
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bacterial pathogens: threat or treat (a review on bioactive natural products from bacterial pathogens)†
Fleurdeliz
Maglangit
 *ab,
Yi
Yu
*c and
Hai
Deng
*b
*ab,
Yi
Yu
*c and
Hai
Deng
*b
aDepartment of Biology and Environmental Science, College of Science, University of the Philippines Cebu, Lahug, Cebu City, 6000, Philippines. E-mail: ffmaglangit@up.edu.ph; Tel: +63 32 232 8185
bDepartment of Chemistry, University of Aberdeen, Aberdeen AB24 3UE, UK. E-mail: h.deng@abdn.ac.uk; Fax: +44 (0)1224 272291; Tel: +44 (0)1224 272953
cKey Laboratory of Combinatorial Biosynthesis and Drug Discovery (MOE), Hubei Province Engineering and Technology Research Centre for Fluorinated Pharmaceuticals, School of Pharmaceutical Sciences, Wuhan University, Wuhan 430071, China. E-mail: yu_yi@whu.edu.cn; Tel: +86 27 68752491
First published on 29th October 2020
Abstract
Covering: up to the second quarter of 2020
Threat or treat? While pathogenic bacteria pose significant threats, they also represent a huge reservoir of potential pharmaceuticals to treat various diseases. The alarming antimicrobial resistance crisis and the dwindling clinical pipeline urgently call for the discovery and development of new antibiotics. Pathogenic bacteria have an enormous potential for natural products drug discovery, yet they remained untapped and understudied. Herein, we review the specialised metabolites isolated from entomopathogenic, phytopathogenic, and human pathogenic bacteria with antibacterial and antifungal activities, highlighting those currently in pre-clinical trials or with potential for drug development. Selected unusual biosynthetic pathways, the key roles they play (where known) in various ecological niches are described. We also provide an overview of the mode of action (molecular target), activity, and minimum inhibitory concentration (MIC) towards bacteria and fungi. The exploitation of pathogenic bacteria as a rich source of antimicrobials, combined with the recent advances in genomics and natural products research methodology, could pave the way for a new golden age of antibiotic discovery. This review should serve as a compendium to communities of medicinal chemists, organic chemists, natural product chemists, biochemists, clinical researchers, and many others interested in the subject.
 Fleurdeliz Maglangit | Dr Fleurdeliz Maglangit finished her Bachelor of Science in Chemistry at the University of the Philippines in the Visayas – Miagao Iloilo. With her continued pursuit for learning, she obtained two Masters degrees, Master in Chemistry and Master of Science in Environmental Studies. She just recently earned her Ph.D. in Chemistry at the University of Aberdeen, Scotland, UK last July 2020. Her PhD work was mainly focused on natural products discovery and biosynthesis. In her research, she was able to isolate novel compounds with excellent antibacterial bioactivities from Streptomyces bacteria. Currently, she is an Assistant Professor in Chemistry at the University of the Philippines Cebu. She hopes to discover novel bioactive natural products with potential for further drug development using local samples. |
 Yi Yu | Dr Yi Yu obtained his Bachelor's and Master's degrees in Biotechnology (2002) from Huazhong Agricultural University. He received his Ph.D. degree in Genetics (2007) at the Institute of Microbiology, Chinese Academy of Sciences (CAS). He then undertook postdoctoral research with Professor Wen Liu at Shanghai Institute of Organic Chemistry CAS. In 2010, he joined the School of Pharmaceutical Sciences, Wuhan University, and was promoted to professor in 2016. Now, he is the Director of the Institute of Traditional Chinese Medicine and Natural Products. His research interests centre around deciphering the biochemical logic and molecular machinery of bacterial and plant natural product biosynthesis. |
 Hai Deng | Dr Hai Deng studied chemistry for his Bachelor's and Masters' degrees in China. In 1999, he came to the UK to pursue his Ph.D. in the field of biochemistry and biotransformations at the University of Wales, Swansea. From 2002–2008, he was a postdoctoral researcher of Professor David O'Hagan in University of St Andrews. He was appointed as a lecturer at the Department of Chemistry, University of Aberdeen in 2008, and promoted to senior lecturer in 2014 and reader in 2018. His current research includes discovery of novel bioactive natural products from various sources, tracing biosynthesis pathways, identifying novel enzyme activities and enzyme mechanism. He became the Fellow of Royal Society of Chemistry in 2017. |
1. Introduction
Antimicrobial resistance (AMR) is amongst the major threats to public health and poses a huge economic burden on global health care. The World Health Organization (WHO) has recently published the priority list of drug-resistant bacteria that pose the greatest danger to human health,1,2 and among these, a majority of Gram-negative bacteria, including Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacteriaceae. Resistance has emerged to all clinically used antibiotics including those of “last-resort” such as colistin and polymyxin B, and continues to rise at alarming rates.3,4Despite the severity of the situation, the number of new chemical entities in the antibiotic development pipeline is in substantial decline. Nearly all the classes of antibiotics currently in clinical use were discovered during the ‘golden era’ (1940s–1960s), with several new drugs that are chemically tailored analogues from existing scaffolds.5 The problem is compounded by the fact that bacteria are evolving resistance at a faster pace than antibiotic development.6,7 The last new class of antibiotics that target the Gram-negative bacteria are the synthetic fluoroquinolones which were introduced into the clinic about 50 years ago.8,9 The high rate of the rediscovery of old known molecules in traditional natural product (NP) screening platforms makes this grim situation even worse. Thus, the research community must find new sources of NPs to cope with the looming antibiotic crisis.
Pathogenic bacteria have shown to be rich sources of novel compounds, yet they remained untapped and understudied.10–13 Virulence factors involved in their pathogenicity have been the subject of extensive study for many decades.14–23 In recent years, however, it has become apparent that entomopathogenic, phytopathogenic and human and animal pathogenic bacteria are prolific sources of structurally novel and highly bioactive druggable molecules.11,12
Threat or treat? While pathogenic bacteria pose a threat to insects, plants, and humans, they also represent gold mines of potential pharmaceuticals to treat various diseases.11,12,24,25 The opportunistic human pathogen, Staphylococcus aureus is a classic example. Despite being a threat, they produce potent bacteriocins (also known as staphylococcins) and several other compounds active against a wide variety of Gram-positive bacteria.25
Microbial genome-level studies and metabolomic approaches have further revealed the untapped biosynthetic potential of the diverse and underexplored group of pathogenic bacteria. Bacterial genomics has shown that they not only encode for virulence factors but also potential leads for drug development.11,12 However, it has been estimated that only a very small portion of this gold mine had just been discovered, and that further drug leads or pharmacophores could be mined given the application of suitable and sufficient resources.11 Thus, this review intends to explore the role that pathogenic bacteria could play in the search for novel compounds and scaffolds. This review should serve as a compendium to communities of medicinal chemists, organic chemists, natural product chemists, biochemists, clinical researchers, and many others interested in the subject.
2. Scope of the review
This review surveys the natural products (NPs) isolated from entomopathogenic, phytopathogenic, human, and animal pathogenic bacteria with antibacterial and/or antifungal activity, highlighting those NPs or NP-modified molecules currently in pre-clinical trials or those with potential for future drug development. These include the polyketides (PKSs), nonribosomal peptides (NRPs), peptide–polyketide hybrid metabolites, and ribosomally-synthesised and post-translationally modified peptides (RiPPs). Selected unique and interesting pathways involved in their biosynthesis and the key roles they play in pathogenesis (where known) are also summarized.Entomopathogenic bacteria such as Photorhabdus spp., Xenorhabdus spp., and Serratia marcescens are the focus of the review. The period from 2017 to the second quarter of 2020 saw a huge rise in the number of bioactive NPs from Photorhabdus spp. and Xenorhabdus spp. that are not covered in previous synopses,12,26 and thus they are the emphasis in our review. It is worth noting that the honeybee pathogen, Paenibacillus larvae also appears as a rich, yet largely understudied source of novel and structurally diverse NPs. The readers are referred to the review by Müller, et al. (2015) which details the metabolites identified from P. larvae.27 Although a rich source, no new metabolite has been identified from this bacterium since 2015.
Phytopathogenic bacteria such as Burkholderia spp., Clostridium puniceum, Dickeya spp., Erwinia amylovora, Pseudomonas syringae, Streptomyces scabies, and Xanthomonas spp. are among the prolific NP producers, and thus they are the topic of this review. The NPs from the diverse genus Burkholderia is summarized in a recent review.28 Another review provided the genomics perspective of NP biosynthesis in phytopathogenic bacteria E. amylovora, Xanthomonas spp., S. scabies, P. syringae, and Dickeya spp.11 Hence, in this review we aim to update and complement previous synopses and cover only those NPs that show the most interesting bioactivities or those that have not been mentioned by Baldeweg, et al. (2019)11 or Kunakom and Eustáquio (2018).28 Furthermore, we included the phytopathogen C. puniceum not mentioned in the above reviews for it produces potent metabolites with antimicrobial activity in nanomolar concentration.
We also explore the human and animal pathogenic bacteria such as Nocardia spp., Staphylococcus spp., Streptococcus mutans, and Yersinia ruckeri as sources of antimicrobials with therapeutic potential. These bacteria have been shown to produce structurally diverse NPs with potent bioactivities.29–33 The antimicrobials from Nocardia spp. and bacteriocins from Staphylococcus spp. have been summarized in recent reviews,25,33 and thus those NPs with remarkable activities from these bacteria were highlighted. Finally, we provide a thorough compilation of the antimicrobial NPs from bacterial pathogens, Burkholderia spp., C. puniceum, Dickeya spp., E. amylovora, Nocardia spp., Photorhabdus spp., P. larvae, Pseudomonas spp., Staphylococcus spp., S. marcescens, S. mutans, Streptomyces spp., Vibrio spp., Xanthomonas spp., Xenorhabdus spp., and Yersinia ruckeri (see Table S1 in the ESI† of this article listed in alphabetical order). We also provide their mode of action (molecular target), activity, and minimum inhibitory concentration (MIC) towards bacteria and fungi (where known), in the pursuit to demonstrate the exceptional biosynthetic ingenuity of the underexplored source of pathogenic bacteria for the production of novel and druggable chemical entities.
3. Pathogenic bacteria as novel sources of antimicrobial discovery
Pathogenic bacteria are master engineers of highly diverse and biologically active molecules. To thrive and survive in highly competitive and resource-limited microbial communities, pathogenic bacteria have developed an approach to protect themselves by producing a plethora of structurally diverse metabolites that have been fine-tuned by the producing organism to have potent and selective biological activities.25,34 It is believed that pathogenic bacteria exploit these molecules to regulate virulence and persistence during infections. Additionally, the vast array of antibacterial armamentarium is thought to fight off predators, compete for nutrients, and protect their host. Other roles have also been suggested such as signalling and quorum sensing, gene expression, stress response, cellular growth and iron acquisition.12,35Pathogenic bacteria represent exceptionally prolific sources of potential therapeutics as indicated in their genomes, yet they have been largely ignored.11,36 Here, we present an overview of the antimicrobial NPs produced by entomopathogenic, phytopathogenic, and human and animal pathogenic bacteria, and highlight a selection of metabolites with antibiotic activity that show promising potential for future development (Fig. 1).
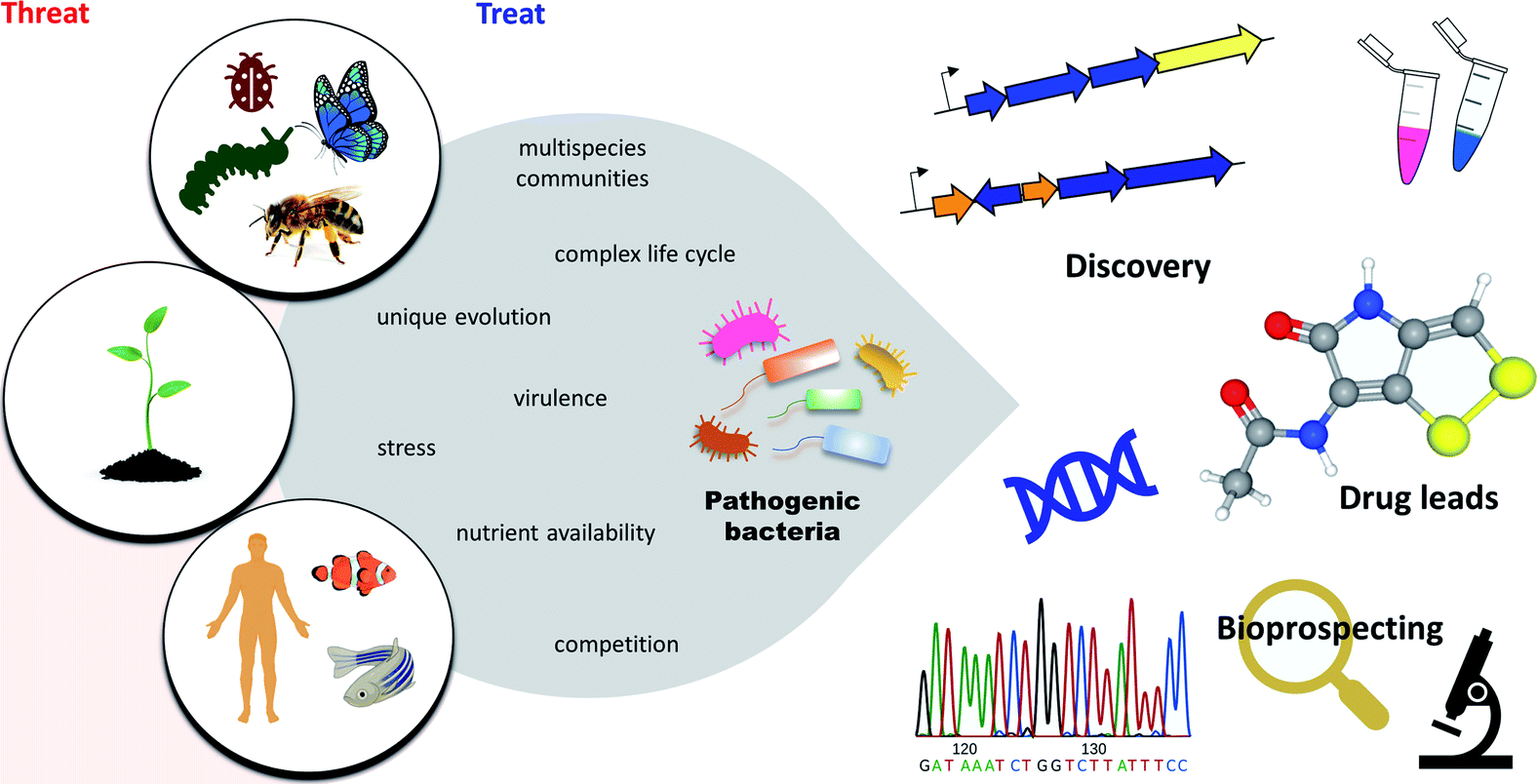 | ||
| Fig. 1 Overview of pathogenic bacteria. Despite a threat to insects, plants, animals, and humans, pathogenic bacteria represent novel sources of potential pharmaceuticals to treat various diseases. | ||
3.1 Entomopathogenic bacteria
Previously regarded as overlooked and neglected sources, the entomopathogenic bacteria have received considerable interest in the last 15 years owing to the novel druggable chemical entities they generate.13,34 Those that have been described recently as prolific NP producers include Photorhabdus spp. and Xenorhabdus spp., S. marcescens, and P. larvae.Members of the genera Photorhabdus and Xenorhabdus (Enterobacteriaceae) produce a wide array of NPs to support a complex life cycle involving insect pathogenesis and nematode symbiosis with Heterorhabditis spp. and Steinernema spp., respectively.37 The antimicrobial compounds produced by these bacteria are non-toxic to the nematode, but lethal to several insect pathogens and other opportunistic microbes that are direct food competitors.24 This indicates the production of antimicrobials with favourable toxicity, good pharmacokinetics, and are likely druggable and safe to eukaryotic organisms. Serratia marcescens is a Gram-negative, facultatively-anaerobic bacterium (Enterobacteriaceae) often associated with insect infection.38 Several insects are susceptible to Serratia species, including crickets, grasshoppers, locusts, cockroach, termites, beetles, butterflies, moths, fruit fly, wasps,39 and recently has been discovered as being pathogenic to bees.40 Some members of S. marcescens also cause opportunistic nosocomial infections of the respiratory tract, urinary tract, brain, meninges, heart, and wounds.39,41 Despite a threat, S. marcescens has been shown to produce not only the characteristic red pigment prodigiosin but also a huge repertoire of antimicrobial compounds.41Paenibacillus larvae is a Gram-positive bacterium that causes fatal intestinal infection of honeybee larvae, called American Foulbrood (AFB). This pathogen spreads very rapidly and poses various threats of different severity leading to massive losses of entire bee colonies. P. larvae secretes a broad spectrum of antibacterial compounds that are critical virulence factors and also, relevant in the quest for new bioactive compounds for drug development. Readers are referred to the recent review by Müller, et al.27
It should be mentioned that several other entomopathogenic bacteria such as Bacillus thuringiensis and Pseudomonas entomophila have the capacity to produce NPs based on their genome sequences but have not been mined further for NP production.42,43
3.2 Phytopathogenic bacteria
Plant pathogenic bacteria can have detrimental effects on plant growth, productivity, and yield. They affect a wide range of crops posing a threat to global food production. Hundreds of phytopathogenic bacteria have been identified to date,44–46 but only a few have been explored for natural product discovery.11Clostridium puniceum, the only known plant pathogenic bacterium from the diverse genus Clostridium to date,44–46 causes potato slimy rot, manifested by the formation of pink pigments by the bacterium.47 All Dickeya species (formerly Erwinia chrysanthemi) cause economically important diseases on different plant hosts worldwide.14,48D. zeae causes soft rot in a variety of plants (e.g. potato, chicory, maize, banana, rice). Erwinia amylovora is the causative agent of fire blight, a destructive disease of Rosaceae plants such as apple and pear trees49 that is typically accompanied by the development of black necrosis.50 Historically, E. amylovora is the first characterised bacterial plant pathogen.51Pseudomonas spp. produce a wide spectrum of phytotoxic compounds. P. syringae pathovars are the topmost phytotoxic-producing bacteria among all Pseudomonas, and all phytopathogens identified to date.14,52Streptomyces species are particularly renowned for their ability to produce numerous bioactive NPs.53–58 Several Streptomyces strains, however, are phytopathogenic and can cause potato common scab diseases such as S. caviscabies, S. acidiscabies, S. turgidiscabies, and S. scabies.21,59 Among the most notable pathogens of the genus Xanthomonas are X. albilineans, the causative agent of leaf scald disease on sugar cane60 and X. campestris, the causal agent of black rot of crucifers that affects all cultivated brassicas.14 Members of the genus Burkholderia include strains that can either be beneficial or harmful. Some strains are pathogenic to plants such as B. glumae, which causes rice rot, while others cause opportunistic human infections such as the strains of Burkholderia cepacia complex (Bcc), which include B. pseudomallei and B. mallei. For detailed information on the diverse Burkholderia genus, refer to the recent review.28
Virulence-mechanisms of plant pathogenic bacteria have been the subject of several different reviews.14,21,28,52,59 Despite being a threat to agriculture, phytopathogens C. puniceum, Dickeya spp., E. amylovora, Pseudomonas spp., Streptomyces spp., Xanthomonas spp., Burkholderia spp. – some of which belong to the top 10 most important plant pathogenic bacteria14 – also serve as huge arsenals for potent drug leads. Genome analyses disclosed that their biosynthetic machinery encodes not only for virulence factors but also for antibiotic-like metabolites with no plant disease-associated function.11 Furthermore, some phytotoxins were found to exhibit potent antimicrobial properties.11,28,47
3.3 Human and animal pathogenic bacteria
While the antimicrobials from non-pathogenic strains are studied in-depth, knowledge of the structural and mechanistic diversity of antibiotics particularly from human and animal pathogenic bacteria is limited. Here, we provide an overview of the potential chemistry to be uncovered from the opportunistic pathogens, Nocardia spp., Staphylococci, S. mutans, Vibrio spp., and Y. ruckeri.Many different species of Nocardia have been identified, and many of these are pathogenic to humans and animals. To date, more than 50 Nocardia species are clinically significant.61 Of these, N. brasiliensis, N. abscessus, N. transvalensis, N. terpenica, and N. pseudobrasiliensis have been identified to be prolific microbial sources of bioactive novel compounds.33
Staphylococci represent the normal flora of the skin and mucous membrane of human and animals.62 There are more than 40 species, but few are important human pathogens such as S. aureus, S. epidermidis, S. haemolyticus, S. lugdunensis, and S. saprophyticus implicated in various infections, especially in immunocompromised patients.18 Though they pose a threat, they are also prolific producers of potent bacteriocins (also known as staphylococcins) exhibiting antibacterial activity against closely related species and a wide variety of Gram-positive bacteria.25,63
Streptococcus mutans is the major causative agent of human dental caries (tooth decay).64 In addition to caries, S. mutans is also implicated in infective endocarditis, a lethal infection, and inflammation of heart valves.65 Bacterial sequence analysis of S. mutans discloses a small genome (about 2 Mb) yet surprisingly harbours rich and diverse biosynthetic gene clusters (BGC) for the production of PKS, NRPS, hybrid PKS–NRPS, and RiPP metabolites.66,67 Several bioactive NPs have recently been isolated from S. mutans.30,68–78
Vibrionaceae includes several species that cause intestinal (diarrhoea, cholera) and extra-intestinal (septicaemia, skin infection) illnesses in both humans and aquatic animals. Among the opportunistic Vibrio pathogens, V. parahaemolyticus has been shown to produce metabolites with remarkable bioactivity.79
Yersinia ruckeri is the etiological agent of yersiniosis or enteric redmouth (ERM) disease in marine and freshwater fish, particularly salmonids.17 Infections due to Y. ruckeri cause high mortalities in fish, contributing to substantial economic losses in the aquaculture industry.80Y. ruckeri has also been isolated from human wound infection, however, it remains unclear whether Y. ruckeri or another bacterium caused the infection.81 Interestingly, Y. ruckeri has been shown to produce the dithiolopyrrolone natural product, holomycin.31,32
4. Chemical diversity of antimicrobials produced by pathogens
Pathogenic bacteria produce numerous NPs with highly diverse structures made up of a handful of simple building blocks, usually derived from one or more primary metabolic pathways. These NPs can be classified into five different groups according to their biosynthetic origin: polyketides, nonribosomal peptides, polyketide–nonribosomal peptide hybrid metabolites, ribosomal peptides, and others. Since numerous NPs from pathogenic bacteria are known, only selected compounds with promising therapeutic potential are presented.4.1 Polyketides
Polyketides, assembled by polyketide synthases (PKS), are among the largest classes of chemically diverse NPs, encompassing molecules such as macrolides, aromatics, and polyenes. The structural diversity exhibited by polyketides is exemplified by the broad spectrum of biological activities they possess, such as antibacterial, antifungal, and anticancer among others (Fig. 2 and Table S1†). PKSs occurring in bacteria are classified into three types (type I, II, and III) depending upon their structure and biochemistry. Type I PKSs are large multifunctional enzymes comprised of multiple functional domains as exemplified by borrelidin 1, gladiolin 2, erythromycin 3, and brasilinolide A 4. Type II PKSs are formed by discrete catalytic domains and are responsible for the biosynthesis of bacterial aromatic polyketides such as clostrubins 5–6 and nocardicyclin A 7. Type III PKSs are simpler chalcone synthase-like proteins that catalyse the formation of the product within a single active site. Examples include chalcones, resorcinol, pyrones, and stilbenes (Fig. 2 and Table S1†).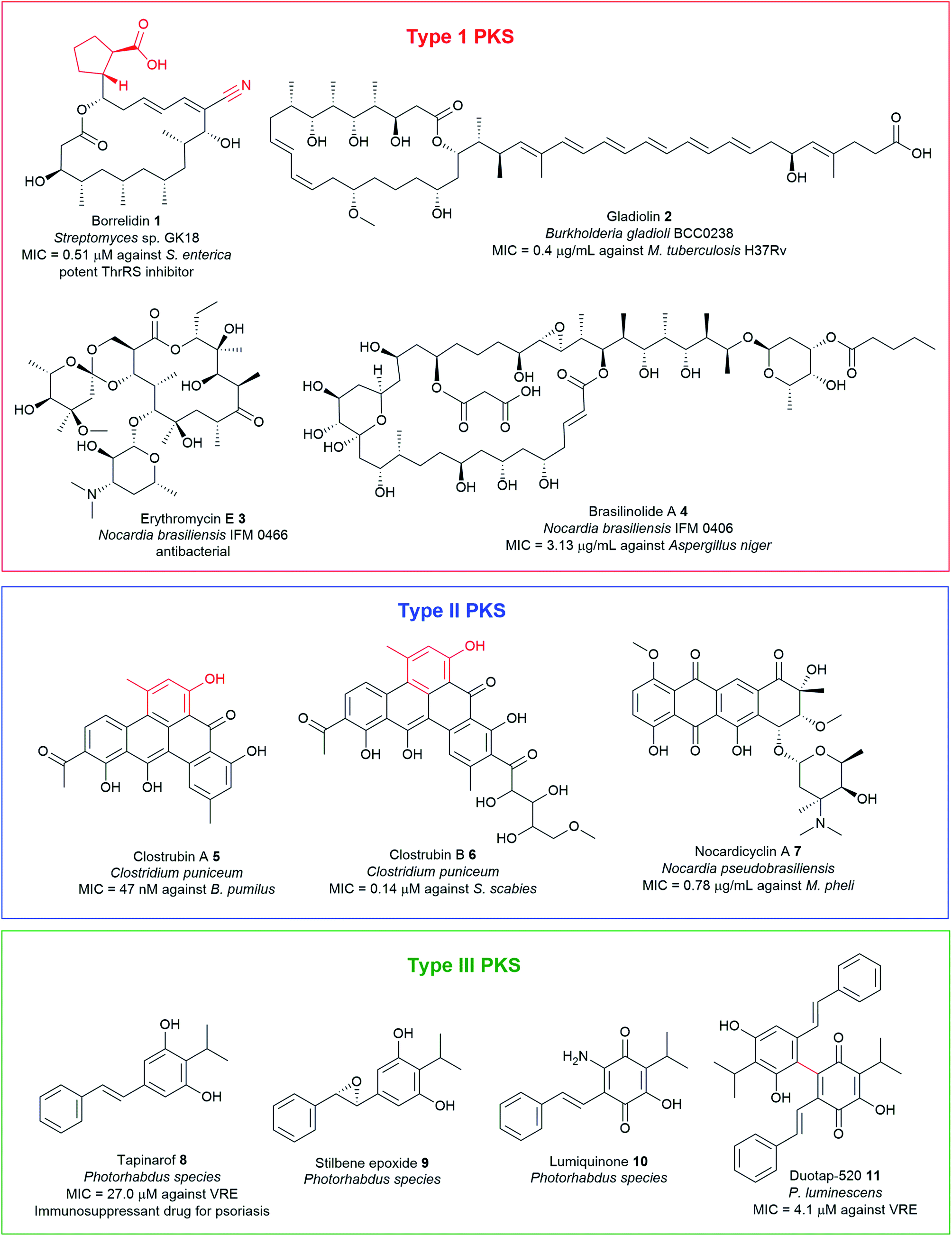 | ||
| Fig. 2 Examples of antimicrobial polyketide natural products with unusual chemical motifs highlighted in red, isolated from pathogenic bacteria. | ||
Polyketides are biosynthesised from two-carbon acetate units derived from activated acetyl-CoA and malonyl-CoA in successive decarboxylative Claisen condensation reactions, in a manner analogous to fatty acid biosynthesis. Typically, this process involves the core domains comprising of the ketosynthase (KSα and KSβ), malonyl/acyl transferase (AT), and a phosphopantethienylated acyl carrier protein (ACP) which serves as an anchor for the growing PK chain.82 A series of post-PKS tailoring enzymes such as ketoreductase (KR), methyltransferase (MT), enoyl reductase (ER), and dehydratase (DH) can variously modify the polyketide backbone, either while the intermediates are still bound to the assembly line or after they are released. Installation of different polyketide starter and extender units also represents a significant route to add unusual moieties such as nitrile functionality, carboxylates, and branched-alkyl chains into polyketide scaffolds to generate mature final products with a high degree of chemical complexity and activity. The mechanistic enzymology of diverse polyketide assembly lines has been the subject of comprehensive reviews.82,83 This section covers some representatives of interesting polyketide antimicrobials containing unusual chemical functionalities from pathogenic bacteria such as PKSI borrelidin 1, PKSII clostrubins 5 and 6, and stilbene-containing PKSIII metabolites 8–11.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 85 and other Streptomyces species86–90 as well as marine-derived microorganisms (Fig. 2 and Table S1†).91–94 Borrelidin 1 features an 18-membered macrolide with a nitrile functionality.95,96 To date, numerous analogues have been discovered including borrelidins B–O,87,90,91,93,94 acetyl-borrelidin89 as well as amide containing congeners, borrelidin CR1 and CR2.92,93,97
85 and other Streptomyces species86–90 as well as marine-derived microorganisms (Fig. 2 and Table S1†).91–94 Borrelidin 1 features an 18-membered macrolide with a nitrile functionality.95,96 To date, numerous analogues have been discovered including borrelidins B–O,87,90,91,93,94 acetyl-borrelidin89 as well as amide containing congeners, borrelidin CR1 and CR2.92,93,97
More than 30 nitrile-containing pharmaceuticals are currently marketed for a wide range of medical indications, including vildagliptin for diabetes and anastrozole for breast cancer treatment.98 The nitrile functionality renders the molecule more water-soluble and less susceptible to oxidative metabolism in the liver.98 Furthermore, nitrile moiety is rare in natural products, hence the biosynthetic mechanism of borrelidin, particularly the nitrile group has attracted significant interest. The biosynthesis of borrelidin proceeds through the typical pathway known for type 1 PKS to form the macrolide ring except for the unique trans-cyclopentane-(1R-2R)-dicarboxylic acid (CDPA) starter unit (Fig. 3). CDPA is likely derived from tyrosine or 4-hydroxyphenyl acetic acid (4-HPA) catabolism.86 The nitrile formation in 1 may start from oxidation of the pendant methyl group in pre-borrelidin 1c to an aldehyde 1e catalysed by cytochrome P450, BorI, and alcohol dehydrogenase, BorK. This is followed by the conversion of the aldehyde to aminomethyl group (borrelidin B) 1b catalysed by the putative aminotransferase, BorJ.94 BorJ is related to CynN1 and CyaN1 aminotransferases in nitrile-containing cyanosporasides that typically act upon carbonyl groups, catalysing conversion to amines.99 The aminomethyl intermediate 1b is finally converted to the nitrile catalysed by the putative BorI and BorK enzymes via a series of oxidation and dehydration reactions. Mutants obtained by inactivation of either BorI or BorJ failed to generate any borrelidin but led to the production of pre-borrelidin 1c, suggesting that BorI/J are responsible for nitrile biosynthesis.86,94 Furthermore, the isolation of borrelidin B 1b from a marine-derived Streptomyces strain supports the plausible mechanism of nitrile formation.94
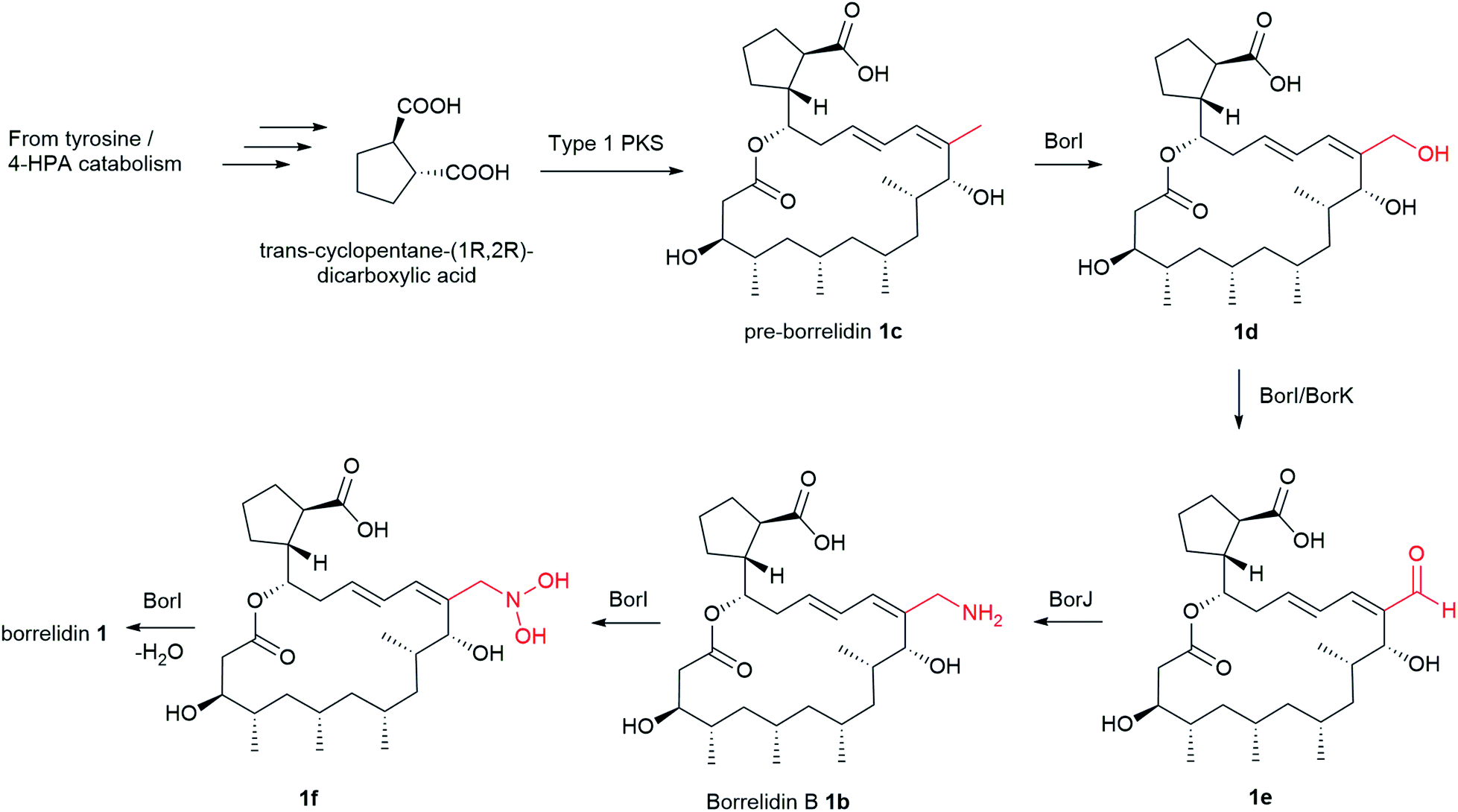 | ||
| Fig. 3 Proposed nitrile formation in borrelidin biosynthesis. | ||
Borrelidin is a potent threonyl-tRNA synthetase inhibitor.100 Borrelidin 1 is active against a wide range of bacteria, including Enterococcus faecalis, Micrococcus luteus, Enterococcus faecium, Proteus hauseri, and Klebsiella pneumoniae (MIC = 0.5–65 μM).90,91,93 Additionally, borrelidin exhibits 3× potent activity against Salmonella enterica (MIC = 0.51 μM), the causative agent of foodborne salmonellosis than the antibiotic ampicillin (MIC = 1.4 μM).91 This remarkable activity has received considerable clinical interest in the search for privileged scaffolds that selectively target S. enterica. On the other hand, borrelidin C and D analogues with an additional hydroxy moiety in the cyclopentane ring are inactive against the tested bacteria and show reduced activity in S. enterica (MIC = 16–63 μM). SAR investigation of the borrelidin scaffold has indicated that the vinylic nitrile and the carboxylic acid moieties are essential for the activity.87,90,93,94,101
The biosynthesis of clostrubins in the anaerobic C. puniceum is proposed to originate from type II PKS (clr) with high homology to the pentacyclic resistomycin (rem) PKS in aerobic bacteria, Streptomyces resistomycificus (Fig. 4).103 Type II PKSs are very common in actinomycetes; only two examples of type II polyketides have been identified in non-actinomycete bacteria so far. Stable-isotope labelling experiments indicated that the striking perifused ring feature of clostrubin is formed from a noncanonical polyketide folding which delineates from the conserved cyclization patterns of typical angucylic decaketides from aerobic bacteria. Numerous tailoring enzymes catalyse diverse post-modification reactions, such as cyclodehydration steps and decarboxylation leading to a loss of one C1 carbon to afford 5. Furthermore, labelling experiments suggest that the polycyclic core undergoes acetylation at ring A, and that ring E could be formed by condensation with an activated aceto-acetyl building block.102 The benzo[a]tetraphene scaffold has also recently been identified in borolithochromes from the specimens of the Jurassic putative macroalgae Solenopora jurassica that has been preserved for over 150 million years, illustrating the evolutionary significance of clostrubin-type polyketides.104
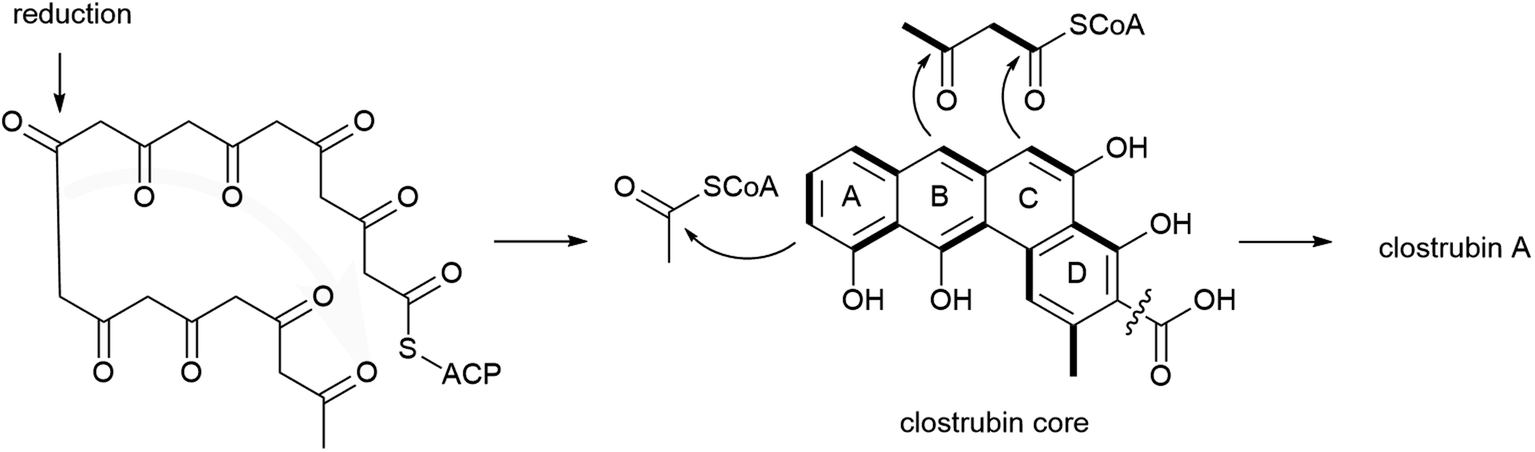 | ||
| Fig. 4 Noncanonical polyketide cyclisation folding in clostrubin biosynthesis. | ||
Clostrubin A 5 displayed nanomolar potency against Bacillus subtilis (MIC = 75 nm) and superior antibacterial activity against several nosocomial pathogens, methicillin-resistant S. aureus, MRSA (MIC = 0.12 μM), vancomycin-resistant Enterococcus, VRE (MIC = 0.97 μM), and Mycobacterium including M. smegmatis, M. aurum, M. vaccae, and M. fortuitum (MIC = 0.12–0.48 μM) than the antibiotic ciprofloxacin.102 Furthermore, when tested against some common potato disease-causing microbial pathogens like Clavibacter michiganensis subsp. sepedonicus (ring rot), Bacillus pumilus (soft rot), and S. scabies (common scab), clostrubin A 5 displayed nanomolar activity with MIC values of 47 nM, 95 nM, and 95 nM, respectively. Likewise, clostrubin B 6 displayed activity but weaker than clostrubin A 5 against the potato pathogens (MIC = 0.14–0.27 μM).47
Clostrubins 5–6 are not virulence factors but rather play dual roles beneficial to the anaerobic bacteria.47,102 First, being potent antibiotics, they act as chemical arsenals to inhibit other microbial competitors in a resource-limited niche.47,102 Second, clostrubins promote the survival of the anaerobic C. puniceum and C. beijerinckii in an oxygen-rich plant environment.47 Taken together, clostrubins represent promising leads for the development of antibacterial agents for use in fighting off potato infections. Furthermore, the total synthesis of clostrubin was achieved,105 which may provide insight into structure–activity relationships (SAR) to guide the development of novel antibiotics.
Although the carbon framework of stilbene monomers consists only of 1,2-diphenylethylene units, they demonstrate an enormous structural diversity because they are easily polymerized by oxidative coupling to produce diverse oligomers with intricate structures.110,111 Since stilbenes possess strong antioxidant/radical scavenging properties,106 their production in Photorhabdus spp. can be induced by supplementation of redox stress that generates reactive oxygen species. Feeding of paraquat (1,1′-dimethyl-4,4′-bipyridinium dichloride) to P. luminescens and P. asymbiotica cultures under aerobic conditions produced tapinarof 8 and its stilbene epoxide 9,107 lumiquinone 10112 and two novel tapinarof dimers, duotap-520 11 and carbocyclinone-534 12 (Fig. 5B).113 Duotap-520 11 contains a resorcinol–benzoquinone C–C bond linkage whereas carbocyclinone-534 12 features a novel hexacyclic core with a cyclopropane bridge. The complex structure of 12 was elucidated by nuclear magnetic resonance (NMR) experiments, X-ray crystallographic analysis, and electronic circular dichroism (ECD) spectral measurements and characterised as a racemic mixture of (+)-carbocyclinone-534 and (−)-carbocyclinone-534 12.
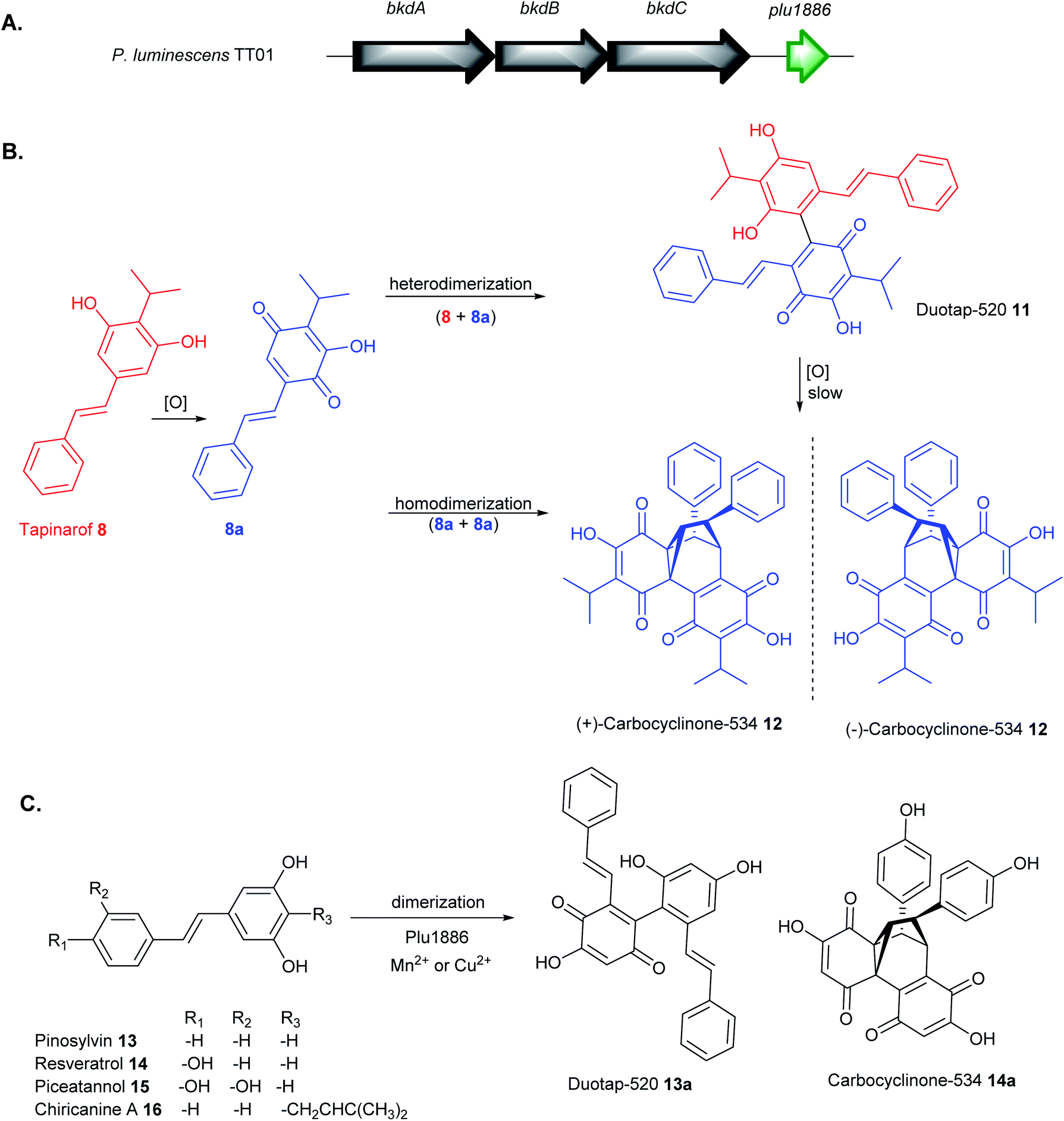 | ||
| Fig. 5 (A) Annotation of Plu1886, which encodes a cupin enzyme, adjacent to known tapinarof biosynthetic genes in P. luminescens TT01 (B) proposed pathway for regioselective oxidative dimerization of tapinarof 8 to duotap-520 11 and carbocyclinone-534 12, and (C) activity of Plu1886 enzyme with plant-derived stilbenes 13–16 in the presence of Mn2+ or Cu2+. | ||
Stilbene monomers such as resveratrol, isorhapontigenin, and piceatannol can undergo spontaneous oxidation and dimerization into an assortment of oxidized oligomers.110,111 Likewise, it has been shown that the formation of tapinarof-derived products, duotap 11, and carbocyclinone 12 involved similar oxidation, Diels–Alder cyclization, and dimerization mechanism (Fig. 5B). Under aerobic conditions, duotap 11 was shown to undergo slow spontaneous conversion into 12. Furthermore, an orphan cupin-type protein, Plu1886 adjacent to tapinarof bkd BGC in P. luminescens TT01 was identified to enhance the transformation of tapinarof 8 to 11 or 8 to 12in vitro (Fig. 5A).113 Cupin superfamily of enzymes are widespread in plants and are known to catalyse numerous diverse oxidation reactions, often requiring metal cofactors (e.g. Ni2+, Ca2+, Fe2+, Cu2+, Zn2+, Co2+, Mg2+, Mn2+) for the activity.114,115In vitro enzymatic tapinarof conversion to carbocyclinone-534 12 is highest in the presence of Mn2+ and to 11 in Cu2+. Microaerobic cultures of Δplu1886 mutant showed a substantial decrease in carbocylinone 12 production relative to the WT, supporting its role to enhance tapinarof dimerization reactions.113
The bacterial Plu1886 enzyme shows substrate promiscuity towards plant-derived stilbenes such as pinosylvin 13, resveratrol 14 (Fig. 5C). The cupin catalysed the robust conversion of pinosylvin 13 to the novel duotap 13a and resveratrol 14 into its new carbocyclinone 14a scaffold in the presence of Mn2+ or Cu2+. The no-enzyme controls only showed a trace amount of dimer 14a and an undetectable level of 13a. The new enzyme-derived products 13a and 14a were purified and structurally confirmed by 2D NMR experiments. No duotap production from 14 or carbocyclinone production from 13 was observed and no derivatives corresponding to dimerization of piceatannol 15 or chiricanine 16.113
Stilbenes are prolific sources of lead molecules in the search for new drugs and medicines. Even slight structural modifications of monomeric stilbenes dramatically alter their chemical complexity and improve their overall pharmacokinetic properties.106 Duotap-520 11 exhibited much higher potency against MRSA (MIC = 6.5 μM) and VRE (MIC = 4.1 μM) compared to tapinarof 8 with MIC values of 50.5 μM and 27.0 μM in MRSA and VRE, respectively. Carbocyclinone-534 12 did not show any significant antimicrobial activity but exhibited antimycobacterial activity against M. smegmatis.113 Duotap 11 showed stronger activity than tapinarof 8 in its ability to regulate the Nrf2 antioxidant reporter gene. Furthermore, dimers 11 and 12 showed little to no efficacy in a colitis mouse model, whereas the monomer reduces disease symptoms. Although 8, 11 and 12 were only produced in the pathogenic P-form of Photorhabdus spp., their varying bioactivity data suggest that the bacterium employs a regulatory mechanism to attain its desired functional outcomes required for symbiosis and pathogenesis.113 The much weaker antimicrobial activity of tapinarof relative to duotap-520 is probably a means of cellular detoxification by the bacteria to support its symbiosis with the nematode, whereas the more potent duotap-520 presumably support its pathogenic lifestyle.107,110,113 The promiscuity of Plu1886 biosynthetic enzyme in vitro represents a significant cornerstone towards the development of an efficient system to generate novel stilbene dimers with specific activity.
4.2 Nonribosomal peptides
Non-ribosomal peptide synthetases (NRPSs) are multi-modular enzymes that catalyse the synthesis of numerous peptide and peptide-like natural products that have wide applications in medicine, agriculture, and biotechnology among other fields (Fig. 6 and Table S1†). These mega enzyme complexes are not limited to the 22 proteinogenic amino acids; a large breadth of substrates is now known to be integrated and modified by post-synthesis action. NRPSs can incorporate a wide variety of nonproteinogenic amino acids, such as D-isomers, α-hydroxy/keto acids, carboxylic acids, and N-methylated residues, as well as several other building blocks such heterocyclic rings and fatty acids. Other common post-synthetic modifications associated with the NRPS machinery include glycosylation and oxidative cross-linking giving rise to diverse molecules with precise functionality for a particular molecular target.116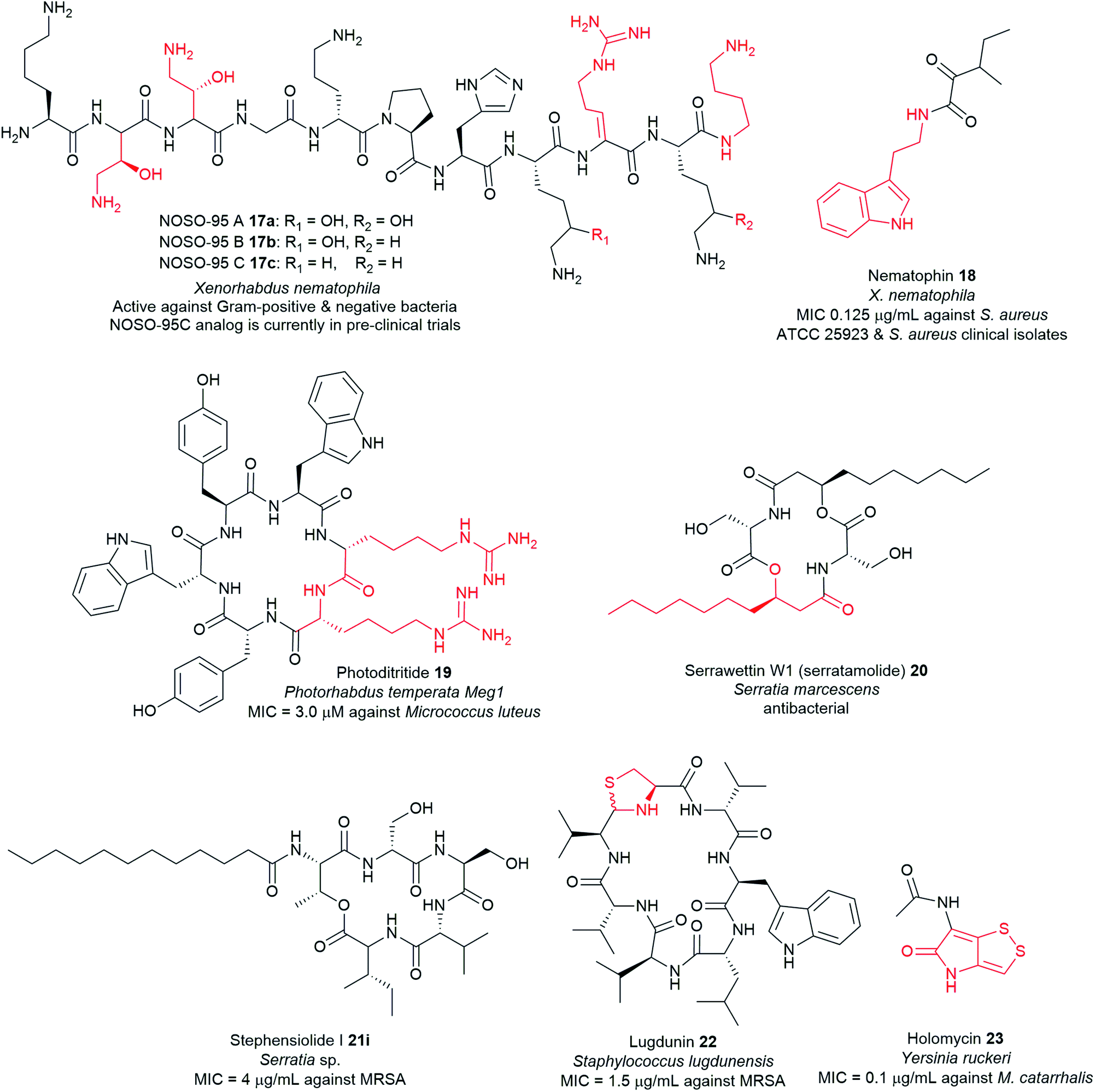 | ||
| Fig. 6 Examples of antimicrobial nonribosomal peptides with unusual motifs highlighted in red, isolated from pathogenic bacteria. | ||
Typical NRPS modules feature an adenylation (A) domain that selects and activates an amino acid monomer (and sometimes other carboxylic acids) as an adenylate followed by acyl transfer to a peptidyl carrier protein (PCP; also known as thiolation domain, T). This thiolation domain loads the activated amino acid on a 4′-phosphopantetheine (4′-Ppant) arm and covalently tethers it to form a peptide bond with an amino acid on the succeeding module, a reaction catalysed by the condensation (C) domain. Together, these three core domains (C, A, T) comprise a minimal NRPS module. In addition to these essential domains, each module may contain an epimerase (E) for the conversion of an L to D-configuration of amino acid, methyltransferase (MT) for N-methylation of the amide nitrogen, oxidase (Ox) for the conversion of a thiazoline to a thiazole or for α-hydroxylation of the incorporated amino acid, and reductase (R) for reductive release of an aldehyde product. The C domain replaced by the cyclization (Cy) domain catalyses both condensation and the intramolecular heterocyclisation of Ser, Cys, or Thr to afford thiazoline or oxazaline heterocycles. The release of the final peptide product from the NRPS is catalysed by a C-terminal reductase (R), thioesterase (TE), or a cyclizing C domain to yield linear, cyclic, or branched peptide chain topologies. The structural biology and enzymology of NRPSs have been the subject of several reviews.83,117,118 This section covers some of the interesting linear and cyclic nonribosomal peptide antimicrobials from pathogenic bacteria such as odilorhabdins 17a–c, nematophin 18, photoditritide 19, serrawettins 20, stephensiolides 21, lugdunin 22, and holomycin 23 (Fig. 6 and Table S1†).
Lead optimization strategies identified a synthetic analogue, NOSO-95179 24 (Fig. 7)120 with improved antibacterial properties over the natural compound NOSO-95C 17c.120,121 NOSO-95179 24 differs from NOSO-95C 17c by the replacement of Dab(βOH)3 by alanine and the removal of the lateral lysine10 and putrescine at the C-terminus. Further structural modification at Ala3 and His7 positions of 24 led to the selection of NOSO-502 25 as the first odilorhabdin clinical candidate (Fig. 7).9,122,123 NOSO-502 25 exhibits potent activity to all classes (Ambler A, B, C, and D classification) of carbapenem-resistant Enterobacteriaceae (CRE) strains (MIC = 0.5–4 μg mL−1). Furthermore, 25 shows excellent in vivo efficacy in several CRE murine infection models, exhibits good in vitro safety profile, and has a low potential for resistance development.119,120,122,123 Notably, 25 exhibits good stability in plasma, microsomes, and hepatocytes.123 Taken together, NOSO-502 25 represents a promising drug candidate.
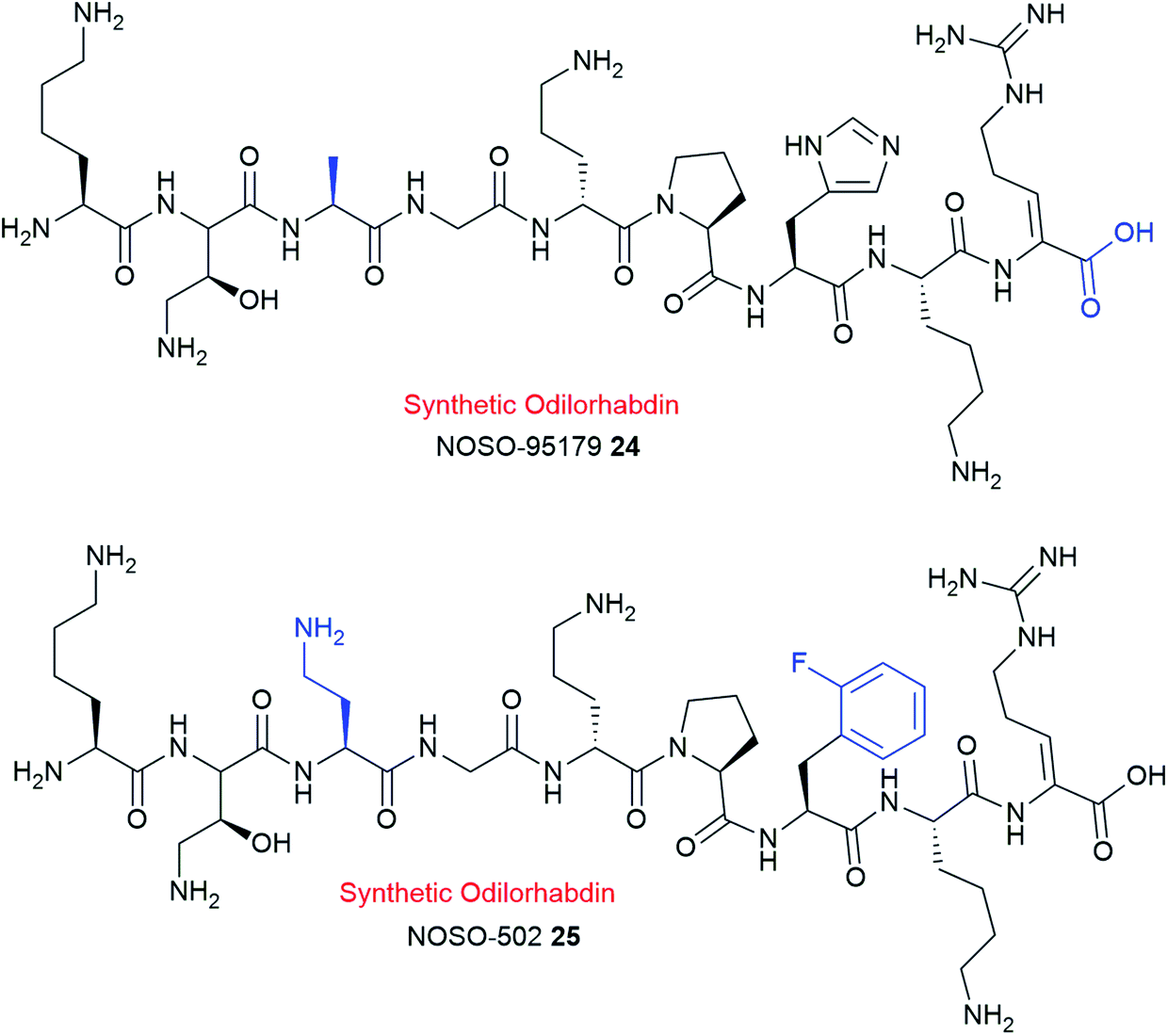 | ||
| Fig. 7 Chemical structures of synthetic analogues NOSO-95179 24 and NOSO-502 25. | ||
Antimicrobial peptides that interfere with bacterial ribosomes are rare.124,125 Nine classes of ribosome-targeting antibiotics are known, five of which, including odilorhabdins target the 30S subunit.125 However, the specific binding site of ODLs on the ribosome and its bactericidal mechanism is distinct from the other four classes.119 ODLs bind to the decoding centre of the 30S small ribosomal subunit119 that has never been exploited by any other known ribosome targeting antibiotics such as negamycin, tetracycline, streptomycin and paromomycin.125–128 ODLs display concentration-dependent bactericidal activity similar to the mechanism described for aminoglycosides and negamycin antibiotics.126,127,129 At lower concentrations, ODLs induce miscoding of the genetic code, likely by increasing the affinity of aminoacyl-tRNAs to the ribosome,128 whereas at higher concentrations they inhibit translocation.119
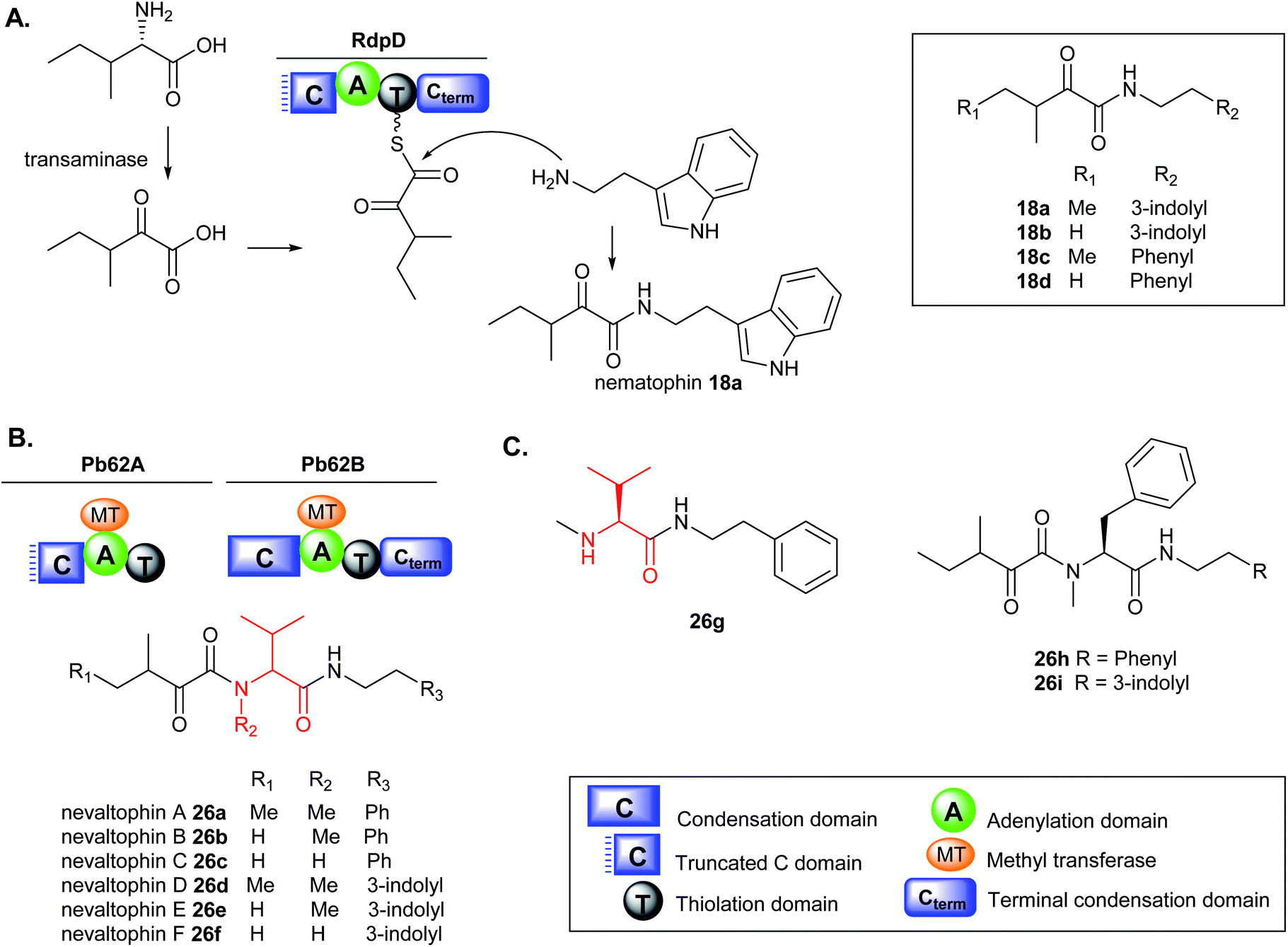 | ||
| Fig. 8 (A) Nematophin 18a and analogues 18b–d from heterologous expression of the rdpD gene in E. coli and proposed biosynthesis, (B) nevaltophin and analogues 26a–f from heterologous expression of the pb62 gene cluster in E. coli, and (C) chemical structures of 26g–i. | ||
The biosynthesis of nematophin is proposed to originate from the monomodular NRPS, RdpD, which is closely related to the RXP-producing NRPS, RdpABC but differs in the incorporation of α-keto carboxylic acid as the starting unit.131 Heterologous expression of the rdpD gene from X. nematophila ATCC 19601 strain in Escherichia coli fed with either phenylethylamine (PEA) or tryptamine (TRA), resulted in the production of new nematophin congeners, 18b–d (Fig. 8A). In contrast, the wild type (WT) X. nematophila strain only produced nematophin 18a even when fed with PEA or TRA and the presence of the amine compounds did not enhance its production level.131
Very few non-ribosomal peptides containing α-keto acid building blocks have been described to date.131,132 The α-keto acid precursors in nonribosomal cereulide from Bacillus cereus and valinomycin from Streptomyces spp. occur via deamination of α-amino acids such as valine, isoleucine or alanine.132–134 A similar deamination mechanism to the corresponding acids is proposed in RdpD biosynthesis which is activated by the A domain and subsequently loaded onto the adjacent T domain. Nucleophilic attack by the free amine via the Cterm generates nematophin 18a and analogues (18b–d). The Cterm domains in RXP-NRPS and RdpD-NRPS indicate that various amines such as TRA and PEA commonly found in Xenorhabdus strains can be used as substrates to access the production of TRA- (18b) and PEA-containing nematophin derivatives (18c–d). The PEA analogues are produced in minor amounts, implying that the substrate preference of the Cterm domain in RdpD is likely tryptamine over phenylethylamine.131
A similar BGC was identified in Xenorhabdus PB62.4 containing two monomodular NRPS, Pb62A resembling RdpD with a broken Cstarter domain, and Pb62B like the RXP RdpC terminal module with a complete C domain. Heterologous expression of the pb62 gene cluster in E. coli fed with either PEA or TRA has permitted to unlock the production of new elongated nematophin derivatives containing an additional valine motif in the structure which was assigned the name nevaltophins 26a–f. The structures of 26a–f suggest a biosynthetic pathway very similar to that of 18a–d but with the incorporation of a valine subunit with α-keto acid building blocks (Fig. 8B).131 The production of 26a–f was abolished in the Ser1303Ala mutation on the conserved Ser of the PCP domain in PB62A and led to the accumulation of 26g, further supporting the proposed biosynthesis (Fig. 8C). Furthermore, when Pb62A was used as a starting module in XndB involved in xenortide biosynthesis,135 nevaltophins with phenylalanine motif 26h–i were produced.131 The results provided a platform for engineered biosynthesis further expanding the nematophin chemical space.
While the crude extracts containing nematophins displayed zone of inhibition against the Gram-positive bacteria M. luteus, the nevaltophins containing-extracts did not exhibit activity.131 The authors, however, only tested the antibacterial activity of nevaltophins against M. luteus;131 and the results may not provide conclusive evidence that the valine unit incorporation in the nematophin core structure may enhance or decrease its bioactivity. In stark contrast, another study indicated that nematophin 18a has no activity against M. luteus at the highest concentration tested (100 μg mL−1). Nematophin, however, showed potent activity against other Gram-positive bacteria such as S. aureus (MIC = 0.125 μg mL−1),130,136 MRSA (MIC = 1.5 μg mL−1) and fungal pathogen, Botrytis cinerea (MIC = 12 μg mL−1).130 Furthermore, the δ-keto amide functionality in nematophin is essential for its anti-staphylococcal activity;136 and the activity is substantially enhanced by N-substitution of the indole ring with an alkyl or a phenyl group.131,136,137 The synthetic N-methyl substituted nematophin analogue displayed nanomolar activity towards several strains of S. aureus (15 ng mL−1), Staphylococcus hyicus (60 ng mL−1), and Staphylococcus intermedius 9503 (50 ng mL−1)136 including MRSA ATCC 43300 (31 ng mL−1) and methicillin-susceptible S. aureus, MSSA ATCC 29213 (125 ng mL−1).137 Conversely, incorporation of azaindole moieties in the nematophin scaffold significantly reduced the antibiotic activity (MIC = 16–128 μg mL−1).137 Nematophin 18a and nevaltophin 26a showed weak activity against parasites, Trypanosoma brucei rhodesiense, Trypanosoma cruzi, Leishmania donovani, and Plasmodium falciparum.131 Phenylethylamide-containing compounds such as nematophin were found to specifically inhibit an insect serotonin receptor facilitating its role in insect pathogenesis.138
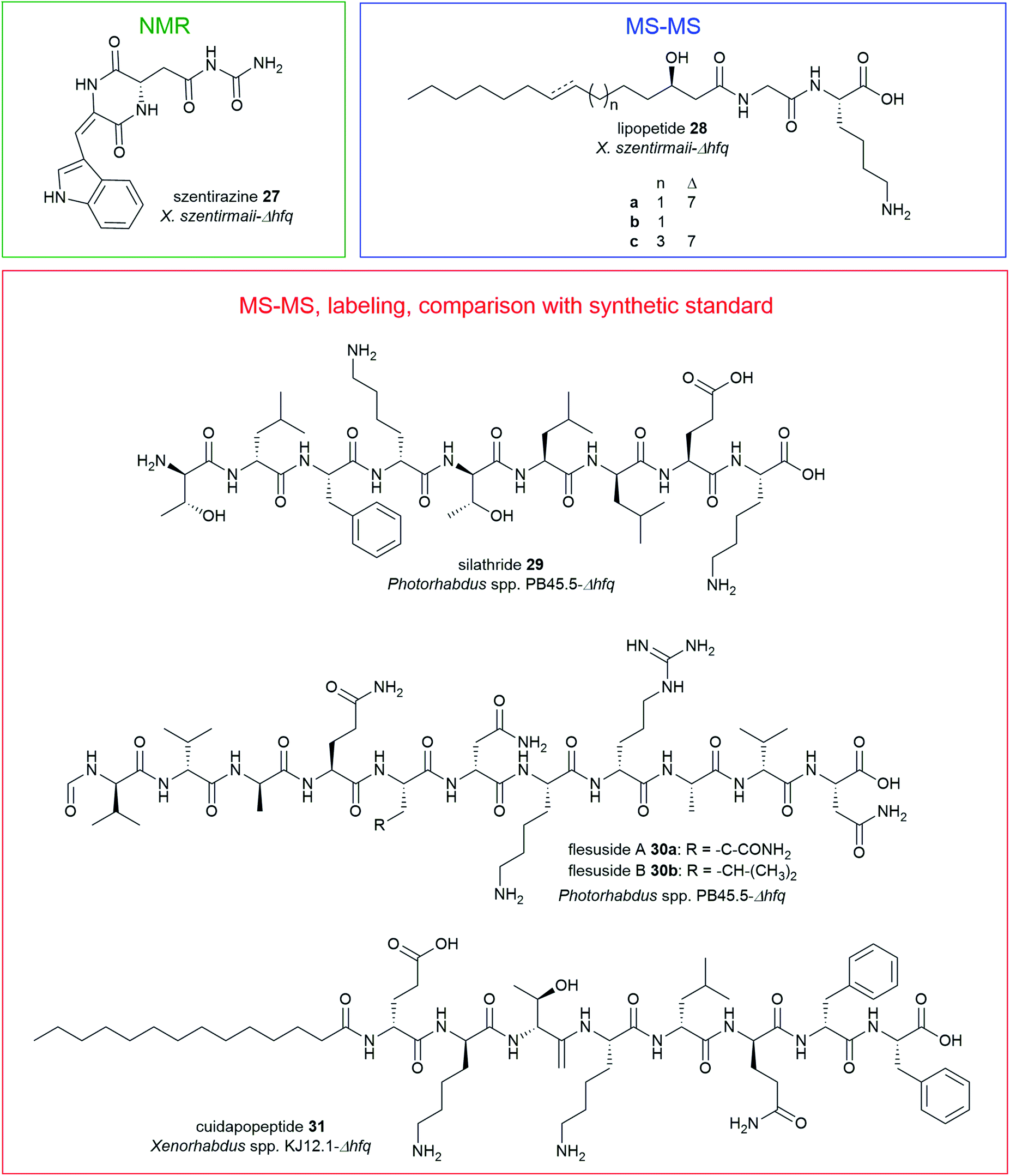 | ||
| Fig. 9 Structures of nonribosomal peptides identified from Δhfq mutants of X. szentirmaii (szentirazine 27, lipopeptides 28a–c), Photorhabdus PB45.5 (silathride 29, flesusides A and B 30a–b), Xenorhabdus KJ12.1 (cuidadopeptide 31) via promoter exchange. | ||
The promoter exchange strategy resulted in overproducing mutants with significantly higher production titres relative to the WT strains.151 In X. szentirmaii-Δhfq, two silent BGCs were activated that encode for the known depsipeptides, xenobactin152 and szentiamide.153 Additionally, a new oxidized diketopiperazine (DKP), szentirazine 27, and three new shortened PAX-peptides (28a–c) were produced. The new compounds 27–28 were exclusively produced by the induced Δhfq mutant. The structures of the lipopeptides (28a–c) were elucidated by detailed MS-MS analysis while szentirazine 27 was isolated from a large-scale culture, and its structure was characterized by NMR spectroscopy.151 Furthermore, new peptides silathride 29 and flesusides A 30a and B 30b were identified from Photorhabdus PB45.5-Δhfq and the new lipopeptide cuidadopeptide 28 from Xenorhabdus KJ12.1-Δhfq via a similar approach. The structures of 29–31 were elucidated by detailed MS/MS fragmentation analysis, labeling experiments and by comparison with synthetic compounds.151 All new NPs 27–31 showed weak to moderate antimicrobial activity against several Gram-positive and Gram-negative bacteria, and fungi.151
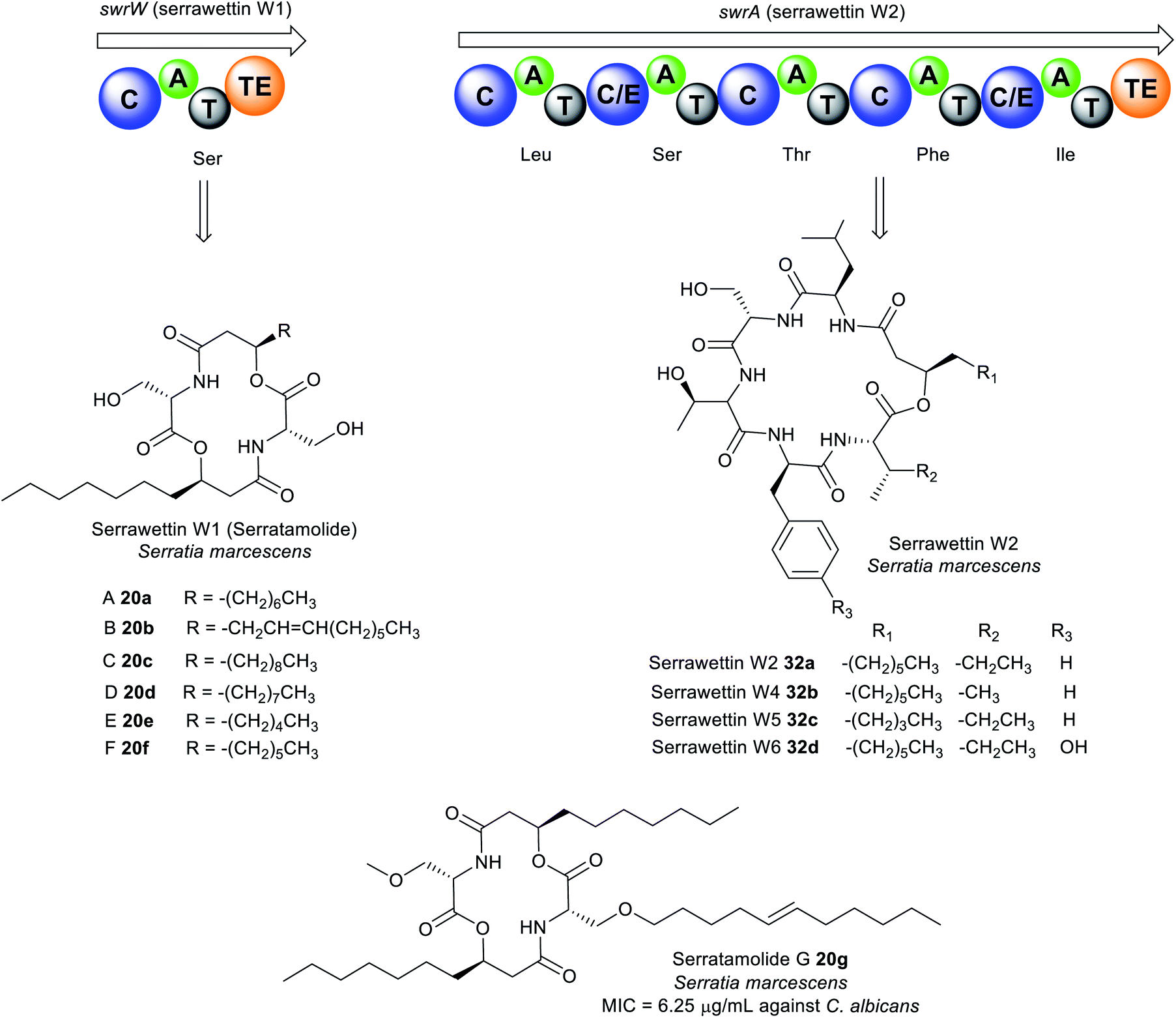 | ||
| Fig. 10 Analogues of serratamolide (serrawettin W1) A–G (20a–g) and serrawettin W2 32a–d identified in Serratia sp. | ||
The general chemical structure of serrawettin W2 consists of five amino acid residues (D-Leu–L-Ser–L-Thr–D-Phe–L-Ile) attached to a β-hydroxy fatty acid moiety (Fig. 10 and Table S1†).162,163 Four analogues of serrawettin W2 32b–d were recently isolated from Serratia sp. which differs based on the amino acids present (Ile or Val, Phe or Tyr) or the length of the fatty acid chain (C5 or C7).162 Further putative analogues (W7–W8) were tentatively identified in Serratia surfactantfaciens sp. nov. YD25 by MS/MS fragmentation analysis.164 The structure of serrawettin W3 described in 1986 is still yet to be determined.165 It is partially characterised and is composed of five amino acid residues (Thr, Ser, Val, Leu, Ile) and one dodecanoic fatty acid.163
The dilactone serrawettin W1 is believed to be formed solely by the action of the monomodular NRPS, SwrW encoding for aminolipid synthetase (Fig. 10). Initially, the biosynthesis of 20a was thought to occur via condensation of two serratamic acid molecules. However, mutational studies indicate the absence of the presumed precursors, suggesting the involvement of NRPS machinery in 16a production. Consequently, the presence of SwrW was identified in S. marcescens 274 by transposon mutagenesis. SwrW exhibits a C–A–T–TE domain architecture specific for only L-serine, and is presumed to be the simplest enzyme in the NRPS family. This simple NRPS system features an unusual dimerization, most likely via two following transesterification steps to assemble the symmetric and cyclic product, serrawettin W1 with no peptide bonds.166 Biosynthesis of serrawettin W1 presumably starts with the adenylation of the L-serine, after which the activated L-serine binds as a thioester to the thiolation domain which has been phosphopantetheinylated through the action of the PPTase, PswP.167 The amino group of the L-serine bound to the thiolation domain forms a bond with the 3-D-hydroxydecanoyl fatty acid which is speculated to come from a yet unknown ACP domain to form the first serratamic acid intermediate, and then subsequently transferred to the TE active site.166 Thereafter, biosynthesis of the second serratamic acid occurs and follows similar dimerization and cyclization processes to the ones catalysed by the multi-modular synthetase in the biosynthesis of the symmetric decapeptide gramicidin S from Brevibacillus brevis.168
Biosynthesis of serrawettin W2 in S. surfactantfaciens sp. YD25T is proposed to be catalysed by the NRPS peptide synthetase, SwrA consisting of five modules (Fig. 10). The unusual feature of SwrA (like SwrW) stems from the assembly of the starter unit. Typical NRPS contains A domains at the initiation site, but the SwrA NRPS harbours a C domain at its N-terminus suggesting that the initiation of peptide synthesis may form from the condensation of a fatty acid rather than an amino acid. It is presumed that a fatty acid adenylate, acyl-ACP, or acyl-CoA is likely the substrate for this C domain, catalysing the N-acylation of leucine. The fatty acid precursor in serrawettin W2 is speculated to be synthesised by the putative PKS SwrEFG gene cluster and other unknown enzymes. Chain elongation then occurs via the action of the other domains by successive incorporation of serine, threonine, phenylalanine, and isoleucine. Finally, cyclisation and chain release of the oligopeptide is catalysed by the TE domain to yield serrawettin W2.164
Serrawettin W1 20a exhibits antimycobacterial activity against M. tuberculosis, M. diernhoferi, and M. avium (MIC = 25 μg mL−1),155,161 and antibacterial and antifungal activities towards S. aureus, B. subtilis, M. luteus, Trichophyton spp., and MRSA (MIC = 6.25–50 μg mL−1).155,169,170 Likewise, serrawettin W2 32a is active against Gram-positive (e.g. S. aureus, Rhodococcus sp. and Micrococcus spp.) and Gram-negative bacteria (e.g. Pseudomonas spp., Shigella spp.) including drug-resistant S. aureus clinical isolates.164 Serrawettin W2 32a is a potent biofilm inhibitor of Candida albicans (IC50 = 7.7 μM), while the W2 analogues 32b–f are moderately active (IC50 = 13.4–60.0 μM).162 Furthermore, 32a is cytotoxic towards Hela (IC50 = 20.9 μM) and Caco2 (IC50 = 54.1 μM) cell lines.
The cyclic lipodepsipeptides, stephensiolides A–K 21a–k were produced by a Serratia strain that was isolated from the midgut and salivary glands of Anopheles stephensi mosquitoes (Fig. 11).171 Stephensiolides were also isolated from the fungal endophyte, Lecanicillium sp. (Hypocreales) obtained from the latex of Sandwithia guyanensis plant.172 Stephensiolides 21a–k mimic the core structure of serrawettin W2 32a as both are cyclic pentapeptides162,163 but differ in the sequence of the amino acid constituents.171 The peptide sequence in stephensiolides is Thr–Ser–Ser–Val/Ile–Ile/Val while serrawettin W2 is Leu–Ser–Thr–Phe–Ile. Furthermore, the lactone in stephensiolides is cyclized through the hydroxy group of the threonine, whereas serrawettin W2 is cyclized via a 3-hydroxy group of the fatty acid chain. Stephensiolide congeners (A to K) 21a–k vary in the length of the alkyl chain, amino acid residues (Ile or Val) or the presence of a double bond in the lipid side chain.171
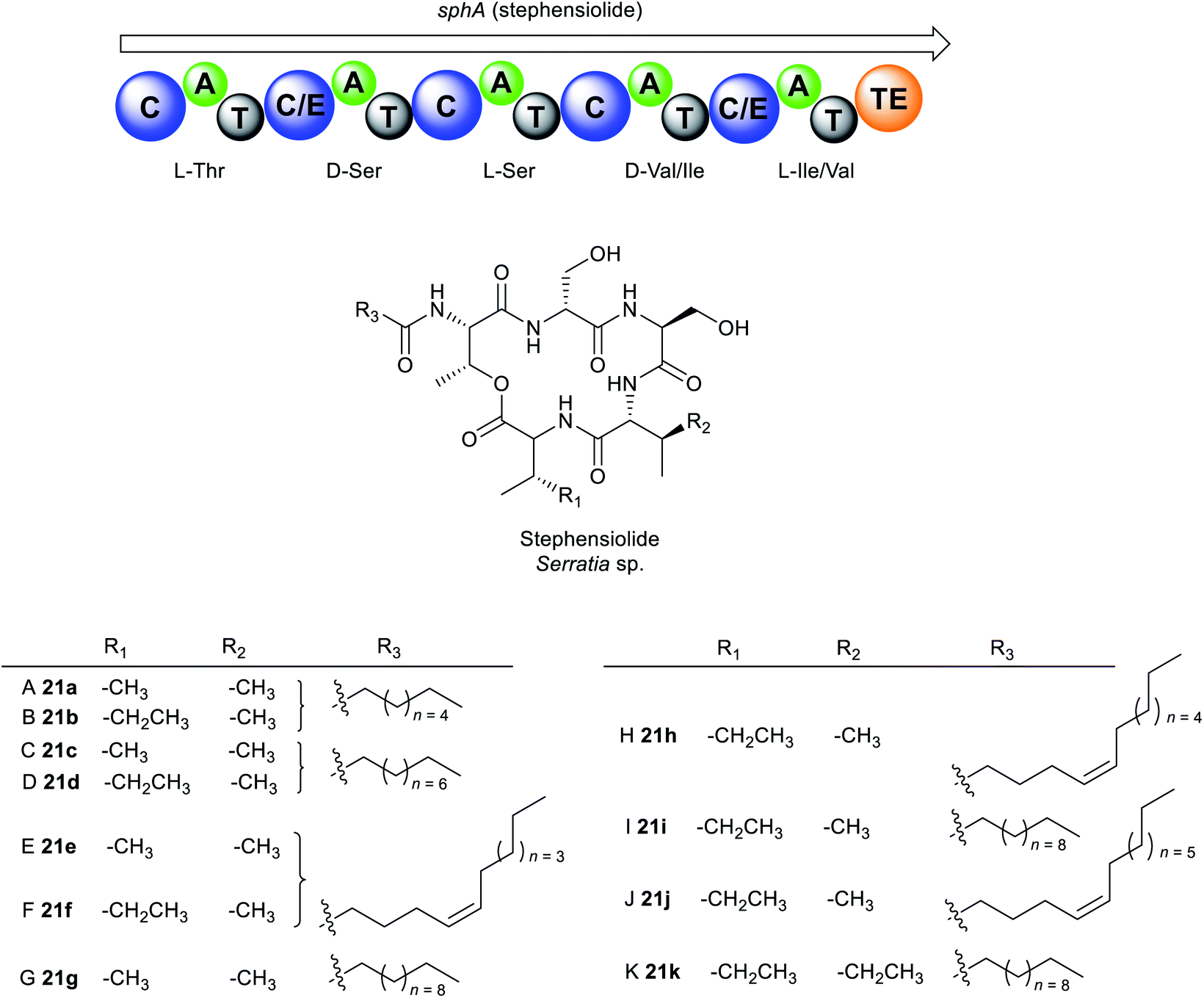 | ||
| Fig. 11 Structures of stephensiolides A–K (21a–k) from Serratia sp. | ||
Like serrawettin W1 20a and serrawettin W2 32a, stephensiolides are biosynthesised by a similar NRPS machinery (Fig. 11). Bioinformatics analysis identified the penta-modular NRPS, sphA which is presumed to be responsible for the incorporation of five amino acids, threonine, serine, serine, valine/isoleucine, and isoleucine/valine.171 SphA contains a unique initial C domain that is homologous to the lipopeptide-loading C module of EndA in the enduracidin biosynthesis,173 which is probably responsible for the incorporation of the fatty acid in 21a–k from an ACP.171
Antimicrobial testing of the stephensiolide mixture (A to K) revealed activity against B. subtilis 3610 (IC50 = 15 μg mL−1), P. falciparum Dd2 (IC50 = 14 μg mL−1), and the human hepatocytes, HepG2 (IC50 = 21 μg mL−1).171 Stephensiolides also demonstrated antibacterial activity against MRSA with stephensiolide I 21i as the most active (MIC = 4 μg mL−1).172 Like serrawettins, stephensiolides facilitate bacterial surface motility as biosurfactants.171 The primary role of swarming motility within mosquitoes is not fully understood, however, it is speculated that an enhanced swarming ability enables the bacteria to colonize and migrate in the different tissues within the insect host. A close relative to S. marcescens, Serratia strain AS1 colonizes diverse anopheline species and infect multiple different tissues within mosquitoes, including the midgut, female ovaries, and male accessory glands.174
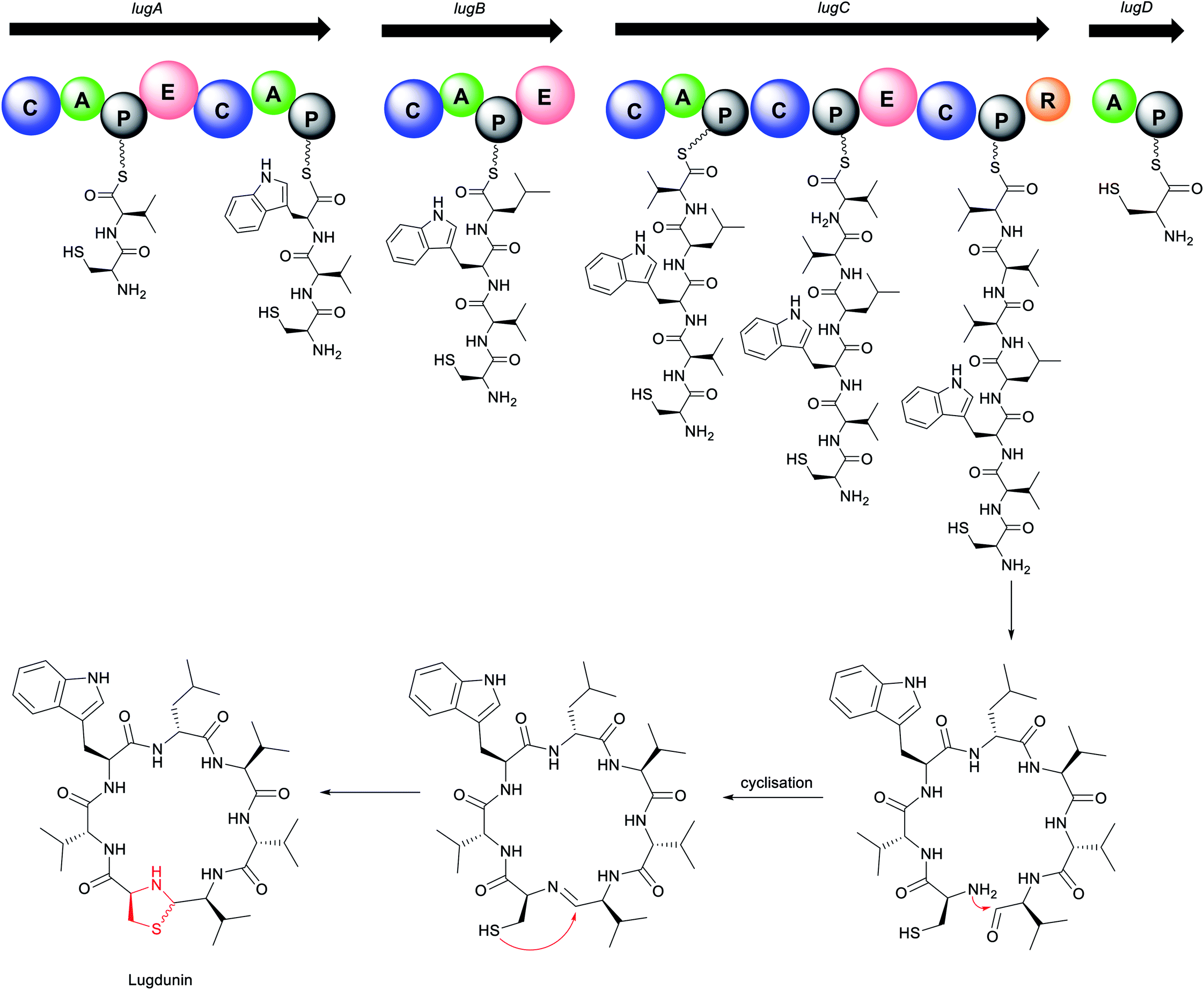 | ||
| Fig. 12 Proposed lugdunin biosynthesis. | ||
The biosynthetic mechanism for lugdunin production features several unusual aspects of the domains and their overall organization (Fig. 12). Four NRPS genes, lugA, B, C, and D, are proposed to direct the biosynthesis of lugdunin. Interestingly for a heptapeptide, the gene cluster encodes adenylation domains for only five amino acids. Biosynthesis presumably starts at the characteristic initiation module of LugD specific for L-cysteine, followed by sequential addition of D-valine and L-tryptophan by LugA, and D-leucine by LugB. The modules encoded in LugC exhibit a very peculiar organization, featuring a single valine-incorporating A domain but two downstream condensation and three PCP domains for peptide bond formation and amino acid transfer, respectively.29 This suggests an iterative biosynthetic logic similar to that of koranimine176 and yersiniabactin,177 where the single LugC adenylation domain activates three successive valine residues for subsequent installation in alternating L- and D-configurations. Chain release of the thioester-bound heptapeptide is catalysed by the terminal reductase of LugC, followed by subsequent cyclisation. Finally, the nucleophilic attack of the cysteine thiol group at either the re or si face of the imine yields two thiazolidine-containing structural diastereomers (depicted with wavy bond). The thiazolidine heterocycle is present in some linear NRPS compounds, such as watasemycins178 and yersiniabactin,177 but is yet unreported in macrocyclic peptides. Lugdunin is the first thiazolidine-containing macrocyclic peptide. Interestingly, production of lugdunin in ample amounts for chemical characterisation and biological profiling was only obtained via substitution of the native tetR-like regulatory gene, lugR, with a xylose-based expression approach.29
Lugdunin 22 exhibits potent bactericidal activities against a wide range of Gram-positive bacteria, including B. subtilis, Listeria monocytogenes, S. aureus, Streptococcus pneumoniae, and opportunistic pathogens MRSA, VRE, and glycopeptide-intermediate resistant S. aureus (GISA) (MIC = 1.5–12 μg mL−1).29 In contrast to rifampicin, S. aureus did not show any resistance to lugdunin even under prolonged exposure to sub-optimal doses of the compound for over 30 days. Furthermore, it shows no toxicity in primary human erythrocytes, neutrophils, or human monocytic cell line HL60, and demonstrates good in vivo efficacy in the mouse model of S. aureus skin infection. In vivo tests show significant reduction and even total eradication of viable S. aureus on the skin surface and in the mouse tissue indicating that the compound can penetrate the deeper layers of the skin.29 This inhibitory mechanism is achieved by the bactericidal activity of lugdunin as well as by the increased innate defence of epithelial cells resulting in efficient protection against S. aureus skin colonization. Lugdunin offers the host three layers of protection. Firstly, it can directly inhibit and kill S. aureus. Secondly, it can work synergistically with the antimicrobial peptides produced naturally by the host as part of the immune response (for example, hCAP18/LL-37 and the dermcidin-derived peptides DCD-1L), enhancing their ability to kill S. aureus. Finally, it can induce an immune response within the skin, thus enabling it to recruit phagocytic immune cells to aid with the clearing of the competing pathogen. Other factors derived from the skin commensal S. epidermidis may serve to amplify this response, increasing efficacy.179
SAR studies indicate that the cyclic structure of the peptide, the N-unsubstituted thiazolidine “clasp”, two amino acids tryptophan and leucine, and an alternating D- and L-amino acid backbone are integral to the activity.175 The nonpolar tryptophan and leucine residues interact with the hydrophobic regions of the bacterial cell membranes similar to the activity of poly-(Trp–Leu)-octapeptides.180 Fibupeptides like lugdunin carry electronically charged particles across the membrane and consequently disintegrate the membrane potential, thereby killing the bacteria. Incorporation of an additional tryptophan motif in the peptide backbone intensifies this membrane interaction and further strengthens the antibacterial effect, exhibiting two-fold increased activity over the parent compound.175 Lugdunin or analogues thereof are promising candidates for the treatment of multi-drug resistant Gram-positive infections. However, it may be challenging to develop these into systemic therapeutics considering that they are membrane-targeting antibiotics. Such compounds also tend to perturb mammalian plasma membranes.181
182 and later was reported to be produced by several other Streptomyces species183–188 and other bacteria, including the marine Gram-negative bacterium Photobacterium halotolerans79 and the fish pathogen Y. ruckeri (Fig. 6 and Table S1†).31,32 Structurally, holomycin belongs to a class of dithiolopyrrolone (DTP) natural products189 which contains a unique heterobicyclic core with a disulfide bridge and a variety of N-alkyl and N-acyl substituents.31,79,182–184
Dithiolopyrrolones possess broad-spectrum inhibitory activity against bacteria, fungi, and cancer cell lines.189–191 Holomycin 23 is potent against several Gram-positive and Gram-negative bacteria including E. coli (MIC = 0.2–2 μg mL−1), S. aureus (MIC = 2–4 μg mL−1), S. epidermidis (MIC = 1 μg mL−1), S. pneumoniae (MIC = 0.1–0.3 μg mL−1), Haemophilus influenzae (MIC = 0.3 μg mL−1), and Moraxella catarrhalis (MIC = 0.1–0.3 μg mL−1),192 as well as rifampicin-resistant S. aureus (RRSA) mutants containing modified RNA polymerase β-subunit (MIC = 4–8 μg mL−1).193 Despite this attractive biological activity, holomycin is toxic, so it may need to be modified for possible future antibiotic use. Chemical synthesis of DTP analogues with modifications at the N-positions has attracted significant interest by several groups.194–199N-Aryl DTP analogues have been shown antitumor activity198 and antileukopenia activity.194,197N-Aryl DTP with 2,4-dimethoxyphenyl moiety displayed potent antibacterial activity against clinical isolates of MRSA, RRSA, vancomycin-resistant S. aureus (VRSA), and moderately penicillin-resistant S. pneumoniae (MPRSP) with MIC values in the range of 0.125–2 μg mL−1 comparable to the antibiotic rifampin.195 Previous works also showed that the biosynthetic pathway of DTPs is susceptible to be manipulated by feeding different organic acids or fatty acids to the cultures to modify the lateral acyl chain.200–202 Another approach involved the generation of hybrid-type antimicrobials by incorporating the holomycin antibiotic into the myxopyronin core.203 The holomycin nucleus has also been more recently identified in the marine hybrid antibiotic thiomarinol 35, in which it is joined to a pseudomonic acid motif, an analogue of the FDA-approved topical antibiotic mupirocin (Bactroban®) (Fig. 13).190,204 The biosynthetic hybridity of thiomarinol may have advantageous effects; when one antibiotic fragment is modified by inactivating enzymes, the other constituent might remain functionally active.205,206 Attempts to stimulate holomycin production have also received considerable interest. Holomycin-high producing variants of S. clavuligerus were obtained via competition-based adaptive evolution against MRSA N315 (ref. 207) as well as manipulation of the regulatory gene, argR which regulates the expression of arginine biosynthesis.208
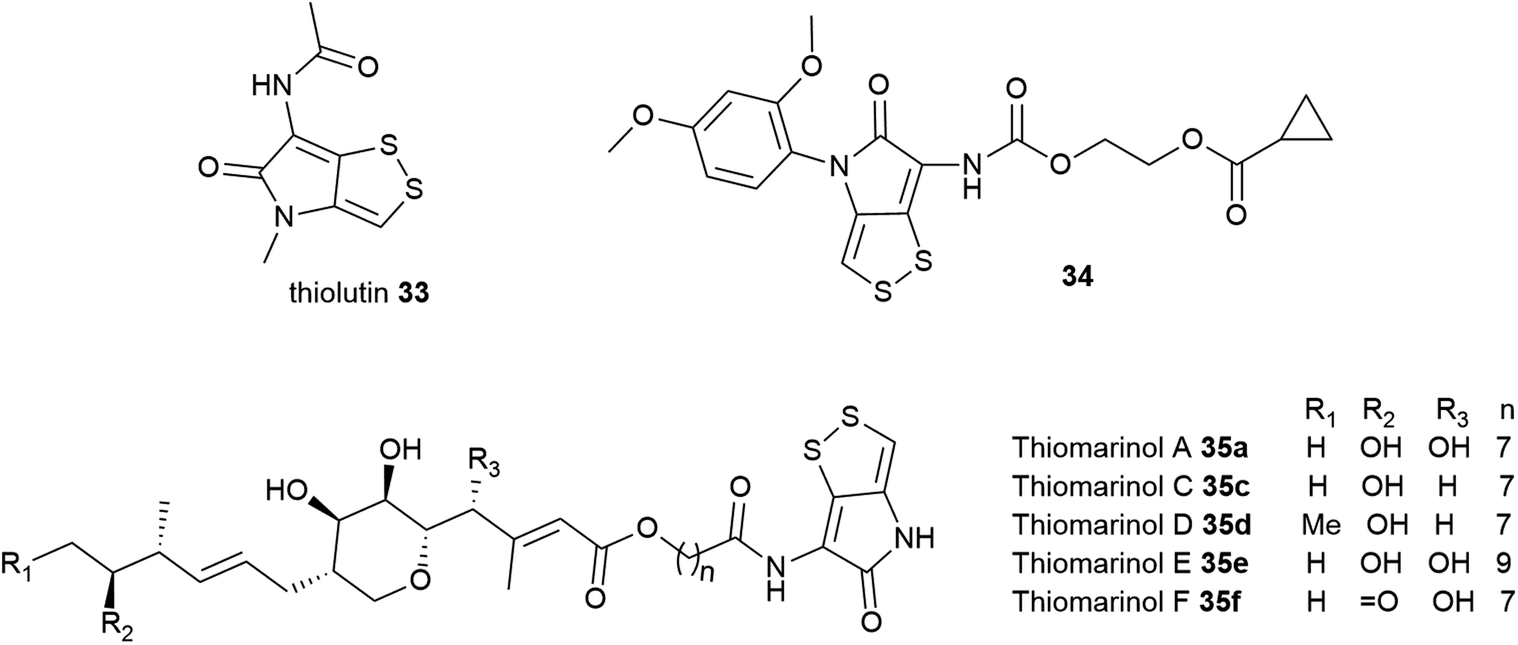 | ||
| Fig. 13 Structures of thiolutin 33, thiomarinols 35, and synthetic holomycin analogue 31 that exhibits notable bacterial RNA polymerase inhibition. | ||
Owing to the promising antimicrobial activity of DTPs, several studies into their mode of action (MOA) have been conducted using some of the more well-studied group members. Two opposing plausible mechanisms of action have been proposed. The first one identifies DTPs as inhibitors of bacterial RNA polymerase (RNAP). Thiolutin 33, a holomycin variant, has been shown to reversibly inhibit RNA and protein synthesis of Saccharomyces cerevisiae at a concentration of 2–4 μg mL−1 in the whole-cell and spheroplasts assays and inactivates yeast RNA transcription in vitro.191,209–211 However, subsequent studies of holomycin or thiolutin in E. coli RNA synthesis inhibition have indicated that although both exhibit activity in vivo, they show weak (or no activity) in vitro. Furthermore, it was also not clear which step of RNA synthesis thiolutin inhibits. Induction of β-galactosidase in E. coli has suggested both RNA transcription initiation and chain elongation as possible targets of thiolutin. These opposing results cast doubt as to whether RNAP is the main target of the antibiotic in E. coli.186,192,212–214 To uncover the intriguing aspects of DTP mechanisms, Tan and co-workers synthesised various N-aryl DTP analogues and investigated their in vitro inhibitory against E. coli RNAP. Among all the tested compounds, synthetic 34 inhibited the most potent RNAP activity in vitro and is also the least cytotoxic. Additionally, molecular docking studies (Fig. 13) of 34 revealed interaction and high binding affinity with the amino acid residues in the switch region of the E. coli RNAP in the same manner as myxopyronin A, indicating that DTP and analogues are bacterial RNA inhibitors.195,199
The second alternative mechanism is proposed by Li and co-workers in which holomycin 23 is considered as an intracellular metal-chelating antibiotic that sequesters free metal ions and selectively targets E. coli metalloenzymes, and not RNA polymerase in vitro.215 The proposed model suggested holomycin acts as a prodrug192,216 whose activation involves the conversion of the ene-disulfide in the cytoplasm to the active ene-dithiol, reduced holomycin (red-holomycin) with high affinity for zinc ions.215,216 The mechanism by which the cyclic disulfide 23 is reduced in the cells is as yet unknown. After entering the cells, the red-holomycin 23a is proposed to exert its metallophoric activity via two different routes (Fig. 14): (1) red-holomycin 23a sequesters essential metals, especially zinc, thereby limiting zinc availability in the bacterial cell, and (2) red-holomycin 23a removes zinc from a subset of zinc-dependent metalloproteins (i.e. E. coli class II fructose bisphosphate aldolase, FbaA), thereby disrupting the cell's metal homeostasis and potentially interfering the essential metabolic processes such as glucose utilization, RNA synthesis, and respiration. Although both routes contribute to the inhibitory effect of holomycin, route two may play a more prominent role in the MOA, consistent with the findings that an increased zinc concentration renders no enhanced effect on the E. coli growth inhibition. Disruption of the zinc import machinery involved in the maintenance of metal homeostasis, such as ZnuABC restricts zinc uptake and further sensitizes E. coli to holomycin.215 This MOA is unique amongst antibiotics and may be further explored to understand the specificity of holomycin and other DTPs against metalloenzymes for the development of novel potent chelators.
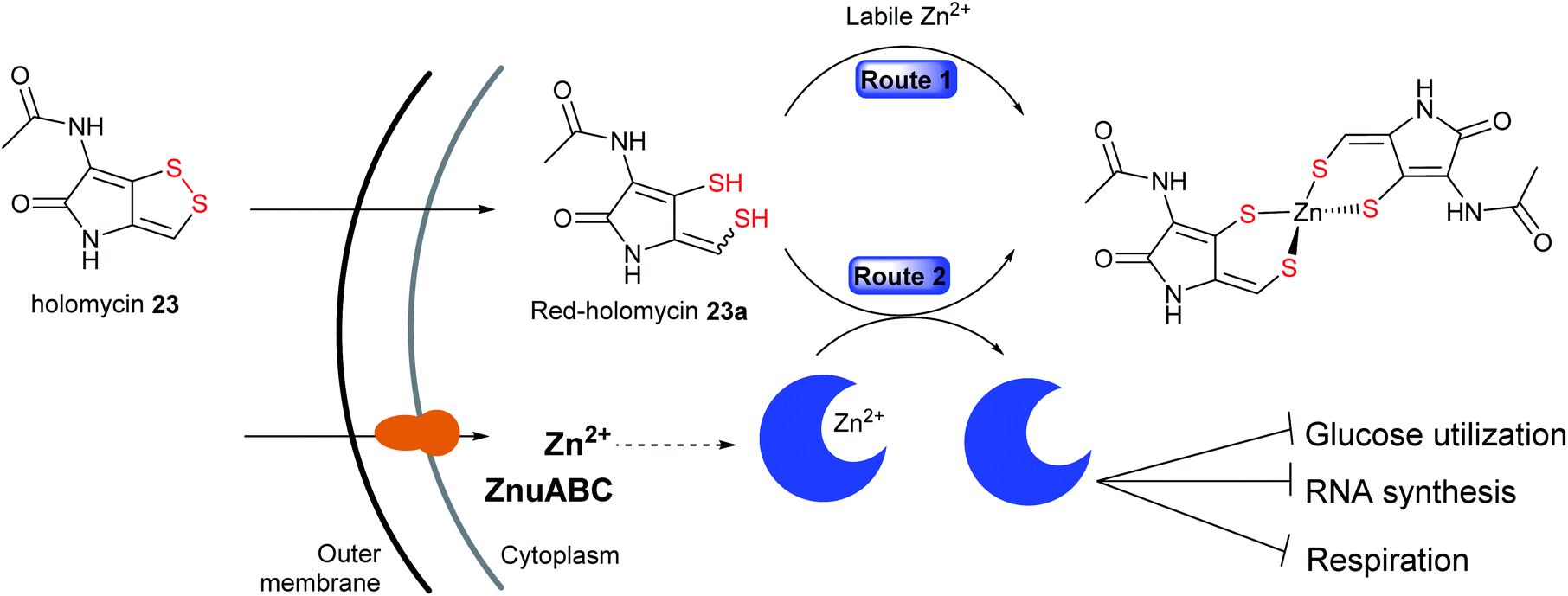 | ||
| Fig. 14 A model for the mechanism of action of holomycin in which it acts as a prodrug which undergoes intracellular reduction to the active red-holomycin that sequester free metal ions, particularly zinc (route 1) or removes zinc from metalloproteins (route 2). | ||
4.3 Hybrid polyketide–nonribosomal peptide natural products
Owing to the structural and catalytic resemblances between PKS and NRPS, they have evolved the ability to communicate with each other and combine modules to form hybrid assembly lines. During the transfer of the growing peptide or polyketide intermediate across NRPS/PKS interfaces, ketosynthase (KS) and condensation (C) domains facilitate chain elongation by accepting upstream PCP-bound peptidyl thioesters and ACP-bound polyketide thioesters, respectively, thereby switching efficiently between C–C bond and C–N bond formation. Together, the biosynthetic versatility of PKS machinery and the substrate flexibility of NRPS modules that can incorporate almost 500 different proteinogenic and nonproteinogenic amino acids coalesce to yield hybrid natural products with astounding structural and biological diversity (Fig. 15 and Table S1†). This biosynthetic machinery has been described extensively elsewhere.83,217 Examples of antimicrobial hybrid polyketide–peptide metabolites produced by pathogenic bacteria include the red-pigment prodigiosin, the broad-spectrum antibiotic althiomycin, the DNA-gyrase inhibitor albicidin, and the antibacterial metabolite associated with dental caries reutericyclin.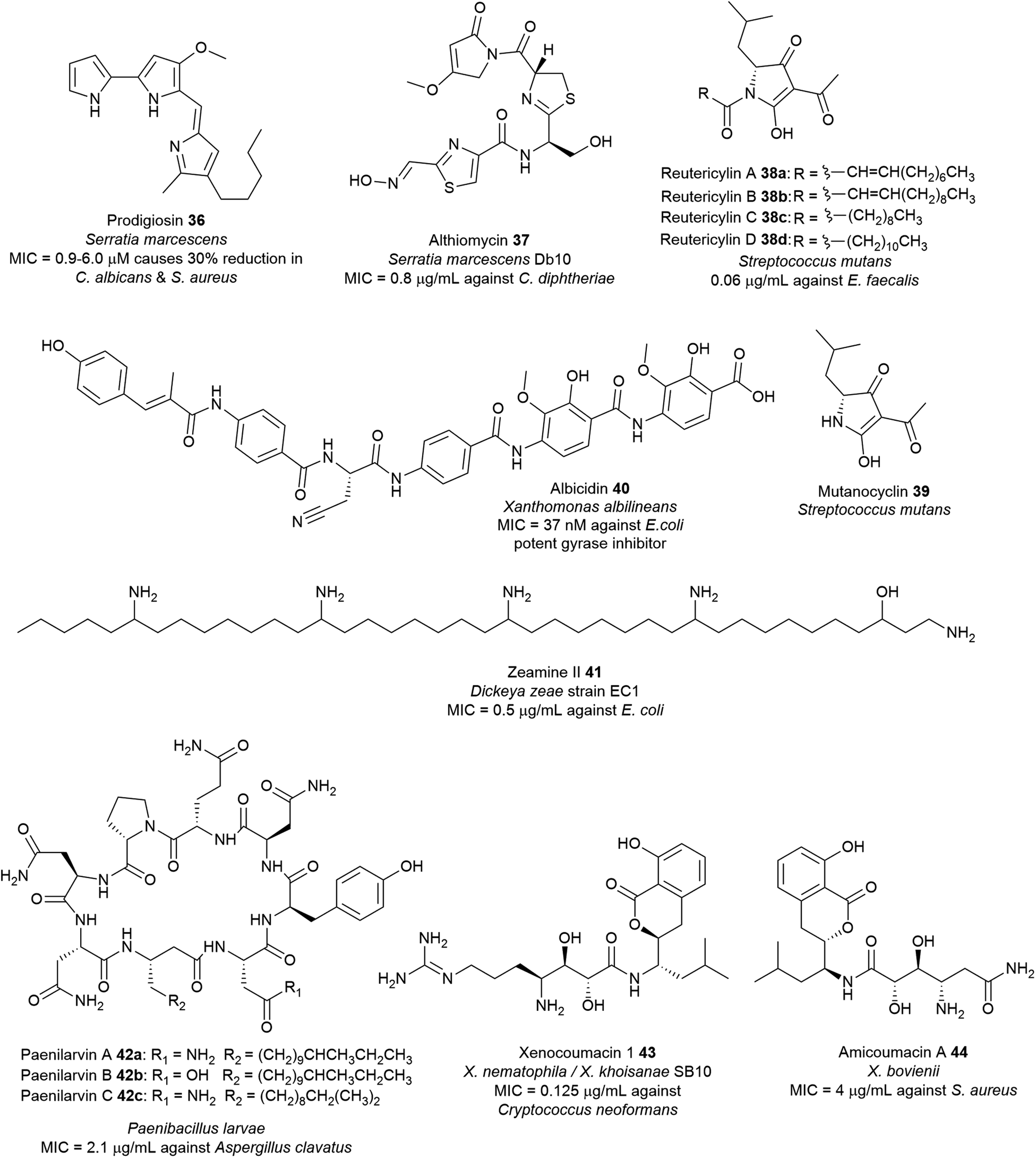 | ||
| Fig. 15 Examples of hybrid polyketide–nonribosomal peptide natural products with antimicrobial activity from pathogenic bacteria. | ||
Prodigiosin 36 has numerous potential beneficial properties such as antibacterial,229 antifungal,223 antimalarial,230 antiprotozoal,231 anticancer,232 immunosuppressant,233 and as natural colourants for the dyeing of silk and wool.227,234 It is active against a wide range of Gram-positive bacteria including S. aureus and B. subtilis,221 and Gram-negative E. coli, Erwinia carotovora, S. enterica, as well as drug-resistant strains such as MRSA and oxacillin-resistant S. aureus (ORSA).223 Prodigiosin targets the bacterial plasma membrane and causes disruption and loss of vital intracellular substances (K+ ions, sugars, amino acids, proteins) via a chaotropicity-mediated mode-of-action.235 Bacterial prodigiosin and related analogues exhibit in vitro antiproliferative activity against over 60 human cancer cell lines with an average inhibitory concentration of 2 μM. Furthermore, they are also potent inhibitors of T lymphocyte proliferation.223 Findings associated with anticancer and immunosuppressive properties of prodiginines and their possible modes of action have been subject to several reviews.236–238 Prodigiosin has also been used as inspiration to develop potent analogues such as obatoclax mesylate (GX15-070) which is currently in clinical trials for the treatment of various types of cancer including lymphoma, myelofibrosis, leukaemia, and mastocytosis.236,239–241
The physiological and ecological function of prodigiosin remains elusive. Its ubiquitous nature suggests that it may be ecologically beneficial to the producer organism. However, the precise role of the pigment remains elusive due to the diversity of prodiginine producers.228 In S. marcescens, prodigiosin 36 is not an essential virulence factor.218 Some reports have suggested potential roles of the pigment which is likely a mode of defence against microbial competitors in a continuously dynamic environment or as a response to natural stressors.223,228 Apart from its protective function against predators, prodigiosin may also serve as a metabolic sink (energy overflow) through the consumption of the excess NAD(P)H or proline from primary metabolism.242S. marcescens colonizes and propagates in the environment via swarming, swimming, and air dispersal. It is speculated that prodigiosin contributes to Serratia's cell surface hydrophobicity and consequently its improved motility facilitates bacterial dispersion through the air.223,243
Althiomycin 36 displays wide spectrum antibiotic activity against several Gram-positive bacteria including strains of S. aureus (MIC = 16–25 μg mL−1),250,251E. faecalis (MIC = 16 μg mL−1),251Corynebacterium diphtheriae (MIC = 0.8 μg mL−1)250 and Gram-negative bacteria including E. coli (MIC = 1 μg mL−1),251K. pneumoniae (MIC = 6.3 μg mL−1)250 and Shigella flexneri (MIC = 25 μg mL−1)250 but exhibits no such effects in mammalian cells.252 Althiomycin 36 blocks the action of the peptidyl transferase by binding to the 50S ribosomal subunit, thus inhibiting prokaryotic protein synthesis.244,252 Althiomycin and derivatives have been chemically synthesised (albeit with low efficiency).251,253 The synthetic de(hydroxymethyl) althiomycin analogue showed comparable antibiotic activity to that of the parent compound. SAR studies indicated that the 4-methoxy-3-pyrrolin-2-one moiety, and the configuration of the oxime group and thiazoline ring are relevant to its bioactivity.250 This methoxypyrrolinone pharmacophoric feature in althiomycin is also present in other bioactive natural products such as malyngamide A,254 sintokamide A,255 and mirabimide E.256 To date, the difficulties encountered in chemical synthesis have hampered further investigations into the potential of althiomycin-based compounds as antibacterial drugs.249
The structure determination of 40 paved the way for chemical synthesis providing multigram quantities of albicidin and enabling SAR studies of the albicidin scaffold.262 Albicidin 40 targets the GyrA subunit of the DNA gyrase (topoisomerase II),263 an essential enzyme that catalyses and modulates the extent of supercoiling of double-stranded DNA.264 Albicidin inhibits this supercoiling activity of E. coli DNA gyrase with half-maximal inhibitory concentrations (∼40 nM) lower than those of most coumarins and quinolones.263 Albicidin is bactericidal against a wide range of Gram-positive and Gram-negative bacteria with nanomolar potency particularly against fluoroquinolone-resistant strains of E. coli (MIC = 0.031–0.5 μg mL−1), Salmonella enteritidis (MIC = 0.5 μg mL−1), and P. aeruginosa DSM 117 (MIC = 1.0 μg mL−1).265 Structural modifications of 40 such as the substitution of the central amino acid β-cyanoalanine with polar threonine residue266 or azahistidine leads to analogues with increased bioactivity over the natural albicidin.267 Replacement of the N-terminal methylcoumaric acid moiety with benzoyl or acyl residues leads to inactivity towards the E. coli gyrase268,269 whereas carbamoylation of the N-terminus motif, which is most likely a post-NRPS reaction gives rise to a more potent bacterial gyrase inhibitor (IC50 ∼ 8 nM).270 Synthetic azahistidine–albicidin variants with ethoxy group substitution on the C-terminal dipeptide motif exhibits increased potency against Gram-positive B. subtilis, Mycobacterium phlei and ciprofloxacin-sensitive (MIC = 0.031 μg mL−1) and -resistant S. aureus (MIC = 0.063 μg mL−1).267 Variation in the molecule's stereocenter has minimal effect on the activity as indicated by ent-albicidin containing the D-Cya exhibiting comparable gyrase activity (IC50 ∼ 40 nM) with the natural product albicidin.265 Furthermore, replacing the central amide bond with a triazole moiety leads to a novel albicidin analogue that can overcome the serine endopeptidase AlbD resistance while preserving biological activity.267,271
The reutericyclin BGC in S. mutans comprises 9 genes (mucA–I) that encode a hybrid modular PKS–NRPS assembly line, as well as enzymes involved in transport and regulation. Reutericyclin 38a–c are proposed to be assembled from C10 or C11 fatty acids as starter units through elongation with leucine, which is subsequently extended via a malonyl-CoA unit (Fig. 16). The reutericyclin genomic island does not code for enzymes related to fatty acid metabolism,277 thus the C10 or C11 lipid chain in 38a–c may come from the general metabolism through the action of hydroxyacyl-ACP dehydratases to generate trans-2-decenoyl-ACP, decanoyl-ACP, and trans-2-dodecenoyl-ACP.30
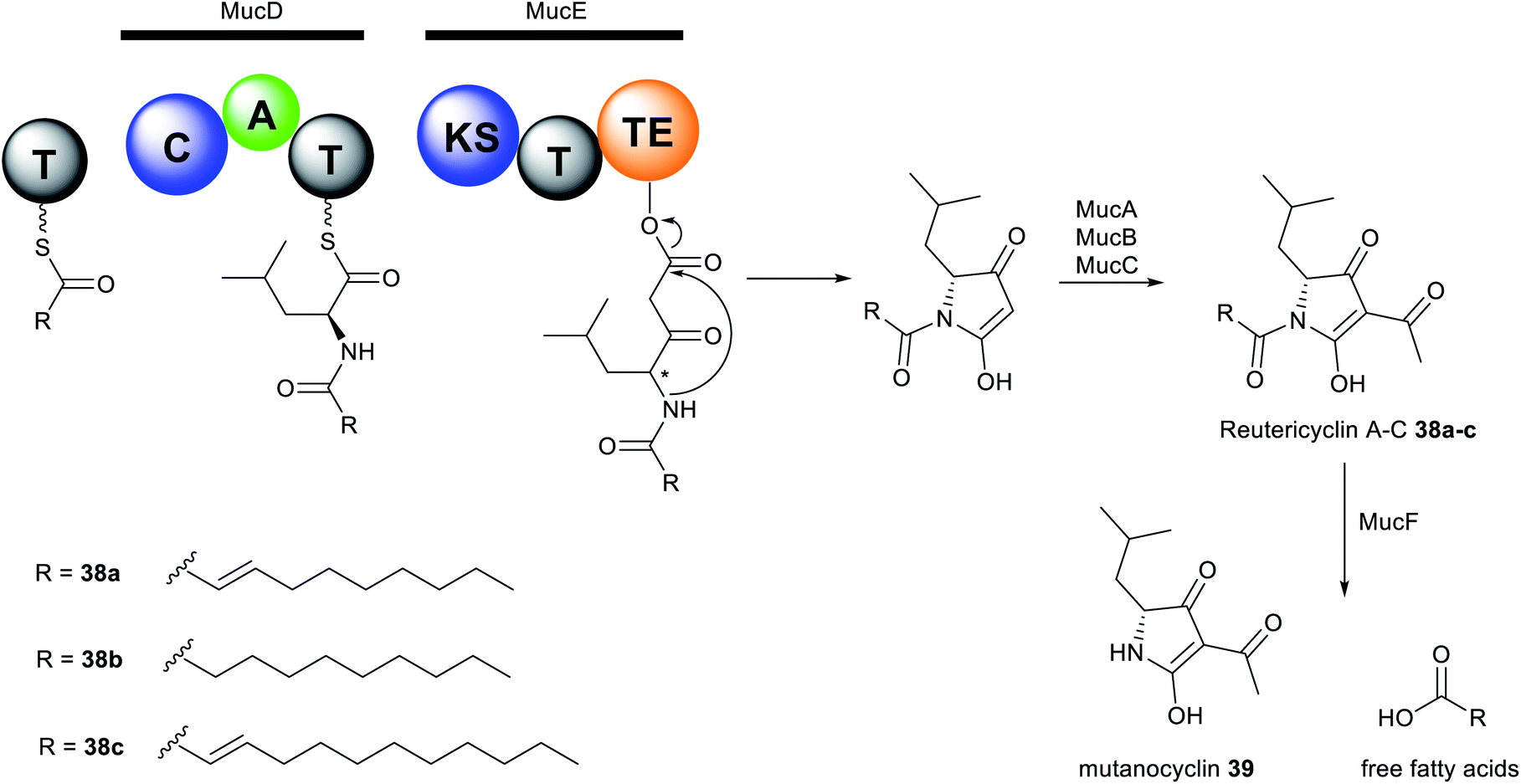 | ||
| Fig. 16 Proposed reutericyclin and mutanocyclin biosynthesis. | ||
Another interesting feature of the muc assembly line is the lack of an epimerase (E) domain or dual functioning C/E domains required in the conversion of L-to D-leucine residue. The A domain in MucD appears to incorporate the D-leucine building block in 38a–c.30 Most Gram-positive bacteria have the ability to synthesise D-alanine and D-glutamic acid as components of the peptidoglycan cell wall, however, the synthesis of other D-amino acids is less common.278 Feeding of [13C1] L- and D-leucine to fermentation cultures of S. mutans and L. reuteri revealed incorporation of only [13C1] L-leucine.30 Conversely, an isoleucine 2-epimerase with leucine epimerase activity has been characterised in lactobacilli,279 and L. reuteri strains have been reported to produce D-leucine.277 Presumably, S. mutans may also contain isoleucine 2-epimerase homologues responsible for D-leucine synthesis. Additionally, the muc TE domain may also act as epimerase as exemplified by the NocTE domain in nocardicin biosynthesis.280 However, MucTE exhibits very low homology to the dual functioning NocTE domain.30 It is currently unclear which enzyme is responsible for the epimerization reaction in reutericyclin biosynthesis. The first three genes, mucABC are homologous to the phloroglucinol biosynthetic proteins PhlABC, and are believed to catalyse the acetylation of the pyrrolidine ring in 38a–c. Expression of the MucA–E in E. coli BAP1 strain resulted in the production of 38a–c and a new analogue reutericyclin D 38d containing an N-dodecanoyl substituent, indicating that genes mucA–E indeed compose the minimal BGC for 38a–c production. Furthermore, heterologous expression and deletion experiments characterised MucF as a new deacylase responsible for converting reutericyclin 38a–c to the tetramic acid 38d lacking the lipid chain.
Reutericyclin exhibits potent activity against a broad range of Gram-positive bacteria, including B. cereus, B. subtilis, E. faecalis, S. aureus, Lactobacillus spp., Weissella confusa and clinical isolates of E. faecium (MIC = 0.06–6.5 μg mL−1)71 as well as pathogens associated with topical infections such as mupirocin-resistant MRSA (MIC = 0.8–3.12 μg mL−1),72 macrolide-resistant Streptococcus pyogenes (MIC = 0.012–0.4 μg mL−1)72 and Clostridium difficile (MIC = 0.09–0.38 μg mL−1).71,281 Gram-negative bacteria, yeast, and fungi are resistant to reutericyclin.71 The natural reutericyclin exhibits slightly higher antibacterial activity compared to the synthetic reutericyclin racemate, indicating that the stereochemistry is vital to the compound's bioactivity.282 Reutericyclin is an amphiphilic molecule consisting of a hydrophilic negatively charged group and two hydrophobic side chains. Thus, it acts as a proton ionophore and targets the cytoplasmic membrane causing dissipation of the transmembrane proton potential (ΔpH) in sensitive cells.72,73,283 SAR revealed that substitution of these hydrophobic groups with polar or charged substituents diminishes the antibacterial activity. The loss of activity in polar-substituted reutericyclins is probably due to the decreased interaction with the hydrophobic regions of the bacterial membrane.283 Although the in vitro profile of reutericyclin 38a is comparable to the antibiotic mupirocin, it's in vivo activity in S. aureus murine infection model is 5-fold weaker compared to the antibiotic. The primary factor that may decrease the efficacy of 38ain vivo is likely the slow partitioning of the aqueous dermis by the highly lipophilic reutericyclin molecules.72 Reutericyclin is cytotoxic towards Vero epithelial cells and causes hemolysis in mammalian cells.283 Conversely, modifications of the substituents in the N-substituted position has shown to modulate the cytopathic effects of this class of compounds.284 Mutanocyclin 39 consisting mainly of the tetramic acid core lacks antibacterial activity, demonstrating that the presence of the appropriate ring moieties plays a critical role in the bioactivity.30,275,276,284 Taken together, reutericyclins appear to be potent candidates for controlling recalcitrant skin infections caused by Gram-positive pathogens. Further medicinal chemistry optimization efforts are necessary to discover reutericyclin-based chemotypes with reduced toxicity whilst retaining or increasing antibacterial activity.
The production of reutericyclin 38a in sourdough is thought to inhibit other competing Gram-positive competitor L. sanfranciscensis while enabling the stable persistence of the producing organism L. reuteri. A wide variety of food-related spoilage pathogens is inhibited by reutericyclin. Hence, reutericyclin-producing strains may find application in food preservation and fermentations.68
In S. mutans, the tetramic acids reutericyclins 38a–d and mutanocyclin 39 are found to inhibit the growth of healthy oral microbes, suggesting that the pathogen likely use these molecules to remove the bacteria that block its growth to further cause severe dental caries.30 The findings lay a foundation for the continued exploration of antibiotic-producing strains within the complex competing microbial niche of the human microbiota.
4.4 Ribosomally synthesized and post-translationally modified peptides (RiPPs)
Ribosomally synthesised and post-translationally modified peptides (RiPPs) are a large class of structurally diverse natural products (Fig. 17 and Table S1†). RiPPs are produced from a short precursor peptide comprising of a leader peptide and a core peptide. Biosynthesis begins with the synthesis of a precursor peptide by the ribosome. Then, the core peptide is subject to post-translational modifications (PTMs) beyond the 20 canonical amino acids; many of which are guided by leader peptides and recognition sequences to install a wide variety of unusual structural features onto the peptide backbone. Such PTMs can often render significant advantages over unmodified linear peptides, including enhanced target affinity and stability, as well as resistance to proteolytic degradation. Following modifications, the leader peptide and recognition sequences are cleaved by proteolysis, sometimes concomitant with cyclisation of the polypeptide chain, to produce the mature active product. In some cases, additional post-translational modifications occur after cleavage of the flanking sequences. For further information and perspectives on RiPP biosynthesis, we direct the readers to several recent reviews.285,286 Numerous ribosomally-synthesised bacteriocins have been isolated from pathogenic bacteria, and they have been the subject of several different reviews.25,287,288 In this section, we highlight those interesting antibiotic RiPPs with unusual PTMs from bacterial pathogens such as darobactin 45, bottromycin 46, and nocardithiocin 47 (Fig. 17 and Table S1†).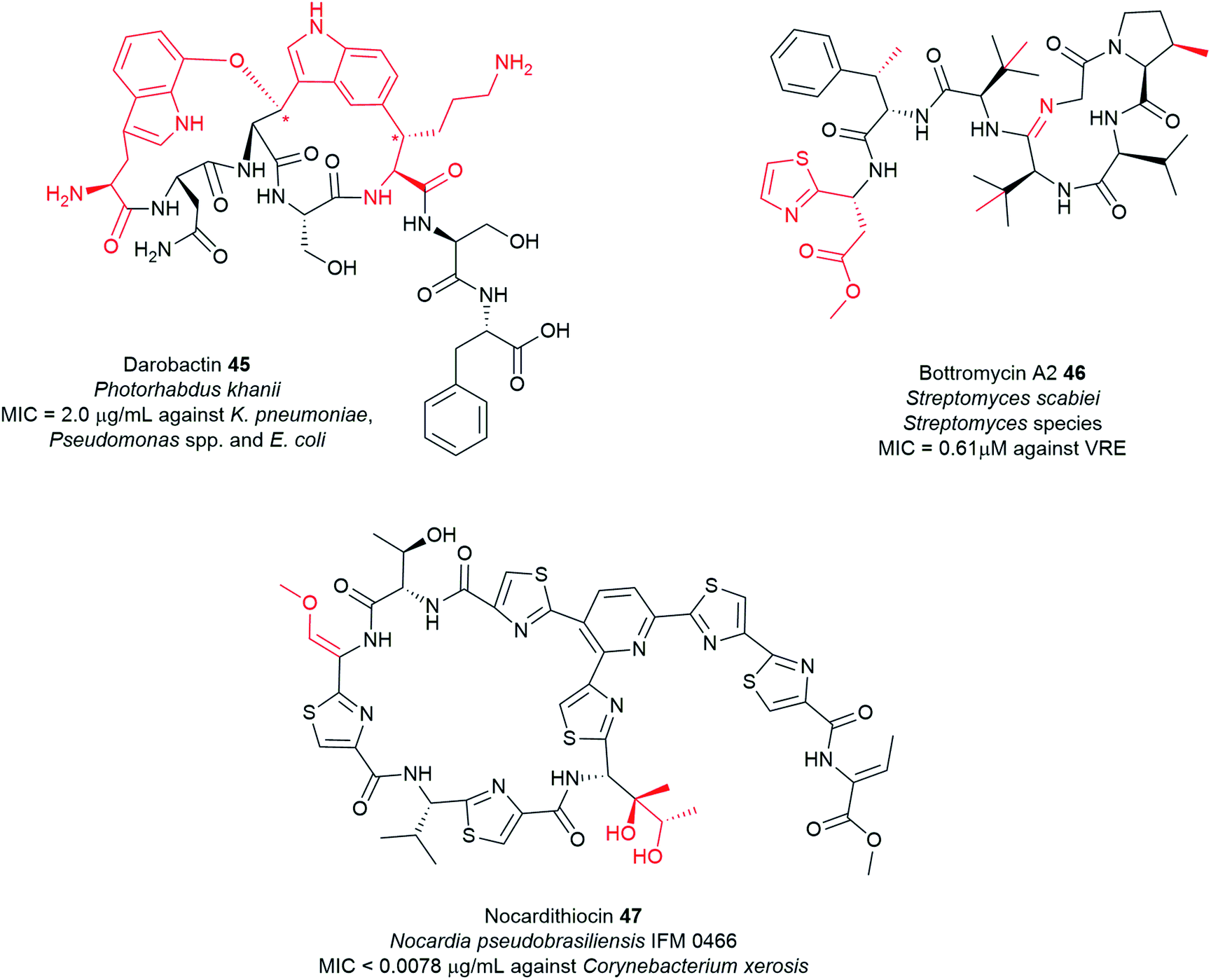 | ||
| Fig. 17 Examples of antimicrobial ribosomal peptides with unusual motifs highlighted in red from pathogenic bacteria. The stereogenic centres of 45 labelled in star (*) were deduced from DFT calculations and ROESY correlations. | ||
The putative BGC involved in darobactin 45 biosynthesis consists of a propeptide DarA, transporters DarB and DarD, membrane fusion protein DarC, and a RaS enzyme DarE. Deletion of the dar operon in P. khanii DSM3369 by double crossover abrogated production of 45. Notably, heterologous expression of the dar BGC into E. coli produced the peptide suggesting that the dar is sufficient for darobactin production. DarE showed little homology to StrB, nonetheless, DarE harbours the rSAM and SPASM/Twitch domains that are characteristic of this diverse protein superfamily. It is speculated that DarE catalyses the versatile formation of Lys-to-Trp macrocyclic crosslink in darobactin. Recently, a novel rSAM enzyme TqqB has been shown to install a C–O–C Thr–Gln ether cross-link.291 The dar operon does not encode a separate putative enzyme that incorporates the C–O–C Trp–Trp ether bond. It was speculated that DarE may not only catalyse the linkage of the Trp–Lys C–C bond but also the formation of the aromatic–aliphatic ether linkage in darobactin. RiPP operons often encode a protease that cleaves out the mature peptide; however, this is not present in the dar operon. Generic proteolysis is presumed to be involved in the maturation of the propeptide.289 It is anticipated that structural and biochemical investigations of the novel darobactin enzymatic system will further expand the repertoire of rSAM enzymes and will aid future engineering efforts of RiPP natural products.
Darobactin 45 is effective against multiple Gram-negative bacteria in vitro, including drug-resistant human pathogens such as polymyxin-resistant P. aeruginosa and extended-spectrum β-lactam-resistant K. pneumoniae and E. coli and carbapenem-resistant clinical isolates (MIC = 2–64 μg mL−1). It exhibits better efficacy in several mouse septicaemia infection models than the antibiotic gentamicin. Darobactin, however, showed little to no activity on Gram-positive bacteria, gut commensals including Bacteroides and human cell lines (HepG2, FaDu, HEK293) up to 128 μg mL−1 concentration.289
Gram-negative bacteria are difficult to treat due to their double-membrane cell wall, which forms a protective barrier from antibiotics.293 The outer membrane contains a layer of negatively charged lipopolysaccharides in addition to proteins and phospholipids that blocks the entry of large and hydrophobic molecules.9 The cut-off size for compounds that can penetrate the membrane is about 600 Da. Given the size of darobactin (966 Da), it cannot breach this permeability barrier but instead acts on the surface of the cell. Darobactin binds to the β-barrel assembly machine (BAM) A protein and induces the closed-gate conformation, thereby preventing the normal protein folding and membrane insertion necessary for bacterial survival.289 The discovery of darobactin 45 offers a promising lead in the dwindling pipeline of antibiotics that selectively target the Gram-negatives. Currently, darobactin 45 is in pre-clinical stage.9
The production of darobactin 45 in large amounts remains a challenge. Heterologous expression in different Photorhabdus species yielded highest in P. khanii DSM 3369 strain (3 mg L−1),289 yet the production titre is still low. The complexity of the structure and the stereochemistry make it difficult to be obtained by chemical synthesis. The poor yield complicates both drug development and further biosynthetic studies of the molecule. Nonetheless, bacterial genome sequences identified further tentative analogues, darobactins B–E from Yersinia, and Photorhabdus species.289 Expression of these putative darobactins may provide insight into the structure–activity relationships (SAR) and determine the pharmacophoric regions of the molecule. The identification of the biosynthetic route of 45 should facilitate the generation of a library of darobactin-like antibiotics that selectively targets the Gram-negatives.
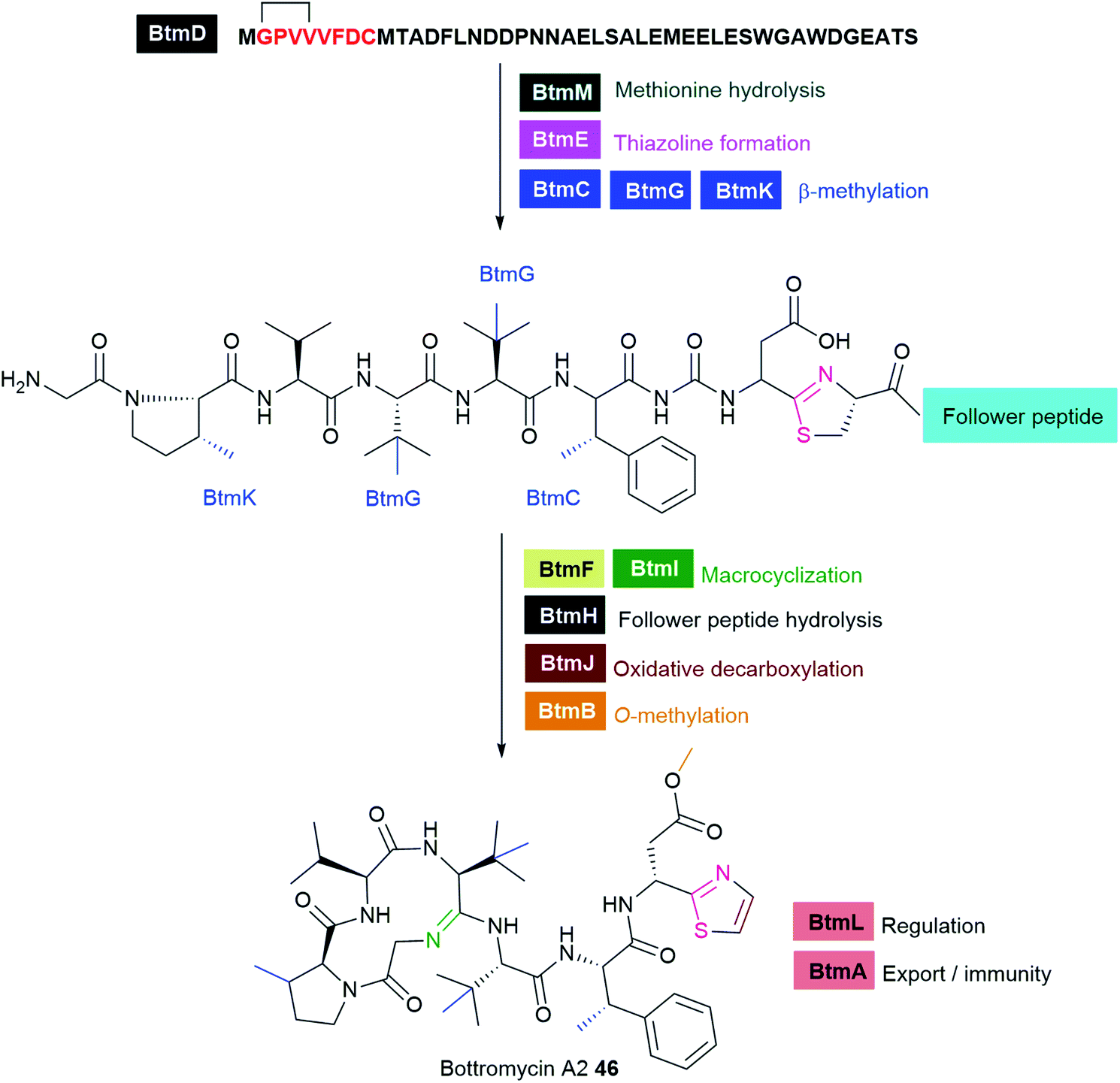 | ||
| Fig. 18 Biosynthesis of bottromycin A2 in S. scabies. | ||
Total synthesis of bottromycins and analogues enabled evaluation of their antibacterial activity.302,305–307 Bottromycin inhibits the growth of a wide range of microorganisms by blocking the binding of aminoacyl tRNAs to the A-site on the 50S ribosome, ultimately leading to inhibition of bacterial protein synthesis.308,309 The bottromycins, particularly bottromycin A2 46, display potent antibacterial activity against Gram-positive bacteria including clinically-isolated MRSA and VRE strains (MIC = 0.5–2.0 μg mL−1)305,306,310 and mycoplasma.306 A natural de-methyl analogue of 46, bottromycin B2, exhibits slightly reduced antibacterial activity (MIC = 4 μg mL−1).305 The three-dimensional structure of bottromycin is essential for the antibacterial activity while the thiazole and methyl ester moieties are not required.305
The development of bottromycin as an antibacterial drug is impeded by the reduced in vivo efficacy in MRSA-infected mice. This reduced efficacy is mainly due to the instability of the terminal methyl ester moiety, which undergoes rapid hydrolysis to carboxylic acid in blood plasma rendering it inactive. Notably, the substitution of this ester with a ketone functionality resulted in potent and stable analogues with improved pharmacological properties and superior in vivo efficacy in the mouse infection model than 46.306 Therefore, further structural optimization of bottromycin-based compounds via engineering of the biosynthetic pathway or chemical synthesis offers promising leads for the development of new bottromycin-based anti-infectives. Recently, yeast-mediated pathway engineering of the bottromycin BGC through an inducible, theophylline-controlled riboswitch system led to an overall 120-fold increase in pathway productivity in a heterologous Streptomycete host.311 Another approach involved promoter exchange that resulted in 5–50 fold higher productivity of a suite of new bottromycin-related compounds compared to the wild type strain.312 Application of these strategies to turn-up or upregulate biosynthetic pathways that are involved in controlling metabolic yields will undoubtedly facilitate the discovery of known NPs and new bioactive NPs in Actinobacteria.
Biosynthesis of nocardithiocin 47 is proposed to be directed by a 12 gene-cluster (notA–L), with NotG as the precursor peptide (Fig. 19).315 The characteristic pyridine core in 47 is likely formed by macrocyclization of the precursor peptide at sites Ser1 and Ser10. The structure of 47 features isoleucine (Ile8) bearing two hydroxy moieties resembling PTMs observed in thiostrepton that is probably catalysed by a cytochrome P450.316 A second P450 is predicted to hydroxylate a dehydroalanine (Dha4), similar to those observed in berninamycin compounds,317 which is subsequently methylated by a putative methyltransferase (NotC or NotE). Another methyltransferase (NotC or NotE) likely installs a methyl group at the C-terminus.315
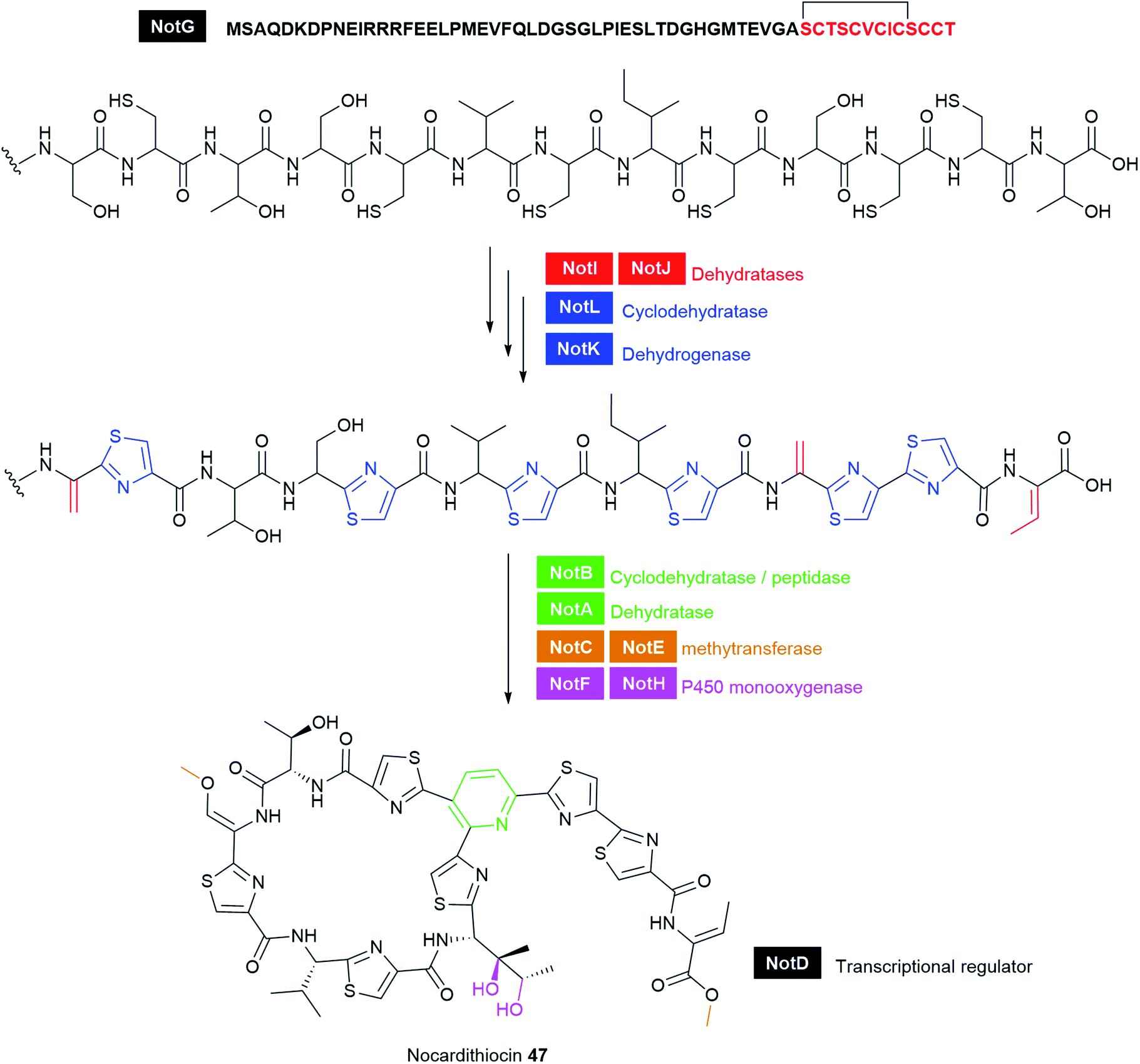 | ||
| Fig. 19 Proposed biosynthetic pathway of nocardithiocin 47. | ||
Nocardithiocin 47 exhibits potent bacteriostatic activity against a variety of bacteria including Corynebacterium xerosis (MIC < 0.0078 μg mL−1), M. smegmatis (MIC = 0.062 μg mL−1), Nocardia asteroides (MIC = 0.062 μg mL−1), and Gordonia bronchialis (MIC = 0.03 μg mL−1). It is also highly active against rifampicin-resistant bacteria as well as -sensitive M. tuberculosis strains, and most of the resistant strains were inhibited at concentrations ranging from 0.025 to 1.56 μg mL−1.313 Despite the impressive antibiotic activity of 47, its clinical use is hampered by poor aqueous solubility and light instability.313,314 The identification of the nocardithiocin BGC expands the possibility for further structural modifications to generate stable analogues with improved pharmacokinetic properties. Genetic modification of the nocardithiocin scaffold via substitution of Val6 of the core peptide by ten mostly hydrophobic amino acids yielded nocardithiocin analogues, two of which showed improved MIC against a panel of Gram-positive bacteria. Furthermore, nocardithiocin and all analogues were stable to light. However, they remained poorly water-soluble.318 Introduction of polar groups at the tail end of the nocathiacin thiopeptide enhanced its water-solubility while retaining its potent in vitro and in vivo antibacterial activity.319 Similar tail modifications could also improve the solubility of nocardithiocin without diminishing antibacterial potency.
4.5 Other categories of metabolites from pathogens
Most of the PKS and peptidyl compounds discussed in this review can be classified based on their biosynthetic class. Other antimicrobial metabolites, including nucleosides, indoles, guanine, β-lactams, and carbapenems will be covered in this section, highlighting those with remarkable activity.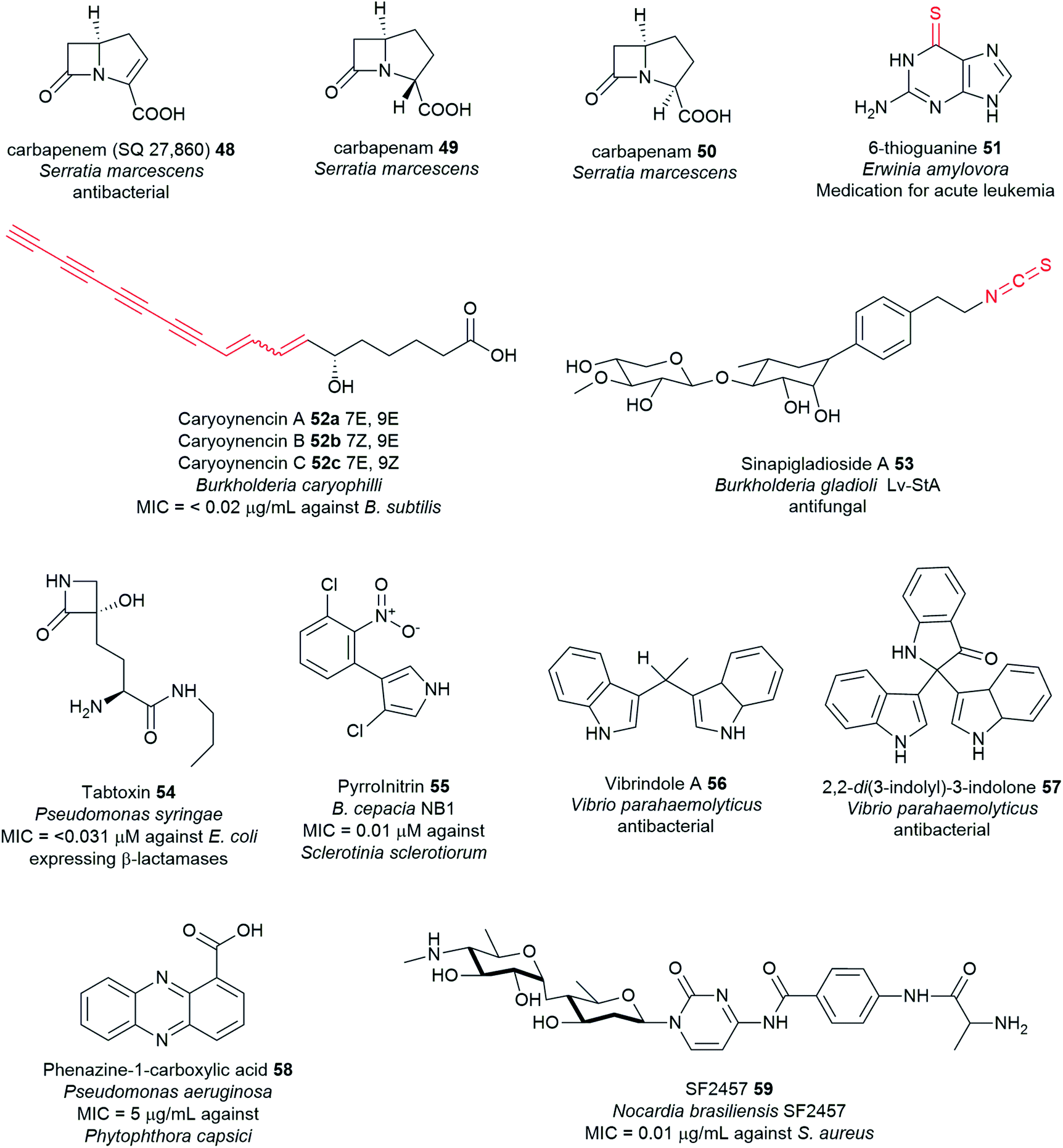 | ||
| Fig. 20 Other categories of metabolites discovered from pathogens. | ||
Carbapenems have broad-spectrum activity against important Gram-positive and Gram-negative pathogens, particularly nosocomial multidrug-resistant bacteria.321,328 Furthermore, they exhibit potent antibacterial and β-lactamase-inhibitory activity.328,329S. marcescens makes the simplest known β-lactam antibiotic containing only the bicyclic nucleus, 1-carbapen-2-em-3-carboxylic acid (SQ 27860) 48 and two saturated diastereomers, (3R,5R)- and (3S,5R)-carbapenam-3-carboxylic acids 49–50 (Fig. 20 and Table S1†).320,330–332 Gram-negative enteric Erwinia strains also produce carbapenem 48 and carbapenams 49–50, and later 48 was identified as a metabolite of the entomopathogen, P. luminescens.330–332 Unlike 48, these carbapenams 49–50 lack antibacterial activity but are resistant to β-lactamases I and II from B. cereus.332 Carbapenem 48 is a potent antibacterial, but it is highly unstable and requires initial derivatization to the p-nitrobenzyl ester for isolation. Carbapenem 48 is active against several strains of S. aureus, E. coli, and Enterobacter cloacae.331
The biosynthesis of thioguanine 51 has been recently elucidated in E. amylovora and is encoded by the ycf gene cluster (Fig. 21A). The rare thioamide moiety in thioguanine is likely derived from the action of two key enzymes YcfA and YcfC which constitutes a bipartite enzyme system, unique from those previously described thionation in RNA systems (Fig. 21B).337,339 The ATP-dependent YcfA enzyme catalyses the transfer of sulfur onto the guanine backbone339 and uses a pyridoxal phosphate (PLP)-dependent specialised sulfur shuttle enzyme, YcfC that functions independently from the general sulfur mobilization pathways.337 While the sulfur source in universal RNA-systems often originates from L-cysteine through the action of cysteine desulfurases (IscS),340 the cysteine-derived sulfur nucleophile in thioguanine biosynthesis is provided by YcfC and then, transferred and bound onto one of the cysteine residues (Cys113) of the YcfA active site.337,339 Meanwhile, no thionated products were detected using the IscS homologue (Ea-IscS)-catalysed reaction in E. amylovora.337 YcfA initially activates the guanine backbone by adenylating the carbonyl oxygen prior to thionation.339 Subsequent YcfA-mediated sulfur transfer to the activated substrate generates the thioamides with concomitant release of adenosine monophosphate (AMP).337,339
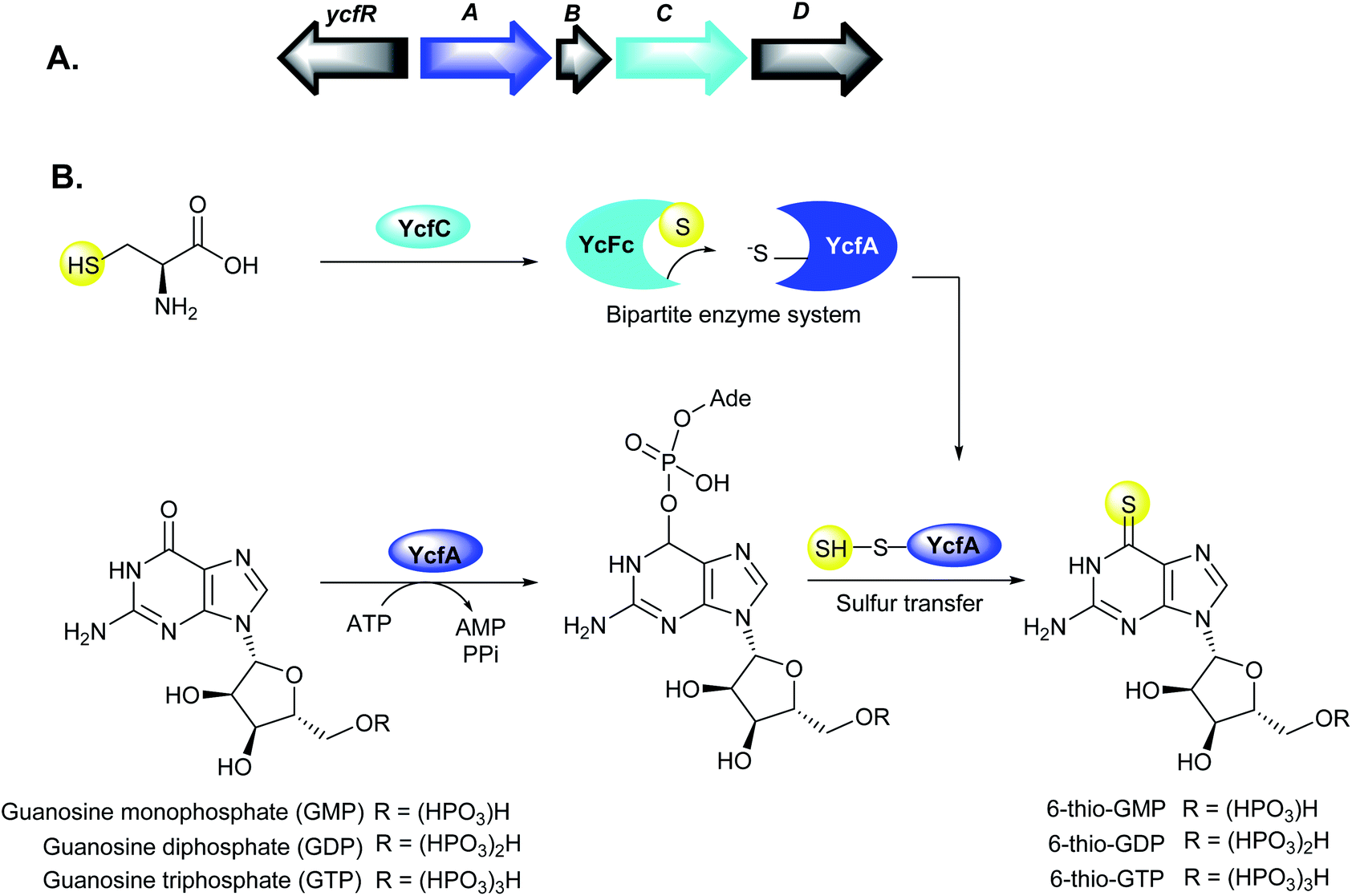 | ||
| Fig. 21 (A) Organization of the ycf gene cluster in Erwinia amylovora, and (B) model for the bipartite enzyme system, YcfA and YcfC for the oxygen-to-sulfur substitution in thioamide formation. | ||
In addition to its anticancer properties,334 thioguanine 51 is bacteriostatic towards E. coli338 and strains of Salmonella typhimurium and Pantoea agglomerans,341 with the resumption of cell growth after prolonged incubation. At 0.25 μM concentration, thioguanine completely inhibited the growth of E. coli strains B and K12.341 This inhibition did not occur when either adenine or guanine was present in the assay medium.338 Growth inhibition was also reported for B. subtilis,342,343 and the abolishment of flagella formation for B. cereus.344 Other organisms like Streptococcusfaecium strain, S. cerevisiae, Rahnella aquatilis, and Gibbsiella sp. (strain BK1) were insensitive to thioguanine.50,341,345
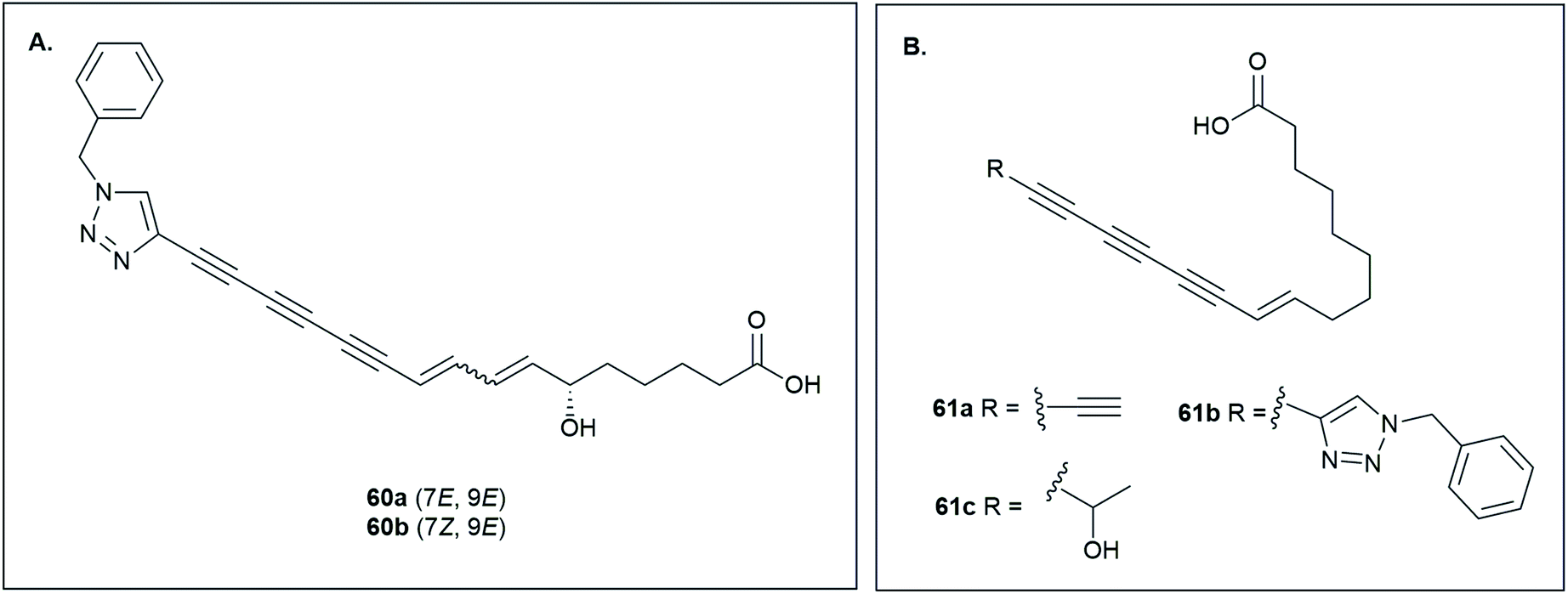 | ||
| Fig. 22 Chemical trapping of the tetraynes by an in situ copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) click reaction. (A) Structures of triazoles 60a–b produced from B. caryophylli wild type after click reaction, and (B) structures of 61a from B. caryophylli ΔcayG and 61b after click reaction, and 61c shunt product. | ||
Transposon mutagenesis and genome sequencing of B. caryophilli (DSM50341) have provided the first insight into the unusual polyyne biosynthesis in bacteria which involves novel desaturases and a cytochrome P450 monooxygenase (Fig. 23A). Disruption of the transposon site points to a Δ9 desaturase-like gene, orthologs of which were identified in the genomes of the plant pathogens Burkholderia gladioli BSR3 and B. gladioli pv. cocovenenans. Comparative genomics analyses further revealed that the caryoynencin (cay) locus is conserved in several Burkholderia strains, and homologous gene clusters were also identified in various other bacteria.347 Metabolomic analyses also revealed that strain BSR3 and B. gladioli Lv-StA are capable of caryoynencin production.347,349 The characterisation of the cay BGC will thus facilitate the discovery of numerous polyyne-bearing NPs from bacteria and lead to the expansion of the polyyne biosynthetic machinery capable of producing polyacetylenes.
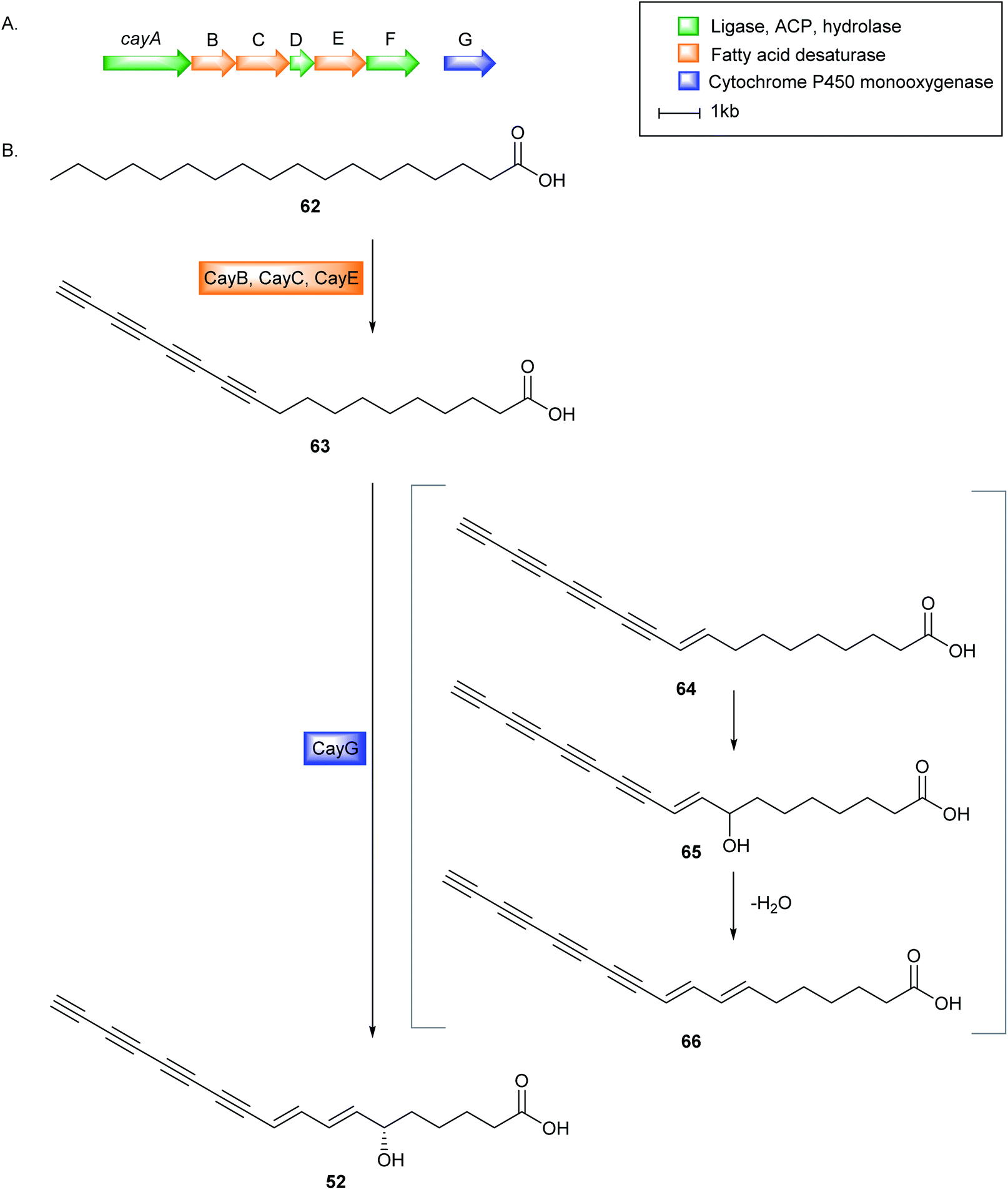 | ||
| Fig. 23 (A) Biosynthetic gene cluster of caryoynencin (cay) in B. caryophilli, and (B) Proposed biosynthetic pathway of caryoynencin. | ||
Caryoynencin is likely derived from fatty acid-ACP, followed by desaturation to yield the alkyne motifs, 62 (Fig. 23B). Three putative desaturase genes (cayB, cayC, cayE) were implicated to be responsible for the incorporation of the triple bonds. Phylogenetic analyses have revealed that the CayBCE desaturases are unique and have probably evolved independently from those found in fungi, plants, and insects.347 Notably, CayB and CayC form a separate clade with the closest desaturase homologue JamB,350 which has been suggested to introduce the terminal alkyne functionality in the jamaicamide pathway of Moorea producens. Subsequent hydroxylation and elimination reactions of 62 catalysed by the putative cytochrome P450 (CayG) generates the allylic alcohol moiety in caryoynencin 52. Formation of the triazole 61b lacking a hydroxyl group in ΔcayG mutant after an in situ click reaction supports the plausible function of CayG. Compound 61c with an alcohol moiety instead of a triple bond is likely a shunt product of terminal alkyne formation.347
Caryoynencin has been shown to possess outstanding activity than the antibiotic kanamycin A. It is active against a wide range of bacteria and fungi including E. coli (MIC = 0.63 μg mL−1), K. pneumoniae (MIC = 0.04 μg mL−1), S. aureus (MIC = 0.02 μg mL−1), B. subtilis (MIC = <0.02 μg mL−1), C. albicans (MIC = 0.05 μg mL−1), and several Trichophyton species (MIC = 0.02–0.05 μg mL−1).346,351 Owing to their high instability, synthetic approaches have been developed to gain more insight into their structure and function.352,353 The terminal alkyne and the hydroxy group were crucial for the antibacterial activity while the diene motif and the butanoic acid were not essential.351,353 The triazole 60b is active against B. subtilis (MIC = 3.12 μg mL−1) and MRSA (12.5 μg mL−1),346 consistent with earlier studies that the tetrayne molecules were more potent than the corresponding triyne or diyne analogues.351 Remarkably, the introduction of a trimethylsilyl motif generates stable polyacetylene derivatives with potent antibacterial activity. Hydrophilicity also plays an important role in the bioactivity, and thus conjugates of polyynes and sugars, amino acids, and nucleic acids are attractive molecules.353
5. Conclusions and future perspective
Pathogenic bacteria have an enormous yet unexploited potential for natural product drug discovery. Entomopathogenic, phytopathogenic, and human and animal pathogenic bacteria produce a repertoire of novel potential therapeutics, with an assortment of unprecedented structures, activities, and modes of action. Some of them are in pre-clinical trials (darobactin, NOSO-95C analogue) or have huge potential for drug development. Darobactin, odilorhabdin, and albicidin are promising candidates in the dwindling pipeline of antibiotics that selectively target the Gram-negatives.Although darobactin is a potent drug lead for Gram-negative bacteria, the greater bottleneck is to produce it in large amounts for pre-clinical and clinical development. Moreover, the complexity and stereochemistry of darobactin make it difficult to be obtained by chemical synthesis, and thus SAR studies to determine the key bioactive moieties remain a challenge. Nevertheless, modifications of the complex vancomycin antibiotic have been achieved by several groups to produce potent analogues with less propensity to antibiotic resistance.354 Lead optimisation of the odilorhabdin scaffold was also achieved and identified NOSO-502 as a clinical candidate for carbapenem-resistant Enterobacteriaceae.128 Several other metabolites that possess potent in vitro activity can be chemically-modified to increase in vivo efficacy and further enhance pharmacokinetic properties without diminishing activity such as bottromycin, althiomycin, caryoynencin, nocardithiocin, lugdunin, nematophin, holomycin, and reutericyclin. The NPs covered in this review could be clinical leads or could provide structural templates for further medicinal chemistry optimisation efforts.
The ecological functions of currently known NPs in pathogenic bacteria remain to be deciphered. Understanding this role might be key to determining their potential use. For example, clostrubins which serve dual functions – kills potential microbial competitors and permits survival of the pathogenic anaerobe in an oxygen-rich potato niche – represent promising leads for the design and development of antibacterial therapeutics and plant protection agents.47 Impairing clostrubin production in C. puniceum could also help prevent potatoes from “soft rot”.
Given that some pathogenic bacteria are a threat to humans, how can one prioritize natural products discovery from these huge untapped resources? Developments in culture-independent meta-omics approaches have provided greater access to underinvestigated taxa that contain unique metabolic profiles that probably encode novel chemistry. The exploitation of these metagenomic data has proven to be beneficial in the characterisation and isolation of cryptic metabolites from the complex human microbiota.355–357 For example, the thiopeptide antibiotic lactocillin was discovered via a sequence-based metagenomic analysis.358 Such techniques can be exploited to identify potential genetic markers of disease in NP producing bacterial pathogens, thereby circumventing the threat of opportunistic infections caused by pathogenic bacteria. Metagenomic approaches also provide a means to access novel bioactive molecules with diverse structures. Integrating other emerging techniques in these NP discovery efforts, such as elicitation of cryptic biosynthetic pathways and refactoring of silent BGCs should help illuminate the chemical “dark matter” in bacterial pathogens. The substitution of native promoters with strong constitutive promoters in cryptic gene clusters has been one such productive strategy in activating biosynthesis and improving antibiotic expression. For example, promoter exchange in Photorhabdus spp. and Xenorhabdus spp. led to the expression of several cryptic nonribosomal peptides. Additionally, promoter exchange in Δhfq mutants resulted in the production of desired metabolites for further bioactivity testing.151 The development of high-throughput next-generation sequencing methods, together with the development of new bioinformatics tools that can assemble nearly complete genomes, will continue to revolutionize microbial “dark matter” exploration. Furthermore, improvements in analytical platforms (mass spectrometry, NMR) coupled with recent advancements in metabolomics enable the detection and identification of compounds in minute quantities from complex biological samples.359–361 Application of recent machine learning tools for structure recognition, bioactivity prediction, drug–target interactions362 such as the NMR-based Small Molecule Accurate Recognition Technology (SMART 2.0)363 further accelerates the drug discovery process.
Taken together, access to the immense repertoire of novel cryptic metabolites encoded in pathogenic bacteria is only achievable through improvements in, and integration of, various approaches and available methods from multiple disciplines. Further exploitation of the untapped chemical diversity of pathogenic bacteria will undoubtedly yield many more novel bioactive molecules and might reboot the antibiotic pipeline. Soon, we can predict a second “Golden Era of Antibiotics” discovery.
6. Conflicts of interest
The authors declare no conflicts of interest.7. Acknowledgements
F. M. thanks the University of the Philippines for the Faculty, Reps and Staff Development Program (FRASDP) for the Ph.D. grant fellowship. H. D. thanks the financial supports of Biotechnology and Biological Sciences Research Council UK (BB/P00380X/1, BB/R00479X/1 and BB/R50547X/1). H. D. and Y. Y. thank the Royal Society-NSFC Newton Mobility Grant Award (IEC\NSFC\170617). Y. Y. thanks the National Natural Science Foundation of China (31870035, 31570033, and 31811530299). We thank Dr Kirstie Rickaby (University of Aberdeen) for help with collection of literatures in the beginning of the manuscript preparation.8. References
- Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics, World Health Organization, Geneva Switz., 2017, pp. 1–7 Search PubMed.
- C. Willyard, Nature, 2017, 543, 15 CrossRef CAS.
- A. P. Zavascki, L. Z. Goldani, J. Li and R. L. Nation, J. Antimicrob. Chemother., 2007, 60, 1206–1215 CrossRef CAS.
- Y. Y. Liu, Y. Wang, T. R. Walsh, L. X. Yi, R. Zhang, J. Spencer, Y. Doi, G. Tian, B. Dong, X. Huang, L. F. Yu, D. Gu, H. Ren, X. Chen, L. Lv, D. He, H. Zhou, Z. Liang, J. H. Liu and J. Shen, Lancet Infect. Dis., 2016, 16, 161–168 CrossRef CAS.
- A. R. Coates, G. Halls and Y. Hu, Br. J. Pharmacol., 2011, 163, 184–194 CrossRef CAS.
- J. L. Martínez and F. Baquero, Upsala J. Med. Sci., 2014, 119, 68–77 CrossRef.
- R. A. Bonomo, Cold Spring Harbor Perspect. Med., 2017, 7, 1–15 Search PubMed.
- A. Luther, M. Urfer, M. Zahn, M. Müller, S. Y. Wang, M. Mondal, A. Vitale, J. B. Hartmann, T. Sharpe, F. Lo Monte, H. Kocherla, E. Cline, G. Pessi, P. Rath, S. M. Modaresi, P. Chiquet, S. Stiegeler, C. Verbree, T. Remus, M. Schmitt, C. Kolopp, M. A. Westwood, N. Desjonquères, E. Brabet, S. Hell, K. LePoupon, A. Vermeulen, R. Jaisson, V. Rithié, G. Upert, A. Lederer, P. Zbinden, A. Wach, K. Moehle, K. Zerbe, H. H. Locher, F. Bernardini, G. E. Dale, L. Eberl, B. Wollscheid, S. Hiller, J. A. Robinson and D. Obrecht, Nature, 2019, 576, 452–458 CrossRef CAS.
- K. Lewis, Cell, 2020, 181, 29–45 CrossRef CAS.
- E. M. Molloy and C. Hertweck, Curr. Opin. Microbiol., 2017, 39, 121–127 CrossRef.
- F. Baldeweg, D. Hoffmeister and M. Nett, Nat. Prod. Rep., 2019, 36, 307–325 RSC.
- Y. M. Shi and H. B. Bode, Nat. Prod. Rep., 2018, 35, 309–335 RSC.
- S. J. Pidot, S. Coyne, F. Kloss and C. Hertweck, Int. J. Med. Microbiol., 2014, 304, 14–22 CrossRef CAS.
- J. Mansfield, S. Genin, S. Magori, V. Citovsky, M. Sriariyanum, P. Ronald, M. A. X. Dow, V. Verdier, S. V Beer, M. A. Machado, I. A. N. Toth, G. Salmond, G. D. Foster, I. P. Lipm and F.-C. Tolosan, Mol. Plant Pathol., 2012, 13, 614–629 CrossRef.
- Y. Ichinose, F. Taguchi and T. Mukaihara, J. Gen. Plant Pathol., 2013, 79, 285–296 CrossRef CAS.
- J. A. Banas, Front. Biosci., 2004, 9, 1267–1277 CrossRef CAS.
- A. Wrobel, J. C. Leo and D. Linke, Genes, 2019, 10, 1–20 CrossRef.
- K. Becker, C. Heilmann and G. Peters, Clin. Microbiol. Rev., 2014, 27, 870–926 CrossRef.
- K. R. Soumya, S. Philip, S. Sugathan, J. Mathew and E. K. Radhakrishnan, 3 Biotech, 2017, 7, 1–10 Search PubMed.
- S. J. Hinchliffe, M. C. Hares, A. J. Dowling and R. H. Ffrench-Constant, Open Toxicol. J., 2010, 3, 83–100 CAS.
- D. R. D. Bignell, J. K. Fyans and Z. Cheng, J. Appl. Microbiol., 2014, 116, 223–235 CrossRef CAS.
- A. Rodou, D. O. Ankrah and C. Stathopoulos, Toxins, 2010, 2, 1250–1264 CrossRef CAS.
- J. W. Wilson, M. J. Schurr, C. L. LeBlanc, R. Ramamurthy, K. L. Buchanan and C. A. Nickerson, Postgrad. Med. J., 2002, 78, 216–224 CrossRef CAS.
- N. J. Tobias, Y. M. Shi and H. B. Bode, Trends Microbiol., 2018, 26, 833–840 CrossRef CAS.
- L. L. Newstead, K. Varjonen, T. Nuttall and G. K. Paterson, Antibiotics, 2020, 9, 1–19 CrossRef.
- J. Dreyer, A. P. Malan and L. M. T. Dicks, Front. Microbiol., 2018, 9, 1–14 CrossRef.
- S. Müller, E. Garcia-Gonzalez, E. Genersch and R. D. Süssmuth, Nat. Prod. Rep., 2015, 32, 765–778 RSC.
- S. Kunakom and A. S. Eustáquio, J. Nat. Prod., 2019, 82, 2018–2037 CrossRef CAS.
- A. Zipperer, M. C. Konnerth, C. Laux, A. Berscheid, D. Janek, C. Weidenmaier, M. Burian, N. A. Schilling, C. Slavetinsky, M. Marschal, M. Willmann, H. Kalbacher, B. Schittek, H. Brötz-Oesterhelt, S. Grond, A. Peschel and B. Krismer, Nature, 2016, 535, 511–516 CrossRef CAS.
- X. Tang, Y. Kudo, J. L. Baker, S. Labonte, P. A. Jordan, S. M. K. McKinnie, J. Guo, T. Huan, B. S. Moore and A. Edlund, ACS Infect. Dis., 2020, 6, 563–571 CrossRef CAS.
- Z. Qin, A. T. Baker, A. Raab, S. Huang, T. Wang, Y. Yu, M. Jaspars, C. J. Secombes and H. Deng, J. Biol. Chem., 2013, 288, 14688–14697 CrossRef CAS.
- H. Deng and C. J. Secombes, Virulence, 2014, 5, 9–11 CrossRef.
- D. Dhakal, V. Rayamajhi, R. Mishra and J. K. Sohng, J. Ind. Microbiol. Biotechnol., 2019, 46, 385–407 CrossRef CAS.
- V. L. Challinor and H. B. Bode, Ann. N. Y. Acad. Sci., 2015, 1354, 82–97 CrossRef.
- Z. Hu and W. Zhang, ACS Infect. Dis., 2020, 6, 25–33 CrossRef CAS.
- H. B. Bode, Curr. Opin. Chem. Biol., 2009, 13, 224–230 CrossRef CAS.
- S. Forst, B. Dowds, N. Boemare and E. Stackebrandt, Annu. Rev. Microbiol., 1997, 51, 47–72 CrossRef CAS.
- M. L. Pineda-Castellanos, Z. Rodríguez-Segura, F. J. Villalobos, L. Hernández, L. Lina and M. Eugenia Nuñez-Valdez, Pathogens, 2015, 4, 210–228 CrossRef.
- P. A. D. Grimont and F. Grimont, Annu. Rev. Microbiol., 1978, 32, 221–248 CrossRef CAS.
- A. Fünfhaus, J. Ebeling and E. Genersch, Curr. Opin. Insect. Sci., 2018, 26, 89–96 CrossRef.
- A. Soenens and J. Imperial, Phytochem. Rev., 2020, 19, 577–587 CrossRef CAS.
- N. Vodovar, D. Vallenet, S. Cruveiller, Z. Rouy, V. Barbe, C. Acosta, L. Cattolico, C. Jubin, A. Lajus, B. Segurens, B. Vacherie, P. Wincker, J. Weissenbach, B. Lemaitre, C. Médigue and F. Boccard, Nat. Biotechnol., 2006, 24, 673–679 CrossRef CAS.
- J. F. Challacombe, M. R. Altherr, G. Xie, S. S. Bhotika, N. Brown, D. Bruce, C. S. Campbell, M. L. Campbell, J. Chen, O. Chertkov, C. Cleland, M. Dimitrijevic, N. A. Doggett, J. J. Fawcett, T. Glavina, L. A. Goodwin, L. D. Green, C. S. Han, K. K. Hill, P. Hitchcock, P. J. Jackson, P. Keim, A. R. Kewalramani, J. Longmire, S. Lucas, S. Malfatti, D. Martinez, K. McMurry, L. J. Meincke, M. Misra, B. L. Moseman, M. Mundt, A. C. Munk, R. T. Okinaka, B. Parson-Quintana, L. P. Reilly, P. Richardson, D. L. Robinson, E. Saunders, R. Tapia, J. G. Tesmer, N. Thayer, L. S. Thompson, H. Tice, L. O. Ticknor, P. L. Wills, P. Gilna and T. S. Brettin, J. Bacteriol., 2007, 189, 3680–3681 CrossRef CAS.
- C. T. Bull, S. H. De Boer, T. P. Denny, G. Firrao, M. F. Saux, G. S. Saddler, M. Scortichini, D. E. Stead, Y. Takikawa, T. P. Denny, G. Firrao, M. F. Saux, G. S. Saddler, M. Scortichini, D. E. Stead and Y. Takikawa, J. Plant Pathol., 2010, 92, 551–592 Search PubMed.
- C. T. Bull, S. H. De Boer, T. P. Denny, G. Firrao, M. F. Saux, G. S. Saddler, M. Scortichini, D. E. Stead and Y. Takikawa, J. Plant Pathol., 2012, 94, 21–27 Search PubMed.
- C. T. Bull, T. A. Coutinho, T. P. Denny, G. Firrao, M. F. Saux, X. Li, G. S. Saddler, M. Scortichini, D. E. Stead and Y. Takikawa, J. Plant Pathol., 2014, 96, 223–226 Search PubMed.
- G. Shabuer, K. Ishida, S. J. Pidot, M. Roth, H.-M. Dahse and C. Hertweck, Plant Sci., 2015, 350, 670–675 CAS.
- I. K. Toth, J. M. van der Wolf, G. Saddler, E. Lojkowska, V. Hélias, M. Pirhonen, L. Tsror (Lahkim) and J. G. Elphinstone, Plant Pathol., 2011, 60, 385–399 CrossRef.
- M. Malnoy, S. Martens, J. L. Norelli, M.-A. Barny, G. W. Sundin, T. H. M. Smits and B. Duffy, Annu. Rev. Phytopathol., 2012, 50, 475–494 CrossRef CAS.
- G. Feistner and C. M. Staub, Curr. Microbiol., 1986, 13, 95–101 CrossRef CAS.
- C. S. Oh and S. V. Beer, FEMS Microbiol. Lett., 2005, 253, 185–192 CrossRef CAS.
- X. F. Xin, B. Kvitko and S. Y. He, Nat. Rev. Microbiol., 2018, 16, 316–328 CrossRef CAS.
- Y. Mast and E. Stegmann, Antibiotics, 2019, 8, 10–13 CrossRef.
- F. Maglangit, Q. Fang, V. Leman, S. Soldatou, R. Ebel and H. Deng, Molecules, 2019, 24, 3384 CrossRef CAS.
- R. E. de Lima Procópio, I. R. da Silva, M. K. Martins, J. L. de Azevedo and J. M. de Araújo, Braz. J. Infect. Dis., 2012, 16, 466–471 CrossRef.
- F. Maglangit, Q. Fang, K. Kyeremeh, J. M. Sternberg, R. Ebel and H. Deng, Molecules, 2020, 25, 256 CrossRef CAS.
- Q. Fang, F. Maglangit, L. Wu, R. Ebel, K. Kyeremeh, J. H. Andersen, F. Annang, G. Pérez-Moreno, F. Reyes and H. Deng, Molecules, 2020, 25, 460 CrossRef CAS.
- Q. Fang, F. Maglangit, M. Mugat, C. Urwald, K. Kyeremeh and H. Deng, Molecules, 2020, 25, 1108 CrossRef CAS.
- Y. Li, J. Liu, G. Díaz-Cruz, Z. Cheng and D. R. D. Bignell, Microbiology, 2019, 165, 1025–1040 CrossRef CAS.
- I. Pieretti, A. Pesic, D. Petras, M. Royer, R. D. Süssmuth and S. Cociancich, Front. Plant Sci., 2015, 6, 1–7 CrossRef.
- P. S. Conville, B. A. Brown-Elliott, T. Smith and A. M. Zelazny, J. Clin. Microbiol., 2018, 56, 1–10 Search PubMed.
- J. N. O'Sullivan, M. C. Rea, P. M. O'Connor, C. Hill and R. P. Ross, FEMS Microbiol. Ecol., 2019, 95, 1–10 CrossRef.
- A. F. d. S. Duarte, H. Ceotto, M. L. V. Coelho, M. A. V. d. P. Brito and M. d. C. d. F. Bastos, Food Control, 2013, 32, 313–321 CrossRef CAS.
- J. A. Lemos, S. R. Palmer, L. Zeng, Z. T. Wen, J. K. Kajfasz, I. A. Freires, J. Abranches and L. J. Brady, Microbiol. Spectrum, 2019, 7, 1–18 Search PubMed.
- K. Nakano, R. Nomura and T. Ooshima, Jpn. Dent. Sci. Rev., 2008, 44, 29–37 CrossRef.
- L. Liu, T. Hao, Z. Xie, G. P. Horsman and Y. Chen, Sci. Rep., 2016, 6, 1–10 CrossRef.
- S. S. Momeni, S. M. Beno, J. L. Baker, A. Edlund, T. Ghazal, N. K. Childers and H. Wu, J. Dent. Res., 2020, 99, 969–976 CrossRef CAS.
- M. G. Gänzle, Appl. Microbiol. Biotechnol., 2004, 64, 326–332 CrossRef.
- X. Wang, L. Du, J. You, J. B. King and R. H. Cichewicz, Org. Biomol. Chem., 2012, 10, 2044–2050 RSC.
- F. Qi, P. Chen and P. W. Caufield, Appl. Environ. Microbiol., 2001, 67, 15–21 CrossRef CAS.
- M. G. Ganzle, A. Holtzel, J. Walter, G. Jung and W. P. Hammes, Appl. Environ. Microbiol., 2000, 66, 4325–4333 CrossRef CAS.
- J. G. Hurdle, R. Yendapally, D. Sun and R. E. Lee, Antimicrob. Agents Chemother., 2009, 53, 4028–4031 CrossRef CAS.
- M. G. Gänzle and R. F. Vogel, Appl. Environ. Microbiol., 2003, 69, 1305–1307 CrossRef.
- D. Dufour, A. Barbour, Y. Chan, M. Cheng, T. Rahman, M. Thorburn, C. Stewart, Y. Finer, S. G. Gong and C. M. Lévesque, J. Bacteriol., 2020, 202, 1–14 CrossRef.
- G. Nicolas and M. Lavoie, Genome and Genomics, 2007, 1, 193–208 Search PubMed.
- M. Mota-Meira, G. LaPointe, C. Lacroix and M. C. Lavoie, Antimicrob. Agents Chemother., 2000, 44, 24–29 CrossRef CAS.
- M. Mota-Meira, H. Morency and M. C. Lavoie, J. Antimicrob. Chemother., 2005, 56, 869–871 CrossRef CAS.
- P. M. Joyner, J. Liu, Z. Zhang, J. Merritt, F. Qi and R. H. Cichewicz, Org. Biomol. Chem., 2010, 8, 5486–5489 RSC.
- M. Wietz, M. Mansson, C. H. Gotfredsen, T. O. Larsen and L. Gram, Mar. Drugs, 2010, 8, 2946–2960 CrossRef CAS.
- G. Kumar, S. Menanteau-Ledouble, M. Saleh and M. El-Matbouli, Vet. Res., 2015, 46, 1–10 CrossRef.
- S. de Keukeleire, A. de Bel, Y. Jansen, M. Janssens, G. Wauters and D. Piérard, New Microbes New Infect., 2014, 2, 134–135 CrossRef CAS.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS.
- J. Berger, L. M. Jampolsky and M. W. Goldberg, Arch. Biochem., 1949, 22, 476–478 CAS.
- Z. Cao, G. Khodakaramian, K. Arakawa and H. Kinashi, Biosci., Biotechnol., Biochem., 2012, 76, 353–357 CrossRef CAS.
- C. Olano, B. Wilkinson, C. Sanchez, S. J. Moss, R. Sheridan, V. Math, A. J. Weston, A. F. Brana, C. J. Martin, M. Oliynyk, C. Mendez, P. F. Leadlay and J. A. Salas, Chem. Biol., 2004, 11, 87–97 CAS.
- J. Sun, J. Shao, C. Sun, Y. Song, Q. Li, L. Lu, Y. Hu, C. Gui, H. Zhang and J. Ju, Bioorg. Med. Chem., 2018, 26, 1488–1494 CrossRef CAS.
- X. Gao, Y. Jiang, L. Han, X. Chen, C. Hu, H. Su, Y. Mu, P. Guan and X. Huang, RSC Adv., 2017, 7, 44401–44409 RSC.
- A. Hamed, A. S. Abdel-Razek, M. Frese, D. Wibberg, A. F. El-Haddad, T. M. A. Ibrahim, J. Kalinowski, N. Sewald and M. Shaaban, Z. Naturforsch., C: J. Biosci., 2018, 73, 49–57 CAS.
- L. Zhang, J. Shi, C. L. Liu, L. Xiang, S. Y. Ma, W. Li, R. H. Jiao, R. X. Tan and H. M. Ge, Tetrahedron Lett., 2018, 59, 4517–4520 CrossRef CAS.
- J. Kim, D. Shin, S. H. Kim, W. Park, Y. Shin, W. K. Kim, S. K. Lee, K. B. Oh, J. Shin and D. C. Oh, Mar. Drugs, 2017, 15, 1–11 Search PubMed.
- A. Sidhu, J. R. Miller, A. Tripathi, D. M. Garshott, A. L. Brownell, D. J. Chiego, C. Arevang, Q. Zeng, L. C. Jackson, S. A. Bechler, M. U. Callaghan, G. H. Yoo, S. Sethi, H. S. Lin, J. H. Callaghan, G. Tamayo-Castillo, D. H. Sherman, R. J. Kaufman and A. M. Fribley, ACS Med. Chem. Lett., 2015, 6, 1122–1127 CrossRef CAS.
- Z. Zhou, Q. Wu, Q. Xie, C. Ling, H. Zhang, C. Sun and J. Ju, Chem. Biodiversity, 2020, 17, e1900560 CAS.
- C. J. Schulze, W. M. Bray, F. Loganzo, M. Lam, T. Szal, A. Villalobos, F. E. Koehn and R. G. Linington, J. Nat. Prod., 2014, 77, 2570–2574 CrossRef CAS.
- M. Kuo, D. A. Yurek and D. A. Kloosterman, J. Antibiot., 1989, 42, 1006–1007 CrossRef CAS.
- B. F. Anderson, A. J. Herit, R. W. Rickards and G. B. Robertson, Aust. J. Chem., 1989, 42, 717–730 CrossRef CAS.
- E. M. Novoa, N. Camacho, A. Tor, B. Wilkinson, S. Moss, P. Marín-García, I. G. Azcárate, J. M. Bautista, A. C. Mirando, C. S. Francklyn, S. Varon, M. Royo, A. Cortés and L. R. De Pouplana, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5508–E5517 CrossRef CAS.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS.
- A. L. Lane, S. J. Nam, T. Fukuda, K. Yamanaka, C. A. Kauffman, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2013, 135, 4171–4174 CrossRef CAS.
- D. Habibi, N. Ogloff, R. B. Jalili, A. Yost, A. P. Weng, A. Ghahary and C. J. Ong, Invest. New Drugs, 2012, 30, 1361–1370 CrossRef CAS.
- C. Hu, H. Su, J. Luo, L. Han, Q. Liu, W. Wu, Y. Mu, P. Guan, T. Sun and X. Huang, Bioorg. Med. Chem., 2018, 26, 6035–6049 CrossRef CAS.
- S. Pidot, K. Ishida, M. Cyrulies and C. Hertweck, Angew. Chem., Int. Ed., 2014, 53, 7856–7859 CrossRef CAS.
- K. Jakobi and C. Hertweck, J. Am. Chem. Soc., 2004, 126, 2298–2299 CrossRef CAS.
- K. Wolkenstein, H. Sun, H. Falk and C. Griesinger, J. Am. Chem. Soc., 2015, 137, 13460–13463 CrossRef CAS.
- M. Yang, J. Li and A. Li, Nat. Commun., 2015, 6, 6–11 Search PubMed.
- T. Shen, X. N. Wang and H. X. Lou, Nat. Prod. Rep., 2009, 26, 916–935 RSC.
- H. B. Park, P. Sampathkumar, C. E. Perez, J. H. Lee, J. Tran, J. B. Bonanno, E. A. Hallem, S. C. Almo and J. M. Crawford, J. Biol. Chem., 2017, 292, 6680–6694 CrossRef CAS.
- S. H. Smith, C. Jayawickreme, D. J. Rickard, E. Nicodeme, T. Bui, C. Simmons, C. M. Coquery, J. Neil, W. M. Pryor, D. Mayhew, D. K. Rajpal, K. Creech, S. Furst, J. Lee, D. Wu, F. Rastinejad, T. M. Willson, F. Viviani, D. C. Morris, J. T. Moore and J. Cote-Sierra, J. Invest. Dermatol., 2017, 137, 2110–2119 CrossRef CAS.
- Q. Ma, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 401–426 CrossRef CAS.
- R. H. Cichewicz, S. A. Kouzi and M. T. Hamann, J. Nat. Prod., 2000, 63, 29–33 CrossRef CAS.
- X. Wan, X. B. Wang, M. H. Yang, J. S. Wang and L. Y. Kong, Bioorg. Med. Chem., 2011, 19, 5085–5092 CrossRef CAS.
- H. B. Park and J. M. Crawford, J. Nat. Prod., 2015, 78, 1437–1441 CrossRef CAS.
- H. B. Park, T. N. Goddard, J. Oh, J. Patel, Z. Wei, C. E. Perez, B. Q. Mercado, R. Wang, T. P. Wyche, G. Piizzi, R. A. Flavell and J. M. Crawford, Angew. Chem., Int. Ed., 2020, 59, 7871–7880 CrossRef CAS.
- J. M. Dunwell, A. Purvis and S. Khuri, Phytochemistry, 2004, 65, 7–17 CrossRef CAS.
- S. Khuri, F. T. Bakker and J. M. Dunwell, Mol. Biol. Evol., 2001, 18, 593–605 CrossRef CAS.
- C. T. Walsh, ACS Chem. Biol., 2014, 9, 1653–1661 CrossRef CAS.
- M. A. Martínez-Núñez and V. E. L. y. López, Sustainable Chem. Processes, 2016, 4, 1–8 CrossRef.
- M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield, Nat. Prod. Rep., 2016, 33, 317–347 RSC.
- L. Pantel, T. Florin, M. Dobosz-Bartoszek, E. Racine, M. Sarciaux, M. Serri, J. Houard, J. M. Campagne, R. M. de Figueiredo, C. Midrier, S. Gaudriault, A. Givaudan, A. Lanois, S. Forst, A. Aumelas, C. Cotteaux-Lautard, J. M. Bolla, C. Vingsbo Lundberg, D. L. Huseby, D. Hughes, P. Villain-Guillot, A. S. Mankin, Y. S. Polikanov and M. Gualtieri, Mol. Cell, 2018, 70, 83–94 CrossRef CAS.
- M. Sarciaux, L. Pantel, C. Midrier, M. Serri, C. Gerber, R. Marcia De Figueiredo, J. M. Campagne, P. Villain-Guillot, M. Gualtieri and E. Racine, J. Med. Chem., 2018, 61, 7814–7826 CrossRef CAS.
- S. C. Blanchard, Mol. Cell, 2018, 70, 3–5 CrossRef CAS.
- M. Zhao, A. J. Lepak, K. Marchillo, J. Vanhecker and D. R. Andes, Antimicrob. Agents Chemother., 2018, 62, 1–9 Search PubMed.
- E. Racine, P. Nordmann, L. Pantel, M. Sarciaux, M. Serri, J. Houard and P. Villain-guillot, Antimicrob. Agents Chemother., 2018, 62, 1–16 CrossRef.
- D. N. Wilson, G. Guichard and C. Axel Innis, Oncotarget, 2015, 6, 16826–16827 CrossRef.
- Y. S. Polikanov, N. A. Aleksashin, B. Beckert and D. N. Wilson, Front. Mol. Biosci., 2018, 5, 1–21 CrossRef.
- N. B. Olivier, R. B. Altman, J. Noeske, G. S. Basarab, E. Code, A. D. Ferguson, N. Gao, J. Huang, M. F. Juette, S. Livchak, M. D. Miller, D. B. Prince, J. H. D. Cate, E. T. Buurman and S. C. Blanchard, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16274–16279 CrossRef CAS.
- Y. S. Polikanov, T. Szal, F. Jiang, P. Gupta, R. Matsuda, M. Shiozuka, T. A. Steitz, N. Vázquez-laslop and A. S. Mankin, Mol. Cell, 2014, 56, 541–550 CrossRef CAS.
- E. Racine and M. Gualtieri, Front. Microbiol., 2019, 10, 1–10 CrossRef.
- L. Wang, A. Pulk, M. R. Wasserman, M. B. Feldman, R. B. Altman, J. H. Doudna Cate and S. C. Blanchard, Nat. Struct. Mol. Biol., 2012, 19, 957–963 CrossRef CAS.
- J. Li, G. Chen and J. M. Webster, Can. J. Microbiol., 1997, 43, 770–773 CrossRef CAS.
- X. Cai, V. L. Challinor, L. Zhao, D. Reimer, H. Adihou, P. Grün, M. Kaiser and H. B. Bode, Org. Lett., 2017, 19, 806–809 CrossRef CAS.
- A. Stanišić and H. Kries, ChemBioChem, 2019, 20, 1347–1356 CrossRef.
- N. A. Magarvey, M. Ehling-Schulz and C. T. Walsh, J. Am. Chem. Soc., 2006, 128, 10698–10699 CrossRef CAS.
- J. Jaitzig, J. Li, R. D. Süssmuth and P. Neubauer, ACS Synth. Biol., 2014, 3, 432–438 CrossRef CAS.
- D. Reimer, F. I. Nollmann, K. Schultz, M. Kaiser and H. B. Bode, J. Nat. Prod., 2014, 77, 1976–1980 CrossRef CAS.
- T. Himmler, F. Pirro and N. Schmeer, Bioorg. Med. Chem. Lett., 1998, 8, 2045–2050 CrossRef CAS.
- F. Wesche, H. Adihou, T. A. Wichelhaus and H. B. Bode, Beilstein J. Org. Chem., 2019, 15, 535–541 CrossRef CAS.
- A. Hasan, H. S. Yeom, J. Ryu, H. B. Bode and Y. Kim, Sci. Rep., 2019, 9, 1–18 CrossRef CAS.
- E. Bode, A. O. Brachmann, C. Kegler, R. Simsek, C. Dauth, Q. Zhou, M. Kaiser, P. Klemmt and H. B. Bode, ChemBioChem, 2015, 16, 1115–1119 CrossRef CAS.
- A. O. Brachmann, F. Kirchner, C. Kegler, S. C. Kinski, I. Schmitt and H. B. Bode, J. Biotechnol., 2012, 157, 96–99 CrossRef CAS.
- H. B. Bode, A. O. Brachmann, K. B. Jadhav, L. Seyfarth, C. Dauth, S. W. Fuchs, M. Kaiser, N. R. Waterfield, H. Sack, S. H. Heinemann and H. D. Arndt, Angew. Chem., Int. Ed., 2015, 54, 10352–10355 CrossRef CAS.
- L. Zhao, R. M. Awori, M. Kaiser, J. Groß, T. Opatz and H. B. Bode, J. Nat. Prod., 2019, 82, 3499–3503 CrossRef CAS.
- L. S. Bonnington, J. Tanaka, T. Higa, J. Kimura, Y. Yoshimura, Y. Nakao, W. Y. Yoshida and P. J. Scheuer, J. Org. Chem., 1997, 62, 7765–7767 CrossRef CAS.
- H. Ishii, M. Nishijima and T. Abe, Water Res., 2004, 38, 2667–2676 CrossRef CAS.
- K. Saito, A. Konno, H. Ishii, H. Saito, F. Nishida, T. Abe and C. Chen, J. Nat. Prod., 2001, 64, 139–141 CrossRef CAS.
- J. W. Cha, J. S. Park, T. Sim, S. J. Nam, H. C. Kwon, J. R. Del Valle and W. Fenical, J. Nat. Prod., 2012, 75, 1648–1651 CrossRef CAS.
- N. De Lay, D. J. Schu and S. Gottesman, J. Biol. Chem., 2013, 288, 7996–8003 CrossRef CAS.
- J. Vogel and B. F. Luisi, Nat. Rev. Microbiol., 2015, 9, 578–589 CrossRef.
- M. Dietrich, R. Munke, M. Gottschald, E. Ziska, J. P. Boettcher, H. Mollenkopf and A. Friedrich, FEBS J., 2009, 276, 5507–5520 CrossRef CAS.
- N. J. Tobias, A. K. Heinrich, H. Eresmann, P. R. Wright, N. Neubacher, R. Backofen and H. B. Bode, Environ. Microbiol., 2017, 19, 119–129 CrossRef CAS.
- E. Bode, A. K. Heinrich, M. Hirschmann, D. Abebew, Y. Shi, T. D. Vo, F. Wesche, Y. Shi, P. Grün, S. Simonyi, N. Keller, Y. Engel, S. Wenski, R. Bennet, S. Beyer, I. Bischoff, A. Buaya, S. Brandt, I. Cakmak, H. Çimen, S. Eckstein, D. Frank, R. Fürst, M. Gand, G. Geisslinger, S. Hazir, M. Henke, R. Heermann, V. Lecaudey, W. Schäfer, S. Schiffmann, A. Schüffler, R. Schwenk, M. Skaljac, E. Thines, M. Thines, T. Ulshöfer, A. Vilcinskas, T. A. Wichelhaus and H. B. Bode, Angew. Chem., Int. Ed., 2019, 131, 19133–19139 CrossRef.
- F. Grundmann, M. Kaiser, M. Kurz, M. Schiell, A. Batzer and H. B. Bode, RSC Adv., 2013, 3, 22072–22077 RSC.
- B. Ohlendorf, S. Simon, J. Wiese and J. F. Imhoff, Nat. Prod. Commun., 2011, 6, 1247–1250 CrossRef CAS.
- T. Clements, T. Ndlovu and W. Khan, Microbiol. Res., 2019, 229, 126329 CrossRef CAS.
- H. H. Wasserman, J. J. Keggi and J. E. McKeon, J. Am. Chem. Soc., 1962, 84, 2978–2982 CrossRef CAS.
- H. H. Wasserman, J. J. Keggi and J. E. McKeon, J. Am. Chem. Soc., 1961, 83, 4107–4108 CrossRef CAS.
- T. Matsuyama, M. Fujita and I. Yano, FEMS Microbiol. Lett., 1985, 28, 125–129 CrossRef CAS.
- M. A. Kroteń, M. Bartoszewicz and I. Święcicka, Pol. J. Microbiol., 2010, 59, 3–10 Search PubMed.
- A. A. Sy-Cordero, C. J. Pearce and N. H. Oberlies, J. Antibiot., 2012, 65, 541–549 CrossRef CAS.
- L. Zhu, C. Pang, L. Chen and X. Zhu, Nat. Prod. Chem. Res., 2018, 6, 312 Search PubMed.
- D. Dwivedi, R. Jansen, G. Molinari, M. Nimtz, B. N. Johri and V. Wray, J. Nat. Prod., 2008, 71, 637–641 CrossRef CAS.
- J. L. Motley, B. W. Stamps, C. A. Mitchell, A. T. Thompson, J. Cross, J. You, D. R. Powell, B. S. Stevenson and R. H. Cichewicz, J. Nat. Prod., 2017, 80, 598–608 CrossRef CAS.
- T. Matsuyama, K. Kaneda, Y. Nakagawa, K. Isa, H. Hara-Hotta and I. Yano, J. Bacteriol., 1992, 174, 1769–1776 CrossRef CAS.
- C. Su, Z. Xiang, Y. Liu, X. Zhao, Y. Sun, Z. Li, L. Li, F. Chang, T. Chen, X. Wen, Y. Zhou and F. Zhao, BMC Genomics, 2016, 17, 1–19 CrossRef.
- T. Matsuyama, T. Murakami and M. Fujita, J. Gen. Microbiol., 1986, 132, 865–875 CAS.
- H. Li, T. Tanikawa, Y. Sato, Y. Nakagawa and T. Matsuyama, Microbiol. Immunol., 2005, 49, 303–310 CrossRef CAS.
- S. Sunaga, H. Li, Y. Sato, Y. Nakagawa and T. Matsuyama, Microbiol. Immunol., 2004, 48, 723–728 CrossRef CAS.
- J. W. Trauger, R. M. Kohli, H. D. Mootz, M. A. Marahiel and C. T. Walsh, Nature, 2000, 407, 215–218 CrossRef CAS.
- D. E. Kadouri and R. M. Q. Shanks, Res. Microbiol., 2013, 164, 821–826 CrossRef CAS.
- T. Clements, T. Ndlovu, S. Khan and W. Khan, Appl. Microbiol. Biotechnol., 2019, 103, 589–602 CrossRef CAS.
- J. G. Ganley, G. Carr, T. R. Ioerger, J. C. Sacchettini, J. Clardy and E. R. Derbyshire, ChemBioChem, 2018, 19, 1590–1594 CrossRef CAS.
- P.-Y. Mai, M. Levasseur, D. Buisson, D. Touboul and V. Eparvier, Plants, 2020, 9, 1–11 CrossRef.
- X. Yin and T. M. Zabriskie, Microbiology, 2006, 152, 2969–2983 CrossRef.
- S. Wang, A. L. A. Dos-santos, W. Huang, K. C. Liu, M. A. Oshaghi, G. Wei, P. Agre and M. Jacobs-lorena, Science, 2017, 357, 1399–1402 CrossRef CAS.
- N. A. Schilling, A. Berscheid, J. Schumacher, J. S. Saur, M. C. Konnerth, S. N. Wirtz, J. M. Beltrán-Beleña, A. Zipperer, B. Krismer, A. Peschel, H. Kalbacher, H. Brötz-Oesterhelt, C. Steinem and S. Grond, Angew. Chem., Int. Ed., 2019, 58, 9234–9238 CrossRef CAS.
- B. S. Evans, I. Ntai, Y. Chen, S. J. Robinson and N. L. Kelleher, J. Am. Chem. Soc., 2011, 133, 7316–7319 CrossRef CAS.
- B. A. Pfeifer, C. C. C. Wang, C. T. Walsh and C. Khosla, Appl. Environ. Microbiol., 2003, 69, 6698–6702 CrossRef CAS.
- Y. Inahashi, S. Zhou, M. J. Bibb, L. Song, M. M. Al-Bassam, M. J. Bibb and G. L. Challis, Chem. Sci., 2017, 8, 2823–2831 RSC.
- K. Bitschar, B. Sauer, J. Focken, H. Dehmer, S. Moos, M. Konnerth, N. A. Schilling, S. Grond, H. Kalbacher, F. C. Kurschus, F. Götz, B. Krismer, A. Peschel and B. Schittek, Nat. Commun., 2019, 10, 2730 CrossRef.
- M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef CAS.
- J. G. Hurdle, A. J. O'Neill, I. Chopra and R. E. Lee, Nat. Rev. Microbiol., 2011, 9, 62–75 CrossRef CAS.
- L. Ettlinger, E. Gäumann, R. Hütter, W. Keller-Schierlein, F. Kradolfer, L. Neipp, V. Prelog and H. Zähner, Helv. Chim. Acta, 1959, 42, 563–569 CrossRef CAS.
- K. Okamura, K. Soga, Y. Shimauchi, T. Ishikura and J. Lein, J. Antibiot., 1977, 30, 334–336 CrossRef CAS.
- M. Kenig and C. Reading, Biochem. Biophys. Res. Commun., 1979, 91, 498–501 CrossRef.
- S. Huang, Y. Zhao, Z. Qin, X. Wang, M. Onega, L. Chen, J. He, Y. Yu and H. Deng, Process Biochem., 2011, 46, 811–816 CrossRef CAS.
- P. Liras, Appl. Microbiol. Biotechnol., 2014, 98, 1023–1030 CrossRef CAS.
- Z. Wei, C. Xu, J. Wang, F. Lu, X. Bie and Z. Lu, PeerJ, 2017, 5, 1–19 CrossRef.
- H. Ding, J. N. Wang, D. S. Zhang and Z. J. Ma, Chem. Biodiversity, 2017, 14, 2–9 CrossRef.
- Z. Qin, S. Huang, Y. Yu and H. Deng, Mar. Drugs, 2013, 11, 3970–3997 CrossRef CAS.
- B. Li, W. J. Wever, C. T. Walsh and A. A. Bowers, Nat. Prod. Rep., 2014, 31, 905–923 RSC.
- S. Huang, M. H. Tong, Z. Qin, Z. Deng, H. Deng and Y. Yu, Anticancer Agents Med. Chem., 2015, 15, 277–284 CrossRef CAS.
- B. Oliva, A. O'Neill, J. M. Wilson, P. J. O'Hanlon and I. Chopra, Antimicrob. Agents Chemother., 2001, 45, 532–539 CrossRef CAS.
- A. O'Neill, B. Oliva, C. Storey, A. Hoyle, C. Fishwick and I. Chopra, Antimicrob. Agents Chemother., 2000, 44, 3163–3166 CrossRef.
- X. Tan, C. Li, Z. Yu, P. Wang, S. Nian, Y. Deng, W. Wu and G. Wang, Chem. Pharm. Bull., 2013, 61, 351–357 CrossRef CAS.
- J. Meng, B. Kong, J. Wang, X. Yang, Y. Lv, L. Lyu, Z. Jiang and X. Tan, Med. Chem. Res., 2020, 29, 1376–1386 CrossRef CAS.
- J. E. Ellis, J. H. Fried, I. T. Harrison, E. Rapp and C. H. Ross, J. Org. Chem., 1977, 42, 2891–2893 CrossRef CAS.
- C. Li, Y. Sun, G. Wang and X. Tan, Bull. Korean Chem. Soc., 2014, 35, 3489–3494 CrossRef CAS.
- B. Li, M. P. A. Lyle, G. Chen, J. Li, K. Hu, L. Tang, M. A. Alaoui-Jamali and J. Webster, Bioorg. Med. Chem., 2007, 15, 4601–4608 CrossRef CAS.
- X. Tan, M. Huang, S. Nian, Y. Peng, J. Qin, B. Kong and X. Duan, Bioorg. Med. Chem. Lett., 2020, 30, 127146 CrossRef CAS.
- N. Bouras, R. Merrouche, L. Lamari, F. Mathieu, N. Sabaou and A. Lebrihi, Process Biochem., 2008, 43, 1244–1252 CrossRef CAS.
- N. Bouras, F. Mathieu, N. Sabaou and A. Lebrihi, Process Biochem., 2007, 42, 925–933 CrossRef CAS.
- A. C. Chorin, L. Bijeire, M. C. Monje, G. Baziard, A. Lebrihi and F. Mathieu, J. Appl. Microbiol., 2009, 107, 1751–1762 CrossRef CAS.
- F. Yakushiji, Y. Miyamoto, Y. Kunoh, R. Okamoto, H. Nakaminami, Y. Yamazaki, N. Noguchi and Y. Hayashi, ACS Med. Chem. Lett., 2013, 4, 220–224 CrossRef CAS.
- A. C. Murphy, S. S. Gao, L. C. Han, S. Carobene, D. Fukuda, Z. Song, J. Hothersall, R. J. Cox, J. Crosby, M. P. Crump, C. M. Thomas, C. L. Willis and T. J. Simpson, Chem. Sci., 2014, 5, 397–402 RSC.
- T. A. Wencewicz, J. Mol. Biol., 2019, 431, 3370–3399 CrossRef CAS.
- Z. D. Dunn, W. J. Wever, D. N. J. Economou and P. B. Li, Angew. Chem., Int. Ed., 2015, 54, 5137–5141 CrossRef CAS.
- P. Charusanti, N. L. Fong, H. Nagarajan, A. R. Pereira, H. J. Li, E. A. Abate, Y. Su, W. H. Gerwick and B. O. Palsson, PLoS One, 2012, 7, e33727 CrossRef CAS.
- H. Yin, S. Xiang, J. Zheng, K. Fan, T. Yu, X. Yang, Y. Peng, H. Wang, D. Feng, Y. Luo, H. Bai and K. Yang, Appl. Environ. Microbiol., 2012, 78, 3431–3441 CrossRef CAS.
- A. Jimenez, D. J. Tipper and J. Davies, Antimicrob. Agents Chemother., 1973, 3, 729–738 CrossRef CAS.
- G. G. Khachatourians and D. J. Tipper, Antimicrob. Agents Chemother., 1974, 6, 304–310 CrossRef CAS.
- D. J. Tipper, J. Bacteriol., 1973, 116, 245–256 CrossRef CAS.
- N. Sivasubramanian and R. Jayaraman, Mol. Genet. Genomics, 1976, 145, 89–96 CrossRef CAS.
- G. G. Khachatourians and D. J. Tipper, J. Bacteriol., 1974, 119, 795–804 CrossRef CAS.
- L. Lauinger, J. Li, A. Shostak, I. A. Cemel, N. Ha, Y. Zhang, P. E. Merkl, S. Obermeyer, N. Stankovic-Valentin, T. Schafmeier, W. J. Wever, A. A. Bowers, K. P. Carter, A. E. Palmer, H. Tschochner, F. Melchior, R. J. Deshaies, M. Brunner and A. Diernfellner, Nat. Chem. Biol., 2017, 13, 709–714 CrossRef CAS.
- A. N. Chan, A. L. Shiver, W. J. Wever, S. Z. A. Razvi, M. F. Traxler and B. Li, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2717–2722 CrossRef CAS.
- B. Li, F. Ry R., B. Albert A., S. Frank C. and W. Christopher T., ChemBioChem, 2013, 13, 2521–2526 CrossRef.
- C. T. Walsh, R. V. O'Brien and C. Khosla, Angew. Chem., Int. Ed., 2013, 52, 7098–7124 CrossRef CAS.
- W. Zhou, J. H. Li, J. Chen, X. Y. Liu, T. T. Xiang, L. Zhang and Y. J. Wan, J. Invertebr. Pathol., 2016, 136, 92–94 CrossRef CAS.
- J. S. Lee, Y. S. Kim, S. Park, J. Kim, S. J. Kang, M. H. Lee, S. Ryu, J. M. Choi, T. K. Oh and J. H. Yoon, Appl. Environ. Microbiol., 2011, 77, 4967–4973 CrossRef CAS.
- H. N. A. Do and T. H. K. Nguyen, J. Appl. Pharm. Sci., 2014, 4, 21–24 CrossRef.
- T. Danevcic, M. B. Vezjak, M. Tabor, M. Zorec and D. Stopar, Front. Microbiol., 2016, 7, 1–10 Search PubMed.
- H. Zhang, H. Wang, W. Zheng, Z. Yao, Y. Peng and S. Zhang, Front. Microbiol., 2017, 8, 1–12 Search PubMed.
- N. Stankovic, L. Senerovic, T. Ilic-Tomic, B. Vasiljevic and J. Nikodinovic-Runic, Appl. Microbiol. Biotechnol., 2014, 98, 3841–3858 CrossRef CAS.
- S. Shahitha and K. Poornima, J. Appl. Pharm. Sci., 2012, 2, 138–140 Search PubMed.
- R. Sakuraoka, T. Suzuki and T. Morohoshi, Genome Biol. Evol., 2019, 11, 931–936 CrossRef CAS.
- N. R. Williamson, H. T. Simonsen, R. A. A. Ahmed, G. Goldet, H. Slater, L. Woodley, F. J. Leeper and G. P. C. Salmond, Mol. Microbiol., 2005, 56, 971–989 CrossRef CAS.
- N. R. Williamson, P. C. Fineran, F. J. Leeper and G. P. C. Salmond, Nat. Rev. Microbiol., 2006, 4, 887–899 CrossRef CAS.
- F. E. Sakai-Kawada, C. G. Ip, K. A. Hagiwara and J. D. Awaya, Front. Microbiol., 2019, 10, 1–9 CrossRef.
- H. Okamoto, Z. Sato, M. Sato, Y. Koiso, S. Iwasaki and M. Isaka, Jpn. J. Phytopathol., 1998, 64, 294–298 CrossRef CAS.
- H.-S. Kim, M. Hayashi, Y. Shibata, Y. Wataya, T. Mitamura, T. Horii, K. Kawauchi, H. Hirata, S. Tsuboi and Y. Moriyama, Biol. Pharm. Bull., 1999, 22, 532–534 CrossRef CAS.
- P. Azambuja, D. Feder and E. S. Garcia, Exp. Parasitol., 2004, 107, 89–96 CrossRef CAS.
- D. Li, J. Liu, X. Wang, D. Kong, W. Du, H. Li, C. Y. Hse, T. Shupe, D. Zhou and K. Zhao, Int. J. Mol. Sci., 2018, 19, 3465 CrossRef.
- K. Kawauchi, K. Tobiume, S. Kaneko, K. Kaneshiro, S. Okamoto, E. Ueda, H. Kamata, Y. Moriyama and H. Hirata, Biol. Pharm. Bull., 2007, 30, 1792–1795 CrossRef CAS.
- N. Darshan and H. K. Manonmani, J. Food Sci. Technol., 2015, 52, 5393–5407 CrossRef CAS.
- R. K. Suryawanshi, C. D. Patil, S. H. Koli, J. E. Hallsworth and S. V. Patil, Nat. Prod. Res., 2017, 31, 572–577 CrossRef CAS.
- B. Díaz De Greñu, P. I. Hernández, M. Espona, D. Quiñonero, M. E. Light, T. Torroba, R. Pérez-Tomás and R. Quesada, Chem.–Eur. J., 2011, 17, 14074–14083 CrossRef.
- A. Fürstner and E. J. Grabowski, ChemBioChem, 2001, 2, 706–709 CrossRef.
- R. Pérez-Tomás, B. Montaner, E. Llagostera and V. Soto-Cerrato, Biochem. Pharmacol., 2003, 66, 1447–1452 CrossRef.
- A. Goy, F. J. Hernandez-Ilzaliturri, B. Kahl, P. Ford, E. Protomastro and M. Berger, Leuk Lymphoma, 2014, 55, 1–17 CrossRef.
- S. Cournoyer, A. Addioui, A. Belounis, M. Beaunoyer, C. Nyalendo, R. Le Gall, P. Teira, E. Haddad, G. Vassal and H. Sartelet, BMC Cancer, 2019, 19, 1–14 CrossRef CAS.
- C. A. Goard and A. D. Schimmer, Core Evidence, 2013, 8, 15–26 CrossRef CAS.
- D. W. Hood, R. Heidstra, U. K. Swoboda and D. A. Hodgson, Gene, 1992, 115, 5–12 CrossRef CAS.
- S. R. Burger and J. W. Bennett, Appl. Environ. Microbiol., 1985, 50, 487–490 CrossRef CAS.
- H. Yamaguchi, Y. Nakayama, K. Takeda, K. Tawara, K. Maeda, T. Takeuchi and H. Umezawa, J. Antibiot., 1957, 10, 195–200 CAS.
- H. Sakakibara, H. Naganawa, M. Ohno, K. Maeda and H. Umezawa, J. Antibiot., 1974, XXVII, 897–899 CrossRef.
- H. A. Kirst, E. F. Szymanski, D. E. Dorman, J. L. Occolowitz, N. D. Jones, M. O. Chaney, R. L. Hamill and M. M. Hoehn, J. Antibiot., 1975, XXVIII, 286–291 CrossRef.
- B. Kunze, H. Reichenbach, H. Augustiniak and G. Höfle, J. Antibiot., 1982, 35, 635–636 CrossRef CAS.
- N. S. Cortina, O. Revermann, D. Krug and R. Müller, ChemBioChem, 2011, 12, 1411–1416 CrossRef CAS.
- G. L. Challis, A. J. Gerc, N. R. Stanley-Wall, S. J. Coulthurst and L. Song, PLoS One, 2012, 7, e44673 CrossRef.
- K. Inami and T. Shiba, Bull. Chem. Soc. Jpn., 1986, 59, 2185–2189 CrossRef CAS.
- P. Zarantonello, C. P. Leslie, R. Ferritto and W. M. Kazmierski, Bioorg. Med. Chem. Lett., 2002, 12, 561–565 CrossRef CAS.
- H. Fujimoto, T. Kinoshita, H. Suzuki and H. Umezawa, J. Antibiot., 1970, 23, 271–275 CrossRef CAS.
- K. Inami and T. Shiba, Bull. Chem. Soc. Jpn., 1985, 58, 352–360 CrossRef CAS.
- J. H. Cardellina, F. J. Marner and R. E. Moore, J. Am. Chem. Soc., 1979, 101, 240–242 CrossRef CAS.
- C. A. Banuelos, I. Tavakoli, A. H. Tien, D. P. Caley, N. R. Mawji, Z. Li, J. Wang, Y. C. Yang, Y. Imamura, L. Yan, J. G. Wen, R. J. Andersen and M. D. Sadar, J. Biol. Chem., 2016, 291, 22231–22243 CrossRef CAS.
- S. Paik, J. Cullingham, R. E. Moore, G. M. L. Patterson, M. A. Tius and S. Carmeli, J. Am. Chem. Soc., 1994, 116, 8116–8125 CrossRef CAS.
- R. G. Birch and S. S. Patil, J. Gen. Microbiol., 1985, 131, 1069–1075 CAS.
- R. G. Birch and S. S. Patil, Physiol. Mol. Plant Pathol., 1987, 30, 199–206 CrossRef CAS.
- R. G. Birch and S. S. Patil, Antibiotic and Process for the Production Thereof, US Pat., 4,525,354, 1985, 1–3.
- S. Cociancich, A. Pesic, D. Petras, S. Uhlmann, J. Kretz, V. Schubert, L. Vieweg, S. Duplan, M. Marguerettaz, J. Noëll, I. Pieretti, M. Hügelland, S. Kemper, A. Mainz, P. Rott, M. Royer and R. D. Süssmuth, Nat. Chem. Biol., 2015, 11, 195–197 CrossRef CAS.
- E. Vivien, D. Pitorre, S. Cociancich, I. Pieretti, D. W. Gabriel, P. C. Rott and M. Royer, Antimicrob. Agents Chemother., 2007, 51, 1549–1552 CrossRef CAS.
- R. D. Süssmuth, D. Kerwat, S. Grätz, I. Behroz, L. von Eckardstein, P. M. Durkin, M. Morkunas and J. Weston, WO2019/015794, 2017, pp. 1–73.
- S. M. Hashimi, M. K. Wall, A. B. Smith, A. Maxwell and R. G. Birch, Antimicrob. Agents Chemother., 2007, 51, 181–187 CrossRef CAS.
- J. J. Champoux, Annu. Rev. Biochem., 2001, 70, 369–413 CrossRef CAS.
- J. Kretz, D. Kerwat, V. Schubert, S. Grätz, A. Pesic, S. Semsary, S. Cociancich, M. Royer and R. D. Süssmuth, Angew. Chem., Int. Ed., 2015, 54, 1969–1973 CrossRef CAS.
- S. Grätz, D. Kerwat, J. Kretz, L. von Eckardstein, S. Semsary, M. Seidel, M. Kunert, J. B. Weston and R. D. Süssmuth, ChemMedChem, 2016, 11, 1499–1502 CrossRef.
- I. Behroz, P. Durkin, S. Grätz, M. Seidel, L. Rostock, M. Spinczyk, J. B. Weston and R. D. Süssmuth, Chem.–Eur. J., 2019, 25, 16538–16543 CrossRef CAS.
- D. Kerwat, S. Grätz, J. Kretz, M. Seidel, M. Kunert, J. B. Weston and R. D. Süssmuth, ChemMedChem, 2016, 11, 1899–1903 CrossRef CAS.
- L. von Eckardstein, D. Petras, T. Dang, S. Cociancich, S. Sabri, S. Grätz, D. Kerwat, M. Seidel, A. Pesic, P. C. Dorrestein, M. Royer, J. B. Weston and R. D. Süssmuth, Chem.–Eur. J., 2017, 23, 15316–15321 CrossRef CAS.
- D. Petras, D. Kerwat, A. Pesic, B. F. Hempel, L. Von Eckardstein, S. Semsary, J. Arasté, M. Marguerettaz, M. Royer, S. Cociancich and R. D. Süssmuth, ACS Chem. Biol., 2016, 11, 1198–1204 CrossRef CAS.
- L. Vieweg, J. Kretz, A. Pesic, D. Kerwat, S. Grätz, M. Royer, S. Cociancich, A. Mainz and R. D. Süssmuth, J. Am. Chem. Soc., 2015, 137, 7608–7611 CrossRef CAS.
- A. Höltzel, M. G. Gänzle, G. J. Nicholson, W. P. Hammes and G. Jung, Angew. Chem., Int. Ed., 2000, 39, 2766–2768 CrossRef.
- M. G. Gänzle and R. F. Vogel, Int. J. Food Microbiol., 2003, 80, 31–45 CrossRef.
- R. Boehme, G. Jung and E. Breitmaier, Helv. Chim. Acta, 2005, 88, 2837–2841 CrossRef CAS.
- T. Hao, Z. Xie, M. Wang, L. Liu, Y. Zhang, W. Wang, Z. Zhang, X. Zhao, P. Li, Z. Guo, S. Gao, C. Lou, G. Zhang, J. Merritt, G. P. Horsman and Y. Chen, Nat. Commun., 2019, 10, 1–13 CrossRef CAS.
- Y. C. Jeong and M. G. Moloney, Synlett, 2009, 15, 2487–2491 Search PubMed.
- X. B. Lin, C. T. Lohans, R. Duar, J. Zheng, J. C. Vederas, J. Walter and M. Gänzle, Appl. Environ. Microbiol., 2015, 81, 2032–2041 CrossRef CAS.
- J. T. Park and J. L. Strominger, Science, 1957, 125, 99–101 CrossRef CAS.
- Y. Mutaguchi, T. Ohmori, T. Wakamatsu, K. Doi and T. Ohshima, J. Bacteriol., 2013, 195, 5207–5215 CrossRef CAS.
- M. Gunsior, S. D. Breazeale, A. J. Lind, J. Ravel, J. W. Janc and C. A. Townsend, Chem. Biol., 2004, 11, 927–938 CrossRef CAS.
- J. G. Hurdle, A. E. Heathcott, L. Yang, B. Yan and R. E. Lee, J. Antimicrob. Chemother., 2011, 66, 1773–1776 CrossRef CAS.
- U. Marquardt, D. Schmid and G. Jung, Synlett, 2000, 8, 1131–1132 Search PubMed.
- P. T. Cherian, X. Wu, M. M. Maddox, A. P. Singh, R. E. Lee and J. G. Hurdle, Sci. Rep., 2014, 4, 1–9 Search PubMed.
- R. Yendapally, J. G. Hurdle, E. I. Carson, R. B. Lee and R. E. Lee, J. Med. Chem., 2008, 51, 1487–1491 CrossRef CAS.
- G. A. Hudson and D. A. Mitchell, Curr. Opin. Microbiol., 2018, 45, 61–69 CrossRef CAS.
- M. A. Ortega and W. A. Van Der Donk, Cell Chem. Biol., 2016, 23, 31–44 CrossRef CAS.
- I. Holtsmark, V. G. H. Eijsink and M. B. Brurberg, FEMS Microbiol. Lett., 2008, 280, 1–7 CrossRef CAS.
- E. Meade, M. A. Slattery and M. Garvey, Antibiotics, 2020, 9, 1–18 CrossRef.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. J. Guo, A. Makriyannis, L. Linares-Otoya, N. Böhringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O'Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schäberle, A. D'Onofrio and K. Lewis, Nature, 2019, 576, 459–464 CrossRef CAS.
- K. R. Schramma, L. B. Bushin and M. R. Seyedsayamdost, Nat. Chem., 2015, 7, 431–437 CrossRef CAS.
- K. A. Clark, L. B. Bushin and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2019, 141, 10610–10615 CrossRef CAS.
- A. Benjdia, C. Balty and O. Berteau, Front. Chem., 2017, 5, 1–13 Search PubMed.
- R. M. Epand, C. Walker, R. F. Epand and N. A. Magarvey, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 980–987 CrossRef CAS.
- J. M. Waisvisz, M. G. Van Der Hoeven, J. Van Peppen and W. C. M. Zwennis, J. Am. Chem. Soc., 1957, 79, 4520–4521 CrossRef CAS.
- Y. Hou, M. D. Tianero, J. Kwan, T. P. Wyche, C. R. Michel, G. A. Ellis, E. Varquez-Rivera, D. Braun, W. E. Rose, E. W. Schmidt and T. S. Bugni, Org. Lett., 2012, 14, 5050–5053 CrossRef CAS.
- W. J. K. Crone, F. J. Leeper and A. W. Truman, Chem. Sci., 2012, 3, 3516–3521 RSC.
- J. P. Gomez-Escribano, L. Song, M. J. Bibb and G. L. Challis, Chem. Sci., 2012, 3, 3522–3525 RSC.
- Y. Takahashi, H. Naganawa, T. Takita, H. Umezawa and S. Nakamura, J. Antibiot., 1976, 29, 1120–1123 CrossRef CAS.
- S. Nakamura, T. Yajima, Y. Lin and H. Umezawa, J. Antibiot., 1967, 20, 1–5 CAS.
- S. Nakamura, N. Tanaka and H. Umezawa, J. Antibiot., 1966, 19, 10–12 CAS.
- D. Schipper, J. Antibiot., 1983, 36, 1076–1077 CrossRef CAS.
- H. Shimamura, H. Gouda, K. Nagai, T. Hirose, M. Ichioka, Y. Furuya, Y. Kobayashi, S. Hirono, T. Sunazuka and S. Omura, Angew. Chem., Int. Ed., 2009, 48, 914–917 CrossRef CAS.
- W. J. K. Crone, N. M. Vior, J. Santos-Aberturas, L. G. Schmitz, F. J. Leeper and A. W. Truman, Angew. Chem., Int. Ed., 2016, 55, 9639–9643 CrossRef CAS.
- N. M. Vior, E. Cea-torrescassana, T. H. Eyles, G. Chandra and A. W. Truman, Front. Microbiol., 2020, 11, 1–16 CrossRef.
- T. Yamada, M. Yagita, Y. Kobayashi, G. Sennari, H. Shimamura, H. Matsui, Y. Horimatsu, H. Hanaki, T. Hirose, S. Omura and T. Sunazuka, J. Org. Chem., 2018, 83, 7135–7149 CrossRef CAS.
- Y. Kobayashi, M. Ichioka, T. Hirose, K. Nagai, A. Matsumoto, H. Matsui, H. Hanaki, R. Masuma, Y. Takahashi, S. Omura and T. Sunazuka, Bioorg. Med. Chem. Lett., 2010, 20, 6116–6120 CrossRef CAS.
- S. Ackermann, H. G. Lerchen, D. Häbich, A. Ullrich and U. Kazmaier, Beilstein J. Org. Chem., 2012, 8, 1652–1656 CrossRef CAS.
- T. Otaka and A. Kaji, J. Biol. Chem., 1976, 251, 2299–2306 CrossRef CAS.
- T. Otaka and A. Kaji, FEBS Lett., 1983, 153, 53–59 CrossRef CAS.
- Y. Inouye, M. Hashimoto, M. Sugiyama, Y. Takesue, T. Santo and T. Yokoyama, Hiroshima J. Med. Sci., 1994, 43, 87–92 CAS.
- T. H. Eyles, N. M. Vior and A. W. Truman, ACS Synth. Biol., 2018, 7, 1211–1218 CrossRef CAS.
- L. Horbal, F. Marques, S. Nadmid, M. V. Mendes and A. Luzhetskyy, Metab. Eng., 2018, 49, 299–315 CrossRef CAS.
- A. Mukai, T. Fukai, Y. Hoshino, K. Yazawa, K. I. Harada and Y. Mikami, J. Antibiot., 2009, 62, 613–619 CrossRef CAS.
- A. A. Vinogradov and H. Suga, Cell Chem. Biol., 2020, 27, 1–20 CrossRef.
- K. Sakai, H. Komaki and T. Gonoi, PLoS One, 2015, 10, e0143264 CrossRef.
- W. L. Kelly, L. Pan and C. Li, J. Am. Chem. Soc., 2009, 131, 4327–4334 CrossRef CAS.
- S. J. Malcolmson, T. S. Young, J. G. Ruby, P. Skewes-Cox and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8483–8488 CrossRef CAS.
- K. Sakai, Y. Hara, M. Ishibashi, M. Sakai, S. Kawahara, S. Imanishi, K. Harada, Y. Hoshino, H. Komaki, A. Mukai and T. Gonoi, Int. J. Pept. Res. Ther., 2020, 26, 281–290 CrossRef CAS.
- L. Xu, A. K. Farthing, J. F. Dropinski, P. T. Meinke, C. McCallum, E. Hickey and K. Liu, Bioorg. Med. Chem. Lett., 2013, 23, 366–369 CrossRef CAS.
- R. Li, A. Stapon, J. T. Blanchfield and C. A. Townsend, J. Am. Chem. Soc., 2000, 122, 9296–9297 CrossRef CAS.
- K. M. Papp-Wallace, A. Endimiani, M. A. Taracila and R. A. Bonomo, Antimicrob. Agents Chemother., 2011, 55, 4943–4960 CrossRef CAS.
- J. F. Martín, J. Casqueiro, K. Kosalková, A. T. Marcos and S. Gutiérrez, Antonie van Leeuwenhoek, 1999, 75, 21–31 CrossRef.
- E. K. Schmitt, B. Hoff and U. Kück, Adv. Biochem. Eng./Biotechnol., 2004, 88, 1–43 CrossRef CAS.
- J. L. Ott, C. W. Godzeski, D. Pavey, J. D. Farran and D. R. Horton, Appl. Microbiol., 1962, 10, 515–523 CrossRef CAS.
- J. O'Sullivan, A. M. Gillum, C. A. Aklonis, M. L. Souser and R. B. Sykes, Antimicrob. Agents Chemother., 1982, 21, 558–564 CrossRef.
- S. E. Jensen, J. Ind. Microbiol. Biotechnol., 2012, 39, 1407–1419 CrossRef CAS.
- P. C. Fineran, H. Slater, L. Everson, K. Hughes and G. P. C. Salmond, Mol. Microbiol., 2005, 56, 1495–1517 CrossRef CAS.
- S. J. Coulthurst, A. M. L. Barnard and G. P. C. Salmond, Nat. Rev. Microbiol., 2005, 3, 295–306 CrossRef CAS.
- K. Bush and P. A. Bradford, Cold Spring Harbor Perspect. Med., 2016, 6, a025247 CrossRef.
- S. J. McGowan, M. Sebaihia, L. E. Porter, G. S. A. B. Stewart, P. Williams, B. W. Bycroft and G. P. C. Salmond, Mol. Microbiol., 1996, 22, 415–426 CrossRef CAS.
- W. L. Parker, M. L. Rathnum, J. Scott Wells, W. H. Trejo, P. A. Principe and R. B. Sykes, J. Antibiot., 1982, 35, 653–660 CrossRef CAS.
- B. W. Bycroft and C. Maslen, J. Antibiot., 1988, XLI, 1231–1241 CrossRef.
- G. B. Elion and G. H. Hitchings, J. Am. Chem. Soc., 1955, 77, 1676 CrossRef CAS.
- P. N. Munshi, M. Lubin and J. R. Bertino, Oncologist, 2014, 19, 760–765 CrossRef CAS.
- M. Stanulla and H. J. Schünemann, Lancet, 2006, 368, 1304–1305 CrossRef.
- G. B. Elion, Biosci. Rep., 1989, 9, 509–529 CrossRef CAS.
- A. Litomska, K. Ishida, K. L. Dunbar, M. Boettger, S. Coyne and C. Hertweck, Angew. Chem., Int. Ed., 2018, 57, 11574–11578 CrossRef CAS.
- D. L. Pruess, M. Kellett and T. C. Demny, J. Antibiot., 1971, 24, 328–329 CrossRef.
- S. Coyne, C. Chizzali, M. N. A. Khalil, A. Litomska, K. Richter, L. Beerhues and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 10564–10568 CrossRef CAS.
- K. A. Black and P. C. Dos Santos, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 1470–1480 CrossRef CAS.
- A. Wensing, M. Gernold, S. Jock, R. Jansen and K. Geider, Mol. Genet. Genomics, 2014, 289, 215–223 CrossRef CAS.
- B. A. Harris, D. A. Weigent and J. A. Nelson, Biochem. Pharmacol., 1979, 28, 1169–1173 CrossRef CAS.
- J. E. Heinze, T. Mitani, K. E. Rich and E. Freese, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1978, 521, 16–26 CrossRef CAS.
- H. G. Mandel, R. G. Latimer and M. Riis, Biochem. Pharmacol., 1965, 14, 66–682 CrossRef.
- B. M. Mehta and D. J. Hutchison, Ann. N. Y. Acad. Sci., 1975, 255, 559–563 CrossRef CAS.
- T. Kusumi, I. Ohtani, K. Nishiyama and H. Kakisawa, Tetrahedron Lett., 1987, 28, 3981–3984 CrossRef CAS.
- C. Ross, K. Scherlach, F. Kloss and C. Hertweck, Angew. Chem., Int. Ed., 2014, 53, 7794–7798 CrossRef CAS.
- E. Haldón, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC.
- L. V. Flórez, K. Scherlach, P. Gaube, C. Ross, E. Sitte, C. Hermes, A. Rodrigues, C. Hertweck and M. Kaltenpoth, Nat. Commun., 2017, 8, 1–9 CrossRef.
- X. Zhu, J. Liu and W. Zhang, Nat. Chem. Biol., 2015, 11, 115–120 CrossRef CAS.
- M. Yamaguchi, H. J. Park, S. Ishizuka, K. Omata and M. Hirama, J. Med. Chem., 1995, 38, 5015–5022 CrossRef CAS.
- A. L. K. Shi Shun and R. R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034–1057 CrossRef.
- M. Yamaguchi, H. J. Park and M. Hirama, Chem. Lett., 1997, 535–536 CrossRef CAS.
- E. Mühlberg, F. Umstätter, C. Kleist, C. Domhan, W. Mier and P. Uhl, Can. J. Microbiol., 2020, 66, 11–16 CrossRef.
- R. S. Mandal, S. Saha and S. Das, Genomics, Proteomics Bioinf., 2015, 13, 148–158 CrossRef.
- M. R. Wilson, L. Zha and E. P. Balskus, J. Biol. Chem., 2017, 292, 8546–8552 CrossRef CAS.
- W. K. Mousa, B. Athar, N. J. Merwin and N. A. Magarvey, Nat. Prod. Rep., 2017, 34, 1302–1331 RSC.
- M. S. Donia, P. Cimermancic, C. J. Schulze, L. C. W. Brown, J. Martin, M. Mitreva, J. Clardy, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 1402–1414 CrossRef CAS.
- A. Bouslimani, L. M. Sanchez, N. Garg and P. C. Dorrestein, Nat. Prod. Rep., 2014, 31, 718–729 RSC.
- D. Krug and R. Müller, Nat. Prod. Rep., 2014, 31, 768–783 RSC.
- J. N. Tabudravu, L. Pellissier, A. J. Smith, K. Subko, C. Autréau, K. Feussner, D. Hardy, D. Butler, R. Kidd, E. J. Milton, H. Deng, R. Ebel, M. Salonna, C. Gissi, F. Montesanto, S. M. Kelly, B. F. Milne, G. Cimpan and M. Jaspars, J. Nat. Prod., 2019, 82, 211–220 CrossRef CAS.
- R. Zhang, X. Li, X. Zhang, H. Qin and W. Xiao, Nat. Prod. Rep., 2020 10.1039/d0np00043d.
- R. Reher, H. W. Kim, C. Zhang, H. H. Mao, M. Wang, L. F. Nothias, A. M. Caraballo-Rodriguez, E. Glukhov, B. Teke, T. Leao, K. L. Alexander, B. M. Duggan, E. L. Van Everbroeck, P. C. Dorrestein, G. W. Cottrell and W. H. Gerwick, J. Am. Chem. Soc., 2020, 142, 4114–4120 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0np00061b |
| This journal is © The Royal Society of Chemistry 2021 |