
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAb initio nanofluidics: disentangling the role of the energy landscape and of density correlations on liquid/solid friction†
Gabriele
Tocci
 *a,
Maria
Bilichenko
a,
Laurent
Joly
bc and
Marcella
Iannuzzi
a
*a,
Maria
Bilichenko
a,
Laurent
Joly
bc and
Marcella
Iannuzzi
a
aDepartment of Chemistry, Universität Zürich, 8057 Zürich, Switzerland. E-mail: gabriele.tocci@chem.uzh.ch
bUniv Lyon, Univ Claude Bernard Lyon 1, CNRS, Institut Lumière Matière, F-69622, Villeurbanne, France
cInstitut Universitaire de France (IUF), France
First published on 5th May 2020
Abstract
Despite relevance to water purification and renewable energy conversion membranes, the molecular mechanisms underlying water slip are poorly understood. We disentangle the static and dynamical origin of water slippage on graphene, hBN and MoS2 by means of large-scale ab initio molecular dynamics. Accounting for the role of the electronic structure of the interface is essential to determine that water slips five and eleven times faster on graphene compared to hBN and to MoS2, respectively. Intricate changes in the water energy landscape as well as in the density correlations of the fluid provide, respectively, the main static and dynamical origin of water slippage. Surprisingly, the timescales of the density correlations are the same on graphene and hBN, whereas they are longer on MoS2 and yield a 100% slowdown in the flow of water on this material. Our results pave the way for an in silico first principles design of materials with enhanced water slip, through the modification of properties connected not only to the structure, but also to the dynamics of the interface.
Nanofluidics has attracted increasing attention over the years for its potential to remedy future shortages of clean water and for the generation of renewable energy.1–3 The miniaturization of devices and the accurate characterization of nanoscale transport have led to the development of extremely efficient nanotube and Å-scale slits membranes for water desalination, purification and osmotic energy conversion membranes.4–7 At the nanoscale specific materials properties, pore chemistry and confinement geometry may all have a strong impact on nanofluidic transport, and therefore on the performance of desalination and osmotic energy conversion membranes. In the case of water transport through nanotubes, there is mounting evidence that slippage is qualitatively different between carbon and boron nitride materials,8 a result that hints at the role of the electronic structure on flow.9 Despite fundamental insights provided by transport experiments across single pores, connecting the electronic structure of materials to water slippage under confinement remains an open challenge. Computer simulations, and in particular molecular dynamics based on empirical force fields (FFMD) are instrumental to provide insights into the atomistic mechanisms underlying transport in nanofluidics.6,10–17 Yet, the role of the electronic structure is not accounted for in these studies and FFMD results are often sensitive to the choice of the interaction parameters. Additionally, the lack of transferability of force-field parameters between different interfaces poses a problem when comparing slippage on different materials. The development of force fields capable of accurately describing the structure and energetics of water interacting on two-dimensional materials is an active area of research, where recently machine learning is playing a prominent role.18,19 Nevertheless, to be able to compare water slippage between different nanofluidic interfaces on equal footing, it is essential to resort to ab initio molecular dynamics (AIMD), where the electronic structure is accounted for with density functional theory (DFT). Due to the formidable challenges associated with carrying out AIMD simulations of nanofluidic transport, these types of studies have been scarce.20–22
Here, we tackle this challenge in full strength by using AIMD to investigate the slippage of water confined between two-dimensional materials. We concentrate our efforts on connecting the atomistic features and electronic structure of the interface to the friction of water on graphene, hBN and MoS2 sheets, motivated by a number of experimental works on nanofluidic transport at such interfaces.8,23–25 We find that the friction on graphene is about five and eleven times smaller than on hBN and MoS2, respectively. The structure of water in the direction perpendicular to the sheets is very similar across the three materials. Yet, slight changes in the corrugation of the water energy landscape of at most a few kBT‡ in the direction parallel to the sheets impact the friction dramatically. Interestingly, slippage is also controlled by the density correlations of the contact layer, and their time relaxation provides the main dynamical origin of the friction. These results illustrate how a delicate interplay of interfacial forces, which are governed by the electronic structure of materials, can have a tremendous impact on nanofluidic transport. Additionally, favourable comparison with experimental slip lengths on carbon nanotubes with small curvature and with BN nanotubes illustrates the relevance of the electronic structure on nanofluidic transport.
We performed a series of extensive AIMD simulations of water confined between graphene, hBN and MoS2 sheets for different confinement widths, as well as with water films supported on the three types of sheets for systems containing up to 2000 atoms. AIMD simulations were carried out in the Born–Oppenheimer approximation with the electronic structure computed at the DFT level using the optB88-vdW exchange–correlation functional26,27 and the dynamics was propagated in the canonical ensemble for about 100 ps at 330 K. Snapshots of the simulated systems for MoS2 with different confinements are shown in Fig. 1.
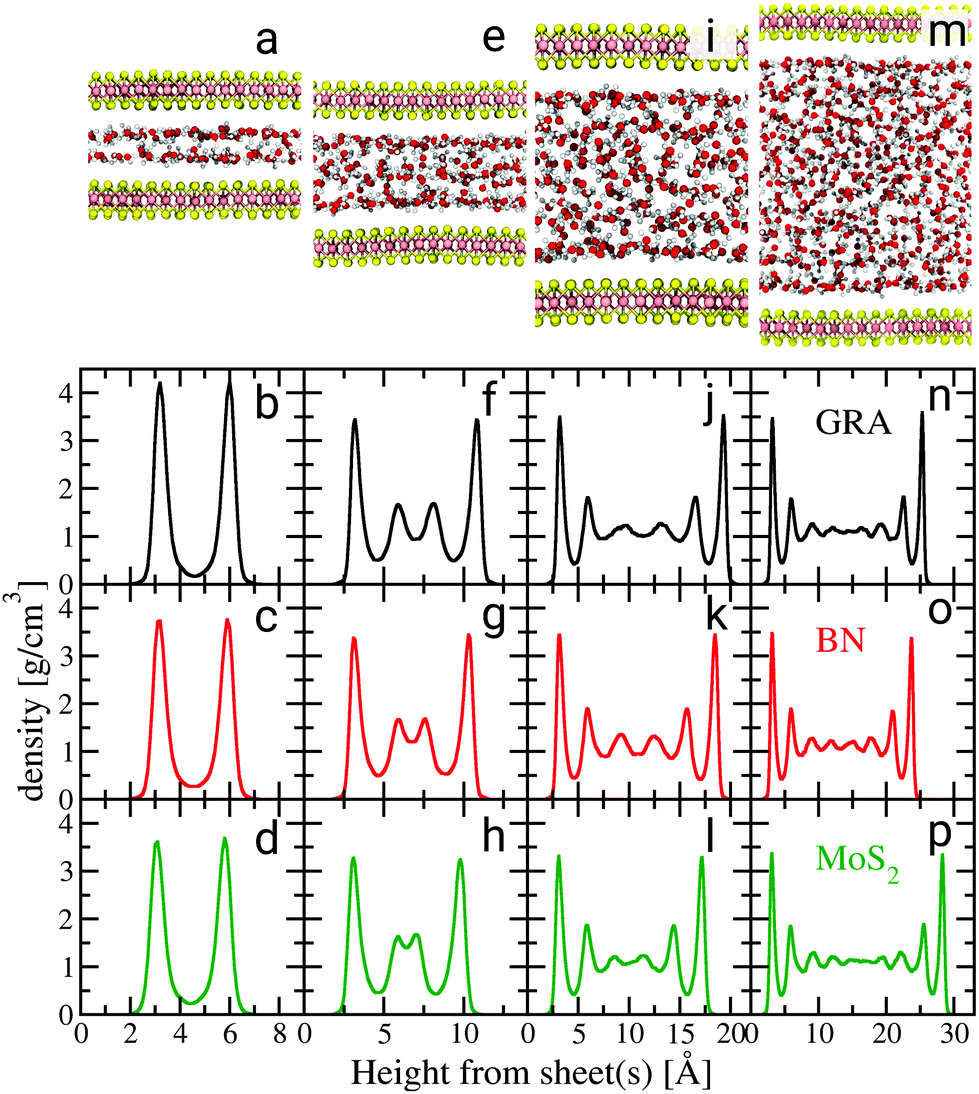 | ||
| Fig. 1 Water density profiles for different confinement heights. Black, red and green graphs are density profiles for graphene (GRA), hBN and MoS2, respectively. The snapshots in (a), (e), (i) and (m) illustrate the configurations of water confined between MoS2 sheets for different confinements. The density profiles for the supported water films are shown in the ESI.† | ||
We start our discussion on the structural properties of water under confinement. A key property of confined fluids is their density profile as a function of the distance from the sheets,28 which is shown in Fig. 1. The water density profiles were computed with respect to the instantaneous height between each fluid molecule and the nearest sheet's atom, in the spirit of ref. 29 (ESI†). There is a strong similarity in the layering of water between the three materials for each confinement height. The properties of the first water layer, so-called contact layer, are most relevant to understand nanofluidic water slip.30 Therefore, particular emphasis is placed on the understanding of its structure. Except for the smallest confinement width, the contact layer density peaks at approximately 3.5 g cm−3, regardless of the type of material. For confinement heights between 1.2 and 1.5 nm (Fig. 1f–h) two additional smaller peaks appear beyond the contact layer. Gradually less pronounced density oscillations are also visible beyond the first two layers for confinement widths between 2.0 and 3.0 nm (Fig. 1j–p). A bulk liquid region is almost recovered in systems whose confinement width reaches about 3.0 nm (Fig. 1n–p). The differences observed in the first density peaks seen in Fig. 1(b)–(d) and in the second peaks in Fig. 1(f)–(h) result from commensurability between the size of the water molecules and the confinement width of each system,31 along with differences in the magnitude of the water/sheet interactions.§
We now turn to water slip, as the most relevant contribution of this work. At liquid–solid interfaces where the major contribution to the water/surface interaction comes from dispersion forces – such as those considered here – the tangential velocity of the fluid in contact with the solid, so-called slip velocity vslip, is non-zero and the friction force parallel to the flow is proportional to the slip velocity via the so-called friction coefficient λ, following the relation 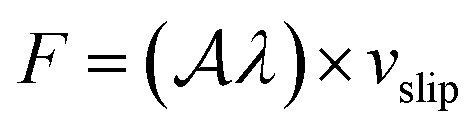 ,32,33 with
,32,33 with 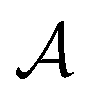 the surface area. Whereas the relation of proportionality between the total force acting on the liquid and the slip velocity can be used to calculate λ from nonequilibrium molecular dynamics simulations,34 here we apply the following Green–Kubo relation and compute the friction coefficient from the equilibrium fluctuations of the force acting on the liquid extracted from our equilibrium AIMD trajectories:33,35
the surface area. Whereas the relation of proportionality between the total force acting on the liquid and the slip velocity can be used to calculate λ from nonequilibrium molecular dynamics simulations,34 here we apply the following Green–Kubo relation and compute the friction coefficient from the equilibrium fluctuations of the force acting on the liquid extracted from our equilibrium AIMD trajectories:33,35
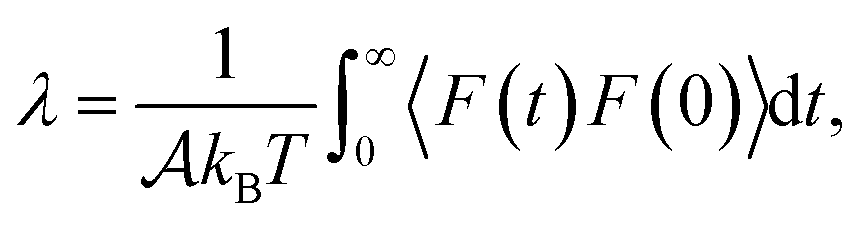 | (1) |
 is the surface area of the sheets. For finite size systems, the integral in eqn (1) decays to zero as t → ∞, but the equilibrium value of the friction coefficient can be obtained when the integral reaches a plateau value at intermediate times22,36 (ESI†). Fig. 2 shows the friction coefficient for different confinement widths and also for the liquid film configurations on graphene, hBN and MoS2.
is the surface area of the sheets. For finite size systems, the integral in eqn (1) decays to zero as t → ∞, but the equilibrium value of the friction coefficient can be obtained when the integral reaches a plateau value at intermediate times22,36 (ESI†). Fig. 2 shows the friction coefficient for different confinement widths and also for the liquid film configurations on graphene, hBN and MoS2.
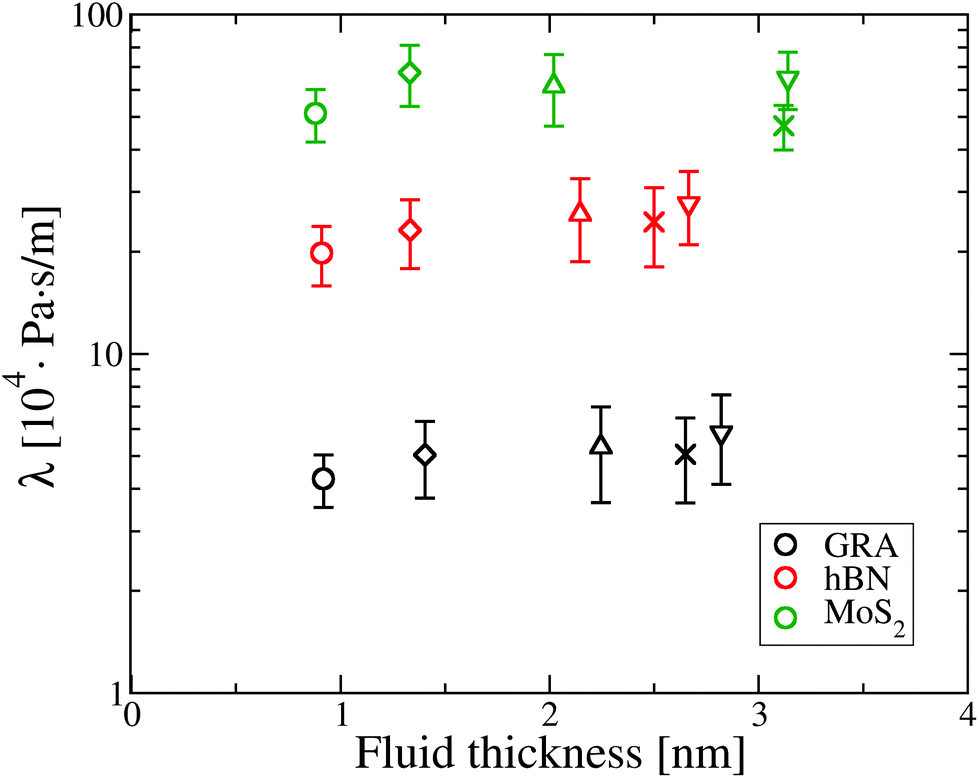 | ||
| Fig. 2 Friction coefficient λ for different fluid thicknesses. ○ refers to a fluid thickness of 0.9 nm, ◊ is for 1.5 nm, △ for 2 nm, ∇ for a confinement between 2.5 and 3.0 nm and × is for the water film configurations which have a thickness between 2.5 and 3.2 nm. In this and subsequent plots the error bars correspond to the statistical error within a 95% confidence level, computed over about 20 blocks of data along the armchair and zigzag axis, for a total of 40 blocks. | ||
It can be seen that water flow is fastest on graphene, as λ on graphene is about 5 times smaller compared to hBN and about 11 times smaller than MoS2. Additionally, there is no apparent effect of confinement on friction, as λ is approximately constant as a function of the fluid thickness for all materials.
To provide mechanistic insights into the observed trends of the friction coefficient, eqn (1) is formulated in terms of the second moment of the force 〈F2〉 and the force correlation time τF:
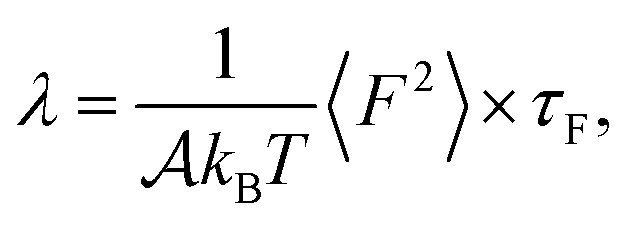 | (2) |
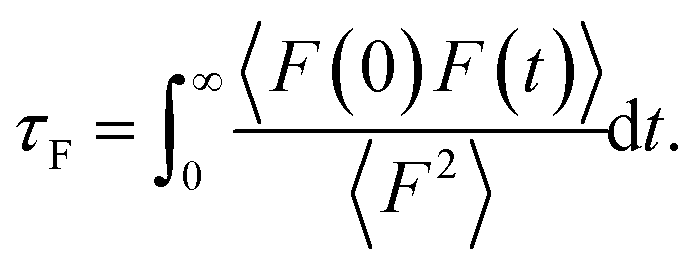 | (3) |
From eqn (2) it is possible to disentangle the dependence of the friction coefficient on static and dynamical properties of the interface, where 〈F2〉 and τF entail a static and dynamical contribution, respectively. We first illustrate the dependence of λ on 〈F2〉 as shown in Fig. 3 for the systems considered. A clear correlation between the friction and the mean squared force is observed: upon going from graphene to hBN to MoS2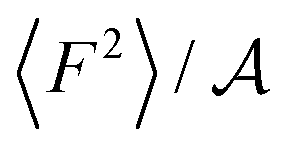 increases, and so too does λ, for all confinement widths. Based on dimensional grounds, the dependence of λ on
increases, and so too does λ, for all confinement widths. Based on dimensional grounds, the dependence of λ on 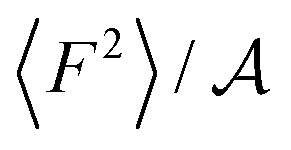 can be further expressed as
can be further expressed as 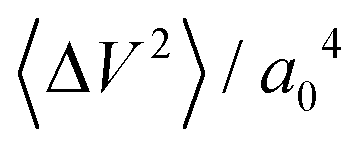 , where 〈ΔV2〉 is a measure of the corrugation of the potential energy of the fluid in the contact layer and a0 is the lattice constant of the sheets.11,30 The corrugation of the water energy landscape can be estimated from the calculation of the free energy profile defined as ΔG(x,y) = −kBT
, where 〈ΔV2〉 is a measure of the corrugation of the potential energy of the fluid in the contact layer and a0 is the lattice constant of the sheets.11,30 The corrugation of the water energy landscape can be estimated from the calculation of the free energy profile defined as ΔG(x,y) = −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log[PO(x,y)/PO(xmax,ymax)], where PO(x,y) is the two-dimensional probability distribution of an oxygen atom in a water molecule in the contact layer and the subscript “max” indicates the point where the probability is at the maximum. The free energy profile thus computed is shown in Fig. 3(b)–(d).
log[PO(x,y)/PO(xmax,ymax)], where PO(x,y) is the two-dimensional probability distribution of an oxygen atom in a water molecule in the contact layer and the subscript “max” indicates the point where the probability is at the maximum. The free energy profile thus computed is shown in Fig. 3(b)–(d).
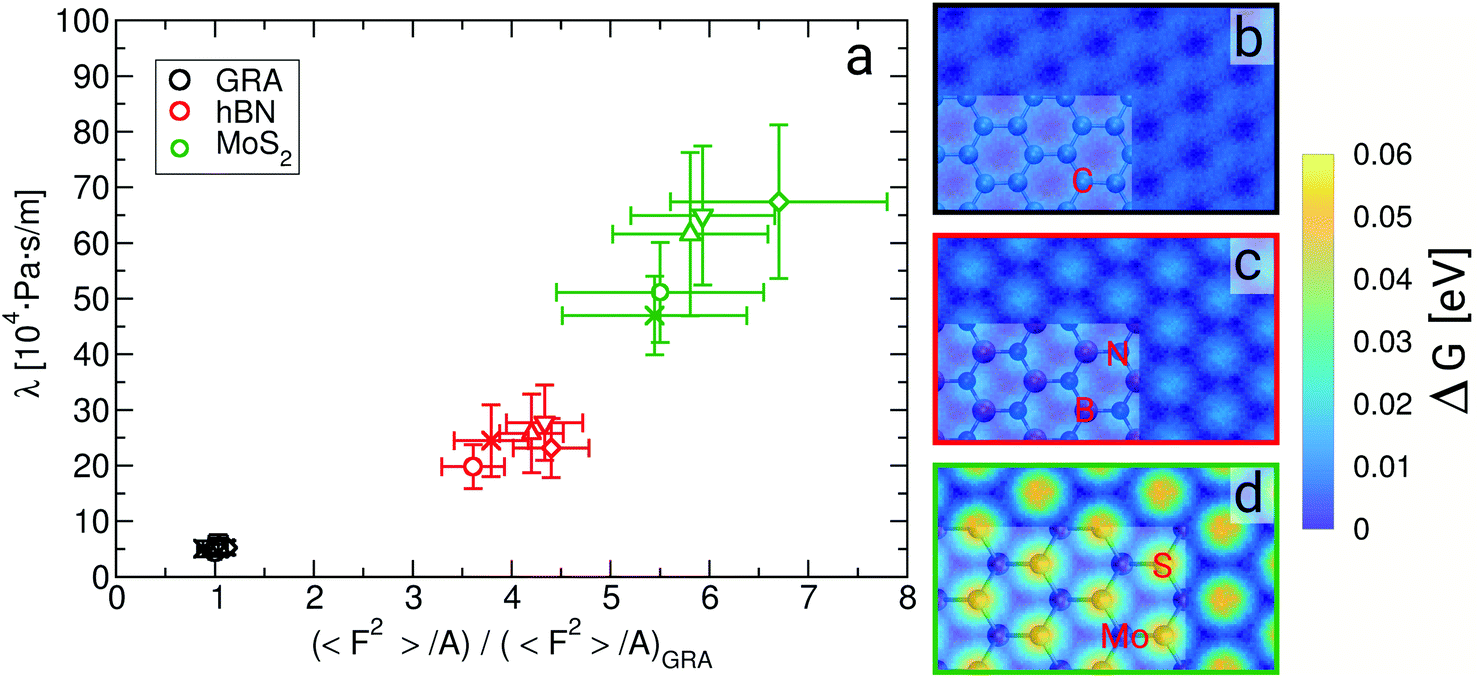 | ||
| Fig. 3 Friction coefficient λ as a function of 〈F2〉/A (a), and free energy landscape of water in the contact layer for (b) graphene, (c) hBN and (d) MoS2, where snapshots of the sheets are superimposed on part of the graphs. In (a) 〈F2〉/A is normalized by its value on graphene at a confinement width of about 0.9 nm, and the different symbols refer to systems with different fluid thicknesses, as per Fig. 2. | ||
The level of corrugation can be extracted from the free energy barrier, obtained as the maximum in ΔG, which is of about 0.01 eV, 0.02 eV and 0.05 eV for graphene, hBN and MoS2, respectively. Whereas the free energy barriers in graphene and hBN are smaller than the thermal energy at room temperature (about 0.025 eV), the free energy barrier on MoS2 is about 0.05 eV, and therefore larger than kBT, indicating definitely a more corrugated overlayer. The increased level in the corrugation of the free energy explains the observed relation between λ and 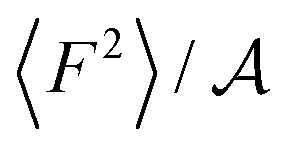 and it provides a static and structural origin for the observed behaviour of the friction coefficient. Looking closely at Fig. 3, some changes in the dynamics can be anticipated by noting that, while λ and 〈F2〉 appear to be proportional for graphene and hBN, the friction of water on MoS2 is too large or it to be explained simply based on its dependence on 〈F2〉.
and it provides a static and structural origin for the observed behaviour of the friction coefficient. Looking closely at Fig. 3, some changes in the dynamics can be anticipated by noting that, while λ and 〈F2〉 appear to be proportional for graphene and hBN, the friction of water on MoS2 is too large or it to be explained simply based on its dependence on 〈F2〉.
Indeed, we provide evidence of an intriguing mechanism underlying friction, inherent to the dynamics of the water contact layer. To this end we plot λ as a function of the force correlation time τF for the systems discussed so far, see Fig. 4. The most apparent feature is that τF is approximately 50 fs for graphene and hBN, considering all fluid thicknesses, whereas it is about 100 fs for MoS2. The larger value of τF indicates a slower relaxation of the lateral force acting on the fluid at the water–MoS2 interface compared to the other two interfaces. The dependence of friction on τF can be tuned by changing the amplitude of the bond length oscillations of the sheets,15 and by modifying the magnitude of the electrostatic and dispersion interactions (see ESI†). Because depositing two-dimensional materials on different substrates may lead to a change in the electrostatic and/or van der Waals interactions at the interface, it is possible to test experimentally this dependence.25,37,38
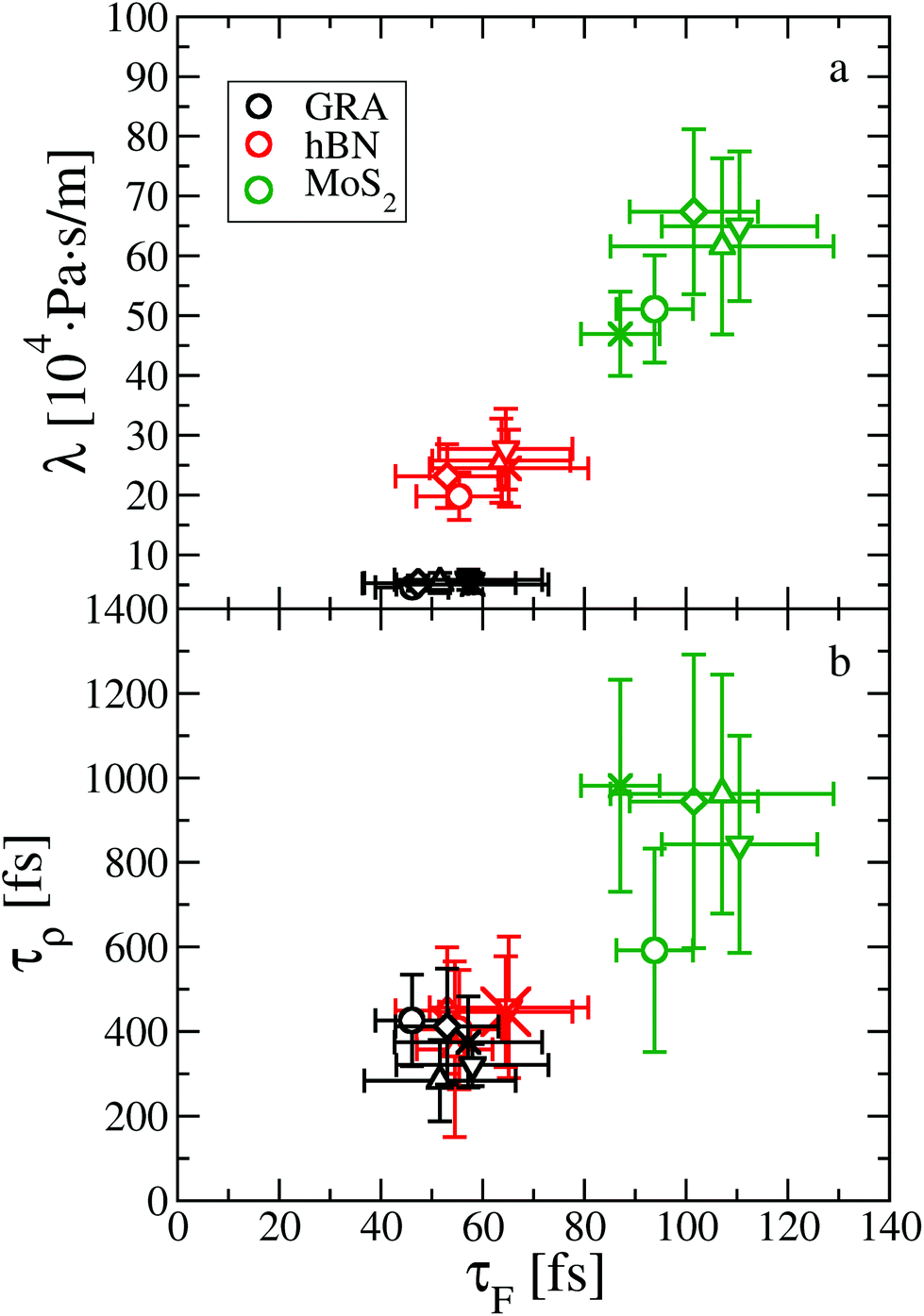 | ||
| Fig. 4 Dynamical origin of water slip on two-dimensional materials, as shown by the dependence (a) of the friction coefficient λ on the force correlation time τF, and (b) of the density correlation time τρ on τF. The same symbols are used as in Fig. 2. | ||
In order to provide further insights into the molecular origin of this phenomenon, eqn (1) is approximated in terms of the structure and dynamics of the liquid contact layer (ESI†):30,39
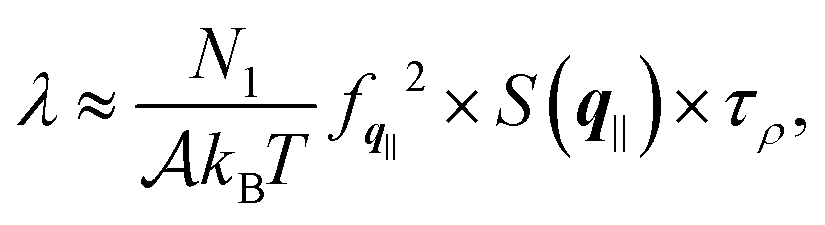 | (4) |
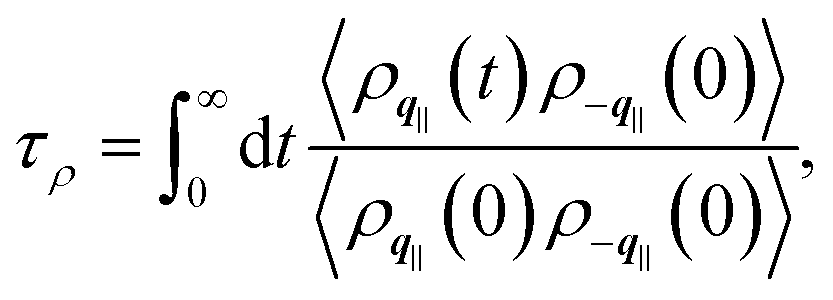 | (5) |
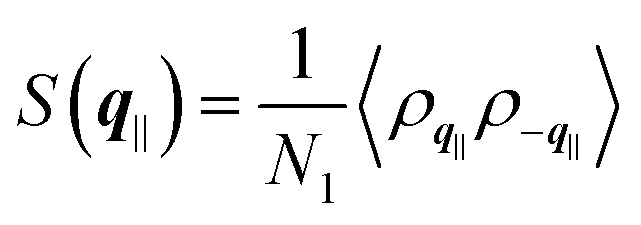 the static structure factor, and τρ the density correlation time, both defined in terms of the Fourier component of the fluid density
the static structure factor, and τρ the density correlation time, both defined in terms of the Fourier component of the fluid density 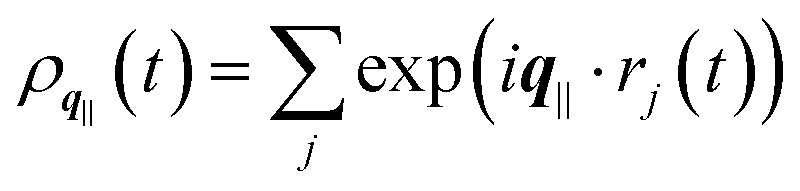 .
.
Altogether, eqn (2) and (4) imply a correlation between the force correlation time and the density correlation time, i.e. τF ∼ τρ. As it can be seen in Fig. 4(b), both τF and τρ are approximately twice as small in graphene and hBN compared to MoS2. The apparent correlation between τF and τρ provides the main explanation for the different dynamical contribution to the friction. The density correlation time on graphene and hBN is smaller than on MoS2 because (i) the density modes on the former materials are associated with a smaller lattice constant a0 and therefore decay more rapidly40 and (ii) the collective diffusion of the fluid probed at a specific wave-vector (Dq∥) is larger on graphene and hBN compared to MoS2. This can be seen by expressing τρ in terms of the wave-vector and of Dq∥ as τρ = 1/(Dq∥q2).30 By taking τρ for graphene (or hBN) and MoS2 to be about 400 fs and 1000 fs, respectively, we can extract Dq∥ to be about 0.38 Å2 ps−1 for graphene and hBN and 0.25 Å2 ps−1 for MoS2. As a result, we find that the differences in the lattice constant (i.e. the q−2 term), and in the collective diffusion coefficient  equally contribute to the observed behaviour of τρ. In the ESI (see Fig. S13†) we also show that stretching and compressing the underlying lattice has a strong effect on τρ, but the larger MoS2 lattice constant is not sufficient to explain alone the larger value of τρ on MoS2. Instead, there is a remaining contribution to τρ on MoS2 beyond the obvious q−2 dependence, which arises from the slower collective diffusion of water on MoS2 compared to the other two materials, as it can be seen by compressing MoS2 to approximately the same lattice constant of hBN and graphene. Despite the evident correlation between τF and τρ, the latter is smaller by almost a factor of 10. A possible reason for the discrepancy is that the density correlation time results from an average over the density modes for the whole contact layer, whereas the force correlation time is more sensitive to those modes closer to the contact layer.30 Note also that the derivation of eqn (4) is based on truncating the Fourier expansion of the potential energy landscape of the wall, but higher order modes could contribute.
equally contribute to the observed behaviour of τρ. In the ESI (see Fig. S13†) we also show that stretching and compressing the underlying lattice has a strong effect on τρ, but the larger MoS2 lattice constant is not sufficient to explain alone the larger value of τρ on MoS2. Instead, there is a remaining contribution to τρ on MoS2 beyond the obvious q−2 dependence, which arises from the slower collective diffusion of water on MoS2 compared to the other two materials, as it can be seen by compressing MoS2 to approximately the same lattice constant of hBN and graphene. Despite the evident correlation between τF and τρ, the latter is smaller by almost a factor of 10. A possible reason for the discrepancy is that the density correlation time results from an average over the density modes for the whole contact layer, whereas the force correlation time is more sensitive to those modes closer to the contact layer.30 Note also that the derivation of eqn (4) is based on truncating the Fourier expansion of the potential energy landscape of the wall, but higher order modes could contribute.
Our results can be used in practice to test experimentally a possible dependence of the viscosity on confinement, which remains still an open question.31,41–44 Indeed, liquid–solid slip is most often characterized experimentally through the slip length b, defined as the ratio between the slip velocity vslip and the shear rate ![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) at the interface: b = vslip/. Because at the interface the viscous shear stress η (with η the shear viscosity) is equal to the wall shear stress λvslip, the slip length can be expressed as the ratio between the viscosity and the friction coefficient:45b = η/λ. Using the experimental viscosity of bulk water (η = 10−3 Pa s), we obtain on average a slip length of 19.6 ± 1.9 nm, 4.0 ± 0.4 nm and 1.8 ± 0.2 nm for graphene, hBN and MoS2, respectively. As we report little to no effect of friction on confinement in all three materials, any dramatic change in the slip length measured in experiments can be ascribed to a change in the viscosity, via the expression b = η/λ. The surface force apparatus, atomic force microscopy as well as scanning tunneling microscopy are some of the most promising techniques to investigate changes in the slip length and in the viscosity under confinement.25,46
at the interface: b = vslip/. Because at the interface the viscous shear stress η (with η the shear viscosity) is equal to the wall shear stress λvslip, the slip length can be expressed as the ratio between the viscosity and the friction coefficient:45b = η/λ. Using the experimental viscosity of bulk water (η = 10−3 Pa s), we obtain on average a slip length of 19.6 ± 1.9 nm, 4.0 ± 0.4 nm and 1.8 ± 0.2 nm for graphene, hBN and MoS2, respectively. As we report little to no effect of friction on confinement in all three materials, any dramatic change in the slip length measured in experiments can be ascribed to a change in the viscosity, via the expression b = η/λ. The surface force apparatus, atomic force microscopy as well as scanning tunneling microscopy are some of the most promising techniques to investigate changes in the slip length and in the viscosity under confinement.25,46
Additionally, measurements of the slip length in nanotubes using optical microscopy have shown that water essentially does not slip inside BN nanotubes, whereas it does slip inside carbon nanotubes, the slip length being larger in nanotubes with smaller radii.8 In the carbon nanotubes with the lowest curvature (100 nm in diameter), the reported slip length of about 23 nm compares favorably with our predicted value for graphene, 19.6 ± 1.9 nm. For BN nanotubes, no slip is detected above the detection limit of 5 nm, consistent with our prediction of 4.0 ± 0.4 nm. This comparison indicates that the difference in the slip length between carbon and BN nanomaterials may originate to a large extent from the different underlying corrugation of the energy landscapes of these materials. Nevertheless, the proclivity for adsorption of hydroxide ions may provide an additional reason for the smaller slip length on BN nanotubes.8,22,47–49 Carrying out experiments at different levels of pH would likely provide a definitive answer to this question.
It has been pointed out before that there is a large uncertainty over the simulated slip length of water on graphene or graphite, with values between 1 and 80 nm.50 In this work we have contributed to reduce this uncertainty, also given the good agreement with recent experiments for large carbon nanotubes, stressing the relevance of ab initio methods for studies of liquid/solid friction. Instead, using previously reported water/sheet interaction potentials, the slip length on the three materials obtained with FFMD may be up to five times larger compared to obtained AIMD (ESI†). Moreover, the mechanisms involved in water slippage may critically depend on the particular choice of the force field, as seen in the strong dependence of τF and λ on the values of the Lennard-Jones interaction and in the values of the partial charges (ESI†). This indicates that empirical interaction potentials, usually developed to describe bulk systems and sometimes combined with ad hoc mixing rules to describe interfaces, may be unreliable for the comparison of slippage mechanisms across different systems.
Overall, in this work we have unraveled the dependence of water slippage on the energy landscape corrugation and on the correlation time of the density fluctuations of water on three prototypical two-dimensional materials. Obtaining such fundamental mechanistic insights from 100 ps-long AIMDs through a systematic investigation of water slippage under different confinement regimes on different materials was not possible just a few years ago, when a first AIMD study of liquid/solid friction appeared,21 and has been made possible thanks to recent advances in computational power and algorithm developments.51 With the aim of designing materials with faster water slip one can envision different mechanisms to further reduce the friction, not only by decreasing the corrugation of the energy landscape but also by reducing the force correlation time. The recent discovery of a few thousands “exfoliatable” two-dimensional materials and the rise of van der Waals layered heterostructures may present a number of additional routes to further improve water slip.52–55 So far graphene is the two-dimensional material which exhibits the lowest friction to water. It remains to be seen whether water slip on newly proposed two-dimensional materials or van der Waals heterostructures can be faster than on graphene. Furthermore, inclusion of non-additive many-body effects may reveal new routes to enhance slp, as a result of repulsive dispersion forces arising from confinement.56,57 It will be interesting to extend AIMD studies to other domains of nanofluidics including problems in osmotic energy conversion, in a time where also ionic transport is accessible experimentally under extreme confinement regimes.
Materials and methods
Electronic structure and ab initio molecular dynamics
The electronic structure problem was solved using DFT with the CP2K code58 with the optB88-vdW density functional. The optB88-vdW functional has been shown to predict accurate interlayer binding and interlayer distances in graphite and bulk hBN,59 to give a sufficiently good description of the structure of bulk liquid water60,61 and also to accurately capture the relative stability of a water monomer on graphene and hBN on different adsorption sites compared to reference diffusion monte carlo data.62,63 Further details on the electronic structure are reported in the ESI.†AIMD simulations within the Born–Oppenheimer approximation were performed in the canonical ensemble for about 100 ps at 330 K to partly compensate for the overstructuring of water with the optB88-vdW functional.60,61 The initial atomic configurations for each system were obtained from pre-equilibrated FFMD trajectories. Additional AIMD and FFMD simulations ensured that our results were not affected by the finite size and time-scales accessible to AIMD and by the type of ensemble chosen (ESI†).
Structural details of the water/sheet interface
The graphene/water and hBN/water interfaces have been modeled using 10 × 6 orthorhombic cells about 2.5 × 2.5 nm2 large and the MoS2/water interface has been modelled using 8 × 4 orthorhombic cells approximately 2.5 × 2.2 nm2 large. The graphene, hBN and MoS2 lattice constants are about 2.46, 2.50 and 3.15 Å, respectively. The number of confined water molecules was varied between 100 and 500, and the systems spontaneously equilibrated at a given confinement width, between about 0.9 nm and 3 nm, as all atoms were allowed to move. Additionally, simulations on up to ca. 3 nm-thick supported water films were carried out, where water is at the interface with the sheets on one side and with vacuum on the other side. Further details on the structure of supported water films and of water under confinement, as well as tests on the size of our simulations are included in the ESI.† These tests also include a comparison on the friction coefficient computed for thinner supported water films, as reported in ref. 21. Overall, they confirm that the results presented so far on water slippage are not altered by changing the lateral cell dimensions and the number of water molecules present in our simulations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for discussions with Yasmine Al-Hamdani and Vladimir Rybkin. GT is supported by the SNSF project PZ00P2_179964. LJ is supported by the ANR through Project ANR-16-CE06-0004-01 NECtAR, and by the Institut Universitaire de France. This research was also supported by the NCCR MARVEL, funded by the SNSF through the “Inspire Potentials” fellowship. We also thank the Swiss National Supercomputer Centre (CSCS) and PRACE for awarding us access to Piz Daint, Switzerland, through projects s826 and pr66, respectively.References
- L. Bocquet and E. Charlaix, Chem. Soc. Rev., 2010, 39, 1073–1095 RSC.
- A. Siria, M.-L. Bocquet and L. Bocquet, Nat. Rev. Chem., 2017, 1, 0091 CrossRef CAS.
- M. Macha, S. Marion, V. V. R. Nandigana and A. Radenovic, Nat. Rev. Mater., 2019, 1 Search PubMed.
- F. Wang, Y. You, X. Jin and R. Joshi, Nanoscale, 2018, 10, 21625–21628 RSC.
- H. Park, J. Kamcev, L. Robeson, M. Elimelech and B. Freeman, Science, 2017, 356, eaab0530 CrossRef.
- L. Wang, M. S. Boutilier, P. R. Kidambi, D. Jang, N. G. Hadjiconstantinou and R. Karnik, Nat. Nanotechnol., 2017, 12, 509–522 CrossRef CAS PubMed.
- A. Esfandiar, B. Radha, F. Wang, Q. Yang, S. Hu, S. Garaj, R. Nair, A. Geim and K. Gopinadhan, Science, 2017, 358, 511–513 CrossRef CAS PubMed.
- E. Secchi, S. Marbach, A. Niguès, D. Stein, A. Siria and L. Bocquet, Nature, 2016, 537, 210–213 CrossRef CAS PubMed.
- A. Michaelides, Nature, 2016, 537, 171–172 CrossRef CAS PubMed.
- A. Striolo, A. Michaelides and L. Joly, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 533–556 CrossRef CAS PubMed.
- D. M. Huang, C. Sendner, D. Horinek, R. R. Netz and L. Bocquet, Phys. Rev. Lett., 2008, 101, 226101 CrossRef PubMed.
- A. Govind Rajan, V. Sresht, A. A. Pàdua, M. S. Strano and D. Blankschtein, ACS Nano, 2016, 10, 9145–9155 CrossRef CAS PubMed.
- A. Govind Rajan, M. S. Strano and D. Blankschtein, Nano Lett., 2019, 19, 1539–1551 CrossRef CAS.
- H. Yoshida, V. Kaiser, B. Rotenberg and L. Bocquet, Nat. Commun., 2018, 9, 1496 CrossRef PubMed.
- M. Ma, F. Grey, L. Shen, M. Urbakh, S. Wu, J. Z. Liu, Y. Liu and Q. Zheng, Nat. Nanotechnol., 2015, 10, 692–695 CrossRef CAS PubMed.
- M. Ma, G. Tocci, G. Aeppli and A. Michaelides, Nat. Mater., 2015, 15, 66–71 CrossRef PubMed.
- W. Cao, J. Wang and M. Ma, J. Phys. Chem. Lett., 2019, 10, 7251–7258 CrossRef CAS PubMed.
- V. Quaranta, J. Behler and M. Hellström, J. Phys. Chem. C, 2018, 123, 1293–1304 CrossRef.
- P. Rowe, G. Csányi, D. Alfè and A. Michaelides, Phys. Rev. B, 2018, 97, 054303 CrossRef.
- S. Seiler, C. E. Halbig, F. Grote, P. Rietsch, F. Börrnert, U. Kaiser, B. Meyer and S. Eigler, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- G. Tocci, L. Joly and A. Michaelides, Nano Lett., 2014, 14, 6872 CrossRef CAS PubMed.
- L. Joly, G. Tocci, S. Merabia and A. Michaelides, J. Phys. Chem. Lett., 2016, 7, 1381–1386 CrossRef CAS PubMed.
- J. Feng, M. Graf, K. Liu, D. Ovchinnikov, D. Dumcenco, M. Heiranian, V. Nandigana, N. R. Aluru, A. Kis and A. Radenovic, Nature, 2016, 536, 197–200 CrossRef CAS PubMed.
- Q. Xie, M. A. Alibakhshi, S. Jiao, Z. Xu, M. Hempel, J. Kong, H. G. Park and C. Duan, Nat. Nanotechnol., 2018, 13, 238 CrossRef CAS PubMed.
- S. F. Mertens, A. Hemmi, S. Muff, O. Gröning, S. De Feyter, J. Osterwalder and T. Greber, Nature, 2016, 534, 676 CrossRef CAS PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- G. Cicero, J. C. Grossman, E. Schwegler, F. Gygi and G. Galli, J. Am. Chem. Soc., 2008, 130, 1871–1878 CrossRef CAS PubMed.
- A. P. Willard and D. Chandler, J. Phys. Chem. B, 2010, 114, 1954–1958 CrossRef CAS PubMed.
- J.-L. Barrat and L. Bocquet, Faraday Discuss., 1999, 112, 119–128 RSC.
- M. Neek-Amal, F. M. Peeters, I. V. Grigorieva and A. K. Geim, ACS Nano, 2016, 10, 3685–3692 CrossRef CAS PubMed.
- C. J. Mundy, S. Balasubramanian, J. I. Siepmann and M. L. Klein, Faraday Discuss., 1996, 104, 17–36 RSC.
- L. Bocquet and J. L. Barrat, Phys. Rev. E, 1994, 49, 3079–3092 CrossRef CAS PubMed.
- J. Ewen, D. Heyes and D. Dini, Friction, 2018, 6, 349–386 CrossRef CAS.
- L. Bocquet and J. L. Barrat, J. Chem. Phys., 2013, 139, 044704 CrossRef PubMed.
- H. Oga, Y. Yamaguchi, T. Omori, S. Merabia and L. Joly, J. Chem. Phys., 2019, 151, 054502 CrossRef.
- C. C. Silva, M. Iannuzzi, D. A. Duncan, P. T. Ryan, K. T. Clarke, J. T. Küchle, J. Cai, W. Jolie, C. Schlueter and T.-L. Lee, J. Phys. Chem. C, 2018, 122, 18554–18561 CrossRef CAS.
- D. Golze, M. Iannuzzi, M.-T. Nguyen, D. Passerone and J. Hutter, J. Chem. Theor. Comput., 2013, 9, 5086–5097 CrossRef CAS PubMed.
- K. Falk, F. Sedlmeier, L. Joly, R. R. Netz and L. Bocquet, Nano Lett., 2010, 10, 4067–4073 CrossRef CAS PubMed.
- J.-P. Hansen and I. R. McDonald, Theory of simple liquids, Elsevier, 1990 Search PubMed.
- U. Raviv, P. Laurat and J. Klein, Nature, 2001, 413, 51–54 CrossRef CAS PubMed.
- J. Gao, R. Szoszkiewicz, U. Landman and E. Riedo, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 115415 CrossRef.
- D. Ortiz-Young, H.-C. Chiu, S. Kim, K. Voïtchovsky and E. Riedo, Nat. Commun., 2013, 4, 24282 Search PubMed.
- A. Zaragoza, M. Gonzalez, L. Joly, I. López-Montero, M. Canales, A. Benavides and C. Valeriani, Phys. Chem. Chem. Phys., 2019, 21, 13653–13667 RSC.
- L. Bocquet and J. L. Barrat, Soft Matter, 2007, 3, 685–693 RSC.
- H. Weiss, H.-W. Cheng, J. Mars, H. Li, C. Merola, F. U. Renner, V. Honkimäki, M. Valtiner and M. Mezger, Langmuir, 2019, 35(51), 16679–16692 CrossRef CAS PubMed.
- A. Siria, P. Poncharal, A.-L. Biance, R. Fulcrand, X. Blase, S. T. Purcell and L. Bocquet, Nature, 2013, 494, 455 CrossRef CAS PubMed.
- B. Grosjean, C. Pean, A. Siria, L. Bocquet, R. Vuilleumier and M.-L. Bocquet, J. Phys. Chem. Lett., 2016, 7, 4695–4700 CrossRef CAS PubMed.
- B. Grosjean, M.-L. Bocquet and R. Vuilleumier, Nat. Commun., 2019, 10, 1656 CrossRef PubMed.
- S. K. Kannam, B. D. Todd, J. S. Hansen and P. J. Daivis, J. Chem. Phys., 2013, 138, 094701 CrossRef PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 4, 15–25 Search PubMed.
- N. Mounet, M. Gibertini, P. Schwaller, D. Campi, A. Merkys, A. Marrazzo, T. Sohier, I. E. Castelli, A. Cepellotti, G. Pizzi and N. Marzari, Nat. Nanotechnol., 2018, 13, 246 CrossRef CAS PubMed.
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419–425 CrossRef CAS PubMed.
- W. Ouyang, I. Azuri, D. Mandelli, A. Tkatchenko, L. Kronik, M. Urbakh and O. Hod, J. Chem. Theory Comput., 2020, 16(1), 666–676 CrossRef PubMed.
- J. Wang, W. Cao, Y. Song, C. Qu, Q. Zheng and M. Ma, Nano Lett., 2019, 19, 7735–7741 CrossRef CAS PubMed.
- J. Hermann, R. A. DiStasio Jr. and A. Tkatchenko, Chem. Rev., 2017, 117, 4714–4758 CrossRef CAS PubMed.
- M. Sadhukhan and A. Tkatchenko, Phys. Rev. Lett., 2017, 118, 210402 CrossRef PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103 CrossRef CAS.
- G. Graziano, J. Klimeš, F. Fernandez-Alonso and A. Michaelides, J. Phys.: Condens. Matter, 2012, 24, 424216 CrossRef PubMed.
- C. Zhang, J. Wu, G. Galli and F. Gygi, J. Chem. Theory Comput., 2011, 7, 3054 CrossRef CAS PubMed.
- M. Del Ben, J. Hutter and J. VandeVondele, J. Chem. Phys., 2015, 143, 054506 CrossRef PubMed.
- J. G. Brandenburg, A. Zen, M. Fitzner, B. Ramberger, G. Kresse, T. Tsatsoulis, A. Grüuneis, A. Michaelides and A. Dario, J. Phys. Chem. Lett., 2019, 10, 358–368 CrossRef PubMed.
- Y. S. Al-Hamdani, M. Ma, D. Alfè, O. A. von Lilienfeld and A. Michaelides, J. Chem. Phys., 2015, 142, 181101 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Further computational and theoretical details, as well as tests on the structure and dynamics of water at the interface with graphene, hBN and MoS2 from force field and ab initio simulations. See DOI: 10.1039/D0NR02511A |
| ‡ kB being Boltzmann constant and T room temperature. |
| § The magnitude of the peaks for the smallest confinement widths (Fig. 1b–d) and the presence of one or two additional peaks beyond the contact layer (Fig. 1f–h) may be marginally affected by the number of water molecules present in our simulations, and as such care must be taken while interpreting these differences in the density profiles. |
| This journal is © The Royal Society of Chemistry 2020 |