
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Constructing a library of metal and metal–oxide nanoparticle heterodimers through colloidal assembly†
Tina A.
Gschneidtner
 a,
Sarah
Lerch
a,
Erik
Olsén
b,
Xin
Wen
a,
Amelia C. Y.
Liu
cd,
Alicja
Stolaś
a,
Joanne
Etheridge
*ce,
Eva
Olsson
*b and
Kasper
Moth-Poulsen
*a
a,
Sarah
Lerch
a,
Erik
Olsén
b,
Xin
Wen
a,
Amelia C. Y.
Liu
cd,
Alicja
Stolaś
a,
Joanne
Etheridge
*ce,
Eva
Olsson
*b and
Kasper
Moth-Poulsen
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-412-96 Göteborg, Sweden. E-mail: kasper.moth-poulsen@chalmers.se
bDepartment of Physics, Chalmers University of Technology, SE-412-96 Göteborg, Sweden. E-mail: eva.olsson@chalmers.se
cMonash Centre for Electron Microscopy, Monash University, VIC 3800, Australia. E-mail: joanne.etheridge@monash.edu
dSchool of Physics and Astronomy, Monash University, VIC 3800, Australia
eDepartment of Materials Science and Metallurgy, Monash University, VIC 3800, Australia
First published on 5th May 2020
Abstract
Nanoparticle dimers composed of different metals or metal oxides, as well as different shapes and sizes, are of wide interest for applications ranging from nanoplasmonic sensing to nanooptics to biomedical engineering. Shaped nanoparticles, like triangles and nanorods, can be particularly useful in applications due to the strong localized plasmonic hot-spot that forms at the tips or corners. By placing catalytic, but traditionally weakly- or non-plasmonic nanoparticles, such as metal oxides and metals like palladium, in these hot-spots, an enhanced function for sensing, photocatalysis or optical use is predicted. Here, we present an electrostatic colloidal assembly strategy for nanoparticles, incorporating different sizes, shapes and metal or metal oxide compositions into heterodimers with smaller gaps than are achievable using nanofabrication techniques. This versatile method is demonstrated on 14 combinations, including a variety of shaped gold nanoparticles as well as palladium, iron oxide, and titanium oxide nanoparticles. These colloidal nanoparticles are stabilized with traditional surfactants, such as citrate, CTAB, PVP and oleic acid/oleylamines, indicating the wide applicability of our approach. Heterodimers of gold and palladium are further analyzed using cathodoluminescence to demonstrate the tunability of these “plasmonic molecules”. Since systematically altering the absorption and emission of the plasmonic nanoparticles dimers is crucial to extending their functionality, and small gap sizes produce the strongest hot-spots, this method indicates that the electrostatic approach to heterodimer assembly can be useful in creating new nanoparticle dimers for many applications.
1. Introduction
Nanoparticles (NPs) have been a topic of significant interest since the beginning of the 19th century, when Faraday began to investigate the optical properties of metal nanoparticles.1 These studies led to the discovery that certain metal NPs exhibit unique optical properties, specifically the scattering and absorption of light ranging from the ultraviolet (UV)2 to the near-infrared (IR), including visible light.3 This phenomenon results when the NP is of comparable size, or smaller than, the wavelength of an incident light which excites the conduction electrons into a predictable resonance. This localized surface plasmon resonance (LSPR) oscillation leads to unique scattering properties for each NP, dependent on the size, shape, composition and surrounding environment of the individual NPs.4–7 Furthermore, the LSPR generates enhanced electromagnetic fields (E-fields) around the NP, which can be further enhanced and isolated through the presence of corners or tips on shaped NPs.8,9 When several NPs are in close proximity, typically with gaps on the order of a few nanometres (nm), the LSPR can concentrate the E-field in this gap between the NP surfaces with a distance-dependent magnitude that also depends on the surrounding media and the composition of the NPs themselves.10–12 This behaviour has been used in applications ranging from LSPR-enhanced sensing13,14 to molecular electronics15–17 to biomedical labelling.18–21In addition to the interesting optical properties, many metallic and semiconducting NPs can also be used in more traditional applications, such as catalysis (e.g. palladium and platinum),22,23 cell labelling in magnetic resonance techniques (e.g. iron),24 or gas storage (H2 storage in metal hydrides).25,26 These potential applications are promising to investigate but also introduce some potential difficulties, as many of the catalytically or magnetically active NPs do not support sharp or significant LSPR modes. One method to counter this challenge is to take advantage of the coupling phenomenon in plasmonic materials, allowing for the combination of plasmonic NPs (sensing NPs) with catalytic or magnetic NPs (active NPs). Coupling of the sensing and active NPs is primarily accomplished through proximity of the NPs, necessitating structures with increasingly small gap sizes to fully attain the ideal coupled LSPR. Once achieved, this coupling greatly increases the applications of plasmonic systems,27–30 since it allows for optical monitoring of the active NP through changes to the plasmon of the sensing NP. One example is the use of a gold (Au) NP plasmon to monitor the shifts in the environment due to the adsorption of gas in the adjacent palladium (Pd) nanoparticle.7,31,32
Since the combination of multiple NPs of different sizes, shapes, or materials is an important aspect to engineering new applications, the assembly of NPs into discrete, well-defined units, such as dimers, is a key topic. Assembly of NPs into dimeric and trimeric units has been achieved through a variety of generalized methods, including through electrostatics33–35 or molecular linkers,36 such as DNA27,37–41 or organic molecules.15,42,43 While many of these techniques are applicable to the formation of heterodimers, with two different NPs, much of the currently published work focuses on the assembly of homodimers, combining NPs of nearly equivalent size, shape and materials. The assembly of heterodimers has been studied for a few specific cases. Vallée et al.11 assembled Au NPs and core–shell Ag@SiO2 NPs using the electrostatic force between the positively charged core–shell NPs and negatively charged Au NPs. Chen et al.44 assembled Au and TiO2 NPs with the help of a Langmuir Blodgett trough to create Janus particles. Other approaches used for heterodimer assembly include the use of nanoscopic phase separation of different metals to form dimers,45 the use of coordination chemistry to link nanoparticles through various molecules,46–48 the use of solid supports during synthesis,49 and the use of electron beam lithography50 but many of these methods are designed for specific combinations of NPs or require significant experimental steps or costs.51 There is not yet a general approach to the formation of heterodimers, like the methods mentioned above for the formation of homodimers, especially for dimers with the small gaps necessary for broader application purposes.
Many of the current applications of assembled homo- or heterodimers rely on specific plasmonic resonances that overlap with the desired function. This necessitates an ability to measure and control the coupled LSPR in dimeric structures. One method for measuring the LSPR with high spatial resolution is cathodoluminescence (CL), which is a technique that raster-scans an electron beam across the specimen using a scanning or transmission electron microscope (SEM or TEM, respectively) and measures the resulting optical emission from the spatially localized electromagnetic excitation of the sample, simultaneously with the structure of the sample.52–54 This method has been extensively utilized for measurements of local doping in semiconductors55–58 and the distribution of materials in geological samples,59,60 but recent work has also extended the use of CL to measure the optical properties of nanostructured materials.52,54,61 For plasmonic materials, hyperspectral CL in the SEM can be employed to map the excitability of LSPRs at high spatial resolution and has been used to characterize the mode distribution in coupled nanostructures grown chemically and assembled electrostatically,35 fabricated using electron beam lithography,62 or milled into surfaces and membranes using ion beams.63,64 When compared to optical microscopy, CL reveals significantly more detailed information about the LSPR, particularly in coupled structures, where multiple LSPRs may overlap in a normal optical spectrum. Additionally, the localized excitation in CL can also be used to measure the angular distribution of the emitted light to further understand the nature of the occurring optically active LSPR modes.65,66
Here we present an extensive investigation into the applicability of an electrostatic-based dimerization method that creates heterodimeric structures with small gaps. Additionally, we identify a variety of potential stabilizers, including metallic nanoparticle stabilizers, like hexadecyltrimethylammonium bromide (CTAB), hexadecyltrimethylammonium chloride (CTAC), polyvinylpyrrolidone (PVP), or sodium methanethiolate (MesNa), as well as semiconductor nanoparticle stabilizers, like oleic acid/oleylamine. By including oleic acid/oleylamine, we are, for the first time with this electostatic method, able to successfully combine colloidal metal NPs with colloidal metal oxide NPs. This is achieved through the intercalation of CTAB in the oleic acid/olelyamine structure, allowing for the typically hydrophobic metal oxide NPs to be phase-transferred to aqueous solution. When in aqueous solution, these NPs can now form heterodimeric structures via our electrostatic approach. This is an interesting development, because it allows us to exploit the hydrophobic colloidal metal oxide NPs, such as iron(III) oxide (Fe2O3) or titanium oxide (TiO2), which are typically synthesized at high temperatures in an oleic acid/oleylamine solution.67–69 Using this approach, it is also possible to dimerize NPs with well-defined composition and crystallinity at potentially sub-nanometre distances, which was not previously achievable with traditional nanofabrication techniques.
Finally, we analyse certain heterodimeric structures using CL to examine the formation of hybrid plasmons in strongly coupled heterodimers. The characterization of these hybrid plasmons is a crucial development, as they allow for the investigation of non-traditional plasmon modes. Additionally, the use of the CL technique allows for the tracking of the plasmon modes across an individual heterodimer, showing the change of the plasmons at the intersection of the two NPs and in the gap between them. This demonstrates the usefulness of the CL technique as well as the potential tunability of the heterodimeric structures due to a broad variety of potential coupled LSPRs.
2. Experimental
2.1 Materials
All starting materials, unless mentioned below and including spherical gold (NPs, nominal diameter 100 nm, DLS diameter 90 nm), were purchased from Sigma-Aldrich and were used as received. Hexadecyltrimethylammonium chloride (CTAC, >95.0%) was purchased from TCI and used without further purification. Au nanostars were purchased from Dianova (NITgold Nanostar, 40 nm).All experiments were done with MilliQ water (18 MΩ cm) provided from a Milli-Q Advantage A10 water purification system from Merck.
2.2 Nanoparticle synthesis and assembly
Pd, Au and Fe2O3 nanoparticles of different sizes and shapes were synthesized with procedures adapted from those previously described in the literature.70–78 Some particles, such as Fe2O3 and TiO2 NPs were further modified, which is described below. All particles were used fresh, since sedimentation was occasionally observed after several hours. However, re-dispersion of these sedimented solutions led to colloidal solutions, where no reshaping or ripening was revealed by TEM inspection. TEM images, concentrations, zeta potentials and hydrodynamic radii are included in the electronic supplementary materials (ESI, S1 and S3†).Assembly of NP heterodimers exploits the electrostatic interactions between NPs with opposite surface charges in order to synthesize material- or shape- selected NP heterodimers, as depicted schematically in Fig. 1. Combining NPs with oppositely charged surfaces typically results in large aggregations due to the electrostatic attractions, so the aggregation process must be carefully controlled to produce the highest possible yield of heterodimers. To assess the charge and stability of each NP and adjust the ratios accordingly, we monitored the surface charge of the NPs via zeta potential measurements (ESI, S1.5 and S2†). NPs were mixed to effectively balance the concentrations (ESI, Table S1-1.†), resulting in solutions that were stable up to a few hours, but were typically used immediately for characterization with TEM and SEM. After the heterodimers were dried on the TEM or SEM supports, they maintained stability for at least one year, which is common for supported or dried NPs, dimers or clusters.5
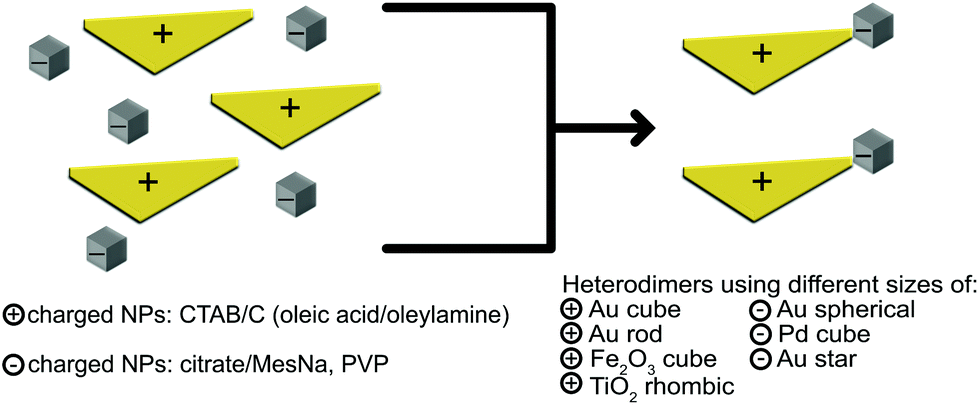 | ||
| Fig. 1 Schematic of the electrostatic assembly to form heterodimeric nanoparticles. Positively charged NPs were stabilized in hexadecyltrimethylammonium bromide/chloride (CTAB/C), while negatively charged NPs were stabilized with citrate/sodium methanethiolate (MesNa) or polyvinylpyrrolidone (PVP). Many colloidal NPs, including Au, Pd and metal oxides such as Fe2O3 or TiO2, can be used. | ||
In order to incorporate colloidal metal oxide NPs, which are often synthesized in solvents other than water, with hydrophobic stabilizers, such as oleic acid/olelyamine, we established a method of intercalating CTAB into the oleic acid/oleylamine layer on the oxide NPs. This intercalation resulted in hydrophilic, water soluble, metal oxide NPs (Fig. 2). This method was applied to both Fe2O3 and TiO2 NPs and resulted in colloidal NP solutions that were successfully transferred to the water phase.
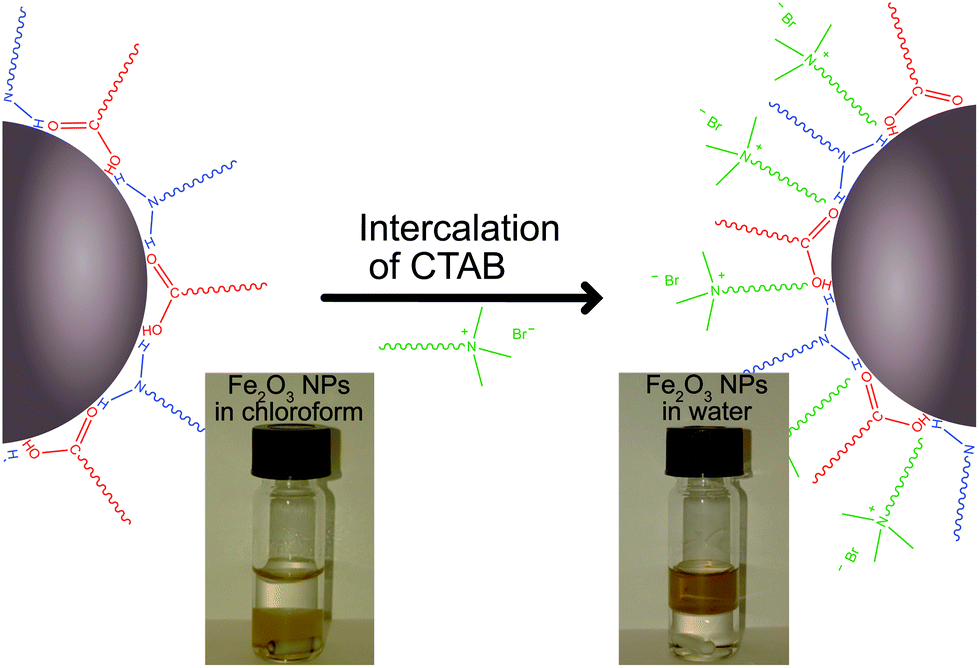 | ||
| Fig. 2 Schematic of the intercalation of hexadecyltrimethylammonium bromide (CTAB, green) into the oleic acid (red)/olelyamine (blue) layer of the metal oxide NPs. Photos show before (left) and after (right) images of the intercalation of the CTAB into the oleic acid/oleylamine stabilization layer. Fe2O3 NPs are originally stable in chloroform (lower phase) but transfer to the aqueous phase (upper phase) during the procedure. | ||
2.3 Characterization of heterodimers
NP heterodimers were characterized with TEM to determine the yield of the various combinations presented in Table 1. TEM samples were prepared by drop-casting heterodimer solutions on copper TEM grids with carbon membranes, and the droplets were immediately allowed to spread and subsequently dry. The samples containing Au rods were placed on holey-carbon TEM grids. This allowed a better separation of the rods.| Particle A | Particle B | Yield | |||||||
|---|---|---|---|---|---|---|---|---|---|
| NP | Shape | Size (nm) | Stabilizer | NP | Shape | Size (nm) | Stabilizer | Dimer (%) | |
| 1 | Au | Triangle | 63 | CTAB | Au | Sphere | 30 | MesNa/citrate | 30 |
| 2 | Au | Rod | 120 | CTAB | Au | Sphere | 30 | MesNa/citrate | 22 |
| 3 | TiO2 | Rhombic | 22 | Oleic acid/CTAB | Au | Sphere | 30 | MesNa/citrate | 22 |
| 4 | Au | Triangle | 63 | CTAB | Au | Sphere | 60 | MesNa/citrate | 25 |
| 5 | Fe2O3 | Cube | 25 | Oleic acid/CTAB | Au | Sphere | 60 | MesNa/citrate | 18 |
| 6 | Au | Rod | 120 | CTAB | Au | Sphere | 60 | MesNa/citrate | 28 |
| 7 | Au | Triangle | 63 | CTAB | Au | Sphere | 95 | MesNa/citrate | 28 |
| 8 | Pd | Cube | 25 | CTAB | Au | Sphere | 95 | MesNa/citrate | 27 |
| 9 | Pd | Cube | 80 | CTAB | Au | Sphere | 95 | MesNa/citrate | 30 |
| 10 | Au | Cube | 55 | CTAC | Au | Sphere | 95 | MesNa/citrate | 24 |
| 11 | Au | Rod | 120 | CTAB | Pd | Cube | 7.5 | PVP | 15 |
| 12 | Au | Triangle | 63 | CTAB | Pd | Cube | 7.5 | PVP | 18 |
| 13 | Au | Rod | 120 | CTAB | Au | Star | 40 | PVP | 23 |
| 14 | Au | Triangle | 63 | CTAB | Au | Star | 40 | PVP | 27 |
Further characterization was carried out using cathodoluminescence (CL) to obtain detailed information about the LSPR peaks for selected heterodimers. Samples were prepared by drop-casting NP heterodimer solutions on a 200 nm silicon nitride film on a silicon substrate. The experiments were performed on the “as-synthesized” NPs, both with and without 30 s plasma cleaning using a mixture of oxygen and hydrogen gas, before the CL measurements, to reduce the effect of surfactants and other potential contaminations. No major difference in the optical properties was observed between the two cases. A detailed explanation of the measurement parameters and data handling, as well as additional CL measurements, are included in the ESI (S6).†
3. Results and discussion
3.1 Electrostatic assembly of oppositely charged nanoparticles
Our goal is to develop a broad, simple method to combine a variety of NPs of specific sizes, shapes or material compositions.Typically, we observed that the concentration of the stabilizer molecules around the NPs must be reduced for dimerization to occur but needs to remain high enough to maintain stable colloidal solutions. Since the dimerization is sensitive to these concentrations, zeta potential measurements are used to monitor the effective salt and stabilizer concentrations. It is possible to adjust the zeta potential after initial synthesis (ESI, Fig. S2-1: S2-4†) through centrifugal washing or adding an oppositely charged stabilizer. For example, a solution of CTAB-stabilized Au triangles (positively charged) can be stabilized by adding a counter NP solution with a high concentration of citrate stabilizer (negatively charged).
Following the successful transfer of metal oxide NPs to the water phase, as outlined in the Experimental section above, we are able to present 14 different electrostatically combined NPs to form a variety of heterodimers, with differing sizes, shapes and NP compositions (Table 1). Fig. 3 shows transmission electron microscope (TEM) images of heterodimers composed of a metal or metal oxide NP stabilized by CTAB or CTAB intercalated into oleic acid/oleylamine and an Au sphere stabilized by MesNa/citrate. The positively charged metal NPs stabilized by CTAB are Au triangles (Fig. 3A, D and G), Au nanorods (Fig. 3B and E) or Pd nanocubes (Fig. 3H and I). The positively charged, metal oxide NPs stabilized through the intercalation of CTAB into oleic acid/oleylamine are TiO2 rhombic NPs (Fig. 3C) and Fe2O3 nanocubes (Fig. 3F). The negatively charged Au “spheres” stabilized by MesNa/citrate are presented in 3 different diameters, 30 nm (Fig. 3A–C), 60 nm (Fig. 3D–F) and 95 nm (Fig. 3G–I). We chose to analyse one of these structures, the Pd cubes with the largest Au spheres, with energy-dispersive X-ray spectroscopy (EDS) in a TEM, a technique that allows for the identification of elements in a sample. As shown in Fig. 4A, the Au and Pd are isolated to the respective NPs, indicating an effective method of forming heterodimers without introducing significant elemental overlap. The sizes and shapes represent a small portion of CTAB-stabilized NPs syntheses reported in the literature, but it encompasses a broad range of features that can affect the formation of larger nanostructures, including the presence of tips or corners (Au triangles, Pd cubes) and the presence of large size variation (small Au nanospheres to large Au nanorods). Additionally, we demonstrate the versatility of our electrostatic techniques across a variety of compositions, including Au, Pd and metal oxides. Small gap sizes are a common feature throughout these heterodimers, indicating the potential for strongly coupled LSPRs in all of these structures.
 | ||
| Fig. 3 TEM images of nanoparticle heterodimers prepared by electrostatic assembly, listed as positively charged NP and negatively charged NP, respectively. All scale bars are 50 nm. (A) Au triangle, 63 nm and Au sphere, 30 nm. (B) Au rod, 120 nm and Au sphere, 30 nm. (C) TiO2 rhombic, 22 nm and Au sphere, 30 nm. (D) Au triangle, 63 nm and Au sphere, 60 nm. (E) Au rod, 120 nm and Au sphere, 60 nm. (F) Fe2O3 cube, 25 nm and Au sphere, 60 nm. (G) Au triangle, 63 nm and Au sphere, 95 nm. (H) Pd cube, 25 nm and Au sphere, 95 nm. (I) Pd cube, 80 nm and Au sphere, 95 nm. | ||
 | ||
| Fig. 4 (A) EDS mapping of Pd cube, 80 nm and Au sphere, 95 nm. Blue represents Au and green represents Pd. (B)–(F) TEM images of nanoparticle heterodimers prepared by electrostatic assembly with alternate stabilizers, listed as positively charged NP and negatively charged NP, respectively. All scale bars are 50 nm. (B) Au rod, 120 nm and Pd cube, 7.5 nm. (C) Au triangle, 63 nm and Pd cube, 7.5 nm. (D) Au cube, 55 nm and Au sphere, 95 nm. (E) Au rod, 120 nm and Au star, 40 nm. (F) Au triangle, 63 nm and Au star, 40 nm. | ||
Other synthesized heterodimers are presented in Fig. 4. These structures indicate the versatility of the electrostatic technique across multiple stabilizing surfactants, polymers and molecules. First, we used the Au nanorods and nanotriangles stabilized by CTAB and dimerized those with PVP-stabilized Pd cubes (Fig. 4B and C). Additionally, we replaced CTAB with CTAC for Au nanocubes (Fig. 4D), while continuing to dimerize with the MesNa/citrate stabilized Au nanospheres. Finally, we also combined the CTAB-stabilized Au nanorods and nanotriangles with PVP-stabilized Au stars (Fig. 4E and F).
As shown in Table 1, we also evaluated the yield of dimers for each combination, with optimal results ranging from 15% (#11, Au nanorods/Pd nanocubes) to 30% (#1, Au nanotriangles/Au spheres and #9, Pd nanocubes/Au spheres), with an average yield of 24% across the entire series. This yield is comparable with previous heterodimer syntheses used for single particle/dimer studies of both resonance and catalysis.7,79
In order to identify potential improvements to the yield, we analysed the entire yield of the dimerization process, revealing several structures other than the expected heterodimers, mainly smaller clusters of trimeric NPs or single NPs. We chose to optimize the assembly conditions to minimize the larger clusters or aggregates, since small aggregations in NP samples can result in the formation of much larger aggregations, while single NPs will not contribute any significant complications to the solution. Optimization routes included adjusting the salt, stabilizer and NP concentrations, as outlined in the experimental methods in the ESI (ESI, S1†). We carefully analysed two samples after optimization, PVP-stabilized Au stars combined with either CTAB-stabilized Au rods (Fig. 5A–D) or CTAB-stabilized Au triangles (Fig. 5E–H). Yields of 23% and 27% dimers are obtained for the stars/rods (Fig. 5A) and stars/triangles (Fig. 5E), respectively. This optimization results in single NP yields at 64% and 51%, and cluster yields of 13% and 22% (Fig. 5D and H). The maximum size of the analysed clusters was limited to 4 NPs (Fig. 5B and F), discounting some larger aggregations that remained in solution and were observed on some TEM grids. Overall views of the typical TEM samples are shown in Fig. 5C and G and ESI Fig. S4-1.†
 | ||
| Fig. 5 TEM analysis of yield for heterodimers. (A)–(C) Au rods, 120 nm and Au stars, 40 nm, showing a dimer (A), a larger cluster (B) and an overview of the sample (C). D) Yield for Au rod-Au star sample is 64% individual NPs, 23% dimers and 13% larger clusters, up to 4 NPs. (E)–(G) Au triangles, 63 nm and Au stars, 40 nm, showing a dimer (E), a larger cluster (F), and an overview of the sample (G). H) Yield for the Au triangle- Au star sample is 51% individual NPs, 27% dimers and 22% larger clusters. Scale bars are 50 nm for (A) and (B) and (E) and (F) and 300 nm for (C) and (G). | ||
Additional optimization of the yield of dimers could be obtained through the incorporation of a filtering step immediately following synthesis. A variety of filtration possibilities for NP clusters have been investigated previously, including the use of gel electrophoresis to separate NP cluster based on size, shape and surface charge,80 the use of cell sorting equipment to filter the NPs and clusters based on weight,81 and the use of encapsulation and centrifugation.46
Careful statistical analysis was performed on all NP heterodimer combinations reported in the paper, resulting in the yields presented in Fig. 5D and H and Table 1. These yields resulted from TEM analysis of more than 3000 NPs from the different experiments (see ESI, S5†). These statistics allow us to compare the dimer yield of a variety of heterodimers, which indicates that those heterodimers composed of NPs of similar sizes produce higher yields of dimers than those with significant size differences. For example, the heterodimers with the smallest NPs, like Fe2O3 cubes (25 nm) or Pd cubes (7.5 nm) had the lowest yields (15%, 18%, 18%). Although this is a general trend, it is not a conclusive trend, since some combinations with small NPs, like the Pd cubes (25 nm) and Au spheres (95 nm), still had higher yields (27%). Additional analysis was performed to determine the geometries of the heterodimers, specifically whether the sharp corners, or short edges of rods, were aligning in any dimer formations. This alignment of the sharpest areas of each NP is important since we expect the strongest plasmonic enhancement in an individual NP to be near the sharp corners, and when these corners are aligned in dimers, it is expected to increase the enhancement associated with the gap. For this method, there were occasional structures that aligned at the corners (Fig. 3A, D, G, 4B, C, D and G), but these were not in the majority throughout the samples (ESI, Table S5.1†).
3.2 Hybridized localized surface plasmon resonance of heterodimers
Due to the tunability of the LSPRs associated with the fine scale material structure of plasmonic particles, it is important to correlate structure to properties in a systematic way. Therefore, in the last part of this work, we investigate the optical properties of the heterodimer assemblies. Dimers consisting of a gold sphere and a palladium cube were used to illustrate the tunability of the heterodimeric system. The optical properties of individual heterodimers were investigated using energy and angle-resolved cathodoluminescence (AR-CL) in a SEM. This allowed the energy and directivity of light, locally excited by an electron beam (e-beam), to be measured and correlated with the NP structure.53Representative CL measurements can be seen in Fig. 6, where each spectrum is well described by two main spectral peaks (analysis methods in ESI†). The size of the investigated Au NP spheres varied between 90 and 110 nm in diameter and the side length of the measured Pd cubes was approximately 85 nm. For a single Pd cube, AR-CL measurements revealed that the two spectral peaks in Fig. 6A both had toroidal emission patterns characteristic of dipoles (ESI, Fig. S6.2†).53,66 Similar observations of two peaks have been made for silver cubes82,83 suggesting that the occurrence of two dipole emission peaks is likely due to the asymmetric dielectric environment surrounding the NP from the presence of a substrate, creating a degeneracy in the dipole modes on the top and bottom of the cubes. For the single Au NPs, in Fig. 6B and C and S6.3,† the low energy peak for the small sphere has an emission pattern characteristic of a vertical dipole, whereas the low energy emission peak for the larger sphere, which occurs at slightly lower energy, has a higher emission directivity and thus shows evidence of higher-order modes (magnetic dipole, electric quadrupole – see S6.3†), in-line with increased retardation effects across the NP as its size increases.5,66 The asymmetry in the AR-CL pattern reflects the asymmetry of the dipole due to the off-axis excitation point. The effect of this is to beam light away from the point of electron beam excitation.66 The high energy peak for both of the single Au NPs show emission patterns characteristic of a combination of dipole emission and electron transitions (see S6.4†).
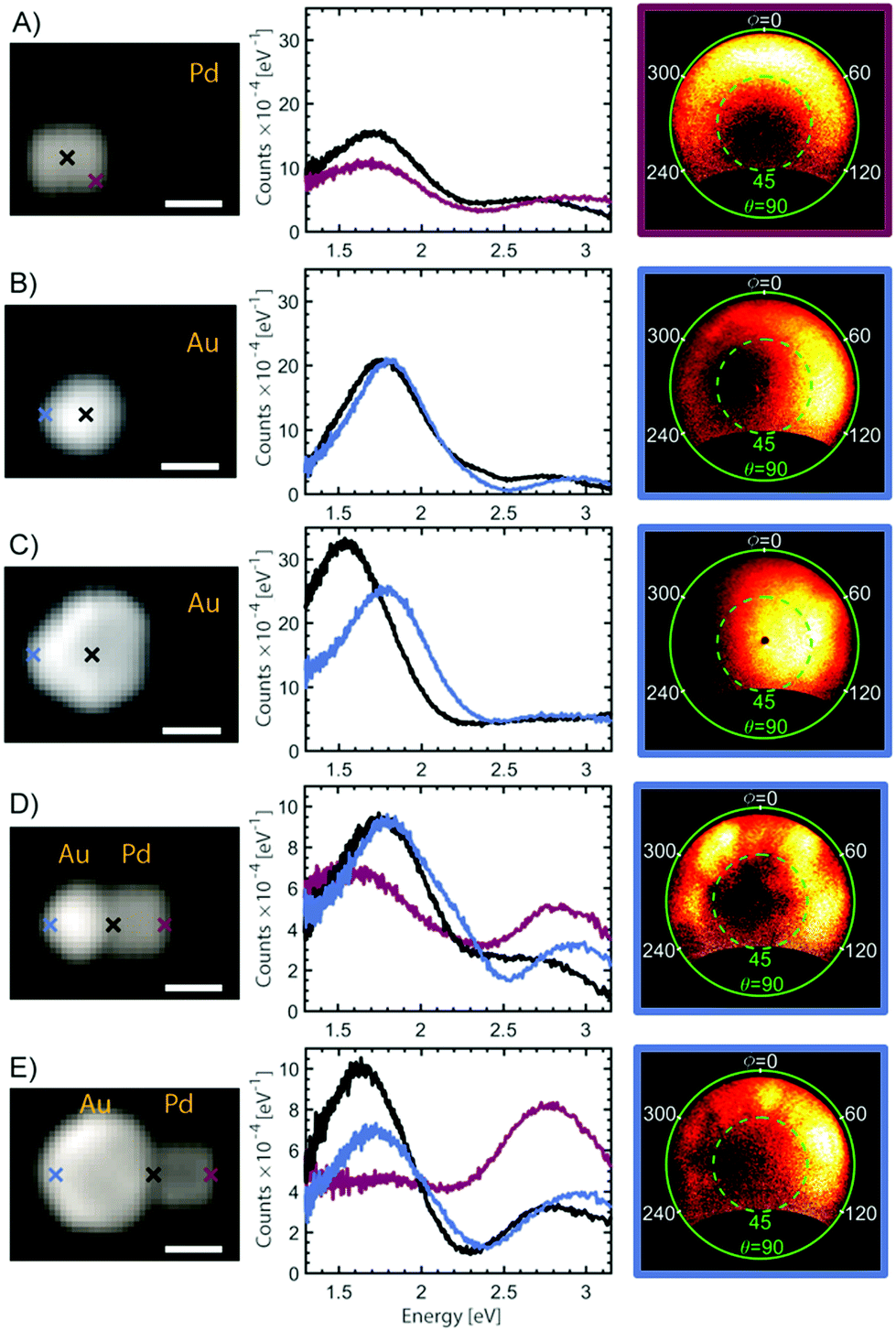 | ||
| Fig. 6 SEM images, cathodoluminescence (CL) spectra of single NPs and heterodimers and AR-CL measurements. The position of the electron beam during CL emission measurements is indicated by the crosses in the SEM image. The frame of the AR-CL images is colour coded according to the colours of the crosses and the spectra. The AR-CL data are shown for the 1.77–2.07 eV energy interval. The AR-CL measurements are presented as polar plots with polar angle θ (green) and azimuthal angle φ (white). All scale bars are 50 nm. (A) Pd cube, 85 nm. (B) Small Au sphere, 90 nm. (C) Large Au sphere, 110 nm. (D) Heterodimer of Pd cube and small Au sphere. (E) Heterodimer of Pd cube and large Au sphere. | ||
When a gold sphere and a palladium cube form a heterodimer, as shown in Fig. 6D and E, each spectrum is still well described by two spectral peaks but the relative intensity of the two spectral peaks now changes depending on the position of the e-beam. For example, the amplitude of the low energy peak for the single NPs and the smaller heterodimer is higher than the high energy peak at every e-beam position, which is not the case for the larger heterodimer. AR-CL measurements from the low energy emission peak of the heterodimers (ESI, Fig. S6.5 and S6.6†) further reveal that both the small and large heterodimer show patterns consistent with an admixture of multiple LSPR modes. The smaller heterodimer has clearer higher-order emission lobes compared with the emission from the large heterodimer which seems to be dominated by the dipole emission of the large sphere, no matter where the system is excited. The difference between the two heterodimers can therefore be understood by the spectral overlap for the LSPRs in the individual NPs.4 The low energy emission peak from the single Pd cube is similar in energy with the low energy peak for the small Au sphere, but overlaps with the larger Au sphere. Since plasmonic hybridization is highly dependent on the overlap between LSPR modes for the individual NPs, the radiant dipole modes on the Pd cube and the small Au sphere are therefore likely to hybridize. For the heterodimer with a larger Au sphere, the dipolar mode on the Pd cube is more likely to hybridize with low-radiance, higher order LSPRs on the Au sphere. This explains the decreased optical emission and increased similarity of the optical emission of the large heterodimer with that of a single Au sphere.
Another explanation for the difference in optical properties between the two heterodimers could be that a considerable amount of the emission of the large heterodimer is shifted outside the investigated spectral region.84,85 However, considering that the emission directivity in the low energy region for the larger heterodimer is similar to that of a single dipole, such an explanation is unlikely. In summary, the spectral and angle-resolved CL show that the distance between the NPs is short enough to get a considerable LSPR hybridization in these heterodimers, and that controlling the size of the individual NPs enables a significant tuning of the resulting optical properties, as desired for a variety of practical applications.
4. Conclusion
In this work, we demonstrated the broad applicability of the electrostatic assembly method for NP heterodimers. Expanding to include a large variety of molecular and polymeric stabilizers, we were able to successfully combine a variety of NPs featuring different shapes, sizes and compositions, including semiconducting NPs like Fe2O3 and TiO2, into dimeric structures with small gaps, ideal for LSPR hybridization. The semiconducting NPs were incorporated by initially intercalating CTAB into the oleic acid/oleylamine stabilizing layer on the NPs, decreasing the hydrophobicity of the NPs and allowing transfer to the water phase for an increased interaction area between the semiconducting NPs and the second NPs. We achieved an average dimer yield of 24% for 14 variations on the heterodimers and showed applicability for NPs from 7.5–120 nm. We included a variety of structures ranging from traditional spherical or rod-like NPs to more interesting triangles, cubes and stars. There exist some limitations to this method, namely that the yield remains lower than is necessary for upscaled applications and that the electrostatic assembly does not provide significant flexibility in the separation of the NPs, limiting the tunability of the resonance for an individual combination. This tunability is alternatively achieved through large variety of potential combinations, which is now limited only by the availability of colloidally stable shaped metal or metal oxide NPs.Finally, we used CL as a proof-of-principle to demonstrate the tunability of the hybridized LSPRs for the heterodimers. These results show that by controlling the size or composition of the NPs, it is possible to adjust the overlapping plasmonic modes to provide a wide range of potential emission spectra and directionality. This tunability, especially combined with the access to the strongest hot spots in these small gaps, is increasingly important for the applications of NPs to encompass a wide variety of topics, from optical sensors to biomedical targeting.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is supported by the European Research Council under the SIMONE project grant. This work is partially supported by the Knut och Alice Wallenberg Foundation. This work is also partially supported by the Swedish Research Council. CL experiments were supported by Australian Research Council (ARC) grant DP160104679 and used instruments at the Monash Centre for Electron Microscopy funded by ARC LE140100104.Notes and references
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- U. Kreibig and M. Vollmer, Optical Properties of Metal Clusters, Springer, Berlin, Germany, 1995 Search PubMed.
- P. Pallavicini, G. Chirico, M. Collini, G. Dacarro, A. Donà, L. D'Alfonso, A. Falqui, Y. Diaz-Fernandez, S. Freddi, B. Garofalo, A. Genovese, L. Sironi and A. Taglietti, Chem. Commun., 2011, 47, 1315–1317 RSC.
- P. Nordlander, C. Oubre, E. Prodan, K. Li and M. I. Stockman, Nano Lett., 2004, 4, 899–903 CrossRef CAS.
- S. A. Maier, Plasmonics: Fundamentals and Applications, Springer, US, 2007 Search PubMed.
- T. Shegai, S. Chen, V. D. Miljković, G. Zengin, P. Johansson and M. Käll, Nat. Commun., 2011, 2, 481 CrossRef PubMed.
- S. Syrenova, C. Wadell, F. A. A. Nugroho, T. A. Gschneidtner, Y. A. Diaz Fernandez, G. Nalin, D. Witlik, F. Westerlund, T. J. Antosiewicz, V. P. Zhdanov, K. Moth-Poulsen and C. Langhammer, Nat. Mater., 2015, 14, 1236–1244 CrossRef CAS PubMed.
- J. P. Kottmann, O. J. F. Martin, D. R. Smith and S. Schultz, Opt. Express, 2000, 6, 213–219 CrossRef CAS PubMed.
- A. Genç, J. Patarroyo, J. Sancho-Parramon, N. G. Bastús, V. Puntes and J. Arbiol, Nanophotonics, 2017, 6, 193 Search PubMed.
- M. Shanthil, R. Thomas, R. S. Swathi and T. K. George, J. Phys. Chem. Lett., 2012, 3, 1459–1464 CrossRef CAS PubMed.
- A. Lombardi, M. P. Grzelczak, A. Crut, P. Maioli, I. Pastoriza-Santos, L. M. Liz-Marzán, N. Del Fatti and F. Vallée, ACS Nano, 2013, 7, 2522–2531 CrossRef CAS PubMed.
- T. Shegai, P. Johansson, C. Langhammer and M. Käll, Nano Lett., 2012, 12, 2464–2469 CrossRef CAS PubMed.
- S. Chen, M. Svedendahl, M. Käll, L. Gunnarsson and A. Dmitriev, Nanotechnology, 2009, 20, 434015 CrossRef CAS PubMed.
- T. a. Gschneidtner, S. Chen, J. B. Christensen, M. Käll and K. Moth-Poulsen, J. Phys. Chem. C, 2013, 117, 14751–14758 CrossRef CAS.
- T. Dadosh, Y. Gordin, R. Krahne, I. Khivrich, D. Mahalu, V. Frydman, J. Sperling, A. Yacoby and I. Bar-Joseph, Nature, 2005, 436, 677–680 CrossRef CAS PubMed.
- T. A. Gschneidtner, Y. A. Diaz Fernandez and K. Moth-Poulsen, J. Mater. Chem. C, 2013, 1, 7127–7133 RSC.
- S. Parveen, R. Misra and S. K. Sahoo, Nanomedicine Nanotechnology, Biol. Med., 2012, 8, 147–166 CAS.
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC.
- D. Cabuzu, A. Cirja, R. Puiu and A. M. Grumezescu, Curr. Top. Med. Chem., 2015, 15, 1605–1613 CrossRef CAS PubMed.
- P. Di Pietro, G. Strano, L. Zuccarello and C. Satriano, Curr. Top. Med. Chem., 2016, 16, 3069–3102 CrossRef CAS PubMed.
- K. Rani, J. Appl. Biotechnol. Bioeng., 2017, 3, 00059 Search PubMed.
- Z. Liu, L. M. Gan, L. Hong, W. Chen and J. Y. Lee, J. Power Sources, 2005, 139, 73–78 CrossRef CAS.
- P. Hernandez-Fernandez, F. Masini, D. N. McCarthy, C. E. Strebel, D. Friebel, D. Deiana, P. Malacrida, A. Nierhoff, A. Bodin, A. M. Wise, J. H. Nielsen, T. W. Hansen, A. Nilsson, I. E. L. Stephens and I. Chorkendorff, Nat. Chem., 2014, 6, 732–738 CrossRef CAS PubMed.
- K. C. Barick, S. Singh, D. Bahadur, M. a. Lawande, D. P. Patkar and P. a. Hassan, J. Colloid Interface Sci., 2014, 418, 120–125 CrossRef CAS.
- M. A. Poyli, V. M. Silkin, I. P. Chernov, P. M. Echenique, R. D. Muiño and J. Aizpurua, J. Phys. Chem. Lett., 2012, 3, 2556–2561 CrossRef CAS.
- R. Jiang, F. Qin, Q. Ruan, J. Wang and C. Jin, Adv. Funct. Mater., 2014, 24, 7328–7337 CrossRef CAS.
- R. C. Mucic, J. J. Storhoff, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 12674–12675 CrossRef CAS.
- A. D. McFarland and R. P. Van Duyne, Nano Lett., 2003, 3, 1057–1062 CrossRef CAS.
- J. Zhao, X. Zhang, C. R. Yonzon, A. J. Hoes and R. P. Van Duyne, Nanomedicine, 2006, 1, 219–228 CrossRef CAS PubMed.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–862 CrossRef CAS.
- C. Langhammer, E. M. Larsson, B. Kasemo and I. Zorić, Nano Lett., 2010, 10, 3529–3538 CrossRef CAS.
- N. Liu, M. L. Tang, M. Hentschel, H. Giessen and A. P. Alivisatos, Nat. Mater., 2011, 10, 631 CrossRef CAS PubMed.
- B. V. Enüstün and J. Turkevich, J. Am. Chem. Soc., 1963, 85, 3317–3328 CrossRef.
- G. Chen, Y. Wang, L. H. Tan, M. Yang, L. S. Tan, Y. Chen and H. Chen, J. Am. Chem. Soc., 2009, 131, 4218–4219 CrossRef CAS PubMed.
- J. A. Lloyd, S. H. Ng, A. C. Y. Liu, Y. Zhu, W. Chao, T. Coenen, J. Etheridge, D. E. Gómez and U. Bach, ACS Nano, 2017, 11, 1604–1612 CrossRef CAS.
- S. Lerch and B. M. Reinhard, Int. J. Mod. Phys. B, 2017, 1740002 CrossRef CAS PubMed.
- A. P. Alivisatos, K. P. Johnsson, X. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez and P. G. Schultz, Nature, 1996, 382, 609–611 CrossRef CAS PubMed.
- C. J. Loweth, W. B. Caldwell, X. Peng, A. P. Alivisatos and P. G. Schultz, Angew. Chem., Int. Ed., 1999, 38, 1808–1812 CrossRef CAS.
- A. J. Mastroianni, S. A. Claridge and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 8455–9 CrossRef CAS.
- L. Guo, A. R. Ferhan, H. Chen, C. Li, G. Chen, S. Hong and D.-H. Kim, Small, 2013, 9, 233 CrossRef CAS.
- J. Chao, Y. Lin, H. Liu, L. Wang and C. Fan, Mater. Today, 2015, 18, 326–335 CrossRef CAS.
- A. K. Boal, F. Ilhan, J. E. Derouchey, T. Thurn-Albrecht, T. P. Russell and V. M. Rotello, Nature, 2000, 404, 746–748 CrossRef CAS PubMed.
- L. C. Brousseau, J. P. Novak, S. M. Marinakos and D. L. Feldheim, Adv. Mater., 1999, 11, 447–449 CrossRef CAS.
- S. Pradhan, D. Ghosh and S. Chen, ACS Appl. Mater. Interfaces, 2009, 1, 2060–2065 CrossRef CAS PubMed.
- Y. Feng, J. He, H. Wang, Y. Y. Tay, H. Sun, L. Zhu and H. Chen, J. Am. Chem. Soc., 2012, 134, 2004–2007 CrossRef CAS PubMed.
- Y. Wang, G. Chen, M. Yang, G. Silber, S. Xing, L. H. Tan, F. Wang, Y. Feng, X. Liu, S. Li and H. Chen, Nat. Commun., 2010, 1, 87 CrossRef CAS PubMed.
- M. R. Dewi, T. A. Gschneidtner, S. Elmas, M. Ranford, K. Moth-Poulsen and T. Nann, ACS Nano, 2015, 9, 1434–1439 CrossRef CAS PubMed.
- A. Ulfkjær, F. W. Nielsen, H. Al-Kerdi, T. Ruβ, Z. K. Nielsen, J. Ulstrup, L. Sun, K. Moth-Poulsen, J. Zhang and M. Pittelkow, Nanoscale, 2018, 10, 9133–9140 RSC.
- M. R. Dewi, G. Laufersky and T. Nann, Microchim. Acta, 2015, 182, 2293–2298 CrossRef CAS PubMed.
- C. Zhang, H. Zhao, L. Zhou, A. E. Schlather, L. Dong, M. J. McClain, D. F. Swearer, P. Nordlander and N. J. Halas, Nano Lett., 2016, 16, 6677–6682 CrossRef CAS PubMed.
- M. Ha, J.-H. Kim, M. You, Q. Li, C. Fan and J.-M. Nam, Chem. Rev., 2019, 119, 12208–12278 CrossRef CAS PubMed.
- F. J. García de Abajo, Rev. Mod. Phys., 2010, 82, 209–275 CrossRef.
- T. Coenen, Angle-resolved Cathodoluminescence Nanoscopy, PhD thesis, University of Amsterdam, 2014, ISBN No. 9789462591622, https://dare.uva.nl/search?identifier=2ab1dd91-ab6e-439a-8b43-739abde4ce25 Search PubMed.
- A. Polman, M. Kociak and F. J. García de Abajo, Nat. Mater., 2019, 18, 1158–1171 CrossRef CAS.
- J. Christen, M. Grundmann and D. Bimberg, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 1991, 9, 2358–2368 CrossRef CAS.
- Z. Jin, T. Fukumura, M. Kawasaki, K. Ando, H. Saito, T. Sekiguchi, Y. Z. Yoo, M. Murakami, Y. Matsumoto, T. Hasegawa and H. Koinuma, Appl. Phys. Lett., 2001, 78, 3824–3826 CrossRef CAS.
- W. Chen, J. Z. Zhang and A. G. Joly, J. Nanosci. Nanotechnol., 2004, 4, 919–947 CrossRef CAS PubMed.
- A. Gustafsson, J. Microsc., 2006, 224, 72–78 CrossRef CAS.
- G. Koschek, J. Microsc., 1993, 171, 223–232 CrossRef CAS.
- G. R. Watt, B. J. Griffin and P. D. Kinny, Am. Mineral., 2000, 85, 1784–1794 CrossRef CAS.
- W. T. Ruane, K. M. Johansen, K. D. Leedy, D. C. Look, H. von Wenckstern, M. Grundmann, G. C. Farlow and L. J. Brillson, Nanoscale, 2016, 8, 7631–7637 RSC.
- T. Coenen, D. T. Schoen, S. A. Mann, S. R. K. Rodriguez, B. J. M. Brenny, A. Polman and M. L. Brongersma, Nano Lett., 2015, 15, 7666–7670 CrossRef CAS PubMed.
- T. Coenen, E. J. R. Vesseur and A. Polman, ACS Nano, 2012, 6, 1742–1750 CrossRef CAS PubMed.
- K. Singh, E. Panchenko, B. Nasr, A. Liu, L. Wesemann, T. J. Davis and A. Roberts, Beilstein J. Nanotechnol., 2018, 9, 1491–1500 CrossRef CAS PubMed.
- T. Coenen, B. J. M. Brenny, E. J. Vesseur and A. Polman, MRS Bull., 2015, 40, 359–365 CrossRef CAS.
- T. Coenen, F. Bernal Arango, A. Femius Koenderink and A. Polman, Nat. Commun., 2014, 5, 3250 CrossRef PubMed.
- A. L. Willis, N. J. Turro and S. O'Brien, Chem. Mater., 2005, 17, 5970–5975 CrossRef CAS.
- X. Liang, X. Wang, J. Zhuang, Y. Chen, D. Wang and Y. Li, Adv. Funct. Mater., 2006, 16, 1805–1813 CrossRef CAS.
- S. Mourdikoudis and L. M. Liz-Marzán, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- W. Niu, Z.-Y. Li, L. Shi, X. Liu, H. Li, S. Han, J. Chen and G. Xu, Cryst. Growth Des., 2008, 8, 4440–4444 CrossRef CAS.
- W. Niu, L. Zhang and G. Xu, ACS Nano, 2010, 4, 1987–1996 CrossRef CAS.
- H.-L. Wu, C.-H. Kuo and M. H. Huang, Langmuir, 2010, 26, 12307–13 CrossRef CAS PubMed.
- X. Ye, C. Zheng, J. Chen, Y. Gao and C. B. Murray, Nano Lett., 2013, 13, 765–771 CrossRef CAS PubMed.
- B. Lim, H. Kobayashi, T. Yu, J. Wang, M. J. Kim, Z.-Y. Li, M. Rycenga and Y. Xia, J. Am. Chem. Soc., 2010, 132, 2506–2507 CrossRef CAS PubMed.
- N. Pinna, J. F. Hochepied, M. Niederberger and M. Gregg, Phys. Chem. Chem. Phys., 2009, 11, 3607 RSC.
- C. Dinh, T. Nguyen, F. Kleitz and T. Do, ACS Nano, 2009, 3, 3737–3743 CrossRef CAS PubMed.
- L. Scarabelli, M. Coronado-Puchau, J. J. Giner-Casares, J. Langer and L. M. Liz-Marzán, ACS Nano, 2014, 8, 5833–5842 CrossRef CAS PubMed.
- M. Pauly, B. P. Pichon, P.-A. Albouy, S. Fleutot, C. Leuvrey, M. Trassin, J.-L. Gallani and S. Begin-Colin, J. Mater. Chem., 2011, 21, 16018 RSC.
- W. Lu, X. Cui, T. H. Chow, L. Shao, H. Wang, H. Chen and J. Wang, Nanoscale, 2019, 11, 9641–9653 RSC.
- M. Hanauer, S. Pierrat, I. Zins, A. Lotz and C. Sönnichsen, Nano Lett., 2007, 7, 2881–2885 CrossRef CAS PubMed.
- J. B. Simonsen, N. E. A. Reeler, A. Fossum, K. A. Lerstrup, B. W. Laursen and K. Nørgaard, Nano Res., 2016, 9, 3093–3098 CrossRef CAS.
- E. Ringe, J. M. McMahon, K. Sohn, C. Cobley, Y. Xia, J. Huang, G. C. Schatz, L. D. Marks and R. P. Van Duyne, J. Phys. Chem. C, 2010, 114, 12511–12516 CrossRef CAS.
- S. Zhang, K. Bao, N. J. Halas, H. Xu and P. Nordlander, Nano Lett., 2011, 11, 1657–1663 CrossRef CAS PubMed.
- O. Pérez-González, N. Zabala and J. Aizpurua, New J. Phys., 2011, 13, 83013 CrossRef.
- J. A. Scholl, A. Garcia-Extarri, A. L. Koh and J. A. Dionne, Nano Lett., 2013, 13, 564–569 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methods, including data handling, and additional characterization. See DOI: 10.1039/d0nr02787a |
| This journal is © The Royal Society of Chemistry 2020 |