
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic factors affecting the stability of methylammonium lead halide perovskite nanocrystals†
Barry
McKenna
a,
Abhinav
Shivkumar
a,
Bethan
Charles
 b and
Rachel C.
Evans
*b
b and
Rachel C.
Evans
*b
aSchool of Chemistry and CRANN, Trinity College, The University of Dublin, Dublin 2, Ireland
bDepartment of Materials Science & Metallurgy, University of Cambridge, UK. E-mail: rce26@cam.ac.uk
First published on 22nd May 2020
Abstract
Lead halide perovskite nanocrystals (PNCs) have emerged as promising candidates for use in optoelectronic devices. Significant focus has been directed towards optimising synthetic conditions to obtain PNCs with tunable emission properties. However, the reproducible production of stable PNC dispersions is also crucial for fabrication and scale-up of these devices using liquid deposition methods. Here, the stability of methylammonium lead halide (MAPbX3 where X = Br, I) PNCs produced via the ligand-assisted reprecipitation process is explored. We have focussed on understanding how different combinations of specific synthetic factors – dilution, halide source and ratio as well as capping-ligand concentration – affect the stability of the resultant PNC dispersion. Photoluminescence spectroscopy, transmission electron microscopy and dynamic light scattering studies revealed that subtle changes in the reaction conditions lead to significant changes in the particle morphology and associated optical properties, often with catastrophic consequences on stability. This study highlights the importance of designing PNC dispersions in order to make more efficient and reliable optoelectronic devices.
Introduction
Perovskite semiconductors have emerged as exciting new materials for the photoactive component in optoelectronic devices. While initial applications have focused on thin film perovskite photovoltaic devices,1,2 developments in the field of perovskite nanocrystals (PNCs)3,4 have revealed them to be promising candidates for light-emitting devices (LEDs),5,6 lasing materials,7 scintillators8 and photocatalysis.9 Archetypical three-dimensional (3D) perovskites adopt an ABX3 structure, where A is a small organic or inorganic cation such as methylammonium (CH3NH3+) or Cs+. The metal cation located at the B-site (usually Pb2+) forms octahedra with the six nearest ions at the X-sites, typically Br− and/or I−.10 An alluring outcome of this site-substitution is the nature of the bandgap, which can be tuned to emit across the entire visible spectrum.11 Optical properties can also be tuned through careful control of nanocrystal size.12 These desirable features, in addition to high photoluminescence quantum yields (PLQYs) have led to an explosion of research in PNCs for applications in optoelectronic devices, such as solar cells,3,13 solar concentrators,14 downconverters15 and photodetectors.16Methylammonium lead halide PNCs (MA-PNCs) can be prepared using low-temperature, solution-based methods; the most common approach is ligand-assisted reprecipitation (LARP).17 Here, the perovskite precursor salts are dissolved in polar solvents, such as N,N-dimethylformamide (DMF) or dimethyl sulfoxide (DMSO). The resulting solution is injected rapidly into an antisolvent – often toluene or hexane – which typically contains long-chain capping ligands. These ligands have multiple functions, including improving the solubility of lead halide salt precursors, controlling the crystallisation kinetics and stabilising the final colloidal dispersion.18,19
The choice of capping ligand has a substantial effect on the behaviour of PNCs. Oleic acid (OA) and oleylamine (OY) are commonly used; however, these form an insulating layer and often need to be removed before application in an optoelectronic device such as an LED leaving the PNC susceptible to degradation.20 Previous work has overcome this issue through replacing OA and OY with a bidentate ligand,20 while use of branched capping ligands has also been shown to improve PNC stability.21
Varying the halide content, X, in MAPbX3 and CsPbX3 nanocrystals is commonly employed to achieve emission in the blue (X = Cl), green (X = Br) or red (X = I) spectral regions.22,23 However, emission can also be tuned across the entire visible spectrum through the use of mixed-halide PNCs, such as MAPbCl3−xBrx or MAPbBr3−xIx (0 ≤ x ≤ 1).24 Synthetically, these may be obtained by mixing the precursor halide salts in the desired ratio prior to injection into the antisolvent,23,25 mixing solutions of pure-halide PNCs,26 or via post-synthesis halide exchange.22,27,28 A tunable bandgap and high colour purity makes these mixed-halide PNC systems promising candidates for LEDs.11,29 However, these desirable properties also have the potential for applications in luminescent downshifting layers, where the emission could be optimised for the chosen solar cell.30
The emission colour also depends on the morphology of the PNCs, which is determined both by the composition and processing conditions.17 The first reported synthesis of MAPbBr3 nanocrystals yielded a mixture of 3D nanoparticles and 2D nanoplatelets.31 Subsequent studies have shown that extremely thin MAPbX3 nanoplatelets can be isolated directly through synthetic control25,32 or obtained indirectly through ligand-assisted exfoliation of the layered morphology.33,34 The capping ligands used in the LARP process significantly impact the crystal growth and dimensions, which can have a profound effect on their optical properties. For example, Sichert et al. showed that as the lateral dimension of 3D MAPbBr3 nanocrystals is gradually reduced to quasi-2D and 2D nanoplatelets with a thickness of a single unit cell, with the emission maximum shifting to higher energy due to quantum confinement effects.34 The precise composition and concentration of capping ligands and/or alkyl ammonium salt used in the LARP process may also have a profound effect on the emission colour and PLQY of the resultant PNCs.18,23
Interestingly, the dilution of PNC dispersions has an unpredictable effect on both nanocrystal size and emission properties. For example, Tong et al. showed that dilution of stock solutions of MAPbBr3 or MAPbI3 PNCs with the LARP antisolvent resulted in fragmentation and exfoliation into 2D nanoplatelets. The resulting emission spectrum was blue shifted, with a decrease in PLQY.35 A separate study investigating the effect of solvent environment on the nucleation and growth mechanism of CsPbX3 PNCs revealed that while the initial shape and size was controlled by the solvent polarity, subsequent dilution led to self-assembly and growth of the nanocrystals.36
As for all colloidal systems, the stability of the resultant PNC dispersion is highly sensitive to the solvent type and concentration.37 Instability may arise due to the retention of residual highly-coordinating polar solvents used in the LARP process to the particle surface.37 Recent studies have shown that a mixture of methylamine in acetonitrile is a good non-coordinating polar solvent for metal halide salts,38 which was later exploited to prepare red-emitting MAPbI3 nanocrystals with a PLQY of >93%.39
The size and morphology of MA-PNCs are significantly affected by synthetic factors such as reaction medium, capping ligands, solvent system and dilution. These factors have been discussed more generally in several excellent reviews.3,4,17,40 However, this sensitivity of MA-PNCs to synthetic conditions results in marked differences in both their stability and optical properties. These results are rarely reported in detail, perhaps because they are unlikely to produce a champion system. Yet, this information is crucial both for new researchers attempting to enter a competitive field, and also for the development of realistic pathways to scale-up. To address this gap, we systematically examine three key areas of PNC synthesis: (1) the effect of dilution of the parent dispersion, (2) choice of the halide source and ratio and (3) choice of capping ligand concentration. We examine the mechanism for particle nucleation and growth and provide insight into the dramatic changes in optical properties that may result upon introducing seemingly minor changes to the synthetic conditions.
Experimental
Materials
Lead(II) iodide (PbI2, 99.999% trace metal basis), oleylamine (tech. 70%), hydroiodic acid (57 wt% in H2O), methylamine solution (33 wt% in ethanol), chloroform (98%) and N,N-dimethyl formamide (99.8% anhydrous) were purchased from Sigma-Aldrich. Toluene (anhydrous, 99.8%) and oleic acid (90%) were purchased from Alfa Aesar. Lead(II) bromide (PbBr2, 98%) was purchased from Acros Organics. Hydrobromic acid (47%) was purchased from VWR. All chemicals were used as received without further purification.Synthesis of methyl ammonium halide (MAX (X = Br, I))
Vigorously stirred methylamine (24 mL, 33 wt% in ethanol) was further diluted with ethanol (50 mL) and placed under nitrogen in an ice bath. HBr (9 mL, 7.6 × 10−2 moles) or HI (10 mL, 7.6 × 10−2 moles) was then slowly added. The reaction solution was kept in the ice bath and stirred for 2 h until a precipitate formed. The precipitate was isolated by vacuum filtration, washed three times with diethyl ether (ca. 100 mL) and dried in an oven at 60 °C.Synthesis and dilution of MAPbX3 PNCs
MAPbX3 PNCs were synthesised using the LARP method shown schematically in Fig. 1a. Initially, mixed halide MAPbBr3−xIx PNCs were targeted using an appropriate mixture of PbBr2 and MAI precursors. In the standard synthesis, PbBr2 (73.4 mg, 0.2 mmol) and methylammonium iodide (25.4 mg, 0.16 mmol) were dissolved in anhydrous DMF (5 mL). Oleylamine (50 μL, 0.15 mmol) and oleic acid (500 μL, 1.6 mmol) were then added to the mixture under ultrasonic agitation to form the precursor solution. An aliquot (500 μL) of this solution was quickly injected into anhydrous toluene (5 mL) at 40 °C under vigorous stirring. Upon mixing, the solution turned bright yellow/green in colour. After stirring for 15 min, a cloudy orange precipitate formed. The suspension was centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 6 min and the precipitate discarded. The yellow/green supernatant was retained. Variations on this synthetic procedure, which included increasing the concentration of the capping ligand between 0.18–0.30 mmol (OY sample series) and changing the ratio of bromide to iodide by replacing PbBr2/MAI with PbI2/MABr (R sample series), are further detailed in the Results and discussion section. A full break-down of the composition of the reaction medium for each sample can be found in Table S1 (see ESI†). As-prepared PNC suspensions were diluted with anhydrous toluene. Samples are denoted according to the dilution factor, for example sample D20 has a dilution factor of 20, i.e. 1 mL of the precursor solution was added to 19 mL of toluene.
000 rpm for 6 min and the precipitate discarded. The yellow/green supernatant was retained. Variations on this synthetic procedure, which included increasing the concentration of the capping ligand between 0.18–0.30 mmol (OY sample series) and changing the ratio of bromide to iodide by replacing PbBr2/MAI with PbI2/MABr (R sample series), are further detailed in the Results and discussion section. A full break-down of the composition of the reaction medium for each sample can be found in Table S1 (see ESI†). As-prepared PNC suspensions were diluted with anhydrous toluene. Samples are denoted according to the dilution factor, for example sample D20 has a dilution factor of 20, i.e. 1 mL of the precursor solution was added to 19 mL of toluene.
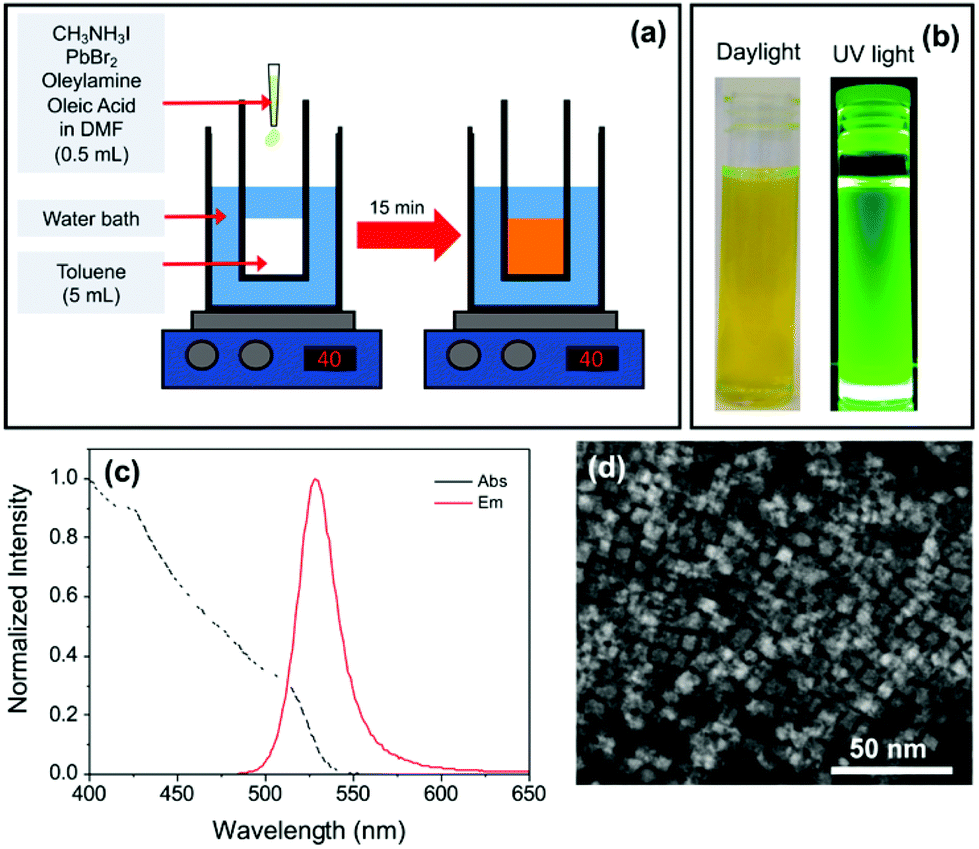 | ||
| Fig. 1 Fabrication and characterisation of a standard sample of MAPbBr3−xIx PNCs in toluene. (a) Scheme of the LARP synthesis protocol. (b) Photographs of the PNC suspension obtained after centrifugation under white (left) and UV (right) illumination. (c) UV/Vis absorption (black dashed line) and steady-state photoluminescence (red solid line) spectra (λex = 400 nm). (d) STEM image revealing the formation of platelets. | ||
Characterisation
All measurements were performed at room temperature. UV/Vis absorption spectroscopy was carried out using a Lambda 35 (PerkinElmer) spectrometer. Steady-state photoluminescence (PL) spectroscopy measurements were performed on a FluoroMax-4 spectrophotometer (Horiba). PL spectra were corrected for the wavelength response of the system using correction factors supplied by the manufacturer. Slit widths were 2 nm for absorption and for PL measurements the excitation and emission slits and integration time were varied according to the emission intensity of the sample. PLQY measurements were performed using an F-3018 integrating sphere accessory mounted on a Fluorolog FL 3-22 (Horiba). The reported values are the mean of three repeat measurements. This method is accurate to within 10%.PL lifetime measurements were carried out using a Horiba Fluorolog FL 3-22 equipped with a FluoroHub v2.0 single photon controller using the time-correlated single photon counting method (TCSPC), run in reverse mode. Samples were excited at 458 nm with a pulsed nanosecond light-emitting diode (NanoLED®). The instrument response function (IRF) was recorded prior to lifetime measurements using a scattering solution (Ludox, Aldrich) and the pulse width was 1.3 ns. Emission decay curves were analysed using IBH DAS6 software. The data were fit as a sum of exponentials and the errors in these fits are accounted for by considering non-linear least squares analysis for error minimisation.
Dynamic light scattering (DLS) measurements were performed on a Zetasizer Nano–ZS (Malvern Instruments) which uses a He–Ne laser (4.0 mW, 633 nm) light source and detection at a backscattering angle of 175°.
Transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), energy dispersive X-ray spectroscopy (EDX) and electron energy loss spectroscopy (EELS) were performed on a FEI Titan operating at 300 kV. A fixed volume (2 × 15 μL) of the PNC dispersion was drop-cast onto a carbon type B on 200 mesh Cu grid. Imaging was performed in TEM and STEM modes using the high-angle annular dark-field (HAADF) detector. EDX and EELS analysis was performed in STEM mode.
Results and discussion
Synthesis of mixed halide PNCs
Our initial aim was to synthesise mixed halide MAPbBr3−xIx PNCs using the LARP method, as illustrated in Fig. 1a, for potential use in luminescent downshifting layers. A precursor solution containing a non-stoichiometric mixture of methylammonium iodide (MAI) and PbBr2 (0.8:1 molar ratio) in DMF was first prepared. Long-chain capping ligands oleic acid (OA) and oleylamine (OY) were dissolved in the mixed halide precursor solution. Upon fast injection of an aliquot of this mixture into toluene (40 °C), an orange precipitate formed, which was removed by centrifugation. The retained supernatant was yellow/green in colour and exhibited bright green photoluminescence under UV light (Fig. 1b).
The PL spectrum (Fig. 1c) is characteristic of MAPbBr3 PNCs (λem ∼ 530 nm), suggesting negligible incorporation of iodide into the structure. TEM revealed the sample was composed of platelets with a lateral diameter of ca. 10 nm (Fig. 1d). EDX analysis showed the absence of iodide peaks, suggesting that, despite the inclusion of MAI in the reaction mixture, the resultant PNCs are predominantly all-bromide in composition. It is noted that the concentration of iodide present could be beyond the detection limit of the technique (Fig. S1†).
We believe that a combination of surface area, charge and ligand effects are responsible for the failed uptake of I− into the mixed halide MAPbBr3−xIx PNCs. PNC formation has been shown to proceed through a seed-mediated nucleation step, with seed Pb0 nanoparticles and the polarity of the environment determining the shape and size of resultant PNCs.36 The small particle size implies that the 1:3 ratio of Pb2+ to X− observed in bulk perovskites does not hold for the nanoparticles, due to the presence of an anion-rich surface, which has previously been shown for CdSe quantum dots41 and PNCs.18
Incorporation of the halide is also dependent on the halide source and ratios, since halide ions are important surface-active species which influence colloidal growth.42 Our precursor solution used a non-stoichiometric ratio of MAI and PbBr2 (0.8:1 molar ratio), it is possible this favoured incorporation of bromide into the structure, considering the structural distortion induced by the larger iodide ion.22 Moreover, the lower electronegativity of the iodide ion implies specific adsorption to the PNC surface is less likely than for the bromide,42 while hydrogen bonding between free I− ions and excess OY capping ligand may prevent competitive uptake.43
Nevertheless, the complete absence of iodide was unexpected. Previous studies have shown that components of PbX2 and MAX can be readily exchanged during crystallisation, both in solution and the solid state for bulk MAPbX3.27 The synthesis of mixed-halide MAPbBr3−xClx and MAPbBr3−xIx PNCs through reversible bromide exchange has also been shown.22 However, several key distinctions can be identified in the synthetic conditions of the latter study and ours, namely solvent, type of capping ligand, and sequential addition of the MAI precursor. In contrast, Zhang et al. reported the formation of MAPbBr3−xXx (X = Cl or I) quantum dots using very similar reaction conditions to those used here, differing only in the amine capping ligand and the halide source.18
These results highlight the sensitivity of the LARP procedure to subtleties in the reaction medium and demonstrate the importance of understanding the effect each step in the synthetic process can have on the final product. From this initial experiment, three key synthetic conditions (halide source, capping ligand concentration and dilution in a solvent) were identified as having significant effects on final PNC properties and were subsequently investigated. These initial findings motivated the work presented in the following sections which set out to assess the effect of (1) dilution of the parent dispersion (2) the choice of the halide source and ratio and (3) the choice of capping ligand concentration on PNC properties.
Effect of dilution
It is common practice to dilute PNC dispersions; however, an important observation from the mixed-halide synthesis reported above, were the significant changes in PL seen depending on the dilution factor, Dn, of the standard MAPbBr3−xIx PNC dispersion. While the absorption spectrum remains effectively unchanged, the emission maximum is red-shifted by ∼20 nm for dilution between D0 and D10, see Fig. 2a. Further dilution yields no additional spectral change. The PLQY also decreases significantly upon dilution, from 54 ± 4% in the parent sample, to 10 ± 0.5% upon dilution (D20). Dilution stimulates aggregation of PNCs which subsequently reduces PLQY through efficient exciton diffusion by the Förster resonance energy transfer (FRET) mechanism.44,45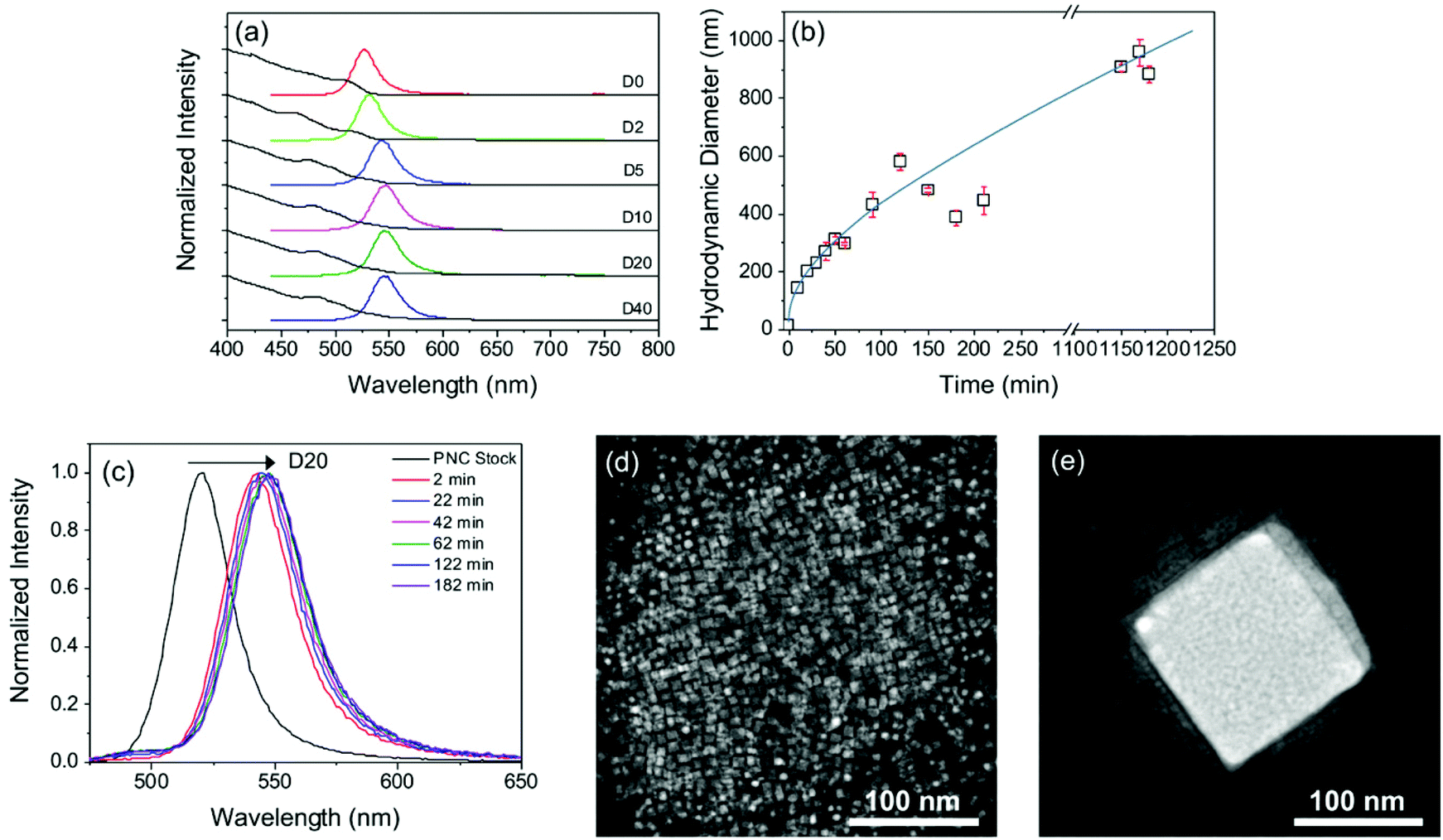 | ||
| Fig. 2 Effect of dilution on the time-dependent PL properties, size and morphology of MAPbBr3−xIx PNCs. (a) UV/Vis absorption (black line) and steady-state PL (coloured lines, λex = 400 nm) spectra for dilution factors of D0 to D40. (b) Variation in the hydrodynamic diameter, dh, upon dilution (D20) with time. The blue line serves only to guide the eye. The error bars indicate the standard deviation of three measurements. (c) Steady-state PL spectra of the parent PNC suspension before dilution (D20, black line) and at timed intervals after dilution (λex = 400 nm). STEM images of MAPbBr3 PNCs (d) before and (e) after dilution with toluene. | ||
As expected, a corresponding increase in the PL lifetime was also observed upon dilution (Fig. S3†). Both samples exhibit biexponential decay curves, whose components are assigned to delocalised (τ1) and localised (τ2) exciton recombination;46 see Table S2† for a summary of the fitting parameters. Upon dilution, both lifetime components increased, from τ1 = 10.7 ns to 12.4 ns and τ2 = 24.2 ns to 28.8 ns. The associated amplitude (A2) of τ2 can provide an estimate for the quantity of defects in the perovskite lattice.46 Interestingly, upon dilution, A2 decreased, suggesting that as the PNCs aggregate, a loss of defect sites occurs.
In order to relate the PL properties upon dilution to changes in the nanocrystal dimensions, we next performed DLS studies on both the parent and diluted PNC samples over time. In the parent sample (D0), the mean hydrodynamic diameter, dh, was ∼14 nm (Fig. S2†). Upon dilution, dh, increased with time until values of >1 μm were obtained after about 16 hours (see Fig. 2b for D20). This behaviour indicates that, in our system, dilution results in particle growth rather than in the fragmentation and exfoliation of layers, as observed previously for diluted MAPbBr3 and MAPbI3 PNCs prepared via a very similar LARP procedure by Tong et al.35 We note that the only difference in reaction conditions used compared to the previous dilution study35 and ours is the use of mixed halide sources (i.e. MAI/PbBr2), hinting that the iodide ion may play a role in promoting particle growth. PL measurements were performed concurrently with the time-dependent DLS study (Fig. 2c). Interestingly, the red-shift in the PL maximum occurs very quickly after dilution (2 min), suggesting that once particle growth exceeds ∼500 nm, the optical properties are dominated by the bulk material.
Fig. 2d and e compare STEM images of MAPbBr3−xIx PNCs before and after dilution (D20). In the parent sample (see also Fig. 1d), square nanoplatelets are observed with a diameter of ∼10 nm (in good agreement with the dh ∼ 14 nm). Following dilution, the nanoplatelets aggregate into a larger cubic structure which reaches dimensions of ∼150 nm in size. EDX analysis of the D20 sample revealed detectable iodide peaks (Fig. S4†), confirming that particle growth results in incorporation of iodide ions from the reaction medium. The peak ratios in the EDX spectrum suggest a Br− to I− mass ratio of 4:1, which corresponds to a molar ratio of ca. 2.5:1. The experimental data suggest that dilution stimulates particle growth. It is also clear that iodide ions present in the dispersion become incorporated into the larger particles formed in the growth process. The combination of these factors leads to the observed redshift in the emission.
We will now consider the potential mechanism for particle growth following dilution. Under our reaction conditions, we consider that the formation of apparent single halide MAPbBr3 PNCs in the parent sample is preferred due to the presence of excess bromide ions and the preferred retention of the cubic phase. Unused iodide ions are thus present in the bulk solution, and/or associated with the surface capping ligands via hydrogen bonding. Dilution of the sample decreases the effective concentration of the capping ligand in the surrounding dispersion. We propose that this provides a driving force for partial dissolution of the capping ligand from the surface of the nanocrystal, reducing the colloidal stability, leading to rapid aggregation of the PNCs. As the particles agglomerate, iodide ions are incorporated into the aggregated structure, moving towards the stoichiometric ratio present in the reaction mixture. Particle growth will lead to a decrease in surface area, and therefore a reduction in the number of surface trap sites, which is supported by the increased in the amplitude of the τ2 component attributed to localised exciton recombination. Moreover, the decrease in the PLQY and increase in PL lifetime are indicative of a transition to more bulk-like behaviour; for example, bulk MAPbBr3−xIx films have shown lifetimes greater than 100 ns.47
To confirm that the dilution-induced changes in the optical properties can be explained by partial desorption of the capping ligands, a sample was prepared using chloroform as the antisolvent instead of toluene. The increased polarity of chloroform (dielectric constant, ε = 4.81, versus ε = 2.38 for toluene)48 should facilitate improved ionic dissociation, and thus prevent excessive agglomeration of particles on mixing with DMF (ε = 37.8). The Hansen solubility parameters of oleic acid (δt = 16.81 MPa1/2) indicates lower solubility in chloroform (δt = 18.94 MPa1/2) than toluene (δt = 17.95 MPa1/2).49 We note that the Hansen solubility parameter for OY should be comparable to that of OA due to the structural similarities.49 As such, if the proposed mechanism is valid, aggregation should be reduced upon dilution of the parent solution with chloroform. Fig. 3 shows the emission properties before and after dilution (D20). While the parent sample exhibits bright green emission (λem = 545 nm), following dilution an intense blue emission with λem = 475 nm is observed.
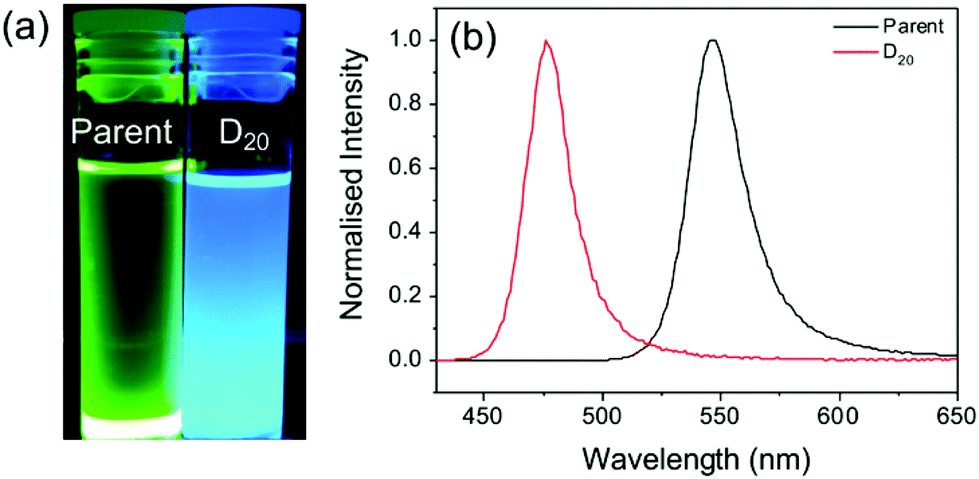 | ||
| Fig. 3 MAPbBr3−xIx PNC dispersions show different optical properties upon dilution in a more polar solvent (chloroform). (a) Photographs of the dispersion before and after dilution D20 under illumination at 365 nm and (b) corresponding PL spectra (λex = 400 nm). | ||
The blue-shifted emission of the diluted solution compared with the parent may be attributed to solvent exfoliation and thinning of the PNCs into 2D layers,34 or anion doping of chloride from the chloroform solvent molecules to the PNCs.35 However, the observed emission behaviour confirms the effect that the solvent can have on the dilution-induced behaviour of PNCs. Since toluene is a better solvent for the capping ligands, ligand dissociation and particle growth are favourable, whereas, chloroform, a poor ligand solvent, hinders this process.
Effect of halide source and ratio
We also found that the emission properties varied depending on which reactant provides the halide. To demonstrate this, precursor solutions containing the standard Br− to I− ratio of ca. 2:1 were prepared using different mixtures of PbX2 (PbBr2 + PbI2 = 0.2 mmol) and MAX (MABr + MAI = 0.16 or 0.2 mmol), see Table S1† for detailed compositions.
All parent samples exhibited the characteristic green emission indicative of bromide-rich MAPbBr3−xIx PNCs (Fig. 4a), indicating lack of incorporation of iodide ions into the crystal structure. Upon dilution (D20) in toluene, the emission maximum red-shifted for all samples, although the extent of this shift depended on the specific mixture of halide sources used. For example, the largest emission shift (∼28 nm) was obtained using a 1:1 ratio of PbX2 and MAX, and from a combination of all four reagents. In comparison, a much smaller red-shift of ∼12 nm was observed for the sample prepared from a 2:1.6 ratio of PbX2 to MAX (Fig. 4c and d). The most significant change occurs when MAI is the primary iodide source (rather than PbI2) (Fig. 4d). Since MA cations are associated with the particle surface, this supports the hypothesis that iodide bound to surface ligands is favourably incorporated upon dilution and subsequent growth. These results demonstrate that the optical properties of PNCs can vary significantly when using different halide precursors.
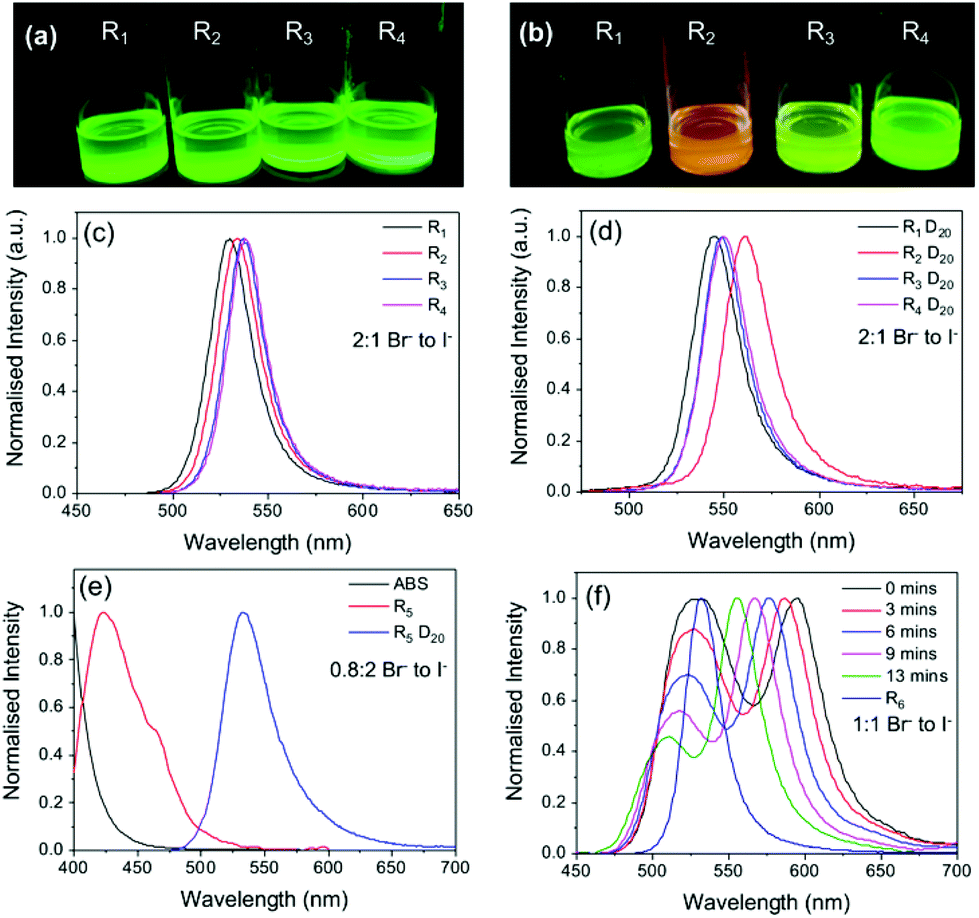 | ||
| Fig. 4 The halide source and ratio have a significant effect on the optical properties of MAPbBr3−xIx PNC dispersions. Photographs under UV illumination and corresponding PL spectra of PNC dispersions made with the standard Br:I ratio of ca. 2:1 using different halide sources (a, c) before and (b, d) after dilution D20. (e) and (f) show the PL spectra of PNC dispersions made using different halide ratios, before and after dilution D20, and as a function of time after dilution for sample R6. Full sample compositions given in Table S1, ESI† (λex = 400 nm). | ||
The effect of halide ratio in conjunction with different halide sources was also explored. PNC dispersions with different Br:I ratios (0.8:2 (R5), 1:1 (R6) and 6:1 (R7)) and different halide sources were also prepared (see Table S1†). After centrifugation, R5 was a transparent solution with a weak blue emission, whose spectrum suggested a composition of primarily PbBr2 seed particles. Upon dilution, the emission maximum changed from ca. 420 to 530 nm, and the sample exhibited the bright green emission characteristic of the standard parent MAPbBr3 PNC sample (∼2:1 Br− to I− ratio in precursor), see Fig. 4a. Sample R6 (1:1 Br− to I−) showed similar emission behaviour, with an emission red-shift of ∼25 nm upon dilution D20 (Fig. 4f); however, for R6, this occurred via a slow stepwise process. Initially upon dilution, two emission peaks of equal intensity were observed at 528 nm and 595 nm. A short time after (ca. 13 min), the relative intensity of the peak at 528 nm had decreased, while the peak at 595 nm had reverse blue shifted back to 555 nm, becoming the primary emission peak. The observed behaviour suggests that upon dilution (D20), two populations with different halide compositions are initially present. Subsequent agglomeration and/or growth results in a single population of micrometre-sized particles comprised primarily of MAPbBr3 with trace amounts of iodide impurities. Sample R7 (6:1 Br− to I−) gave a green emission and notably, upon dilution, no change in the optical properties were observed (Fig. S6†). Given the low iodide content of the precursor solution, this result further indicates that the change in the spectral properties upon dilution is partially due to iodide uptake as the particles grow.
Role of capping ligand concentration
Assuming the mechanism described above, in that particle growth is directly related to a decrease in the effective concentration of the capping ligand upon dilution, a similar behaviour should be observed if different capping ligand concentrations are used in the parent sample. Previous studies have shown that the thickness of PNCs can be tailored by varying the capping ligand content.12,33 Typically, increased capping ligand concentration results in thinner nanocrystals, with excess capping ligand stabilising the increased surface area of smaller particles.12 A series of samples was prepared in which the volume of the stock solution of the OY capping ligand was varied. Samples are designated as OYn, where n = 50–100 (μl) and OY50 corresponds to the standard fabrication procedure. A modest increase in the OY concentration (OY60–OY70) compared to the standard OY50 sample resulted in a slight blue-shift in emission maximum (Fig. S7†), while upon dilution (D20), these samples displayed an emission red-shift of 20–30 nm. Conversely, the samples OY80 and OY90, containing a higher concentration of OY, were observed to turn from pale yellow to colourless with time (Fig. S7†). While these samples initially showed green emission, the PL was lost as the solution became transparent.Interestingly, for the samples containing the highest capping ligand concentration (OY100), the initial yellow solution was not observed, but rather a colourless, transparent solution was obtained immediately after centrifugation (Fig. 5a). This solution was only weakly emissive, with several additional peaks in the spectrum, indicating that a variety of particle sizes and compositions were present (Fig. 5b). Dilution (D20) of this sample in toluene results in an immediate red-shift in the emission maximum (λem ∼ 480 nm), corresponding to the blue–green emission observed (Fig. 5a and b).
 | ||
| Fig. 5 Dilution effects in PNC dispersions under high capping concentrations. (a) Photographs of MAPbBr3−xIx PNCs prepared with excess capping ligand (OY100) under illumination with daylight or UV light, before and after dilution (D20). Following dilution, the emission turns from blue–green (t = 0 h) to red (t = 24 h). (b) Steady-state PL spectra of the OY100 sample, before and after dilution D20 (λex = 400 nm). STEM images of OY100 samples, (c) before and (d) after dilution. | ||
STEM images of the samples before and after dilution reveal no change in particle size, with spherical dot-like structures with dimensions <10 nm retained (Fig. 5c and d). Similar behaviour has previously been attributed to the formation of one-dimensional PbBr2 nanoparticles upon solvation of excess MAI and Br− by the capping ligands, leading to a similarly structured emission spectrum as observed here for OY100.50 Since PbBr2 nanoparticles act as seeds for the growth of PNCs,18 it follows that upon dilution, the relative concentration of the capping ligand in the solution decreases, decreasing the solubility of any excess MAI and Br− and facilitating particle growth from these seeds. However, the emergence of a single emission peak upon dilution and the small particle dimensions (<10 nm), suggest further growth into a single population of quantum-confined MAPbBr3 PNCs.34 The dilute OY100 sample is unstable, as after 24 h a bright red fluorescence was observed (Fig. 5a). The corresponding emission spectrum contained an additional emission peak at 580 nm, of similar intensity to the primary peak at 480 nm (Fig. S8†). Upon centrifugation the red colour was lost, and the dispersion became non-photoluminescent, suggesting that large PNC particles (removed by centrifugation) were the origin of the red emission.
Conclusions
This study highlights some of the most pressing issues facing scale-up and application of PNC dispersions: reproducibility and stability. We have explored the effect of subtle changes in the synthetic conditions used in the LARP process on the morphology, photoluminescence and stability of the resultant PNC dispersions. An initial investigation targeting MAPbBr3−xIx PNCs, using standard LARP conditions, revealed a lack of iodide incorporation into the parent dispersions, as well as a strong tendency to aggregate into larger structures whose emission properties were more characteristic of the bulk material. This motivated subsequent work assessing the effect of dilution, choice of halide source and ratio and choice of capping ligand concentration. These systematic investigations supported the hypothesis that aggregation proceeds via desorption of surface capping ligands, accompanied by iodide incorporation into the bulk aggregate, suggested by a low, but detectable, iodide content in PNC aggregates. This study demonstrates that accurate reporting of reaction conditions is crucial for reproducibility in colloidal synthesis. The conditions used here are remarkably similar to those commonly reported in the literature; yet there are subtle differences which have a profound influence on the final PNC dispersion. We hope that this work will stimulate transparent reports related to the challenges associated with reproducible synthesis of PNC dispersions. It is our belief that this will both accelerate commercialisation of PNC-based applications and facilitate the entry of new researchers to the field, as a greater understanding of these minor changes has significant implications for the reproducibility and performance of PNC based optoelectronic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science Foundation Ireland under Grant No. 12/IP/1608. R. C. E. acknowledges funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 818762 - SPECTRACON).References
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- D. Chen and X. Chen, J. Mater. Chem. C, 2019, 7, 1413–1446 RSC.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- L. Polavarapu, B. Nickel, J. Feldmann and A. S. Urban, Adv. Energy Mater., 2017, 7, 1–9 Search PubMed.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- L. J. Chen, J. H. Dai, J. De Lin, T. S. Mo, H. P. Lin, H. C. Yeh, Y. C. Chuang, S. A. Jiang and C. R. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 33307–33315 CrossRef CAS PubMed.
- Y. Li, W. Shao, X. Ouyang, X. Ouyang, Z. Zhu, H. Zhang, B. Liu and Q. Xu, J. Phys. Chem. C, 2019, 123, 17449–17453 CrossRef CAS.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- I. Levchuk, P. Herre, M. Brandl, A. Osvet, R. Hock, W. Peukert, P. Schweizer, E. Spiecker, M. Batentschuk and C. J. Brabec, Chem. Commun., 2017, 53, 244–247 RSC.
- Q. A. Akkerman, M. Gandini, F. Di Stasio, P. Rastogi, F. Palazon, G. Bertoni, J. M. Ball, M. Prato, A. Petrozza and L. Manna, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- F. Meinardi, Q. A. Akkerman, F. Bruni, S. Park, M. Mauri, Z. Dang, L. Manna and S. Brovelli, ACS Energy Lett., 2017, 2, 2368–2377 CrossRef CAS PubMed.
- J. Zhao, M. Liu, L. Fang, S. Jiang, J. Zhou, H. Ding, H. Huang, W. Wen, Z. Luo, Q. Zhang, X. Wang and C. Gao, J. Phys. Chem. Lett., 2017, 8, 3115–3121 CrossRef CAS PubMed.
- J. Zhang, Q. Wang, X. Zhang, J. Jiang, Z. Gao, Z. Jin and S. (Frank) Liu, RSC Adv., 2017, 7, 36722–36727 RSC.
- K. Chen, S. Schünemann, S. Song and H. Tüysüz, Chem. Soc. Rev., 2018, 47, 7045–7077 RSC.
- F. Zhang, H. Zhong, C. Chen, X. G. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- Z. Yuan, Y. Shu, Y. Xin and B. Ma, Chem. Commun., 2016, 52, 3887–3890 RSC.
- J. Pan, Y. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-Zohry, M. N. Hedhili, A. H. Emwas, O. F. Mohammed, Z. Ning and O. M. Bakr, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS PubMed.
- B. Luo, Y. C. Pu, S. A. Lindley, Y. Yang, L. Lu, Y. Li, X. Li and J. Z. Zhang, Angew. Chem., Int. Ed., 2016, 55, 8864–8868 CrossRef CAS PubMed.
- D. M. Jang, K. Park, D. H. Kim, J. Park, F. Shojaei, H. S. Kang, J. P. Ahn, J. W. Lee and J. K. Song, Nano Lett., 2015, 15, 5191–5199 CrossRef CAS PubMed.
- S. Pathak, N. Sakai, F. Wisnivesky Rocca Rivarola, S. D. Stranks, J. Liu, G. E. Eperon, C. Ducati, K. Wojciechowski, J. T. Griffiths, A. A. Haghighirad, A. Pellaroque, R. H. Friend and H. J. Snaith, Chem. Mater., 2015, 27, 8066–8075 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- M. C. Weidman, M. Seitz, S. D. Stranks and W. A. Tisdale, ACS Nano, 2016, 10, 7830–7839 CrossRef CAS PubMed.
- D. J. Freppon, L. Men, S. J. Burkhow, J. W. Petrich, J. Vela and E. A. Smith, J. Mater. Chem. C, 2017, 5, 118–126 RSC.
- D. T. Moore, H. Sai, K. W. Tan, L. A. Estroff and U. Wiesner, APL Mater., 2014, 2, 081802 CrossRef.
- C. Jia, H. Li, L. Tan, X. Meng, J. Gao and H. Li, Nanoscale, 2019, 11, 3248–3260 RSC.
- S. D. Stranks and H. J. Snaith, Nat. Nanotechnol., 2015, 10, 391–402 CrossRef CAS PubMed.
- Y. C. Kim, H. J. Jeong, S. T. Kim, Y. H. Song, B. Y. Kim, J. P. Kim, B. K. Kang, J. H. Yun and J. H. Jang, Nanoscale, 2020, 12, 558–562 RSC.
- L. C. Schmidt, A. Pertegas, S. Gonzalez-Carrero, O. Malinkiewicz, S. Agouram, G. Minguez Espallargas, H. J. Bolink, R. E. Galian and J. Perez-Prieto, J. Am. Chem. Soc., 2014, 136, 850–853 CrossRef CAS PubMed.
- M. C. Weidman, A. J. Goodman and W. A. Tisdale, Chem. Mater., 2017, 29, 5019–5030 CrossRef CAS.
- V. A. Hintermayr, A. F. Richter, F. Ehrat, M. Döblinger, W. Vanderlinden, J. A. Sichert, Y. Tong, L. Polavarapu, J. Feldmann and A. S. Urban, Adv. Mater., 2016, 28, 9478–9485 CrossRef CAS PubMed.
- J. A. Sichert, Y. Tong, N. Mutz, M. Vollmer, S. Fischer, K. Z. Milowska, R. García Cortadella, B. Nickel, C. Cardenas-Daw, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6521–6527 CrossRef CAS PubMed.
- Y. Tong, F. Ehrat, W. Vanderlinden, C. Cardenas-Daw, J. K. Stolarczyk, L. Polavarapu and A. S. Urban, ACS Nano, 2016, 10, 10936–10944 CrossRef CAS PubMed.
- T. Udayabhaskararao, M. Kazes, L. Houben, H. Lin and D. Oron, Chem. Mater., 2017, 29, 1302–1308 CrossRef CAS.
- F. Zhang, S. Huang, P. Wang, X. Chen, S. Zhao, Y. Dong and H. Zhong, Chem. Mater., 2017, 29, 3793–3799 CrossRef CAS.
- N. K. Noel, S. N. Habisreutinger, B. Wenger, M. T. Klug, M. T. Horantner, M. B. Johnston, R. J. Nicholas, D. T. Moore and H. J. Snaith, Energy Environ. Sci., 2017, 10, 145–152 RSC.
- Y. Hassan, O. J. Ashton, J. H. Park, G. Li, N. Sakai, B. Wenger, A.-A. Haghighirad, N. K. Noel, M. H. Song, B. R. Lee, R. H. Friend and H. J. Snaith, J. Am. Chem. Soc., 2019, 3, 1269–1279 CrossRef PubMed.
- H. Huang, L. Polavarapu, J. A. Sichert, A. S. Susha, A. S. Urban and A. L. Rogach, NPG Asia Mater., 2016, 8, e328–e328 CrossRef CAS.
- J. Jasieniak and P. Mulvaney, J. Am. Chem. Soc., 2007, 129, 2841–2848 CrossRef CAS PubMed.
- S. Ghosh and L. Manna, Chem. Rev., 2018, 118, 7804–7864 CrossRef CAS PubMed.
- J. Liu, X. Yang, K. Wang, X. He, Q. Wang, J. Huang and Y. Liu, ACS Nano, 2012, 6, 4973–4983 CrossRef CAS PubMed.
- J. K. Stolarczyk, A. Deak and D. F. Brougham, Adv. Mater., 2016, 28, 5400–5424 CrossRef CAS PubMed.
- J. Jagielski, S. Kumar, M. Wang, D. Scullion, R. Lawrence, Y. T. Li, S. Yakunin, T. Tian, M. V. Kovalenko, Y. C. Chiu, E. J. G. Santos, S. Lin and C. J. Shih, Sci. Adv., 2017, 3, eaaq0208 CrossRef PubMed.
- J. Zhao, M. Liu, L. Fang, S. Jiang, J. Zhou, H. Ding, H. Huang, W. Wen, Z. Luo, Q. Zhang, X. Wang and C. Gao, J. Phys. Chem. Lett., 2017, 8, 3115–3121 CrossRef CAS PubMed.
- Y. Yamada, T. Nakamura, M. Endo, A. Wakamiya and Y. Kanemitsu, J. Am. Chem. Soc., 2014, 136, 11610–11613 CrossRef CAS PubMed.
- I. M. Smallwood, Handbook of organic solvent properties, Arnold, London, 1966 Search PubMed.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, Boca Raton, 2007 Search PubMed.
- R. Nishikubo, N. Tohnai, I. Hisaki and A. Saeki, Adv. Mater., 2017, 29, 1–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Sample composition details, additional TEM, DLS, and photoluminescence spectra. See DOI: 10.1039/d0nr03227a |
| This journal is © The Royal Society of Chemistry 2020 |