
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLong-armed hexapod nanocrystals of cesium lead bromide†
Shangkun
Li
 ,
Julie
Probst
,
Philip D.
Howes
* and
Andrew J.
deMello
*
,
Julie
Probst
,
Philip D.
Howes
* and
Andrew J.
deMello
*
Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir Prelog Weg 1, 8093 Zürich, Switzerland. E-mail: andrew.demello@chem.ethz.ch; philip.howes@chem.ethz.ch
First published on 7th July 2020
Abstract
Colloidal lead halide perovskite nanocrystals (LHP NCs) assume a variety of morphologies (e.g. cubes, sheets, and wires). Their labile structural and surface characters allow them to undergo post-synthetic evolution of shape and crystallographic characters. Such transformations can be advantageous or deleterious, and it is therefore vital to both understand and exert control over these processes. In this study, we report novel long-armed hexapod structures of cesium lead bromide nanocrystals. These branched structures evolve from quantum-confined CsPbBr3 nanosheets to Cs4PbBr6 hexapods over a period of 24 hours. Time-resolved optical and structural characterization reveals a post-synthesis mechanism of phase transformation, oriented attachment and branch elongation. More generally, the study reveals important processes associated with LHP NC aging and demonstrates the utility of slow reaction kinetics in obtaining complex morphologies.
Introduction
The photophysical properties of colloidal nanocrystals (NCs) can be tuned as a function of shape, size, composition, crystalline phase, and surface character,1–3 with structures in the quantum-confined regime showing a particularly strong dependence on crystal morphology. The ability to control these physical parameters is critical for applications in multifunctional nanodevices.4–6 This is particularly prescient in the case of lead halide perovskite (LHP) NCs, which whilst showing tremendous promise as solar energy converters, light emitters, and in other optoelectronic applications, still present many mysteries, not least regarding how to stabilize them for long-term applications.7To date, a large number of different NC morphologies have been reported, with researchers being able to exert exquisite control over both growth and post-synthetic evolution of crystalline nanostructures.8 The motivation for these investigations comes from, on the one hand trying to understand how and why anisotropic structures form, and on the other trying to create novel structures with new or enhanced properties. Important examples include the widely tunable plasmonic and catalytic character of anisotropic noble metal nanoparticles,9 controlled long-range assembly of anisotropic NCs into superlattices,10,11 inorganic nanowires as charge transporters in flexible electronics,12 and the combined strong confinement and free carrier transport in low-dimensional lead halide perovskite nanostructures.13
Branched NCs are an important and diverse class of anisotropic structures.14 Addition of branches onto a central core allows complex structures, with tunable characteristics, to be synthesized.15–17 Branching is an important approach to controlling the morphology of noble metal nanostructures, for example imparting control over catalytic activity by exposure of specific facets,14 or for tuning absorption into the near-infrared range for photothermal therapy.18 Additionally, branched hexapod metal–organic framework colloids have been shown to exhibit increased catalytic activity in a model Knoevenagel reaction.19 For fluorescent inorganic semiconductors, branched tetrapod NCs have found an important application as strain gauges in flexible materials, as the force applied to the CdS arms transmits to the CdSe core, resulting in widening of CdSe bands and a proportional red-shift in photoluminescence.20 Furthermore, there is great interest in the construction of ordered mesostructures (superlattices) from branched NCs, since such structures have been shown to have tunable photonic, plasmonic, electronic, magnetic and catalytic properties.21–23
Several phases of cesium lead halide NCs have been reported, including CsPbX3 (X = Cl, Br, or I) and Cs4PbX6, with each possessing distinct properties. Although control over the phase can be exerted by modulating the ratio of Cs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb in the precursors, the phase can still be readily transformed by post-synthesis treatments, for example through addition of metal and/or halide sources,24,25 or by modulating surface ligands in solution. For example, the transition between cubic phase CsPbX3 NCs and rhombohedral phase Cs4PbX6 NCs can be controlled through variations in the surface ligand concentration and ratio,26,27 and it has been revealed that the ligand shells of CsPbBr3 and Cs4PbBr6 have completely different characters.27 Moreover, there has been some debate over the optical properties of the Cs4PbBr6 phase, with some parties claiming it has intrinsic green fluorescence,28–30 while others attributing this to CsPbBr3 impurities in the Cs4PbBr6 structure.24,31–33 There have also been some key observations made of cesium lead halide NCs undergoing slow crystal reformation processes over extended time periods. For example, Mehetor et al. observed slow crystal reformation (over 3–5 days) of purified ultrathin CsPbBr3 nanowires into wide-area 2D self-assemblies of monodisperse quantum rods, which they ascribed to a self-digestive conversion, whilst noting that the Pb-rich precursor nanowires and solvents with higher dielectric constants drive crystal transformation.34 Furthermore, Chen and co-workers have reported the solvothermal synthesis of CsPbBr3 dodecapods, whilst Peng et al. recently reported halide-controlled facet growth leading to dimension-tunable multipod nanostructures for perovskite NCs.35,36 However, both the arm length and arm–core ratio obtained using this method are low, with 10 nm arms on 50 nm cores yielding an arm:core ratio of 0.2. To facilitate further applications, for example in self-assembled mesostructures, it is imperative to exert control over arm growth, and to achieve extended arm growth of branched colloidal NCs.21
:Pb in the precursors, the phase can still be readily transformed by post-synthesis treatments, for example through addition of metal and/or halide sources,24,25 or by modulating surface ligands in solution. For example, the transition between cubic phase CsPbX3 NCs and rhombohedral phase Cs4PbX6 NCs can be controlled through variations in the surface ligand concentration and ratio,26,27 and it has been revealed that the ligand shells of CsPbBr3 and Cs4PbBr6 have completely different characters.27 Moreover, there has been some debate over the optical properties of the Cs4PbBr6 phase, with some parties claiming it has intrinsic green fluorescence,28–30 while others attributing this to CsPbBr3 impurities in the Cs4PbBr6 structure.24,31–33 There have also been some key observations made of cesium lead halide NCs undergoing slow crystal reformation processes over extended time periods. For example, Mehetor et al. observed slow crystal reformation (over 3–5 days) of purified ultrathin CsPbBr3 nanowires into wide-area 2D self-assemblies of monodisperse quantum rods, which they ascribed to a self-digestive conversion, whilst noting that the Pb-rich precursor nanowires and solvents with higher dielectric constants drive crystal transformation.34 Furthermore, Chen and co-workers have reported the solvothermal synthesis of CsPbBr3 dodecapods, whilst Peng et al. recently reported halide-controlled facet growth leading to dimension-tunable multipod nanostructures for perovskite NCs.35,36 However, both the arm length and arm–core ratio obtained using this method are low, with 10 nm arms on 50 nm cores yielding an arm:core ratio of 0.2. To facilitate further applications, for example in self-assembled mesostructures, it is imperative to exert control over arm growth, and to achieve extended arm growth of branched colloidal NCs.21
In this paper, we report a new class of long-armed multipod NCs—hexapod Cs4PbBr6 NCs—formed by slow post-synthesis phase and morphological transformation. Synthesis proceeds over ca. 24 hours, where the as-synthesized CsPbBr3 nanosheets undergo phase transformation (towards Cs4PbBr6) and oriented attachment, yielding hexapods with highly elongated arms and arm:core ratios up to 6.0. We are not aware of any previous reports of post-synthesis transformation of LHP NCs into long-arm branched nanostructures. As a result of the complex evolution process, the exact reaction mechanism for full shape transformation was hard to clarify. Nevertheless, this work paves the way for improved understanding and engineering of the size, shape, composition, and crystalline phase in anisotropic LHP NCs, and strengthens the understanding of phase-dependent optical properties.
Results
CsPbBr3 nanosheets were synthesized in a segmented-flow microfluidic reactor (Scheme S1†) as described previously,37 with oleylamine and octanoic acid surface ligands (molar ratio 1.2:1). After mixing the reagents and ligands in flow, in octadecene, the reaction mixture was heated to 180 °C for 30 seconds, before collecting the product. The crude solution (separated from the carrier oil) was then stirred at room temperature for 36 hours.
Morphology and elemental composition
Transmission electron microscopy (TEM) revealed that the final NCs took the form of long-armed multipods after 24 hours of incubation. Common among these were hexapods, possessing a core with six arms distributed around it. These hexapods exhibited an arm length of 360 ± 76 nm (Fig. 1a and b), with an average arm:core ratio of 6. Scanning electron microscopy (SEM) (Fig. 1c and Fig. S1†) and scanning transmission electron microscopy (STEM) (Fig. 1d) images suggest that the hexapods adopt a three-dimensional branch distribution. The angles between the arms are difficult to confirm from the images obtained; however the varied angles seen in the branched structures lying flat on the substrate offer some clues. Branching angles of ca. 60° and 120° are seen throughout the different electron microscopy images (Fig. 1c and Fig. S1†). Furthermore, Fig. S2† shows TEM images of multi-armed structures found in the same reaction product, where 90° branching is clearly visible. Combining these observations, we surmise that the arms of hexapods grow with a good correspondence to the rhombohedral Cs4PbBr6 phase (Fig. 2p), which has the angle γ = 120° (the angle between positive a and positive b) and the angle α = β = 90° (the angle between positive b and positive c, and the angle between positive a and positive c).
 | ||
| Fig. 1 Characterization of the final hexapod-branched NCs prepared by a two-step reaction. (a and b) TEM images of the hexapod-branched NCs with different magnifications. (c) SEM and (d) STEM images of the NCs. (e–g) STEM-EDS elemental mappings of a single hexapod NC. (h) The EDS spectrum of the NCs and the tabulated Cs, Pb, and Br contents. All scale bars are 200 nm. | ||
 | ||
| Fig. 2 Crystal structure analysis of the hexapod NCs. (a) HRTEM image of a hexapod NC, highlighting the four selected areas 1, 2, 3, and 4 with their corresponding FFT images (b–e); 10 nm scale bars. (f–i) High magnification HRTEM images of the selected areas in image (a). (j–m) The crystal model of the selected areas. (n) Time-resolved XRD patterns of the NCs. Crystal structure of orthorhombic CsPbBr3 (o) and Cs4PbBr6 (p). Dark green = Cs1, light green = Cs2, purple = Pb, and brown = Br. | ||
Elemental analysis by STEM-EDS (energy dispersive spectrometry) mapping (Fig. 1e–g) confirmed that Cs, Pb and Br were homogeneously distributed throughout the structure. From the EDS spectrum (Fig. 1h), we extracted an atomic ratio of Cs:Pb:Br = 3.95:1:5.3, which is close to that of Cs4PbBr6. The discrepancy between the measured and expected ratios for Cs4PbBr6 is likely due to a small amount of residual CsPbBr3, with the copper signal originating from the TEM grid.
Crystal structure
High-resolution transmission electron microscopy (HRTEM) was used to determine the crystal structure at various points within the hexapod structure (Fig. 2a). Four zones from the arms and core were selected for a detailed examination of the structure based on high magnification HRTEM images (Fig. 2f–i), and their corresponding fast Fourier transform (FFT) (Fig. 2b–e). The FFT data were used to calculate both the lattice spacing and crystal structure. In zone 1 from arm I, the observed planes with a d-spacing of 0.71 nm (Fig. 2f) correspond to the (012) planes of Cs4PbBr6.38 For zones 2 and 3, within the core area (Fig. 2g and h), the observed lattice fringes of 0.69 nm and 0.40 nm can be indexed to the (110) and (300) planes. The (110) plane shows a strong FFT signal (Fig. 2c and d) in both areas, which implies that the core faces the [001] direction. For zone 4, the d-spacing of 0.32 nm and 0.44 nm (Fig. 2i) matches well with the (-3-11) and (113) planes of Cs4PbBr6. The corresponding FFT pattern is shown in Fig. 2e. For all the investigated areas, HRTEM measurements are in good agreement with the rhombohedral phase (R 3c) crystal model as depicted in Fig. 2j–m. Importantly, the CsPbBr3 phase was not detected, suggesting that any CsPbBr3 phase is either in the interior of the crystal, or not present at all.Time-resolved XRD was subsequently used to determine the phase proportions during hexapod growth (Fig. 2n). The XRD pattern of the product obtained directly after synthesis (red trace, time 0 hours) matches well with the pattern of the orthorhombic CsPbBr3 perovskite. The peaks at 15.21, 21.50, 30.38, 30.70, 34.33, 37.89, 43.81 and 49.38° can be indexed to the (101), (121), (040), (202), (222), (321), (242), and (204) lattice planes, respectively, of the orthorhombic CsPbBr3 phase (Powder Diffraction File Card – 00-018-0364). After 4 hours of post-synthesis incubation, peaks at 22.41, 27.50, 28.60 and 38.98° with small amplitudes can be observed in the XRD pattern. As the incubation time increases between 8 and 24 hours, these peaks become more prominent, while peaks associated with the CsPbBr3 phase decrease until they disappear from the XRD pattern. Peaks at 12.63, 12.88, 20.07, 22.41, 25.42, 27.50, 28.60, 30.26, 30.94, 34.53, 38.98 and 45.74° correspond to diffractions from (012), (110), (113), (030), (204), (131), (214), (223), (006), (140), (324) and (600) lattice planes of the Cs4PbBr6 phase (PDF Card – 04-015-9683). This demonstrates that the product experiences a slow phase transformation spanning several hours, specifically, from the orthorhombic CsPbBr3 phase (Fig. 2o) to the rhombohedral Cs4PbBr6 phase (Fig. 2p). The Cs4PbBr6 phase is not present in the initial product, but originates solely from the phase transformation.
Optical property evolution
Absorbance and photoluminescence (PL) measurements were performed over a period of 34 hours during incubation. Evolution of the absorption spectra is shown in Fig. 3a. CsPbBr3 exhibits two excitonic peaks at 473 and 486 nm (Fig. 3b). The absorbance of both excitonic peaks gradually decreases, reaching parity at 16 hours and background by 26 hours, reflecting a progressive reduction in the concentration of CsPbBr3.40 The PL spectra contain two primary peaks, at 491 nm and 522 nm, which map to 5 unit cell quantum-confined CsPbBr3 and bulk CsPbBr3.39 As shown in Fig. 3c and d, photoluminescence remains stable over the first 4 hours, and then shows a slow decrease between 4 and 8 hours before declining to near zero by 26 hours.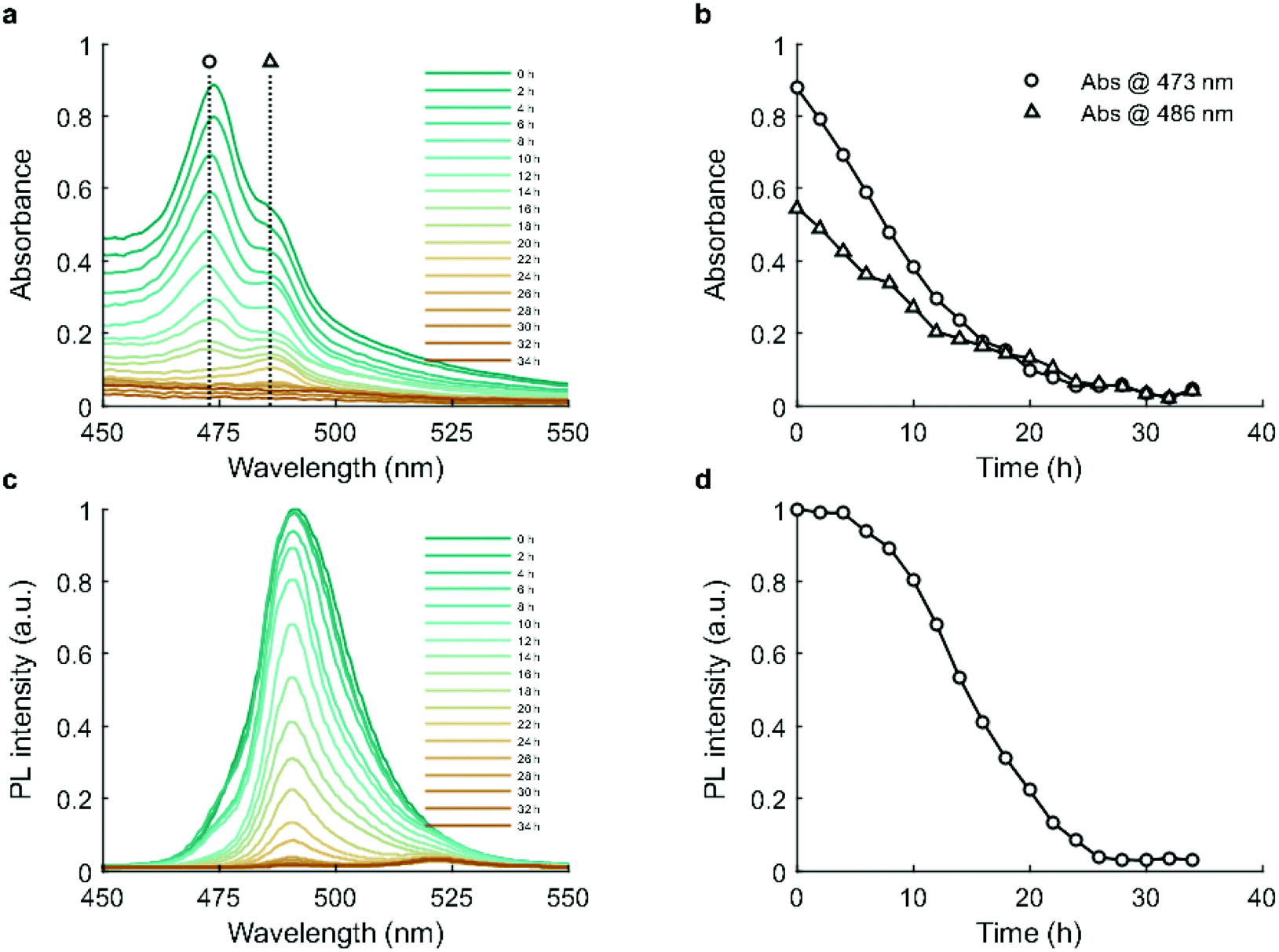 | ||
| Fig. 3 Optical monitoring of the transformation of CsPbBr3 nanosheets to Cs4PbBr6 hexapods NCs over time (2 hours intervals). (a) Time-resolved absorption spectra. (b) Temporal evolution of the absorbance at two spectral features: 473 nm and 486 nm. (c) Time-resolved PL spectra, with excitation at 365 nm. (d) Temporal evolution of PL intensity at 491 nm. | ||
The decrease in absorbance during the first 4 hours suggests that the decline of the CsPbBr3 phase commences immediately,41 which is in good agreement with the XRD results (Fig. 2n). However, as CsPbBr3 decreases one might expect a concurrent PL reduction, but this was not observed. This phenomenon has been observed previously, where a small amount of Cs4PbBr6 phase exhibited a photobrightening effect due to its effective passivation of the CsPbBr3 surface.40,42 Beyond 4 hours however, the continuing decline of CsPbBr3 and growth of Cs4PbBr6 led to a drastic reduction in PL, reaching near zero at 26 hours. Time-resolved photoluminescence measurements (Fig. S8a†) showed that average PL lifetime (Fig. S8b†) remained relatively stable over 24 hours. This observation is consistent with those of Quan et al.,42 who reported that the passivation effect of a Cs4PbBr6 matrix has no influence on the total fluorescence lifetime of CsPbBr3 due to the quantum yield increasing concurrently with the non-radiative lifetime.
As Cs4PbBr6 NCs have potential applications in deep ultraviolet detectors,43 we isolated the product after 36 hours and compared its deep ultraviolet absorption to the as-synthesized CsPbBr3 nanosheets. Fig. S9† shows that the final Cs4PbBr6 product developed a distinct peak at 309 nm, while CsPbBr3 is without a peak at that position.
Shape evolution
TEM imaging of aliquots taken from an incubating reaction solution over 24 hours revealed the distinct morphology changes that occur during hexapod formation. The as-synthesized NCs take the form of nanosheets (48.5 ± 14 nm, Fig. 4a). After 4 hours, a mixed population of sheets and rhombic prism NCs was observed (with side lengths of 40.7 ± 25 nm, Fig. 4b). This rhombic prism morphology is consistent with previous studies on Cs4PbBr6 NCs.26 After 8 hours, large (413 ± 110 nm) short-armed multipod structures were visible (Fig. 4c), formed by rhombic NCs with an average side length of 144 ± 42 nm (Fig. S6a†). At 12 hours, visible necking and extension of the arms have occurred (Fig. 4d), before the final long-armed hexapod structure is formed at 24 hours (Fig. 4e). The evolving nanostructures would gradually sediment during incubation, which necessitated stirring of the reaction solution. We observed that this agitation had a distinct effect on NC transformation. In a static solution, growth proceeded up to the formation of hexapods, but without arm elongation after oriented attachment (Fig. S7a†). However, with stirring, the final product exhibited high aspect ratio arms (Fig. S7b†). | ||
| Fig. 4 Shape evolution and phase transformation of cesium lead bromide NCs. TEM images show (a) as-synthesised CsPbBr3 nanosheets, (b) mixed nanosheets and rhombic prism NCs after 4 hours, (c) multipods forming by oriented attachment, (d) necking between the multipod nucleus and arms, and (e) the final hexapod nanostructure. (f) Schematic illustration of the proposed growth mechanism from CsPbBr3 nanosheets to Cs4PbBr6 hexapods. All the TEM images have a scale bar of 200 nm. | ||
Next, we sought to probe the growth mechanism by performing purification, dilution, and reagent additions to the fresh reaction solutions, and studying the resultant NC morphology post-incubation. Purified CsPbBr3 nanosheets (Fig. S3†) were stored in toluene for 108 hours in total, and they did not undergo shape evolution or phase transformation. Next, a crude reaction solution was diluted using ODE to four different degrees of dilution (0.5/1, 1/1, 5/1, and 10/1, Fig. S4†). In the 0.5/1 diluted sample (i.e. ODE:crude solution = 0.5:1), multipod structures were still visible in the sample after 24 hours (Fig. S4a†), but most of the products were still nanosheets without transformation. With further incubation to 108 hours (Fig. S4i†), shape evolution proceeded and more multipods were formed. Thus, with this degree of dilution, the rate of shape evolution and phase transformation was reduced, but the formation of hexapods was still possible with a longer incubation time. The nanosheets in the 1/1 diluted solution were without obvious change in the initial 24 hours (Fig. S4b†), and exhibited some rhombic NCs after 48 hours (Fig. S4f†). Finally, at 108 hours, hexapod structures had formed and coexisted with a population of nanosheets (Fig. S4j†). When the dilution degree was increased to 5/1 (Fig. S4c, g and k†) and 10/1 (Fig. S4d, h and l†), the phase transformation and shape evolution were even slower, with rhombic nanocrystals only appearing after 108 h.
Finally, octanoic acid, oleylamine, the Cs precursor and the PbBr2 precursor were all added separately into crude CsPbBr3 nanosheet solutions, and then stirred for 36 hours (Fig. S5a–d†). All samples were non-fluorescent by the end of the incubation period. Addition of excess octanoic acid (Fig. S5a†) and oleylamine (Fig. S5b†) prevented the formation of multipods and elongation of the rhombic nanostructures. Addition of either the cesium precursor (Fig. S5c†) or lead bromide precursor (Fig. S5d†) allowed the formation of varied branched structures; however, these lacked the symmetry of the hexapods of the standard reaction, instead showing random branching and elongation.
Discussion
The structural and optical characterization performed herein revealed a slow transformation of the as-synthesized CsPbBr3 nanosheets into Cs4PbBr6 long-armed hexapods, over the course of 24 hours. The initial formation of the CsPbBr3 nanosheets proceeds according to| 2Cs(OOC8) + 3PbBr2 → 2CsPbBr3 + Pb(OOC8)2 |
Besides containing CsPbBr3 nanosheets, the crude reaction solution comprises excess lead octanoate, oleylammonium bromide, octanoic acid and oleylamine, all of which can undergo extensive and dynamic interactions with the NCs in solution.44 Extended incubation of such mixtures gives ample time for the reaction to move towards thermodynamic equilibrium, and the dynamic ligand binding and release typical of the binary organic acid and amine ligand system likely allow significant material diffusion and transfer.44 Therefore, we would expect the nanoparticles to undergo significant morphological changes as a result.
Optical measurements (Fig. 3) revealed a steady decrease of excitonic absorbance and emission throughout incubation, reaching background by 26 hours. This suggests a steady loss of CsPbBr3. XRD results revealed the same loss of CsPbBr3, with a concurrent increase in Cs4PbBr6 (Fig. 2n). Accordingly, we have observed a direct conversion of CsPbBr3 to Cs4PbBr6, or dissolution of the CsPbBr3 followed by recrystallization in the Cs4PbBr6 phase, or both. Since the reaction was carried out in a slight excess of oleylamine (1.2:1 versus octanoic acid), it is possible that PbBr2 could be slowly extracted from CsPbBr3 leading to the formation of the Cs4PbBr6 phase,45,46 according to
| 4 CsPbBr3 → Cs4PbBr6 + 3PbBr2 |
However, the fact that the subsequent rhombic Cs4PbBr6 NCs (Fig. 4b and c) were larger (by mass) than the parent nanosheets suggests that extraction of PbBr2 did not lead directly to Cs4PbBr6 without the addition of new material. The growth of larger NCs without an injection of additional monomers necessitates that the total number of NCs must reduce. Previous studies have suggested that CsPbBr3 nanostructures can undergo a complete dissolution–recrystallization process giving rise to new Cs4PbBr6 nanostructures;45–47 however this phenomenon alone does not explain the photo-brightening effect observed during the first 4 hours, which we ascribe to the passivation of CsPbBr3 crystals by Cs4PbBr6.40 We believe it is more likely that Ostwald ripening occurs in this system, whereby smaller CsPbBr3 nanosheets completely dissolve and larger nanosheets grow, with the Cs4PbBr6 phase forming over CsPbBr3 sheets. Hybrid structures of these two phases of cesium lead bromide have been previously reported.32 Over the course of incubation, photoluminescence reduces to background levels, suggesting that the CsPbBr3 phase is almost completely lost due to the dissolution of free nanosheets and the conversion of the CsPbBr3 material to Cs4PbBr6 phase inside the hybrid structures. The fact that the position of the PL peak does not (blue) shift with nanosheet etching suggests that the material is lost from the side faces and not from the top and bottom, which is consistent with the observation that alkylammonium ligands bind more strongly to the basal planes than the side faces of CsPbBr3 nanosheets.48
During the slow dissolution of CsPbBr3 nanosheets, the reaction is unable to reach equilibrium, but is kept under thermodynamic control, i.e. there is ample time for monomers/ions to diffuse into their equilibrium positions to yield a minimum Gibbs free energy of the entire system.49 This process is assisted by the soft and dynamic nature (high ion mobility and dynamic surface ligand binding) of the perovskite lattice, which favors ionic rearrangement.7 But the question remains whether crystallographic rearrangement is sufficient to allow the large rhombic Cs4PbBr6 NCs to evolve into multipods and eventually into long-armed hexapods. Besides ionic diffusion and crystallographic transformation, another way for the system to minimize Gibbs free energy is via fusion of nanostructures. Although nanoparticle surface ligands generally inhibit this, it can occur that the differences in ligand binding between different facets lead to preferential fusion of specific facets, whilst avoiding uncontrolled aggregation. Such oriented attachment is a well-established mechanism in the formation of anisotropic NCs,50–53 for example in PbSe,54 PbS,55 ZnS,56 CdSe,57 and MnO51 nanostructures, and it has also been observed numerous times with LHP NCs.58–60 TEM images obtained at the 8 hour timepoint (Fig. 4c, Fig. S6†) suggest the formation of large (413 ± 110 nm) multipod structures by the coalescence of parent rhombic NCs, which indicates that the multipods form by oriented attachment. Importantly, the oriented attachment appears to show the parent NCs fusing at their six faces. Subsequent growth outwards from this starting point could yield branching angles of 90° and 60/120° due to the rhombic Cs4PbBr6 NCs possessing both angles, as observed in the full hexapod structure (Fig. 4f). When additional octanoic acid (Fig. S5a†) or oleylamine (Fig. S5b†) was added to the incubation solution (post-heating), oriented attachment was no longer observed, with rhombic structures persisting to the end of the incubation period. This suggests that the ligand ratio and/or concentration is an important factor for inducing oriented attachment. As the 0.5/1 (Fig. S4a, e and i†) and 1/1 (Fig. S4b, f and j†) diluted crude reaction solutions still achieve hexapod formation, but only after extended incubation times, we see that the ligand ratio is more influential on the final nanostructure, while the concentration in more influential on the rate.
Having formed the hexapods from rhombic components, further incubation saw progressive necking of the arms from 12 hours (Fig. 4d), followed by elongation and thinning to form the final long-armed extended hexapod structure at 24 hours (Fig. 4e). This suggests that the nanostructure is moving towards its equilibrium morphology by maximizing the prevalence of certain facets over others. Although it is difficult to conclude the exact mechanism from our data, the fact that once the CsPbBr3 dissolution stops (at ca. 26 hours, Fig. 4) so does the growth of the hexapods suggests that monomer availability is required to stimulate arm extension, and that we are not simply observing reformation of the crystal without material exchange with the surrounding medium. Furthermore, as the incubating solution must be stirred to stimulate arm growth (Fig. S7†), it appears that diffusion of the material between the surface and the surrounding solution is critical. The unstirred reaction products in Fig. S7a† show rounding of the vertices but not arm extension. Such rounded Cs4PbBr6 structures (after crystallographic transformation from CsPbBr3) have been observed previously.45 This suggests that the unstirred structures undergo crystal reorganization, but that arm growth does not proceed due to slow kinetics in the static solution. An explanation for the observed thinning and elongation is that the facets on the sides of the hybrid CsPbBr3/Cs4PbBr6 arms are etched, possibly with extraction of PbBr2, followed by both crystal reformation and direct growth of the Cs4PbBr6 material on the arm ends. The limiting factor in the direct growth of cesium-rich Cs4PbBr6 on the arm ends would be the availability of cesium; therefore we see that once the dissolution of the CsPbBr3 material is complete, the arms stop growing.
After incubation, we observed that although the product sediments, the hexapod structures are stable in the crude reaction solution for several weeks (data not shown), indicating that the reaction had indeed attained equilibrium. Since nanostructure thinning and elongation were seen after post-synthesis additions of the Cs precursor (Fig. S5c†) and PbBr2 precursor (Fig. S5d†), it seems that this process is insensitive to the absolute balance between the precursor and ligand concentration. However, the fact that they had not formed symmetric multipods suggests that the oriented attachment process was perturbed. This again points to a dependence of oriented attachment on the ligand ratio and concentration.
Conclusions
We have reported the synthesis and characterization of a new class of anisotropic cesium lead bromide NCs, namely Cs4PbBr6 crystalline hexapods. The arm length of hexapods extends up to 360 nm, with arm:core ratios up to 6.0 (i.e. 360 nm arms around 60 nm cores). Hexapod structures form with slow kinetics (24 hours) during a post-synthetic incubation of nanosheets. These first develop into rhombic NCs and then form hexapods by oriented attachment. This is followed by necking, elongation and thinning of the six arms. Due to the slow reaction kinetics, the reaction could in theory be stopped at any point during the shape evolution and phase transformation by separation of the NCs from the crude solution, making all of the intermediate-state nanostructures available for further characterization and application. Ultimately, the complex equilibrium between the molecular precursors (lead octanoate, cesium octanoate, oleylammonium bromide), the molecular ligands (octanoic acid, octanoate, oleylamine, oleylammonium), in both solution and on the NC surfaces, and between the different crystal phases of the NCs, and even between the different facets of these NCs, makes the exact reaction mechanism hard to elucidate. Nevertheless, such a growth process shows the possible utility of slow reaction kinetics in obtaining novel NC structures, which could find significant applications in creating complex functional LHP nanostructures as components in superlattices, or as deep ultraviolet detectors.
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors acknowledge partial financial support from ETH Zürich. S. L. acknowledges support from the China Scholarship Council (CSC), Grant No. 201608140097. P. D. H. would like to thank the Swiss National Science Foundation (SNSF) for support via a Spark Grant (Project No. CRSK-2_190750). We thank Tian Liu for help with the XRD, ScopeM (Scientific Center for Optical and Electron Microscopy) at ETH for their support, Léonard Bezinge for assistance in data plotting, and Hiren Kotadia for helpful discussions regarding the manuscript.References
- Y. Yin and A. P. Alivisatos, Colloidal nanocrystal synthesis and the organic–inorganic interface, Nature, 2004, 437(7059), 664 CrossRef PubMed.
- L. Manna, D. J. Milliron, A. Meisel, E. C. Scher and A. P. Alivisatos, Controlled growth of tetrapod-branched inorganic nanocrystals, Nat. Mater., 2003, 2(6), 382 CrossRef CAS PubMed.
- S. J. Lim, A. Schleife and A. M. Smith, Optical determination of crystal phase in semiconductor nanocrystals, Nat. Commun., 2017, 8, 14849 CrossRef CAS PubMed.
- Y. W. Jun, J. S. Choi and J. Cheon, Shape control of semiconductor and metal oxide nanocrystals through nonhydrolytic colloidal routes, Angew. Chem., Int. Ed., 2006, 45(21), 3414–3439 CrossRef CAS PubMed.
- Z. L. Wang, Z. Dai and S. Sun, Polyhedral shapes of cobalt nanocrystals and their effect on ordered nanocrystal assembly, Adv. Mater., 2000, 12(24), 1944–1946 CrossRef CAS.
- S. M. Lee, S. N. Cho and J. Cheon, Anisotropic shape control of colloidal inorganic nanocrystals, Adv. Mater., 2003, 15(5), 441–444 CrossRef CAS.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Genesis, challenges and opportunities for colloidal lead halide perovskite nanocrystals, Nat. Mater., 2018, 17(5), 394–405 CrossRef CAS PubMed.
- Z. Wu, S. Yang and W. Wu, Shape control of inorganic nanoparticles from solution, Nanoscale, 2016, 8(3), 1237–1259 RSC.
- G. Paramasivam, N. Kayambu, A. M. Rabel, A. K. Sundramoorthy and A. Sundaramurthy, Anisotropic noble metal nanoparticles: Synthesis, surface functionalization and applications in biosensing, bioimaging, drug delivery and theranostics, Acta Biomater., 2017, 49, 45–65 CrossRef CAS PubMed.
- F. Li, D. P. Josephson and A. Stein, Colloidal assembly: the road from particles to colloidal molecules and crystals, Angew. Chem., Int. Ed., 2011, 50(2), 360–388 CrossRef CAS PubMed.
- Y. Taniguchi, M. A. B. Sazali, Y. Kobayashi, N. Arai, T. Kawai and T. Nakashima, Programmed self-assembly of branched nanocrystals with an amphiphilic surface pattern, ACS Nano, 2017, 11(9), 9312–9320 CrossRef CAS PubMed.
- Z. Liu, J. Xu, D. Chen and G. Shen, Flexible electronics based on inorganic nanowires, Chem. Soc. Rev., 2015, 44(1), 161–192 RSC.
- E. Shi, Y. Gao, B. P. Finkenauer, A. H. Coffey and L. Dou, Two-dimensional halide perovskite nanomaterials and heterostructures, Chem. Soc. Rev., 2018, 47(16), 6046–6072 RSC.
- E. Ye, M. D. Regulacio, S.-Y. Zhang, X. J. Loh and M.-Y. Han, Anisotropically branched metal nanostructures, Chem. Soc. Rev., 2015, 44(17), 6001–6017 RSC.
- H. Li, A. G. Kanaras and L. Manna, Colloidal branched semiconductor nanocrystals: state of the art and perspectives, Acc. Chem. Res., 2013, 46(7), 1387–1396 CrossRef CAS PubMed.
- D. J. Milliron, S. M. Hughes, Y. Cui, L. Manna, J. Li, L.-W. Wang and A. P. Alivisatos, Colloidal nanocrystal heterostructures with linear and branched topology, Nature, 2004, 430(6996), 190 CrossRef CAS PubMed.
- J. Wang, A. Singh, P. Liu, S. Singh, C. Coughlan, Y. Guo and K. M. Ryan, Colloidal synthesis of Cu2SnSe3 tetrapod nanocrystals, J. Am. Chem. Soc., 2013, 135(21), 7835–7838 CrossRef CAS PubMed.
- E. Ye, K. Y. Win, H. R. Tan, M. Lin, C. P. Teng, A. Mlayah and M.-Y. Han, Plasmonic gold nanocrosses with multidirectional excitation and strong photothermal effect, J. Am. Chem. Soc., 2011, 133(22), 8506–8509 CrossRef CAS PubMed.
- Z. Ou, X. Song, W. Huang, X. Jiang, S. Qu, Q. Wang, P. V. Braun, J. S. Moore, X. Li and Q. Chen, Colloidal Metal–Organic Framework Hexapods Prepared from Postsynthesis Etching with Enhanced Catalytic Activity and Rollable Packing, ACS Appl. Mater. Interfaces, 2018, 10(48), 40990–40995 CrossRef CAS PubMed.
- S. N. Raja, X. Ye, M. R. Jones, L. Lin, S. Govindjee and R. O. Ritchie, Microscopic mechanisms of deformation transfer in high dynamic range branched nanoparticle deformation sensors, Nat. Commun., 2018, 9(1), 1155 CrossRef PubMed.
- K. Miszta, J. de Graaf, G. Bertoni, D. Dorfs, R. Brescia, S. Marras, L. Ceseracciu, R. Cingolani, R. van Roij, M. Dijkstra and L. Manna, Hierarchical self-assembly of suspended branched colloidal nanocrystals into superlattice structures, Nat. Mater., 2011, 10(11), 872–876 CrossRef CAS PubMed.
- A. Castelli, J. de Graaf, M. Prato, L. Manna and M. P. Arciniegas, Tic-Tac-Toe Binary Lattices from the Interfacial Self-Assembly of Branched and Spherical Nanocrystals, ACS Nano, 2016, 10(4), 4345–4353 CrossRef CAS PubMed.
- E. Sutter, P. Sutter, A. V. Tkachenko, R. Krahne, J. de Graaf, M. Arciniegas and L. Manna, In situ microscopy of the self-assembly of branched nanocrystals in solution, Nat. Commun., 2016, 7, 7 Search PubMed.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nearly monodisperse insulator Cs4PbX6 (X=Cl, Br, I) nanocrystals, their mixed halide compositions, and their transformation into CsPbX3 nanocrystals, Nano Lett., 2017, 17(3), 1924–1930 CrossRef CAS.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao and B. Sun, From nonluminescent Cs4PbX6 (X=Cl, Br, I) nanocrystals to highly luminescent CsPbX3 nanocrystals: water-triggered transformation through a CsX-stripping mechanism, Nano Lett., 2017, 17(9), 5799–5804 CrossRef CAS.
- Z. Liu, Y. Bekenstein, X. Ye, S. C. Nguyen, J. Swabeck, D. Zhang, S.-T. Lee, P. Yang, W. Ma and A. P. Alivisatos, Ligand mediated transformation of cesium lead bromide perovskite nanocrystals to lead depleted Cs4PbBr6 nanocrystals, J. Am. Chem. Soc., 2017, 139(15), 5309–5312 CrossRef CAS PubMed.
- T. Udayabhaskararao, L. Houben, H. Cohen, M. Menahem, I. Pinkas, L. Avram, T. Wolf, A. Teitelboim, M. Leskes and O. Yaffe, A mechanistic study of phase transformation in perovskite nanocrystals driven by ligand passivation, Chem. Mater., 2017, 30(1), 84–93 CrossRef.
- M. I. Saidaminov, J. Almutlaq, S. Sarmah, I. Dursun, A. A. Zhumekenov, R. Begum, J. Pan, N. Cho, O. F. Mohammed and O. M. Bakr, Pure Cs4PbBr6: highly luminescent zero-dimensional perovskite solids, ACS Energy Lett., 2016, 1(4), 840–845 CrossRef CAS.
- Y. H. Song, S. H. Choi, W. K. Park, J. S. Yoo, S. B. Kwon, B. K. Kang, S. R. Park, Y. S. Seo, W. S. Yang and D. H. Yoon, Innovatively Continuous Mass Production Couette-Taylor Flow: Pure Inorganic Green-Emitting Cs4PbBr6 Perovskite Microcrystal for Display Technology, Sci. Rep., 2018, 8(1), 2009 CrossRef PubMed.
- H. Zhang, Q. Liao, Y. Wu, J. Chen, Q. Gao and H. Fu, Pure zero-dimensional Cs4PbBr6 single crystal rhombohedral microdisks with high luminescence and stability, Phys. Chem. Chem. Phys., 2017, 19(43), 29092–29098 RSC.
- N. Riesen, M. Lockrey, K. Badek and H. Riesen, On the origins of the green luminescence in the “zero-dimensional perovskite” Cs4PbBr6: conclusive results from cathodoluminescence imaging, Nanoscale, 2019, 11(9), 3925–3932 RSC.
- X. Chen, F. Zhang, Y. Ge, L. Shi, S. Huang, J. Tang, Z. Lv, L. Zhang, B. Zou and H. Zhong, Centimeter-Sized Cs4PbBr6 Crystals with Embedded CsPbBr3 Nanocrystals Showing Superior Photoluminescence: Nonstoichiometry Induced Transformation and Light-Emitting Applications, Adv. Funct. Mater., 2018, 28(16), 1706567 CrossRef.
- B. Kang and K. Biswas, Exploring polaronic, excitonic structures and luminescence in Cs4PbBr6/CsPbBr3, J. Phys. Chem. Lett., 2018, 9(4), 830–836 CrossRef CAS PubMed.
- S. K. Mehetor, H. Ghosh and N. Pradhan, Blue Emitting CsPbBr3 Perovskite Quantum Rods and their Wide Area 2D Self-Assembly, ACS Energy Lett., 2019, 4(6), 1437–1442 CrossRef CAS.
- M. Chen, H. Hu, Y. Tan, N. Yao, Q. Zhong, B. Sun, M. Cao, Q. Zhang and Y. Yin, Controlled growth of dodecapod-branched CsPbBr3 nanocrystals and their application in white light emitting diodes, Nano Energy, 2018, 53, 559–566 CrossRef CAS.
- L. C. Peng, S. K. Dutta, D. Mondal, B. Hudait, S. Shyamal, R. Xie, P. Mahadevan and N. Pradhan, Arm growth and facet modulation in perovskite nanocrystals, J. Am. Chem. Soc., 2019, 141, 16160–16168 CrossRef CAS PubMed.
- I. Lignos, S. Stavrakis, G. Nedelcu, L. Protesescu, A. J. Demello and M. V. Kovalenko, Synthesis of Cesium Lead Halide Perovskite Nanocrystals in a Droplet-Based Microfluidic Platform: Fast Parametric Space Mapping, Nano Lett., 2016, 16(3), 1869–1877 CrossRef CAS PubMed.
- Cs4PbBr6 Crystal Structure: Datasheet from “PAULING FILE Multinaries Edition – 2012” in SpringerMaterials, Springer-Verlag Berlin Heidelberg & Material Phases Data System (MPDS), Switzerland & National Institute for Materials Science (NIMS), Japan Search PubMed.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, Highly luminescent colloidal nanoplates of perovskite cesium lead halide and their oriented assemblies, J. Am. Chem. Soc., 2015, 137(51), 16008–16011 CrossRef CAS.
- F. Palazon, C. Urso, L. De Trizio, Q. Akkerman, S. Marras, F. Locardi, I. Nelli, M. Ferretti, M. Prato and L. Manna, Postsynthesis Transformation of insulating Cs4PbBr6 nanocrystals into bright perovskite CsPbBr3 through physical and chemical extraction of CsBr, ACS Energy Lett., 2017, 2(10), 2445–2448 CrossRef CAS.
- J. Maes, L. Balcaen, E. Drijvers, Q. Zhao, J. De Roo, A. Vantomme, F. Vanhaecke, P. Geiregat and Z. Hens, Light absorption coefficient of CsPbBr3 perovskite nanocrystals, J. Phys. Chem. Lett., 2018, 9(11), 3093–3097 CrossRef CAS PubMed.
- L. N. Quan, R. Quintero-Bermudez, O. Voznyy, G. Walters, A. Jain, J. Z. Fan, X. Zheng, Z. Yang and E. H. Sargent, Highly emissive green perovskite nanocrystals in a solid state crystalline matrix, Adv. Mater., 2017, 29(21), 1605945 CrossRef PubMed.
- G. Q. Tong, H. Li, Z. F. Zhu, Y. Zhang, L. W. Yu, J. Xu and Y. Jiang, Enhancing Hybrid Perovskite Detectability in the Deep Ultraviolet Region with Down-Conversion Dual-Phase (CsPbBr3-Cs4PbBr6) Films, J. Phys. Chem. Lett., 2018, 9(7), 1592–1599 CrossRef CAS PubMed.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, Highly Dynamic Ligand Binding and Light Absorption Coefficient of Cesium Lead Bromide Perovskite Nanocrystals, ACS Nano, 2016, 10(2), 2071–2081 CrossRef CAS PubMed.
- F. Palazon, G. Almeida, Q. A. Akkerman, L. De Trizio, Z. Dang, M. Prato and L. Manna, Changing the dimensionality of cesium lead bromide nanocrystals by reversible postsynthesis transformations with amines, Chem. Mater., 2017, 29(10), 4167–4171 CrossRef CAS PubMed.
- G. Almeida, L. Goldoni, Q. Akkerman, Z. Dang, A. H. Khan, S. Marras, I. Moreels and L. Manna, Role of acid–base equilibria in the size, shape, and phase control of cesium lead bromide nanocrystals, ACS Nano, 2018, 12(2), 1704–1711 CrossRef CAS.
- Z. Liu, Y. Bekenstein, X. Ye, S. C. Nguyen, J. Swabeck, D. Zhang, S. T. Lee, P. Yang, W. Ma and A. P. Alivisatos, Ligand Mediated Transformation of Cesium Lead Bromide Perovskite Nanocrystals to Lead Depleted Cs4PbBr6 Nanocrystals, J. Am. Chem. Soc., 2017, 139(15), 5309–5312 CrossRef CAS.
- J. Cho, H. Jin, D. G. Sellers, D. F. Watson, D. H. Son and S. Banerjee, Influence of Ligand Shell Ordering on Dimensional Confinement of Cesium Lead Bromide (CsPbBr3) Perovskite Nanoplatelets, J. Mater. Chem. C, 2017, 5(34), 8810–8818 RSC.
- S. Ghosh and L. Manna, The Many “Facets” of Halide Ions in the Chemistry of Colloidal Inorganic Nanocrystals, Chem. Rev., 2018, 118(16), 7804–7864 CrossRef CAS PubMed.
- R. L. Penn and J. F. Banfield, Imperfect oriented attachment: Dislocation generation in defect-free nanocrystals, Science, 1998, 281(5379), 969–971 CrossRef CAS PubMed.
- D. Zitoun, N. Pinna, N. Frolet and C. Belin, Single crystal manganese oxide multipods by oriented attachment, J. Am. Chem. Soc., 2005, 127(43), 15034–15035 CrossRef CAS PubMed.
- Y. Tong, B. J. Bohn, E. Bladt, K. Wang, P. Müller-Buschbaum, S. Bals, A. S. Urban, L. Polavarapu and J. Feldmann, From precursor powders to CsPbX3 perovskite nanowires: One-pot synthesis, growth mechanism, and oriented self-assembly, Angew. Chem., Int. Ed., 2017, 56(44), 13887–13892 CrossRef CAS PubMed.
- L. Manna, E. C. Scher and A. P. Alivisatos, Synthesis of soluble and processable rod-, arrow-, teardrop-, and tetrapod-shaped CdSe nanocrystals, J. Am. Chem. Soc., 2000, 122(51), 12700–12706 CrossRef CAS.
- K. S. Cho, D. V. Talapin, W. Gaschler and C. B. Murray, Designing PbSe nanowires and nanorings through oriented attachment of nanoparticles, J. Am. Chem. Soc., 2005, 127(19), 7140–7147 CrossRef CAS PubMed.
- C. Schliehe, B. H. Juarez, M. Pelletier, S. Jander, D. Greshnykh, M. Nagel, A. Meyer, S. Foerster, A. Kornowski, C. Klinke and H. Weller, Ultrathin PbS Sheets by Two-Dimensional Oriented Attachment, Science, 2010, 329(5991), 550–553 CrossRef CAS PubMed.
- J. H. Yu, J. Joo, H. M. Park, S. I. Baik, Y. W. Kim, S. C. Kim and T. Hyeon, Synthesis of quantum-sized cubic ZnS nanorods by the oriented attachment mechanism, J. Am. Chem. Soc., 2005, 127(15), 5662–5670 CrossRef CAS PubMed.
- N. Pradhan, H. F. Xu and X. G. Peng, Colloidal CdSe quantum wires by oriented attachment, Nano Lett., 2006, 6(4), 720–724 CrossRef CAS PubMed.
- T. Udayabhaskararao, M. Kazes, L. Houben, H. Lin and D. Oron, Nucleation, growth, and structural transformations of perovskite nanocrystals, Chem. Mater., 2017, 29(3), 1302–1308 CrossRef CAS.
- P. Li, D. Yang, Y. Tan, M. Cao, Q. Zhong, M. Chen, H. Hu, B. Sun, Y. Xu and Q. Zhang, Consecutive Interfacial Transformation of Cesium Lead Halide Nanocubes to Ultrathin Nanowires with Improved Stability, ACS Appl. Mater. Interfaces, 2018, 11(3), 3351–3359 CrossRef PubMed.
- J. Pan, X. Li, X. Gong, J. Yin, D. Zhou, L. Sinatra, R. Huang, J. Liu, J. Chen and I. Dursun, et al., Halogen vacancies enable ligand-assisted self-assembly of perovskite quantum dots into nanowires, Angew. Chem., Int. Ed., 2019, 131(45), 16223–16227 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr02985h |
| This journal is © The Royal Society of Chemistry 2020 |