
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePairwise semi-hydrogenation of alkyne to cis-alkene on platinum-tin intermetallic compounds†
Yuchen
Pei‡
 a,
Minda
Chen‡
a,
Xiaoliang
Zhong
b,
Tommy Yunpu
Zhao
c,
Maria-Jose
Ferrer
c,
Raghu V.
Maligal-Ganesh
a,
Tao
Ma
d,
Biying
Zhang
a,
Zhiyuan
Qi
a,
Lin
Zhou
d,
Clifford R.
Bowers
*c,
Cong
Liu
*e and
Wenyu
Huang
*ad
a,
Minda
Chen‡
a,
Xiaoliang
Zhong
b,
Tommy Yunpu
Zhao
c,
Maria-Jose
Ferrer
c,
Raghu V.
Maligal-Ganesh
a,
Tao
Ma
d,
Biying
Zhang
a,
Zhiyuan
Qi
a,
Lin
Zhou
d,
Clifford R.
Bowers
*c,
Cong
Liu
*e and
Wenyu
Huang
*ad
aDepartment of Chemistry, Iowa State University, Ames, IA 50011, USA. E-mail: whuang@iastate.edu
bHuazhong University of Science and Technology, Wuhan, Hubei, China
cDepartment of Chemistry, University of Florida, Gainesville, FL 32611, USA. E-mail: bowers@chem.ufl.edu
dAmes Laboratory, the U.S. Department of Energy, Ames, IA 50011, USA
eChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 S Cass Ave., Lemont, IL 60439, USA. E-mail: congliu@anl.gov
First published on 25th March 2020
Abstract
The molecular basis for the high cis-alkene selectivity over intermetallic PtSn for alkyne semi-hydrogenation is demonstrated. Unlike the universal assumption that the bimetallic surface is saturated with atomic hydrogen, molecular hydrogen has a higher barrier for dissociative adsorption on intermetallic PtSn due to the deficiency of Pt three-fold sites. The resulting molecular behavior of adsorbed hydrogen on intermetallic PtSn nanoparticles leads to pairwise-hydrogenation of three alkynes to the corresponding cis-alkenes, satisfying both high stereoselectivity and high chemoselectivity.
Introduction
The semi-hydrogenation of alkynes to alkenes using rationally designed catalysts continues to benefit industries and academia. An important example is the selective hydrogenation of acetylene to ethylene in order to remove trace acetylene impurity from ethylene feed, which is essential for the long lifetime of polymerization catalysts and the production of high-quality polyethylene.1–5 Also, in pharmaceutical and fine chemical industries, designing new catalysts to achieve high chemoselectivity and stereoselectivity of cis-/trans-alkene products is indispensable to synthetic methodology development.6cis-Alkenes are kinetically controlled products in this reaction, and thus are more difficult to be stabilized and isolated than the thermodynamically favored trans-alkenes. Lindlar catalyst is a commonly used heterogeneous catalyst for selective hydrogenation of alkyne to cis-alkene. In this reaction, alkyne molecules adopt the syn geometry on the Lindlar catalyst surface, where the adsorbed hydrogen atoms approach from the same side of the carbon–carbon triple bond to form the cis-alkene products.3,7 However, over-hydrogenation to alkane by-products can prevail in the syn addition, and minimizing such reaction requires deliberate deactivation of Lindlar catalyst.Stabilizing cis-alkenes while inhibiting trans-isomerization and over-hydrogenation is challenging for heterogeneous catalysts due to the heterogeneity of the active sites and the lack of understanding of the structure–property relationship. Previous studies have attempted to address this challenge from the perspectives of either stereoselectivity or chemoselectivity. Lee and Zaera et al. have demonstrated that Pt(111) can be a specific facet to stabilize cis-alkenes compared to trans-isomers.8 Furukawa and Komatsu et al. have reported that chemoselective hydrogenation can be performed over Pd-based bimetallic alloys, and hydrogen accessibility to the adsorbed alkyne is critical to the chemo- and stereo-selectivity for alkene production.9,10 Hu and Li et al. have also revealed the strong structure–property relationships in hollow Ni3Ga multishell catalysts and single-atom Pd supported catalysts with high ethylene selectivity in acetylene semi-hydrogenation.11–13 These studies indicate that both the catalyst structure and the hydrogenation pathway are crucial for controlling the selectivity in alkyne semi-hydrogenation.
Here, we envision the pairwise-addition as a special hydrogenation pathway that could ensure both high stereoselectivity and high chemoselectivity in alkyne semi-hydrogenation to cis-alkene. Pairwise-addition refers to the transfer of both hydrogen atoms in a H2 molecule to one carbon–carbon triple bond in an alkyne molecule. During this reaction, adsorbed H2 molecules do not dissociate into hydrogen atoms on the catalyst surface. The weak H2 adsorption14 and catalytic addition of H2 to propene with high pairwise selectivity over the PtSn intermetallic nanoparticles (iNPs)15 have been studied in our group. In this work, we demonstrate that the pairwise-hydrogenation of alkynes over PtSn leads to high chemo- and stereo-selectivity to cis-alkenes.
Results and discussion
PtSn iNPs have atomic-level homogeneity and high surface stability, differentiating them from random alloys prepared by the impregnation method. These PtSn iNPs are specially designed to be encapsulated in a mesoporous silica shell (Fig. 1a) that endows PtSn iNPs with accurate structures after synthesis and allows reactants to access the PtSn intermetallic surface during catalysis.14,16–19 We have also used selective hydrogenations to probe and illustrate the structure of these PtSn iNPs and its correlation to catalytic behaviors.14,16,17 Additional details of the structural characterizations of PtSn iNPs and impregnated catalysts are summarized in the ESI (Fig. S1, S2 and Tables S1, S2†). The surface uniformity and stability of PtSn iNPs affords structural clarification that renders this material an ideal platform for studying fundamental structure–property correlations. As an example, Fig. 1b and c show the atomically ordered PtSn structure as confirmed by aberration-corrected scanning transmission electron microscopy (STEM). The bulk and surface structure of the PtSn iNPs are also stable under a reducing environment up to 500 °C (ESI, section 2†).14,17 | ||
| Fig. 1 (a) TEM image of PtSn iNPs encapsulated in mesoporous silica at lower magnification showing clear core–shell structure, with its model illustration. Inset: TEM image of a PtSn iNP at higher magnification and a model structure of PtSn iNP encapsulated in a mesoporous silica shell. (b) Atomic resolution aberration-corrected STEM image of a PtSn iNP displaying the PtSn(110) facet. (c) Aberration-corrected STEM image of PtSn(110) facet placed aside the theoretical model of PtSn(110) with an adsorbed alkyne. c views part of the same particle, as shown in b. Pearl (yellow) dots represent Pt (Sn) atoms. Blue (orange) dots represent carbon(hydrogen) atoms in alkyne, and red dots represent H2 undergoing pairwise-hydrogenation. | ||
We evaluated the stereoselectivity and chemoselectivity of PtSn iNPs in the semi-hydrogenation of three alkyne substrates: diphenylacetylene, 1-phenyl-1-butyne, and dimethyl acetylenedicarboxylate (Fig. 2 and Tables S3–S5†). The three substrates have different degrees of bulky substituent groups in the vicinity of the alkyne group. PtSn iNPs exhibit high chemoselectivity and stereoselectivity to cis-alkene products for all substrates. Chemoselectivity of 99%, 98% and 88% were obtained with respect to diphenylacetylene, 1-phenyl-1-butyne, and dimethyl acetylenedicarboxylate. For comparison, the Pt NPs yielded >60% over-hydrogenated product for the same three substrates. The PtSn iNPs also yielded remarkably high cis-alkene selectivities of 91%, 86%, and >99% for diphenylacetylene, 1-phenyl-1-butyne, and dimethyl acetylenedicarboxylate, respectively. In particular, PtSn iNPs maintained high cis-alkene selectivity at the high conversion of 79.8% at 110 °C for dimethyl acetylenedicarboxylate (Table S3†). In contrast, trans-alkenes dominated on the Pt NPs (trans/cis = 2/1 for dimethyl acetylenedicarboxylate), at only 50 °C. Hence, the hydrogenation pathway leading to cis-stereoselectivity is exclusively preferred for PtSn iNPs. Due to the dramatic difference in activity between Pt and PtSn, it is a challenge to compare their catalytic behavior at the same conversion and reaction conditions. Nevertheless, PtSn iNPs show much higher cis-selectivity compared to Pt NPs, likely due to the inhibition of both trans-isomerization and over-hydrogenation pathways. A further recycle study of PtSn iNPs shows that the catalyst can be recycled 5 times without a significant loss of activity and selectivity (Fig. S5a†). X-ray diffraction (XRD) and transmission electron microscopy (TEM) characterization of the used catalyst shows the stability of the intermetallic phase and mesoporous shell (Fig. S5b and c†).
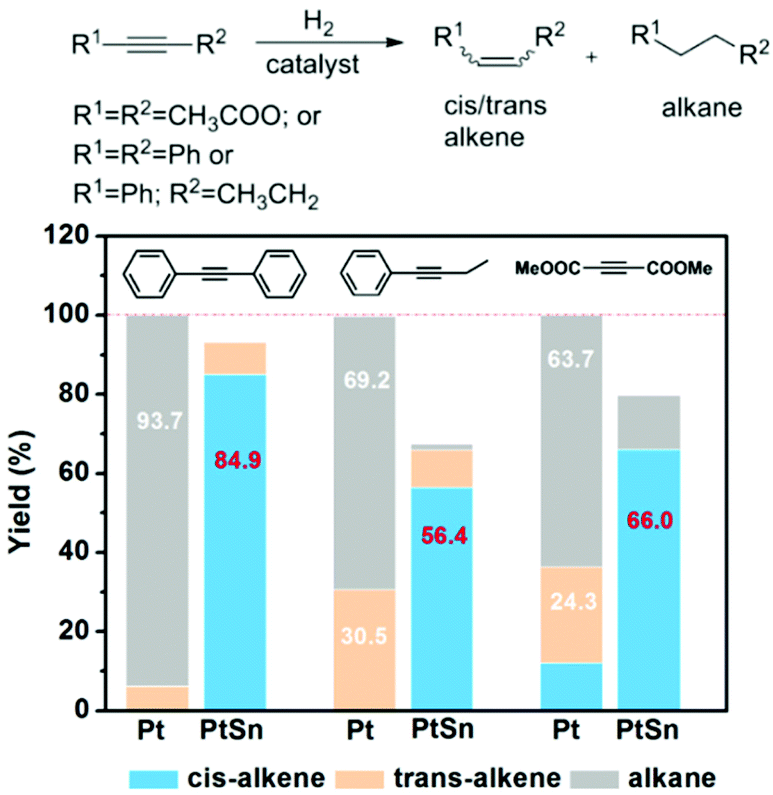 | ||
| Fig. 2 The product distribution of alkynes hydrogenation with Pt and PtSn catalysts. | ||
Generally, alkynes approach the surface of a heterogeneous catalyst (e.g., Pd and Pt) in syn geometry. However, syn addition cannot solely explain both the high stereoselectivity and chemoselectivity over PtSn. Our previous studies of PtSn-promoted pairwise-hydrogenation can shed light on the molecular understanding of the hydrogenation mechanism on PtSn iNPs: molecular H2 adds to alkene (propene) in a pairwise fashion rather than undergoing dissociation into H atoms, which was experimentally confirmed via operando Nuclear Magnetic Resonance (NMR) studies.15 We envision that the pairwise-hydrogenation could be a key mechanism to ensure cis-alkene stereoselectivity over PtSn iNPs for alkyne semi-hydrogenation.
As we propose in Fig. 3a, alkyne first adopts the syn geometry upon adsorption onto the PtSn surface. Due to the high barrier to dissociate H2 molecules on PtSn, the H atoms in molecular H2 approach the carbon–carbon triple bond from the same side, thereby favoring a concerted, pairwise-hydrogenation leading to cis-alkene. The dilution effect of PtSn iNPs explains the molecular behavior of adsorbed H2, where the Pt ensemble sites are eliminated on the PtSn surface, thus inhibiting H2 dissociation (generally achieved by Pt ensembles).14,20–22 The weak adsorption of alkene on the PtSn surface prevents its over-hydrogenation to alkane. In contrast, the Pt surface is saturated with atomic H due to its barrierless H2 dissociation over Pt ensemble sites (Fig. 3b).14 The alkyne would still prefer the syn geometry on Pt; however, the abundant surface H atoms could attack the triple bond with equal probability from the same or opposite side of the bond regardless of whether the H addition is sequential or simultaneous. Therefore, the Pt surface cannot ensure exclusive cis-alkene stereoselectivity. Moreover, atomic hydrogen could add to one end of the as-formed double bonds allowing the rotation of this bond, resulting in the production of both cis- and trans-alkene.8,23 The abundant excess atomic hydrogen on Pt surface also explains the over-hydrogenation of alkynes to alkanes. It has not been commonly realized that pairwise-hydrogenation can lead to the kinetically controlled cis-alkene rather than the thermodynamically favored trans-alkene, except in the case of very small (<1 nm) Pt nanoparticles in alkyne hydrogenation at low H2 partial pressure.23 However, the assumption of the atomic H saturated bimetallic surface does not apply to well-defined intermetallic PtSn, a model catalyst presenting high energy barrier for H2 dissociation.14,17,24
 | ||
| Fig. 3 The suggested hydrogenation pathways for (a) PtSn and (b) Pt. | ||
This hypothesis of pairwise-hydrogenation on PtSn iNPs was strongly evident in the PASADENA (Parahydrogen And Synthesis Allow Dramatically Enhanced Nuclear Alignment) NMR experiments, where intense antiphase NMR signals occur exclusively through the pairwise-addition of parahydrogen. The stereoselectivity of pairwise H2 addition to 1-phenyl-1-butyne is evidenced by the PASADENA NMR spectra presented in Fig. 4. The hydrogenation reaction was carried out at 100 °C by bubbling 50% enriched parahydrogen (p-H2) through the solution in a 5 mm NMR tube at 7 T. Fig. 4a shows the three different products obtained in 1-phenyl-1-butyne semi-hydrogenation. As shown in Fig. 4b, the PASADENA spectrum (top trace) exhibits intense antiphase multiplets centered at 6.41 ppm and 5.67 ppm, corresponding to the chemical shifts of the CH groups in cis-1-phenyl-1-butene. The formation of a small fraction of trans-1-phenyl-1-butene was evidenced by the CH peaks at 6.43 and 6.35 ppm observed in thermally polarized spectrum acquired at 600 MHz spectrometer (Fig. 4c), demonstrating the high stereoselectivity of alkyne hydrogenation over PtSn iNPs. A small peak due to the fully hydrogenated butylbenzene is noticeable in the PASADENA and thermally polarized spectrum (Fig. 4b, c, and S7†). A maximum signal enhancement factor of 126 was observed (after correction for destructive interference in the antiphase multiplets, Fig. S6†) on the third cycle of p-H2 bubbling for 20 s. In a complementary experiment, the batch reaction of 1-phenyl-1-butyne semi-hydrogenation also shows 97.6% chemoselectivity to alkenes and 85.8% stereoselectivity to the cis-alkene (Table S5†).
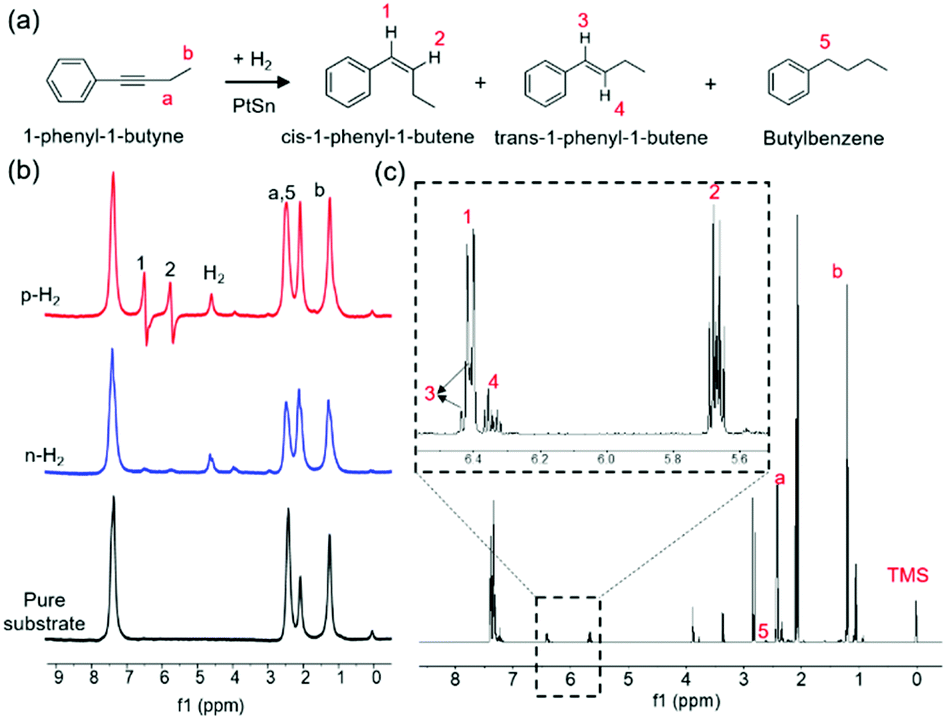 | ||
| Fig. 4 (a) Products of the hydrogenation of 1-phenyl-1-butyne and the labeling scheme for the protons. (b) 300 MHz NMR spectra collected at 100 °C in d6-acetone. The bottom spectrum is that of the pure substrate before any H2 bubbling. The PASADENA and thermally polarized spectra (top and middle, respectively), were each obtained after three cycles of 20 s of bubbling of p-H2 or normal H2 (n-H2), respectively, through the substrate/catalyst suspension. All the spectra were acquired in a single scan and are plotted on the same vertical scale. (c) Thermally polarized spectrum after bubbling p-H2 for 140 s. Catalysts are removed prior to acquisition. Spectrum is acquired at 14.1 T and room temperature. | ||
We also observed that on PtSn iNPs, the high cis-stilbene selectivity in diphenylacetylene hydrogenation is not significantly dependent on solvent, temperature, and reaction time (Table S4†), strongly suggesting the pairwise-hydrogenation pathway. We also confirmed that the isomerization of cis-stilbene to trans-stilbene could not proceed on PtSn iNPs in the semi-hydrogenation of diphenylacetylene (Table S6†). To further demonstrate the structure–property relationship of PtSn, we prepared Pt-rich PtSn-1.1 (Pt/Sn = 1.1). We had confirmed in our previous work that Pt-rich Pt–Sn iNPs have Pt surface domains.17 On PtSn-1.1, we observed more cis- to trans-stilbene isomerization after full conversion, in sharp contrast to the high cis-stilbene selectivity on the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PtSn iNPs (Table S7†).
:1 PtSn iNPs (Table S7†).
The three alkyne semi-hydrogenations suggest that pairwise-hydrogenation on PtSn could be induced by its surface structure, where the adsorbed atomic H atoms are rare. In our previous work, we ascribed the absence of H ad-atoms on intermetallic PtSn to its intrinsic structure where Pt three-fold sites are eliminated.15 The three-fold Pt or Pd sites (denoted M3) are known to be highly active for H2 dissociation.25,26,32 Although the surface reconstruction cannot be ruled out in the reactions, we endeavored to examine if the scope of this structure–property relationship can be extended to other intermetallic catalysts, including PdCu (w/Pd3) and Pd3Sn2 (w/o Pd3) bimetallic NPs. Their structural and compositional analysis are shown in Table S2 and Fig. S3–S4.† As shown in Fig. 5, only Pd3Sn2 shows both cis-stereoselectivity and chemoselectivity coinciding with the absence of Pd3 site (Table S8†). PtSn iNPs also appear as highly selective catalysts among the literature under similar reaction conditions (Table S9†).
 | ||
| Fig. 5 The role of the three-fold M3 sites to pairwise-hydrogenation exemplified by semi-hydrogenation of diphenylacetylene over Pt and intermetallic catalysts. | ||
The pairwise-hydrogenation on M3-deficient intermetallic compounds was further supported by the results of a kinetic H/D isotope study. First, a gas-phase H/D exchange experiment using a co-feed of 1:1 H2/D2 over PtSn shows that the onset of exchange occurs at a temperature (150 °C) that is higher than the alkyne hydrogenation reaction temperature (80–110 °C) as reported in our previous work.14 Second, we also observed a strong D2 inhibition effect on PtSn, where the reaction hardly proceeds at all when D2 was supplied for diphenylacetylene hydrogenation. When supplied with 1:1 mixture of H2/D2, H-substituted trans-alkene is still dominant with a negligible D-substituted product. The strong D2 inhibition is consistent with pairwise-hydrogenation that likely proceeds by a tunneling mechanism27 involving the molecular-like H2 on PtSn because the PtSn surface lacks Pt ensemble sites where H2 molecule cannot undergo dissociation to H atoms. The kinetic isotope effect is reported to be particularly high when involving tunneling mechanism.28–30 Thus, we propose that the high energy barrier for H2 dissociation leads to pairwise-hydrogenation over PtSn.
Density functional theory (DFT) calculations were performed to provide molecular insights into the stereoselectivity on Pt(111) vs. PtSn(110). In general, calculations show that diphenylacetylene (Ph2C2) binds with these metal surfaces more strongly (−3.2 eV on Pt; −1.3 eV on PtSn) than H2 (−0.022 eV on Pt; −0.052 eV on PtSn), as shown in Tables S10, S11 and Fig. S8–S11.† This binding energy difference indicates that H2 could only be adsorbed within the interspace between Ph2C2 molecules on the almost fully Ph2C2-covered metal surface. The weak binding interactions between the surfaces and H2 could be one of the factors explaining the higher selectivity of the cis-alkene products observed experimentally. Hydrogenation to cis-products could occur right after the adsorption and pairwise-addition of H2 on either side of the carbon–carbon triple bond. The trans-alkene is suppressed during hydrogenation because H ad-atoms are not present on both sides of the carbon triple bond. In addition, the catalyst surfaces also show interesting features in the apparent conversion rates and chemoselectivity. At similar catalyst amounts, Pt (w/M3) exhibits high conversion rates and a strong tendency to yield fully hydrogenated alkanes. On the other hand, PtSn (w/o M3) showed low conversion rates with high chemoselectivity (Tables S3–S5†). Our calculations suggest that the conversion rate could be correlated with the barrier for H–H bond dissociation. As shown in Table S11,† Pt easily dissociates H2 with no barrier, yielding an abundant atomic H supply that is responsible for a fast reaction rate and over-hydrogenation. On the contrary, PtSn has larger H2 dissociation barriers (0.3 eV) than Pt (∼0 eV), leading to slower hydrogenation kinetics with lower conversion rates.
Conclusions
In summary, we have studied the structure–property relationship of intermetallic PtSn, where the elimination of Pt three-fold sites leads to high cis-alkene selectivity in alkyne semi-hydrogenations. Higher hydrogen dissociation barrier and alkyne-dominated surface suggest the exclusive pairwise-hydrogenation over PtSn iNPs. We anticipate engineering intermetallic structures for satisfying high chemoselectivity and stereoselectivity in broader catalytic reactions. A complete understanding of these correlations in selectivity would also enable the synthesis of heterogeneous catalysts offering improved parahydrogen enhanced polarization for more important applications such as hyperpolarized biomedical magnetic resonance imaging.31Author contributions
Y. P., M. C., C. L, C. R. B., W. H. conceived the idea, designed the project, and wrote the manuscript. Y. P., M. C., R. V. M., B. Z., Z. Q., W. H. initiated the projects, conducted material synthesis, and obtained catalytic results. X. Z. and C. L. conducted DFT calculations. T. Y. Z., M.-J. F., and C. R. B. performed PASADENA NMR experiments. T. M. and L. Z acquired aberration-corrected scanning transmission electron microscopy images.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSF grant CHE-1808239 (C. R. B. and W. H.) and the National High Magnetic Field Laboratory's User Collaborative Grant Program, which is supported by the National Science Foundation Cooperative Agreement No. DMR-1644779* and the State of Florida. The computational studies were supported by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, under Contract No. DE-AC02-06CH11357, with U Chicago Argonne, LLC, the operator of Argonne National Laboratory (ANL). The DFT calculations were performed using the computational resources at the Laboratory Computing Resource Center (LCRC) at ANL. The use of APS, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. We thank Gordon J. Miller and Matthew Besser for the use of X-ray diffractometer. We thank Tianpin Wu, Chengjun Sun, and Lu Ma for XAFS measurement at station 9-BM-C and 20-BM-B of Advanced Photon Source (APS).Notes and references
- N. Lopez and C. Vargas-Fuentes, Chem. Commun., 2012, 48, 1379–1391 RSC.
- A. Molnar, G. V. Smith and M. Bartok, J. Catal., 1986, 101, 67–72 CrossRef CAS.
- J. Rajaram, A. P. S. Narula, H. P. S. Chawla and S. Dev, Tetrahedron, 1983, 39, 2315–2322 CrossRef CAS.
- C. Panja, N. A. Saliba and B. E. Koel, J. Phys. Chem. B, 2001, 105, 3786–3796 CrossRef CAS.
- B. Chen, U. Dingerdissen, J. G. E. Krauter, H. G. J. Lansink Rotgerink, K. Mobus, D. J. Ostgard, P. Panster, T. H. Riermeier, S. Seebald, T. Tacke and H. Trauthwein, Appl. Catal., A, 2005, 28, 17–46 CrossRef.
- W. Y. Siau, Y. Zhang and Y. Zhao, Top. Curr. Chem., 2012, 327, 1–275 CrossRef PubMed.
- H. Lindlar, Helv. Chim. Acta, 1952, 35, 446–450 CrossRef CAS.
- I. Lee, F. Delbecq, R. Morales, M. A. Albiter and F. Zaera, Nat. Mater., 2009, 8, 132–138 CrossRef CAS PubMed.
- S. Furukawa, K. Ochi, H. Luo, M. Miyazaki and T. Komatsu, ChemCatChem, 2015, 7, 3472–3479 CrossRef CAS.
- S. Furukawa and T. Komatsu, ACS Catal., 2016, 6, 2121–2125 CrossRef CAS.
- M. Hu, W. Yang, S. Liu, W. Zhu, Y. Li, B. Hu, Z. Chen, R. Shen, W.-C. Cheong, Y. Wang, K. Zhou, Q. Peng, C. Chen and Y. Li, Chem. Sci., 2019, 10, 614–619 RSC.
- M. Hu, J. Zhang, W. Zhu, Z. Chen, X. Gao, X. Du, J. Wan, K. Zhou, C. Chen and Y. Li, Nano Res., 2018, 11, 905–912 CrossRef CAS.
- M. Hu, S. Zhao, S. Liu, C. Chen, W. Chen, W. Zhu, C. Liang, W.-C. Cheong, Y. Wang, Y. Yu, Q. Peng, K. Zhou, J. Li and Y. Li, Adv. Mater., 2018, 30, 1801878 CrossRef PubMed.
- Y. Pei, Z. Qi, T. W. Goh, L. L. Wang, R. V. Maligal-Ganesh, H. L. MacMurdo, S. R. Zhang, C. Xiao, X. Li, F. Tao, D. D. Johnson and W. Huang, J. Catal., 2017, 356, 307–314 CrossRef CAS.
- E. W. Zhao, R. V. Maligal-Ganesh, C. Xiao, T. W. Goh, Z. Qi, Y. Pei, H. E. Hagelin-Weaver, W. Huang and C. R. Bowers, Angew. Chem., Int. Ed., 2017, 56, 3925–3929 CrossRef CAS PubMed.
- R. V. Maligal-Ganesh, C. Xiao, T. W. Goh, L. L. Wang, J. Gustafson, Y. Pei, Z. Qi, D. D. Johnson, S. Zhang, F. Tao and W. Huang, ACS Catal., 2016, 6, 1754–1763 CrossRef CAS.
- Y. Pei, B. Zhang, R. V. Maligal-Ganesh, P. J. Naik, T. W. Goh, H. L. MacMurdo, Z. Qi, M. Chen, R. K. Behera, I. I. Slowing and W. Huang, J. Catal., 2019, 374, 136–142 CrossRef CAS.
- Y. Pei, R. V. Maligal-Ganesh, C. Xiao, T. W. Goh, K. Brashler, J. A. Gustafson and W. Huang, Nanoscale, 2015, 7, 16721–16728 RSC.
- M. Chen, Y. Han, T. W. Goh, R. Sun, R. V. Maligal-Ganesh, Y. Pei, C. K. Tsung, J. W. Evans and W. Huang, Nanoscale, 2019, 11, 5336–5345 RSC.
- H. S. Wei, X. Y. Liu, A. Q. Wang, L. L. Zhang, B. T. Qiao, X. F. Yang, Y. Q. Huang, S. Miao, J. Y. Liu and T. Zhang, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- J. Prinz, R. Gaspari, C. A. Pignedoli, J. Vogt, P. Gille, M. Armbrüster, H. Brune, O. Gröning, D. Passerone and R. Widmer, Angew. Chem., Int. Ed., 2012, 51, 9339–9343 CrossRef CAS PubMed.
- Q. C. Feng, S. Zhao, Y. Wang, J. C. Dong, W. X. Chen, D. S. He, D. S. Wang, J. Yang, Y. M. Zhu, H. L. Zhu, L. Gu, Z. Li, Y. X. Liu, R. Yu, J. Li and Y. D. Li, J. Am. Chem. Soc., 2017, 139, 7294–7301 CrossRef CAS PubMed.
- R. Zhou, W. Cheng, L. M. Neal, E. W. Zhao, K. Ludden, H. E. Hagelin-Weaver and C. R. Bowers, Phys. Chem. Chem. Phys., 2015, 17, 26121–26129 RSC.
- E. W. Zhao, R. V. Maligal-Ganesh, Y. Du, T. Y. Zhao, J. Collins, T. Ma, L. Zhou, T. W. Goh, W. Huang and C. R. Bowers, Chem, 2018, 4, 1387–1403 CAS.
- Y. L. Tsai, C. Xu and B. E. Koel, Surf. Sci., 1997, 385, 37–59 CrossRef CAS.
- P. Samson, A. Nesbitt, B. E. Koel and A. Hodgson, J. Chem. Phys., 1998, 109, 3255–3264 CrossRef CAS.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–800 CrossRef CAS PubMed.
- E. S. Lewis and L. H. Funderburk, J. Am. Chem. Soc., 1978, 89, 2322–2327 CrossRef.
- S. G. Christov and Ž. L. Georgiev, J. Phys. Chem., 1971, 75, 1748–1756 CrossRef.
- T. Miyazaki, Atom Tunneling Phenomena in Physics, Chemistry and Biology, Springer-Verlag, Berlin Heidelberg, 2004 Search PubMed.
- J. B. Hovener, A. N. Pravdivtsev, B. Kidd, C. R. Bowers, S. Gloggler, K. V. Kovtunov, M. Plaumann, R. Katz-Brull, K. Buckenmaier, A. Jerschow, F. Reineri, T. Theis, R. V. Shchepin, S. Wagner, P. Bhattacharya, N. M. Zacharias and E. Y. Chekmenev, Angew. Chem., Int. Ed., 2018, 57, 11140–11162 CrossRef PubMed.
- J. Prinz, C. A. Pignedoli, Q. S. Stöckl, M. Armbrüster, H. Brune, O. Gröning, R. Widmer and D. Passerone, J. Am. Chem. Soc., 2014, 136, 11792–11798 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr00920b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |