
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ X-ray emission and high-resolution X-ray absorption spectroscopy applied to Ni-based bimetallic dry methane reforming catalysts†
Abbas
Beheshti Askari
a,
Mustafa
al Samarai
a,
Nozomu
Hiraoka
b,
Hirofumi
Ishii
b,
Lukas
Tillmann
c,
Martin
Muhler
 *ac and
Serena
DeBeer
*a
*ac and
Serena
DeBeer
*a
aMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34-36, D-45470 Mülheim an der Ruhr, Germany. E-mail: serena.debeer@cec.mpg.de
bNational Synchrotron Radiation Research Center, Hsinchu Science Park, Hsinchu 30076, Taiwan
cLaboratory of Industrial Chemistry, Ruhr-University Bochum, Universitätsstr. 150, D-44780 Bochum, Germany. E-mail: muhler@techem.rub.de
First published on 24th June 2020
Abstract
The promoting effect of cobalt on the catalytic activity of a NiCoO Dry Methane Reforming (DMR) catalyst was studied by a combination of in situ Kβ X-ray Emission Spectroscopy (XES) and Kβ-detected High Energy Resolution Fluorescence Detected X-ray absorption spectroscopy (HERFD XAS). Following the calcination process, Ni XES and Kβ-detected HERFD XAS data revealed that the NiO coordination in the NiCoO catalyst has a higher degree of symmetry and is different than that of pure NiO/γ-Al2O3. Following the reductive activation, it was found that the NiCoO/γ-Al2O3 catalyst required a relatively higher temperature compared to the monometallic NiO/γ-Al2O3 catalyst. This finding suggests that Co is hampering the reduction of Ni in the NiCoO catalyst by modulation of its electronic structure. It has also been previously shown that the addition of Co enhances the DMR activity. Further, the Kβ XES spectrum of the partly reduced catalysts at 450 °C reveals that the Ni sites in the NiCoO catalyst are electronically different from the NiO catalyst. The in situ X-ray spectroscopic study demonstrates that reduced metallic Co and Ni are the primary species present after reduction and are preserved under DMR conditions. However, the NiCo catalyst appears to always be somewhat more oxidized than the Ni-only species, suggesting that the presence of cobalt modulates the Ni electronic structure. The electronic structural modulations resulting from the presence of Co may be the key to the increased activity of the NiCo catalyst relative to the Ni-only catalyst. This study emphasizes the potential of in situ X-ray spectroscopy experiments for probing the electronic structure of catalytic materials during activation and under operating conditions.
1. Introduction
The development of catalysts that enable environmentally friendly means for the production of fuels is currently a subject of intense research.1–3 These efforts are motivated not only by the depletion of fossil fuels, but also the emission of greenhouse gases, which are known to have a devastating impact on the environment.4–6 One reaction that has been receiving increasing attention due to its high potential for converting CH4 and CO2 to syngas (H2 and CO) (reaction 1) is the Dry Methane Reforming (DMR) process.7,8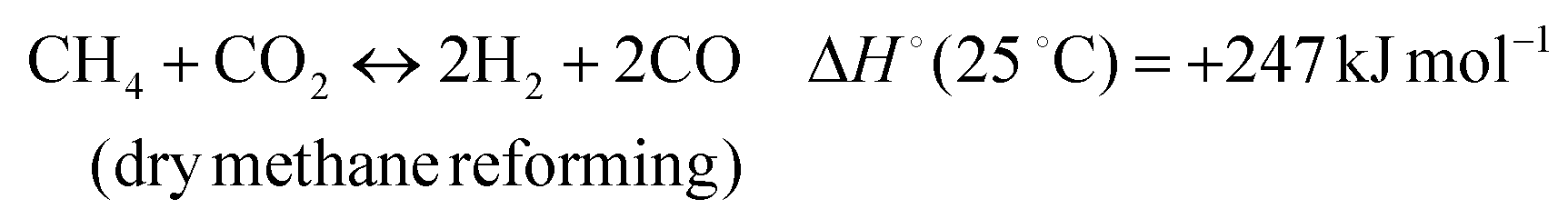 | (1) |
In addition, various undesired side reactions (reactions (2)–(5)) can occur during the DMR reaction that eventually lead to carbon deposition on the active phase.
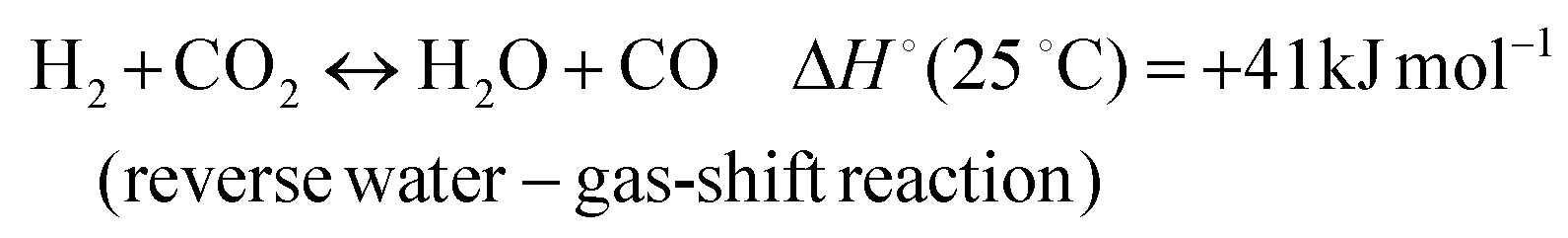 | (2) |
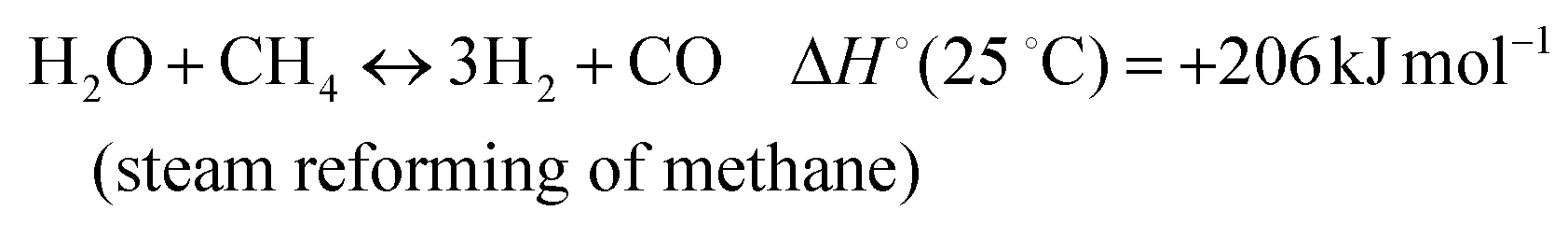 | (3) |
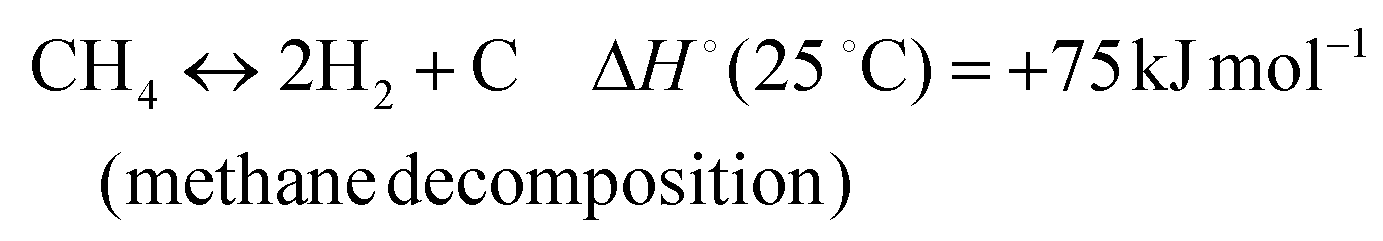 | (4) |
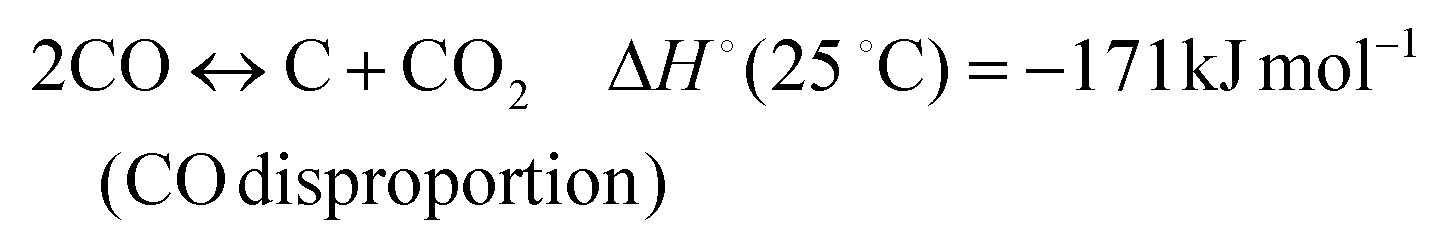 | (5) |
Due to the carbon deposition side reactions, the active sites are gradually blocked, leading to catalyst deactivation.9 In the literature, numerous studies aimed at preventing coke formation on the DMR catalysts have been reported in which the catalyst structure, reaction temperature and the DMR gas-mixture ratios during the operating conditions have been varied.10–13 Presently, the best strategy to reduce carbon deposition on DMR catalysts is based on operating at high temperatures (>700 °C) and selected CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH4 ratios e.g. 1:1. Among the various classes of DMR catalysts, the highest DMR activity, selectivity, and stabilities are achieved by noble-metals based systems.14 However, due to their scarcity, they are not considered as sustainable candidates for the large scale industrial DMR catalysis.15 Alternatively, nickel-based catalysts with comparable activities have been pursued. However, a major disadvantage for the Ni-based catalyst is their relatively high carbon deposition rate when compared to noble-metals based catalysts.16,17 Various approaches have been reported in literature which aim to reduce coke formation on the Ni-based catalysts. Chang and co-workers illustrated that the activation of CO2 is promoted by supporting the active phase on basic supports/promotors of alkali metals e.g. CaO, K2O and MgO.18,19 Separate studies have shown that alloying Ni-based catalysts with other metals, such as Co, Fe, Mo, Mn, Sn, Ce and Cu, increases the catalytic activity, selectivity and coke resistivity as compared to monometallic Ni catalysts.20–22 Furthermore, Zhang et al. reported that the synthesis of bimetallic catalysts results in a decreased Ni domain size, which results in a lower sensitivity to coking.22 It was also found that among 3d transition metals, Co most efficiently promotes the DMR activity of Ni-based catalysts.23,24
:CH4 ratios e.g. 1:1. Among the various classes of DMR catalysts, the highest DMR activity, selectivity, and stabilities are achieved by noble-metals based systems.14 However, due to their scarcity, they are not considered as sustainable candidates for the large scale industrial DMR catalysis.15 Alternatively, nickel-based catalysts with comparable activities have been pursued. However, a major disadvantage for the Ni-based catalyst is their relatively high carbon deposition rate when compared to noble-metals based catalysts.16,17 Various approaches have been reported in literature which aim to reduce coke formation on the Ni-based catalysts. Chang and co-workers illustrated that the activation of CO2 is promoted by supporting the active phase on basic supports/promotors of alkali metals e.g. CaO, K2O and MgO.18,19 Separate studies have shown that alloying Ni-based catalysts with other metals, such as Co, Fe, Mo, Mn, Sn, Ce and Cu, increases the catalytic activity, selectivity and coke resistivity as compared to monometallic Ni catalysts.20–22 Furthermore, Zhang et al. reported that the synthesis of bimetallic catalysts results in a decreased Ni domain size, which results in a lower sensitivity to coking.22 It was also found that among 3d transition metals, Co most efficiently promotes the DMR activity of Ni-based catalysts.23,24
However, to date the synergistic interaction between Co and Ni in optimizing DMR activity is not fully understood. In a previous study, we utilized in situ Scanning Transmission X-ray Microscopy (STXM) to follow the changes in the morphology and electronic structure of multiple NiCoO/γ-Al2O3 DMR catalyst particles during activation and under DMR reaction conditions.24 It was found that through the activation step, Co and Ni oxides were gradually reduced by heating the catalyst under 5% H2 in Ar. Further, the formation of a segregated structure with a Ni-rich shell was observed. This structure was also conserved after exposure to DMR conditions, thus hinting at the significance of the segregated structure for the DMR catalysis. Despite these findings, no direct comparison to the monometallic Ni-based methane reforming catalyst and the effect of Co on both the reduction of Ni site and the DMR activity was made.
In the present study, both the Ni and Co species in NiCoO/γ-Al2O3 and NiO/γ-Al2O3 DMR catalysts are investigated under the reduction/activation and DMR operating conditions by using hard X-ray spectroscopies. Specifically, we utilize a combination of Ni K-Beta X-ray emission spectroscopy (XES) and Ni and Co K-Beta detected High Energy Resolution Fluorescence Detected X-ray absorption spectroscopy (HERFD XAS) in order to obtain insight in the local geometric and electronic structural changes that occur at both the Ni and the Co sites.25–33 In addition, Scanning Transmission Electron Microscopy-Energy Dispersive X-ray spectroscopy (STEM-EDX), Temperature Programmed Reduction (TPR) and activity measurements were performed to elucidate the differences in the morphology, reducibility and activity of both catalysts. Taken together, these data allow us to draw structure–function correlations and to better understand the role of Co in optimizing NiO-based catalysts for DMR.
2. Experimental section
2.1 NiO/γ-Al2O3 catalyst synthesis
For the synthesis of the NiCoO/γ-Al2O3 and NiO/γ-Al2O3 catalyst, the following chemicals were used without any purification: cobalt(II) acetate tetrahydrate (>98%, Sigma Aldrich), nickel(II) acetate tetrahydrate (99.998%, Sigma Aldrich), ammonium oxalate monohydrate 98% (Alfa Aesar), methanol 99% (Alfa Aesar), 1-hexanol 99% (Alfa Aesar), n-hexane 99%, and γ-alumina support (Alfa Aesar).The NiCoO/γ-Al2O3 catalyst was synthesized according to a previously reported protocol.24,34 The NiO/γ-Al2O3 catalyst was prepared by an adaptation of the reverse micellar method.35 In this approach, two different aqueous mixtures were prepared by dissolving 2.3 g ammonium oxalate monohydrate and 3 g nickel acetate tetrahydrate in 15 mL demineralized water and stirring for 15 minutes. A third mixture was prepared by mixing 0.9 g cetyltrimethylammonium bromide, 9 mL hexanol and 12.6 mL hexane and stirring for 20 minutes. This mixture was split into two equal portions and consequently added to the aqueous solutions and stirred for additional 20 minutes. Next, the resulting three solutions were mixed and stirred for additional 48 hours. The sample was purified by centrifuging the mixture at 5500 rpm for 5 minutes and washed with a mixture of 30 mL methanol and 30 mL chloroform. This procedure was followed by centrifugation at 5500 rpm for 15 minutes and drying at 50 °C for hours. Finally, to deposit the catalyst on the γ-Al2O3 support, a mixture of the prepared sample and 2.5 mL demineralized water was mixed under stirring with 0.8 g of the support and dried at 90 °C for 12 hours. Drying was performed by transferring the sample to a quartz boat positioned in a tubular quartz tube of an electrical furnace. To convert the metal acetates to their respective oxides, the samples were calcined at 400 °C for 10 hours under dynamic air flow. A gas stream of 60 mL min−1 synthetic air (20% O2 in He) was applied while heating the oven with a rate of 2 K min−1 up to the final temperature of 400 °C. After calcining the sample for 10 hours, the temperature was reduced to room temperature at a rate of 2 K min−1.
2.2 STEM-EDX
The STEM measurements for the NiO/γ-Al2O3 and NiCoO/γ-Al2O3 catalyst were performed by depositing small quantity of the respective catalyst powder on a polymer coated Cu TEM grid. The Hitachi HD-2700 spherical aberration corrected Scanning Transmission Electron Microscope (STEM) was used to get information about the morphology of the catalyst. EDX measurements were recorded with an EDAX Octane T Ultra W 200 mm2 SDD that was connected to the Hitachi HD-2700 Microscope.2.3 TPR
In an earlier study, the details of the TPR experimental setup and the reduction profile for the NiCoO/γ-Al2O3 catalyst were reported.24 Additionally, the TPR profile of the NiO/γ-Al2O3 was measured by filling a quartz cell with 64 mg of the calcined catalysts. In this experiment, the temperature was increased to 850 °C, with a temperature ramp of 6 °C min−1 in a flow of 4.6% H2 in Ar, 84.1 mL min−1 total. The H2 consumption was measured by a thermal conductivity detector (Hydros, Rosemount).2.4 DMR activity tests
The DMR activity for the NiCoO/γ-Al2O3 sample was reported in an earlier publication.24 The experimental setup for measuring the activity of the NiO/γ-Al2O3 DMR catalysts has been described in detail elsewhere by Tillmann et al.,24,36 Fig. S1.† Based on both the obtained data from STXM study,24 on the NiCoO/γ-Al2O3 catalyst and the limitation of the hard X-ray reactor, additional DMR catalytic activity data were collected at 750 °C, 600 °C, 500 °C, and 450 °C.2.5 Synchrotron based experiments
In situ XES and HERFD data for the NiCoO/γ-Al2O3 and NiO/γ-Al2O3 catalyst were obtained at the Taiwan beamline BL12XU at the SPring-8 synchrotron facility (8 GeV, 99 mA).37 The beamline has four main optical elements, a high heat-load Si (111) double-crystal pre-monochromator, a four-bounce channel-cut Si (220) monochromator, a cylindrical Si collimating mirror and a toroidal Pt-coated focusing mirror. A Pt-coated mirror was used to focus the beam both vertically and horizontally to ∼80 (V) × 120 (H) μm2 at the sample position. The incident energy was calibrated with references to the first inflection points of Ni and Co foils at 8333 eV and 7709 eV, respectively. A crystal spectrometer aligned in a Rowland geometry was utilized to detect the fluorescence from the sample. The Ni Kβ (1s3p) emission spectra using a 1 m radius spherically bent Si (551) analyzer. For the Co Kβ measurements, a Ge (444) analyzer was used. A He-filled path was used between the sample and spectrometer to reduce signal attenuation and emitted X-rays. The signal selected by the spectrometer crystals was collected using an energy-resolving Si detector. A set of four Ni Kβ XES spectra were collected in the emission energy region of 8220 to 8335 eV using an incident energy of 8450 eV. The Co Kβ emission was collected from 7615 to 7700 eV energy range using a 7800 eV excitation energy (Fig. S2 and S3†). All XES spectra were normalized to the energy maximum of the Kβ1,3 peak. Co and Ni XES data were calibrated by setting the Kβ maximum of Co3O4 and NiO to 7649.1 and 8264.8 eV,37 respectively.The HERFD spectra were measured by setting the spectrometer to the maximum of the Kβ1,3 XES peak. For Co and Ni HERFD data, four sets of scans in the region from 7700 to 7900 eV and 8320 to 8450 eV where selected, respectively. This protocol was repeated for each step of the in situ experiment.
To validate the quality of the collected data, radiation damage studies were conducted by measuring multiple XES and HERFD spectra during 3 h exposure. To avoid beam damage, filters were inserted before the sample to attenuate the incident beam until no observable damage in the pre-edge or edge region was observed.
3. Results
The STEM data in Fig. 1a show the formation of small coalesced nanoparticles for the calcined NiCoO/γ-Al2O3 sample, as previously demonstrated.24 The elemental distributions for the Ni (green) and oxygen (blue) for the NiCoO sample are shown in Fig. 1b and c. Based on the Co-EDX image (in Fig. 1d) and the Ni-EDX image (1b), rather unequal distributions for Ni and Co on the γ-Al2O3 support are seen. These observations are also consistent with STEM-EDX and STXM results from an earlier study which demonstrated the existence of Ni-rich domains and a variable distribution of Co through the NiCoO/γ-Al2O3 particle.24 These data also suggest a similar morphology for both NiO/γ-Al2O3 and NiCoO/γ-Al2O3 samples (Fig. 1a and e). | ||
| Fig. 1 STEM-EDX images of the; (a–d) NiCoO/γ-Al2O3 and (e–g) NiO/γ-Al2O3 samples after the calcination step at 400 °C. (b and f) nickel (green); (c and g) oxygen (blue); (d) cobalt (purple). | ||
The STEM-EDX images for the NiO/γ-Al2O3 sample indicates rather homogeneous distributions for nickel (green) and oxygen (blue) on the support (Fig. 1f and g).
To follow the reduction and conversion of the oxides to the metallic active phase, TPR profiles were obtained for the NiO/γ-Al2O3 (Fig. 2) and NiCoO/γ-Al2O3 (Fig. SI.5 of ref. 24) DMR catalysts. For the NiCoO sample, there are two prominent reduction peaks. There is a relatively small peak at 265 °C which is assigned to the reduction of Co3+ species to Co2+ (ref. 38 and 39) followed by the main reduction peak at 325 °C where both Ni2+ and Co2+ are reduced to their metallic states.40,41 This result is consistent with in situ Ni and Co L-edges spectra for this catalyst.24 However, from the TPR profile of the NiO catalyst in Fig. 2, a single peak reduction is observed at 317 °C which corresponds to the reduction of Ni2+ to the metallic nickel.42,43 By comparing the TPR profiles for both samples, it appears that the reduction of Ni2+ in the NiO sample initiates at a relatively lower temperature. These data suggest that due to the presence of cobalt in the NiCoO catalyst the reduction temperature is modulated and therefore Ni2+ is reduced at relatively higher temperature as compared to the monometallic NiO catalyst.
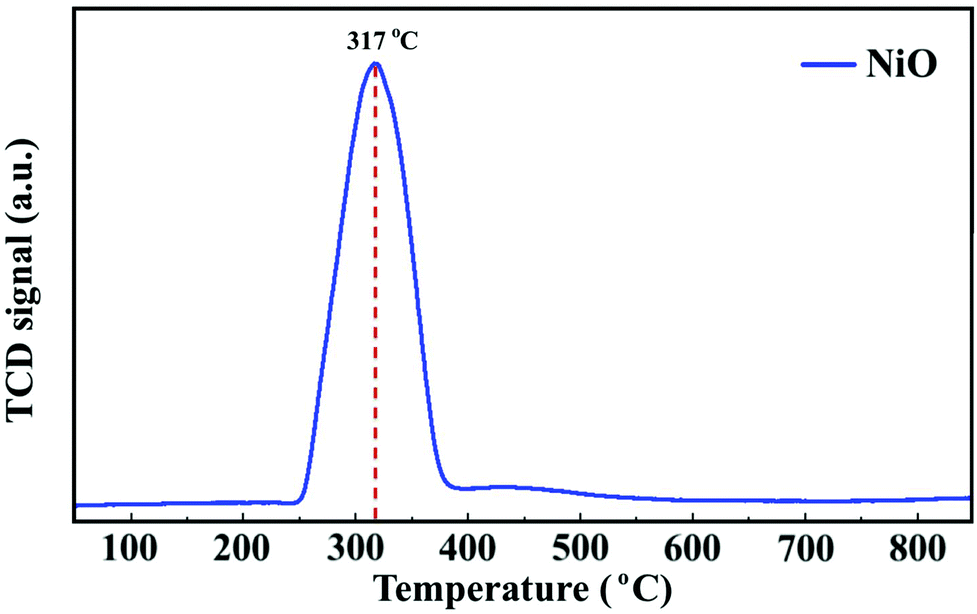 | ||
| Fig. 2 H2 TPR profile of the NiO/γ-Al2O3 catalyst. | ||
3.1. DMR activity experiments
Prior to the DMR activity measurements, the catalysts were activated by reducing the oxides at 850 °C for 20 minutes under 10% H2 in N2. The DMR activities of the NiCoO/γ-Al2O3,24 and NiO/γ-Al2O3 catalysts (Fig. 3) were obtained at 800 °C under dynamic flow of the DMR gas mixture (7% CH4 & 9.5% CO2 diluted in N2). The conversion values for NiO and NiCoO systems vary substantially with NiCoO at ∼90%,24 and NiO at only ∼50%. The initial activity of the NiCoO catalyst is similar to other recently reported highly active DMR catalyst systems.44,45 Table S1† summarizes the stabilized conversion/yield values for the NiCoO and NiO catalysts under DMR conditions for 15 hours. These data suggest that following the DMR experiment, the activity of both catalysts has stabilized and thus suggests a high resistivity against coke formation.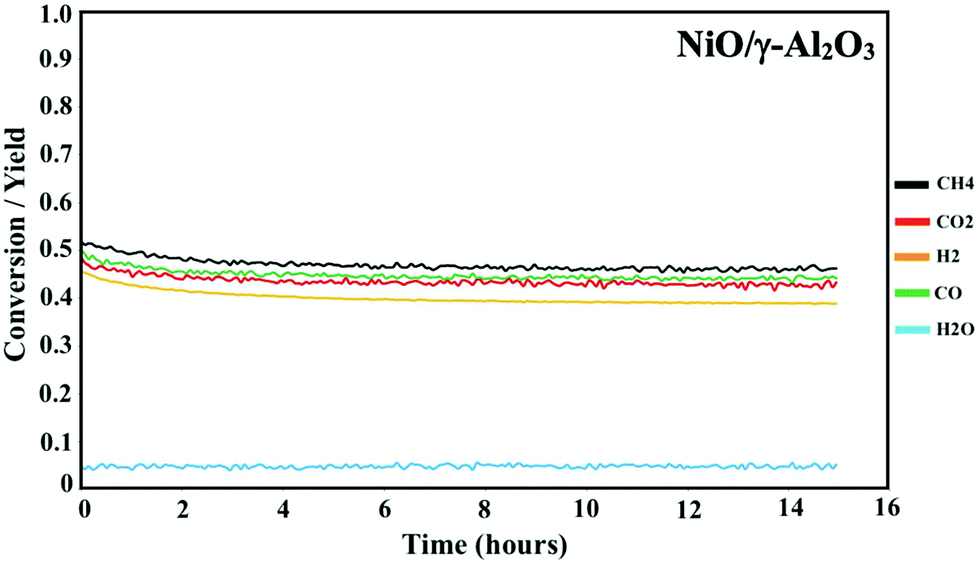 | ||
| Fig. 3 Activity profile of the NiO/γ-Al2O3 DMR catalysts for 15 hours under methane reforming conditions. Prior to the DMR activity measurements, the catalyst was activated for 20 minutes under 10% H2 in N2 with a flow rate of 100 mL min−1. The DMR experiment was performed at 800 °C under 7% CH4 and 9.5% CO2 in N2 and a flow rate of 490 mL min−1. DMR activity profiles for the NiCoO/γ-Al2O3 catalysts were measured in our previous study.24 | ||
The DMR activity data shows that the presence of cobalt in the NiCoO DMR catalysts has a promoting effect on the catalytic activity, resulting in an almost two-fold higher conversion when compared to the monometallic NiO catalyst. In addition, to understand the effect of temperature on the catalytic activity of the NiCoO catalyst additional DMR activity tests at 750 °C, 600 °C, 500 °C and 450 °C under a continuous flow of DMR gas mixture (7% CH4, 9.5% CO2 in N2) for 35 hours were performed (Fig. S6†). This data shows that decreasing the DMR reaction temperature dramatically reduces the achieved conversion which may be enhanced by the increase in the rate of coke deposition on the active phase.46 Nonetheless the activity data for the NiCoO catalyst collected at 600 °C shows modest DMR activity, indicating that the X-ray spectroscopic data correspond to an operating catalyst.
3.2. Ni Kβ XES of NiCoO/γ-Al2O3vs. NiO/γ-Al2O3
Fig. 4 shows the Ni Kβ mainline XES spectra (Ni 3p to 1s) for both the NiO/γ-Al2O3 and NiCoO/γ-Al2O3 catalysts in their calcined form, during the reductive activation step, and during the DMR reaction. The black and grey spectra correspond to the Ni XES for the freshly calcined NiO and NiCoO samples, respectively. The spectra exhibit subtle differences with the Kβ1,3 maxima occurring at 8264.5 eV and 8264.4 eV, for the NiO and NiCoO, respectively. Both calcined samples also exhibit a Kβ′ feature at ∼8253.1 eV, which is typical for a triplet (S = 1) Ni2+ oxide species (Fig. S7†).47 The slight differences in the mainline position could reflect modulations in site symmetry and/or covalency of the Ni site.33,48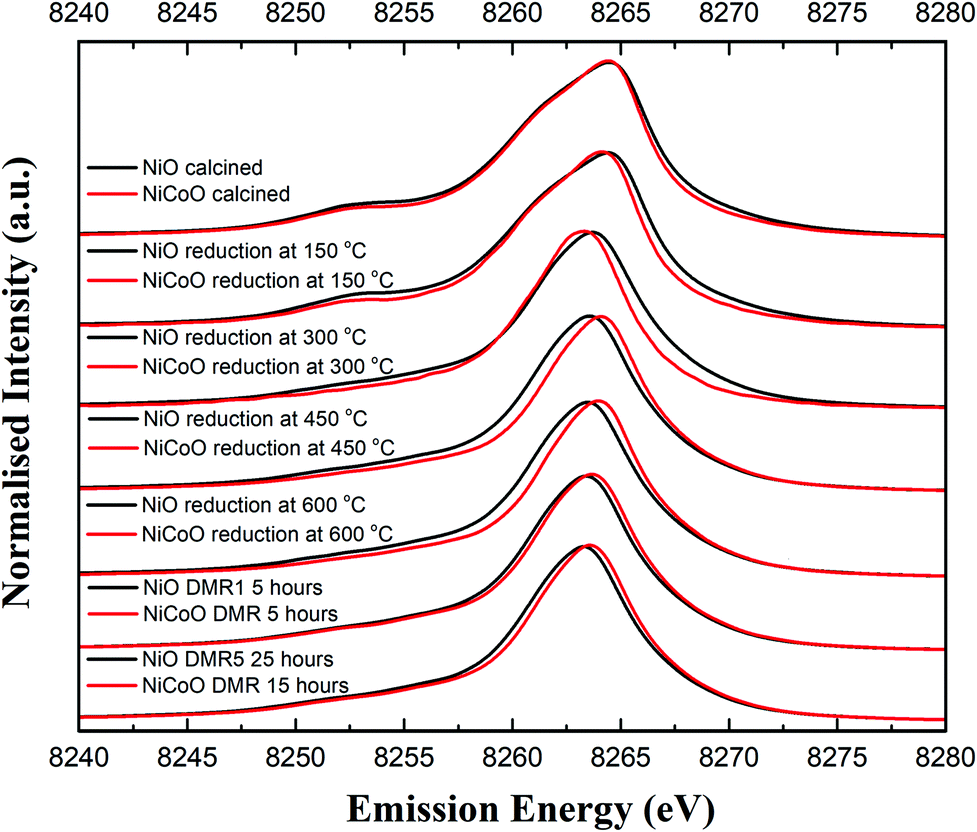 | ||
| Fig. 4 Ni Kβ XES spectra of the NiO/γ-Al2O3 and NiCoO/γ-Al2O3 samples obtained under ambient, reductive activation, and DMR conditions. | ||
The samples were activated by flowing 5% H2 in Ar gas and by gradually increasing the temperature from 25 °C to 600 °C. The selected temperature range for the reduction is based on the previously collected TPR data (Fig. 2), which revealed a full reduction of the active phase at T < 500 °C. To follow the reduction of the Ni sites and make a correlation with the TPR data, XES spectra were measured in a selected temperature range. First, following the reduction of the metal oxides at 150 °C and 300 °C, the Ni XES peak for the NiO sample shifts by −0.05 and −0.78 eV, respectively to lower emission energies and the Kβ′ peak at 8253 eV has largely vanished (Fig. 4 and Table S2†). This is consistent with the reduction of Ni2+ to Ni0. Upon complete reduction a d10 configuration is reached and the p–d exchange, which gives rise to the Kβ′ feature, is no longer present. Next, by increasing the temperature to 450 °C and 600 °C, the Ni XES Kβ1,3 peak position for NiO shifts to lower emission energies, which indicated the full reduction of NiO–Al2O4 at T > 400 °C (Fig. 2).49 This is consistent with in situ STXM data for the NiCoO sample which showed that full reduction occurs at 500 °C.24 Finally, due to the reduction of NiO–Al2O4 sites to metallic Ni at 600 °C, the Ni Kβ1,3 emission peak shifts by ∼−1 eV relative to the emission spectrum for the calcined sample to 8263.48 eV.
Fig. 4 shows that the Ni Kβ1,3 XES peak for the NiCoO sample at 150 °C and 300 °C shifts by −0.30 eV and −1.12 eV, respectively, relative to the emission spectrum of the calcined sample. Based on a previous study, the full reduction of Co ions in NiCoO is proposed to proceed by a two-step process whereby first Co3+ (Oh) is reduced to Co2+ and its electronic and spin configurations changes from a 3d6, S = 2 to 3d7, S = 3/2. Subsequently at elevated temperatures the remaining Co2+ (Td) and Co2+ (Oh) are fully reduced to metallic Co.24 Following the reduction at T ≤ 300 °C the XES spectra of the NiO vs. the NiCoO samples have different spectral shape and peak positions (Fig. 4 and S8, 9†). At 450 °C the shift in the emission spectra of NiCoO is reversed and moves towards higher emission energies. Further, by increasing the reduction temperature to 450 °C the Ni XES peak position for the NiO is shifted by additional −0.53 eV to lower energies with respect to the NiCoO spectrum and its Kβ′ peak intensity is slightly higher, thus suggesting the formation of distinct Ni species (Fig. S10†). To understand the origin of this behavior and test if the sample at 300 °C is simply a mixture of both the oxide phase and reduced Ni, the emission spectra for the calcined and reduced NiO sample at 450 °C were averaged in different ratios (Fig. S11†). The Kβ spectrum of the reduced NiCo catalyst at 450 °C is clearly different from any of other spectra presented in Fig. S11,† indicating that this is not simply a partially converted species, but rather that the NiCoO sample has a Ni site that is electronically distinct from the Ni only catalyst. Further, upon full reduction of the Ni ions in NiCoO at 600 °C the Kβ1,3 XES peak position for the sample at 600 °C is shifted by −0.45 eV relative to the calcined sample. This energy shift is smaller when compared to the NiO case and thus suggests a different reduction mechanism for the Ni ions in NiCoO. Further, the Kβ1,3 XES peak position for the fully reduced monometallic catalyst is shifted by ∼−0.5 eV relative to the bimetallic catalyst at 600 °C (Fig. S12 and Table S2†). These results confirm that the fully reduced NiO and NiCoO are not identical at 600 °C.
Finally, the XES spectra collected under dynamic flow of DMR gas mixture at 600 °C for both NiCoO and NiO indicate that a dominantly metallic Ni phase remains during the course of catalysis (Fig. 4). This observation is further supported by the XES data analysis in Fig. S13 and S14.† However, it is important to note that even during the DMR reaction the spectra of both catalysts remain somewhat different from each other, with the NiCoO catalyst appearing slightly more oxidized than the NiO catalyst at all stages. The changes that occur in the Ni local geometric and electronic structure may be key to understanding the increased activity of the NiCo catalyst relative to the Ni-only catalyst.
3.3. Ni Kβ HERFD of NiCoO/γ-Al2O3vs. NiO/γ-Al2O3
The Ni Kβ-detected HERFD XAS spectra for the calcined NiCoO/γ-Al2O3 and NiO/γ-Al2O3, during the reductive activation, and under DMR conditions are shown in Fig. 5. The Ni K-edge HERFD spectra for the calcined NiO and NiCoO samples are different in terms of their spectral shapes and their K-edge peak positions is at 8350.37 eV and 8350.40 eV, respectively. The pre-edge of the NiO catalyst has a higher intensity than the NiCoO catalyst, which suggests that the former has a more distorted local structure (Fig. S15†).48,50 The sensitivity of the pre-edge intensity to the local geometric structure derives from symmetry allowed Ni p–d mixing, which occurs as the site deviates from ideal centrosymmetry. In addition, based on the pre-edge peak position, rising edge position, and spectral shape of the Ni K-edge HERFD for the calcined NiCoO and NiO samples, the Ni-site in both calcined catalyst is best described as Ni2+.48,51 Ni HERFD XAS data acquired during the catalyst activation in the temperature range from 150 °C to 600 °C are shown in Fig. S16–19.† Due to the induced reduction of the Ni at 150 °C, the K-edge peak positions for both catalysts equally shift by −0.3 eV relative to the spectra for the calcined samples (Fig. 5). In contrast to the NiCoO case, following the reductive activation at 300 °C the pre-edge feature of the NiO sample diminishes and its K-edge peak position shifts to lower energies indicating a further reduction of the Ni and consistent with loss of oxygen from the crystal structure.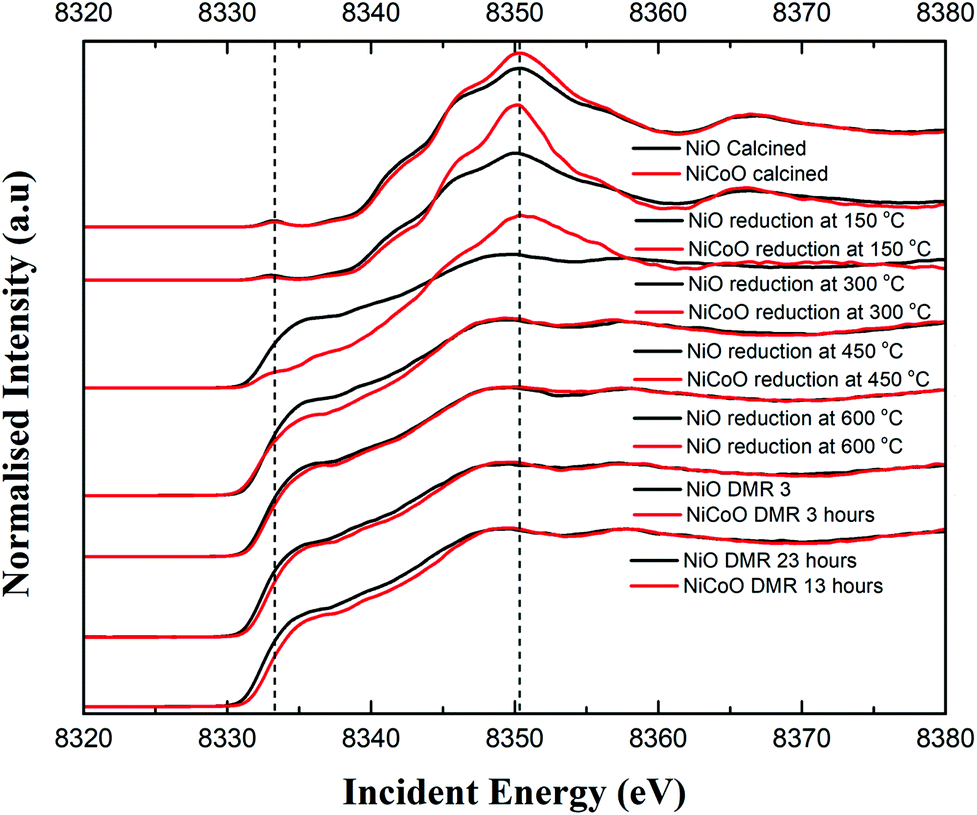 | ||
| Fig. 5 Ni Kβ-detected HERFD XAS spectra of NiO/γ-Al2O3 and NiCoO/γ-Al2O3 collected for the freshly calcined samples, during the reduction/activation step, and under DMR conditions. The pre-edge and main edge peak positions have been indicated with dashed lines. | ||
The most striking difference during the catalyst activation step is the full reduction of the Ni ions in the NiO catalyst at just below 300 °C, while for the NiCoO catalyst the full conversion to the metallic state just occurs at just below 450 °C (Fig. 5). We note that a one-to-one comparison to the TPR temperatures is limited by the 150 °C temperature steps that were taken for the XAS measurements.
To test if the more oxidized appearance of the Ni K-edge HERFD in the NiCoO catalyst (at 300 °C) is simply a result of incomplete conversion, a similar data analysis as in the case of XES spectra was performed by mixing the HERFD spectra of the calcined NiO and reduced sample at 450 °C (Fig. S20†). This analysis reveals that the EXAFS region of the spectrum for the reduced NiCoO samples at 450 °C and any of the obtained mixed spectra are not superimposable with the NiCoO catalyst at 300 °C. Therefore, we propose that this species in NiCoO is very likely a distinct Ni species, which results from the presence of Co in the catalyst. In addition, the overlaid HERFD spectra for the NiO and NiCoO samples at 450 °C indicate a different electronic structure (Fig. S18†). The HERFD spectra for reduced NiO and NiCoO at 600 °C remain distinct from each other. While both catalysts appear to be comprised of dominantly reduced metallic Ni, the NiCo catalysts shows a slightly larger contribution of oxidized Ni (Fig. S19†). Finally, HERFD data were continuously collected during exposure to DMR conditions and revealed the high stability of Ni in its metallic state under the DMR conditions (Fig. 5 and S21, 22†).
3.4. Co Kβ HERFD of NiCoO/γ-Al2O3
Co Kβ HERFD spectra for the NiCoO/γ-Al2O3 catalyst in the calcined form, activated form, and under the DMR conditions are presented in Fig. 6. Based on the HERFD spectrum and previous Co L-edge XAS data,24 the Co species in the freshly calcined sample is consistent with Co3O4.52 At 150 °C, the reduction of Co3+ (Oh) sites to most probably Co2+ oxide initiates, and the Co K-edge HERFD white line position dramatically shifts from 7729.8 eV to 7726.96 eV (ΔE = 2.8 eV). In the following step, Co HERFD data collected at 300 °C shows an additional shift of +1.6 eV to higher energies which is assigned to the reduction of the remaining Co2+ species to metallic cobalt. Following the full reduction to metallic Co at 450 °C and 600 °C, the Co HERFD spectrum white line peak position is shifted from 7730 eV to 7726.5 eV (Fig. S23†). Finally, upon exposing the NiCo catalyst to DMR conditions for 8 h the HERFD spectra remain unchanged and confirms the stability of metallic Co under the DMR conditions.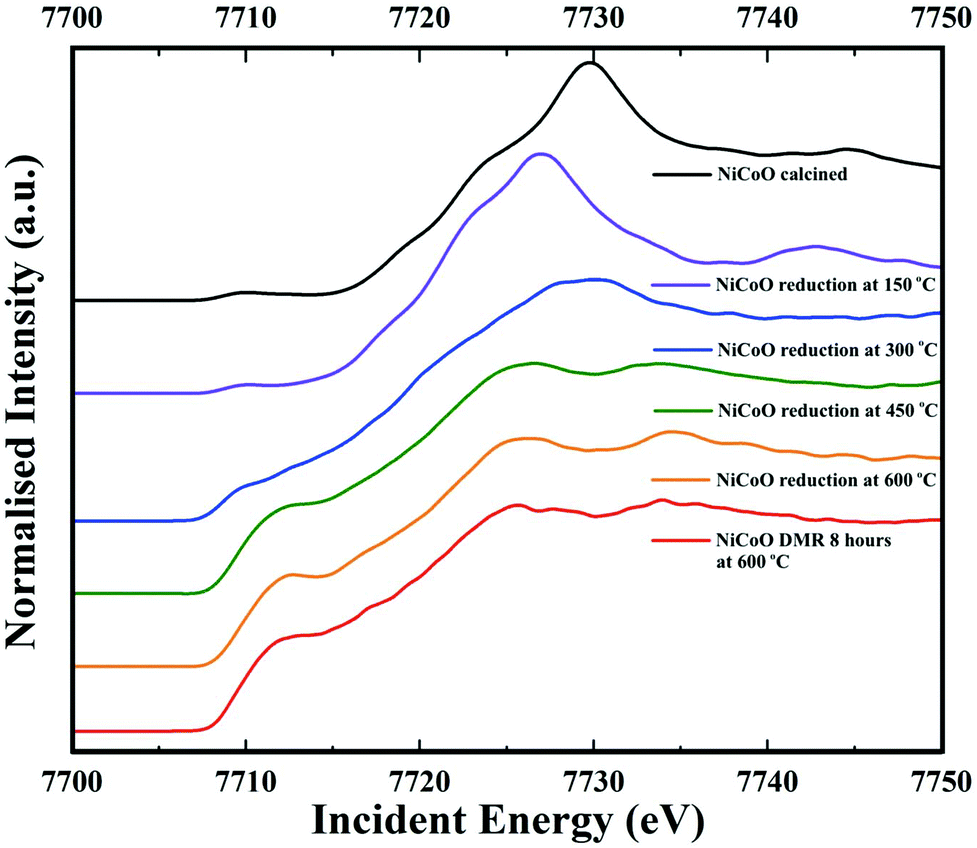 | ||
| Fig. 6 Co Kβ-detected HERFD spectra of the NiCoO/γ-Al2O3 sample measured under ambient, reduction activation (5% H2 in Ar), and DMR (7% CH4 and 9.5% CO2 diluted in N2) conditions. | ||
4. Conclusion
We studied the structural and electronic properties of NiO/γ-Al2O3 and NiCoO/γ-Al2O3 DMR catalysts in the calcined and reduced states and in situ under DMR conditions. DMR activity measurements revealed a significantly higher activity for the bimetallic NiCoO catalyst compared to the pure NiO catalyst. In situ XES spectroscopy for the calcined DMR catalysts showed subtle differences in the Ni Kβ mainline spectral shape and positions, which are attributed to differences in site symmetry and/or covalency of the Ni sites upon addition of Co. Further, during the course of the reduction/activation of the catalyst, and also during the DMR reaction, the NiCoO catalyst appears to be slightly more oxidized when compared to the NiO catalyst. Kβ-detected HERFD XAS data revealed that the structure of the NiCoO/γ-Al2O3 catalyst is less distorted (based on the pre-edge intensities) relative to the NiO/γ-Al2O3 catalyst. In addition, Co in NiCoO was identified as being structural analogous to Co3O4. Based on both Ni Kβ XES and HERFD spectra, it was shown that due to the presence of Co in the NiCoO sample, the Ni ions under reductive activation conditions are reduced at relatively higher temperatures as compared to the NiO catalyst. Following the reduction of the NiCoO catalyst at 300 °C, a Ni species with a distinct geometric and electronic structure is formed. At 600 °C, both catalysts are activated and the metal oxides are largely reduced to the metallic phase. Both catalysts remain unchanged during DMR, but distinct differences remain between the Ni-only and the NiCo catalyst, which provide experimental evidence that the presence of Co modulates the Ni geometric and electronic structure.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the Max Planck Society and the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013) ERC Grant Agreement No. 615414 (S.D.). We acknowledge F.M.F de Groot and A. van der Eerden from Utrecht University for providing the hard X-ray reactor and Christian W. Lehmann from the Max-Planck-Institut für Kohlenforschung for supporting the STEM measurements. Open Access funding provided by the Max Planck Society.References
- A. Lee, J. Bennett, J. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43, 7887–7916 RSC.
- C. Descorme, P. Gallezot, C. Geantet and C. George, ChemCatChem, 2012, 4, 1897–1906 CrossRef CAS.
- P. Anastas and J. Warner, Green Chemistry, Oxford University Press, New York, 1998 Search PubMed.
- P. Mohanty, K. Pant, S. Naik, J. Parikh, A. Hornung and J. Sahu, Renewable Sustainable Energy Rev., 2014, 38, 131–153 CrossRef CAS.
- R. de Richter, T. Ming, P. Davies, W. Liu and S. Caillol, Prog. Energy Combust. Sci., 2017, 60, 68–96 CrossRef.
- W. Chen, T. Suzuki and M. Lackner, Handbook Of Climate Change Mitigation And Adaptation; Springer, Cham, 2016, pp. 2827–2880 Search PubMed.
- J. Lavoie, Front. Chem., 2014, 2, 81 Search PubMed.
- Y. Wang, L. Yao, S. Wang, D. Mao and C. Hu, Fuel Process. Technol., 2018, 169, 199–206 CrossRef CAS.
- B. Hua, N. Yan, M. Li, Y. Sun, J. Chen, Y. Zhang, J. Li, T. Etsell, P. Sarkar and J. Luo, J. Mater. Chem. A, 2016, 4, 9080–9087 RSC.
- J. Han, C. Kim, J. Park and H. Lee, ChemSusChem, 2014, 7(2), 451–456 CrossRef CAS PubMed.
- P. Djinović, I. Osojnik Črnivec, B. Erjavec and A. Pintar, Appl. Catal., B, 2012, 125, 259–270 CrossRef.
- S. Arora and R. Prasad, RSC Adv., 2016, 6(110), 108668–108688 RSC.
- O. Muraza and A. Galadima, Int. J. Energy Res., 2015, 39, 1196–1216 CrossRef.
- A. Şener, M. Günay, A. Leba and R. Yildirim, Catal. Today, 2018, 299, 289–302 CrossRef.
- M. Usman, W. Wan Daud and H. Abbas, Renewable Sustainable Energy Rev., 2015, 45, 710–744 CrossRef CAS.
- A. Budiman, S. Song, T. Chang, C. Shin and M. Choi, Catal. Surv. Asia, 2012, 16, 183–197 CrossRef CAS.
- J. Rostrupnielsen and J. Hansen, J. Catal., 1993, 144, 38–49 CrossRef CAS.
- J. Chang, S. Park and H. Chon, Appl. Catal., A, 1996, 145(1–2), 111–124 CrossRef CAS.
- J. Juan-Juan, M. Román-Martínez and M. Illán-Gómez, Appl. Catal., A, 2006, 301(1), 9–15 CrossRef CAS.
- M. Fan, A. Abdullah and S. Bhatia, ChemSusChem, 2011, 4(11), 1643–1653 CrossRef CAS PubMed.
- T. Huang, W. Huang, J. Huang and P. Ji, Fuel Process. Technol., 2011, 92(10), 1868–1875 CrossRef CAS.
- J. Zhang, H. Wang and A. Dalai, J. Catal., 2007, 249, 300–310 CrossRef CAS.
- J. Zhang, H. Wang and A. Dalai, Appl. Catal., A, 2008, 339, 121–129 CrossRef CAS.
- A. Beheshti Askari, M. al Samarai, B. Morana, L. Tillmann, N. Pfänder, A. Wandzilak, B. Watts, R. Belkhou, M. Muhler and S. DeBeer, ACS Catal., 2020, 6223–6230 CrossRef CAS PubMed.
- P. Glatzel and U. Bergmann, Coord. Chem. Rev., 2005, 249, 65–95 CrossRef CAS.
- P. Glatzel, G. Smolentsev and G. Bunker, J. Phys.: Conf. Ser., 2009, 190, 012046 CrossRef.
- F. de Groot and A. Kotani, Core Level Spectroscopy Of Solids, CRC Press, Boca Raton, 2008 Search PubMed.
- C. Natoli, M. Benfatto, S. Della Longa and K. Hatada, J. Synchrotron Radiat., 2002, 10, 26–42 CrossRef PubMed.
- R. Castillo, R. Banerjee, C. Allpress, G. Rohde, E. Bill, L. Que, J. Lipscomb and S. DeBeer, J. Am. Chem. Soc., 2017, 139(49), 18024–18033 CrossRef CAS PubMed.
- M. Baker, M. Mara, J. Yan, K. Hodgson, B. Hedman and E. Solomon, Coord. Chem. Rev., 2017, 345, 182–208 CrossRef CAS PubMed.
- C. Pollock and S. DeBeer, Acc. Chem. Res., 2015, 48(11), 2967–2975 CrossRef CAS PubMed.
- J. Kowalska, F. Lima, C. Pollock, J. Rees and S. DeBeer, Isr. J. Chem., 2016, 56(9–10), 803–815 CrossRef CAS.
- C. Pollock, M. Delgado-Jaime, M. Atanasov, F. Neese and S. DeBeer, J. Am. Chem. Soc., 2014, 136(26), 9453–9463 CrossRef CAS PubMed.
- M. al Samarai, A. Hahn, A. Beheshti Askari, Y. Cui, K. Yamazoe, J. Miyawaki, Y. Harada, O. Rüdiger and S. DeBeer, ACS Appl. Mater. Interfaces, 2019, 11(42), 38595–38605 CrossRef CAS PubMed.
- P. Menezes, A. Indra, O. Levy, K. Kailasam, V. Gutkin, J. Pfrommer and M. Driess, Chem. Commun., 2015, 51, 5005–5008 RSC.
- L. Tillmann, J. Schulwitz, A. van Veen and M. Muhler, Catal. Lett., 2018, 148, 2256–2226 CrossRef CAS.
- M. Al Samarai, M. Delgado-Jaime, H. Ishii, N. Hiraoka, K. Tsuei, J. Rueff, B. Lassale-Kaiser, B. Weckhuysen and F. de Groot, J. Phys. Chem. C, 2016, 120, 24063–24069 CrossRef CAS.
- M. Cesário, C. Gennequin, E. Abi-Aad and D. de Macedo, Catalytic Materials for Hydrogen Production and Electro-Oxidation Reactions, Bentham Science Publishers, 2018, p. 140 Search PubMed.
- D. Wang, S. Li, Y. Du, X. Wu and Y. Chen, Catalysts, 2019, 9, 352 CrossRef CAS.
- Y. Ji, Z. Zhao, A. Duan, G. Jiang and J. Liu, J. Phys. Chem. C, 2009, 113, 7186–7199 CrossRef CAS.
- T. Nyathi, N. Fischer, A. York and M. Claeys, Faraday Discuss., 2017, 197, 269–285 RSC.
- X. Zhang, J. Liu, Y. Jing and Y. Xie, Appl. Catal., A, 2003, 240, 143–150 CrossRef CAS.
- Y. Kobayashi, J. Horiguchi, S. Kobayashi, Y. Yamazaki, K. Omata, D. Nagao, M. Konno and M. Yamada, Appl. Catal., A, 2011, 395, 129–137 CrossRef CAS.
- J. Horlyck, C. Lawrey, E. Lovell, R. Amal and J. Scott, Chem. Eng. J., 2018, 352, 572–580 CrossRef CAS.
- K. Mette, S. Kühl, H. Düdder, K. Kähler, A. Tarasov, M. Muhler and M. Behrens, ChemCatChem, 2013, 6, 100–104 CrossRef.
- H. Düdder, K. Kähler, B. Krause, K. Mette, S. Kühl, M. Behrens, V. Scherer and M. Muhler, Catal. Sci. Technol., 2014, 4, 3317–3328 RSC.
- J. Sá, Y. Kayser, C. Milne, D. Abreu Fernandes and J. Szlachetko, Phys. Chem. Chem. Phys., 2014, 16, 7692 RSC.
- S. Hugenbruch, H. Shafaat, T. Krämer, M. Delgado-Jaime, K. Weber, F. Neese, W. Lubitz and S. DeBeer, Phys. Chem. Chem. Phys., 2016, 18(16), 10688–10699 RSC.
- X. You, X. Wang, Y. Ma, J. Liu, W. Liu, X. Xu, H. Peng, C. Li, W. Zhou, P. Yuan and X. Chen, ChemCatChem, 2014, 6(12), 3377–3386 CrossRef CAS.
- E. Krüger , arXiv:1911.08190v1.
- S. Bare, F. Modica and A. Ringwelski, J. Synchrotron Radiat., 1999, 6, 436–438 CrossRef CAS PubMed.
- A. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107(5), 1692–1744 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr01960g |
| This journal is © The Royal Society of Chemistry 2020 |