
Quasi-hydrophilic black silicon photocathodes with inverted pyramid arrays for enhanced hydrogen generation†
Shuai
Zhao
 ab,
Guodong
Yuan
*ab,
Qi
Wang
ab,
Wenqiang
Liu
ab,
Ru
Wang
c and
Shenghua
Yang
c
ab,
Guodong
Yuan
*ab,
Qi
Wang
ab,
Wenqiang
Liu
ab,
Ru
Wang
c and
Shenghua
Yang
c
aState Key Laboratory for Superlattices and Microstructures, Institute of Semiconductors, Chinese Academy of Sciences, Beijing 100083, China. E-mail: gdyuan@semi.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cSchool of Electronic Information Engineering, Hebei University of Technology, Tianjin 300130, China
First published on 16th November 2019
Abstract
Micro-/nanostructured silicon (Si) photoelectrodes are promising for efficient solar-driven water splitting. In this work, an elaborate study on textured Si photocathodes is reported. Compared to conventional textured Si photocathodes, the well-designed Si photocathode with randomly-distributed inverted pyramid arrays (SiIPs) generates a larger photovoltage of 440 mV for its higher effective minority carrier density, and produces a higher photocurrent density at a high reverse bias voltage due to its quasi-hydrophilicity. With the help of cobalt disulfide (CoS2) nanocrystals, sluggish charge kinetics of SiIP photocathodes can be further improved. The optimal SiIP/CoS2 photocathode yields an onset potential of 0.22 V vs. reversible hydrogen electrode (RHE) and a saturated photocurrent density of 10.4 mA cm−2 at −0.45 V (vs. RHE). Besides, this cathode produces a stable photocurrent density of ∼6.60 mA cm−2 at 0 V (vs. RHE) for 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s in acidic media. Notably, our work presents a facile and inexpensive method to fabricate efficient Si photoelectrodes, which may promote the evolution of textured Si-based electrodes for potential photoelectrochemical and photocatalytic applications.
000 s in acidic media. Notably, our work presents a facile and inexpensive method to fabricate efficient Si photoelectrodes, which may promote the evolution of textured Si-based electrodes for potential photoelectrochemical and photocatalytic applications.
1. Introduction
Converting solar energy to carbon-free chemical fuels is a promising way to ease the increasingly serious energy crisis without any greenhouse gas emission.1 Given this, tremendous efforts were made to realize efficient solar-driven water splitting for ultraclean and storable hydrogen within the last few decades.2 In a conventional photoelectrochemical (PEC) water splitting system, photo-induced electron–hole pairs generated by semiconductor photoelectrodes inject into electrolytes and react with protons or hydroxyls to fulfil water reduction and oxidation.2,3 Hence, there exist three ways to attain high-performance photoelectrodes: (i) enhancement of light trapping in semiconductor photoelectrodes, (ii) acceleration of charge transfer at solid–liquid interfaces, and (iii) promotion of chemical reactions on electrode surfaces. Since the first discovery of PEC water oxidation at a TiO2 electrode in the 1970s,4 stable and cost-effective semiconducting metal oxide photoelectrodes, such as Cu2O, Fe2O3, WO3 and BiVO4, have been widely studied by many groups.5–9 However, only a small fraction of solar energy can be absorbed by them due to their wide band gaps. Thus, narrow-band-gap semiconductor photoelectrodes, including Si, GaAs, InP and CuInGaSe, are attracting increasing research interest to maximize the utilization of the solar spectrum.10–15 Among them, silicon (Si), the extensively used material in the photovoltaic industry,16,17 is one of the most promising materials for efficient water splitting due to its optimum energy band structure, technical maturity and Earth-abundance.Many studies have confirmed the feasibility and superiority of Si photoelectrodes previously,18 nevertheless, the high light reflectivity of planar Si excludes it from practical PEC electrodes. To minimize light reflections at the Si/electrolyte interfaces, various micro-/nanostructures were built on Si surfaces. Black Si with porous surfaces is the most common material for Si-based photoelectrodes, but its large surface area results in serious carrier recombination.19 Moreover, its hydrophobic high-aspect-ratio structures severely decelerate the mass transfer process during reactions. To overcome these shortcomings, hydrophilic passivation layers such as Al2O320 and TiO219,21 thin films were introduced via atomic layer deposition (ALD). Despite their excellent performances, it is difficult for these electrodes to be applied in mass production due to the high cost and low productivity of ALD techniques. On the other hand, improving the etched morphology of Si electrodes, a facile and inexpensive method, was adopted to solve the above problems. Some researchers presented macroporous Si photoelectrodes fabricated through a two-step etching method,21 while others demonstrated alkali-textured Si photoelectrodes with upright pyramids.22,23 Both the carrier lifetime and the wetting ability of the Si electrodes may be improved on this occasion,24,25 but the insufficient light absorption limits their PEC performances.
It is crucial to synthesize novel micro-/nanostructures on Si photoelectrodes to maximize their PEC efficiencies. Herein, we design and introduce black Si photoelectrodes with inverted pyramid arrays for enhanced solar-hydrogen conversion. We controllably synthesized randomly-distributed Si inverted pyramid arrays (SiIPs) on p-type Si (p-Si) wafers via facile copper-assisted chemical etching (Cu-ACE). These wafers exhibit a mean reflectivity of ∼6.55% and meanwhile a quasi-hydrophilic nature. The SiIP photocathode, compared with the conventional Si upright pyramid array (SiUP) and Si nanowire array (SiNW) counterparts, produces a larger photovoltage of 440 mV and a higher photocurrent density at a bias voltage ranging from 0.2 V to −1 V vs. reversible hydrogen electrode (RHE). Furthermore, we synthesized intrinsically metallic cobalt disulfide (CoS2) nanocrystals as hydrogen evolution reaction (HER) cocatalysts on SiIP photocathodes through a two-step method to improve the sluggish charge kinetics of Si. With an optimal loading amount of CoS2, the SiIP photocathode attains an onset potential of 0.22 V vs. RHE and a high photocurrent density of 6.60 mA cm−2 at 0 V vs. RHE, which is a record for p-Si/CoS2 photocathodes to the best of our knowledge.
2. Results and discussion
In this work, randomly-distributed inverted pyramid arrays were synthesized on Si(100) wafers with an aqueous Cu2+/H2O2/HF system, in which copper ions act as oxidants while silicon serves as the reductant and hydrofluoric acid works as an auxiliary solution to dissolve silicon oxide. The main reactions occurring in the Cu-ACE process can be described as follows:| Cu2+ → Cu(s) + 2h+E^{0} = +0.337 V vs. RHE, | (1) |
| H2O2 + 2H+ → 2H2O + 2h+E^{0} = +1.776 V vs. RHE, | (2) |
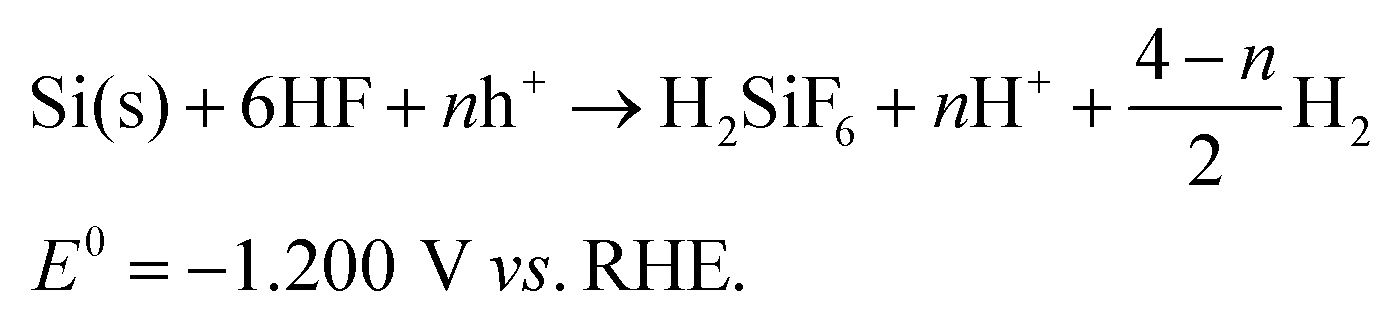 | (3) |
In virtue of the low redox potential of the Cu2+/Cu couple, Si can be anisotropically oxidized by Cu2+ and transformed into soluble matters with the help of HF. As a result, active Si crystallographic planes with lower atomic packing densities and more dangling bonds are etched away, while stable Si{111} planes remain and small inverted pyramids take shape. Meanwhile, the strong oxidant H2O2 facilitates the dissolution of Cu nanoparticles to realize a successive etching process so that inverted pyramids are enlarged. However, H2O2 molecules can also occupy some active Si atoms and lead to a slow isotropic stain etching result. Thus, it is necessary to regulate the relationships between [Cu2+] and [H2O2]. According to our previous work,26 Si{111}-like crystallographic planes will be preferentially exposed after Cu-ACE under certain conditions; hence, SiIP wafers for PEC electrodes can be controllably prepared via facile Cu-ACE by managing the etch end-points. Typical scanning electron microscopy (SEM) images of SiIPs are shown in Fig. 1a–d. Varisized but closely connected inverted pyramids, whose top side lengths are in the range of 1–4 μm, come into being after etching. For comparison, conventional SiUPs and SiNWs were synthesized on Si(100) wafers through alkaline texturing24 and silver-assisted chemical etching,27 respectively. As shown in Fig. 1e and Fig. S1,† the bottom side lengths of upright pyramids are in the range of 1–5 μm. And the lengths of nanowires are about 8 μm, as shown in Fig. 1f. To further reveal the morphological differences between SiIPs and SiUPs, we established three-dimensional models for these two arrays based on their actual geometries. Sunk inverted-pyramid-like etching pits of SiIPs and projecting upright-pyramid-like bulges of SiUPs can be clearly distinguished from Fig. S2a and b,† respectively.
 | ||
| Fig. 1 Microscopy of micro-/nanostructured Si wafers. (a) Plane view, (b) 25°-tilted plane view, and (c and d) cross-sectional SEM images of SiIP wafers. (e and f) Cross-sectional SEM images of SiUP and SiNW wafers, respectively. | ||
As shown in Fig. 2a, the mean reflectivities are 13.71% for dark gray SiUP wafers, 3.89% for tawny SiNW wafers and 6.55% for black SiIP wafers over the wavelength range of 300 to 1100 nm, respectively. Textured p-Si wafers exhibit effective anti-reflection nature, nevertheless, the nuances between etched morphologies determine the vast differences in their light-trapping abilities. Thanks to the multiple light reflections in the above graded-index optical structures,28 more photons can be effectively captured in these textured wafers. It is noted that inverted pyramids enable more rays to suffer a triple bounce29 than upright pyramids, and thus enhance the light-trapping ability of Si wafers. As for SiNW wafers, unique sub-wavelength features result in an ultralow reflectivity. However, their large surface area leads to intense surface recombination and the enhanced carrier density induced by quantum effects gives rise to serious Auger recombination.24 Hence, as can be found in Table S1,† the effective minority carrier lifetime of SiNW wafers is very short (∼0.61 μs). In contrast, the effective minority carrier lifetimes are 0.99 and 0.93 μs for SiUP and SiIP wafers, respectively, in which carrier recombination is more sluggish on account of their smaller surface areas.
 | ||
| Fig. 2 Optical and PEC properties of textured p-Si. (a) Reflectance spectra of planar Si, SiUP, SiNW and SiIP wafers. (b) J–V curves for textured Si photocathodes (under one sun illumination). The black dashed line in the inset is the result of a planar n+-Si cathode tested in the dark. | ||
Current density–potential (J–V) curves of Si photocathodes with the above three micro-/nanostructures were obtained through linear sweep voltammetry. As shown in Fig. 2b, textured Si photocathodes show obvious cathodic current characteristics under one sun illumination and the SiIP photocathode reaches the highest PEC efficiency. Chopped current density–time (J–t) curves for Si photocathodes at −0.85 V vs. RHE (Fig. S3†) further verify their negligible dark current densities. The photovoltages generated by SiUP, SiNW, and SiIP cathodes are about 340, 390, and 440 mV, respectively, as defined by the positive HER onset shifts between textured p-Si photocathodes (under illumination) and a metallic heavily-doped (0.001–0.005 Ω cm) n-type Si cathode (in the dark, the black dashed line in the inset of Fig. 2b).30,31 The largest photovoltage of the SiIP photocathode may stem from its higher effective electron density, that is, low reflectivity and meanwhile a moderate effective minority carrier lifetime. Furthermore, the SiIP photocathode produces the highest photocurrent density at a bias voltage ranging from 0.2 V to −1 V (vs. RHE), as shown in Fig. 2b. As for the other two counterparts, the SiNW photocathode leads to a secondary photocurrent density at a low reverse bias voltage (0.2 V to −0.46 V vs. RHE) while the SiUP photocathode induces a secondary photocurrent density at a high reverse bias voltage (−0.46 V to −1 V vs. RHE). It can be inferred that different limiting factors determine the photocurrent densities for textured Si cathodes at different ranges of the bias voltage.
Fig. 3a and c show the simplified energy band diagrams for p-Si/electrolyte junctions at a low and a high reverse bias voltage under illumination, respectively, where Ec, Ev, and Ef represent the energy level of the conduction band minimum, the energy level of the valence band maximum, and the Fermi level for p-Si, respectively. The height of an electron potential well (ΦW) can be determined by the contact potential difference between p-Si and the electrolyte (ΦSE), the Helmholtz double-layer potential drop (VH), the photovoltage of p-Si (VL) and the applied reverse bias voltage (VR):  . To determine the differences of VH values between these textured Si electrode/electrolyte junctions, we measured their flat-band potentials (EFB) from Mott–Schottky (MS) plots obtained via impedance–potential methods (Fig. S4†). The EFB values are −0.02, −0.01, 0 V vs. RHE for SiUP, SiNW and SiIP cathode/electrolyte junctions, respectively, close to those obtained in the previous reports.32,33 It can be legitimately assumed that VH, which varies with the potential and surface area of electrodes, is a fixed value for these three textured Si electrodes at a certain VR according to their nearly identical EFB values
. To determine the differences of VH values between these textured Si electrode/electrolyte junctions, we measured their flat-band potentials (EFB) from Mott–Schottky (MS) plots obtained via impedance–potential methods (Fig. S4†). The EFB values are −0.02, −0.01, 0 V vs. RHE for SiUP, SiNW and SiIP cathode/electrolyte junctions, respectively, close to those obtained in the previous reports.32,33 It can be legitimately assumed that VH, which varies with the potential and surface area of electrodes, is a fixed value for these three textured Si electrodes at a certain VR according to their nearly identical EFB values  .34 Hence, the ΦW values are controlled by (VR − VL). Fig. 3b and d show the Nyquist plots obtained by electrochemical impedance spectroscopy (EIS) experiments of p-Si photocathodes at −0.05 V and −0.65 V (vs. RHE) under one sun illumination, and the frequency range is from 100 kHz to 5 Hz. EIS data were fitted using an equivalent circuit with two time-related components, which is shown in the insets of Fig. 3b and d and Fig. S5.† The Cdl component models the double-layer capacitance, while Cj represents the Si/electrolyte junction capacitance. Resistive components Rct and Rj are the charge-transfer resistance and the junction resistance, respectively, while Re is related to the bulk Si resistance, the electrical contact resistance and the electrolyte resistance. The fitting results are listed in Table S2,† and the corresponding Rct values for cathodes, derived from the second impedance semicircles, are labeled in Fig. 3b and d as well.
.34 Hence, the ΦW values are controlled by (VR − VL). Fig. 3b and d show the Nyquist plots obtained by electrochemical impedance spectroscopy (EIS) experiments of p-Si photocathodes at −0.05 V and −0.65 V (vs. RHE) under one sun illumination, and the frequency range is from 100 kHz to 5 Hz. EIS data were fitted using an equivalent circuit with two time-related components, which is shown in the insets of Fig. 3b and d and Fig. S5.† The Cdl component models the double-layer capacitance, while Cj represents the Si/electrolyte junction capacitance. Resistive components Rct and Rj are the charge-transfer resistance and the junction resistance, respectively, while Re is related to the bulk Si resistance, the electrical contact resistance and the electrolyte resistance. The fitting results are listed in Table S2,† and the corresponding Rct values for cathodes, derived from the second impedance semicircles, are labeled in Fig. 3b and d as well.
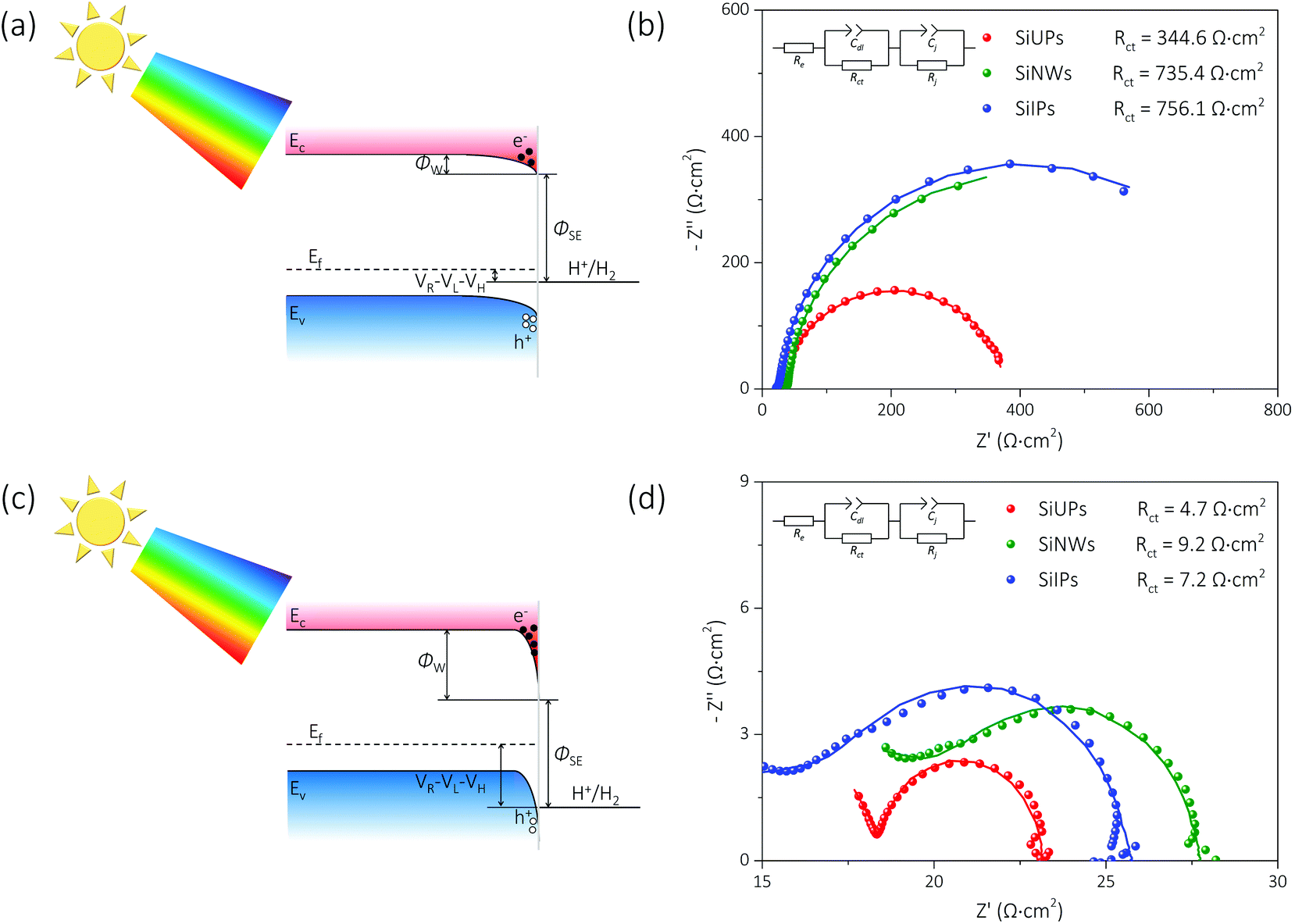 | ||
| Fig. 3 Characterization of charge transfer between the textured Si photocathodes and the electrolyte. (a and c) Energy band diagrams of p-Si/electrolyte junctions at a low and a high reverse bias voltage (under illumination), respectively. (b and d) Nyquist plots of textured Si photocathodes for the EIS experiments in the frequency range of 100 kHz to 5 Hz at −0.05 V and −0.65 V (vs. RHE) under one sun illumination, respectively. | ||
As shown in Fig. 3a, the ΦW values for p-Si photocathodes are small, and the electric-induced electrons are insufficient at a low reverse bias voltage. Thus, the photo-induced electron density is a key factor here. The SiIP photocathode attains the best PEC performance for its high effective minority carrier density as mentioned. However, the SiUP photocathode produces the lowest current density on account of its poor light-trapping ability. In addition, the low electric field intensity for electron acceleration leads to a slow charge separation process, and thus large Rct values of p-Si electrodes are observed at −0.05 V (vs. RHE). The Rct values are 344.6, 735.4 and 756.1 Ω cm2 for SiUP, SiNW and SiIP photocathodes, respectively. Therefore, the current density of each p-Si photocathode is very low here due to the intrinsic properties of Si.3
At a high reverse bias voltage, large ΦW values yield more electric-induced electrons, and thus the photo-induced electron density is less dominant here. Besides, the larger energy band bending promotes the charge separation process, and carrier recombination is no longer severe. The Rct values are 4.7, 9.2 and 7.2 Ω cm2 for SiUP, SiNW and SiIP photocathodes at −0.65 V vs. RHE, respectively, and faster charge transfer immensely lifts the current densities of p-Si photocathodes. Carrier recombination is not crucial as mentioned, but we found that the PEC performance of the SiNW photocathode is extremely poor at a high reverse bias voltage. We attribute the poor HER activity to its hydrophobicity. The measured water droplet contact angles are 84° ± 1.5°, 98° ± 3°, 120° ± 1° and 94° ± 2.5° for planar Si, SiUP, SiNW and SiIP wafers, respectively (Fig. 4a), consistent with the previous literature studies.21,33,35 The wide and open geometry enables SiUPs to contact with water closely (Fig. 4b), while the high-aspect-ratio SiNWs hinder wafers from being wetted (Fig. 4c). As for SiIP wafers, quasi-hydrophilic properties were observed because of the sunken and open structures (Fig. 4d). Moreover, it was proved that hydrophobic surfaces will become aerophilic under water by Yong's work;36 hence, we can infer that hydrophobic SiNWs severely inhibit the underwater gas evolution reactions. In contrast, the quasi-hydrophilic SiIP photocathode exhibits a high HER activity because it can promote the mass transfer (proton adsorption) process and may also enhance the depinning and release of the produced hydrogen bubbles.37 Additionally, this quasi-hydrophilic properties of SiIP cathodes are stable in acidic media. As can be seen from Fig. S6,† the water droplet contact angle and morphological features of SiIPs are scarcely changed after an 8000 s-long bulk electrolysis at −0.5 V (vs. RHE) in aqueous H2SO4 solution (pH = 1).
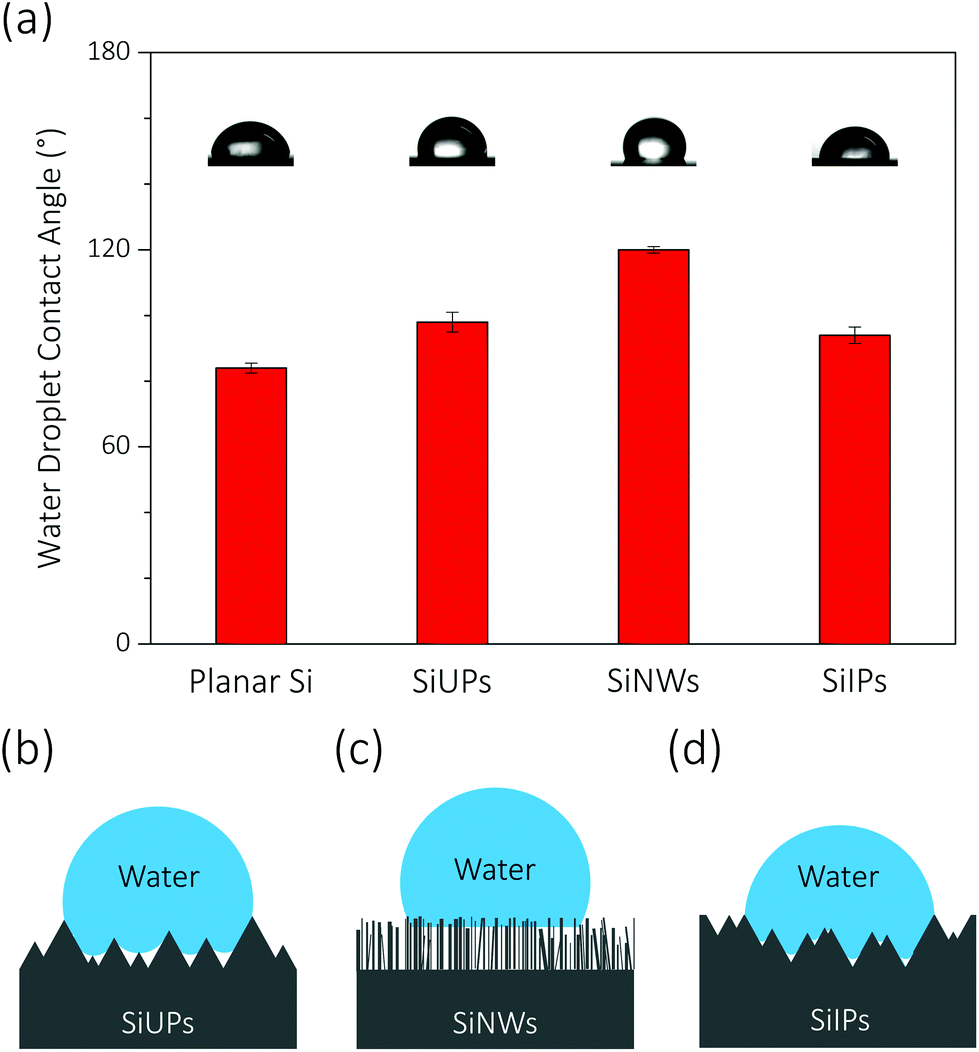 | ||
| Fig. 4 Characterization of surface wettability for textured Si wafers. (a) Contact angles and shapes of water droplets on various Si surfaces in air. (b–d) Schematic depictions for the shapes of water droplets on SiUP, SiNW and SiIP wafers, respectively. | ||
Quasi-hydrophilic black silicon photocathodes with inverted pyramid arrays realize enhanced hydrogen generation for their low reflectivity, moderate minority carrier lifetime and quasi-hydrophilicity, as compared with conventional silicon photocathodes with upright pyramid arrays or nanowire arrays. The bare-SiIP photocathode generates a large photovoltage of 440 mV and reaches a photocurrent density of 10 mA cm−2 at −0.55 V (vs. RHE). However, its large Rct value, which may be ascribed to the long path for carrier transportation and the sluggish HER kinetics of Si, limits the onset potential and the photocurrent density at a low reverse bias voltage. To further accelerate the charge transfer process and enhance the PEC efficiency, we synthesized metallic CoS2 nanocrystals as HER cocatalysts on SiIP photocathodes via a two-step method. Cobalt (Co) nanosheets or nanoflowers were electrodeposited on SiIP wafers as precursors and then sulfurated into CoS2 nanocrystals. A typical J–t curve during potentiostatic Co depositions is shown in Fig. S7,† and it can be seen that the deposition current density firstly decreases and then stabilizes. After a deposition duration of 20, 43, 92 and 197 s, Co precursors with a deposition charge density of 50, 100, 200, and 400 mC cm−2 form on SiIP wafers. Co nanosheets grow and aggregate into nanoflowers over time, as shown in Fig. S8a, c, e and g.† After thermal sulfidation, Co nanostructures collapse and cohere into spherical CoS2 crystals (Fig. S8b, d, f and h†). Note that SiIP photocathodes loaded with different doses of CoS2 cocatalysts are labeled as “SiIPs/CoS2-C”, where C is the corresponding deposition charge density of Co precursors.
Transmission electron microscopy (TEM) observation was carried out to characterize the detailed crystalline structure of CoS2. Compact crystalline grains with a diameter of tens of nanometers are revealed by the TEM image (Fig. 5a), and the well-demarcated lattice fringes manifest the good crystallinity of CoS2 nanocrystals as shown in the high-resolution transmission electron microscopy (HRTEM) image (Fig. 5b). Besides, the phase structure of CoS2 is identified as a cubic phase (Pa3, a = 5.538 Å) based on the distinctive interplanar spacings. The discrete diffraction spots in the selective area electron diffraction (SAED) patterns of CoS2 crystals, as shown in Fig. 5c, further confirm the monocrystalline nature of the synthesized cubic-phase CoS2 large grains.
 | ||
| Fig. 5 Investigation of the morphology and crystalline structure of CoS2 cocatalysts. (a) TEM image, (b) lattice-resolved HRTEM image, and (c) SAED patterns of synthesized CoS2 nanocrystals on the SiIP/CoS2-200 photocathode. | ||
X-ray diffraction (XRD) patterns show the crystallographic structure of the as-prepared SiIP/CoS2-200 photocathode, as shown in Fig. 6a. A broad diffraction peak centered at 69.1°, corresponding to the Si(100) planes, confirms the curved sidewalls of the etched Si inverted pyramids. However, the Si substrate, whose SAED patterns are shown in Fig. S9,† remains monocrystalline after etching. The other detected XRD peaks at 27.9°, 32.3°, 36.2°, 39.8°, 46.3°, 54.9°, 60.2°, 62.7° and 92.6° are matched with the diffraction peaks for the (111), (200), (210), (211), (220), (311), (230), (321) and (511) planes of cubic-phase cobalt disulfide (JCPDS No. 41-1471). No impurity phase can be observed, and thus we can infer that Co precursors are converted into CoS2 after thermal sulfidation. The narrow and distinct peaks confirm the good crystallinity and large grains of the as-synthesized CoS2 cocatalysts. Energy-dispersive X-ray spectroscopy (EDX) elemental mappings reveal the homogeneous distribution of Co and S elements in the CoS2 cocatalysts (Fig. S10c and d†). Moreover, the sulfur-rich CoS2 crystal surfaces are indicated by the Co/S atomic ratio of ∼1:3,38 as can be observed in the EDX spectrum (Fig. S10e†).
 | ||
| Fig. 6 Characterization of SiIP/CoS2 photocathodes. (a) XRD patterns of the as-prepared SiIP/CoS2-200 photocathode. (b and c) XPS spectra of the as-prepared SiIP/CoS2-200 photocathode in the Co 2p and S 2p regions, respectively. | ||
The electronic structures of Co and S elements in the as-prepared SiIP/CoS2-200 photocathode were analyzed by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 6b, four peaks at binding energies of 801.4, 794.2, 780.6 and 778.9 eV are found in the Co 2p region, and the major peaks at 794.2 and 778.9 eV correspond to the Co 2p1/2 and Co 2p3/2 levels of Co–S bonds, respectively. Three peaks at binding energies of 167.5, 163.9 and 162.7 eV are observed in the S 2p region (Fig. 6c), among which the two major peaks at 163.9 and 162.7 eV are proved to be the signature peaks for the S 2p1/2 and S 2p3/2 levels of Co–S bonds, respectively. Therefore, it can be concluded that Co metals were transformed into CoS2 crystals.38–41 Besides, the satellite peaks at 801.4 and 780.6 eV originate from the Co–O bonds, while that at 167.5 eV stems from the S–O bonds.39 A small lattice oxygen peak centered at 529.6 eV in the O 1s region (Fig. S11†) further verifies the appearance of a handful of CoOx substances. The cause of oxygen incorporation may be the possible oxygen contamination during thermal processing and also the inevitable oxidation of CoS2 when exposed to air.
The charge transfer capacity of each CoS2-coated Si photocathode was evaluated by EIS experiments. As shown in Fig. 7a and Table S3,† the Rct values are 756.1, 453.0, 294.4, 169.4 and 110.3 Ω cm2 for the bare-SiIP, SiIP/CoS2-50, SiIP/CoS2-100, SiIP/CoS2-200, and SiIP/CoS2-400 photocathodes at −0.05 V vs. RHE, respectively. Sluggish kinetics of the bare-SiIP photocathode can be understood from its largest Rct, while the reaction-active CoS2 nanocrystals endow SiIP photocathodes with better charge transfer abilities. With an increasing loading amount of CoS2, electrons transfer to the electrolyte with a higher speed. The PEC performances of SiIP/CoS2 photocathodes were examined by linear sweep voltammetry (Fig. 7b). The photovoltages generated by SiIP/CoS2-50, SiIP/CoS2-100, SiIP/CoS2-200, and SiIP/CoS2-400 photocathodes are about 340, 770, 960 and 950 mV, respectively. At 0 V vs. RHE, the cathodic photocurrent density reaches up to 0.32, 1.09, 6.60 and 3.10 mA cm−2 for the SiIP/CoS2-50, SiIP/CoS2-100, SiIP/CoS2-200, and SiIP/CoS2-400 photocathode, respectively. Compared to the bare-SiIP photocathode, each CoS2-coated photocathode gains a higher PEC performance at a small overpotential. Nevertheless, the light-blocking effects30,42 induced by the CoS2 cocatalysts limit the amount of photocarriers generated by the underlying Si photocathodes; thus, the bare-SiIP cathode shows a higher current density at a more cathodic potential than −0.55 V vs. RHE, and the SiIP/CoS2-400 photocathode exhibits a lower PEC efficiency than the SiIP/CoS2-200 photocathode. With an optimal loading amount of CoS2 nanocrystals, the SiIP photocathode attains an onset potential of 0.22 V vs. RHE and a photocurrent density of 6.60 mA cm−2 at 0 V vs. RHE (nearly twice the photocurrent density realized by the optimal core–shell SiNW/CoS2 photocathode43), which is promising for potential unbiased water splitting.
 | ||
| Fig. 7 PEC measurement and interface characterization of SiIP/CoS2 photocathodes. (a) Nyquist plots of CoS2-coated SiIP photocathodes for the EIS experiments in the frequency range of 100 kHz to 5 Hz at −0.05 V (vs. RHE) under one sun illumination. (b) J–V curves for SiIP/CoS2 photocathodes (under one sun illumination). (c and d) TEM and (e) lattice-resolved HRTEM images for Si/CoS2 interfaces of the SiIP/CoS2-200 photocathode. (f) MS plots obtained via impedance–potential methods for the bare-SiIP and SiIP/CoS2-200 photocathodes. The dashed lines are linear fitting results for the corresponding measured data. (g) A typical J–t curve (HER) of the SiIP/CoS2-200 photocathode at 0 V vs. RHE under one sun illumination. | ||
We ascribe the high photocurrent density to the good performance of the SiIP cathode and also the catalysis of CoS2 crystals. As shown in Fig. 7c and d, CoS2 cocatalysts contact with Si inverted pyramids closely, and scarcely any voids at the Si/CoS2 interface can be found. Atoms of CoS2 nanocrystals are precisely inlaid into the Si etching pits, as revealed in the HRTEM image for the Si/CoS2 interface (Fig. 7e), ensuring the formation of a fine heterojunction electrode. Compared with the bare-SiIP photocathode, the SiIP/CoS2-200 photocathode realizes an anodic shift (∼0.28 V) of EFB as can be observed from the MS plots (Fig. 7f). It means that the deposition of CoS2 crystals drives a larger downward energy band bending (ΦW)44 for less carrier recombination, faster electron extraction and perhaps a higher electron density. On the other hand, intrinsically metallic CoS2, whose excellent HER catalytic activity (reducing overpotential) has been demonstrated in the previous reports,41,45 guarantees faster chemical reactions by providing more active sites. Meanwhile, a further slow carrier recombination process is realized due to the rapid charge consumption.46 Hence, copious gas bubbles evolve from the CoS2-coated Si photocathode during measurements, demonstrating a further enhanced HER performance. Besides, the SiIP/CoS2-200 photocathode is proved to be stable in acidic aqueous solutions. As presented by Faber et al.,45 the electronic structures of CoS2 remain essentially unchanged after HER characterization; thus, CoS2 cocatalysts are of high chemical stability. Combined with the effective passivation realized by the CoS2 nanocrystals, only a slight degradation can be observed (Fig. 7g) in the photocurrent density during the 12000 s-long HER measurements at 0 V vs. RHE in aqueous H2SO4 solutions (pH = 1).
3. Conclusions
In summary, we demonstrate a quasi-hydrophilic black silicon photocathode with inverted pyramid arrays for enhanced hydrogen generation. The as-fabricated bare-SiIP photocathode generates a large photovoltage of 440 mV and reaches a high photocurrent density. The large photovoltage can be attributed to its low reflectivity and long minority carrier lifetime, while the high cathodic current density at a high reverse bias voltage can be attributed to its quasi-hydrophilic nature. Besides, we synthesized cubic-phase CoS2 nanocrystals on SiIP photocathodes, and the catalytic but light-blocking effects of CoS2 were observed. By optimizing the loading amount of CoS2 cocatalysts, the SiIP photocathode gains a high onset potential of 0.22 V (vs. RHE) and a saturated photocurrent density of 10.4 mA cm−2 at −0.45 V (vs. RHE). Furthermore, the optimal SiIP/CoS2 photocathode exhibits a stable and high photocurrent density of ∼6.60 mA cm−2 at 0 V (vs. RHE) for 12000 s, which is the best performance for the p-Si/CoS2 counterparts. It is noted that the HER activities of our SiIP photocathodes may be further boosted by using higher-quality crystalline Si wafers or introducing other efficient cocatalysts. This work presents the feasibility and superiority of quasi-hydrophilic black silicon materials with inverted pyramid arrays as efficient photoelectrodes, bringing a new promise for solar-driven overall water splitting.
4. Experimental
4.1. Fabrication of (i) SiIP, (ii) SiUP, and (iii) SiNW wafers
180 μm-thick solar-grade p-type c-Si(100) wafers with a resistivity of 1–3 Ω cm were cut into little pieces and then a standard RCA cleaning process was employed to remove all the contaminants on the wafer surfaces. A ten-minute etching process in 20 wt% NaOH solution at 80 °C was conducted to remove saw damage before RCA cleaning when preparing the SiUP and SiNW wafers. After that, Si wafers were soaked in the etchant (i) containing 18 mM CuSO4, 0.43 M H2O2 and 4 M HF at 40 °C for 12 minutes to synthesize SiIPs, (ii) containing 2 wt% NaOH, 2 wt% Na2SiO3, and 5 vol% IPA at 75 °C for 30 minutes to synthesize SiUPs, or (iii) containing 0.02 M AgNO3 and 5 M HF at 60 °C for 30 minutes to synthesize SiNWs. As for the SiIP and SiNW wafers, a following treatment in concentrated HNO3 solutions for 30 minutes was adopted to remove the residual metal nanoparticles.4.2. Electrodeposition of Co on SiIP wafers
The electrodeposition of Co precursors was carried out in a typical three-electrode setup with a CHI-660e electrochemical workstation (CH Instruments), where the SiIP wafers act as the working electrodes, while Pt foil (2.25 cm2 area) and a KCl-saturated Ag/AgCl electrode serve as the counter and reference electrodes, respectively. Prior to Co depositions, the SiIP wafers were immersed in buffered oxide etch solutions for 2 h to remove the oxide layers. To establish a reliable ohmic contact, the back side of the Si wafers was firstly scratched with a diamond scribe, then coated using a Ga/In eutectic and finally attached to a copper wire. Conductive silver paint was then applied to affix the wire. After drying, edges, some parts of the front side and the whole back side of the Si wafers were covered with insulative tapes to expose an active area of ∼1 cm2. The electrodeposition was conducted in an aqueous solution of 0.1 M CoSO4 under one sun illumination (100 mW cm−2) simulated by using a 300 W Xe lamp (PerfectLight) with an AM 1.5G filter. A constant bias voltage of −1.2 V vs. Ag/AgCl was applied for different durations to deposit Co nanostructures on SiIP wafers.4.3. Synthesis of CoS2 on SiIP wafers
The Co-coated SiIP wafers were sulfurated in a quartz tube inside a chemical vapor deposition reactor to synthesize the CoS2 nanocrystals after removing the tapes and ohmic electrodes. The SiIP/Co wafers were placed in a quartz boat at the center of the tube reactor, and another quartz boat filled with 2 g of S powder was placed at the upstream position inside the tube (∼5 cm away from the center). The tube was purged under an Ar flow of 30 sccm. The furnace was heated to 500 °C and held for 1 h. Thereafter, the furnace was naturally cooled down under the Ar flow.4.4. PEC measurements
All PEC measurements were carried out with a CHI-660e electrochemical workstation in a three-electrode setup, with Si wafers as the working electrodes, and a graphite rod (6 mm diameter) and a KCl-saturated Ag/AgCl electrode as the counter and reference electrodes, respectively. To establish a reliable ohmic contact, the back side of Si wafers was firstly scratched with a diamond scribe, then coated using a Ga/In eutectic and finally attached to a copper wire. Conductive silver paint was then applied to affix the wire. After drying, edges, some parts of the front side and the whole back side of the Si wafers were sealed with epoxy to expose an active area of ∼0.8 cm2. Aqueous H2SO4 solutions (pH = 1) were used as electrolytes during the PEC measurements, and the applied potentials vs. Ag/AgCl have been converted to the potentials vs. RHE following the relationship: E(vs. RHE) = E(vs. Ag/AgCl) + 0.197 V + pH × 0.059 V. EIS experiments were performed at a frequency range of 100 kHz to 5 Hz using an A.C. voltage of 10 mV under one sun illumination, and the obtained Nyquist plots were fitted using an equivalent circuit including two time-related components. MS plots were obtained via the impedance–potential measurements at an A.C. voltage of 10 mV and a frequency of 1 kHz in the dark, and the step value of applied bias voltages is 5 mV. All of the J–V and J–t curves were obtained under one sun illumination or in the dark. Besides, J–V curves were recorded at a scan rate of 10 mV s−1.4.5. Characterization
Plane-view and cross-sectional morphologies, together with element distributions were examined by using an S-4800 scanning electron microscope (Hitachi) equipped with an energy-dispersive X-ray spectrometer. Reflectance spectra were recorded by using an Agilent Cary-7000 UV-Vis-IR spectrophotometer equipped with an integrating sphere. Effective carrier lifetime measurements were performed using a WT-2000 lifetime tester (SemiLab). Water droplet contact angles and shapes were acquired using a JC2000D1 contact angle meter (Powereach). TEM observations were performed using a FEI Technai G2 transmission electron microscope, while ultrathin TEM samples were fabricated using focused ion beams integrated on a Helios FEI 600i scanning electron microscope. XRD experiments were carried out using a Rigaku MiniFlex600 powder X-ray diffractometer with Cu Kα radiation. And XPS spectra were recorded using a Thermo Scientific Escalab-250Xi X-ray photoelectron spectrometer with an Al Kα source.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China under Grant No. 51472229, 61422405 and 11574301, the National Natural Science Foundation of Tianjin under Grant No. 14JCQNJC01000, and the National Science Foundation for Post-doctoral Scientists of China (No. 2016M600231).References
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- I. Oh, J. Kye and S. Hwang, Nano Lett., 2012, 12, 298–302 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. Huang, Y. Wang, X. Liu, Y. Li, X. Hu, B. He, Z. Shu, Z. Li and Y. Zhao, Nano Energy, 2019, 59, 33–40 CrossRef CAS.
- N. Nasori, T. Y. Dai, X. H. Jia, A. Rubiyanto, D. W. Cao, S. C. Qu, Z. G. Wang, Z. J. Wang and Y. Lei, J. Semicond., 2019, 40, 052701 CrossRef CAS.
- H. Zhang, W. Y. Noh, F. Li, J. H. Kim, H. Y. Jeong and J. S. Lee, Adv. Funct. Mater., 2019, 29, 1805737 CrossRef.
- F. Zhan, Y. Liu, K. Wang, Y. Liu, X. Yang, Y. Yang, X. Qiu, W. Li and J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 15467–15477 CrossRef CAS PubMed.
- A. H. Jia, M. Kan, J. P. Jia and Y. X. Zhao, J. Semicond., 2017, 38, 053004 CrossRef.
- X. Wang, K. Q. Peng, X. J. Pan, X. Chen, Y. Yang, L. Li, X. M. Meng, W. J. Zhang and S. T. Lee, Angew. Chem., Int. Ed., 2011, 50, 9861–9865 CrossRef CAS PubMed.
- Z. Huang, Z. Chen, Z. Chen, C. Lv, H. Meng and C. Zhang, ACS Nano, 2014, 8, 8121–8129 CrossRef CAS PubMed.
- V. M. Nikale, S. S. Shinde, C. H. Bhosale and K. Y. Rajpure, J. Semicond., 2011, 32, 033001 CrossRef.
- D. Kang, J. L. Young, H. Lim, W. E. Klein, H. M. Chen, Y. Xi, B. Gai, T. G. Deutsch and J. Yoon, Nat. Energy, 2017, 2, 17043 CrossRef CAS.
- N. Kornienko, N. A. Gibson, H. Zhang, S. W. Eaton, Y. Yu, S. Aloni, S. R. Leone and P. Yang, ACS Nano, 2016, 10, 5525–5535 CrossRef CAS PubMed.
- M. Chen, Y. Liu, C. Li, A. Li, X. Chang, W. Liu, Y. Sun, T. Wang and J. Gong, Energy Environ. Sci., 2018, 11, 2025–2034 RSC.
- Y. H. Tang, C. L. Zhou, W. J. Wang, S. Zhou, Y. Zhao, L. Zhao, H. L. Li, B. J. Yan, J. W. Chen, J. M. Fei and H. B. Cao, J. Semicond., 2012, 33, 064007 CrossRef.
- H. H. Yue, R. Jia, C. Chen, W. C. Ding, D. Q. Wu and X. Y. Liu, J. Semicond., 2011, 32, 084005 CrossRef.
- Z. Luo, T. Wang and J. Gong, Chem. Soc. Rev., 2019, 48, 2158–2181 RSC.
- Y. Yu, Z. Zhang, X. Yin, A. Kvit, Q. Liao, Z. Kang, X. Yan, Y. Zhang and X. Wang, Nat. Energy, 2017, 2, 17045 CrossRef CAS.
- M. J. Choi, J. Y. Jung, M. J. Park, J. W. Song, J. H. Lee and J. H. Bang, J. Mater. Chem. A, 2014, 2, 2928–2933 RSC.
- L. Santinacci, M. W. Diouf, M. K. Barr, B. Fabre, L. Joanny, F. Gouttefangeas and G. Loget, ACS Appl. Mater. Interfaces, 2016, 8, 24810–24818 CrossRef CAS PubMed.
- R. Fan, S. Cheng, G. Huang, Y. Wang, Y. Zhang, S. Vanka, G. A. Botton, Z. Mi and M. Shen, J. Mater. Chem. A, 2019, 7, 2200–2209 RSC.
- C. J. Chen, C. W. Liu, K. C. Yang, L. C. Yin, D. H. Wei, S. F. Hu and R. S. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 37142–37149 CrossRef CAS PubMed.
- M. Hong, G. D. Yuan, Y. Peng, H. Y. Chen, Y. Zhang, Z. Q. Liu, J. X. Wang, B. Cai, Y. M. Zhu, Y. Chen, J. H. Liu and J. M. Li, Appl. Phys. Lett., 2014, 104, 253902 CrossRef.
- Y. Xiu, L. Zhu, D. W. Hess and C. P. Wong, Nano Lett., 2007, 7, 3388–3393 CrossRef CAS PubMed.
- S. Zhao, G. Yuan, Q. Wang, W. Liu, S. Zhang, Z. Liu, J. Wang and J. Li, Appl. Surf. Sci., 2019, 489, 776–785 CrossRef CAS.
- G. Yuan, R. Mitdank, A. Mogilatenko and S. F. Fischer, J. Phys. Chem. C, 2012, 116, 13767–13773 CrossRef CAS.
- J. Oh, H. C. Yuan and H. M. Branz, Nat. Nanotechnol., 2012, 7, 743–748 CrossRef CAS PubMed.
- A. W. Smith and A. Rohatgi, Sol. Energy Mater. Sol. Cells, 1993, 29, 37–49 CrossRef CAS.
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef CAS PubMed.
- J. C. Hill, A. T. Landers and J. A. Switzer, Nat. Mater., 2015, 14, 1150–1155 CrossRef CAS PubMed.
- S. Lee, S. Cha, Y. Myung, K. Park, I. H. Kwak, I. S. Kwon, J. Seo, S. A. Lim, E. H. Cha and J. Park, ACS Appl. Mater. Interfaces, 2018, 10, 33198–33204 CrossRef CAS PubMed.
- W. Cai, H. Xiong, X. Su, H. Zhou, M. Shen and L. Fang, Appl. Phys. Lett., 2017, 111, 203902 CrossRef.
- T. W. Kim and K. S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- M. Hsiao, K. Y. Chen and C. Y. Chen, Sci. Rep., 2019, 9, 1579 CrossRef PubMed.
- J. Yong, F. Chen, Y. Fang, J. Huo, Q. Yang, J. Zhang, H. Bian and X. Hou, ACS Appl. Mater. Interfaces, 2017, 9, 39863–39871 CrossRef CAS PubMed.
- H. Li, S. Chen, Y. Zhang, Q. Zhang, X. Jia, Q. Zhang, L. Gu, X. Sun, L. Song and X. Wang, Nat. Commun., 2018, 9, 2452 CrossRef PubMed.
- L. Zhu, D. Susac, M. Teo, K. C. Wong, P. C. Wong, R. R. Parsons, D. Bizzotto, K. A. R. Mitchell and S. A. Campbell, J. Catal., 2008, 258, 235–242 CrossRef CAS.
- W. Kong, X. Luan, H. Du, L. Xia and F. Qu, Chem. Commun., 2019, 55, 2469–2472 RSC.
- H. van der Heide, R. Hemmel, C. F. van Bruggen and C. Haas, J. Solid State Chem., 1980, 33, 17–25 CrossRef CAS.
- Y. Sun, C. Liu, D. C. Grauer, J. Yano, J. R. Long, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 17699–17702 CrossRef CAS PubMed.
- H. M. Chen, C. K. Chen, C. J. Chen, L. C. Cheng, P. C. Wu, B. H. Cheng, Y. Z. Ho, M. L. Tseng, Y. Y. Hsu, T. S. Chan, J. F. Lee, R. S. Liu and D. P. Tsai, ACS Nano, 2012, 6, 7362–7372 CrossRef CAS PubMed.
- C. J. Chen, P. T. Chen, M. Basu, K. C. Yang, Y. R. Lu, C. L. Dong, C. G. Ma, C. C. Shen, S. F. Hu and R. S. Liu, J. Mater. Chem. A, 2015, 3, 23466–23476 RSC.
- A. J. Nozik, Annu. Rev. Phys. Chem., 1978, 29, 189–222 CrossRef CAS.
- M. S. Faber, R. Dziedzic, M. A. Lukowski, N. S. Kaiser, Q. Ding and S. Jin, J. Am. Chem. Soc., 2014, 136, 10053–10061 CrossRef CAS.
- D. Li, J. Shi and C. Li, Small, 2018, 14, 1704179 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr06635g |
| This journal is © The Royal Society of Chemistry 2020 |