
Tuning the catalytic properties of P22 nanoreactors through compositional control†
Jhanvi
Sharma
 and
Trevor
Douglas
*
and
Trevor
Douglas
*
Department of Chemistry, Indiana University, 800 E. Kirkwood Avenue, Bloomington, Indiana 47405, USA. E-mail: trevdoug@indiana.edu
First published on 3rd December 2019
Abstract
Enzymes are biomacromolecular protein catalysts that are widely used in a plethora of industrial-scale applications due to their high selectivity, efficiency and ability to work under mild conditions. Many industrial processes require the immobilization of enzymes to enhance their performance and stability. Encapsulation of enzymes in protein cages provides an excellent immobilization platform to create nanoreactors with enhanced enzymatic stability and desired catalytic activities. Here we show that the catalytic activity of nanoreactors, derived from the bacteriophage P22 viral capsids, can be finely-tuned by controlling the packaging stoichiometry and packing density of encapsulated enzymes. The packaging stoichiometry of the enzyme alcohol dehydrogenase (AdhD) was controlled by co-encapsulating it with wild-type scaffold protein (wtSP) at different stoichiometric ratios using an in vitro assembly approach and the packing density was controlled by selectively removing wtSP from the assembled nanoreactors. An inverse relationship was observed between the catalytic activity (kcat) of AdhD enzyme and the concentration of co-encapsulated wtSP. Selective removal of the wtSP resulted in the similar activity of AdhD in all nanoreactors despite the difference in the volume occupied by enzymes inside nanoreactors, indicating that the AdhD enzymes do not experience self-crowding even under high molarity of confinement (Mconf) conditions. The approach demonstrated here not only allowed us to tailor the activity of encapsulated AdhD catalysts but also the overall functional output of nanoreactors (enzyme–VLP complex). The approach also allowed us to differentiate the effects of crowding and confinement on the functional properties of enzymes encapsulated in an enclosed system, which could pave the way for designing more efficient nanoreactors.
Introduction
In the past few decades biology has greatly influenced the process by which functional materials are designed and constructed.1 The complex organization of natural biological systems, at almost all length scales, has inspired the rapid growth of biomimetic approaches to tailor the synthesis of materials having defined structure–function relationships.2,3 Cells frequently use self-assembled molecular complexes and supramolecular compartments to organize and regulate complex biological functions. Sequestration of enzymatic reactions into the confined environment of compartments, self-assembled from lipid molecules or protein subunits such as membrane organelles or bacterial micro-compartments, exerts spatial and temporal control over the pathways.4 This is believed to optimize the efficiency by enhancing local concentration of reacting species involved in the pathway, by regulating transport of molecules into and out of the compartment, and by isolating toxic or reactive intermediates from the rest of the cell.4–6 The precise control over biochemical reactions, selectivity, and efficient catalysis that these compartments afford has led to a growing interest in mimicking such systems for materials applications by encapsulating enzymes in well-defined self-assembled macromolecular compartments such as virus-like particles (VLPs). Encapsulation of enzymes in such compartments confers enzyme stability, ease of purification and reusability, and offers the potential to create useful designer nanoreactors with very high local concentration of enzymes, which would otherwise be impossible to attain without significant aggregation.7–37 The local concentration of enzymes can be increased further by condensing enzyme filled protein compartments into 3D superlattices in order to realize accelerated catalytic conversion.38 The high local concentration of enzymes in the nanoreactors can lead to modulation of the enzymatic activity.14–16,26 The alteration in activity has been ascribed to slower diffusion of enzyme–VLP capsule, or to the combined effects arising from the crowded and confined microenvironment.14–16,26 While crowding and confinement have often been used in conjunction to rationalize the outcome of encapsulated systems, statistical-thermodynamics models suggest that confinement and crowding can have different effects on the structural and functional properties of enzymes.39–42 Biological systems utilize confinement as a basic strategy to increase the local concentration of any encapsulated molecule, which can lead to enhanced reaction rates and increased intermolecular interactions between the encapsulated species.43 Crowding, on the other hand, is an intracellular phenomenon where the concentration of macromolecules, such as proteins, carbohydrates and nucleic acids can reach up to 300 mg ml−1.44,45 At such high concentrations macromolecules experience excluded volume effects due to the inaccessibility of space occupied by other macromolecules of comparable size. These effects are such that, even in the absence of direct interactions between macromolecules, they can profoundly alter the stability, structural dynamics and kinetics of enzyme catalyzed reactions.46–48Approaches such as physical entrapment of enzyme molecules during cage assembly, or directed encapsulation involving the covalent (e.g. coat protein fusion, SpyTag/SpyCatcher, or Sortase A enzyme mediated) and non-covalent interactions (e.g. protein–protein, nucleic acid-protein) have been used to achieve the packaging of a variable number of enzymes in VLPs both in vivo and in vitro.14,15,26,49–56In vivo encapsulation is a simple and efficient approach as the entire packaging process takes place in a host bacterial cell. However, it relies on the transcriptional-translational machinery of the host cell for encapsulation and can result in little to no control over the amount of cargo encapsulation with the added possible complication of co-encapsulation of (unwanted) host macromolecules. In contrast, in vitro assembly, requiring separate purification of VLP and enzyme, offers better control over cargo encapsulation. Despite recent advances in in vitro encapsulation, controlling the composition (i.e. protein–protein interactions) and packing density (crowding) of guest molecules in the interior volume remains difficult to achieve. Here we have engineered nanoreactors utilizing VLPs derived from the P22 bacteriophage and have controlled the composition and packing density of enzymatic cargo within the interior of the VLP affording fine-tuning of the activity of the resulting nanoreactors. The P22 VLP assembles from 420 subunits of coat protein (CP) and 100–300 subunits of scaffolding protein (SP) to form a T = 7 icosahedral capsid, 58 nm in diameter.57 The SP templates the assembly of the CP into the P22 capsid through a positively charged helix-turn-helix domain at its C-terminus and gets encapsulated during the assembly process.57,58 Treatment of the P22 with 0.5 M guanidine hydrochloride (GuHCl) disrupts the non-covalent interactions between the CP and the SP helix-turn-helix domain, and the SP freely diffuses out to generate an empty shell.59 The SP can be extensively truncated to a minimal assembly domain and be modified at the N- or C-terminus without affecting its templating ability for capsid assembly.49,60,61 This truncated scaffold, fused to a protein cargo (cargo-SP), has been used to direct the P22 assembly and encapsulation of a number of cargo proteins through heterologous expression of both the components (CP and cargo-SP) in a host bacterial cell in an in vivo approach.49 We have previously shown encapsulation of many different gene products, including enzymes, with very high efficiency within the P22 VLPs. This in vivo directed encapsulation approach does not allow control over the stoichiometry and packing density of the encapsulated cargoes and suffers from some limitations such as encapsulation of misfolded proteins, or partial inactivation of proteins due to inherently high confinement molarity (Mconf ∼6–7 mM) of P22 SP-directed encapsulation system.16,49,62,63 However, using an in vitro assembly approach it is possible to overcome some of these limitations and control packaging stoichiometry and the packing density of protein cargos inside P22.64–66 Premixing of wild type (wt) SP and cargo-SP in a range of defined stoichiometric ratios and then adding them to CP subunits in a fixed stoichiometric ratio results in capsid assembly having an average composition determined by the input stoichiometry of SP and cargo-SP.63 The subsequent selective removal of wtSP from the series of co-assembled nanoreactors, upon mild treatment with a chaotrope, enables control over packing density.64 The pore size (∼2.5 nm)67 of the P22 capsid shell allows the escape of wtSP but not the bulky folded cargo-SP.16,49,63
This methodology was used to design and construct P22 nanoreactors with controlled composition using the enzyme alcohol dehydrogenase (AdhD) as a model system (Fig. 1). The AdhD is a monomeric thermostable, metal-ion independent enzyme from the aldo-keto reductase (AKR) family and presents a useful class of enzymes with considerable interest in the production of biofuels, agrochemicals and pharmaceutical products.68,69
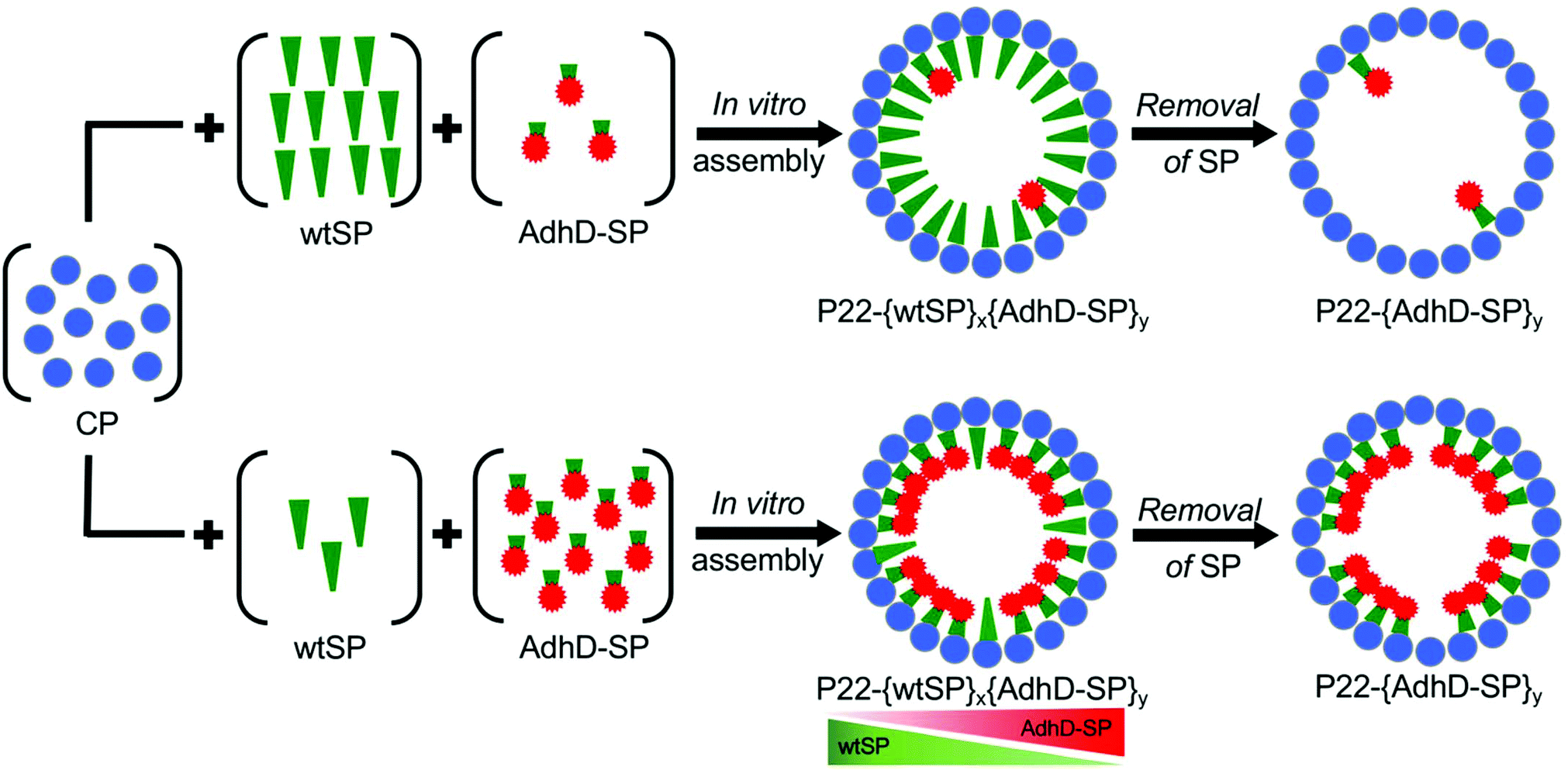 | ||
| Fig. 1 Schematic showing the methodology used to control the composition of P22 nanoreactors. Nanoreactors were co-assembled in vitro with AdhD-SP and wtSP to control the loading of cargo macromolecules. A constant amount of CP was added to variable ratios of wtSP and AdhD-SP and allowed to self-assemble in vitro. Subsequently, the wtSP was selectively removed from these co-assembled nanoreactors to alter the packing density of enzymes inside P22. | ||
Our results demonstrate that the compositional control, achieved through the encapsulation of variable amounts of AdhD and wtSP, influences the functional output at two levels: the molecular level (i.e. individual AdhD catalysts) and nanoreactor level (VLP nanoparticles encapsulating these catalysts). The wtSP not only acts as scaffold to co-template the assembly but exerts macromolecular crowding in a concentration dependent manner, leading to the tuning of enzymic output. Further, similar enzymic output of wtSP depleted nanoreactors despite differential packing density of AdhD suggested that the AdhD enzymes do not experience self-crowding even at high Mconf and that the resultant activity is influenced largely by the confinement inside P22 as the activities were lower than the free (unencapsulated) enzyme. Although the local environment of co-assembled and wtSP depleted nanoreactors impacted the functional output of AdhD differently, the overall activity of nanoreactors (with or without SP) increased with the increase in copy number of AdhD inside these nanoreactors. Thus, the ability to control composition has allowed us to create a range of nanoreactors that are modelled on a cell-like system for mimicking the crowding and confinement experienced by catalytic enzymes. To our knowledge this is the first experimental investigation where the underlying effects of crowding and confinement have been used in the design, construction, and modulation of the behavior of functional catalytic nanomaterials. These findings broaden the scope for experimental and theoretical work aimed at developing methodologies to realize better control over the catalytic activity of biomimetic nanomaterials.
Results and discussion
Controlling the loading of AdhD in P22 nanoreactors using in vitro assembly
To control the loading of AdhD in P22 nanoreactors, the AdhD was genetically fused to the N-terminus of truncated SP (amino acids 142-303). The fusion protein (AdhD-SP) was heterologously expressed in E. coli BL21(DE3) cells and purified by Ni-NTA column chromatography. The purified protein was characterized by sodium-dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and mass-spectrometry (Fig. S1†). The nanoreactors were self-assembled in vitro, where AdhD-SP and wtSP were mixed together at molar ratios of 0.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0, 0.1:0.9, 0.25:0.75, 0.5:0.5, 0.75:0.25, 0.9:0.1, 1.0:0.0 and added to a molar equivalent of CP. The co-assembled nanoreactors (P22-[AdhD-SP]x[wtSP]y, where x and y represents the input molar ratios) were purified using ultracentrifugation, and analyzed by SDS-PAGE (Fig. 2A). The gel electrophoresis confirmed the presence of both the components in co-assembled nanoreactors. Densitometric profiling of each lane in SDS-PAGE was used to calculate the average amount of AdhD-SP and wtSP encapsulated into the co-assembled nanoreactors. The densitometric profiles were analyzed with respect to known standards of each component and the area under each peak was determined for CP, wtSP and AdhD-SP (Fig. S2†). A calibration curve from the standards was used to calculate the mole ratios of wtSP, and AdhD-SP (relative to CP) encapsulated in the purified co-assembled nanoreactors. The output mole ratios encapsulated in the purified co-assembled nanoreactors, as a function of input stoichiometry used for assembly, is shown in Fig. 2B. The data indicate that both the components, AdhD-SP and wtSP, were incorporated into the capsids in proportion to the input stoichiometric ratios. When imaged by transmission electron microscopy (TEM), the co-assembled nanoreactors were found to be homogeneous and monodispersed, showing an average size of 56.1 ± 3.1 nm (≈100 particles analyzed) and intact spherical morphology, indicating that in vitro co-assembly did not alter the P22 VLP structure (Fig. 2C). The particles were further analyzed by size exclusion chromatography (SEC) coupled with multi-angle light scattering (MALS)/quasi-elastic light scattering (QELS) to determine their molecular weight and sizes (Table 1 and Fig. S3†). P22 empty shell (ES) particles, having no SP present, were also analyzed to determine the molecular weight of the capsid alone, which was then subtracted from the observed molecular weight of co-assembled nanoreactors to calculate the total amount of cargo (AdhD-SP and wtSP) encapsulated. The number of copies of each component was calculated from the ratio of the two SP components, as determined by the densitometric analysis of the SDS-PAGE (Fig. 2D and Table 1). The radius of gyration (Rrms) and hydrodynamic radius (Rh) of the co-assembled nanoreactors, determined from the angular dependence of the scattered light and their diffusion coefficients, respectively, were found to be similar to those previously reported for encapsulated P22 materials (Table 1).16,17,49 The value of Rrms/Rh reflects the distribution of densities from the center of the particle to radially outward; values around 0.78 indicate filled particles while values closer to 1.0 represent spherical shells with infinitely thin walls.70,71 The empty shell (ES) was found to have value of 1.0 and the co-assembled nanoreactors exhibited Rrms to Rh values ranging from 0.82–0.89, indicating cargo filled capsids. These findings show that in vitro assembly is a robust and generic approach for controlling the loading of cargo without affecting the integrity of P22 VLPs. The co-assembled nanoreactors were also analyzed by dynamic light scattering (DLS) to investigate the stability of capsids at 50 °C as the kinetics measurements were performed at that temperature and were found to have the same size distribution as P22 measured at room temp (Table S1†).63 After assembly and characterization, the kinetics of the co-assembled nanoreactors were measured at 50 °C.
:1.0, 0.1:0.9, 0.25:0.75, 0.5:0.5, 0.75:0.25, 0.9:0.1, 1.0:0.0 and added to a molar equivalent of CP. The co-assembled nanoreactors (P22-[AdhD-SP]x[wtSP]y, where x and y represents the input molar ratios) were purified using ultracentrifugation, and analyzed by SDS-PAGE (Fig. 2A). The gel electrophoresis confirmed the presence of both the components in co-assembled nanoreactors. Densitometric profiling of each lane in SDS-PAGE was used to calculate the average amount of AdhD-SP and wtSP encapsulated into the co-assembled nanoreactors. The densitometric profiles were analyzed with respect to known standards of each component and the area under each peak was determined for CP, wtSP and AdhD-SP (Fig. S2†). A calibration curve from the standards was used to calculate the mole ratios of wtSP, and AdhD-SP (relative to CP) encapsulated in the purified co-assembled nanoreactors. The output mole ratios encapsulated in the purified co-assembled nanoreactors, as a function of input stoichiometry used for assembly, is shown in Fig. 2B. The data indicate that both the components, AdhD-SP and wtSP, were incorporated into the capsids in proportion to the input stoichiometric ratios. When imaged by transmission electron microscopy (TEM), the co-assembled nanoreactors were found to be homogeneous and monodispersed, showing an average size of 56.1 ± 3.1 nm (≈100 particles analyzed) and intact spherical morphology, indicating that in vitro co-assembly did not alter the P22 VLP structure (Fig. 2C). The particles were further analyzed by size exclusion chromatography (SEC) coupled with multi-angle light scattering (MALS)/quasi-elastic light scattering (QELS) to determine their molecular weight and sizes (Table 1 and Fig. S3†). P22 empty shell (ES) particles, having no SP present, were also analyzed to determine the molecular weight of the capsid alone, which was then subtracted from the observed molecular weight of co-assembled nanoreactors to calculate the total amount of cargo (AdhD-SP and wtSP) encapsulated. The number of copies of each component was calculated from the ratio of the two SP components, as determined by the densitometric analysis of the SDS-PAGE (Fig. 2D and Table 1). The radius of gyration (Rrms) and hydrodynamic radius (Rh) of the co-assembled nanoreactors, determined from the angular dependence of the scattered light and their diffusion coefficients, respectively, were found to be similar to those previously reported for encapsulated P22 materials (Table 1).16,17,49 The value of Rrms/Rh reflects the distribution of densities from the center of the particle to radially outward; values around 0.78 indicate filled particles while values closer to 1.0 represent spherical shells with infinitely thin walls.70,71 The empty shell (ES) was found to have value of 1.0 and the co-assembled nanoreactors exhibited Rrms to Rh values ranging from 0.82–0.89, indicating cargo filled capsids. These findings show that in vitro assembly is a robust and generic approach for controlling the loading of cargo without affecting the integrity of P22 VLPs. The co-assembled nanoreactors were also analyzed by dynamic light scattering (DLS) to investigate the stability of capsids at 50 °C as the kinetics measurements were performed at that temperature and were found to have the same size distribution as P22 measured at room temp (Table S1†).63 After assembly and characterization, the kinetics of the co-assembled nanoreactors were measured at 50 °C.
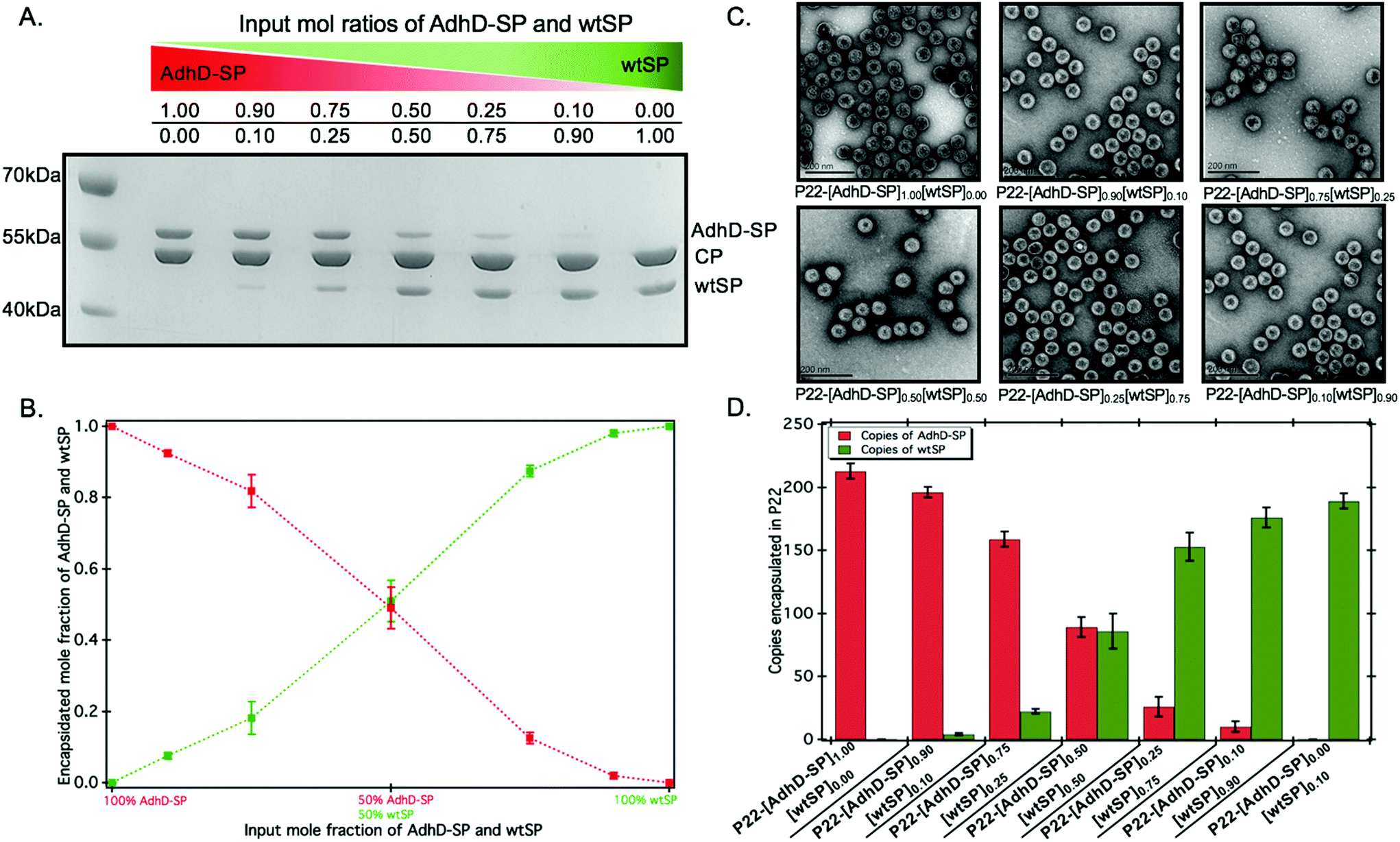 | ||
| Fig. 2 Characterization of co-assembled nanoreactors (P22-[AdhD-SP]x[wtSP]y, where, x and y represents the input molar ratios). (A) SDS-PAGE analysis of isolated co-assembled nanoreactors shows gradations in the band intensities of AdhD-SP and wtSP, as the input molar ratios are changed. (B) Graph of the encapsulated AdhD-SP and wtSP over a range of input stoichiometric ratios of each component, generated from densitometric analysis of SDS-PAGE. (C) TEM images show the sizes and intact spherical morphology of co-assembled nanoreactors. (D) Number of copies of AdhD-SP and wtSP encapsulated in P22 calculated using SEC-MALS data. | ||
| Sample P22-[AdhD-SP]x[wtSP]y | Molar mass (MDa) | Copies of AdhD-SP | Copies of wtSP | R rms (nm) | R h (nm) | R rms/Rh |
|---|---|---|---|---|---|---|
| Empty shell (ES) | 19.4 ± 0.4 | — | — | 25.9 ± 0.1 | 25.8 ± 0.2 | 1 ± 0.004 |
| P22-[AdhD-SP]1.00 [wtSP]0.0 | 31.10 ± 0.96 | 219 ± 6 | — | 20.9 ± 2.1 | 25.4 ± 1.1 | 0.82 ± 0.05 |
| P22-[AdhD-SP]0.90 [wtSP]0.10 | 30.22 ± 0.92 | 200 ± 4 | 3 ± 1 | 20.9 ± 2.3 | 25.4 ± 1.1 | 0.82 ± 0.05 |
| P22-[AdhD-SP]0.75 [wtSP]0.25 | 28.50 ± 0.74 | 154 ± 6 | 22 ± 2 | 21.6 ± 1.9 | 25.4 ± 0.8 | 0.85 ± 0.05 |
| P22-[AdhD-SP]0.50 [wtSP]0.50 | 26.94 ± 1.33 | 98 ± 8 | 74 ± 14 | 22.3 ± 1.7 | 25.4 ± 0.5 | 0.88 ± 0.05 |
| P22-[AdhD-SP]0.25 [wtSP]0.75 | 26.55 ± 1.03 | 43 ± 8 | 143 ± 11 | 22.5 ± 1.7 | 25.3 ± 0.5 | 0.89 ± 0.05 |
| P22-[AdhD-SP]0.10 [wtSP]0.90 | 26.34 ± 1.01 | 14 ± 4 | 183 ± 8 | 22.4 ± 1.7 | 25.5 ± 0.6 | 0.88 ± 0.05 |
| P22-[AdhD-SP]0.00 [wtSP]1.00 | 26.08 ± 0.83 | — | 194 ± 6 | 22.6 ± 1.8 | 25.6 ± 0.5 | 0.88 ± 0.05 |
The composition dictates the activity of co-assembled nanoreactors
The activity of AdhD was measured by monitoring the rate of disappearance of the cofactor, NADH, upon reduction of acetoin at 50 °C under slightly acidic pH (6.2) conditions to minimize any interference of the oxidation (reverse) reaction that takes place at alkaline pH (8.8).68 The activity of nanoreactors encapsulating variable amounts of AdhD-SP and wtSP (Fig. 2B) was measured under conditions where the AdhD concentration was constant for all samples. The dependence of the reaction rate on acetoin followed Michaelis–Menten kinetics. The AdhD kinetic data from all the nanoreactors were fit to a Michaelis–Menten equation and the kinetic parameters kcat and KM were calculated from the equation. The data indicated that the increase in concentration of co-encapsulated wtSP significantly reduces the kcat of AdhD in these nanoreactors (Fig. 3A), but the overall activity (kcat × number of copies of AdhD) of nanoreactor particles was increased as the packaging amount of AdhD was increased inside the nanoreactors (Fig. 3B). The concentration of encapsulated wtSP ranges from 0.1–5.5 mM (4–193 mg ml−1) inside these nanoreactors. At such high concentrations, macromolecules can affect the conformation and kinetics of enzymes either through macromolecular crowding (excluded volume effects) and/or through soft (weak non-covalent) interactions.44,72–76 Soft interactions between the SP and AdhD are less likely given their similar pIs (AdhD-SP1–266: 6.01 and wtSP1–266: 5.13), and thus overall charge at the working pH conditions. Similar activity exhibited by both free (AdhD) and SP-fused enzyme (AdhD-SP) as a function of their increasing concentrations further suggests that the wtSP do not interact chemically with the enzyme and therefore do not influence kinetic parameters (Fig. S4†). Some researchers have reported that macromolecular crowding increases enzymatic rate because of the higher activity coefficients of reacting molecules, potentially overcoming the slowed diffusion in a crowded medium.77–79 A decrease in enzymatic rate is normally associated with the changes in conformational dynamics of the active site of enzyme induced by crowded surroundings.80–83 Coarse-grained simulations of HIV-1 protease revealed that the presence of crowder significantly reduced the fraction of protease with open-state conformation through the continuous collisions, leading to slowing down of enzymatic activity.83 The observed reduction in reaction rates of AdhD in these nanoreactors with the increase in wtSP is suggestive of a process inhibiting conformational dynamics, leading to either perturbation of the docking site or product release. Studies also show that a crowded medium generally favors molecular compaction, such as the adoption of globular forms as opposed to expanded conformations that occupy larger volumes, and molecular association to reduce the excluded volume and increase the entropy of the system.40,72,76,84 Thus, if AdhD adopts an expanded conformation during its catalytic cycle, as reported in the literature for some members of the AKR family that undergo a conformational change upon cofactor binding and release,85 then we would expect to see a reduction in the activities as a consequence of the macromolecular crowding induced by wtSP, which would stabilize compact states of the enzyme in comparison to expanded conformations. The stabilization of compact states would thus lead to a reduction in activity of AdhD in nanoreactors as the concentration of co-encapsulated wtSP was increased. The crowders can affect the kinetics by slowing down the diffusion, however the catalytic efficiency of AdhD enzyme (kcat/KM) is on the order of 102 M−1 s−1 (Fig. S8†), which is far lower than the enzymatic reactions that has kcat/KM on the order of 108 M−1 s−1 or greater and are susceptible to diffusional limitations.86 At such a low catalytic efficiency, the kinetics is unlikely to be affected by substrate diffusion, especially at a higher temperatures (50 °C) where the kinetic measurements were done and a previous study showed that the P22 capsid does not present a barrier to the diffusion of small molecules used in this study and therefore does not influence the rate of reaction.16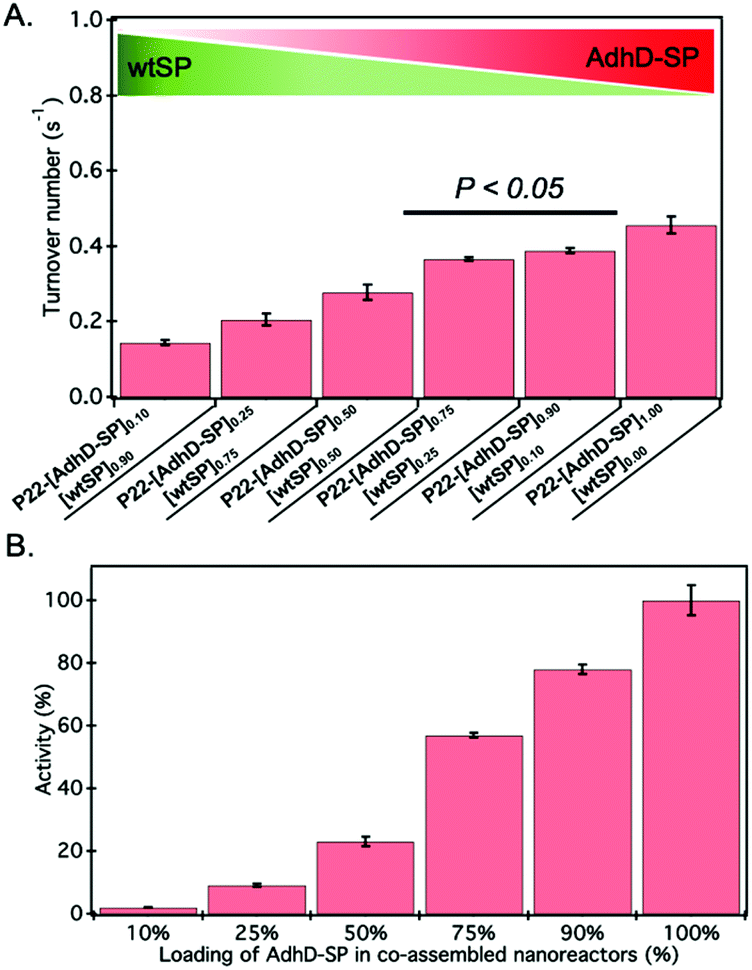 | ||
| Fig. 3 (A) The turnover number (kcat) of AdhD in co-assembled nanoreactors. Error bars represent standard deviations of triplicate measurements. The P-value (<0.05) indicates the statistically significant difference between the activities of those two data sets. (B) Graph showing the increase in overall activity of nanoreactor particle as the input packaging stoichiometry of AdhD-SP was increased in co-assembled nanoreactors. | ||
We tested the behaviour of the free (unencapsulated) AdhD enzyme under macromolecular crowding conditions generated using poly-ethylene glycol (PEG) (Fig. S5†). PEG 8k was chosen because it is an elongated crowding agent with no significant secondary structure and thus mimics the wtSP, which is partially disordered (37 ± 3% α-helix, 30 ± 6% random coil, 33 ± 6% β-turns)87,88 at the N-terminus region and elongated structurally with estimated dimensions 24.7 nm in length and 2.2 nm in diameter.89 The free AdhD activity was measured with PEG present at three different concentrations (5%, 15%, and 25% w/v) and a decrease in kcat was observed with increasing PEG concentration. This is likely due to the adoption of compact conformation or the formation of self-associated forms of the AdhD under the influence of excluded volume effects. This correlates well with our data from the co-assembled nanoreactors and suggests that wtSP, inside the P22 nanoreactor, can act analogously to PEG as a molecular crowding agent to AdhD resulting in a reduction of activity of the enzyme inside co-assembled nanoreactors.
Removal of wtSP resulted in the restoration of activities of AdhD in the nanoreactors
The approach utilized here for the removal of wtSP takes advantage of the fact that wtSP, but not bulky cargo-fused SP, can escape across the capsid shell upon treatment with 0.5 M GuHCl.49,63 Thus, the wtSP was selectively removed from co-assembled nanoreactors containing AdhD-SP and wtSP (P22-[AdhD-SP]x[wtSP]y) encapsulated at different stoichiometric ratios and the resulting materials were analyzed by SDS-PAGE to determine the degree of wtSP removal. As shown in Fig. 4A, we were able to remove all of the wtSP from all of the co-assembled nanoreactors without losing AdhD. TEM of these samples showed no change in morphology after the treatment (Fig. 4B). The capsids were further analyzed by SEC-MALS/QELS to determine the size and number of enzymes in each capsid (Table 2). Molecular weight determined by SEC-MALS was used to calculate the number of copies of enzyme after subtracting the molecular weight of the P22 empty shell. Both, the radius of gyration and the hydrodynamic radius of the GuHCl treated capsids were comparable to non-treated capsids, indicating no change in size after the GuHCl treatment (Table 2 and Fig. S6†). Our results are supported by the literature that shows only very weak density differences in the areas surrounding hexons in three-dimensional reconstruction (3DR) images obtained from the cryo-EM of P22 capsids and the capsids that were treated with GuHCl to remove wtSP.90 Further analysis by DLS confirmed the integrity of particles at 50 °C and the particle sized matched well with the size of co-assembled nanoreactors (Table S1†). The enzymes, in GuHCl treated nanoreactors, followed Michaelis–Menten kinetic behavior and the data were fit to obtain kcat and KM (Fig. 4C and Fig. S7†). Removal of wtSP from all the co-assembled nanoreactors resulted in an increase in kcat to a level similar to P22 encapsulating only the AdhD enzymes (Fig. 4C). A similar trend was observed with KM in co-assembled and wtSP depleted capsids (Fig. S7†). We found that the increase in concentration of wtSP resulted in a concentration-dependent decrease in substrate affinity (increased KM), but when the wtSP was selectively removed the KM approached similar values for all nanoreactors. This suggests that the crowding induced by wtSP may have affected the conformational dynamics of the active site of the enzymes to carry out catalysis, thereby effectively increasing the KM. Interestingly, the kcat of AdhD in all the wtSP depleted nanoreactors approached a similar value, despite differences in their packing density, but remained lower than the kcat of the free enzyme (Fig. S8†). These results strongly suggest that the activity of enzymes in wtSP depleted nanoreactors is influenced by the enzymatic confinement in P22 and that the AdhD molecules do not undergo self-crowding even at local concentrations as high as 6.3 mM (∼334 mg ml−1, Table S2†) present inside the nanoreactors assembled with AdhD only. Despite the similar kcat values of the encapsulated AdhD, the overall activity of SP depleted nanoreactors increased as the loading of AdhD-SP was increased, thus recapitulating the trend observed with co-assembled nanoreactors, but the activities were slightly enhanced at each loading in comparison to co-assembled nanoreactors as shown in the Fig. 4D.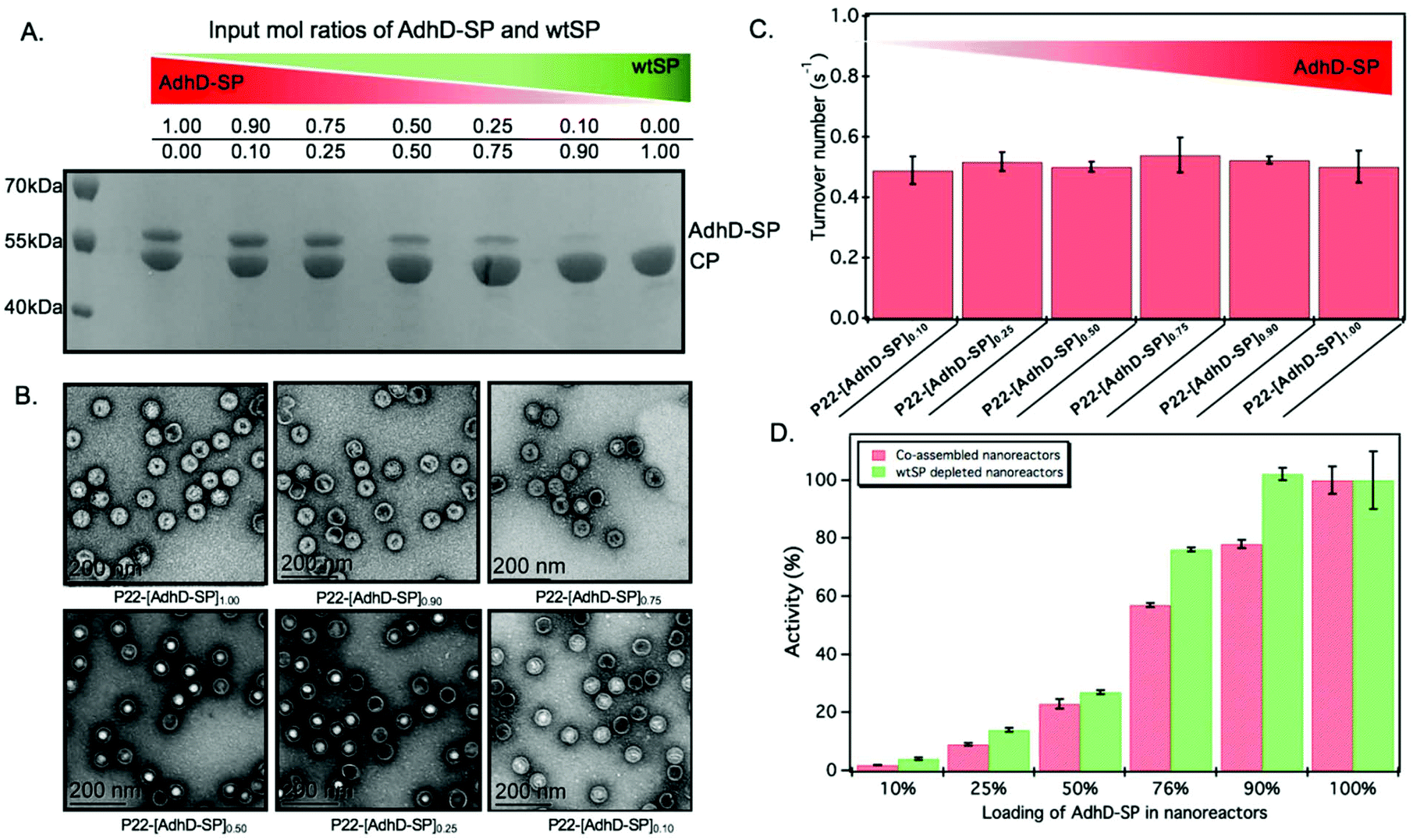 | ||
| Fig. 4 Characterization and activity of AdhD in wtSP depleted nanoreactors. (A) SDS-PAGE analysis shows selective removal of wtSP from all the co-assembled nanoreactors. (B) TEM images show that morphology is maintained after chaotrope treatment. (C) Graph showing the similar activities of AdhD in nanoreactors after removing wtSP from co-assembled nanoreactors. (D) Graph comparing the activity of co-assembled and wtSP depleted nanoreactors. The nanoreactors containing AdhD only (100 percent loading level) showed similar activities before and after GuHCl treatment, indicating that GuHCl does not affect the enzymatic activity and the capsid porosity. | ||
| Sample P22-[AdhD-SP]x | Copies of AdhD-SP | R rms (nm) | R h (nm) |
|---|---|---|---|
| Empty shell (ES) | — | 25.9 ± 0.1 | 25.8 ± 0.2 |
| P22-[AdhD-SP]1.00 | 175 ± 13 | 20.5 ± 2.3 | 25.4 ± 0.9 |
| P22-[AdhD-SP]0.90 | 152 ± 6 | 20.5 ± 2.7 | 25.0 ± 0.4 |
| P22-[AdhD-SP]0.75 | 108 ± 6 | 21.5 ± 2.4 | 24.9 ± 0.8 |
| P22-[AdhD-SP]0.50 | 42 ± 13 | 22.3 ± 2.8 | 25.2 ± 0.8 |
| P22-[AdhD-SP]0.25 | 21 ± 7 | 23.5 ± 2.1 | 24.9 ± 0.7 |
| P22-[AdhD-SP]0.10 | 7 ± 1 | 22.2 ± 1.0 | 25.2 ± 0.3 |
Concentration dependent activity of the free (unencapsulated) AdhD enzyme
The confinement molarity (Mconf), or the local concentration of the encapsulated AdhD enzyme inside these wtSP depleted nanoreactors, ranges from 0.2–5.8 mM (10–285 mg ml−1). High local concentration in an enclosed system can lead to enhanced molecular associations.91–93 Therefore, to probe the effects of high local concentration on the AdhD enzyme (by analogy to Mconf), we measured the activity of the free enzyme (AdhD-SP and AdhD), as a function of bulk concentration. The concentration of substrate and cofactor used were at least 30-fold over the highest concentration of enzyme used in the experiment. The kcat for the reaction exhibited a strong concentration dependence and decreased markedly with increasing enzyme concentration (Fig. 5). Free AdhD, without the SP fusion, also showed concentration dependent activity with a similar trend (Fig. S4†). This suggests that some self-association interactions between individual AdhD monomers could be responsible for the diminished activity. The association may have led to the formation of clusters in which the buried enzymes may not be involved in catalysis due to restricted substrate accessibility or the dynamic movement of the active site, whereas the surface exposed enzymes and some freely diffusing enzymes may still be contributing towards the activity.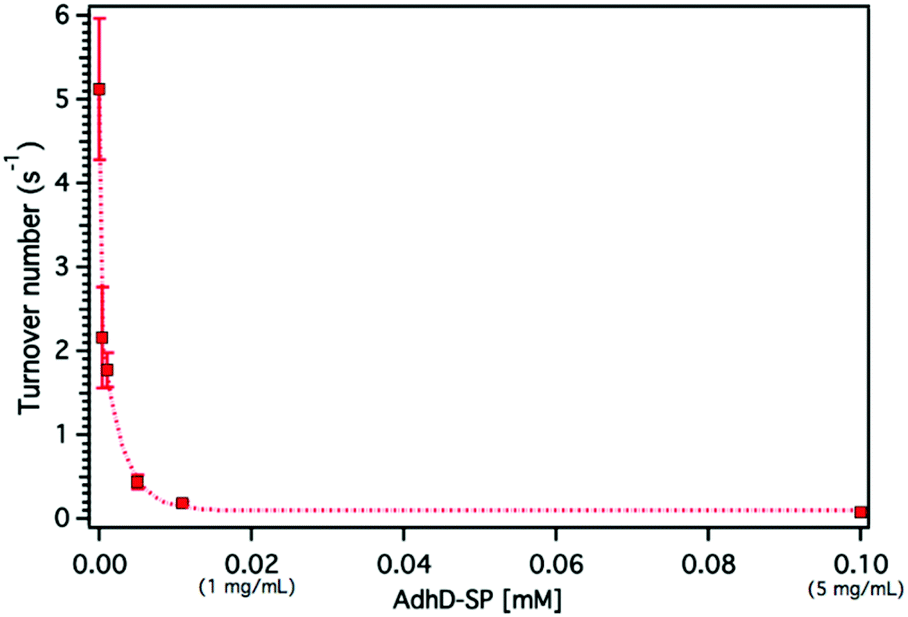 | ||
| Fig. 5 Graph showing the concentration dependence of free AdhD enzyme activity. | ||
Dynamic light scattering (DLS) measurements revealed the formation of higher order clusters of the AdhD enzymes with increasing concentration, supporting the self-association hypothesis (Fig. S9†). The fit of the activity versus concentration data indicates that the kinetic behavior of the free AdhD plateaus at high concentrations. This behavior of the free enzyme at high concentration potentially allows us to interpret the role of confinement in the AdhD encapsulated in the P22 nanoreactors. Under the high local concentrations of enzymes inside the P22 there could be significant self-association resulting in confinement effects that contribute to the lowered activity of the encapsulated AdhD when compared to the free enzyme at low concentrations. Just as observed in the free AdhD, where the activity plateaus at higher concentrations, the encapsulated AdhD shows the same activity at all loadings (after wtSP removal) where the Mconf for all nanoreactors is at or above the highest concentrations measured for the free AdhD where the decrease in activity has already plateaued.
These experiments show that crowding and confinement can influence the outcome of an encapsulation system independently. The experiments are, however, ensemble averages, and previously Dashti et al.52 showed a wide distribution in the amount of co-encapsulated cargoes in single particle measurements of mixed in vitro assemblies despite a clear trend observed in ensemble measurements. Therefore, complementing ensemble-averaged measurements with single-particle analysis will enable appropriate interpretation of the data and the insight into the structural dynamics of enzymes with better sensitivity and resolution under crowded and confined conditions.
Conclusion
Previously we have shown that a wide range of cargo molecules can be encapsulated in P22 using genetically programmed in vivo assembly approach. This approach, however, suffers from some limitations such as the lack of ability to control packaging amount of a cargo (or multiple cargos), encapsulation of misfolded proteins, or partial inactivation of proteins upon encapsulation in P22. To overcome some of these challenges, an in vitro assembly approach was developed for cargo encapsulation in P22. Using this approach, we have designed and constructed P22 nanoreactors, whose catalytic properties could be fine-tuned by controlling the composition of encapsulated cargos. A variety of nanoreactors were assembled in the presence of AdhD-SP and wtSP mixed at different stoichiometric ratios. The composition of assembled nanoreactors followed the input stoichiometry, and contained AdhD-SP and wtSP encapsulated in well-regulated and variable amounts, which was then translated into the control over functional output of AdhD catalyst molecules and the nanoreactors (i.e. enzyme–VLP complex). The selective removal of wtSP from the co-assembled nanoreactors allowed us to gain further control over the catalytic performance of the nanoreactors and independently investigate the effects of microenvironment on the functional properties of encapsulated AdhD enzyme. The insights gained from this research could prove useful in designing new materials with cell-like complexity using bio-inspired and biomimetic approaches.Materials and methods
Materials
Reagents for buffer preparation were purchased from Fisher Scientific, unless otherwise specified. DNA primers were purchased from Eurofins MWG Operon. E. coli BL21(λDE3) were purchased from Lucigen. Protease and phosphatase inhibitor mini tablets were purchased from Roche. For gene assembly, NEBuilder® HiFi DNA assembly master mix was purchased from New England Biolabs. Gene block were purchased from IDT.Cloning of AdhD into pBad vector
The AdhD gene from Pyrococcus furiosus was ordered from the IDT after codon optimization for BL21(DE3). The AdhD gene was cloned into a pBad vector containing gene for truncated scaffold protein (SPt) using Gibson assembly and the primers 5′ CGCAGCAATGCCGTAGCAGAACAGGGC 3′ and 5′ GCCGCTGTGA TGGTGATGATGGGC 3′ for linearization of SPt vector. The Gibson assembly was carried out using HiFi DNA assembly master mix. After verifying the DNA sequence from Eurofins Genomics, the assembled vector was transformed into BL21(DE3) cells for protein expression.Expression and purification of free AdhD-SP
Transformed BL21(DE3) cells were grown in LB medium at 37 °C in the presence of ampicillin to maintain selection for the vector containing cells. The expression of AdhD-SP was induced by addition of L-arabinose to a final concentration of 13 mM at OD600 = 0.6. The cultures were grown for 4 h post induction. The cells were harvested at 4500g in a benchtop centrifuge and re-suspended in PBS (50 mM phosphate, 100 mM sodium chloride, pH 7.8) with lysozyme, DNase, RNase and protease mini tablets for 30 min. The cell suspension was lysed by sonication and centrifuged at 12000g for 45 min at 4 °C. The supernatant was removed and filtered through 0.45-micron filter. For purification, the filtered supernatant was loaded on to Ni-NTA column at 1 mL min−1, followed by washing of the column with wash buffer (50 mM phosphate, 100 mM sodium chloride, pH 7.8) at 1 ml min−1. The protein was eluted with a linear gradient of imidazole (20–500 mM imidazole) at 2 ml min−1. The protein was dialyzed into an assembly buffer (50 mM Tris-HCl, 25 mM NaCl, 2 mM EDTA, 3 mM β-mercaptoethanol, 1% glycerol, and pH 7.4) for in vitro assembly.
Preparation of coat protein (CP) subunits for in vitro assembly
CP subunits were obtained by dissociating the ES in 3 M GuHCl (final concentration) for 30 min at room temperature. To obtain ES, the P22 procapsids were incubated with 0.5 M GuHCl for 1.5 h at 4 °C so as to extract SP. Capsids were pelleted in ultracentrifuge at 45000 rpm for 50 min followed by resuspension and incubation in 0.5 M GuHCl for 1 h. This extraction process was repeated until the supernatant was free from SP. The ES were then purified over an S-500 Sephadex size exclusion column. Assembly buffer was used in the background to make GuHCl solution for dissociation of ES and removal of SP.
In vitro assembly
CP subunits (∼2 mg ml−1), as prepared above in 3 M GuHCl, were mixed with purified AdhD-SP (or pre-mix of AdhD-SP and wtSP) subunits in a 1:1 molar ratio. The volume of the AdhD-SP was adjusted such that the final concentration of GuHCl was 1.5 M in all assembly reaction mixtures. The mixture was dialyzed into assembly buffer for 24 h at room temperature with a buffer exchange in between.
Removal of wtSP from co-assembled nanoreactors
To remove wtSP, the co-assembled nanoreactors were dialyzed into the assembly buffer containing 0.5 M GuHCl using 100 kDa MWCO dialysis membrane. The dialysis buffer was exchanged three times with a dialysis time of 4 h each time. The final dialysis was done with the PBS buffer with no GuHCl added.SDS-PAGE
Protein samples were mixed with 4× loading buffer containing 100 mM DTT, heated in a boiling water bath for 10 min, removed, and spun down on a benchtop centrifuge. Samples were separated on a 15% acrylamide gel at a constant current of 35 mA for approximately 1 h. Gels were stained with an Instant Blue protein stain, rinsed with water, and imaged. Images were recorded on a UVP MultDoc-IT Digital Imaging System. A 10–180 kDa PageRuler prestained ladder was used for reference.Transmission electron microscopy
Samples (4 μL, 0.3 mg mL−1 protein) were applied to glow-discharged carbon coated grids and incubated for 45 s. Excess liquid was wicked away with filter paper. The sample loaded grids were then washed on a droplet of water. Grids were stained with 4 μL 2% uranyl acetate for 10 s and excess stain was wicked away with filter paper. Images were taken on a JEOL 1010 transmission electron microscope at accelerating voltage of 80 kV.Size exclusion chromatography with multiangle light scattering and refractive index detection
Samples were separated over a WTC-0200S (Wyatt Technologies) size exclusion column utilizing an Agilent 1200 HPLC system at a flow rate 0.7 ml min−1 of MALS buffer (50 mM phosphate, 100 mM NaCl and 200 ppm NaN3, pH 7.2). Sample volume of 25 μL per injection was loaded onto the column, and the column was run for 35 min. Eluted peaks were detected using a UV-Vis detector (Agilent), a Wyatt HELEOS multi-angle laser light scattering (MALS) detector, and an Optilab rEX differential refractometer (Wyatt Technology Corporation). The number-average particle molecular weight, Mn, was measured across FWHM of each peak with Astra 6.0.3.16 (Wyatt Technology Corporation) using a previously calculated dn/dc value of 0.185 mL g−1.Kinetics assay of free and P22 encapsulated enzyme
The activity assays were carried out on an Agilent's 8453 UV-Visible spectrophotometer fitted with an 8 position multicell transport and a VWR temperature controller. All assays were conducted at 50 °C in triplicates and an assay volume of 110 μL was used for measurement. Kinetic assays were performed in 100 mM sodium phosphate, pH 6.2 buffer. Reaction mixtures containing acetoin substrate (ranging from 0.3 mM–100 mM), NADH (290 μM) and buffer were preheated in a quartz cuvette for 2 minutes prior to the addition of free or encapsulated enzyme. NADH thermal degradation was monitored for 80–100 seconds then enzyme was added to the solution. Reactions were mixed thoroughly by pipetting and the enzyme activity was measured by monitoring the rate of disappearance of NADH at A340 nm. For activity of the free enzyme, a 1 cm pathlength cuvette and an NADH concentration of up to 2.90 mM was used. Concentrations were calculated using extinction coefficients of NADH, P22 coat protein, AdhD-SP, and wtSP as 6220 M−1 cm−1, 44920 mM−1 cm−1, 61310 M−1 cm−1, 17420 M−1 cm−1, respectively (ExPASy's ProtParam tool). Total concentration of the encapsulated enzyme was calculated using densitometry and molar mass data from SEC-MALS. From the densitometry, ratio of AdhD-SP to wtSP was determined, which was then applied to SEC-MALS data to calculate number of copies of AdhD-SP and wtSP encapsulated inside the capsids. From the number of copies and A280, the concentration of encapsulated enzyme was calculated using the set of equations given in the ESI.† Kinetics plots were fit to Michaelis–Menten kinetics or substrate inhibition model using Igor pro 6.37.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by grants from the National Science Foundation (BMAT DMR-1507282) and the National Institutes of Health (R01 AI104905). We thank the Indiana University Electron Microscopy Center, Physical Biochemistry Instrumentation facility and Biological mass-spectrometry lab for access to instrumentation used in this work.Notes and references
- M. A. Meyers, P. Y. Chen, A. Y. M. Lin and Y. Seki, Prog. Mater. Sci., 2008, 53, 1–206 CrossRef CAS.
- R. R. Naik and S. Singamaneni, Chem. Rev., 2017, 117, 12581–12583 CrossRef CAS PubMed.
- N. F. Lepora, P. Verschure and T. J. Prescott, Bioinspiration Biomimetics, 2013, 8, 013001 CrossRef PubMed.
- T. W. Giessen and P. A. Silver, J. Mol. Biol., 2016, 428, 916–927 CrossRef CAS PubMed.
- A. H. Chen and P. A. Silver, Trends Cell Biol., 2012, 22, 662–670 CrossRef CAS PubMed.
- C. A. Kerfeld, C. Aussignargues, J. Zarzycki, F. Cai and M. Sutter, Nat. Rev. Microbiol., 2018, 16, 277–290 CrossRef CAS PubMed.
- Y. Azuma, T. G. W. Edwardson and D. Hilvert, Chem. Soc. Rev., 2018, 47, 3543–3557 RSC.
- T. W. Giessen, Curr. Opin. Chem. Biol., 2016, 34, 1–10 CrossRef CAS PubMed.
- E. Y. Kim and D. Tullman-Ercek, Curr. Opin. Biotechnol., 2013, 24, 627–632 CrossRef CAS PubMed.
- D. M. Vriezema, M. C. Aragones, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS PubMed.
- C. M. Jakobson, Y. Chen, M. F. Slininger, E. Valdivia, E. Y. Kim and D. Tullman-Ercek, J. Mol. Biol., 2016, 428, 2989–2996 CrossRef CAS PubMed.
- H. J. Wagner, C. C. Capitain, K. Richter, M. Nessling and J. Mampel, Eng. Life Sci., 2017, 17, 36–46 CrossRef CAS.
- S. Tetter and D. Hilvert, Angew. Chem., 2017, 56, 14933–14936 CrossRef CAS PubMed.
- J. E. Glasgow, S. L. Capehart, M. B. Francis and D. Tullman-Ercek, ACS Nano, 2012, 6, 8658–8664 CrossRef CAS PubMed.
- J. D. Fiedler, S. D. Brown, J. L. Lau and M. G. Finn, Angew. Chem., Int. Ed., 2010, 49, 9648–9651 CrossRef CAS PubMed.
- D. P. Patterson, P. E. Prevelige and T. Douglas, ACS Nano, 2012, 6, 5000–5009 CrossRef CAS PubMed.
- D. P. Patterson, B. Schwarz, R. S. Waters, T. Gedeon and T. Douglas, ACS Chem. Biol., 2014, 9, 359–365 CrossRef CAS PubMed.
- B. Worsdorfer, Z. Pianowski and D. Hilvert, J. Am. Chem. Soc., 2012, 134, 909–911 CrossRef CAS PubMed.
- J. P. Colletier, B. Chaize, M. Winterhalter and D. Fournier, BMC Biotechnol., 2002, 2, 9 CrossRef PubMed.
- Z. Zhao, J. Fu, S. Dhakal, A. Johnson-Buck, M. Liu, T. Zhang, N. W. Woodbury, Y. Liu, N. G. Walter and H. Yan, Nat. Commun., 2016, 7, 10619 CrossRef CAS PubMed.
- C. Dulieu, M. Moll, J. Boudrant and D. Poncelet, Biotechnol. Prog., 2000, 16, 958–965 CrossRef CAS PubMed.
- U. T. Bornscheuer, Angew. Chem., Int. Ed., 2003, 42, 3336–3337 CrossRef CAS PubMed.
- S. Datta, L. R. Christena and Y. R. Rajaram, 3 Biotech, 2013, 3, 1–9 CrossRef PubMed.
- L. Sanchez-Sanchez, R. D. Cadena-Nava, L. A. Palomares, J. Ruiz-Garcia, M. S. Koay, J. J. L. M. Cornelissen and R. Vazquez-Duhalt, Enzyme Microb. Technol., 2014, 60, 24–31 CrossRef CAS PubMed.
- M. Comellas-Aragones, H. Engelkamp, V. I. Claessen, N. A. J. M. Sommerdijk, A. E. Rowan, P. C. M. Christianen, J. C. Maan, B. J. M. Verduin, J. J. L. M. Cornelissen and R. J. M. Nolte, Nat. Nanotechnol., 2007, 2, 635–639 CrossRef CAS PubMed.
- I. J. Minten, V. I. Claessen, K. Blank, A. E. Rowan, R. J. M. Nolte and J. J. L. M. Cornelissen, Chem. Sci., 2011, 2, 358–362 RSC.
- L. Schoonen, R. J. Nolte and J. C. van Hest, Nanoscale, 2016, 8, 14467–14472 RSC.
- B. Maity, K. Fujita and T. Ueno, Curr. Opin. Chem. Biol., 2015, 25, 88–97 CrossRef CAS PubMed.
- J. W. Wilkerson, S. O. Yang, P. J. Funk, S. K. Stanley and B. C. Bundy, New Biotechnol., 2018, 44, 59–63 CrossRef CAS PubMed.
- R. Frey, S. Mantri, M. Rocca and D. Hilvert, J. Am. Chem. Soc., 2016, 138, 10072–10075 CrossRef CAS PubMed.
- M. Brasch, R. M. Putri, M. V. de Ruiter, D. Luque, M. S. Koay, J. R. Caston and J. J. L. M. Cornelissen, J. Am. Chem. Soc., 2017, 139, 1512–1519 CrossRef CAS PubMed.
- D. P. Patterson, K. McCoy, C. Fijen and T. Douglas, J. Mater. Chem. B, 2014, 2, 5948–5951 RSC.
- H. Li, G. Zheng and S. Zhu, Microb. Cell Fact., 2018, 17, 26 CrossRef PubMed.
- H. Ren, S. Zhu and G. Zheng, Int. J. Mol. Sci., 2019, 20, pii: E592 CrossRef PubMed.
- V. A. Kickhoefer, Y. Garcia, Y. Mikyas, E. Johansson, J. C. Zhou, S. Raval-Fernandes, P. Minoofar, J. I. Zink, B. Dunn, P. L. Stewart and L. H. Rome, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4348–4352 CrossRef CAS PubMed.
- E. Selivanovitch and T. Douglas, Curr. Opin. Virol., 2019, 36, 38–46 CrossRef CAS PubMed.
- H. Choi, B. Choi, G. J. Kim, H. U. Kim, H. Kim, H. S. Jung and S. Kang, Small, 2018, 14, e1801488 CrossRef PubMed.
- M. Uchida, B. LaFrance, C. C. Broomell, P. E. Prevelige Jr. and T. Douglas, Small, 2015, 11, 1562–1570 CrossRef CAS PubMed.
- S. Long, J. Kunkel and P. Asuri, Biochem. Anal. Biochem., 2018, 7, 355 Search PubMed.
- H. X. Zhou, G. Rivas and A. P. Minton, Annu. Rev. Biophys., 2008, 37, 375–397 CrossRef CAS PubMed.
- H. X. Zhou, J. Mol. Recognit., 2004, 17, 368–375 CrossRef CAS PubMed.
- G. Ping, J. M. Yuan, Z. Sun and Y. Wei, J. Mol. Recognit., 2004, 17, 433–440 CrossRef CAS PubMed.
- S. Oehler and B. Muller-Hill, J. Mol. Biol., 2010, 395, 242–253 CrossRef CAS PubMed.
- R. J. Ellis, Trends Biochem. Sci., 2001, 26, 597–604 CrossRef CAS PubMed.
- R. J. Ellis and A. P. Minton, Nature, 2003, 425, 27–28 CrossRef CAS PubMed.
- G. B. Ralston, J. Chem. Educ., 1990, 67, 857–860 CrossRef CAS.
- I. M. Kuznetsova, K. K. Turoverov and V. N. Uversky, Int. J. Mol. Sci., 2014, 15, 23090–23140 CrossRef PubMed.
- S. Mittal, R. K. Chowhan and L. R. Singh, Biochim. Biophys. Acta, 2015, 1850, 1822–1831 CrossRef CAS PubMed.
- A. O'Neil, C. Reichhardt, B. Johnson, P. E. Prevelige and T. Douglas, Angew. Chem., Int. Ed., 2011, 50, 7425–7428 CrossRef PubMed.
- M. Comellas-Aragones, H. Engelkamp, V. I. Claessen, N. A. Sommerdijk, A. E. Rowan, P. C. Christianen, J. C. Maan, B. J. Verduin, J. J. L. M. Cornelissen and R. J. Nolte, Nat. Nanotechnol., 2007, 2, 635–639 CrossRef CAS PubMed.
- I. J. Minten, L. J. A. Hendriks, R. J. M. Nolte and J. J. L. M. Cornelissen, J. Am. Chem. Soc., 2009, 131, 17771–17773 CrossRef CAS PubMed.
- N. H. Dashti, R. S. Abidin and F. Sainsbury, ACS Nano, 2018, 12, 4615–4623 CrossRef CAS PubMed.
- Y. Azuma and D. Hilvert, Methods Mol. Biol., 2018, 1798, 39–55 CrossRef CAS PubMed.
- Y. Azuma, R. Zschoche, M. Tinzl and D. Hilvert, Angew. Chem., 2016, 55, 1531–1534 CrossRef CAS PubMed.
- L. Schoonen, R. J. M. Nolte and J. C. M. van Hest, Nanoscale, 2016, 8, 14467–14472 RSC.
- L. Schoonen, J. Pille, A. Borrmann, R. J. M. Nolte and J. C. M. van Hest, Bioconjugate Chem., 2015, 26, 2429–2434 CrossRef CAS PubMed.
- D. Botstein, C. H. Waddell and J. King, J. Mol. Biol., 1973, 80, 669–695 CrossRef CAS PubMed.
- G. P. Padilla-Meier, E. B. Gilcrease, P. R. Weigele, J. R. Cortines, M. Siegel, J. C. Leavitt, C. M. Teschke and S. R. Casjens, J. Biol. Chem., 2012, 287, 33766–33780 CrossRef CAS PubMed.
- B. Greene and J. King, Virology, 1994, 205, 188–197 CrossRef CAS PubMed.
- M. H. Parker, S. Casjens and P. E. Prevelige Jr., J. Mol. Biol., 1998, 281, 69–79 CrossRef CAS PubMed.
- S. Qazi, H. M. Miettinen, R. A. Wilkinson, K. McCoy, T. Douglas and B. Wiedenheft, Mol. Pharm., 2016, 13, 1191–1196 CrossRef CAS PubMed.
- A. O'Neil, P. E. Prevelige, G. Basu and T. Douglas, Biomacromolecules, 2012, 13, 3902–3907 CrossRef PubMed.
- J. Sharma, M. Uchida, H. M. Miettinen and T. Douglas, Nanoscale, 2017, 9, 10420–10430 RSC.
- G. Anand, S. Sharma, A. K. Dutta, S. K. Kumar and G. Belfort, Langmuir, 2010, 26, 10803–10811 CrossRef CAS PubMed.
- M. T. Fuller and J. King, Biophys. J., 1980, 32, 381–401 CrossRef CAS PubMed.
- P. E. Prevelige Jr., D. Thomas and J. King, J. Mol. Biol., 1988, 202, 743–757 CrossRef CAS.
- D. H. Chen, M. L. Baker, C. F. Hryc, F. DiMaio, J. Jakana, W. Wu, M. Dougherty, C. Haase-Pettingell, M. F. Schmid, W. Jiang, D. Baker, J. A. King and W. Chiu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1355–1360 CrossRef CAS PubMed.
- R. Machielsen, A. R. Uria, S. W. M. Kengen and J. van der Oost, Appl. Environ. Microbiol., 2006, 72, 233–238 CrossRef CAS PubMed.
- K. Solanki, W. Abdallah and S. Banta, Biotechnol. J., 2016, 11, 1483–1497 CrossRef CAS PubMed.
- J. Hotz and W. Meier, Langmuir, 1998, 14, 1031–1036 CrossRef CAS.
- S. U. Egelhaaf and P. Schurtenberger, J. Phys. Chem., 1994, 98, 8560–8573 CrossRef CAS.
- A. P. Minton and J. Wilf, Biochemistry, 1981, 20, 4821–4826 CrossRef CAS PubMed.
- A. C. Miklos, M. Sarkar, Y. Q. Wang and G. J. Pieak, J. Am. Chem. Soc., 2011, 133, 7116–7120 CrossRef CAS PubMed.
- M. Sarkar, A. E. Smith and G. J. Pielak, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19342–19347 CrossRef CAS PubMed.
- M. Jiang and Z. H. Guo, J. Am. Chem. Soc., 2007, 129, 730–731 CrossRef CAS PubMed.
- M. G. Norris and N. Malys, Biochem. Biophys. Res. Commun., 2011, 405, 388–392 CrossRef CAS PubMed.
- J. R. Wenner and V. A. Bloomfield, Biophys. J., 1999, 77, 3234–3241 CrossRef CAS PubMed.
- M. T. Moran-Zorzano, A. Viale, F. Munoz, N. Alonso-Casajus, G. Eydallin, B. Zugasti, E. Baroja-Fernandez and J. Pozueta-Romero, FEBS Lett., 2007, 581, 1035–1040 CrossRef CAS PubMed.
- Y. Sasaki, D. Miyoshi and N. Sugimoto, Nucleic Acids Res., 2007, 35, 4086–4093 CrossRef CAS PubMed.
- I. Pastor, E. Vilaseca, S. Madurga, J. L. Garces, M. Cascante and F. Mas, J. Phys. Chem. B, 2011, 115, 1115–1121 CrossRef CAS PubMed.
- S. N. Olsen, Thermochim. Acta, 2006, 448, 12–18 CrossRef CAS.
- S. N. Olsen, H. Ramlov and P. Westh, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2007, 148, 339–345 CrossRef CAS PubMed.
- D. D. Minh, C. E. Chang, J. Trylska, V. Tozzini and J. A. McCammon, J. Am. Chem. Soc., 2006, 128, 6006–6007 CrossRef CAS PubMed.
- S. Hou, N. Ziebacz, T. Kalwarczyk, T. S. Kaminski, S. A. Wieczorek and R. Holyst, Soft Matter, 2011, 7, 3092–3099 RSC.
- G. Sanli, J. I. Dudley and M. Blaber, Cell Biochem. Biophys., 2003, 38, 79–101 CrossRef CAS PubMed.
- H. W. Blanch and D. S. Clark, Biochemical Engineering, Marcel Dekker, New York, 1997 Search PubMed.
- R. Tuma, P. E. Prevelige Jr. and G. J. Thomas Jr., Biochemistry, 1996, 35, 4619–4627 CrossRef CAS PubMed.
- C. M. Teschke, J. King and P. E. Prevelige, Biochemistry, 1993, 32, 10658–10665 CrossRef CAS PubMed.
- B. Greene and J. King, J. Biol. Chem., 1999, 274, 16135–16140 CrossRef CAS PubMed.
- P. A. Thuman-Commike, B. Greene, J. A. Malinski, M. Burbea, A. McGough, W. Chiu and P. E. Prevelige Jr., Biophys. J., 1999, 76, 3267–3277 CrossRef CAS PubMed.
- P. C. Jordan, D. P. Patterson, K. N. Saboda, E. J. Edwards, H. M. Miettinen, G. Basu, M. C. Thielges and T. Douglas, Nat. Chem., 2016, 8, 179–185 CrossRef CAS PubMed.
- G. L. Martin, S. D. Minteer and M. Cooney, Chem. Commun., 2011, 47, 2083–2085 RSC.
- F. Wu and S. D. Minteer, Biomacromolecules, 2013, 14, 2739–2749 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr08348k |
| This journal is © The Royal Society of Chemistry 2020 |