
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single molecule binding of a ligand to a G-protein-coupled receptor in real time using fluorescence correlation spectroscopy, rendered possible by nano-encapsulation in styrene maleic acid lipid particles†
Rachael L.
Grime
 ab,
Joelle
Goulding
bc,
Romez
Uddin
d,
Leigh A.
Stoddart
bc,
Stephen J.
Hill
bc,
David R.
Poyner
d,
Stephen J.
Briddon
bc and
Mark
Wheatley
*be
ab,
Joelle
Goulding
bc,
Romez
Uddin
d,
Leigh A.
Stoddart
bc,
Stephen J.
Hill
bc,
David R.
Poyner
d,
Stephen J.
Briddon
bc and
Mark
Wheatley
*be
aSchool of Biosciences, University of Birmingham, Birmingham, B15 2TT, UK
bCentre of Membrane Proteins and Receptors (COMPARE), University of Birmingham and University of Nottingham, Midlands, UK
cDivision of Physiology, Pharmacology and Neuroscience, School of Life Sciences, University of Nottingham, Nottingham, NG7 2UH, UK
dLife and Health Sciences, Aston University, Birmingham B4 7ET, UK
eCentre for Sport, Exercise and Life Sciences, Faculty of Health and Life Sciences, Alison Gingell Building, Coventry University, Coventry, CV1 2DS, UK. E-mail: mark.wheatley@coventry.ac.uk
First published on 19th May 2020
Abstract
The fundamental importance of membrane proteins in cellular processes has driven a marked increase in the use of membrane mimetic approaches for studying and exploiting these proteins. Nano-encapsulation strategies which preserve the native lipid bilayer environment are particularly attractive. Consequently, the use of poly(styrene co-maleic acid) (SMA) has been widely adopted to solubilise proteins directly from cell membranes by spontaneously forming “SMA Lipid Particles” (SMALPs). G-protein-coupled receptors (GPCRs) are ubiquitous “chemical switches”, are central to cell signalling throughout the evolutionary tree, form the largest family of membrane proteins in humans and are a major drug discovery target. GPCR-SMALPs that retain binding capability would be a versatile platform for a wide range of down-stream applications. Here, using the adenosine A2A receptor (A2AR) as an archetypical GPCR, we show for the first time the utility of fluorescence correlation spectroscopy (FCS) to characterise the binding capability of GPCRs following nano-encapsulation. Unbound fluorescent ligand CA200645 exhibited a monophasic autocorrelation curve (dwell time, τD = 68 ± 2 μs; diffusion coefficient, D = 287 ± 15 μm2 s−1). In the presence of A2AR-SMALP, bound ligand was also evident (τD = 625 ± 23 μs; D = 30 ± 4 μm2 s−1). Using a non-receptor control (ZipA-SMALP) plus competition binding confirmed that this slower component represented binding to the encapsulated A2AR. Consequently, the combination of GPCR-SMALP and FCS is an effective platform for the quantitative real-time characterisation of nano-encapsulated receptors, with single molecule sensitivity, that will have widespread utility for future exploitation of GPCR-SMALPs in general.
Introduction
Membrane proteins have evolved to function within the unique environment of the hydrated membrane bilayer, which locates proteins in close association with lipids and provides lateral pressure. Extracting membrane proteins from this bilayer for purification and characterisation has, until recently, universally required detergents. This detergent solubilisation strips away closely-associated lipids, removes lateral pressure and perturbs the protein conformation, resulting in protein instability in the detergent micelle.1,2 This is particularly problematic for highly dynamic membrane proteins such as G-protein-coupled receptors (GPCRs).3 GPCRs all share a common protein architecture of a bundle of seven transmembrane helices and adopt a wide spectrum of conformational states in executing their cell signalling role.4 They are found in organisms throughout the phylogenetic tree including humans, fish, insects, plants, slime-moulds and viruses (but not bacteria) and form the largest class of ‘chemical switches’ in biology. They transduce signals from chemical messengers acting on a cell, such as hormones and neurotransmitters, into biochemical changes within the cell via activation of intracellular signalling cascades.5 As a result, they regulate almost every physiological process. Consequently, GPCRs are the largest class of membrane proteins in the human genome (with >800 receptors). Furthermore, they are very important to the pharmaceutical industry as they are the therapeutic target of 30–40% of clinically-prescribed drugs.6,7In recent years, a detergent-free method has been developed for solubilising membrane proteins using poly(styrene co-maleic acid) (SMA). SMA spontaneously incorporates into membranes to generate nanoscale sections of the lipid bilayer as discs (∼10 nm in diameter) containing encapsulated membrane protein, referred to as ![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif) tyrene
tyrene ![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) aleic
aleic ![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) cid
cid ![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif) ipid
ipid ![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) articles (SMALPs).8–12 Nanoscale encapsulation of GPCRs in SMALPs (GPCR-SMALPs) has potential utility for facilitating a wide range of downstream approaches such as; supporting biophysical10,11 and structural studies,13,14 a platform for discovery of GPCR-targeted therapeutic antibodies using phage display libraries, and for high-throughput screening in drug discovery following immobilisation of GPCR-SMALPs on surface plasmon resonance (SPR) chips.3 In addition, there is potential for harnessing the exquisite ligand-recognition capability of GPCR-SMALPs into bespoke molecular detection devices using synthetic biology. A pre-requisite to all of these applications is establishing that the ligand-binding capability of the encapsulated GPCR is preserved in the GPCR-SMALP.
articles (SMALPs).8–12 Nanoscale encapsulation of GPCRs in SMALPs (GPCR-SMALPs) has potential utility for facilitating a wide range of downstream approaches such as; supporting biophysical10,11 and structural studies,13,14 a platform for discovery of GPCR-targeted therapeutic antibodies using phage display libraries, and for high-throughput screening in drug discovery following immobilisation of GPCR-SMALPs on surface plasmon resonance (SPR) chips.3 In addition, there is potential for harnessing the exquisite ligand-recognition capability of GPCR-SMALPs into bespoke molecular detection devices using synthetic biology. A pre-requisite to all of these applications is establishing that the ligand-binding capability of the encapsulated GPCR is preserved in the GPCR-SMALP.
It is noteworthy that in recent years there has been a rapid increase in the design of fluorescent ligands for quantitative characterisation of binding to GPCRs.15–17 This development has enabled fluorescence correlation spectroscopy (FCS) to be applied to studying GPCRs in cells.18 FCS is a quantitative technique with single molecule sensitivity which uses confocal optics and a high numerical aperture objective lens to generate a small, defined illumination volume (∼0.25 fL). As a fluorescent species moves through the detection volume, fluctuations in fluorescent intensity are recorded in real time. Statistical analysis of the time-dependency of these fluctuations using autocorrelation analysis allows the average dwell time of the fluorescent moiety within the detection volume to be determined.18 Additionally, the amplitude of the autocorrelation curve is inversely proportional to the average concentration of fluorescent particles in the detection volume. This makes FCS particularly sensitive for investigating low concentrations of particles, a definite advantage for studying low-abundance targets such as GPCRs. A property of FCS is that the dwell time (τD) is proportional to the cube root of the molecular mass, so a doubling of mass only increases τD by 1.3-fold. When two fluorescent species are present in the detection volume simultaneously, they can only be resolved by FCS if the difference in their masses is large enough to yield a difference in τD values of at least 1.6-fold.19 Consequently, this combination of fluorescent probes and FCS has proven to be highly effective for studying GPCRs embedded in the surface of live cells.20–24 In this study we establish that nano-encapsulation of a GPCR within a SMALP provides the required size differential to make this a versatile strategy for quantitative analysis of ligand![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GPCR complex formation by FCS. In addition, it is likely that the utility of FCS is not restricted to SMALPs but could apply more widely to GPCRs embedded in nanoscale discs of membrane irrespective of the method of encapsulation.
:GPCR complex formation by FCS. In addition, it is likely that the utility of FCS is not restricted to SMALPs but could apply more widely to GPCRs embedded in nanoscale discs of membrane irrespective of the method of encapsulation.
The adenosine A2A receptor (A2AR) is a typical GPCR with a well-defined pharmacology. It belongs to a family of four GPCRs (A1R, A2AR, A2BR, A3R) that mediate the actions of adenosine and are attractive drug targets.25 The A2AR regulates the release of the neurotransmitters dopamine and glutamate in the brain, regulates blood flow to cardiac muscle and is the target for the most widely used psychoactive drug – caffeine, which blocks this receptor.26 In this study, utilising the human A2AR as the GPCR, we demonstrate for the first time, the application of FCS to provide quantitative data on the binding characteristics of a GPCR that has been purified embedded within the nanoscale bilayer of the SMALP. This SMALP strategy ensures that the native lipid in close-association with the receptor protein has never been disrupted by detergent at any stage. Our study not only establishes the utility of employing FCS for characterising GPCR-SMALPs, but opens up the possibility of exploiting high-throughput solution-based FCS,27 and highlights the potential for identifying novel fluorescent ligands targeted to specific GPCRs. In recent years, SMA has been widely used to solubilise structurally-diverse membrane proteins from a wide range of species (including bacteria, yeast, plants, insect cells and mammalian cells). The application of SMA for studying GPCRs and other membrane proteins is increasing rapidly as the utility of SMALPs becomes ever more apparent. Furthermore, ‘second-generation’ SMA-like polymers have already been reported that possess different properties to SMA, so the field is still expanding.28–30 The application of FCS as a quantitative, real-time technique, with single molecule sensitivity will have widespread utility for the development of down-stream applications of SMALPs as an investigation platform in this growing field.
Experimental
Materials
CA200645 was supplied by HelloBio (Bristol, UK). SMA2000 anhydride was from Cray Valley (UK). ZM241385 {4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5yl amino]ethyl)phenol} was purchased from Tocris.Human A2AR expression
Human A2AR was expressed in Pichia pastoris as described previously.11 Prior to SMA-extraction, cells were disrupted following suspension in breaking buffer (50 mM sodium-phosphate buffer, 100 mM NaCl, 5% glycerol, EDTA-free protease inhibitor, pH 7.5, 4 °C) by 3–5 passes using an Avestin-C3 cell-disrupter. Unbroken cells and debris were removed by centrifugation (5000g, 10 min, 4 °C). The A2AR-expressing membrane fraction was then sedimented (100000g, 60 min, 4 °C) and re-suspended to 80 mg mL−1 (wet weight) in extraction buffer (300 mM NaCl, 20 mM HEPES, pH 7.5). Membranes were stored at −80 °C until needed.
Generation of A2AR-SMALPs
SMA was prepared from SMA anhydride and used to solubilise A2AR from membranes as described previously.11,12 Briefly, A2AR-expressing membrane preparations were thawed on ice, and an equal volume of 2× SMA buffer (5% w/v SMA, 300 mM NaCl, 20 mM HEPES, EDTA-free protease inhibitor, pH 7.5) added to yield a final concentration of 40 mg mL−1 (wet weight) in 2.5% (w/v) SMA. Following gentle agitation for 1 h at room temperature, non-solubilised material was removed by centrifugation (100000g, 60 min, 4 °C) to yield a supernatant containing A2AR-SMALPs.
Purification of A2AR-SMALPs
All purification steps were carried out at 4 °C. The A2AR-SMALP supernatant was incubated with ∼1 mL Ni2+-NTA resin, overnight on an end-over-end rotator. The column was washed with 20 column volumes (cv) of wash buffer (300 mM NaCl, 20 mM HEPES, 25 mM imidazole, EDTA-free protease inhibitor pH 7.5). Elution of A2AR-SMALP was achieved with 10 cv of elution buffer (300 mM NaCl, 20 mM HEPES, 250 mM imidazole, EDTA-free protease inhibitor pH 7.5). Elution fractions were pooled, buffer-exchanged into assay buffer (300 mM NaCl, 20 mM HEPES, pH 7.5) and concentrated using spin-concentrators (10 kDa cut-off). Concentrations of purified A2AR-SMALP were determined using SDS-PAGE and densiometric analysis against protein standards in ImageJ.31 Final concentrations ranged between 0.2–1 mg mL−1.FCS protocols
Solution-based FCS was performed essentially as described previously.15 FCS measurements were carried out on Nunc Lab-Tek 8-well chambered #1.0 cover-glasses (Thermo Scientific, UK), using a ZEISS LSM510 Confocor3 using 633 nm excitation (0.5–1 kW cm−2), with emission collected through an LP650 filter. The confocal volume was placed 200 μm above the surface of the coverslip. Cy5 NHS ester (Amersham Pharmacia Bioscience) was prepared in high performance liquid chromatography grade water (Chromasolv, Sigma-Aldrich, UK). As Cy5 has a known diffusion coefficient (D = 3.16 × 10−10 m2 s−1), it was employed as the standard fluorescent dye (10 nM and 500 nM) to calculate the confocal volume dimensions using eqn (1), where V = volume (mL),![[scr W, script letter W]](https://www.rsc.org/images/entities/char_e536.gif) 1 is the radius of the confocal volume, determined from 1 = (4·D·τD1)½, where D and τD1 are the diffusion coefficient and dwell time of Cy5, respectively. 2 represents half the height of the confocal volume and is calculated by multiplying 1 by the structural parameter (S). Calibration readings were taken on each day's experiments. FCS measurements were performed in a final assay volume of 200 μL. A2AR-SMALPs were incubated with CA200645 (25 nM), in the presence or absence of varying concentrations of ZM241385 as stated. Equilibrium was established at 30 min and four read-times, each of 10 s, were employed to record time-dependent fluctuations in fluorescent intensity.
1 is the radius of the confocal volume, determined from 1 = (4·D·τD1)½, where D and τD1 are the diffusion coefficient and dwell time of Cy5, respectively. 2 represents half the height of the confocal volume and is calculated by multiplying 1 by the structural parameter (S). Calibration readings were taken on each day's experiments. FCS measurements were performed in a final assay volume of 200 μL. A2AR-SMALPs were incubated with CA200645 (25 nM), in the presence or absence of varying concentrations of ZM241385 as stated. Equilibrium was established at 30 min and four read-times, each of 10 s, were employed to record time-dependent fluctuations in fluorescent intensity.
Data analysis
Autocorrelation analysis was performed using Zeiss AIM 4.2 software. Cy5 calibration data were used to calibrate the detection volume as previously described.15 Experimental data were fitted to eqn (2), where N = particle number, fi is fraction of ith component, τDi is dwell time of ith component, S = structure parameter (ratio of diameter to height of volume). A 1-component or 2-component 3-D diffusion model was used incorporating a triplet state component fitted using a pre-exponential. Fit quality was assessed on residuals to the fit. For 2-component fits, the first component (τD1), representing free ligand, was fixed during the fitting process to the value determined for CA200645 alone, and the second component (τD2) represented bound ligand. The concentrations of free and bound components were then calculated directly from their relative contributions to the amplitude of the autocorrelation function. Specific binding was determined using eqn (3). Total binding was defined as N(τD2) (nM) as calculated from the autocorrelation analysis curve for A2AR-SMALPs in the absence of competing ligand. Non-specific binding was defined as N(τD2) (nM) calculated from the autocorrelation analysis curve for A2AR-SMALPs in the presence of saturating competing ligand. The affinity (pKi) of ZM241385 binding to the A2AR-SMALP was determined from the IC50 value of the ZM241385 competition binding curve following correction for occupancy by the tracer ligand CA200645.32 The Stokes–Einstein equation (eqn (4)) was utilised to probe the relationship between the diffusion time of the SMALP nanoparticle and its’ hydrodynamic radius; where D = diffusion coefficient, kB is Boltzmann's constant, T = temperature (K), η = dynamic viscosity and r = radius of the particle. Pooled data are presented as the mean ± s.e.m. for 3, or 4, independent preparations of A2AR-SMALP. Significance (p < 0.05) was determined by Student's paired t-test.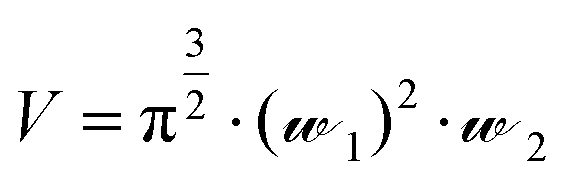 | (1) |
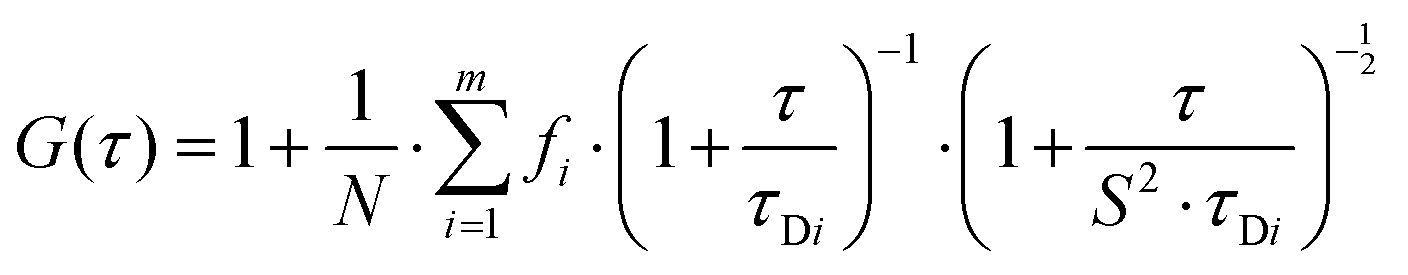 | (2) |
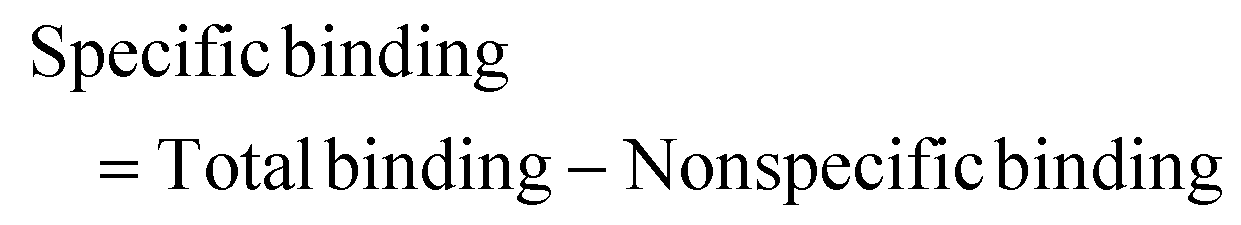 | (3) |
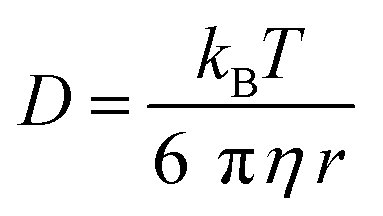 | (4) |
Results and discussion
A2AR-SMALPs provides a platform for FCS
The A2AR-SMALPs were generated using SMA (2.5% w/v) to extract the receptors from A2AR-expressing membranes into SMALPs, followed by purification of the A2AR-SMALPs as previously described (Fig. S1†).11 The current studies utilized the fluorescent ligand CA200645, which comprises the adenosine receptor antagonist xanthine amine congener (XAC) linked to a red BODIPY630/650 fluorophore via a β-alanine linker.33 CA200645 is a high affinity ligand for both the A1R and A3R subtypes33–35 and was used previously as the tracer ligand to investigate dimerization and allostery of A3R in whole live cells using FCS.20 Given that CA200645 is derived from the non-selective adenosine receptor antagonist XAC, it was hypothesized that CA200645 would not be A1R/A3R-selective and would also bind to the A2AR. Subsequent pharmacological characterization established that CA200645 bound to the A2AR with high affinity (42 nM; pA2 = 7.37 ± 0.17, n = 3; Fig. S2†).The initial stage of the investigation was to optimize the concentration of CA200645 for solution-based FCS, to ensure that specific binding to the A2AR encapsulated in the SMALP was high compared to non-specific binding. The CA200645 concentration was varied between 5–300 nM and non-specific binding determined in each case by adding a saturating concentration of a non-fluorescent competing A2AR ligand (ZM241385,36 1 μM). Specific binding varied between 20 ± 11% and 70 ± 12% of total binding (Fig. 1), with 25 nM CA200645 being used in all subsequent experiments. A data-collection time of 10 s was routinely employed which provided ample fluorescent fluctuation data for autocorrelation analysis.
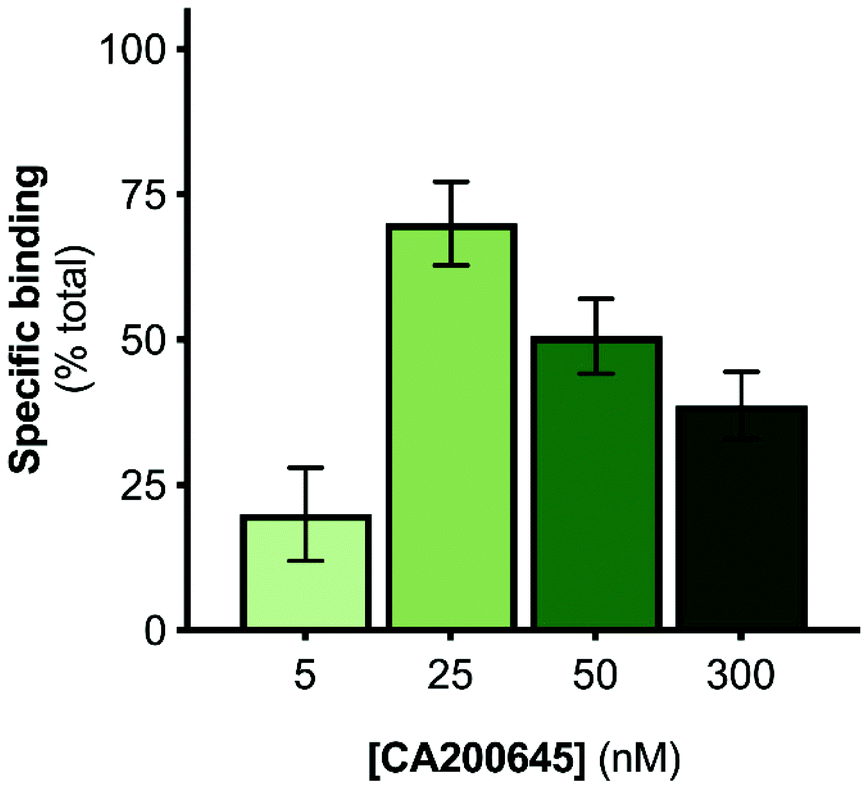 | ||
| Fig. 1 Optimisation of tracer ligand concentration for solution-based FCS with A2AR-SMALPs. The specific binding of CA200645 to A2AR encapsulated in a SMALP was determined at the stated concentrations. Non-specific binding was defined by a saturating concentration of ZM241385 (1 μM). Data are mean ± s.e.m. (n = 3). | ||
The initial FCS experiments were performed with CA200645 alone in assay buffer to determine the characteristics of the free ligand. Fluctuations in fluorescence intensity for CA200645 over time are shown in Fig. 2a. Autocorrelation analysis of these fluctuations revealed a monophasic autocorrelation curve (Fig. 2b) with an average dwell time (τD) of the fluorescent moiety within the detection volume of 68 ± 2 μs and a diffusion coefficient D = 287 ± 15 μm2 s−1, (mean ± s.e.m., n = 4) with residuals (Fig. 2c) confirming that there was no systematic deviation in the fitted curve. The experiments were then repeated using CA200645 in the presence of A2AR-SMALP. The fluorescence intensity fluctuations for CA200645 + A2AR-SMALP are shown in Fig. 2d. Analysis of these fluctuations revealed that the autocorrelation curve for CA200645 was significantly altered by the addition of A2AR-SMALP. In contrast to the monophasic curve observed for ligand alone (Fig. 2b), the autocorrelation curve for CA200645 + A2AR-SMALP was clearly biphasic (Fig. 2e) exhibiting fast and slow diffusing components (τD1 and τD2, respectively) with the residuals (Fig. 2f) confirming that there was no systematic deviation in the fitted curve. The faster diffusing component exhibited a dwell-time (τD1) consistent with the free ligand (Fig. 2b). The slower component had an average dwell-time (τD2) for CA200645 of 625 ± 23 μs and a diffusion coefficient D = 30 ± 4 μm2 s−1 (mean ± s.e.m., n = 4). As a control, A2AR-SMALPs alone were shown to have no detectable auto-fluorescence (Fig. S3†).
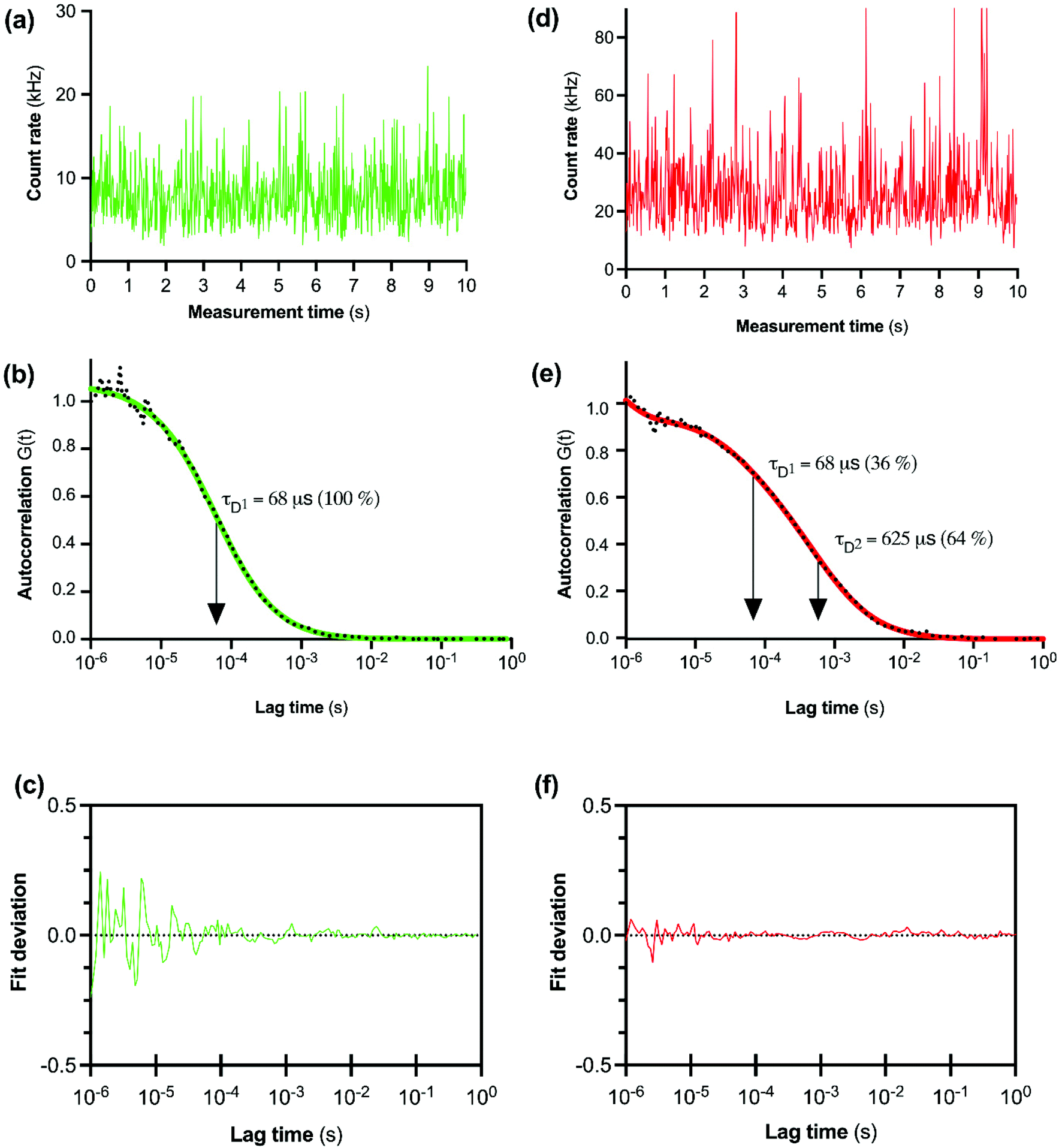 | ||
| Fig. 2 FCS analysis of CA200645 binding to purified A2AR-SMALP. (a) Fluctuations in fluorescent intensity with CA200645 alone. (b) Autocorrelation curve for CA200645 alone, fitted to a single-component diffusion model. (c) Deviation of data for CA200645 alone from the fitted curve. (d) Fluctuations in fluorescent intensity with CA200645 + A2AR-SMALP. (e) Autocorrelation curve for CA200645 + A2AR-SMALP fitted to a two-component diffusion model, in which the first component (τD1) was fixed to a three-dimensional diffusion rate of the free-ligand in solution. (f) Deviation of data for CA200645 + A2AR-SMALP from the fitted curve. | ||
Characterization of the slower diffusing component (τD2)
The logical explanation for the emergence of the slower diffusing component in the presence of A2AR-SMALP is that the ligand was binding to the receptor in the A2AR-SMALP and that both free and bound CA200645 were being detected by FCS simultaneously as τD1 and τD2 respectively (Fig. 2e). Indeed, the simultaneous determination of the free ligand concentration precisely where the binding event is occurring is a particular benefit of FCS for characterising ligand:GPCR complexes. The binding of CA200645 to the A2AR-SMALP could have been specific binding to the encapsulated GPCR or could possibly have been non-specific binding caused by partitioning of CA200645 into the lipid bilayer of the SMALP or interaction of the ligand with the SMA polymer surrounding the SMALP. The pharmacological properties of the receptor were exploited to establish the nature of the binding. A competing A2AR-selective ligand ZM241385 was used at a saturating concentration. This would fully occupy the available A2AR binding sites and prevent binding of CA200645 but would not prevent any non-specific binding. The τD2 component of the autocorrelation curve for CA200645 + A2AR-SMALP in the absence, and presence, of ZM241385 (1 μM) is shown in Fig. 3. In the presence of ZM241385, the τD2 component particle number (N) decreased by 62%, as the ZM241385 competed for the ligand binding site of the receptor and prevented CA200645 binding. In contrast, there was no effect of ZM241385 (1 μM) in control experiments performed using a different membrane protein encapsulated in a SMALP (ZipA, derived from Escherichia coli). ZipA-SMALP showed a lower particle number than A2AR-SMALP consistent with low CA200645 binding expected in the absence of receptor (Fig. 3).
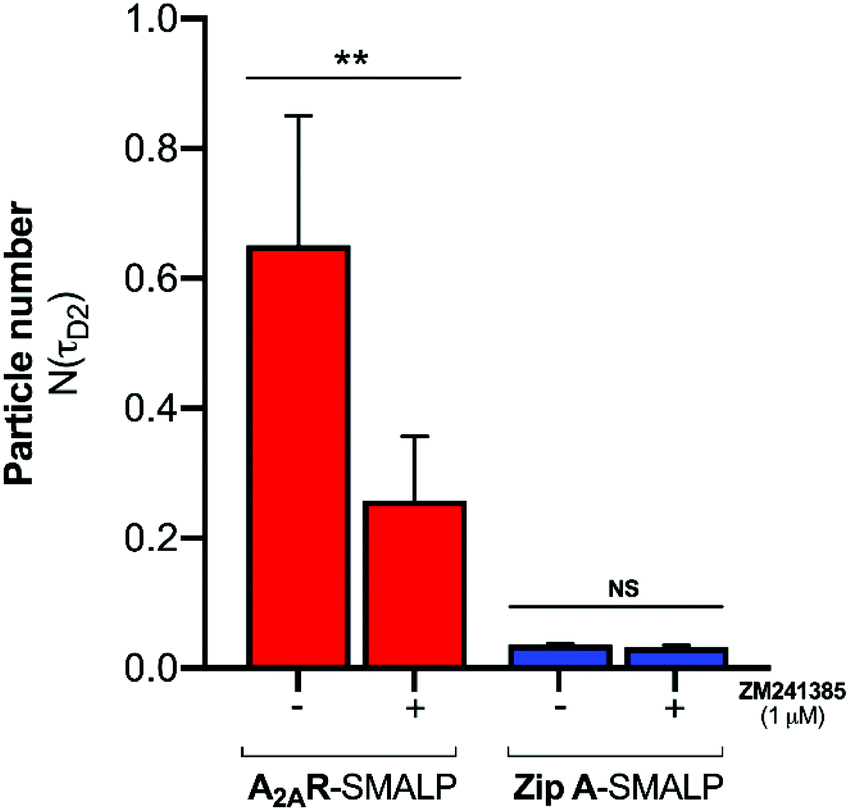 | ||
| Fig. 3 Specific binding of CA200645 to A2AR-SMALP detected with FCS. The particle number of the (τD2) component of the autocorrelation curve is shown for CA200645 (25 nM) binding to A2AR-SMALP or to ZipA-SMALP, in the absence (−) or presence (+) of a saturating concentration of competing ligand ZM241385 (1μM). **p < 0.01, NS = not significant. Data are mean ± s.e.m. (n = 3). | ||
A range of ZM241385 concentrations between 10−12 M–10−6 M was used to construct a competition curve for ZM241385 binding to the nano-encapsulated A2AR using FCS with CA200645 as tracer ligand (Fig. 4). From these data, the affinity (pKi) of ZM241385 for the A2AR-SMALP was calculated as 8.2 ± 0.5 (mean ± s.e.m., n = 3). This is not significantly different to the reported affinity of ZM241385 binding to A2AR in the original membranes (pKi = 7.95 ± 0.45).11
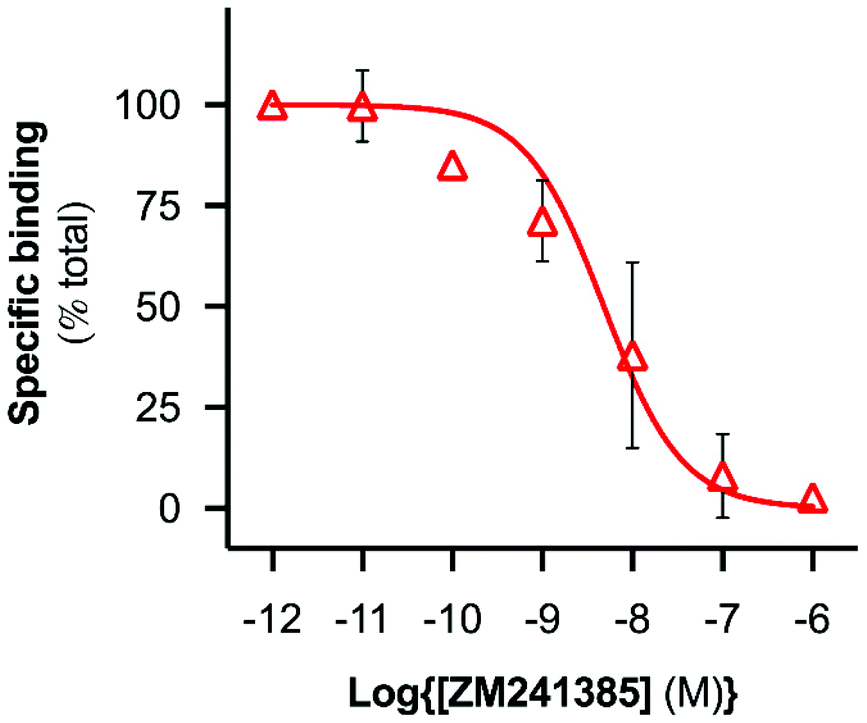 | ||
| Fig. 4 Competition binding curve derived using FCS with A2AR-SMALP. CA200645 (25 nM) was used as fluorescent tracer ligand in FCS experiments with A2AR-SMALP in the presence of ZM421385 at the concentrations indicated. Data are mean ± s.e.m. (n = 3). | ||
The diffusion coefficient of the CA200645:A2AR-SMALP complex (D = 30 ± 4 μm2 s−1) was used to estimate the hydrodynamic radius of the A2AR-SMALP as calculated by the Stokes–Einstein equation (eqn (4)), with the caveat that SMALPs are disc-shaped rather than spherical. The resulting values of 7–9 nm were consistent with the published values for the SMALP diameter of c.10 nm determined by a wide range of biophysical techniques, including small angle neutron scattering (SANS), electron microscopy (EM), attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR), differential scanning calorimetry (DSC) and nuclear magnetic resonance spectroscopy (NMR).10,37
Numerous applications of FCS with GPCR-SMALPs can be envisaged. For example, allosteric modulators of GPCRs bind to sites on the receptor discrete from the ‘classical’ ligand binding site and are of current interest to the pharmaceutical industry as they have therapeutic potential for ‘tuning’ receptor signalling (up or down).38 The size differential between GPCR-SMALPs and fluorescent ligands is sufficiently large that FCS will be a useful tool in the future for investigating potential allosteric modulators as it is compatible with a very wide range of modulator sizes from small molecules to therapeutic antibodies. Of course other approaches for nano-encapsulation of GPCRs, such as reconstituted lipid nanodiscs stabilised by an annulus of membrane scaffolding proteins39 or the use of recently-reported SMA-like polymers,28–30 may also be compatible with characterisation using FCS.
The application of SMALPs to studying membrane proteins is expanding rapidly40 and recently SMALPs have been used to obtain high resolution structures of membrane proteins; including crystallisation of the proton pump bacteriorhodopsin transferred from SMALPs into lipidic cubic phase for in meso crystalisation13 and the cryo-EM structure of bacterial alternative complex III in a super-complex with cytochrome oxidase encapsulated in a SMALP.14 Combining the technologies of fluorescence imaging and SMALP-solubilisation for studying membrane proteins is becoming increasingly versatile. For example, using proteins fluorescently labelled via introduced unnatural amino acids to enable single-molecule fluorescence studies and using FCS to confirm the existence of dimers of the transporter protein ABCG2 and its interaction with substrate.41,42 This expanding utility of SMALPs has been the driver for developing new amphipathic co-polymers that retain the ability of SMA to solubilise membrane proteins but exhibit different physico-chemical characteristics to SMA. For example, styrene-co-maleimide (SMI) is positively-charged and retains functionality at acidic pH, in contrast to the negative charge and alkaline working range of SMA;28 styrene maleimide quarternary ammonium (SMA-QA) forms relatively large nanodiscs (∼30 nm diameter) that remain stable between pH 2.5–pH 10;29 diisobutylene-co-maleic acid (DIBMA) in which the styrene aromatic ring of SMA is replaced by the aliphatic diisobutylene thereby changing how the polymer interacts with the lipid in the nanoparticles30 and thiolated SMA (SMA-SH) which can be derivatised or immobilised via the introduced thiol.43 All of these ‘second generation’ polymers generate lipid nanoparticles in the complete absence of detergent so preserve the native lipid in close association with the membrane protein. This is particularly important for GPCRs, as it has been shown that they can be regulated by the juxtaposition of specific membrane lipids. For example, phosphatidylethanolamine and phosphatidylglycerol favour inactive and active conformational states of the β2-adrenergic receptor (β2-AR), respectively.44 Likewise, cholesterol can modulate receptor conformation45 and function.46,47 Indeed a specific cholesterol binding site incorporating a ‘cholesterol consensus motif’ has been proposed for some GPCRs following the identification of cholesterol in GPCR crystal structures.48 In addition, the selectivity of the β1-AR for signalling via the G-protein Gs, in preference to other G-proteins, is enhanced by the membrane lipid phosphatidylinositol 4,5 bisphosphate (PIP2) but not by structural homologues. Using engineered ‘mini-Gα’ constructs, formation of β1-AR:mini-Gs complex was stabilised by two molecules of PIP2 whereas PIP2 did not stabilise coupling between β1-AR and other mini-Gα subunits.49 Although the plasma membrane of P. pastoris differs from that of mammalian cells, notably in substitution of cholesterol by ergosterol, the major classes of phospholipids are similar and the A2AR was pharmacologically active.50 Furthermore, phospholipids in SMALPS can also be exchanged,51 which offers opportunities in the future for modifying the lipid composition following isolation of GPCRs in SMALPs. Overall, the preservation of native lipid encapsulated in GPCR-SMALPs combined with the advantages of FCS described above, establishes a versatile platform for investigating ligand:GPCR interactions specifically, and membrane protein interactions in general, in the future.
Conclusions
Nanodisc technology is becoming widely adopted as a membrane protein solubilisation strategy due to the benefits provided by SMALPs over other solubilisation approaches. Proteins are directly solubilised from membranes with preservation of the annular lipids thereby maintaining the native environment of the encapsulated protein. This is a notable advantage for research focusing on the large GPCR family of membrane proteins which constitute the primary therapeutic target for drug discovery.6Nano-encapsulation of one of these GPCRs (the A2AR) into SMALPs allowed us to report the first use of solution-based FCS to investigate the ligand binding capability of the receptor within the nanodisc particle in real time. Our study established that GPCR-SMALP used in combination with FCS is a powerful approach for characterising ligand:receptor complexes in a nano-scale native environment. It is anticipated that given the versatility and general utility of our strategy, it can be applied to the quantitative investigation of a wide range of target receptors plus their cognate ligands, modulators and interacting protein partners in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the Biotechnology and Biological Sciences Research Council (BBSRC: BB/R016615/1 and BB/R016755/1) to MW and DRP and the Medical Research Council (MRC: MR/N020081/1) to SJH and SJB. RLG was supported by a BBSRC-MIBTP award to RLG and MW.References
- J.-L. Popot, Annu. Rev. Biochem., 2010, 79, 737–775 CrossRef CAS PubMed.
- M. Le Maire, P. Champeil and J. V. Moller, Biochim. Biophys. Acta, 2000, 1508, 86–111 CrossRef CAS.
- M. Wheatley, J. Charlton, M. Jamshad, S. J. Routledge, S. Bailey, P. J. La-Borde, M. T. Azam, R. T. Logan, R. M. Bill, T. R. Dafforn and D. R. Poyner, Biochem. Soc. Trans., 2016, 44, 619–623 CrossRef CAS PubMed.
- A. J. Venkatakrishnan, X. Deupi, G. Lebon, F. M. Heydenreich, T. Flock, T. Miljus, S. Balaji, M. Bouvier, D. B. Veprintsev, C. G. Tate, G. F. Schertler and M. M. Babu, Nature, 2016, 536, 484–487 CrossRef CAS PubMed.
- M. Wheatley, D. Wootten, M. T. Conner, J. Simms, R. Kendrick, R. T. Logan, D. R. Poyner and J. Barwell, Br. J. Pharmacol., 2012, 165, 1688–1703 CrossRef CAS PubMed.
- A. S. Hauser, M. M. Attwood, M. Rask-Andersen, H. B. Schiöth and D. E. Gloriam, Nat. Rev. Drug Discovery, 2017, 16, 829–842 CrossRef CAS PubMed.
- K. Sriram and P. A. Insel, Mol. Pharmacol., 2018, 93, 251–258 CrossRef CAS PubMed.
- T. J. Knowles, R. Finka, C. Smith, Y. P. Lin, T. Dafforn and M. Overduin, J. Am. Chem. Soc., 2009, 131, 7484–7485 CrossRef CAS PubMed.
- C. Vargas, R. C. Arenas, E. Frotscher and S. Keller, Nanoscale, 2015, 7, 20685–20696 RSC.
- M. Jamshad, V. Grimard, I. Idini, T. J. Knowles, M. R. Dowle, N. Schofield, P. Sridhar, Y. Lin, R. Finka, M. Wheatley, O. R. T. Thomas, R. E. Palmer, M. Overduin, C. Govaerts, J.-M. Ruysschaert, K. J. Edler and T. R. Dafforn, Nano Res., 2015, 8, 774–789 CrossRef CAS PubMed.
- M. Jamshad, J. Charlton, Y.-P. Lin, S. J. Routledge, Z. Bawa, T. J. Knowles, M. Overduin, N. Dekker, T. R. Dafforn, R. M. Bill, D. R. Poyner and M. Wheatley, Biosci. Rep., 2015, 35, e00188 CrossRef PubMed.
- S. C. Lee, T. J. Knowles, V. L. G. Postis, M. Jamshad, R. A. Parslow, Y.-P. Lin, A. Goldman, P. Sridhar, M. Overduin, S. P. Muench and T. R. Dafforn, Nat. Protoc., 2016, 11, 1149–1162 CrossRef CAS PubMed.
- J. Broeker, B. T. Eger and O. P. Ernst, Structure, 2017, 25, 384–392 CrossRef PubMed.
- C. Sun, S. Benlekbir, P. Venkatakrishnan, Y. Wang, S. Hong, J. Hosler, E. Tajkhorshid, J. L. Rubinstein and R. B. Gennis, Nature, 2018, 557, 123–126 CrossRef CAS PubMed.
- S. J. Briddon, B. Kellam and S. J. Hill, Methods Mol. Biol., 2011, 746, 211–236 CrossRef CAS PubMed.
- L. A. Stoddart, L. E. Kilpatrick, S. J. Briddon and S. J. Hill, Neuropharmacology, 2015, 98, 48–57 CrossRef CAS PubMed.
- L. A. Stoddart, L. E. Kilpatrick and S. J. Hill, Trends Pharmacol. Sci., 2018, 39, 136–147 CrossRef CAS PubMed.
- S. J. Briddon, L. E. Kilpatrick and S. J. Hill, Trends Pharmacol. Sci., 2018, 39, 158–174 CrossRef CAS PubMed.
- U. Meseth, T. Wohland, R. Rigler and H. Vogel, Biophys. J., 1999, 76, 1619–1631 CrossRef CAS PubMed.
- R. Corriden, L. E. Kilpatrick, B. Kellam, S. J. Briddon and S. J. Hill, FASEB J., 2014, 28, 4211–4222 CrossRef CAS PubMed.
- S. J. Briddon, R. J. Middleton, Y. Cordeaux, F. M. Flavin, J. A. Weinstein, M. W. George, B. Kellam and S. J. Hill, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4673–4678 CrossRef CAS PubMed.
- L. E. Kilpatrick, S. J. Briddon and N. D. Holliday, Biochim. Biophys. Acta, 2012, 1823, 1068–1081 CrossRef CAS PubMed.
- R. Machan and T. Wohland, FEBS Lett., 2014, 588, 3571–3584 CrossRef CAS PubMed.
- L. E. Kilpatrick and S. J. Hill, Biochem. Soc. Trans., 2016, 44, 624–629 CrossRef CAS PubMed.
- B. B. Fredholm, A. P. IJzerman, K. A. Jacobson, J. Linden and C. E. Müller, Pharmacol. Rev., 2011, 63, 1–34 CrossRef CAS PubMed.
- B. B. Fredholm, J. F. Chen, S. A. Masino and J. M. Vaugeois, Annu. Rev. Pharmacol. Toxicol., 2005, 45, 385–412 CrossRef CAS PubMed.
- M. Wachsmuth, C. Conrad, J. Bulkescher, B. Koch, R. Mahen, M. Isokane, R. Pepperkok and J. Ellenberg, Nat. Biotechnol., 2015, 33, 384–389 CrossRef CAS PubMed.
- S. C. L. Hall, C. Tognoloni, J. Charlton, E. C. Bragginton, A. J. Rothnie, P. Sridhar, M. Wheatley, T. J. Knowles, T. Arnold, K. J. Edler and T. R. Dafforn, Nanoscale, 2018, 10, 10609–10619 RSC.
- T. Ravula, N. Z. Hardin, S. K. Ramadugu, S. J. Cox and A. Ramamoorthy, Angew. Chem., Int. Ed., 2018, 57, 1342–1345 CrossRef CAS PubMed.
- A. O. Oluwole, B. Danielczak, A. Meister, J. O. Babalola, C. Vargas and S. Keller, Angew. Chem., Int. Ed., 2017, 56, 1919–1924 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- Y. Cheng and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS PubMed.
- L. A. Stoddart, A. J. Vernall, J. L. Denman, S. J. Briddon, B. Kellam and S. J. Hill, Chem. Biol., 2012, 19, 1105–1115 CrossRef CAS PubMed.
- A. J. Vernall, L. A. Stoddart, S. J. Briddon, H. W. Ng, C. A. Laughton, S. W. Doughty, S. J. Hill and B. Kellam, Org. Biomol. Chem., 2013, 11, 5673–5682 RSC.
- L. A. Stoddart, E. K. M. Johnstone, A. J. Wheal, J. Goulding, M. B. Robers, T. Machleidt, K. V. Wood, S. J. Hill and K. D. G. Pfleger, Nat. Methods, 2015, 12, 661–663 CrossRef CAS PubMed.
- J. R. Keddie, S. M. Poucher, G. R. Shaw, R. Brooks and M. G. Collis, Eur. J. Pharmacol., 1996, 301, 107–113 CrossRef CAS PubMed.
- S. C. L. Hall, C. Tognoloni, G. J. Price, B. Klumperman, K. J. Edler, T. R. Dafforn and T. Arnold, Biomacromolecules, 2018, 19, 761–772 CrossRef CAS PubMed.
- D. Wootten, A. Christopoulos and P. Sexton, Nat. Rev. Drug Discovery, 2013, 12, 630–644 CrossRef CAS PubMed.
- T. H. Bayburt and S. G. Sligar, FEBS Lett., 2010, 584, 1721–1727 CrossRef CAS PubMed.
- K. S. Simon, N. L. Pollock and S. C. Lee, Biochem. Soc. Trans., 2018, 46, 1495–1504 CrossRef CAS PubMed.
- J.-M. Swiecicki, J. T. Santana and B. Imperiali, Cell Chem. Biol., 2020, 27, 245–251 CrossRef CAS PubMed.
- A. J. Horsey, D. A. Briggs, N. D. Holliday and S. J. Briddon, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183218 CrossRef CAS PubMed.
- V. Schmidt and J. N. Sturgis, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 777–783 CrossRef CAS PubMed.
- R. Dawaliby, C. Trubbia, C. Delporte, M. Masureel, P. Van Antwerpen, B. K. Kobilka and C. Govaerts, Nat. Chem. Biol., 2016, 12, 35–39 CrossRef CAS PubMed.
- S. Muth, A. Fries and G. Gimpl, Biochem. J., 2011, 437, 541–553 CrossRef CAS PubMed.
- G. Gimpl, K. Burger and F. Fahrenholz, Biochemistry, 1997, 36, 10959–10974 CrossRef CAS PubMed.
- L. Pang, M. Graziano and S. Wang, Biochemistry, 1999, 38, 12003–12011 CrossRef CAS PubMed.
- M. A. Hanson, V. Cherezov, M. T. Griffith, C. B. Roth, V. P. Jaakola, E. Y. Chien, J. Velasquez, P. Kuhn and R. C. Stevens, Structure, 2008, 16, 897–905 CrossRef CAS PubMed.
- H. Y. Yen, K. K. Hoi, I. Liko, G. Hedger, M. R. Horrell, W. Song, D. Wu, P. Heine, T. Warne, Y. Lee, B. Carpenter, A. Plückthun, C. G. Tate, M. S. P. Sansom and C. V. Robinson, Nature, 2018, 559, 423–427 CrossRef CAS PubMed.
- S. J. Routledge, M. Jamshad, H. A. Little, Y.-P. Lin, J. Simms, A. Thakker, C. M. Spickett, R. M. Bill, T. R. Dafforn, D. R. Poyner and M. Wheatley, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183235 CrossRef CAS PubMed.
- G. Hazell, T. Arnold, R. D. Barker, L. A. Clifton, N. J. Steinke, C. Tognoloni and K. J. Edler, Langmuir, 2016, 32, 11845–11853 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures (Fig. S1–S3). See DOI: 10.1039/d0nr01060j |
| This journal is © The Royal Society of Chemistry 2020 |