
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Refined construction of antibody-targeted nanoparticles leads to superior antigen binding and enhanced delivery of an entrapped payload to pancreatic cancer cells†
Michelle K.
Greene
a,
João C. F.
Nogueira
b,
Shannon R.
Tracey
a,
Daniel A.
Richards
b,
William J.
McDaid
a,
James F.
Burrows
c,
Katrina
Campbell
d,
Daniel B.
Longley
a,
Vijay
Chudasama
 *b and
Christopher J.
Scott
*a
*b and
Christopher J.
Scott
*a
aThe Patrick G Johnston Centre for Cancer Research, School of Medicine, Dentistry and Biomedical Sciences, Queen's University Belfast, Belfast, UK. E-mail: c.scott@qub.ac.uk
bDepartment of Chemistry, University College London, London, UK. E-mail: v.chudasama@ucl.ac.uk
cSchool of Pharmacy, Queen's University Belfast, Belfast, UK
dInstitute for Global Food Security, School of Biological Sciences, Queen's University Belfast, Belfast, UK
First published on 14th May 2020
Abstract
Antibody-targeted nanoparticles have shown exceptional promise as delivery vehicles for anticancer drugs, although manufacturability challenges have hampered clinical progress. These include the potential for uncontrolled and random antibody conjugation, resulting in masked or inactive paratopes and unwanted Fc domain interactions. To circumvent these issues, we show that the interchain disulfide of cetuximab F(ab) may be selectively re-bridged with a strained alkyne handle, to permit ‘click’ coupling to azide-capped nanoparticles in a highly uniform and oriented manner. When compared to conventional carbodiimide chemistry, this conjugation approach leads to the generation of nanoparticles with a higher surface loading of cetuximab F(ab) and with markedly improved ability to bind to the target epidermal growth factor receptor. Moreover, we show that entrapment of a camptothecin payload within these nanoparticles can enhance drug targeting to antigen-expressing pancreatic cancer cells, resulting in superior cytotoxicity versus the conventional nanoformulation. Collectively, this work highlights the critical need to develop refined methods for the construction of targeted nanoparticles that will accelerate their clinical translation through improved performance and manufacturability.
Introduction
Nanoparticles can enhance the bioavailability of drug agents, particularly if they possess stability or solubility issues. Within the cancer therapeutics field, the ability to exploit nanoparticles as drug delivery vehicles has been extensively explored, where they can passively locate to tumours by diffusing through defective endothelial cell-to-cell junctions in the neovasculature; named the ‘enhanced permeability and retention’ (EPR) effect.1–3 However, reliance on passive targeting may not be sufficient to realise the full clinical application of chemotherapy-loaded nanoparticles, and so surface functionalisation with targeting ligands is an attractive approach to enhance cellular binding and uptake.4 Despite a wealth of preclinical data supporting this conceptual approach,5–7 none have successfully translated through to clinical approval.There are several issues with these modalities that preclude their clinical adoption. For example, one major issue arises from the common conjugation chemistries that lead to random and uncontrolled attachment of the targeting ligands (e.g. antibodies) to the surface of drug-loaded nanoparticles, such as carbodiimide coupling between carboxylic acids and primary amines.8–10 Such coupling approaches are inefficient and afford minimal control over the orientation of antibodies due to their high amine content, leading to heterogeneous nanoparticles with poor batch-to-batch reproducibility. These attributes can obstruct clinical progress, since they do not conform to regulatory standards and are likely to pose challenges during upscaled production at the industrial level.
Furthermore, the nature of the targeting ligand may also explain the limited success of targeted nanomedicines. In formulations containing antibodies as the targeting moiety, most approaches have employed the full immunoglobulin molecule, which enhances the diameter of the nanoparticles and can lead to significant surface crowding that may adversely affect stability and impair target binding through steric hindrance effects.11,12 Moreover, the Fc domain of a fully intact immunoglobulin is susceptible to immune clearance mechanisms, which can markedly reduce the circulation half-life of nanoparticles.13 To overcome some of these drawbacks, researchers have begun to explore the use of antibody fragments and alternative ligand formats.14 By moving away from traditional immunoglobulin G ligands, researchers have been able to infer significant advantages to emerging nanoformulations, including reduced susceptibility to immune clearance, prolonged circulation, increased tumour uptake and the potential for enhanced coverage of paratopes on the surface of nanoparticles due to the comparatively smaller dimensions of fragments.13,15,16 Of the various antibody fragments that have been explored as nanoparticle-directing ligands, F(ab) fragments remain the most commonly employed due to their accessibility and simplicity. These fragments can be readily generated from full antibodies without the need for further engineering that is often required with other scaffold fragments. Additionally, F(ab) fragments retain strong binding to their target and contain a single disulfide bond that provides opportunities for site-selective conjugation technologies.
Here, we outline a next-generation approach for the site-specific functionalisation of nanoparticles with F(ab) fragments derived from the epidermal growth factor receptor (EGFR) antibody cetuximab (CTX 1). We show that these fragments may be chemically manipulated via the selective insertion of a strained alkyne handle, to facilitate ‘click’ conjugation to the surface of complementary azide-capped nanoparticles. Importantly, these nanoparticles display superior EGFR binding compared to those synthesised via conventional carbodiimide chemistry, which we exploit for the enhanced delivery of nanoformulated camptothecin (CPT) to pancreatic cancer cells. Although several issues must yet be resolved before targeted nanoparticles can be adopted into clinical practice, this work outlines a strategy for overcoming one of the most prominent challenges that have obstructed their translation to date.
Experimental
General
All reagents and starting materials were obtained from chemical suppliers, unless specifically stated otherwise, and were used as received. Reactions were monitored by thin layer chromatography using pre-coated SIL G/UV 254 plates (VWR). Flash chromatography was carried out automatically using a BioTage Isolera with KP-Snap columns. NMR spectra were recorded using a Bruker AC600 spectrometer (600 MHz). Chemical shifts (δ) are given in ppm units relative to the solvent reference and coupling constants (J) are measured in Hertz. Proton (1H) NMR multiplicities are shown as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (double doublet), dt (double triplet), etc. HMBC, HSQC and DEPT were employed to aid with accurate assignments. Infrared spectra were recorded on a PerkinElmer Spectrum 100 FTIR spectrometer (ATR mode). High and low resolution mass spectrometry of organic molecules was provided by the EPSRC Mass Spectrometry facility at Swansea using a LTQ Orbitrap XL.UV-Vis spectroscopy
Protein concentrations were determined photometrically using a Nanodrop 2000C (Thermo Fisher Scientific), and UV-Vis spectra were obtained using a Varian Cary 100 Bio UV-Vis spectrophotometer operating at 21 °C.SDS-PAGE gels
Non-reducing 12% acrylamide gels were made using standard procedures. A 4% stacking gel was utilised. Samples (15 μM) were mixed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 with a 5× R-250 Dye SDS-loading buffer, heated for 3 min at 75 °C and loaded onto the gel with a total volume of 5 μL. Samples were run at constant current (30 mA) for 40 min in 1× SDS running buffer and stained with Coomassie Brilliant Blue.
:5 with a 5× R-250 Dye SDS-loading buffer, heated for 3 min at 75 °C and loaded onto the gel with a total volume of 5 μL. Samples were run at constant current (30 mA) for 40 min in 1× SDS running buffer and stained with Coomassie Brilliant Blue.
Protein liquid chromatography-mass spectrometry (LC-MS)
All proteins were prepared for analysis by repeated diafiltration into 50 mM ammonium acetate buffer at pH 6.9 using VivaSpin 10000 MWCO sample concentrators (GE Healthcare) to a concentration of 2 μM. Samples were submitted to the UCL Chemistry Mass Spectrometry Facility at the Chemistry Department, UCL, for analysis on the Agilent 6510 QTOF LC-MS system. 10 μL of each sample was injected onto a PLRP-S, 1000A, 8 mM, 150 mm × 2.1 mm column, which was maintained at 60 °C. Flow rate was set at 0.6 mL min−1. Solvent A was H2O (0.1% formic acid), solvent B was MeCN (0.1% formic acid) and separation was achieved using a gradient elution. The column effluent was continuously electrosprayed into the capillary ESI source of the Agilent 6510 QTOF mass spectrometer and ESI mass spectra were acquired in positive ESI mode using the m/z range 1000–8000 in profile mode. The raw data was converted to zero charge mass spectra using maximum entropy deconvolution algorithms using MassHunter software (version B.07.00).
Formulation of nanoparticles
Nanoparticles were synthesised in 20 mg batches consisting of either (1) poly(lactic-co-glycolic acid) (PLGA) 502H (Sigma), (2) a 3:1 blend of PLGA 502H:PLGA-poly(ethylene glycol) (PEG)-N-hydroxysuccinimide (NHS) (PolySciTech) or (3) a 3:1 blend of PLGA 502H:PLGA-PEG-azide (PolySciTech). The polymers were dissolved in 1 mL of dichloromethane (DCM) and then injected into 7 mL of 2.5% w/v polyvinyl alcohol (PVA) in 50 mM 2-(N-morpholino) ethanesulfonic acid hydrate buffer at pH 5 (MES), under moderate stirring on ice. The resultant emulsion was subjected to probe sonication for 90 s on ice using a Model 120 sonic dismembrator (Fisher Scientific) set at an amplitude of 50% and left stirring overnight at ambient temperature to facilitate DCM evaporation. Nanoparticles were then purified by repeated centrifugation (3×) at 17000g for 20 min at 4 °C. Probe sonication was used to resuspend nanoparticle pellets in MES between centrifugations. Where required, 40 μL of 5 mg mL−1 CPT in dimethyl sulfoxide (DMSO), or 100 μL of 2 mg mL−1 rhodamine 6G in DCM, was added to the organic phase during nanoparticle synthesis.
Surface functionalisation of nanoparticles
Surface functionalisation was achieved by adding 1 nmole of CTX F(ab) 2 or CTX F(ab) 9 to 1 mg of nanoparticles that were suspended in 1 mL of MES. After gentle stirring for 2 h at ambient temperature, nanoparticles were washed by repeated centrifugation (2×) at 12000g for 20 min at 4 °C. Probe sonication was used to resuspend nanoparticle pellets in phosphate buffered saline (PBS) between centrifugations.
Characterisation of nanoparticles
Nanoparticles were suspended at 100–200 μg polymer mL−1 in PBS prior to analysis of size and polydispersity index (PDI) using a NanoBrook Omni (Brookhaven Instruments Corp). To quantify the loading of targeting ligands on nanoparticles, nude NHS NP and nude azide NP (either non-, rhodamine 6G- or CPT-loaded) were suspended at 2 mg polymer mL−1 in PBS and used as a diluent to construct a series of CTX F(ab) 2 and CTX F(ab) 9 standards, respectively. Likewise, native CTX F(ab) NP and modified CTX F(ab) NP [disulfide] (either non-, rhodamine 6G- or CPT-loaded) were also suspended at 2 mg polymer mL−1 in PBS. After adding each sample to replicate wells of a 96-well microtiter plate, a Micro BCA™ kit (Thermo Fisher Scientific) was used to assess protein content as per the manufacturer's instructions. Loading was then quantified by reference to the standard curve. To quantify CPT entrapment within nanoparticles, nude NHS NP and nude azide NP (non-loaded) were dissolved at 4 mg polymer mL−1 in a 1:1 mixture of acetonitrile (ACN):DMSO and used as a diluent to construct a series of CPT standards. Likewise, nude NHS CPT NP and nude azide CPT NP were also dissolved at 4 mg polymer mL−1 in a 1:1 mixture of ACN:DMSO. After adding 50 μL of each sample to replicate wells of a black 96-well microtiter plate, fluorescence was measured at excitation and emission wavelengths of 330 and 460 nm, respectively. Entrapment was then quantified by reference to the standard curve.
Fluorescence-linked immunosorbent assay (FLISA)
High-binding black 96-well microtiter plates (Greiner Bio-One) were initially coated with the target antigen, where 100 μL of 0.5 μg mL−1 human EGFR Fc chimera protein (EGFR-Fc; Sino Biological) in PBS was added to each well and incubated overnight at 4 °C. All subsequent steps were performed at ambient temperature and all concentrations of nanoparticles, EGFR-Fc and CTX 1 are provided in the figure legends. After washing (3×) the plates in 0.1% v/v Tween 20 in PBS (PBST), 150 μL of 1% w/v bovine serum albumin in PBS (block buffer) was added to each well for 1 h. Following further washes (3×) in PBST, 100 μL of fluorescent rhodamine 6G-loaded nanoparticles in block buffer were added to each well for 2 h. Lastly, the plates were washed (6×) in PBST, 50 μL of a 1:1 mixture of ACN:DMSO was added to each well and fluorescence was measured at excitation and emission wavelengths of 516 and 557 nm, respectively, using a Cytation 5 plate reader (Biotek). For studies examining the EGFR targeting specificity of nanoparticles, several modifications were applied to this protocol, including: (1) an extra step prior to nanoparticle addition, where 100 μL of free CTX 1 in block buffer was added to each well for 2 h, followed by washing (3×), (2) pre-incubation of nanoparticles with free EGFR-Fc under gentle agitation for 30 min in block buffer, of which 100 μL was then added to each well for 2 h and (3) pre-mixing of nanoparticles with free CTX 1 in block buffer, of which 100 μL was then added to each well for 2 h.
Surface plasmon resonance (SPR)
Nanoparticle binding to EGFR-Fc was analysed on a Biacore Q instrument (GE Healthcare). All experiments were performed in HBS-EP running buffer (GE Healthcare) at 25 °C. At first, EGFR-Fc was immobilised on a CM5 sensor chip (GE Healthcare) via carbodiimide coupling, where the following solutions were sequentially injected at a flow rate of 10 μL min−1 for 7 min each: (1) 0.4 M EDC and 0.1 M NHS to activate carboxyl groups on the sensor chip surface, (2) 20 μg mL−1 EGFR-Fc in 10 mM sodium acetate buffer at pH 4.5 and (3) 1 M ethanolamine hydrochloride at pH 8.5 to quench residual NHS esters on the sensor chip surface. Thereafter, 10 mg polymer mL−1 nanoparticles in PBS were injected over the EGFR-Fc-coated chip at a flow rate of 20 μL min−1 for 15 s. Between each injection of nanoparticles, the chip surface was regenerated with 25 mM sodium hydroxide at a flow rate of 20 μL min−1 for 15 s. Where SPR data is reported as a relative response, this was calculated as the difference in absolute resonance units (RU) from 10 s before, to 30 s after, each injection.Cell culture
BxPC-3, MIA PaCa-2 and PANC-1 human pancreatic cancer cell lines were purchased from the American Type Culture Collection and maintained in 5% CO2 at 37 °C in a humidified incubator. BxPC-3 cells were cultured in RPMI supplemented with 1 mM sodium pyruvate, 50 units mL−1 penicillin, 50 μg mL−1 streptomycin, 10 mM HEPES and 10% v/v foetal bovine serum (FBS). Both MIA PaCa-2 and PANC-1 cells were cultured in DMEM supplemented with 1 mM sodium pyruvate, 50 units mL−1 penicillin, 50 μg mL−1 streptomycin and 10% v/v FBS.Binding of nanoparticles to EGFR-expressing cells
MIA PaCa-2 and PANC-1 cells were seeded at 10000 per well on black 96-well plates. Following overnight adherence, cells were chilled in culture media for 20 min at 4 °C and maintained under these conditions during subsequent treatment with rhodamine 6G-loaded nanoparticles at 600 μg polymer mL−1 (MIA PaCa-2) or 800 μg polymer mL−1 (PANC-1) for a further 45 min. Cells were then washed (3×) in PBS and lysed by adding 50 μL of 0.5% v/v Triton X-100 in 0.2 M sodium hydroxide to each well. Fluorescence was measured at excitation and emission wavelengths of 516 and 557 nm, respectively, using a Cytation 5 plate reader (BioTek).
Flow cytometry
PANC-1 and BxPC-3 cells were seeded at 400000 per well on 6-well plates. Following overnight adherence, cells were washed in PBS and incubated in 0.1% w/v EDTA in PBS for 10 min at 37 °C. Detached cells from each well were centrifuged at 200g for 5 min and suspended in culture media. After repeating this centrifugation-resuspension wash step, cells were incubated in culture media containing 500 μg polymer mL−1 nanoparticles for 1 h at 4 °C under gentle agitation. Cells were then centrifuged at 200g for 5 min and incubated in 5% v/v FBS in PBS (FACS buffer) containing 5 μg mL−1 FITC-labelled anti-human EGFR or anti-mouse IgG2a isotype control antibodies (Santa Cruz Biotechnology) for 45 min at 4 °C. Following the addition of FACS buffer, cells were centrifuged at 200g for 5 min and this wash cycle was then repeated. Cells were suspended in FACS buffer prior to measurement of FITC fluorescence on a FACSCalibur flow cytometer (Becton Dickinson). A minimum of 30000 events per sample were acquired.
Clonogenic assay
MIA PaCa-2 cells were seeded at 250000 per well on 6-well plates. Following overnight adherence, cells were chilled in culture media for 10 min at 4 °C and maintained under these conditions during subsequent treatments. Initially, cells were treated with PBS or 200 μg mL−1 free CTX 1 for 15 min, followed by the addition of 500 ng mL−1 free CPT, or drug equivalents in nanoencapsulated format, for a further 45 min. Cells were then washed (5×) in PBS and incubated in fresh culture media at 37 °C. On the following day, cells were detached from culture plasticware, seeded at 250 per well on 6-well plates and incubated at 37 °C for several days to allow colony formation. At study endpoint, cells were stained in 0.4% w/v crystal violet solution for 10 min and the number of colonies containing >50 cells were enumerated.
Data analysis
GraphPad Prism software (version 6.0c) was used to graph data and perform statistical analyses. Student's t-test was used to determine statistical significance in datasets comprised of two groups. One-way analysis of variance with Tukey's post hoc test was used to determine statistical significance in datasets comprised of three or more groups. Statistical significance was defined as follows: *P < 0.05, **P < 0.01, ***P < 0.001. All data within bar graphs and dose response curves was presented as mean ± standard error of the mean. FlowJo software (version 10.4.2) was used to construct flow cytometry histograms.Results and discussion
Generation of targeting ligands for downstream conjugation to nanoparticles
CTX 1 is an EGFR-targeted monoclonal antibody approved for the treatment of KRAS wild-type colorectal carcinoma and squamous cell carcinoma of the head and neck. Binding of the antibody to the receptor on the tumour cell can block proliferation, but many tumours harbour mutations in EGFR itself or downstream signalling proteins such as KRAS, rendering the cells resistant to CTX 1. Nonetheless, we hypothesised that EGFR, which is frequently overexpressed on the surface of tumour cells, could still be exploited for targeted drug delivery even in KRAS mutant cancers; thus CTX 1 was chosen as a model ligand for this study. Synthesis of our targeted nanoparticles began with papain digestion of CTX 1 and subsequent protein A purification to generate CTX F(ab) 2 with a 68% yield (Fig. 1A). To impart ‘click’ reactivity onto CTX F(ab) 2, we chose to chemically manipulate the interchain disulfide for several reasons. Firstly, since each F(ab) contains only one interchain disulfide, this would ensure that modification was exclusively confined to the same site within each fragment, providing a single locus for homogeneous attachment to nanoparticles. Secondly, as the interchain disulfide is located distal to the paratope, this would minimise interference with target binding. To achieve this, a heterobifunctional linker composed of a dibromopyridazinedione bearing a strained alkyne was synthesised (PD 8; Fig. 1B). Dibromopyridazinediones are considered an excellent platform for functional re-bridging of disulfide bonds due to their stability profile and exclusive thiol selectivity, thus allowing site-specific installation of the linker across the reduced interchain disulfide of CTX F(ab) 2 (CTX F(ab) 9; Fig. 1C). To confirm the integrity of the strained alkyne functionality after conjugation to the F(ab), CTX F(ab) 9 was incubated with azide-functionalised Alexa Fluor-488 to yield fluorescent CTX F(ab) 10 (Fig. 1C). Successful strain-promoted alkyne–azide cycloaddition (SPAAC) reaction was confirmed via SDS-PAGE, UV-Vis spectroscopy and LC-MS (ESI Fig. S6–S8†).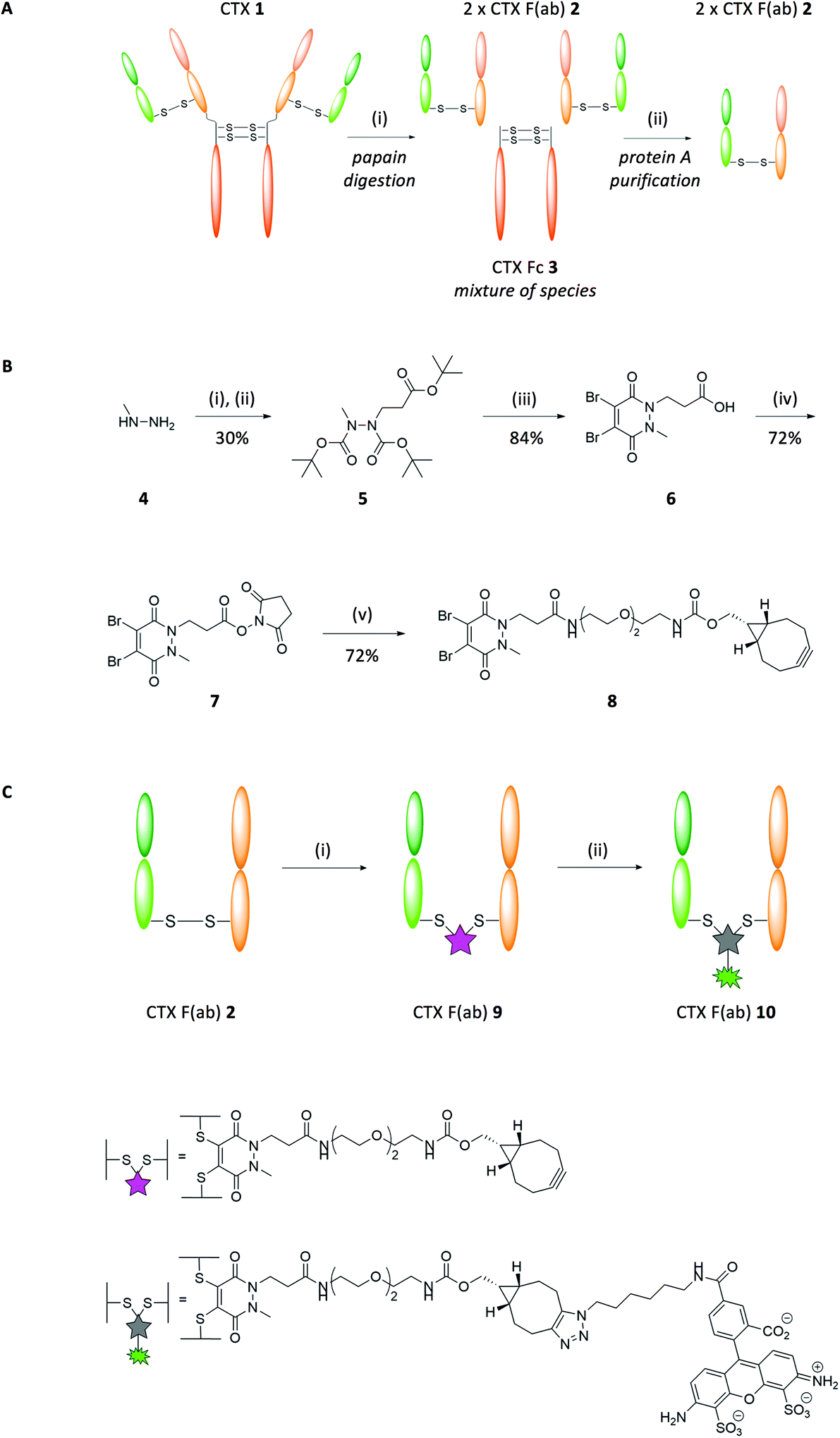 | ||
| Fig. 1 Purification and functional re-bridging of the F(ab) domains of CTX 1. (A) Digestion protocol applied to CTX 1 to yield CTX F(ab) 2. Reagents and conditions: (i) Papain, digest buffer pH 6.8, 37 °C, 5 h. (B) Synthesis route of PD 8. Reagents and conditions: (i) Boc anhydride, i-PrOH, CH2Cl2, 21 °C, 16 h; (ii) tert-butyl acrylate, i-PrOH, 60 °C, 24 h; (iii) dibromomaleic acid, AcOH, reflux, 5 h; (iv) DCC, NHS, THF, 21 °C, 16 h; (v) BCN(endo)-PEG2-NH2, MeCN, 21 °C, 16 h. (C) Modification of CTX F(ab) 2 with PD 8 and subsequent ‘click’ coupling with Alexa Fluor-488-N3. Reagents and conditions: (i) PD 8, TCEP·HCl, borate buffer pH 8 (5 mM EDTA), 21 °C, 16 h; (ii) Alexa Fluor-488-N3, PBS pH 7.4, 21 °C, 2 h. | ||
Nanoparticle formulation and characterisation
Having successfully re-bridged the interchain disulfide of CTX F(ab) 9 with a strained alkyne-functionalised linker, we next explored whether the fragment could be site-selectively conjugated to the surface of polymeric nanoparticles. We prepared PLGA-based nanoparticles incorporating surface accessible azide moieties and a PEG corona to limit non-specific protein adsorption, using a 3:1 polymer blend of PLGA 502H:PLGA-PEG-azide that yielded nanoparticles of approximately 200 nm in diameter with a monodisperse size distribution (Table 1) (nude azide NP). The presence of surface-exposed azide moieties on these nanoparticles facilitated site-specific ‘click’ coupling to the complementary alkyne entity of CTX F(ab) 9, which was enabled by gentle mixing of both components for 2 h at ambient temperature (modified CTX F(ab) NP [disulfide]) (Fig. 2A). These reaction conditions bypass potential toxicity associated with copper catalysis, which has been employed in many similar efforts to functionalise nanoparticles via alkyne-azide cycloaddition.17–19 Characterisation of these nanoparticles revealed a marginal increase in diameter versus the nude azide NP control and a protein content of 16.1 ± 1.5 μg mg−1 polymer, indicating that conjugation had been achieved (Table 1).
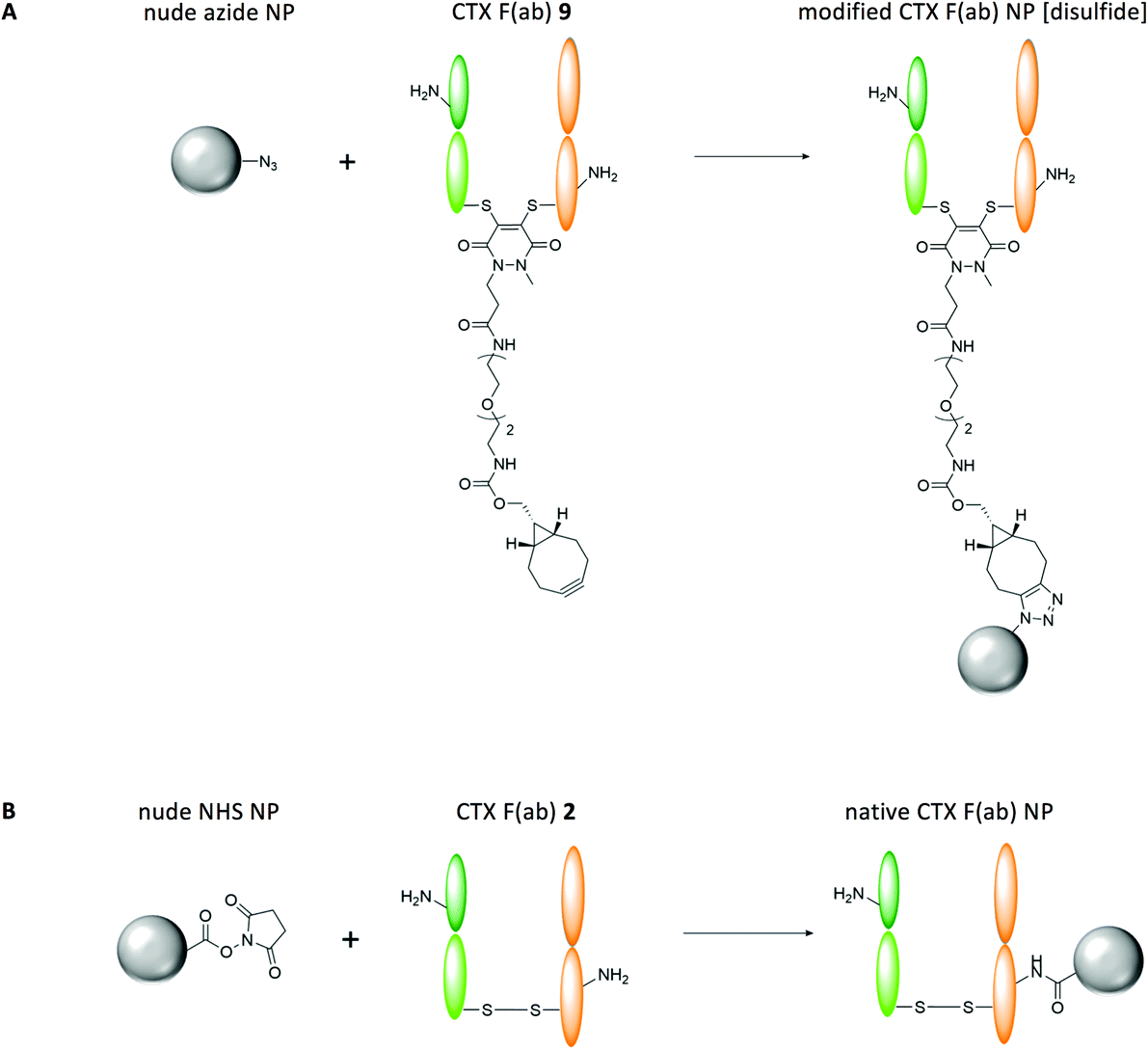 | ||
| Fig. 2 Overview of control and EGFR-targeted nanoformulations. (A) Modified CTX F(ab) NP [disulfide] were synthesised through site-specific conjugation of azide moieties on the surface of nanoparticles to strained alkyne handles within CTX F(ab) 9. The presence of a single ‘click’-reactive alkyne within CTX F(ab) 9, at a position that lies distal to the paratope, facilitated homogeneous coupling of the fragments to the surface of nanoparticles in a highly optimised conformation. (B) Native CTX F(ab) NP were synthesised by reacting NHS esters on the surface of nanoparticles with the amine side chains of lysine residues throughout CTX F(ab) 2. Due to the high abundance of lysines within CTX F(ab) 2, coupling via this conventional approach proceeded in a random and heterogeneous manner, affording minimal control over the orientation of the fragments on the surface of nanoparticles. | ||
| Nanoformulation | Polymer | Diametera (nm) | PDIa | F(ab) conjugateda,b (μg mg−1 polymer) | CPT entrappeda (μg mg−1 polymer) |
|---|---|---|---|---|---|
| a Data expressed as mean ± standard deviation. b Equimolar amounts of each F(ab) domain were initially added to the nanoparticle conjugation reaction. | |||||
| Non-loaded | |||||
| Nude NHS NP | PLGA-PEG-NHS | 207.5 ± 13.9 | 0.12 ± 0.07 | — | — |
| Native CTX F(ab) NP | PLGA-PEG-NHS | 215.0 ± 13.0 | 0.15 ± 0.06 | 10.1 ± 2.7 | — |
| Nude azide NP | PLGA-PEG-azide | 204.7 ± 10.2 | 0.07 ± 0.03 | — | — |
| Modified CTX F(ab) NP [disulfide] | PLGA-PEG-azide | 211.7 ± 5.6 | 0.08 ± 0.05 | 16.1 ± 1.5 | — |
| Rhodamine 6G-loaded | |||||
| Nude NHS NP | PLGA-PEG-NHS | 218.8 ± 20.8 | 0.13 ± 0.06 | — | — |
| Native CTX F(ab) NP | PLGA-PEG-NHS | 226.1 ± 23.7 | 0.15 ± 0.04 | 12.2 ± 5.3 | — |
| Nude azide NP | PLGA-PEG-azide | 215.1 ± 12.2 | 0.08 ± 0.04 | — | — |
| Modified CTX F(ab) NP [disulfide] | PLGA-PEG-azide | 218.8 ± 13.8 | 0.07 ± 0.04 | 16.0 ± 5.6 | — |
| CPT-loaded | |||||
| Native CTX F(ab) CPT NP | PLGA-PEG-NHS | 214.2 ± 12.5 | 0.14 ± 0.05 | 16.6 ± 1.5 | 3.4 ± 1.8 |
| Modified CTX F(ab) CPT NP [disulfide] | PLGA-PEG-azide | 215.7 ± 13.5 | 0.12 ± 0.03 | 14.2 ± 3.4 | 3.9 ± 1.4 |
In parallel, we also prepared control PLGA-PEG nanoparticles incorporating surface NHS moieties to enable comparison of this ‘click’ coupling approach to conventional carbodiimide conjugation. Here, native CTX F(ab) 2 was coupled to PLGA-PEG-NHS nanoparticles (native CTX F(ab) NP) (Fig. 2B). Physicochemical characteristics of native CTX F(ab) NP and the non-functionalised control (nude NHS NP) were comparable to the corresponding azide-based nanoformulations (Table 1). Notably however, native CTX F(ab) 2 loading on NHS-functionalised nanoparticles was lower than that observed previously for modified CTX F(ab) NP [disulfide], despite the addition of equimolar amounts of each fragment to the respective nanoparticle conjugation reactions (Table 1). In agreement with these findings, other publications have also shown that the use of refined bioconjugation strategies can significantly enhance the loading of targeting ligands on nanoparticles compared to carbodiimide coupling.20,21
Nanoparticle binding to recombinant EGFR-Fc
Binding of the nanoformulations to the cognate EGFR antigen was next evaluated, where nanoparticles (fluorescently labelled through incorporation of rhodamine 6G in the polymeric core) were added to microtiter plates coated with recombinant EGFR-Fc, followed by measurement of fluorescence as a readout of target binding. Upon incubation with EGFR-Fc-immobilised wells, binding of modified CTX F(ab) NP [disulfide] was significantly enhanced in comparison to native CTX F(ab) NP (Fig. 3A). These effects were conditional on the F(ab) targeting corona, since minimal binding was observed with both nude control nanoformulations. Further analyses also revealed that binding of native CTX F(ab) NP and modified CTX F(ab) NP [disulfide] was dose-dependent, with each increment in polymer concentration leading to an enhancement in fluorescence (Fig. 3B). These findings suggest that site-specific ‘click’ coupling leads to improved presentation and accessibility of F(ab) paratopes on the surface of nanoparticles, thus maximising their ability to bind to EGFR.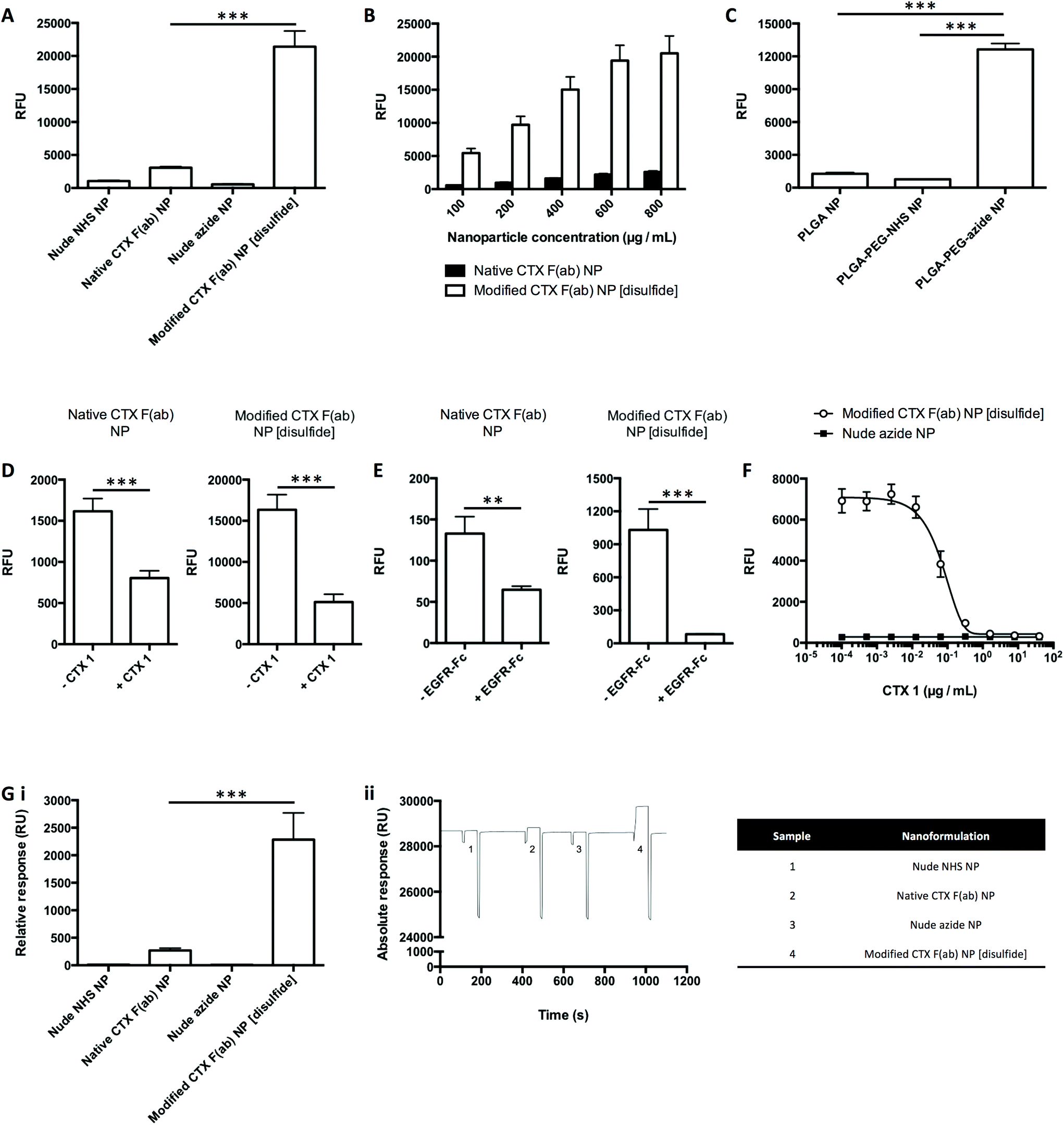 | ||
| Fig. 3 Binding of nanoformulations to recombinant EGFR-Fc. (A–C) Binding of fluorescent nanoformulations to EGFR-Fc was evaluated by FLISA at concentrations of (A) 1 mg polymer mL−1, (B) 100–800 μg polymer mL−1 and (C) 500 μg polymer mL−1. (D) Binding of fluorescent nanoformulations (500 μg polymer mL−1) to EGFR-Fc was evaluated by FLISA ± pre-block with CTX 1 (40 μg mL−1). (E) Binding of fluorescent nanoformulations (50 μg polymer mL−1) to EGFR-Fc was evaluated by FLISA ± pre-incubation with EGFR-Fc (10 μg mL−1). (F) Binding of fluorescent nanoformulations (500 μg polymer mL−1) to EGFR-Fc was evaluated by FLISA in competition with CTX 1 (0.0001024–40 μg mL−1). (G) (i) Binding of non-fluorescent nanoformulations (10 mg polymer mL−1) to EGFR-Fc was evaluated by SPR. (ii) Representative sensorgram from experiment in (i), with details of the annotated samples 1–4 in the accompanying table. | ||
To further confirm that the superior EGFR binding of modified CTX F(ab) NP [disulfide] was due to optimised F(ab) orientation enabled by ‘click’ conjugation to complementary azide-capped nanoparticles, CTX F(ab) 9 was instead coupled to nanoparticles functionalised with either NHS or carboxyl groups. However, only marginal binding of both these nanoformulations to EGFR-Fc was detected, in marked contrast to that synthesised via SPAAC chemistry, indicating that oriented display of CTX F(ab) 9 is necessary for optimal target engagement (Fig. 3C).
Next, modifications of this assay approach were used to confirm that binding of the nanoparticles was via specific engagement of EGFR-Fc. Firstly, the EGFR-Fc-coated wells were pre-incubated with an excess of CTX 1 prior to addition of the nanoparticles. This led to a significant decline in the binding of native CTX F(ab) NP and modified CTX F(ab) NP [disulfide] (Fig. 3D). In an alternative approach, the F(ab) paratopes on the nanoparticles were initially saturated with an excess of free EGFR-Fc. Upon incubating these samples with EGFR-Fc-immobilised wells, a significant reduction in nanoparticle binding was once more observed (Fig. 3E). Finally, CTX 1 was mixed with modified CTX F(ab) NP [disulfide] and both were added simultaneously to EGFR-Fc-coated wells. In this case, nanoparticle binding was progressively inhibited with each concentration increment of competing CTX 1 (Fig. 3F).
SPR was then used to evaluate nanoparticle binding to EGFR-Fc immobilised on a carboxymethylated dextran chip. These studies produced similar trends to those observed via FLISA, where binding of modified CTX F(ab) NP [disulfide] was significantly greater than that of native CTX F(ab) NP (Fig. 3G). Thus, from data generated using two independent techniques, it can be concluded that the EGFR binding activities of CTX F(ab) 2 and CTX F(ab) 9 are retained following conjugation to nanoparticles via distinct chemistries. Notably, however, exploitation of the alkyne–azide ‘click’ reactivity during nanoparticle construction leads to enhanced EGFR binding.
Other common strategies for the site-specific functionalisation of nanoparticles include maleimide-based coupling, or engineering of antibodies with enzyme recognition motifs or non-canonical amino acids.22–26 Whilst these can markedly improve the homogeneity and antigen binding ability of targeted nanoparticles, multiple factors often limit their success, such as the instability of the resultant conjugate, disruption of the native structure of antibodies and the need for a significant investment of time and expense. In contrast, our approach generates a highly stable triazole linkage, preserves the structural integrity of the F(ab) through re-bridging of the reduced disulfide and circumvents the need for engineering, ensuring facile translation to other platforms as we have previously demonstrated.27,28
Nanoparticle binding to EGFR-expressing pancreatic cancer cells
Cell-based assays were next employed to evaluate the EGFR binding activity of the nanoparticles. The human pancreatic cancer cell lines PANC-1, MIA PaCa-2 and BxPC-3 were chosen as suitable models for these studies, due to their surface expression of EGFR.29–33 Nanoparticles were incubated with the cells at 4 °C to limit non-specific endocytosis as previously shown.34 Cellular binding of fluorescent modified CTX F(ab) NP [disulfide] was enhanced when compared to native CTX F(ab) NP (Fig. 4A), consistent with previous FLISA and SPR results. In a subsequent experiment to confirm that the observed binding was due to EGFR engagement, cells were first incubated with non-fluorescently labelled nanoparticles, prior to staining of surface-expressed EGFR with a FITC-tagged antibody to visualise uncomplexed receptor remaining on the cell surface. High basal staining of EGFR was detected on untreated cells and a similar pattern was also apparent following incubation with the nude control nanoparticles (Fig. 4B). In contrast, EGFR staining was markedly reduced upon treatment with native CTX F(ab) NP and, to a more pronounced extent, by modified CTX F(ab) NP [disulfide] (Fig. 4B). These findings suggest that both native CTX F(ab) NP and modified CTX F(ab) NP [disulfide] engage EGFR on the surface of PANC-1 cells, thereby impeding binding of the fluorophore-tagged antibody to the receptor. Upon replicating these studies in other cell lines including MIA PaCa-2 and BxPC-3, the trends were largely comparable to those observed in the PANC-1 model (ESI Fig. S9†). Collectively, this in vitro dataset confirms that the superior EGFR binding activity of modified CTX F(ab) NP [disulfide] translates to a cell-based setting.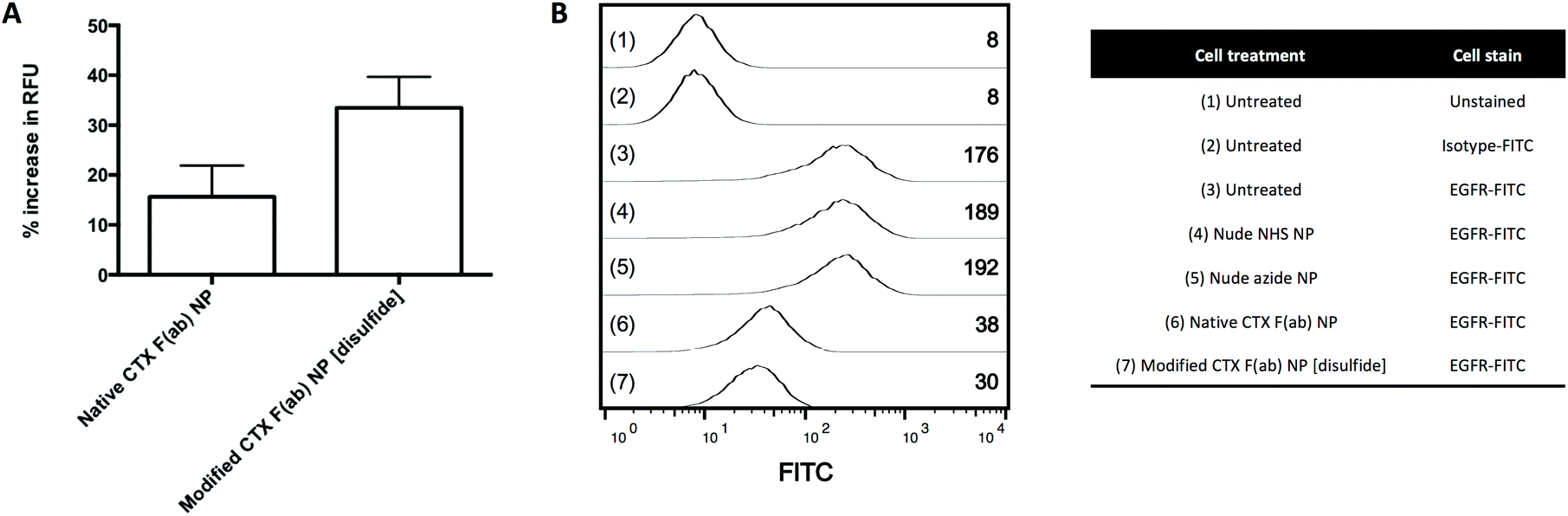 | ||
| Fig. 4 Binding of nanoformulations to EGFR-expressing cells. (A) PANC-1 cells were treated with fluorescent nanoformulations (800 μg polymer mL−1) for 45 min at 4 °C. Cells were then washed and binding of the nanoformulations was assessed through fluorescence measurement. Results for native CTX F(ab) NP and modified CTX F(ab) NP [disulfide] are presented as % increase in relative fluorescence units (RFU) versus the corresponding nude NHS NP and nude azide NP controls, respectively. (B) PANC-1 cells were treated with non-fluorescent nanoformulations (500 μg polymer mL−1) for 1 h at 4 °C. Cells were then washed, stained with FITC-labelled EGFR or isotype control antibodies and analysed by flow cytometry. Representative histograms are shown for each of the numbered treatments 1–7, with inset values denoting the geometric mean fluorescence intensity. | ||
Targeted delivery of nanoformulated CPT to EGFR-expressing pancreatic cancer cells
To date, numerous EGFR-targeted nanoparticles have been examined as drug delivery vehicles in the treatment of pancreatic cancer.6,35–37 However, most of these efforts have involved random bioconjugation of full antibody targeting ligands to the surface of nanoparticles, providing clear scope for further improvement that we wished to address. Thus, following the successful development and validation of native CTX F(ab) NP and modified CTX F(ab) NP [disulfide], their utility as a targeted drug delivery platform was next explored using the topoisomerase 1 inhibitor CPT as a model agent. This drug cargo was selected due to the ease of entrapping it within PLGA-based nanoparticles without loss of activity,7,38 and due to the clinical relevance of topoisomerase inhibitors in pancreatic cancer therapy such as the recently approved liposomal formulation of irinotecan.39 Entrapment was achieved through direct addition of CPT to the organic phase during nanoparticle synthesis, to generate native CTX F(ab) CPT NP and modified CTX F(ab) CPT NP [disulfide] with a comparative drug loading of 3.4 ± 1.8 and 3.9 ± 1.4 μg mg−1 polymer, respectively (Table 1). Furthermore, other physicochemical characteristics of these CPT-loaded nanoformulations were similar, including size, PDI and F(ab) loading (Table 1).The clonogenicity of MIA PaCa-2 cells was then assessed following treatment with the CPT-loaded and control nanoformulations, or an equivalent concentration of free CPT. This cell line was considered the most appropriate model based on its ability to form defined colonies. Whereas free CPT, nude NHS NP and nude azide NP yielded similar results to the untreated control in these assays, colony numbers were reduced by native CTX F(ab) CPT NP and, to a significantly greater extent, by modified CTX F(ab) CPT NP [disulfide] (Fig. 5). Despite the sensitivity of MIA PaCa-2 cells to CPT, uptake of the free drug during the short duration of treatment at 4 °C was not sufficient to exert an impact upon survival. In contrast, however, both native CTX F(ab) CPT NP and modified CTX F(ab) CPT NP [disulfide] were capable of engaging surface EGFR under these conditions, facilitating their internalisation (and thus intracellular delivery of CPT) upon transfer to 37 °C. Moreover, the enhanced ability of modified CTX F(ab) CPT NP to bind to EGFR may potentially explain the superior reduction in colony numbers following treatment with this nanoformulation.
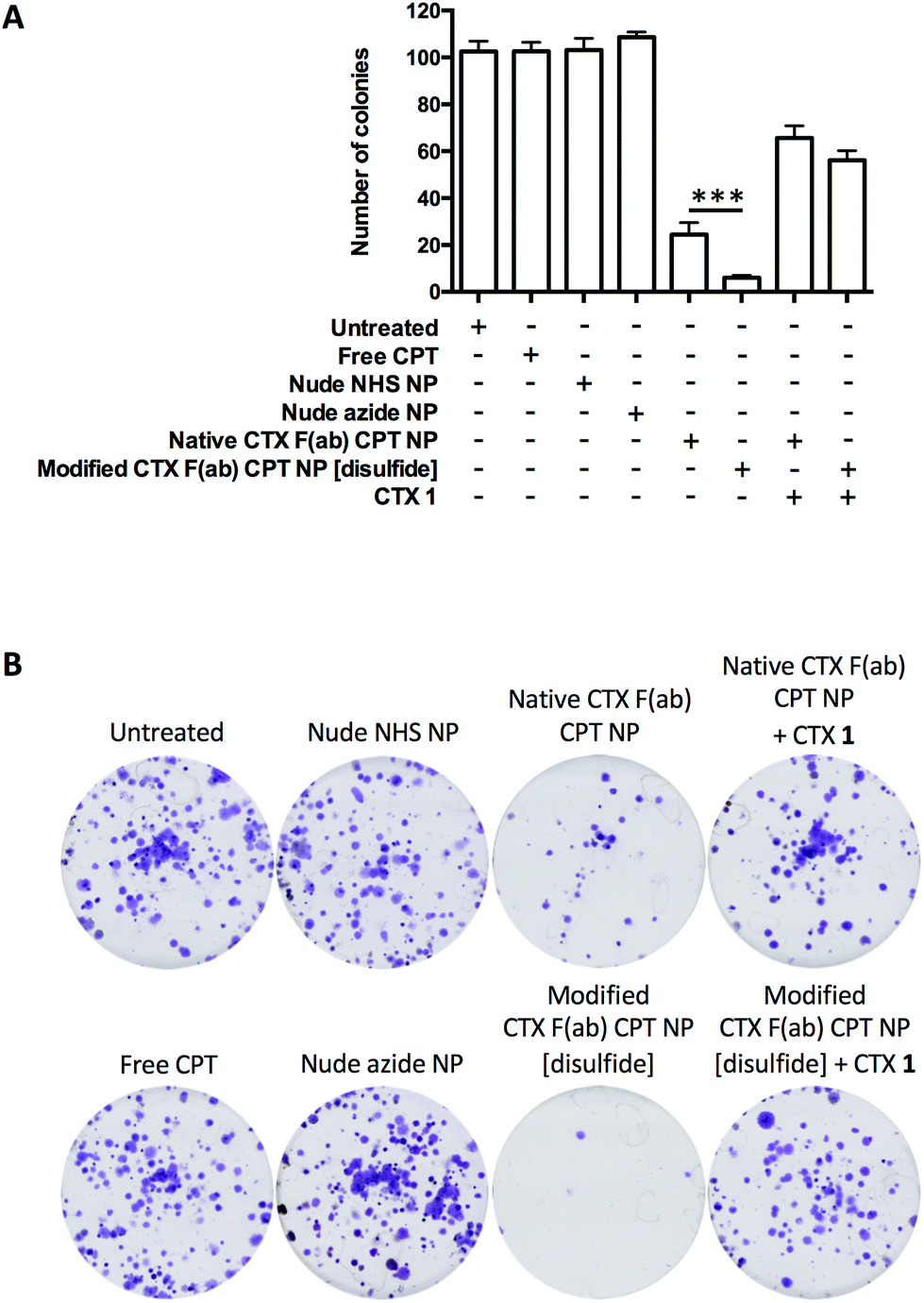 | ||
| Fig. 5 Application of nanoparticles as targeted drug delivery vehicles. (A) MIA PaCa-2 cells were treated with free CTX 1 (200 μg mL−1) for 15 min at 4 °C where appropriate, followed by the addition of free CPT and various nanoformulations for a further 45 min at 4 °C. Both free and nanoencapsulated CPT were added to cells in equivalent concentrations of 500 ng mL−1. Control nanoformulations containing no CPT were added to cells at an equivalent polymer concentration as the corresponding drug-loaded nanoformulations. Following treatment, cells were washed and then maintained at 37 °C to allow colony formation. Colony numbers were quantified for each treatment, with representative images shown in (B). | ||
To confirm that the observed reduction in colony formation was mediated via EGFR-targeted drug delivery, cells were also pre-incubated with an excess of CTX 1 prior to treatment with the CPT-loaded nanoformulations. Clonogenic survival was partially restored under these conditions, demonstrating the EGFR targeting specificity of native CTX F(ab) CPT NP and modified CTX F(ab) CPT NP [disulfide] (Fig. 5). Taken together, these data highlight the enhanced performance of modified CTX F(ab) NP [disulfide] as a targeted drug delivery vehicle and the importance of site-specific functionalisation for optimal EGFR binding.
Similar attempts to repurpose antibodies for targeted drug delivery have yielded a promising class of biotherapeutics known as antibody–drug conjugates (ADCs), where a cytotoxic warhead is directly coupled to an antibody via a synthetic linker.40–42 Whilst a small number of ADCs have gained FDA approval, their full clinical potential has been hampered by issues such as linker instability that can lead to premature drug release and off-target effects.43,44 Moreover, only a small number of drug molecules can be appended to each antibody, so ultrapotent payloads tend to be used that further increase the risk of toxicity. Crucially, these factors may be overcome by our antibody-targeted nanoparticles described herein, which allow for higher drug loading on a per vehicle basis and do not necessitate linker technology since the payload is entrapped within the polymeric core.45,46
Conclusions
To conclude, we have outlined a method for the refined construction of antibody-targeted nanoparticles that may be generically applied to other platforms with minimal time and cost requirements. By exploiting the disulfide reactivity of pyridazinediones, we show that the interchain disulfide of an antibody F(ab) may be selectively re-bridged with a strained alkyne handle to facilitate ‘click’ coupling to cognate azide-capped nanoparticles. This strategy leads to optimised orientation of F(ab) paratopes on the surface of nanoparticles in a conformation that maximises target engagement, whilst also avoiding issues with the use of full antibodies. Crucially, these nanoparticles outperform those synthesised via conventional NHS chemistry, as confirmed through their enhanced ability to bind to EGFR and deliver an entrapped payload to pancreatic cancer cells. Through improving the homogeneity and biological performance of targeted nanoparticles, our approach could potentially overcome regulatory and manufacturing hurdles that are often encountered during development, representing a significant advance amongst efforts to expedite the clinical translation of these platforms.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partly funded by the Medical Research Council UK (MRC grant MC_PC_15013) and through a US-Ireland R&D Partnership grant awarded by HSCNI (STL/5010/14). JCFN was funded by the EU's Horizon 2020 programme under Marie-Curie grant agreement 675007. We also acknowledge UCL Chemistry Mass Spectrometry Facility (Dr K. Karu/Dr X. Yang).References
- A. C. Anselmo and S. Mitragotri, Nanoparticles in the clinic, Bioeng. Transl. Med., 2016, 1(1), 10–29 CrossRef PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date, Pharm. Res., 2016, 33(10), 2373–2387 CrossRef CAS PubMed.
- S. Acharya and S. K. Sahoo, PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect, Adv. Drug Delivery Rev., 2011, 63(3), 170–183 CrossRef CAS PubMed.
- A. D. Friedman, S. E. Claypool and R. Liu, The smart targeting of nanoparticles, Curr. Pharm. Des., 2013, 19(35), 6315–6329 CrossRef CAS PubMed.
- X. Chen, W. Zhou, C. Liang, S. Shi, X. Yu, Q. Chen, T. Sun, Y. Lu, Y. Zhang, Q. Guo, C. Li, Y. Zhang and C. Jiang, Codelivery nanosystem targeting the deep microenvironment of pancreatic cancer, Nano Lett., 2019, 19(6), 3527–3534 CrossRef CAS PubMed.
- W. J. McDaid, M. K. Greene, M. C. Johnston, E. Pollheimer, P. Smyth, K. McLaughlin, S. Van Schaeybroeck, R. M. Straubinger, D. B. Longley and C. J. Scott, Repurposing of Cetuximab in antibody-directed chemotherapy-loaded nanoparticles in EGFR therapy-resistant pancreatic tumours, Nanoscale, 2019, 11(42), 20261–20273 RSC.
- D. Schmid, F. Fay, D. M. Small, J. Jaworski, J. S. Riley, D. Tegazzini, C. Fenning, D. S. Jones, P. G. Johnston, D. B. Longley and C. J. Scott, Efficient drug delivery and induction of apoptosis in colorectal tumors using a death receptor 5-targeted nanomedicine, Mol. Ther., 2014, 22(12), 2083–2092 CrossRef CAS PubMed.
- R. Meir, K. Shamalov, T. Sadan, M. Motiei, G. Yaari, C. J. Cohen and R. Popovtzer, Fast Image-Guided Stratification Using Anti-Programmed Death Ligand 1 Gold Nanoparticles for Cancer Immunotherapy, ACS Nano, 2017, 11(11), 11127–11134 CrossRef CAS PubMed.
- C. Monterrubio, S. Paco, N. G. Olaciregui, G. Pascual-Pasto, M. Vila-Ubach, M. Cuadrado-Vilanova, M. M. Ferrandiz, H. Castillo-Ecija, R. Glisoni, N. Kuplennik, A. Jungbluth, C. de Torres, C. Lavarino, N. K. V. Cheung, J. Mora, A. Sosnik and A. M. Carcaboso, Targeted drug distribution in tumor extracellular fluid of GD2-expressing neuroblastoma patient-derived xenografts using SN-38-loaded nanoparticles conjugated to the monoclonal antibody 3F8, J. Controlled Release, 2017, 255, 108–119 CrossRef CAS PubMed.
- S. T. Jahan and A. Haddadi, Investigation and optimization of formulation parameters on preparation of targeted anti-CD205 tailored PLGA nanoparticles, Int. J. Nanomed., 2015, 10, 7371–7384 CAS.
- N. A. Byzova, I. V. Safenkova, E. S. Slutskaya, A. V. Zherdev and B. B. Dzantiev, Less is More: A Comparison of Antibody-Gold Nanoparticle Conjugates of Different Ratios, Bioconjugate Chem., 2017, 28(11), 2737–2746 CrossRef CAS PubMed.
- B. Saha, T. H. Evers and M. W. Prins, How antibody surface coverage on nanoparticles determines the activity and kinetics of antigen capturing for biosensing, Anal. Chem., 2014, 86(16), 8158–8166 CrossRef CAS PubMed.
- W. W. Cheng and T. M. Allen, Targeted delivery of anti-CD19 liposomal doxorubicin in B-cell lymphoma: a comparison of whole monoclonal antibody, Fab’ fragments and single chain Fv, J. Controlled Release, 2008, 126(1), 50–58 CrossRef CAS PubMed.
- D. A. Richards, A. Maruani and V. Chudasama, Antibody fragments as nanoparticle targeting ligands: a step in the right direction, Chem. Sci., 2017, 8(1), 63–77 RSC.
- K. Kanazaki, K. Sano, A. Makino, Y. Shimizu, F. Yamauchi, S. Ogawa, N. Ding, T. Yano, T. Temma, M. Ono and H. Saji, Development of anti-HER2 fragment antibody conjugated to iron oxide nanoparticles for in vivo HER2-targeted photoacoustic tumor imaging, Nanomedicine, 2015, 11(8), 2051–2060 CrossRef CAS PubMed.
- L. Fiandra, S. Mazzucchelli, C. De Palma, M. Colombo, R. Allevi, S. Sommaruga, E. Clementi, M. Bellini, D. Prosperi and F. Corsi, Assessing the in vivo targeting efficiency of multifunctional nanoconstructs bearing antibody-derived ligands, ACS Nano, 2013, 7(7), 6092–6102 CrossRef CAS PubMed.
- E. B. Glass, S. Masjedi, S. O. Dudzinski, A. J. Wilson, C. L. Duvall, F. E. Yull and T. D. Giorgio, Optimizing mannose ‘click’ conjugation to polymeric nanoparticles for targeted siRNA delivery to human and murine macrophages, ACS Omega, 2019, 4(16), 16756–16767 CrossRef CAS PubMed.
- J. Wei, X. Shuai, R. Wang, X. He, Y. Li, M. Ding, J. Li, H. Tan and Q. Fu, Clickable and imageable multiblock polymer micelles with magnetically guided and PEG-switched targeting and release property for precise tumor theranosis, Biomaterials, 2017, 145, 138–153 CrossRef CAS PubMed.
- C. F. Wang, E. M. Mäkilä, C. Bonduelle, J. Rytkönen, J. Raula, S. Almeida, A. Närvänen, J. J. Salonen, S. Lecommandoux, J. T. Hirvonen and H. A. Santos, Functionalization of alkyne-terminated thermally hydrocarbonized porous silicon nanoparticles with targeting peptides and antifouling polymers: effect on the human plasma protein adsorption, ACS Appl. Mater. Interfaces, 2015, 7(3), 2006–2015 CrossRef CAS PubMed.
- J. Bolley, E. Guenin, N. Lievre, M. Lecouvey, M. Soussan, Y. Lalatonne and L. Motte, Carbodiimide versus click chemistry for nanoparticle surface functionalization: a comparative study for the elaboration of multimodal superparamagnetic nanoparticles targeting αvß3 integrins, Langmuir, 2013, 29(47), 14639–14647 CrossRef CAS PubMed.
- D. L. Thorek, D. R. Elias and A. Tsourkas, Comparative analysis of nanoparticle-antibody conjugations: carbodiimide versus click chemistry, Mol. Imaging, 2009, 8(4), 221–229 CrossRef CAS PubMed.
- K. W. Yong, D. Yuen, M. Z. Chen, C. J. H. Porter and A. P. R. Johnston, Pointing in the right direction: controlling the orientation of proteins on nanoparticles improves targeting efficiency, Nano Lett., 2019, 19(3), 1827–1831 CrossRef CAS PubMed.
- Y. Mi, C. C. Smith, F. Yang, Y. Qi, K. C. Roche, J. S. Serody, B. G. Vincent and A. Z. Wang, A Dual Immunotherapy Nanoparticle Improves T-Cell Activation and Cancer Immunotherapy, Adv. Mater., 2018, 30(25), e1706098 CrossRef PubMed.
- Y. Zhang, N. Li, H. Suh and D. J. Irvine, Nanoparticle anchoring targets immune agonists to tumors enabling anti-cancer immunity without systemic toxicity, Nat. Commun., 2018, 9(1), 6 CrossRef PubMed.
- J. Ahn, Y. Miura, N. Yamada, T. Chida, X. Liu, A. Kim, R. Sato, R. Tsumura, Y. Koga, M. Yasunaga, N. Nishiyama, Y. Matsumura, H. Cabral and K. Kataoka, Antibody fragment-conjugated polymeric micelles incorporating platinum drugs for targeted therapy of pancreatic cancer, Biomaterials, 2015, 39, 23–30 CrossRef CAS PubMed.
- W. Kim, C. Haller, E. Dai, X. Wang, C. E. Hagemeyer, D. R. Liu, K. Peter and E. L. Chaikof, Targeted antithrombotic protein micelles, Angew. Chem., Int. Ed., 2015, 54(5), 1461–1465 CrossRef CAS PubMed.
- M. K. Greene, D. A. Richards, J. C. F. Nogueira, K. Campbell, P. Smyth, M. Fernández, C. J. Scott and V. Chudasama, Forming next-generation antibody-nanoparticle conjugates through the oriented installation of non-engineered antibody fragments, Chem. Sci., 2017, 9(1), 79–87 RSC.
- J. C. F. Nogueira, M. K. Greene, D. A. Richards, A. O. Furby, J. Steven, A. Porter, C. Barelle, C. J. Scott and V. Chudasama, Oriented attachment of VNAR proteins, via site-selective modification, on PLGA-PEG nanoparticles enhances nanoconjugate performance, Chem. Commun., 2019, 55(53), 7671–7674 RSC.
- C. Kratschmer and M. Levy, Targeted Delivery of Auristatin-Modified Toxins to Pancreatic Cancer Using Aptamers, Mol. Ther.–Nucleic Acids, 2018, 10, 227–236 CrossRef CAS PubMed.
- E. L. McMichael, A. C. Jaime-Ramirez, K. D. Guenterberg, E. Luedke, L. S. Atwal, A. R. Campbell, Z. Hu, A. S. Tatum, S. V. Kondadasula, X. Mo, S. Tridandapani, M. Bloomston, E. C. Ellison, T. M. Williams, T. Bekaii-Saab and W. E. Carson, IL-21 Enhances Natural Killer Cell Response to Cetuximab-Coated Pancreatic Tumor Cells, Clin. Cancer Res., 2017, 23(2), 489–502 CrossRef CAS PubMed.
- S. Derer, P. Bauer, S. Lohse, A. H. Scheel, S. Berger, C. Kellner, M. Peipp and T. Valerius, Impact of epidermal growth factor receptor (EGFR) cell surface expression levels on effector mechanisms of EGFR antibodies, J. Immunol., 2012, 189(11), 5230–5239 CrossRef CAS PubMed.
- N. Ioannou, A. G. Dalgleish, A. M. Seddon, D. Mackintosh, U. Guertler, F. Solca and H. Modjtahedi, Anti-tumour activity of afatinib, an irreversible ErbB family blocker, in human pancreatic tumour cells, Br. J. Cancer, 2011, 105(10), 1554–1562 CrossRef CAS PubMed.
- M. S. Pino, M. Shrader, C. H. Baker, F. Cognetti, H. Q. Xiong, J. L. Abbruzzese and D. J. McConkey, Transforming growth factor alpha expression drives constitutive epidermal growth factor receptor pathway activation and sensitivity to gefitinib (Iressa) in human pancreatic cancer cell lines, Cancer Res., 2006, 66(7), 3802–3812 CrossRef CAS PubMed.
- R. M. Straubinger, K. Hong, D. S. Friend and D. Papahadjopoulos, Endocytosis of liposomes and intracellular fate of encapsulated molecules: encounter with a low pH compartment after internalization in coated vesicles, Cell, 1983, 32(4), 1069–1079 CrossRef CAS PubMed.
- Ö. Er, S. G. Colak, K. Ocakoglu, M. Ince, R. Bresolí-Obach, M. Mora, M. L. Sagristá, F. Yurt and S. Nonell, Selective Photokilling of Human Pancreatic Cancer Cells Using Cetuximab-Targeted Mesoporous Silica Nanoparticles for Delivery of Zinc Phthalocyanine, Molecules, 2018, 23(11), 2749 CrossRef PubMed.
- L. Wang, Y. An, C. Yuan, H. Zhang, C. Liang, F. Ding, Q. Gao and D. Zhang, GEM-loaded magnetic albumin nanospheres modified with cetuximab for simultaneous targeting, magnetic resonance imaging, and double-targeted thermochemotherapy of pancreatic cancer cells, Int. J. Nanomed., 2015, 10, 2507–2519 CrossRef CAS PubMed.
- C. R. Patra, R. Bhattacharya, E. Wang, A. Katarya, J. S. Lau, S. Dutta, M. Muders, S. Wang, S. A. Buhrow, S. L. Safgren, M. J. Yaszemski, J. M. Reid, M. M. Ames, P. Mukherjee and D. Mukhopadhyay, Targeted delivery of gemcitabine to pancreatic adenocarcinoma using cetuximab as a targeting agent, Cancer Res., 2008, 68(6), 1970–1978 CrossRef CAS PubMed.
- D. Schmid, G. E. Jarvis, F. Fay, D. M. Small, M. K. Greene, J. Majkut, S. Spence, K. M. McLaughlin, K. D. McCloskey, P. G. Johnston, A. Kissenpfennig, D. B. Longley and C. J. Scott, Nanoencapsulation of ABT-737 and camptothecin enhances their clinical potential through synergistic antitumor effects and reduction of systemic toxicity, Cell Death Dis., 2014, 5, e1454 CrossRef CAS PubMed.
- A. Wang-Gillam, C. P. Li, G. Bodoky, A. Dean, Y. S. Shan, G. Jameson, T. Macarulla, K. H. Lee, D. Cunningham, J. F. Blanc, R. A. Hubner, C. F. Chiu, G. Schwartsmann, J. T. Siveke, F. Braiteh, V. Moyo, B. Belanger, N. Dhindsa, E. Bayever, D. D. Von Hoff and L. T. Chen, Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial, Lancet, 2016, 387(10018), 545–557 CrossRef CAS.
- J. M. Lambert and A. Berkenblit, Antibody-Drug Conjugates for Cancer Treatment, Annu. Rev. Med., 2018, 69, 191–207 CrossRef CAS PubMed.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Strategies and challenges for the next generation of antibody-drug conjugates, Nat. Rev. Drug Discovery, 2017, 16(5), 315–337 CrossRef CAS PubMed.
- A. Thomas, B. A. Teicher and R. Hassan, Antibody-drug conjugates for cancer therapy, Lancet Oncol., 2016, 17(6), e254–e262 CrossRef CAS PubMed.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates, Nat. Biotechnol., 2014, 32(10), 1059–1062 CrossRef CAS PubMed.
- B. Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S. F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates, Nat. Biotechnol., 2012, 30(2), 184–189 CrossRef CAS PubMed.
- M. C. Johnston and C. J. Scott, Antibody conjugated nanoparticles as a novel form of antibody drug conjugate chemotherapy, Drug Discovery Today: Technol., 2018, 30, 63–69 CrossRef PubMed.
- R. Qi, Y. Wang, P. M. Bruno, H. Xiao, Y. Yu, T. Li, S. Lauffer, W. Wei, Q. Chen, X. Kang, H. Song, X. Yang, X. Huang, A. Detappe, U. Matulonis, D. Pepin, M. T. Hemann, M. J. Birrer and P. P. Ghoroghchian, Nanoparticle conjugates of a highly potent toxin enhance safety and circumvent platinum resistance in ovarian cancer, Nat. Commun., 2017, 8(1), 2166 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr02387f |
| This journal is © The Royal Society of Chemistry 2020 |