium Oxide and Polyurethane Surfaces, Biomaterials, 1990, 11(4), 255–257 CrossRef CAS.
- R. Pal and D. Kundu, Sol-Gel Synthesis of Porous and Dense Silica Microspheres, J. Non-Cryst. Solids, 2009, 355(1), 76–78, DOI:10.1016/j.jnoncrysol.2008.03.052.
- B. Schneider, J. Štokr, P. Schmidt, M. Mihailov, S. Dirlikov and N. Peeva, Stretching and Deformation Vibrations of CH2, C (CH3) and O (CH3) Groups of Poly (Methyl Methacrylate), Polymer, 1979, 20(6), 705–712 CrossRef CAS.
- S. Turgman-Cohen and J. Genzer, Simultaneous Bulk-and Surface-Initiated Controlled Radical Polymerization from Planar Substrates, J. Am. Chem. Soc., 2011, 133(44), 17567–17569 CrossRef CAS.
- Y. Cai, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, A Smart Membrane with Antifouling Capability and Switchable Oil Wettability for High-Efficiency Oil/Water Emulsions Separation, J. Membr. Sci., 2018, 555(January), 69–77, DOI:10.1016/j.memsci.2018.03.042.
- L. Hu, S. Gao, Y. Zhu, F. Zhang, L. Jiang and J. Jin, An Ultrathin Bilayer Membrane with Asymmetric Wettability for Pressure Responsive Oil/Water Emulsion Separation, J. Mater. Chem. A, 2015, 3(46), 23477–23482 RSC.
- D. Han, X. Tong, O. Boissière and Y. Zhao, ACS Macro Lett., 2011, 1, 57–61 CrossRef.
- H. Jiang, E. Wang and J. Wang, RSC Adv., 2015, 5, 35622–35630 RSC.
- S. Kumar, X. Tong, Y. L. Dory, M. Lepage and Y. Zhao, Chem. Commun., 2013, 49, 90–92 RSC.
- S. Turgman-Cohen and J. Genzer, J. Am. Chem. Soc., 2011, 133, 17567–17569 CrossRef CAS.
- Y. Cai, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, J. Memb. Sci., 2018, 555, 69–77 CrossRef CAS.
- L. Hu, S. Gao, Y. Zhu, F. Zhang, L. Jiang and J. Jin, J. Mater. Chem. A, 2015, 3, 23477–23482 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr04248j |
| This journal is © The Royal Society of Chemistry 2020 |