
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monitoring the insertion of Pt into Cu2−xSe nanocrystals: a combined structural and chemical approach for the analysis of new ternary phases†
Alberto
Casu‡
 a,
Mariona
Dalmases‡
bc,
Mengxi
Lin
bc,
Yan
Wang
bc,
Narcís
Homs
bcd,
Pilar
Ramírez de la Piscina
bc,
Jordi
Llorca
e,
Albert
Figuerola
*bc and
Andrea
Falqui
*a
a,
Mariona
Dalmases‡
bc,
Mengxi
Lin
bc,
Yan
Wang
bc,
Narcís
Homs
bcd,
Pilar
Ramírez de la Piscina
bc,
Jordi
Llorca
e,
Albert
Figuerola
*bc and
Andrea
Falqui
*a
aNabla Lab, Biological and Environmental Sciences and Engineering (BESE) Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: andrea.falqui@kaust.edu.sa
bDepartament de Química Inorgànica i Orgànica, Secció de Química Inorgànica, Universitat de Barcelona, Martí i Franquès 1-11, 08028 Barcelona, Spain. E-mail: albert.figuerola@ub.edu
cInstitut de Nanociència i Nanotecnologia (IN2UB), Universitat de Barcelona, Martí i Franquès 1-11, 08028 Barcelona, Spain
dCatalonia Institute for Energy Research (IREC), Jardins de les Dones de Negre 1, 08930 Barcelona, Spain
eInstitute of Energy Technologies, Department of Chemical Engineering and Barcelona Research Center in Multiscale Science and Engineering, Universitat Politècnica de Catalunya, EEBE, Eduard Maristany 10-14, 08019 Barcelona, Spain
First published on 30th July 2020
Abstract
The tuning of the chemical composition in nanostructures is a key aspect to control for the preparation of new multifunctional and highly performing materials. The modification of Cu2−xSe nanocrystals with Pt could provide a good way to tune both optical and catalytic properties of the structure. Although the heterogeneous nucleation of metallic Pt domains on semiconductor chalcogenides has been frequently reported, the insertion of Pt into chalcogenide materials has not been conceived so far. In this work we have explored the experimental conditions to facilitate and enhance the insertion of Pt into the Cu2−xSe nanocrystalline lattice, forming novel ternary phases that show a high degree of miscibility and compositional variability. Our results show that Pt is mainly found as a pure metal or a CuPt alloy at high Pt loads (Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu atomic ratio in reaction medium >1). However, two main ternary CuPtSe phases with cubic and monoclinic symmetry can be identified when working at lower Pt:Cu atomic ratios. Their structure and chemical composition have been studied by local STEM-EDS and HRTEM analyses. The samples containing ternary domains have been loaded on graphite-like C3N4 (g-C3N4) semiconductor layers, and the resulting nanocomposite materials have been tested as promising photocatalysts for the production of H2 from aqueous ethanolic solutions.
:Cu atomic ratio in reaction medium >1). However, two main ternary CuPtSe phases with cubic and monoclinic symmetry can be identified when working at lower Pt:Cu atomic ratios. Their structure and chemical composition have been studied by local STEM-EDS and HRTEM analyses. The samples containing ternary domains have been loaded on graphite-like C3N4 (g-C3N4) semiconductor layers, and the resulting nanocomposite materials have been tested as promising photocatalysts for the production of H2 from aqueous ethanolic solutions.
Introduction
The extraordinary variety of nanostructured materials available today is the result of the development of advanced synthetic and fabrication techniques, as well as of the conception of precisely designed materials. Size, shape, structure and obviously chemical composition are the main key parameters defining the physical properties stemming from nanocrystals.1 Depending on the desired performance, the complexity of simple materials should be increased by exploiting one or more such parameters, in order to tune, enhance or add a new property. In the last decade, several advances have been reached for the fine control over the composition of nanostructured materials, leading to either doped, alloyed or heterostructured novel systems, depending on the amount and miscibility of a specific guest element in the host nanocrystals.2–4 The structural and compositional characterization of multinary structures might represent a significant challenge since unknown and/or metastable phases with specific symmetries and lattice parameters can be formed, especially at the nanoscale, with variable composition.5 The stability and homogeneity of such phases could be strongly compromised beyond the nanometric level, or simply it might be hard to obtain sufficiently large single crystals for single crystal X-ray analysis and structure resolution.6 On top of that, alloyed or multinary structures might also show a non-stoichiometric composition, rather than a chemical formula with only integer subindexes.Then, advanced characterization techniques are required to analyze the structure and composition of such complex nanostructured materials on a more local level. This kind of approach permits to appreciate in detail the features of the nanostructures, which is particularly useful to point out smaller compositional and/or structural features (e.g., the formation of smaller-sized secondary phases) that would be hardly observable by more volume-averaged techniques.
Copper chalcogenide semiconductor nanocrystals have experienced a huge growing interest in the last years due to their fascinating optical properties that are unfolded in the shape of an intense absorption band in the near infrared region (NIR), known as the Localized Surface Plasmon Resonance (LSPR).3 Thus, the reactivity of a metal precursor with respect to a colloidal suspension of copper chalcogenide nanoparticles (NPs) has often been studied with the aim of tuning or complementing this optical response. So far, this research field has provided a set of heterostructures with segregated phases, either as metal–semiconductor or as semiconductor–semiconductor composite structures depending on the occurrence of either ion reduction and heterogeneous nucleation of the metal, or alternatively cation exchange of the oxidized metal ions, respectively. As a result of such reactions, Au–Cu2−xSe, Cu2S–CdS, Cu2S–HgS, Cu2−xSe–CdSe, CuS–Au2S and Cu2−xSe–ZnSe have been prepared and characterized.7–14 Alternatively, and depending on the miscibility of the incoming species, copper chalcogenide nanocrystals have been employed as precursors for the synthesis of ternary, quaternary or multinary materials through partial cation exchange. Some examples are AgxCuyEz, CuInE2, Cu2MGeE4, Cu2ZnSnE4, or CuInxGa1−xE2 (where E = S, Se, Te) among others,7,15–22 which show potential applications in catalysis, sensing and as thermoelectric or photovoltaic energy converters.3,23
Nanostructured materials presenting Pt in any form (single atoms, small clusters made of tens of Pt atoms or Pt NPs) are especially interesting due to their excellent catalytic properties.24,25 If the enhanced catalytic performance often observed in non-stoichiometric materials is also considered,26 exploring new ternary systems derived from the incorporation of Pt species into the Cu2−xSe crystal lattice becomes particularly attractive, potentially improving the overall catalytic activity of the semiconductor domain. However, Pt reacts with copper chalcogenides forming a segregated phase, either as a pure metal or as a CuPt alloy, and no examples have been so far reported on Pt incorporation into the binary Cu2−xE semiconductor lattice.27–29
In this work the gradual insertion of Pt into Cu2−xSe NPs is described, which is achieved by defining new reaction conditions under which the segregation of pure metallic phases is minimized. An in-depth structural and chemical analysis has been performed by high resolution transmission electron microscopy (HRTEM) and scanning TEM (STEM) with energy dispersive spectroscopy (EDS)-based chemical mapping in order to assess the crystalline structure, the composition and the distribution of the chemical elements within these complex systems. To conclude, their role as co-catalysts onto g-C3N4 in the photocatalytic production of hydrogen from aqueous ethanolic solutions has been preliminarily assessed with positive results, which are discussed on the light of X-Ray photoelectron spectroscopy (XPS).
Experimental
Chemicals
Copper(I) chloride (CuCl, 99.999%-Cu), platinum(II) acetyl-acetonate (Pt(acac)2, 98%) and selenium powder (Se, 99.99%) were obtained from Strem Chemicals. Oleylamine (OLAm, 70%), 1-octadecene (ODE, 90%), oleic acid (OLAc, ≥99%), 2-propanol (≥99.9%), toluene (99.9%) and ethanol (HPLC, 99.999%) were purchased from Sigma-Aldrich. Melamine (99%) was purchased from Alfa Aesar. All the reagents and solvents were used without further purification, except for the ODE and OLAm which had been previously degassed under vacuum for 3 h at 120 °C.Synthesis of Cu2−xSe NPs
The synthesis of Cu2−xSe NPs was carried out following a method published by Kriegel and co-workers.30 Briefly, 47.4 mg (0.6 mmol) Se, 4.5 mL ODE and 3 mL OLAm were degassed in a three-neck flask for 1 h at 120 °C under vacuum. Meanwhile, Cu precursor solution was prepared in the glovebox: 49.5 mg (0.5 mmol) CuCl were dissolved in 1.5 mL ODE and 1 mL OLAm, the solution was slightly heated to achieve the complete dissolution of the solid. Under N2 atmosphere, the temperature of the system was raised to 310 °C and the Cu precursor solution was injected. After 20 min of reaction, the heating was stopped, and the solution was let cool down naturally. The sample was diluted with 2-propanol, centrifuged 4 min at 4500 rpm and the precipitate was re-dispersed in ODE.Synthesis of Pt–Cu–Se_X samples
The syntheses of Pt–Cu–Se NPs were based on the method developed by Sun and co-workers for the synthesis of metallic platinum nanocubes but using a solution of pre-synthesized Cu2−xSe NPs instead of the Fe(CO)5 catalyst.31 Specifically, 5 mg of Pt(acac)2 (0.013 mmol), 1.25 mL ODE, 125 μL OLAm and 125 μL OLAc were degassed in a three-neck flask for 30 minutes at 120 °C under vacuum. Once under N2, a Cu2−xSe NPs solution ([Cu] = 80 mM) in toluene was injected and the temperature of the system was slowly raised to 200 °C. Once at this temperature, the system was let react for 30 minutes and, subsequently the heating was removed, and the solution was let cool down naturally. The sample was diluted with 2-propanol, centrifuged 4 min at 4500 rpm and the precipitate was redispersed in toluene. Samples are stored as colloidal solutions for several weeks under normal ambient conditions without any appreciable degradation. The exact amounts of Cu2−xSe solution injected in each sample are shown in Table 1.:Cu molar ratio used during each synthesis
| Sample | Volume of Cu2−xSe solution injected | Pt:Cu ratio |
|---|---|---|
| Pt–Cu–Se_A | 1.5 mL | 0.1 |
| Pt–Cu–Se_B | 0.75 mL | 0.2 |
| Pt–Cu–Se_C | 0.25 mL | 0.65 |
| Pt–Cu–Se_D | 0.1 mL | 1.6 |
Synthesis of g-C3N4 nanosheets
g-C3N4 nanosheets were synthesized by a two steps-annealing process.32 In brief, melamine was heated in air at 5 °C min−1 up to 520 °C keeping this temperature for 4 h. After cooling down to room temperature, the yellow agglomerates were grinded to powder and calcined again following the procedure described above. Finally, the g-C3N4 nanosheets with lighter yellow colour were collected.Synthesis of Pt–Cu–Se_X/g-C3N4 nanocomposites
Pt–Cu–Se_X/g-C3N4nanocomposites (Pt–Cu–Se_X 1%, wt/wt) were synthesized by ultrasound-assisted deposition method. Typically, Pt–Cu–Se_X NPs and g-C3N4 were dispersed in ethanol and treated under ultrasound using a SONICS VCX 750, 20 kHz, maximum power 500 W equipment, operating at 50% amplitude at 20 °C for 1 h, then ethanol was carefully evaporated under continuous stirring at 50 °C. At last, the grey powders were transferred to an alumina crucible and heated at 200 °C for 4 h under air to remove any residual of surfactant (both OLAc and/or OLAm).A similar method was used for the preparation of the non-containing Pt sample, CuSe/g-C3N4 (Cu2−xSe 1%, wt/wt), which was employed as reference in the photocatalytic experiments; Cu2−xSe NPs and g-C3N4 were applied in this case.
Characterization methods
Cu2−xSe and Pt–Cu–Se ternary NPs were prepared for observation by conventional TEM by dilution in toluene followed by sonication. A droplet of the solution was then poured in holey carbon-covered copper TEM grids. A JEOL 2000 FX II conventional TEM operating at an accelerating voltage of 80 kV was used.HRTEM structural analysis was performed by a CS-image corrected FEI Titan working at 300 kV and equipped with a FEI CMOS Ceta camera. STEM-EDS elemental mapping was performed in high angle annular dark field (HAADF) geometry on a double spherical corrected FEI Titan Themis cubed microscope working at 80 kV and equipped with a SuperX EDS spectrometer with 0.7 srad of collection angle. Both instruments were equipped with an ultra-bright Schottky (FEI X-FEG) electron source. The lower voltage used for EDS mapping was chosen to maximize the X-Ray signal production and to minimize the possibility of beam damage to the samples, thus improving the S/N ratio during the collection of elemental maps. The elemental maps were also used to perform quantitative analysis and assess the composition of the NPs and of the different phases constituting them. Obviously, given the intrinsic 2-Dimensional nature of TEM related techniques, where 3-Dimensional objects are observed and analyzed according to their 2-D projections, and the irregular shape and distribution of the phases composing the NPs, the quantification conducted on different Regions Of Interest (ROIs) is forcibly the convolution of all the different phases comprised in each ROI. Then, the results of the quantification will be averaged among the phases within the ROI but mostly driven by the main phase (i.e., the most present phase within the ROI).
The structural characterization was conducted by 2-Dimensional Fast Fourier Transform (2D-FFT) analysis. The diffraction peaks of the 2D diffractograms could not be attributed to any known crystal phases in the case of the ternary Pt–Cu–Se NPs, thus a “blind” approach was used: at first the planar and angular relationships occurring between diffraction spots in the 2D-FFTs (numerical diffractograms) were calculated in order to verify the kind of crystal lattice formed and its orientation within the NPs, then the lattice parameters were calculated from the diffraction spots of each orientation using the corresponding formulas for calculating interplanar spacing. This two-step procedure led to lattice parameters that were self-consistent within each 2D-FFT diffractogram, while also resulting in different lattice parameters for each ternary sample. Finally, the structural data were successfully put in relation with the different phases observed by EDS mapping.
X-ray diffraction (XRD) patterns were acquired with a PANalytical X'Pert Pro MPD Alpha1 diffractometer operating in θ/2θ geometry at 45 kV, 40 mA, and λ = 1.5406 Å (Cu Kα1). Thin layers of the samples were prepared by drop casting and evaporation of the solvent on a monocrystalline Si flat substrate. Scans in the range 2θ = 4–100° were run at a step size of 2θ = 0.017° and 100 s per step. The data were treated with X'Pert HighScore Plus software.
X-ray photoelectron spectroscopy (XPS) was performed on a SPECS system equipped with an Al anode XR50 source operating at 150 W and a Phoibos 150 MCD-9 detector. The pass energy of the hemispherical analyzer was set at 25 eV, and the energy step was set at 0.1 eV. The binding energy (BE) values referred to the C 1s peak at 284.8 eV.
Scanning Electron Microscopy (SEM) images were recorded using a Zeiss Neon40 Crossbeam Station instrument equipped with a field emission source. Elemental analysis was carried out using an Oxford energy dispersive X-ray spectrometer (EDS).
The composition and concentration of Cu2−xSe NPs solution was determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES). The measurements were carried out by an Optima 3200 RL PerkinElmer spectrometer. For these measurements, 50 μL of each of the solutions were precipitated in MeOH and redispersed in CHCl3. The solution was evaporated in an oven overnight at 90 °C. Before the vial was sealed, 2.5 mL of aqua regia and 0.7 mL of H2O2 were added to the precipitate and then heated to 90 °C for 72 h. The resulting solution was transferred to a 25 mL volumetric flask and diluted with Milli-Q water.
Thermogravimetric analysis (TGA) was performed by using a METTLER TOLEDO model TGA/DSC1 in the temperature range of 30–400 °C under air flow (50 mL min−1) at a heating rate of 10 °C min−1.
UV-Vis-NIR absorption spectra of all samples were recorded in toluene solution on a PerkinElmer LAMBDA 950 spectrophotometer.
Photocatalytic tests
The photocatalytic tests were carried out at atmospheric pressure in a jacketed reactor of 300 mL capacity designed for continuous gas flow operation and equipped with a condenser (kept at −15 °C) at the outlet. A UV-visible 175 W Hg broad-spectrum lamp (maximum power of 25.6 W at λ = 366 nm) was used. The lamp was placed inside the reactor in a water-cooled jacket that serves as UV cut off filter (λ ≥ 385 nm) for UVA-visible light irradiation. Before the photocatalytic reaction, the Pt–Cu–Se_X/g-C3N4 photocatalysts were degassed under vacuum at 100 °C for 12 h, and then transferred directly to the photoreactor. 200 mg of photocatalyst and 250 mL of an ethanol(aq) (25% v/v) solution, previously purged with Ar, were used. Before irradiation, the suspension was stirred for 30 min under continuous Ar flow to remove all residual air in the reactor, then the system was irradiated and kept at 25 °C. The gas phase products evolved were analyzed on-line using a micro-gas chromatograph Varian CP-4900 equipped with two columns (Molsieve 5 Å and PPQ) and two independent micro-TCD detectors (detection limit for H2 of 50 ppm).Results and discussion
Our synthetic strategy entails the reaction of a platinum molecular precursor with Cu2−xSe pre-synthesized NPs in a highly hydrophobic environment at high temperature. The solubility and colloidal stability of the metallic and nanostructured precursors is achieved by a mixture of amphiphilic surfactants. Due to the presence of a small amount of OLAm as surfactant, the reaction conditions are slightly reducing.33 TEM micrograph in Fig. 1a shows the as-synthesized Cu2−xSe precursor NPs, with spherical shape and an average diameter of 13.9 nm (15.1% standard deviation). The XRD pattern of the sample shown in Fig. 2a indicates the NPs crystallized in the cubic phase of Cu2Se, also known as the mineral berzelianite. The partial oxidation of Cu(I) ions to Cu(II), leading to Cu2−xSe, causes the small shift observed in the XRD peaks.30 | ||
| Fig. 1 TEM micrographs of (a) Cu2−xSe NPs, (b) sample A (ratio Pt:Cu 0.1), (c) sample B (ratio Pt:Cu 0.2), (d) sample C (ratio Pt:Cu 0.65) and, (e) sample D (ratio Pt:Cu 1.6). | ||
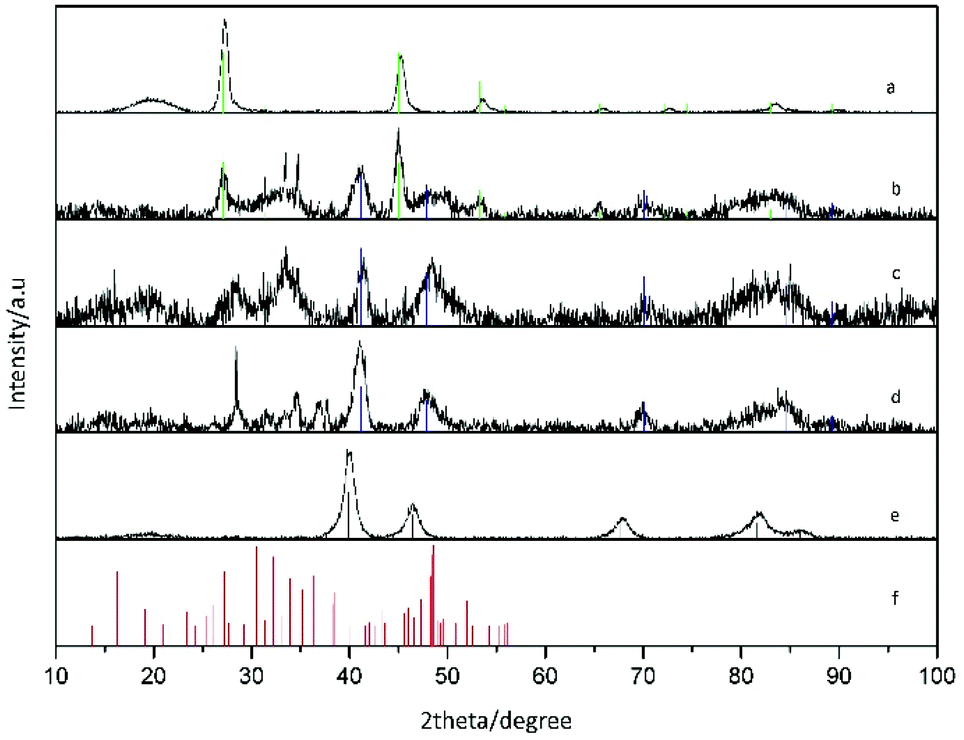 | ||
| Fig. 2 XRD patterns of (a) Cu2−xSe NPs, (b) sample A (ratio Pt:Cu 0.1), (c) sample B (ratio Pt:Cu 0.2), (d) sample C (ratio Pt:Cu 0.65) and, (e) sample D (ratio Pt:Cu 1.6). Reference patterns of cubic Cu2Se (JCPDS 088-2043) in green; cubic CuPt (JCPDS 048-1549) in blue; cubic Pt (JCPDS 087-0640) in black, and monoclinic Pt5Se4 (JCPDS 00-031-0950) in red. | ||
The progressive reaction between the as-prepared Cu2−xSe NPs and the Pt(acac)2 precursor in solution was accompanied by gradual and noticeable changes in the initial sample. The morphology and the composition of the nanocrystals obtained were different depending on the Pt:Cu ratio used in the synthesis. TEM images of the resultant nanostructures are shown in Fig. 1b–e. The Pt:Cu molar ratio was significantly increased, starting from 0.1 and up to a value of 1.6.
At the lowest Pt:Cu molar ratios of 0.1 (sample A) and 0.2 (sample B), (Fig. 1b and c), the NPs lost their sphericity and became more faceted. Additionally, their average size was slightly larger than that of the precursor NPs, and most of them presented a likely second domain, especially in sample B. The addition of more platinum to the reaction medium, a Pt:Cu molar ratio of 0.65 (sample C, Fig. 1d), caused a stronger faceting of the NPs, leading to heterogeneous nanostructures characterized by darker and lighter zones, hinting at the possible presence of multiple domains in the same particle. Finally, when the amount of platinum was larger than the amount of copper (sample D, Pt:Cu molar ratio 1.6, Fig. 1e), star-like NPs were obtained.
XRD characterization shows a significant enlargement and overlapping of XRD peaks suggesting the formation of small-sized and/or highly defective crystal domains upon reaction with Pt. The vanishing of the Cu2−xSe peaks and the appearance of the metallic CuPt or Pt peaks as the amount of platinum was increased are clear in XRD characterization, as well as the presence of additional peaks partially compatible with platinum chalcogenides, like Pt5Se4 (Fig. 2).
More in detail, Pt–Cu–Se_A exhibits the highest number of reflections, suggesting the presence of multiple different phases: reflections derived from the initial Cu2−xSe are still observed in this sample, while well-defined peaks centered at 41° and 48°, together with some other minor peaks at higher angles, are consistent with the cubic phase of CuPt alloy and are preserved along samples B and C. Additional peaks between 25° and 35° and also at angles below 20° could be indicative of platinum chalcogenide materials, like monoclinic Pt5Se4, although they could not be unequivocally assigned to any specific phase, and so further analyses by HRTEM and STEM-EDS were essential to figure out the crystallographic phases and composition of these nanostructures, respectively.
This joint structural and compositional approach allowed investigating on a local level the variations occurring on the structural and chemical features of the intermediate samples due to the introduction of Pt, thus highlighting the spatial distribution of the elements and the consequent formation of multiple phases.
In particular, the elemental maps of the ternary NPs collected by STEM-EDS for samples Pt–Cu–Se_A–C and the elemental line profiles drawn across multiple NPs show the ubiquitous presence of Se and an uneven distribution of Cu and Pt (Fig. 3–5 and ESI Fig. 1–4†). Then, the NPs can be ideally classified according to their local and overall composition by using two general compositional classes: Se-rich zones (i.e., characterized by lower Pt and Cu content) and irregular Pt-rich and Cu-rich zones (characterized by lower Se content), which can be described more in detail by the line profiles. When comparing the elemental profiles with the intensity profile obtained from the STEM-HAADF images, it is apparent that Se is equally distributed along the whole volume of the NPs, since its signal usually follows that of STEM-HAADF along the line profiles of samples A–C. Furthermore, the profile shapes typically associated with core/shell systems (i.e., a dome-shaped element-specific signal smaller than the STEM-HAADF signal for the core region, and an inverse-dome, M-shaped signal that extends along the whole length of the STEM-HAADF signal for the shell) are not observed.
 | ||
| Fig. 3 HAADF-STEM image and corresponding elemental maps by STEM EDS of Pt–Cu–Se_A. | ||
 | ||
| Fig. 4 HAADF-STEM image and corresponding elemental maps by STEM EDS of Pt–Cu–Se_B. | ||
 | ||
| Fig. 5 HAADF-STEM image and corresponding elemental maps by STEM EDS of Pt–Cu–Se_C. | ||
The progressively increased presence of Pt gradually modified the general appearance of the resulting NPs. At first (Pt–Cu–Se_A), the partial loss of shape already observed by TEM (Fig. 1a and b) due to the introduction of Pt corresponds to a highly disordered internal elemental rearranging inside the NPs, characterized by the presence of small and not fully superimposed Pt-rich and Cu-rich zones, as indicated by the partially corresponding bright and very faint zones observed in the elemental maps of the sample presented in Fig. 3 and ESI Fig. 1a† and from the elemental linescan of ESI Fig. 2.† Here, Cu and Pt follow a core/shell distribution, with Cu signal being usually more intense in the central regions of the NPs and almost absent at their borders, and Pt signal following the opposite trend and giving rise to an M-shaped intensity profile. Seldom this distribution is not followed by Cu and Pt, and both signals are paired in terms of intensity, being this configuration of Cu and Pt correspondent to the Cu/Pt rich zones that can be easily observed also from the elemental maps.
Enhancing the presence of platinum in Pt–Cu–Se_B and Pt–Cu–Se_C leads to a higher Pt-content when considering the overall composition of the NPs and to a rearrangement of the elements along similar trends. In particular, the main phase in Pt–Cu–Se_B is constituted by wide Se-rich zones with a Cu-rich core and a Pt-rich shell arrangement characterized by higher intensity in the central “valleys” of the M-shaped Pt shell profiles, which tends to lessen the difference with the wider lateral “peaks” (Fig. 4, ESI Fig. 1b and 3†). Also, the pairing of Cu and Pt signal can still be observed.
A further elemental rearranging in Pt–Cu–Se_C induces a higher degree of homogeneity in terms of elemental distribution, bringing to the formation of wide and well-defined Se-rich and Pt/Cu-rich zones in the NPs (Fig. 5, ESI Fig. 1c and 4†). This evolution coincides with a more pronounced pairing of the intensities of Cu and Pt signals, although Pt is still more intense than Cu in the peripheral regions of the NPs, and at times predominant.
This trend concludes with sample Pt–Cu–Se_D, whose elemental maps highlight the formation of heterostructures heavily based on platinum, with irregular binary cores of platinum and copper and peripheral regions of pure platinum (Fig. 6 and ESI Fig. 5†). Here, the division between core/shell and paired intensities of Cu and Pt previously observed in samples A–C changes once more towards a different arrangement. In fact, both elemental profiles tend to follow closely the intensity profile of HAADF, despite the Cu signal being much weaker or almost absent from the borders of the NPs, while Pt follows precisely the HAADF signal.
 | ||
| Fig. 6 HAADF-STEM image and corresponding elemental maps by STEM EDS of Pt–Cu–Se_D. | ||
Still, the dome/inverse-dome shaped signals of Cu/Pt profiles previously identified in sample A, which are typical of core/shell systems, cannot be observed in the sample Pt–Cu–Se_D. In fact, on one hand Pt is still present in a shell-like fashion at the borders of the NPs, but it is also present in the central regions of the NPs paired with the Cu, likely indicating that Cu and Pt likely form an alloy in the cores of the NPs, enclosed by pure Pt at their borders. Thus, considering that the STEM-HAADF signal is basically related to the Z1.8 of each element, in the current case it is dominated by the Pt signal and this finally explains why their two related profiles show an almost complete superposition. Moreover, no selenium signal could be detected here, indicating that the radical phase variation triggered by the massive introduction of platinum resulted in the expulsion of selenium from the NPs, without the formation of ternary phases observed in the previous samples.
The quantitative elemental analysis of the samples was conducted on the EDS elemental maps by considering the uneven distribution of chemical elements inside the NPs (namely, platinum and copper) to possibly identify the different phases formed therein, while also calculating the chemical composition of the whole NPs. The averaged composition obtained by quantitative analysis of the whole NPs confirms that increasing the quantity of platinum introduced in the reaction medium leads to increasingly higher percentages of platinum in the final NPs. The qualitative division already observed in the EDS maps between those zones rich and poor in metals (Pt and/or Cu) is clearly confirmed by the results of quantitative analysis on Pt–Cu–Se_A–D. In particular, the Pt/Cu-poor zones observed in the elemental maps of Pt–Cu–Se_A–C effectively correspond to the local presence of a Se-rich phase, while the Cu-poor zones observed in Pt–Cu–Se_D indicate the formation of pure platinum shells (Table 2).
| Phase A | Phase A* | Phase B | Phase B* | Full np | |
|---|---|---|---|---|---|
| Pt–Cu–Se_A | |||||
| *Indicates a minor phase; full np indicates the averaged composition of the whole NPs constituted by different phases. | |||||
| Cu | 31 | 26 | 21 | 35 | 27 |
| Se | 21 | 29 | 64 | 53 | 47 |
| Pt | 48 | 45 | 24 | 12 | 26 |
| Pt–Cu–Se_B | |||||
| Cu | 14 | — | 13 | — | 13 |
| Se | 43 | — | 56 | — | 58 |
| Pt | 43 | — | 31 | — | 29 |
| Pt–Cu–Se_C | |||||
| Cu | 36 | — | 14 | — | 29 |
| Se | 5 | — | 47 | — | 23 |
| Pt | 59 | — | 39 | — | 48 |
| Pt–Cu–Se_D | Core | Shell | |||
| Cu | 12 | — | 4 | — | 7 |
| Se | — | — | — | — | — |
| Pt | 88 | — | 96 | — | 93 |
More in detail, four different NPs configurations can be pointed out depending on the availability of platinum and the reorganization of cations within the NPs. At first, the rearrangement of elements leading to the formation of Pt/Cu-rich and Pt/Cu-poor zones in Pt–Cu–Se_A is characterized by an increased compositional variability, which translates in the identification of two “main” phases accompanied by two additional “minor” phases with slightly different composition with respect to their “main” counterparts (* indicate minor phases in Table 2). Pt–Cu–Se_B represents the first “stable” arrangement of the ternary system (Fig. 4), with a clear distinction between Pt/Cu-rich and Se-rich zones. This arrangement gives rise to heterostructures mainly constituted by a Se-rich ternary phase which is generally poor on copper (see Table 2). Pt–Cu–Se_C is constituted by Pt-rich/Se-rich heterostructures that partially share the composition previously observed in Pt–Cu–Se_B, albeit with an overall higher content of platinum: in particular Se-rich zones are still composed by ternary phases, while Pt-rich ones mainly feature platinum and copper. This variation also affects the structure of NPs as a whole, which shift towards that of proper heterostructures, with increasingly well-defined phases.
Finally, further increasing the amount of platinum available to the system leads to the formation of star-shaped binary core/shell NPs featuring platinum/copper cores and pure platinum shells, with the complete expulsion of selenium from the system (Fig. 6).
Following the results of the elemental characterization, which did not allow identifying known crystal phases, the structural analysis of the ternary samples was performed according to the planar and angular relationships occurring between diffraction spots in the 2D-FFTs (numerical diffractograms), and the corresponding lattice parameters were calculated for each orientation (see Methods for full details). Taking into account the indications provided by XRD analysis, the presence of an fcc Cu/Pt crystalline structure was supposed for the metal-rich zones, while a monoclinic Pt5Se4-like phase was taken as initial reference for the analysis of the Se-rich zones.
All the 2D-FFT patterns analyzed were compatible with the formation of the expected crystalline structures and different lattice parameters were identified in the samples, supporting the formation of multiple phases within the NPs, in agreement with what already indicated by the results of elemental analysis.
At first, the structural features of the initial Cu2−xSe sample were analyzed by HRTEM: the planar and angular relationships between diffraction spots in the 2D-FFTs diffractograms indicate the formation of single crystal NPs of Cu2Se (JCPDS card no. 79-1841) (Fig. 7a). From a structural point of view, the introduction of Pt causes a general loss of crystallinity in the final NPs, at first giving rise to polycrystalline NPs (Pt–Cu–Se_A), then to heterostructures (Pt–Cu–Se_B, C) and finally to star-shaped highly disordered polycrystalline NPs (Pt–Cu–Se_D), respectively. The evolution of the system also affects the size of the crystalline domains observed in the ternary NPs, but two main crystal phases can be isolated according to their lattice parameters and successfully identified among the different samples: the first fcc phase (phase A) presents a lattice parameter of 4.0 Å that is consistent in all the samples, while the lattice parameters calculated for the second phase (phase B) are consistent with the presence of a monoclinic phase (a = 7.0 Å, b = 3.9 Å, c = 12.6 Å, β = 102.3°).
 | ||
| Fig. 7 HRTEM images of representative NPs of Cu2Se (panel a), Pt–Cu–Se_A (panels b and c), Pt–Cu–Se_B (panel d), Pt–Cu–Se_C (panel e), Pt–Cu–Se_D (panel f). Different phases in panels B–E are depicted in false colors as indicated: phase A (main) → white; phase B (main) → red, phase A (minor) → green; phase B (minor) → orange. | ||
More in detail, Pt–Cu–Se_A features highly disordered polycrystalline NPs, characterized by the abovementioned two main phases along with two structurally distinct “minor” fcc phases clearly appearing (see Table 3, Fig. 7b and c). Pt–Cu–Se_B and Pt–Cu–Se_C are structurally less disordered and progressively revert from polycrystals to proper heterostructures (see Table 3, Fig. 7d and e), while keeping two distinct phases and maintaining the same lattice parameters and structures (i.e., fcc and monoclinic, respectively), also previously observed in Pt–Cu–Se_A. Finally, structural analysis on the crystalline domains in Pt–Cu–Se_D indicates the formation of just one fcc crystal structure with a lattice parameter of 4.0 Å (see Table 3, Fig. 7f).
| Pt–Cu–Se_A | Pt–Cu–Se_B | Pt–Cu–Se_C | Pt–Cu–Se_D | |
|---|---|---|---|---|
| *Indicates a minor phase. | ||||
| Phase A | 4.0 Å | 4.0 Å | 4.0 Å | 4.0 Å |
| Phase A* | 6.0 Å | — | — | — |
| Phase B | 7.0 Å | 7.0 Å | 7.0 Å | — |
| 3.9 Å | 3.9 Å | 3.9 Å | ||
| 12.6 Å | 12.6 Å | 12.6 Å | ||
| 102.3° | 102.3° | 102.3° | ||
| Phase B* | 5.3 Å | — | — | — |
The results of the structural and compositional analysis can be successfully combined by taking into account the similarities between the formation of crystalline domains with recurring lattice parameters observed by HRTEM and the phases with distinct compositions observed by STEM-EDS. Both approaches identify two main compositional and structural phases among the Pt–Cu–Se samples: at first the higher degree of variability manifests in Pt–Cu–Se_A in the form of two main and two “minor” phases, which subsequently converge to the two well-defined ones observed in Pt–Cu–Se_B and Pt–Cu–Se_C. Finally, only one structural phase can be observed in the Pt-rich core/shell NPs of Pt–Cu–Se_D. Despite the impossibility of discriminating between core and shell in Pt–Cu–Se_D from a structural point of view, the lack of Se provides a clear indication that the crystalline domains with average lattice spacing a = 4.0 Å obtained by HRTEM should be attributed to Pt-based phases (metallic Pt features a fcc crystal structure whose lattice parameter is 3.9 Å). Then, this recurring lattice spacing can also be put in relation to the Pt-rich phases observed in samples Pt–Cu–Se_A–C by STEM-EDS, while the other crystal structure observed in those same samples and characterized by a monoclinic crystal lattice (a = 7.0 Å, b = 3.9 Å, c = 12.6 Å, β = 102.3°) should be attributed to the Se-rich phases identified by compositional analysis and similar to Pt5Se4 (a = 6.6 Å, b = 4.6 Å, c = 11.2 Å, β = 101.6°). Finally, the presence of fcc structures corresponding to the “minor” A and B phases (lattice spacings being a = 6.0 Å and a = 5.3 Å, respectively) only observed in the Pt–Cu–Se_A sample can be put in relation with the presence of a depleting CuxSey phase (lattice spacing of cubic Cu2Se being a = 5.8 Å) and can be explained considering the high structural and compositional disorder caused by the initial introduction of Pt in the system.
Thus, the combination of XRD, STEM-EDS and HRTEM results provides a consistent picture of the evolution observed in the ternary system following the progressive introduction of Pt. At first, the numerous reflections observed in the XRD pattern of Pt–Cu–Se_A (Fig. 2) are matched by the highly disordered compositional and structural rearrangement observed by STEM-EDS and HRTEM, respectively: the initial assessment of Cu2−xSe and CuPt peaks is confirmed by the formation of fcc phases with consistent lattice parameters, while the additional Pt5Se4-like phase, initially suggested by the presence of additional XRD peaks in samples Pt–Cu–Se_A–C, is also compatible with the monoclinic phase individuated by HRTEM. Then, the broadening observed in the XRD peaks can be attributed to the highly disordered polycrystalline character of the NPs, which leads to the formation of small, likely defective crystalline domains.
The compositional analysis of sample Pt–Cu–Se_A by STEM-EDS highlights the presence two metal-rich phases and two Se-rich phases and further confirms the interpretation obtained by XRD and HRTEM, while proving that all the phases formed are indeed ternary. An increased presence of Pt led to an internal rearrangement of the NPs, and a decrease in the number of XRD peaks in Pt–Cu–Se_B and C. In particular, reflections derived from the initial Cu2−xSe are no longer observed for these samples, while those previously associated with cubic metal-rich and monoclinic Se-rich phases are still present. The presence of these cubic and monoclinic phases with lattice parameters close to CuPt and Pt5Se4, respectively, is also in agreement with HRTEM and STEM-EDS results (see Tables 2 and 3).
Finally, the powder XRD diffractogram of sample Pt–Cu–Se_D shows exclusively well-defined reflections clearly corresponding to cubic Pt, as already determined by STEM-EDS (see Table 2), where the atomic percentage of Pt atoms was either 88 or 96 depending on the phase measured, and no Se was detected.
Overall, as indicated in Scheme 1, experimental data suggest that Pt(II) monomer species initially diffuse through the Cu2−xSe lattice forming ternary phases when interacting with both Cu and Se. During the diffusion process, Pt(II) cations might partially exchange with Cu(I/II) cations and get also partially reduced, leading to the nucleation of Pt5Se4-like domains. The formation of ternary phases seems to be optimized in sample Pt–Cu–Se_B, where both A and B main structural phases contain significant amounts of the three elements (see Table 2).
 | ||
| Scheme 1 Simplified view of structural and chemical transformations upon reaction of Cu2−xSe NPs with Pt precursor at different molar ratios (volume ratio between phase A and B in each sample is fully qualitative). | ||
Nevertheless, with increasing Pt concentration, the formation of metal-rich and Se-rich domains is favored. It can be hypothesized that selenide (Se(-II)) anions are acting as reducing agents for the initially partial and later total reduction of Pt(II) and Cu(I/II) cations to metallic Pt and CuPt domains, originating almost pure Pt nanostructures at high Pt loads like those of Pt–Cu–Se_D, while elemental Se is finally dissolved into the solution. Both the metal reduction and the concomitant Se leaching into the solution might be assisted by the presence of a weak reducing amphiphilic surfactant like OLAm.
The optical absorption of these samples was measured in solution in the UV-Visible and NIR range. The sample containing the Cu2−xSe initial NPs showed a broad and intense absorption centered at 976 nm corresponding to its well-known LSPR band, as shown in Fig. 8.3 After the reaction with the Pt precursor, the damping of the LSPR is obvious and no absorption is observed in the NIR region already from sample Pt–Cu–Se_A. At wavelengths below 900 nm, an exponential and continuous absorption profile is displayed for all samples, where no absorption bands are visible.
 | ||
| Fig. 8 UV-Vis-NIR absorption spectra of Cu2−xSe and Pt–Cu–Se_A–D samples. | ||
As suggested in the introduction, the nanostructured ternary materials described in this work are expected to act as effective co-catalysts with regard to the hydrogen evolution reaction (HER). Therefore, the photocatalytic performance towards HER of Pt–Cu–Se_B and Pt–Cu–Se_C supported on g-C3N4 was investigated separately. We choose the g-C3N4 2D material as semiconductor-support because it has been reported to be a promising photocatalyst due to its wide light absorption ability, high chemical and thermal stability, and suitable electronic structure for water splitting.34–37
The same Pt–Cu–Se_X mass fraction of about 1%, wt/wt has been successfully loaded onto the g-C3N4 in both cases. As reported in the Experimental section, the samples were annealed at 200 °C for 4 h under air before performing photocatalytic experiments, in order to ensure the removal of the residual material coming from the surfactant. The annealing temperature has been chosen based on TGA results shown in ESI Fig. 6.† The thermal decomposition of organic components is then expected to increase the active sites available on the surface of nanocrystals.
The photocatalysts were analyzed by XRD. The XRD diffraction peaks at 13° and 27.4°, visible for both Pt–Cu–Se_B/g-C3N4 and Pt–Cu–Se_C/g-C3N4, belong to the (110) and (002) reflections of the g-C3N4 support (ESI Fig. 7†). Additionally, diffraction peaks at 41° and 48° and some minor intensity peaks at higher diffraction angles in the XRD patterns of samples Pt–Cu–Se_B/g-C3N4 and Pt–Cu–Se_C/g-C3N4 are consistent with the experimental patterns of their parental Pt–Cu–Se_X nanostructures, although the low amounts of cocatalyst loaded into the composites lead to very low intensities of their reflections. Characterization of the photocatalysts performed by SEM confirms the flake-like morphology of the g-C3N4 support as observed in Fig. 9, and suggests an homogeneous loading of the Pt–Cu–Se_X NPs over their surfaces. Pt–Cu–Se_X cocatalyst NPs are marked with red circles in Fig. 9b and d and show sizes similar to those of their precursor NPs. They are evenly distributed in the composite, and no appreciable aggregation is perceived, regardless of the annealing treatment. EDX spectrum shown in ESI Fig. 8† confirms the presence of the three elements, Pt, Cu and Se in the composites. The photocatalytic experiments for as-prepared samples were carried out under UVA-visible irradiation (λ ≥ 385 nm) for 4 h and the produced gases were continuously analyzed by on-line gas chromatography. In both cases, besides H2, only acetaldehyde was detected as carbon containing product. Acetaldehyde is expected as the primary oxidation product coming from the ethanol sacrificial agent. As shown in Table 4, both photocatalysts Pt–Cu–Se_B/g-C3N4 and Pt–Cu–Se_C/g-C3N4 show similar H2 production abilities. Despite the total H2 produced with Pt–Cu–Se_C/g-C3N4 photocatalyst being slightly higher than that obtained with Pt–Cu–Se_B/g-C3N4, their difference is less than 10% when this value is referred to the Pt content of the samples. This indicates that the properties of both ternary NPs are similar in terms of co-catalyst effect for the g-C3N4 semiconductor.
 | ||
| Fig. 9 SEM micrographs of (a and b) Pt–Cu–Se_B/g-C3N4 and, (c and d) Pt–Cu–Se_C/g-C3N4 nanocomposites. Red circles indicate the position of Pt–Cu–Se_X NPs. | ||
| Sample | H2 [μmol] | H2 [mmol g−1 of Pt] |
|---|---|---|
| g-C3N4 | 0.0 | — |
| CuSe/g-C3N4 | 22.6 | — |
| Pt–Cu–Se_B/g-C3N4 | 180.8 | 177.3 |
| Pt–Cu–Se_C/g-C3N4 | 233.6 | 162.2 |
On the other hand, when the reference sample CuSe/g-C3N4 containing binary Cu2−xSe NPs, without Pt, was used, the amount of H2 produced was very much lower (Table 4), which further confirms that ternary Pt–Cu–Se systems in Pt–Cu–Se_B/g-C3N4 and Pt–Cu–Se_C/g-C3N4 are efficient co-catalysts for H2 evolution. No H2 could be detected during a photocatalytic test performed under the same conditions using the g-C3N4 support.
For a deeper understanding of the features of the catalysts, XPS analysis was performed for Pt–Cu–Se_B/g-C3N4 and Pt–Cu–Se_C/g-C3N4. In the full XPS spectra, C, N, Cu, Se and Pt elements were detected (ESI Table 1†). More importantly, the deconvolution of the Pt 4f signal, shown in Fig. 10, indicates the simultaneous presence of metal Pt and Pt(II) with similar atomic concentration in both samples, as indicated in Table 5. This could indicate that both samples produced comparative amounts of H2 per Pt on surface. Nevertheless, and considering the complexity of the systems under study, further experiments are necessary in order to elucidate the specific role of Pt and Pt(II) species in the samples, as well as the result of the coexistence of both Pt-based metallic and semiconductor ternary compounds within the cocatalyst and their synergy with g-C3N4.
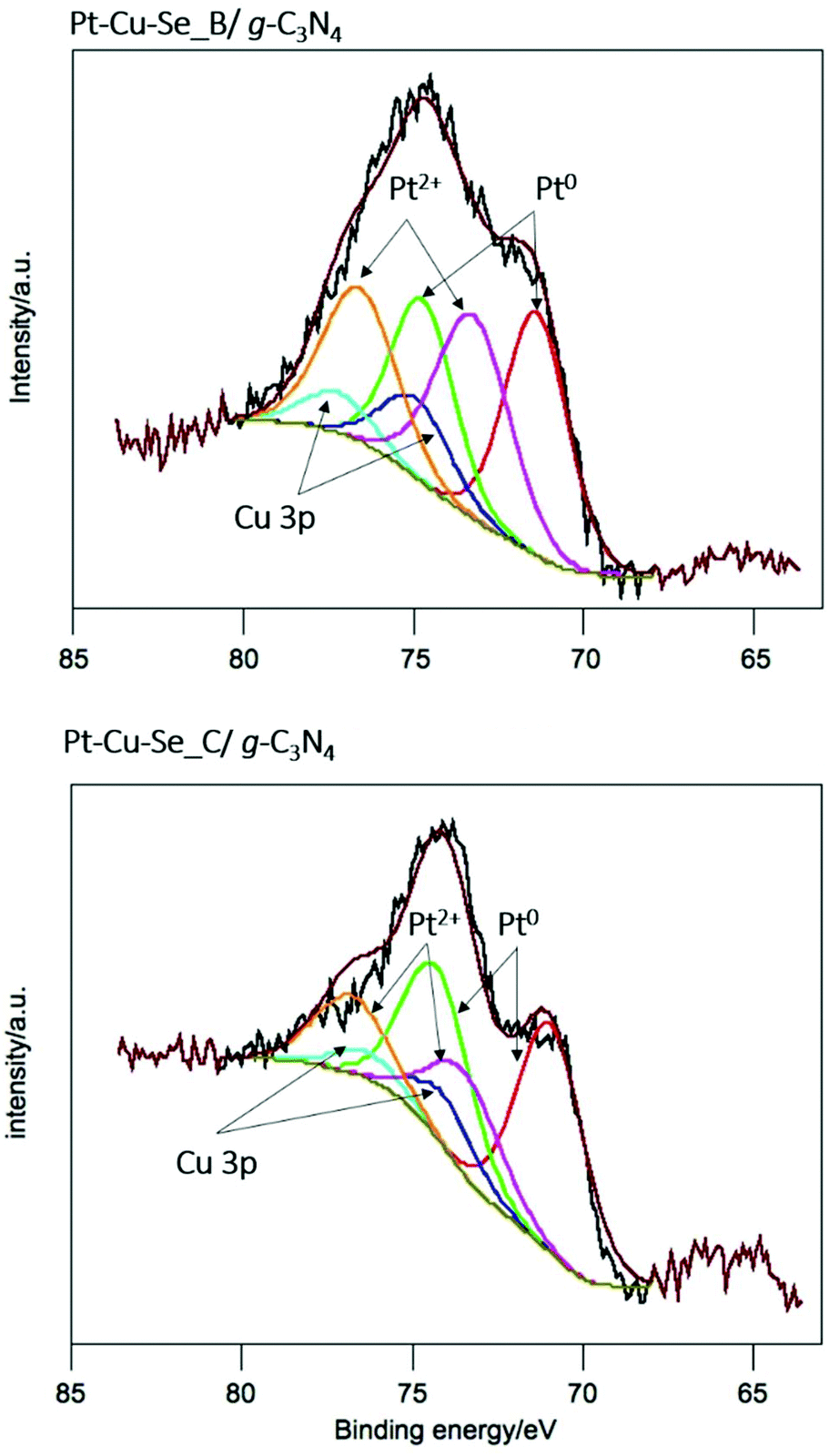 | ||
| Fig. 10 X-ray photoelectron spectra of the Pt 4f core level for Pt–Cu–Se_B/g-C3N4 and Pt–Cu–Se_C/g-C3N4 nanocomposites, and corresponding deconvolutions. | ||
| Sample | Specie | BE/eV | % atomic |
|---|---|---|---|
| BE = binding energy. | |||
| Pt–Cu–Se_B/g-C3N4 | Pt(0) | 71.4 | 0.03 |
| Pt(II) | 73.3 | 0.03 | |
| Pt–Cu–Se_C/g-C3N4 | Pt(0) | 70.9 | 0.04 |
| Pt(II) | 73.4 | 0.02 | |
Conclusions
All in all, we have demonstrated in this work that the insertion of Pt within the Cu2−xSe nanocrystalline lattice is feasible under certain conditions, with the resulting formation of ternary phases with variable composition that appear often segregated into two main structural formats, i.e. a cubic Pt-rich and a monoclinic Se-rich phase, for which the relative composition and lattice parameters have been measured by means of local STEM-EDS and HRTEM analyses. The introduction of Pt causes an immediate damping of the LSPR band characteristic of the starting Cu2−xSe NPs. Nevertheless, the ternary Pt-based nanostructured materials prepared exhibit a synergy with g-C3N4 layered semiconductor-support acting as effective co-catalysts for the HER.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A.Fi. acknowledges financial support from the Spanish Ministerio de Economía y Competitividad (MINECO) through CTQ2015-68370-P and the Spanish Ministerio de Ciencia e Innovación (MICINN) through PID2019-106165GB-C22, and from the regional Generalitat de Catalunya authority (2017 SGR 15). A.Fi. is a Serra Húnter fellow. JL is a Serra Húnter Fellow and is grateful to ICREA Academia program and projects GC 2017 SGR 128 and MICINN/FEDER RTI2018-093996-B-C31. N.H and P.RP acknowledge financial support of MAT2017-87500-P project and Y.W thanks the China Scholarship Council for his PhD grant (CSC 201608460014). This research was also supported by KAUST Baseline funding of A. Fa.References
- L. Cademartiri and G. A. Ozin, Concepts of Nanochemistry, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 1st edn., 2009 Search PubMed.
- R. Buonsanti and D. J. Milliron, Chem. Mater., 2013, 25, 1305–1317 CrossRef CAS.
- W. Van Der Stam, A. C. Berends and C. De Mello Donega, ChemPhysChem, 2016, 17, 559–581 CrossRef CAS PubMed.
- U. Banin, Y. Ben-shahar and K. Vinokurov, Chem. Mater., 2014, 26, 97–110 CrossRef CAS.
- J. M. R. Tan, M. C. Scott, W. Hao, T. Baikie, C. T. Nelson, S. Pedireddy, R. Tao, X. Ling, S. Magdassi, T. White, S. Li, A. M. Minor, H. Zheng and L. H. Wong, Chem. Mater., 2017, 29, 9192–9199 CrossRef CAS.
- H. Chen, P.-F. Liu, B.-X. Li, H. Lin, L.-M. Wu and X.-T. Wu, Dalt. Trans., 2017, 429–437 Search PubMed.
- A. Wolf, T. Kodanek and D. Dorfs, Nanoscale, 2015, 7, 19519–19527 RSC.
- V. Lesnyak, R. Brescia, G. C. Messina and L. Manna, J. Am. Chem. Soc., 2015, 137, 9315–9323 CrossRef CAS PubMed.
- Y. Xie, G. Bertoni, A. Riedinger, A. Sathya, M. Prato, S. Marras, R. Tu, T. Pellegrino and L. Manna, Chem. Mater., 2015, 27, 7531–7537 CrossRef CAS PubMed.
- C. Hu, W. Chen, Y. Xie, S. K. Verma, P. Destro, G. Zhan, X. Chen, X. Zhao, P. J. Schuck, I. Kriegel and L. Manna, Nanoscale, 2018, 10, 2781–2789 RSC.
- X. Liu, C. Lee, W.-C. Law, D. Zhu, M. Liu, M. Jeon, J. Kim, P. N. Prasad, C. Kim and M. T. Swihart, Nano Lett., 2013, 13, 4333–4339 CrossRef CAS PubMed.
- X. Wang, X. Liu, D. Zhu and M. T. Swihart, Nanoscale, 2014, 6, 8852–8857 RSC.
- G. Gariano, V. Lesnyak, R. Brescia, G. Bertoni, Z. Dang, R. Gaspari, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2017, 139, 9583–9590 CrossRef CAS PubMed.
- K. Miszta, G. Gariano, R. Brescia, S. Marras, F. De Donato, S. Ghosh, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2015, 137, 12195–12198 CrossRef CAS PubMed.
- C. Coughlan, A. Singh and K. M. Ryan, Chem. Mater., 2013, 25, 653–661 CrossRef CAS.
- C. Dong, D. Yao, J. Feng, T. Huang, X. Hu, Z. Wu, Y. Liu, B. Yang and H. Zhang, Chem. Mater., 2016, 28, 9139–9149 CrossRef CAS.
- Y. Liu, M. Liu, D. Yin, L. Qiao, Z. Fu and M. T. Swihart, ACS Nano, 2018, 12, 7803–7811 CrossRef CAS PubMed.
- L. De Trizio, H. Li, A. Casu, A. Genovese, A. Sathya, G. C. Messina and L. Manna, J. Am. Chem. Soc., 2014, 136, 16277–16284 CrossRef CAS PubMed.
- Q. A. Akkerman, A. Genovese, C. George, M. Prato, I. Moreels, A. Casu, S. Marras, A. Curcio, A. Scarpellini, T. Pellegrino, L. Manna and V. Lesnyak, ACS Nano, 2015, 9, 521–531 CrossRef CAS PubMed.
- V. Lesnyak, C. George, A. Genovese, M. Prato, A. Casu, S. Ayyappan, A. Scarpellini and L. Manna, ACS Nano, 2014, 8, 8407–8418 CrossRef CAS PubMed.
- Y. Xie, W. Chen, G. Bertoni, I. Kriegel, M. Xiong, N. Li, M. Prato, A. Riedinger, A. Sathya and L. Manna, Chem. Mater., 2017, 29, 1716–1723 CrossRef CAS PubMed.
- Y. Liu, D. Yin and M. T. Swihart, Chem. Mater., 2018, 30, 1399–1407 CrossRef CAS.
- C. Coughlan, M. Ibáñez, O. Dobrozhan, A. Singh, A. Cabot and K. M. Ryan, Chem. Rev., 2017, 117, 5865–6109 CrossRef CAS PubMed.
- M. Rondelli, G. Zwaschka, M. Krause, M. D. Rötzer, M. N. Hedhili, M. P. Högerl, V. D'Elia, F. F. Schweinberger, J. M. Basset and U. Heiz, ACS Catal., 2017, 7, 4152–4162 CrossRef CAS.
- E. C. Tyo and S. Vajda, Nat. Nanotechnol., 2015, 10, 577–588 CrossRef CAS PubMed.
- Y. L. Huang, C. Pellegrinelli and E. D. Wachsman, Angew. Chem., Int. Ed., 2016, 55, 15268–15271 CrossRef CAS PubMed.
- X. Ding, Y. Zou, F. Ye, J. Yang and J. Jiang, J. Mater. Chem. A, 2013, 1, 11880–11886 RSC.
- W. Sang, T. Zheng, Y. Wang, X. Li, X. Zhao, J. Zeng and J. G. Hou, Nano Lett., 2014, 14, 6666–6671 CrossRef CAS PubMed.
- A. Wolf, D. Hinrichs, J. Sann, J. F. Miethe, N. C. Bigall and D. Dorfs, J. Phys. Chem. C, 2016, 120, 21925–21931 CrossRef CAS.
- I. Kriegel, C. Jiang, J. Rodríguez-Fernández, R. D. Schaller, D. V. Talapin, E. Da Como and J. Feldmann, J. Am. Chem. Soc., 2012, 134, 1583–1590 CrossRef CAS PubMed.
- C. Wang, H. Daimon, Y. Lee, J. Kim and S. Sun, J. Am. Chem. Soc., 2007, 129, 6974–6975 CrossRef CAS PubMed.
- X. She, J. Wu, J. Zhong, H. Xu, Y. Yang, R. Vajtai, J. Lou, Y. Liu, D. Du, H. Li and P. M. Ajayan, Nano Energy, 2016, 27, 138–146 CrossRef CAS.
- J. D. S. Newman and G. J. Blanchard, Langmuir, 2006, 22, 5882–5887 CrossRef CAS PubMed.
- J. Wang, Q. Zhou, Y. Shen, X. Chen, S. Liu and Y. Zhang, Langmuir, 2019, 35, 12366–12373 CrossRef CAS PubMed.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- G. Liu, T. Wang, H. Zhang, X. Meng, D. Hao, K. Chang, P. Li, T. Kako and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 13561–13565 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Combined elemental maps and linescans by STEM EDS, UV-Vis-NIR absorption spectra, TGA, XRD and XPS data. See DOI: 10.1039/d0nr02726j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |