
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Natural products as modulators of retinoic acid receptor-related orphan receptors (RORs)
Angela
Ladurner†
,
Patrik F.
Schwarz†
and
Verena M.
Dirsch
 *
*
Department of Pharmacognosy, University of Vienna, Vienna, Austria. E-mail: Verena.Dirsch@univie.ac.at
First published on 29th October 2020
Abstract
Covering: 1994 to 2020
Retinoic acid receptor-related orphan receptors (RORs) belong to a subfamily of the nuclear receptor superfamily and possess prominent roles in circadian rhythm, metabolism, inflammation, and cancer. They have been subject of research for over two decades and represent attractive but challenging drug targets. Natural products were among the first identified ligands of RORs and continue to be of interest to this day. This review focuses on ligands and indirect modulators of RORs from natural sources and explores their roles in a therapeutic context.
 Angela Ladurner | Angela Ladurner studied Molecular Biology at the University of Vienna (Austria) and completed her doctoral studies on natural product research in the group of Verena Dirsch in 2013. She then worked as a postdoctoral scientist at the Medical University of Vienna and at the University of Vienna. Her research focused on the characterization of the mechanism of action of natural products. |
 Patrik F. Schwarz | Patrik Schwarz studied Pharmacy at the University of Vienna (Austria) where he graduated in 2019. Since March 2020, he works as a doctoral student at the Department of Pharmacognosy, University of Vienna, under the supervision of Verena Dirsch. He is interested in finding novel (natural) ligands of the nuclear receptor RORγ. |
 Verena M. Dirsch | Verena Dirsch received her PhD from the University of Munich (1993) and then joined as postdoctoral fellow of the German Research Council the group of Koji Nakanishi at the Columbia University, New York. From 1995 to 2004 she held several academic positions in the group of Prof. Angelika Vollmar at the University of Munich. Since 2004, she is full professor at the University of Vienna and since 2006 head of the Department of Pharmacognosy. She also served as Vice-Dean at the Faculty of Life Sciences (2008–2014). Her main interest is to study the molecular mechanisms of natural products. |
1 Introduction
The retinoic acid receptor-related orphan receptors (ROR) α, β, and γ are a subfamily of nuclear receptors, encoded by the RORA-C (or NR1F1-3) genes. In general, nuclear receptors are ligand-dependent transcription factors that translate endocrine and dietary signals into differential gene expression patterns. An endogenous ligand for RORs has not been unequivocally confirmed, however, intermediates and metabolites of cholesterol metabolism have been suggested.1–3All members of the nuclear receptor superfamily feature a significant sequence homology and conserved structure. The ligand-independent activating function 1 (AF1) is located at the N-terminus, followed by a DNA-binding domain (DBD), hinge region, and ligand-binding domain (LBD). The most conserved region is the DBD, which contains two zinc-finger motifs that mediate binding to response elements located in the promoter region of target genes and are involved in receptor dimerization. ROR response elements (ROREs) consist of the AGGTCA consensus sequence proceeded by an A/T-rich region. Nuclear receptors can either bind DNA as monomers like RORs, homodimers, or heterodimers with a member of the retinoid X receptor subfamily as partner. The LBD consists of twelve α-helices that create a hydrophobic cavity to which ligands can bind. The AF2 domain, also referred to as helix 12 and included in the LBD, provides the structural surface for the interaction with co-activator and co-repressor proteins.4,5
In their unliganded basal state, nuclear receptors are bound to co-repressor proteins and act as transcriptional repressors. Binding of agonistic ligands leads to conformational changes, primarily stabilizing helix 12, which entails the displacement of co-repressor and recruitment of co-activator proteins, ultimately leading to the modulation of target gene expression. Interestingly, inverse ROR agonists like digoxin have been reported to destabilize helix 12, resulting in loss of co-activator interaction, however, without increased recruitment of co-repressors (for more details on digoxin's mechanism of action, see chapter 3.4.1).6 Co-activator proteins are bound to the LBD via their LXXLL interaction domain by a charge clamp (reviewed in ref. 4 and 7). X-ray studies revealed that, in addition, stabilization of the active conformation of RORs is established by formation of a hydrogen bond between a histidine residue in helix 11 and a tyrosine residue in helix 12 (His–Tyr lock). Both, histidine and tyrosine further form π–π interactions with a phenylalanine residue on helix 12, which overall stabilizes the active conformation. If the His–Tyr lock is broken, the aromatic interactions are also terminated and helix 12 is destabilized as a consequence.8–10 Inverse agonistic ligands are characterized by their ability to repress the transcriptional activity of its nuclear receptor below basal level via recruitment of additional co-repressors and have been shown to disrupt the active conformation of helix 12. Co-repressors contain a different interaction motif than co-activators with a similar amphipathic core (φXXφφ; φ is a hydrophobic amino acid) but additional flanking sequences and increased length (reviewed in ref. 11). Other mechanisms include NCoR or SMRT tethering via other transcription factors12 (reviewed in ref. 13) or “water trapping”, which was proposed for two synthetic compounds by Kallen et al. in 2017.9 This mechanism involves a water molecule becoming “trapped” in a partially hydrophobic environment when the inverse agonists are bound to the RORγt-LBD. Subsequent release of the water molecule into bulk solvent leads to destabilization of helix 12.9 However, many more mechanisms are involved in transcriptional regulation by nuclear receptors (reviewed in ref. 14).
RORs have been reported to influence various physiological processes such as circadian rhythm, neuronal cell development, and immune cell differentiation. At the same time, they are implicated in several pathologies like autoimmune, inflammatory, and metabolic diseases. RORα is expressed in many peripheral tissues like the liver, skeletal muscle, skin, lung, and adipose tissue. RORβ expression is restricted to brain, retina, bone, and pineal gland. The RORC gene encodes two isoforms via the use of alternative promoters. RORγ (RORγ1) differs from RORγt (RORγ2) only at the first 100 nucleotides at the N-terminus.15 RORγ1 is expressed in muscle tissue, prostate, pancreas, heart, liver, and testicles, whereas RORγt is exclusively expressed in lymphatic tissues.5,14,16 With the recent discovery of several ligands interacting with ROR receptors, interest for such ligands in drug development has increased.14,17–19
Due to their diversity and still often undiscovered biological potential, natural products are an important source for lead structures in the development of novel drugs.20–23 Even though natural products have been an important source for medicinal preparations, the focus on them in the pharmaceutical industry has diminished over the last decades. With high-throughput screening (HTS) and combinatorial chemistry on the rise, natural products were believed not to fit the requirements of these systems.20,21 While many chemical probes have been discovered by screening, it is not a magic bullet.21,22 Natural products offer a wide range of pharmacophores and a high number of stereocenters, which provides libraries containing such compounds a higher hit rate (reviewed in ref. 22 and 23). As a guide to obtain sufficient oral bioavailability, Lipinski's rule of five is used. As many natural products are substrates for active cellular transporters, they often do not have to fit these rules. This is a big advantage, as it makes such compounds more likely to succeed.23 This is apparent by the fact that between 1981 and 2014, approximately 50% of newly approved drugs are inspired by natural products, be it natural products, natural product analogues, or synthetic mimetics.23
2 Biological roles of RORs and their potential as drug targets
2.1 Biological roles of RORs
As the biological roles of RORs have already been explored in depth earlier (examples: ref. 16, 17 and 24–26), preference will be given to the aspects necessary to understand the studies covered in the present review.The first encounter with the at that time still unknown RORs was made, when a naturally occurring mutant strain of mice was discovered in the 1960s.27 These mice were called “staggerer” because of their staggering gait. Severe cerebellar underdevelopment with a lack of up to 90% of the Purkinje cells compared to wild type (WT)28 and a shorter life span were noticed, among others.27 Over 30 years later, it was discovered that staggerer mice possess a deletion in the Rora gene (thus also referred to as Rorasg/sg mice) that leads to the elimination of the LBD, leaving the nuclear receptor inactive as interactions with co-activators are not possible anymore.29 The phenotype of Rora−/− mice is very similar compared to that of staggerers.30 Interestingly, Rorasg/sg mice experience a variety of metabolic benefits (see chapter 2.2) but also deficiencies like an impaired immune system, increased inflammation, osteopenia, muscle atrophy, atherosclerosis (all reviewed in ref. 31), and irregularities in circadian rhythm.32 Since RORβ is found mainly in the CNS, especially in areas connected to processing of sensory information (retina) or involved in circadian rhythm (suprachiasmatic nucleus),33 it is not surprising that Rorb−/− mice show several related issues including retinal degeneration and blindness in adulthood as well as abnormalities in circadian rhythm and male sexual behavior.34 Lastly, RORγ was proven to be key for the development of lymphatic tissues and reduces apoptosis of CD4+CD8+ cells by up-regulating the anti-apoptotic gene Bcl-XL.35,36
Interestingly, circadian rhythm and RORs have been shown to be directly connected. RORs upregulate the expression of brain and muscle ARNT-like 1 (Bmal1),32,37 a subunit of a key transcription factor in circadian regulation called CLOCK-BMAL1 (CLOCK = circadian locomotor output cycles kaput) (reviewed in ref. 38). Of note, neuronal PAS domain protein 2 (NPAS2), a paralog of CLOCK, can substitute for it39 and was also shown to be under the control of RORs.40 Briefly, CLOCK-BMAL1 controls the expression of cryptochrome (Cry) and period (Per) as well as various other clock-regulated genes. After being expressed in a sufficient amount, the CRY-PER dimer can inhibit CLOCK-BMAL1 and is later subjected to ubiquitination, after which the cycle can start anew.38 On the other hand, RORs themselves experience a rhythmic expression.41 For instance, RORγ was shown to be under the control of CLOCK-BMAL1 in certain tissues (e.g. the liver) while its isoform RORγt exhibits constitutive expression.42 It was proposed that RORs act as “intermediaries” between the circadian clock and the cyclic expression of certain genes, affecting the extent of gene expression rather than rhythmicity itself.43,44 This was proven for metabolic genes like insulin induced gene 2a (Insig2a), elongation of very long chain fatty acids 3 (Elovl3), Cyp8b1, glucose-6-phosphatase (G6pc) and phosphoenolpyruvate carboxykinase (Pepck), among others.43,44 Furthermore, the expression of numerous phase I and II enzymes was shown to be controlled by RORα and RORγ and thus a connection to bile acid synthesis, drug and fatty acid metabolism as well as glutathione conjugation was established, to name a few.45
Several types of cancer are linked to an increase or decrease in the activity of all three RORs (reviewed in ref. 46) as well. In short, RORα showed tumor suppressive activities that were amongst others mediated by p53 (ref. 47–50) and the value of RORγt as a target in tumor therapy is currently under investigation (see chapter 2.2).
RORγ and especially its isoform RORγt are the most researched RORs due to their connection to various inflammatory and autoimmune diseases such as multiple sclerosis,51 rheumatoid arthritis (RA),52 systemic lupus erythematosus (SLE),53 psoriasis,54,55 asthma,56,57 and, again, cancer46,58,59 (also reviewed in ref. 60). This is due to the role of RORγt as a critical regulator of Th17 cell differentiation.61 The underlying mechanisms of Th17 cell differentiation are complex (Fig. 1). The expression of RORγt requires IL-6 and TGF-β.61 Upon activation, both cytokines are secreted by dendritic cells, which promotes differentiation of CD4+ cells into Th17 cells. Then, these cells up-regulate the IL-23 receptor and increase the expression of key cytokines like IL-17A/F.61 Subsequently, IL-17 is able to promote IL-6 production in various cell types (reviewed in ref. 62). While IL-23 is not necessary for Th17 cell differentiation, it is required to maintain their differentiated state.63Via signal transducer and activator of transcription 3 (STAT3), IL-6 also increases the expression of Il21, which henceforth acts in an autocrine manner, promoting Th17 differentiation.64 Of note, both cytokines can increase RORγt protein levels STAT3-dependently (reviewed in ref. 65). However, not only RORγt, but also RORα expression is necessary for Th17 differentiation through the aforementioned cytokines and STAT3.66 It is known that by upregulating forkhead box P3 (Foxp3), TGF-β promotes an immunosuppressive response via Treg differentiation,67 while FOXP3 also inhibits RORγt function and thus Th17 differentiation.68 However, TGF-β is also required for Th17 differentiation via up-regulation of Il23r.69 It was elucidated that these phenomena were dependent on the TGF-β concentration and the presence or absence of certain cytokines during differentiation: high TGF-β concentrations led to an increase in Foxp3 expression and a decrease in Il23r expression, thus favoring Treg differentiation. On the other hand, low concentrations of TGF-β were found to enhance Il23r and inhibit Foxp3 expression in concert with IL-6 and IL-21,64,70,71 thus favoring Th17 differentiation.68,72
 | ||
| Fig. 1 (a) Differentiation of CD4+ cells into Th17 or Treg cells. Activated dendritic cells secrete a variety of cytokines like IL-6, IL-23 and TGF-β. These cytokines at varying concentrations are responsible for either Th17 or Treg differentiation. High concentrations of IL-6 and low concentrations of TGF-β favor Th17 differentiation. STAT3 downstream of IL-6 signaling is responsible for the expression of IL-21, which henceforth acts in an autocrine manner. Via STAT3, IL-6 and IL-21 promote RORα and RORγt expression, the latter being an important regulator of Th17 cell differentiation. Both RORs then drive IL-17A/F and IL-22 expression. Inverse agonists of RORγt like digoxin were shown to inhibit Th17 differentiation. IL-23 is necessary for the maintenance of the Th17 phenotype and TGF-β seems to promote the expression of its receptor. Tregs are created in the presence of higher TGF-β concentrations and in the absence of pro-inflammatory cytokines like IL-6. Via up-regulation of FOXP3, TGF-β inhibits RORγt function, thus favoring Treg differentiation. (b) Inverse RORγ(t) agonists like digoxin can lead to a re-balance of the Th17/Treg ratio in favor of anti-inflammatory Tregs. | ||
2.2 Consequences of ROR (inverse) agonism and RORs as drug targets
Inverse agonists of RORα will face difficulties to succeed as drug targets, mainly due to the aforementioned tumor suppressive capabilities of this nuclear receptor (see chapter 2.1). From a metabolic perspective, however, there are some interesting implications of RORα inverse agonism that were first discovered in Rorasg/sg mice, including drastically reduced triglyceride and apo-CIII levels,73 enhanced breakdown of fatty acids, reduction in lipogenesis, prevention of weight gain74 and elevated glucose uptake in skeletal muscle cells.75 Furthermore, the synthetic RORα inverse agonist SR3335 was shown to decrease the expression of two major gluconeogenic enzymes in mice, G6pc and Pepck, thus lowering blood glucose levels and potentially being useful in type 2 diabetes therapy.76 Of note, the inhibition of G6PC as a therapy strategy for type 2 diabetes has been proposed before,77 although not in the context of RORα inverse agonism. Conversely, RORα agonists could play a role in the therapy of inflammatory diseases (e.g., RORα promotes Ikba expression78), atherosclerosis (e.g., RORα promotes Abca1/Abca8/g1 and Apoa1 expression, thus increasing cholesterol efflux and HDL formation74,79), cancer (e.g. the synthetic RORα agonist SR1078 increased p53 stability49) or possibly even disorders linked to circadian rhythm. Furthermore, RORα was shown to promote Ibsp expression, with the according protein being involved in bone mineralization.80 While the therapeutic potential of RORβ has not been explored much hitherto, in a more recent study, a connection between this nuclear receptor and bone loss was reported via RORβ-dependent inhibition of RUNX2.81 Thus, RORα and RORβ possibly could be targets in osteoporosis therapy. Moreover, in the last few years an aminothiazole compound has been identified as a dual inverse agonists of RORβ and RORγ82 and derivatives thereof were reported to be neutral antagonists of RORβ.83 These findings could benefit further research on RORβ. Both, RORγt agonists and inverse agonists were shown to have the potential to be used as therapeutics. RORγt agonism using the synthetic compound SR0987 showed an increase in IL-17 and a decrease in programmed cell death protein 1 (PD-1) mRNA levels in vitro, which indicates a possible beneficial combination in cancer therapy. More importantly, they found a decline in T cells expressing PD-1 on their surface following SR0987 treatment, although it is unclear how exactly RORγt and PD-1 are connected.84 Still, the mechanisms involved in an antitumor activity of RORγt agonism seem to be far more complex than that, with a wide range of co-stimulatory receptors (e.g. CD137) up-regulated and co-inhibitory receptors (e.g. TIGIT) down-regulated in T17 cells in response to a RORγt agonist.85 Interestingly, the synthetic RORγt agonist cintirorgon (= LYC-55716) was deemed safe for use in various types of cancer in a recent phase I clinical trial.86 Myeloid-derived suppressor cells (MDSC) are known suppressors of the immune system – especially T cells – (reviewed in ref. 87) and can directly exert various pro-tumor effects (reviewed in ref. 88). In a paper published in 2015 by Strauss et al.,89 a connection between MDSC-expansion and RORγ was described. RORγ acts by promoting positive (C/EBPβ) and suppressing negative (Socs3 and Bcl3) transcriptional regulators of myelopoiesis.89 When transplanting bone marrow of Rorc-deficient mice into lethally irradiated WT mice, they saw a significant decrease in tumor growth, metastasis and splenic MDSC in the recipients. Conversely, treating tumor-bearing WT-mice with the RORγ agonist SR1078 increased lung metastatic burden and splenic MDSC.89 RORγ(t) inverse agonists are interesting due to their anti-inflammatory potential. Most of the RORγ(t) ligands currently in clinical development have psoriasis as their target indication (reviewed in ref. 19). This is probably due to the promising results gathered from compounds like A213.90 A213 was successfully used for oral treatment of psoriasis in two different mouse models of this disease.90 Although one of the most promising candidates in this field, the RORγt inverse agonist VTP-43742, failed in phase II, the development of novel compounds is on the rise.19 In 2016, Wang et al. reported that RORγ is overexpressed in tumors of patients suffering from metastatic castration-resistant prostate cancer (mCRPC) and able to increase the expression of the androgen receptor. Consequently, RORγ inverse agonists (e.g. SR2211) were found to inhibit androgen receptor signaling and could therefore represent novel therapy options in mCRPC.91 Recently it was discovered that RORγ is a pivotal regulator of cholesterol biosynthesis in triple-negative breast cancer cells and that its inhibition exhibits antitumor effects, for instance in patient-derived xenografts.92 From a metabolic perspective, Rorc−/− mice displayed a time-dependent decrease in gluconeogenesis and an improvement in glucose tolerance and insulin sensitivity, suggesting a therapeutic potential for RORγ inverse agonism in diabetes type II as well.43 Noteworthy, RORα often exerts its effects in synergy with RORγ. For instance, staggerer-Rorc−/− double knockout mice showed significantly lowered blood glucose levels compared to WT littermates, though these effects could not be observed in either, staggerer or Rorc−/− mice alone.45 Regarding anti-inflammatory capabilities, it was shown that Th17 differentiation is not completely abolished in the absence of RORγt alone,61 but rather by a RORα–RORγ-double deficiency.66 Importantly, mice that lacked both nuclear receptors (Rorasg/sg/c−/−) experienced complete protection against experimental autoimmune encephalomyelitis (EAE),66 an animal model for multiple sclerosis.93 Both examples indicate that in some instances, inhibition of more than one ROR at once is desirable.3 Natural ligands directly binding to RORs
An overview of natural ligands directly binding to RORs is depicted in Fig. 2 and Table 1. | ||
| Fig. 2 Natural products as ligands of RORs. Various natural products were shown to act as ROR (inverse) agonists, thus affecting ROR target gene expression. | ||
| Natural product | Structure | Target(s) | Comment(s) | Reference(s) |
|---|---|---|---|---|
| Cholesterol |

|
RORα agonist | EC50 = 200 nM (co-activator binding to RORγ LBD in AlphaScreen® assay)100 | 1, 8, 96 and 100–103 |
| No activity in RORα/β/γ-LBD:Gal4-DBD luciferase assay,1,100–103 RORγ Pcp2 promoter luciferase co-transfection assay in Cos-7 cells,100 co-activator binding to RORα LBD in AlphaScreen® assay,100 25-[3H]OHC competition assay101 | ||||
| Cholesterol sulfate |

|
RORα agonist | EC50 = 7.1 nM | 2, 8, 9, 96 and 103–105 |
| Activity = 88% (FRET assay)9 | ||||
| 4α-Carboxy, 4β-methyl-zymosterol |

|
RORγ agonist | 1 | |
| Desmosterol |
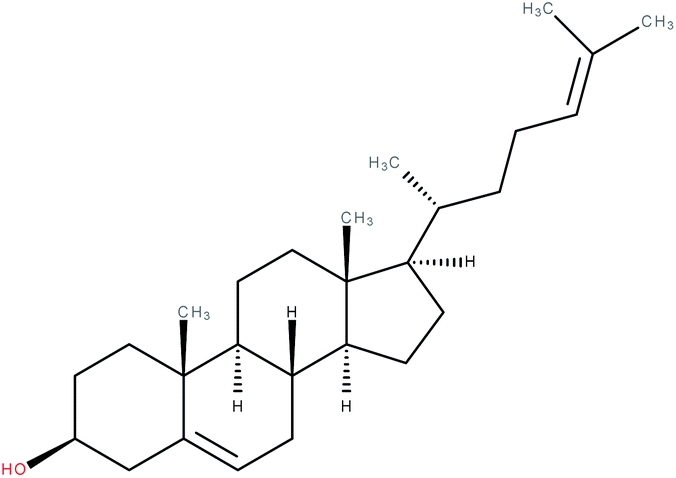
|
RORγ agonist | EC50 = 0.08 μM (co-activator binding to RORγt LBD using a TR-FRET assay) | 2 |
| Zymosterol |

|
RORγ agonist | EC50 = 0.11 μM (co-activator binding to RORγt LBD using a TR-FRET assay) | 2 |
| 25-Hydroxycholesterol |

|
RORγ agonist | EC50 = 20–40 nM (co-activator binding to RORγ LBD in AlphaScreen® assay)100 | 1, 96, 100, 103, 108, 155 and 172 |
| K d of [3H]-25-OHC for RORα LBD = 3.3 ± 0.89 nM (ref. 103) | ||||
| K d of fluorescein labeled 25-OHC for RORγ LBD = 109 nM (ref. 155) | ||||
| 7α-Hydroxycholesterol |

|
RORα/γ inverse agonist | IC50 = 1.3 μM (RORα-LBD:Gal4-DBD luciferase assay) | 103 |
| IC50 = 1.6 μM (RORγ-LBD:Gal4-DBD luciferase assay) | ||||
| K i (RORα LBD) = 12–18 nM (radioligand binding assay vs. [3H]-25-OHC) | ||||
| K i (RORγ LBD) = 17–31 nM (radioligand binding assay vs. [3H]-25-OHC) | ||||
| IC50 = 1.3 μM (RORα G6PC promoter luciferase co-transfection assay in HEK-293 cells) | ||||
| IC50 = 1.7 μM (RORγ G6PC promoter luciferase co-transfection assay in HEK-293 cells) | ||||
| Cerebrosterol (24S-hydroxycholesterol) |

|
RORα/γ inverse agonist | IC50 = 620 nM (RORα-LBD:Gal4-DBD luciferase assay) | 112 |
| IC50 = 1300 nM (RORγ-LBD:Gal4-DBD luciferase assay) | ||||
| K i (RORα LBD) = 27 nM (radioligand binding assay vs. [3H]-25-OHC) | ||||
| K i (RORγ LBD) = 25 nM (radioligand binding assay vs. [3H]-25-OHC) | ||||
| 24R-Hydroxycholesterol |
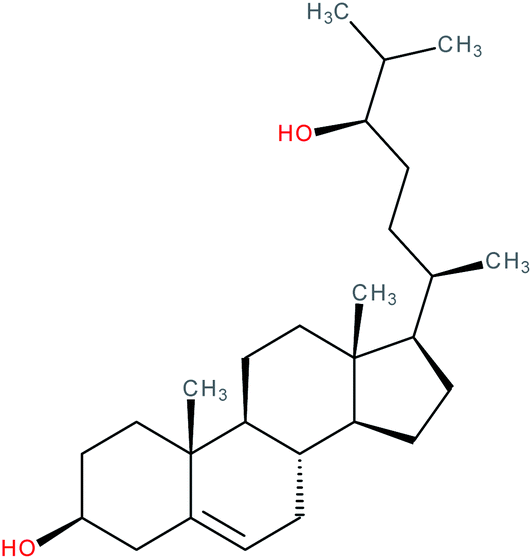
|
RORγ inverse agonist | IC50 = 90 nM (RORγ-LBD:Gal4-DBD luciferase assay) | 112 |
| K i (RORγ LBD) = 102 nM (radioligand binding assay vs. [3H]-25-OHC) | ||||
| Secosteroids e.g. 1,25(OH)2D3 |
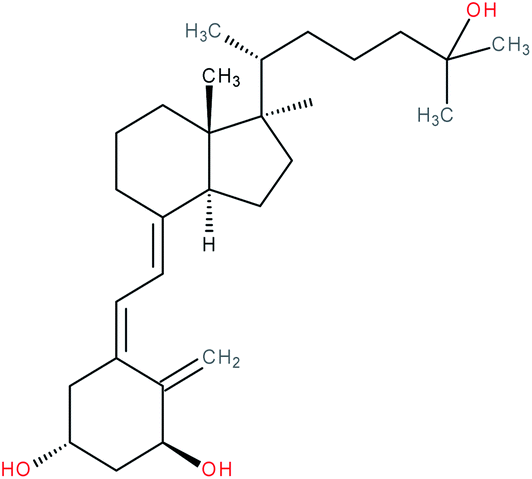
|
RORα/γ inverse agonist | IC50 = 0.1–0.01 nM (luciferase reporter assay in RORα or RORγ stable transfected CHO Tet-on cells co-transfected with 5 × RORE) | 119 |
| Neoruscogenin |
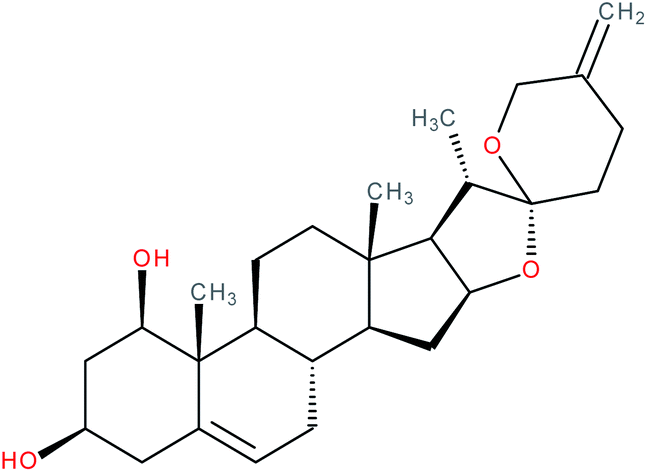
|
RORα agonist | EC50 = 0.11 μM (pull-down assay) | 121 |
| 25S-Ruscogenin |

|
RORα agonist | EC50 = 0.78 μM (pull-down assay) | 121 |
| Ursolic acid |

|
RORγ(t) inverse agonist | IC50 = 0.68 ± 0.1 μM (co-activator binding to RORγt LBD using a TR-FRET assay)123 | 123, 124, 138 and 139 |
| IC50 = 0.56 ± 0.1 μM (Th17 cell differentiation)123 | ||||
| EC50 = 0.25 μM (co-activator release from RORγ LBD in FRET assay)138 | ||||
| IC50 = 1.3 μM (inhibition of co-activator binding to RORγt LBD in AlphaScreen® assay)139 | ||||
| IC50 = 6.5 μM (inhibition of co-repressor binding to RORγt LBD in HTRF assay)139 | ||||
| K d = 3.20 μM (SPR binding assay with RORγt protein)139 | ||||
| Betulinaldehyde |
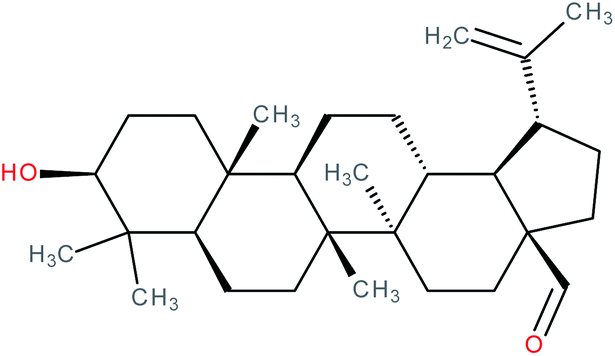
|
RORγt agonist | EC50 = 11.4 μM (co-activator binding to RORγt LBD in AlphaScreen® assay) | 139 |
| IC50 = 15.6 μM (inhibition of co-repressor binding to RORγt LBD in HTRF assay) | ||||
| K d = 2.99 μM (SPR binding assay with RORγt protein) | ||||
| 3β,28-Dihydroxy-lupan-29-oic acid |

|
RORγt inverse agonist | IC50 = 6.8 μM (inhibition of co-activator binding to RORγt LBD in AlphaScreen® assay) | 139 |
| IC50 = 19.8 μM (inhibition of co-repressor binding to RORγt LBD in HTRF assay) | ||||
| K d = 1.47 μM (SPR binding assay with RORγt protein) | ||||
| Methyl corosolate |
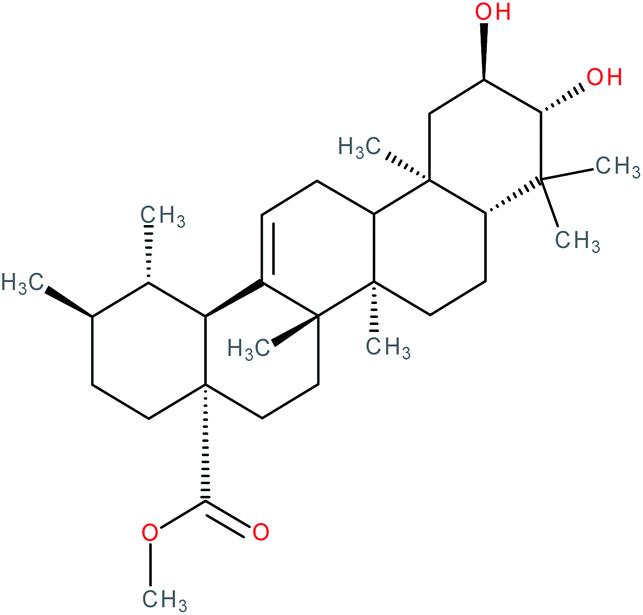
|
RORγt inverse agonist | IC50 = 6.512 μM (RORγ-LBD:Gal4-DBD luciferase reporter assay in Jurkat cells) | 141 |
| Uvaol |
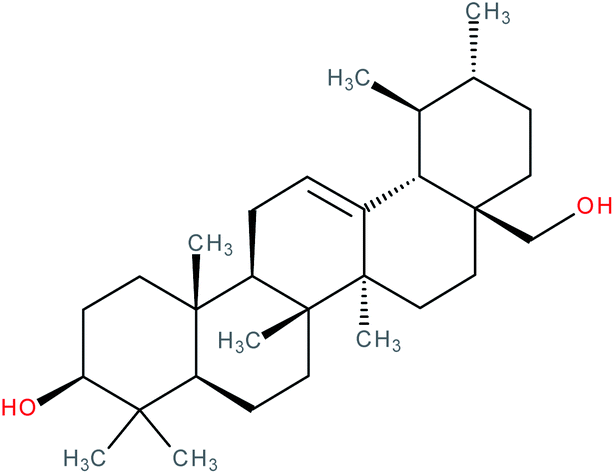
|
RORγt inverse agonist | IC50 = 4.254 μM (RORγ-LBD:Gal4-DBD luciferase reporter assay in Jurkat cells) | 141 |
| Oleanolic acid |

|
RORγt inverse agonist | IC50 = 8.589 μM (RORγ-LBD:Gal4-DBD luciferase reporter assay in Jurkat cells) | 141 |
| Rockogenin |

|
IC50 = 0.2 μM (IL-17 production in Th17 cells)143 | 138 and 143 | |
| IC50 = 5.1 μM (RORγ-LBD:Gal4-DBD luciferase reporter assay)143 | ||||
| EC50 = 2.5 μM (co-activator displacement FRET assay)138,143 | ||||
| All-trans retinoic acid |

|
RORβ inverse agonist | K d = 280 nM (radioligand competition assay)102 | 102 and 144 |
| IC50 = 0.15 nM (RORβ-LBD:Gal4-DBD luciferase reporter assay)102 | ||||
| Amethinol A |

|
RORγ(t) inverse agonist | 145 | |
| Biochanin A |
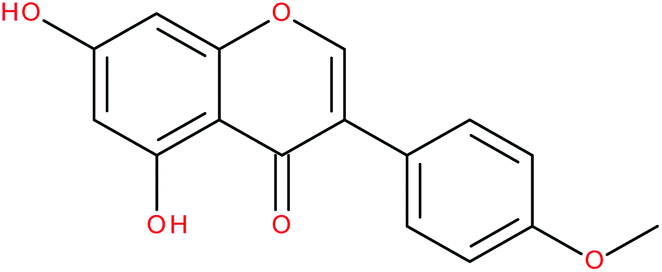
|
RORα/γ-agonist | 147 and 148 | |
| Genistein |

|
RORα/γ-agonist | 147 and 149 | |
| Formonenetin |

|
RORα/γ-agonist | 147 | |
| Daidzein |

|
RORα/γ-agonist | 147 | |
| Nobiletin |

|
RORα/γ-agonist | K i (RORα-LBD) = 53.4 nM (radioligand binding assay vs. [3H]-25-OHC)152 | 152–154 |
| K i (RORγ-LBD) = 8.0 nM (radioligand binding assay vs. [3H]-25-OHC)152 | ||||
| Digoxin |
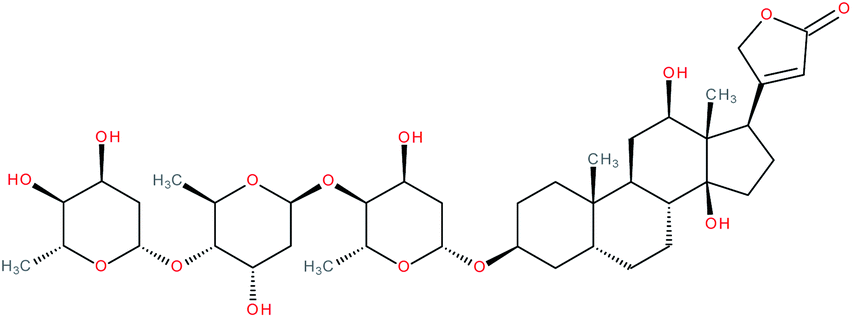
|
RORγ(t) inverse agonist155 and RORγ(t) agonist171 | IC50 = 1.98 μM (RORγ-LBD:Gal4-DBD luciferase reporter assay in S2 cells)155 | 6, 155, 157, 161, 162, 165–167 and 171 |
| IC50 = 4.1 μM (in vitro competition assay with fluorescein-labelled 25-OHC)155 | ||||
| IC50 = 1.8 μM (displacement of NCOA3-1b co-activator peptide)155 | ||||
| IC50 = 3.9 μM (promotion of NCOR2 co-repressor peptide binding)155 | ||||
| Digitoxin |

|
RORγ(t) inverse agonist | IC50 value “similar” to digoxin according to the authors (luciferase reporter assay in S2 cells) | 155 |
| β-Acetyldigoxin |

|
RORγ(t) inverse agonist | IC50 value “similar” to digoxin according to the authors (luciferase reporter assay in S2 cells) | 155 |
3.1 Steroids
When the crystal structure of the human RORα LBD was first elucidated the authors found a ligand within its ligand binding pocket that turned out to be cholesterol.8 This was confirmed using mass spectrometry (MS).8,96 Mutations in the LBD impairing cholesterol binding as well as inhibition of cholesterol synthesis using a statin resulted in a decrease in transcriptional activity in a full-length RORα luciferase reporter assay. Depleting cells of cholesterol using hydroxypropyl-β-cyclodextrin (HPCD) and a statin also led to a decrease in transcriptional activity, which could be reversed by the addition of e.g. cholesterol and, even more effectively, 7-dehydrocholesterol (= provitamin-D3) at 10 μM.8 Although helix 12 in the LBD is in an active conformation when bound to cholesterol, cholesterol does not directly interact with this helix.4,8,97 However, it is known that agonists can function without this ability, for instance by stabilizing the hydrogen bond of the His–Tyr lock10 after binding, which in turn stabilizes helix 12.9,98,99 Hence, after mutating this tyrosine residue to phenylalanine, effectively eliminating the His–Tyr lock, a decrease – but no obliteration – in transcriptional activity was observed by the authors,8 indicating that other interactions must be able to stabilize the active conformation as well. In later studies, cholesterol could increase RORγ co-activator recruitment, while having no effect on co-repressor interaction and on RORα co-activator recruitment, indicating RORγ-specific agonistic properties. The affinity of cholesterol was, however, much lower when compared to other cholesterol metabolites.100 Notably, cholesterol was not able to upregulate transcriptional activity in a RORγ-Gal4, RORα-Gal4, and RORβ-Gal4 reporter assay as well as a full-length RORγ transactivation assay and was not able to displace 25-[3H]OHC in a competition assay.1,100–103
Interestingly, cholesterol sulfate was able to replace cholesterol in the ligand binding pocket of RORα and was predicted to bind the RORα LBD with even higher affinity by docking, which was proven experimentally via electrospray ionization (ESI)-MS and differential scanning calorimetry.8,96,104 The crystal structure of cholesterol sulfate in complex with the RORα LBD104 was structurally very similar to that of cholesterol and showed an agonistic conformation.8 A luciferase reporter assay in cholesterol-depleted and statin-treated cells (as described before8) showed that treatment with 10 μM cholesterol sulfate led to a higher transcriptional activity relative to cholesterol. On the other hand, a critical mutation (RORα Cys 288 → Gln) within the LBD selectively decreased the affinity of cholesterol sulfate while not affecting cholesterol binding and thus resulted in a reduced transcriptional activity relative to cholesterol.104 Moreover, cholesterol sulfate was active in a RORγ co-activator recruitment assay, RORγ-Gal4 and RORα-Gal4 reporter assays in the presence of either the inverse agonist ursolic acid or an azole (CYP51 inhibitor).2 However, another study could not observe an activity of cholesterol sulfate at 500 μM in RORγ-Gal4 or RORα-Gal4 reporter assays where cells were cultivated in lipid depleted and statin and mevalonate supplemented medium, although cholesterol sulfate was active in a 25-[3H]OHC competition assay.103 In a cell-based study, 40 μM cholesterol sulfate increased the mRNA expression levels of RORα and the RORα-regulated epidermal barrier precursor protein profilaggrin in a RORα-dependent manner in normal human epidermal keratinocytes.105
Taken together, due to the low affinity for the LBD of RORs, cholesterol seems unlikely to be a physiological ligand. However, cholesterol sulfate has been shown to be present in Th17 cells and although functional assays have not been performed, data with desmosterol sulfate (see chapter 3.1.2) suggests similar properties for cholesterol sulfate.2
In the same year Hu et al.2 found that endogenous sterol metabolites control Th17 differentiation via RORγ agonism. Notably, the inhibition of the mevalonate-cholesterol synthetic pathway via statins reduced Th17 differentiation and IL-17A production.106,107 Desmosterol as well as zymosterol potently increased co-activator recruitment in the presence of the inverse agonists ursolic acid or digoxin and RORγ-Gal4 transcriptional activity, thus suggesting that these compounds occupy the same binding site. They also both increased IL-17A production and Th17 differentiation in the presence of ursolic acid. Moreover, in Th17 cells desmosterol increased RORγ target gene expression but not RORγt itself. The RORγ-dependence of the IL-17A increase elicited by desmosterol was confirmed via knockdown of RORγ with siRNA during differentiation and the use of T cells from RORγ knockout mice. Quantification of selected sterols in Th17 cells revealed that only cholesterol and desmosterol were detectable. Furthermore, sulfated sterols, especially desmosterol sulfate, were basally or in the presence of ursolic acid more potent agonists of RORγ than the corresponding 3-OH sterols. Higher production of sulfated sterols together with the fact that desmosterol sulfate as well as cholesterol sulfate could be quantified in Th17 cells, suggests that sterol sulfates might act as endogenous RORγ agonists in Th17 cells.
Taken together, upregulation of cholesterol biosynthesis and uptake and simultaneous downregulation of cholesterol metabolism and efflux during Th17 differentiation leads to the accumulation of the cholesterol precursor desmosterol and its sulfate conjugates, which then act as endogenous RORγ agonists in Th17 cells.2
20α-Hydroxycholesterol (20α-OHC), 22(R)-hydroxycholesterol (22R-OHC), and 25-hydroxycholesterol (25-OHC) were all active with similar affinity in RORγ co-activator recruitment assays with EC50 values between 20 and 40 nM, while being inactive in RORγ co-repressor recruitment and RORα co-activator recruitment assays.100 Notably, in another study 22R-OHC and 22S-OHC (10 μM) were both not able to elicit an effect in RORγ- or RORα-Gal4 assays.103 Co-crystal structures with the RORγ LBD, the co-activator peptide NCOA2-2 and the putative ligands 20α-OHC, 22R-OHC, and 25-OHC revealed very similar LBD structure for all of them with the C-terminal AF-2 in the active conformation, suggesting they act as RORγ agonists. Although they take up the same position in the LBD, binding is dependent on unique pocket residues relevant for size and polarity, as could be shown with a mutagenesis approach. Transcriptional activation of RORγ-Gal4 and full-length RORγ confirmed the agonistic properties of these oxysterols and mutation of the unique binding residues in the RORγ LBD abolished the activity of the respective oxysterols. Interestingly, sulfation of cholesterol and oxysterols carried out via SULT2B1 overexpression led to a decrease in transcriptional activity and re-supplementation of these oxysterols partly reversed this effect. It was further revealed that 25-OHC interacts with the two amino acid residues involved in the His–Tyr lock, indirectly with tyrosine on helix 12 and directly with histidine on helix 11.9,10 Although 25-OHC was not active in a RORα co-activator recruitment assay,100 it was shown to directly bind to the LBD of RORα in a MS approach96 and slightly but non-significantly to reduce activity in a RORα-Gal4 assay,103 suggesting inverse agonism. Interestingly, the enzyme CH25H , which is responsible for 25-OHC production, is downregulated in bone marrow-derived macrophages from sg/sg mice.108 Lipid storage is disturbed in these cells, which could be restored by treatment with physiological concentrations of 25-OHC,109 pointing to 25-OHC as endogenous ligand.
Next to the already mentioned compounds, other oxysterols, like 22S-OHC and 27-OHC did not influence RORα or RORγ activity.100,103
Given the role of RORs in bile acid metabolism,45 7α-hydroxycholesterol produced by CYP7A1, the key enzyme in bile acid synthesis,110 and other related 7-oxysterols (7-OS, 7β-hydroxycholesterol and 7-ketocholesterol) were investigated on RORα/γ.103 A competitive radioligand binding assay performed against tightly bound 25-[3H]OHC indeed showed that all these 7-OS bind with high affinity to the RORα/γ LBD. Furthermore, 7-OS decreased transcriptional activity in a RORα/γ-Gal4 and full-length RORα/γ assay with the RORE-containing G6PC promoter43,111 in HEK-293 cells, which suggests an inverse agonistic mechanism of action. When mutating this RORE, the effects of the sterols vanished.103 Furthermore, 7-OS inhibited the mRNA expression of G6PC in HepG2 cells and ChIP experiments revealed a 7-OS-dependent decrease in NCOA-2 recruitment to the G6PC promoter. A reChIP experiment using 7α-OHC confirmed the RORα dependence of the decrease in NCOA-2 recruitment to the promoter. Together with the data obtained by hydrogen deuterium exchange MS, the authors proposed a model of RORα/γ being in a constitutively active (and thus co-activator-bound) state, where inverse agonists such as 7-OS can interfere with this process. Finally, in a more functional setting using RORα/γ siRNA in murine hepatocytes, the metabolic effects of 7-αOS (decrease in G6pc/Pepck gene expression and glucose output) was obliterated, as expected from direct ligands like 7-OS. Additionally, the tested 7-OS showed no affinity towards LXR in a Gal4 luciferase assay. Possible effects on other nuclear receptors were not examined.103 In addition to these three 7-OS, GC-MS revealed that 7-dehydrocholesterol acts as a ligand of RORα.96
Later additional oxysterols were investigated.101 In RORγ-Gal4, full-length RORγ and RORγt reporter assays several oxysterols showed significant agonistic activity in the presence of the inverse agonist ursolic acid, with the highest potency and efficacy observed for 27-OHC and 7β, 27-OHC. Notably, in this assay 7α, 25-OHC was inactive, while 7-keto, 27-OHC was only active in the Gal4 but not in the full-length transcriptional assays. Several oxysterols that were previously reported as RORγ or RORα inverse agonists or agonists101,103,112 including 25-epoxycholesterol and 7-ketocholesterol were only weakly to moderately active. Cholestenoic acid derivatives of 27-hydroxylated sterols were only barely active, emphasizing the importance of the hydroxyl group at carbon 27 for RORγt agonism. Many of the tested oxysterols had activities on other nuclear receptors, however, 7β, 27-OHC and 7α, 27-OHC seemed to be the most selective RORγt agonists. Direct binding of oxysterols was investigated in thermal shift assays with RORα/β/γ LBD and the co-activator peptide NCOA1, where 27-OHC oxysterols bound most potently to the RORγ LBD (7β, 27-OHC > 7-keto, 27-OHC > 27-OHC > 7α, 27-OHC). Notably, 27-OHC binding to the RORγ LBD was significantly lower in the presence of NCOA1, suggesting that this oxysterol is no endogenous RORγ agonist.101 In a functional approach 7β, 27-OHC and 7α, 27-OHC, but not 7α, 25-OHC increased the number of IL-17A producing cells from total or naïve mouse and human CD4+ T cells under Th17 differentiating conditions in the absence or presence of ursolic acid. RORγt dependency was confirmed with RORγt-deficient mouse CD4+ T cells. In addition, it was confirmed that 7β, 27-OHC and 7α, 27-OHC do not activate RORα, as functional RORα is expressed in RORγt knockout cells. The production of 27-OHCs are dependent on the enzyme CYP27A1. Mouse Cyp27a1 knockout-naïve CD4+ cells showed significantly reduced Th17 differentiation and exogenous addition of 7β, 27-OHC restored this effect, suggesting a physiological role of 27-OHC oxysterols in this process. The importance of CYP27A1 and 7β/α, 27-OHCs for Th17 differentiation was confirmed in vivo in mice. Cyp27a1 knockout mice had elevated 25-OHC levels, which led the authors to the conclusion that this oxysterol is unlikely to function as endogenous RORγt agonist.101
Another oxysterol studied in more detail is 24S-OHC (cerebrosterol), mostly found in the brain.112,113 It showed similar effects on RORα and RORγ transcriptional activity in Gal4 assays as 7-oxysterols. Using ChIP-reChIP assays with the coactivator NCOA2, 24S-OHC was demonstrated to reduce recruitment of this peptide to RORα. Notably, also 24(S),25 epoxycholesterol, found in micromolar concentrations in the liver and brain,114,115 and 24R-OHC act as specific partial inverse agonists for RORγ with an IC50 of 280 nM and 90 nM in a Gal4 assay and a Ki value of 20 nM and 102 nM in a competition assay against 25-[3H]OHC, respectively, with no activity on RORα.112 The high abundance of cerebrosterol in young children113 and its RORα agonistic properties led the authors to propose a role for it in the developing brain.112 It is interesting to see that only small structural changes in the molecules can evidently lead to a specificity for a certain ROR protein, which can be explained by the differences in the LBD amongst RORs (reviewed in ref. 24). Noteworthy, all compounds tested in this study were previously identified to be agonists of LXR.116
In differentiated Th17 cells, oxysterols could not be detected2 but in naïve T-cells, 27-OHCs were quantifiable.101 Moreover, the level of 27-OHC has been reported to be 5 times lower than that of desmosterol in human plasma.117,118 The physiological relevance of different sterols thus still seems to be not completely clear and needs further evaluation.
3.2 Terpenoids
In another study124 ursolic acid lessened the incidence and severity of collagen-induced arthritis in mice while decreasing the expression of the proinflammatory cytokines TNF-α, IL-1β, IL-6, IL-21, and IL-17 and the oxidative stress markers iNOS and nitrotyrosine. Ursolic acid moved the balance between Treg and Th17 cells in the spleen of these mice to the Treg side, consistent with a reduced expression of IL-17, IL-21, phosphorylated (p)-STAT3, and RORγt. Moreover, the inhibitory effect of ursolic acid on Th17 cell differentiation was confirmed in an in vitro model. However, in this model the amount of Treg cells was not influenced, as shown via Foxp3 expression.124 Some of these results are in contrast to the findings from Xu et al. as outlined before.123 In their study ursolic acid did not influence STAT3 phosphorylation and RORγt mRNA levels in Th17 cells. The authors argued that these discrepancies might result from different Th17 differentiation cocktails or cell types used.123,124 Moreover, ursolic acid has been reported to inhibit STAT3 activation in many other model systems.125–131 STAT3 has been previously shown to be required for Th17 differentiation in vivo and to act upstream of RORγt.3,61,132 Furthermore, ursolic acid is a known inhibitor of the NF-κB pathway.133 Inhibition of NF-κB leads to decreased expression of IL-6, which in turn acts as activator of STAT3 signaling.134–137 This suggests that ursolic acid modulates the transcriptional activity of RORγ also indirectly via NF-κB inhibition. More mechanistically, ursolic acid was able to displace a co-activator and co-repressor peptide in vitro.138,139 Direct interaction of ursolic acid with the LBD was suggested via an increase in melting temperature (Tm) in a thermal shift assay and via surface plasmon resonance (SPR).139 A co-crystal structure of ursolic acid and the RORγ-LBD suggested a unique mode of action. Ursolic acid was shown to form a hydrogen bond with a histidine residue leading to a flip of helix 11, moving it closer towards helix 12, thereby causing a disordered C-terminus. They hypothesize that this flip leads to the displacement of the co-activator peptide and prevents the recruitment of a co-repressor.138 Lastly, RORγt selectivity (over RORα and RORβ) was evaluated by an AlphaScreen® assay.139
Since ursolic acid has many other targets besides RORγt (for a comprehensive overview, see ref. 140) it must be carefully evaluated to what extent this could become an issue when using it for therapeutic purposes in humans.
3.3 Polyketides
These contradicting results regarding IL-17 for isoflavones might stem from the different model systems used.
3.4 Cardiac glycosides
In contrast to the studies mentioned above, digoxin showed no effect on murine Th17 cell differentiation at 10 μM in a later study.157
After digoxin was established as an inhibitor of RORγ(t),6,155 its value for treating various, mostly inflammatory diseases was examined in several preclinical studies.
3.4.1.1 Preclinical studies performed with digoxin. In a study by Huh et al., digoxin but not its aglycone was able to ameliorate EAE in mice when compared to the vehicle control DMSO.155 The onset of the disease was pushed back by a few days and the clinical scores were consistently (and significantly) lower in the treatment group. A significantly decreased amount of Th17 cells were found in the spinal cords of mice in the treatment group compared to control while Th1 cells were left mostly unaffected,155 which contrasts with other observations.123 Previously, it has been shown that transplant rejection is connected to Th17 cells and IL-17,158,159 and direct antagonism of IL-17 (ref. 160) could suppress this process in rats. Accordingly, in a study investigating digoxin's effect on heart transplant rejection in mice, treatment with the cardenolide doubled the survival time compared to control. Moreover, inflammation and necrosis of allografts were decreased and re-balancing of the Th17/Treg ratio in favor of Tregs was accomplished.161 Another study suggested a benefit when treating abdominal aortic aneurysm (AAA) using digoxin in mice,162 which is in line with the important role of IL-17 in AAA pathophysiology.163 The authors found a reduction of aortic diameter (key for the diagnosis and risk assessment of AAA164), a reduced incidence of AAA and a re-balancing of the Th17/Treg ratio in favor of Tregs. The survival ratio did not change, indicating a rather prophylactic value of digoxin for this indication.162 Furthermore, digoxin was shown to possess prophylactic and therapeutic capabilities in vivo as it was able to suppress and ameliorate collagen-induced arthritis in mice.165 Inflammation and cartilage loss were markedly reduced in the ankles of digoxin treated mice, as were arthritis scores and disease incidence in general. Lower expression levels of certain proinflammatory cytokines (e.g. IL-6, -17 and -21) in arthritic joints were accompanied by a significant decrease in Th17 and a rise in Treg cells in murine spleens.165 Another study examined the therapeutic potential of digoxin regarding atherosclerosis in ApoE−/− mice on a western-type diet.166 Histologically, a reduction of atherosclerosis could be quantified. After 12 weeks, a statistically significant reduction in total cholesterol, triglyceride and LDL levels was found, while HDL levels were unchanged. mRNA levels of three RORγ target genes involved in metabolism (Insig2a, Elovl3, Cyp8b1 (ref. 44)) were examined and found to be decreased significantly in the digoxin groups. In the spleens, flowcytometric measurements saw a significant decrease in Th17 cells and a significant increase in Tregs following digoxin treatment and the same was true for Tregs in atherosclerotic plaques. Also, Th17 cell invasiveness of the plaques was decreased upon digoxin treatment.166 A further study used a murine model in order to examine the possible benefits of digoxin treatment in inflammatory bowel diseases (IBD).167 Weight loss due to colitis and colon colitis scores were reduced significantly in the treatment group and fewer proinflammatory CD3+ T cells were found in the colonic mucosa. Th17 cells (together with IL-17A and IL-23R mRNA levels) were decreased and Tregs (together with IL-10 mRNA levels) were increased significantly in the colon due to digoxin treatment. When colitis was induced in mice using CD4+ cells of Il10 knockout mice, digoxin treatment led to a significant decrease in weight loss, but histological scores were not significantly different between the groups. Therefore, they suggested a beneficial effect of digoxin treatment in disorders like Crohn's disease through direct inhibition of RORγt in a partly IL-10 dependent fashion.167
Importantly, these studies were all performed in mice. Cardiac glycosides are known to bind to the Na+/K+-ATPase α1 subunit of rodents with a significantly decreased affinity. Therefore, concentrations that are toxic in humans can be expected to be tolerated well in mice.168–170 For this reason, the results of such studies cannot simply be applied to humans but give an impression of the clinical values of RORγ(t) inhibition in general.
3.4.1.2 Digoxin as RORγ(t)-agonist?. Surprisingly, another group reported agonism of digoxin on RORγ(t) in a RORγ transactivation assay.171 This contrasts with the results published in 2011 by Huh et al.155 and Fujita-Sato et al.6 who proposed the exact opposite. While Huh et al. and Fujita-Sato et al. worked in the micromolar concentration range using mostly insect or murine cells,6,155 Karaś et al. employed 100 nM, since cytotoxic effects on HepG2 and human Th17 cells were observed at concentrations as low as 200 nM. Interestingly, when using 10 μM of digoxin (as in ref. 66 and 155) the authors observed cell viabilities of approx. 40% and 5% for HepG2 and human Th17 cells, respectively. They also reported increased expression of the RORγ-regulated genes G6PC and NPAS2 in HepG2 cells following digoxin treatment. In agreement with previous studies, a mammalian one-hybrid assays with RORγ-LBD:Gal4-DBD and RORα-LBD:Gal4-DBD constructs showed a RORγ specific activity of digoxin.6,155,171 Interestingly, when overexpressing human and mouse RORγ(t) in (RORE)6-tk-Luc transfected HepG2 reporter cells, digoxin was shown to have a greater effect on the murine compared to the human variants.171 However, this finding was not explored further. Moreover, the authors performed an electrophoretic mobility shift assay, which showed that digoxin treatment increased nuclear protein binding to a RORE-containing DNA probe. A ChIP assay showed increased RORγt and NCOA-1 (but decreased NCOA-2) binding in the promoter region of RORγ-regulated genes (G6PC, NPAS2, IL17).171 It would be interesting to see whether the increase in co-activator occupancy is dependent on RORγ via a sequential ChIP experiment, as conducted in previous studies.103,112 Application of 100 nM digoxin during polarization of CD4+ cells into Th17 cells yielded higher IL-17 mRNA and protein levels and a transcriptome analysis of digoxin-treated Th17 cells derived from human donors showed the induction of certain Th17-related genes like IL17A/F. Additional docking experiments with digoxin gave the best results when the active conformation of the RORγ LBD was used and NCOA-2 was absent. Of note, docking of digoxin to the RORγt LBD domain in the inverse agonistic conformation resulted in the formation of a hydrogen bond to the histidine residue of the His–Tyr lock. Importantly, the hydrogen bond was not established via digitoxose (as seen in the co-crystal structure of Fujita-Sato et al.6) but the carbonyl residue of the butenolide ring instead.171 Why these results drastically differed compared to the earlier studies6,155 is not conclusively clarified. The authors speculate that differences in the protein biosynthesis of the insect cells (see ref. 155) – e.g. with respect to co-activators – could be responsible. However, this does not explain why Huh et al.155 found a decreased co-activator and an increased co-repressor binding to the RORγt LBD in vitro when applying digoxin, although different co-activators were used in both studies. The conversion of digoxin into its aglycone digoxigenin under experimental conditions could be another explanation for the different results, since the agonistic effect of digoxigenin was suggested in an earlier study.156 Assuming that no conversion took place, questions remain to be answered, e.g. why the X-ray analysis by Fujita-Sato et al.6 suggested an inverse agonistic rather than an agonistic mechanism of action of digoxin. Further X-ray or NMR analyses of digoxin in complex with RORγ(t) LBD could potentially provide clarification on this subject. Also, it would be interesting to repeat some key experiments of Huh et al.155 and Fujita-Sato et al.6 at lower digoxin concentrations to see if the results of this study can be reproduced. Lastly, it could indeed be possible that digoxin may function as both, agonist and inverse agonist at different concentrations, as the authors suggested.171
4 Natural products indirectly affecting RORs
4.1 Melatonin
The first proposed endogenous ligand for RORs was the amino acid hormone melatonin (N-acetyl-5-methoxytryptamine).95,173 Melatonin is produced in the pineal gland and regulates circadian rhythm, sleep–wake cycles, and seasonal reproduction in mammals, among others. Although direct binding of melatonin to RORβ has been shown initially, these results were not reproducible and the respective report has been retracted.95 However, a study from the same group showing a direct interaction of melatonin with RORα has not been withdrawn.173 In a more recent study,94 melatonin was shown to decrease RORα transactivation in the human breast cancer cell line MCF-7 in the presence of 10% FCS to 34% of control at a concentration of 10 μM and to decrease the ability of RORα to bind to its response elements, as shown in a transfection assay and a gel mobility shift assay, respectively, without affecting RORα protein levels. It has been shown before that increased [Ca2+]i levels and enhanced Ca2+/calmodulin (CaM)-dependent protein kinase IV activity stimulates RORα transcriptional activity.174 CaM kinase IV may influence the phosphorylation status of ROR co-factors, thereby modulating its activity. Accordingly, a calmodulin antagonist, calmidazole, decreased RORα transactivation similarly than melatonin.94 Melatonin is known to modulate [Ca2+]i levels via G-protein coupled membrane receptors and act as CaM antagonist.175–177 In MCF-7 cells, melatonin had no direct effect on [Ca2+]i levels, suggesting that melatonin influences RORα activity via CaM antagonism.94 In in vitro cultured goat spermatids, 0.1 μM melatonin increased RORα mRNA and protein levels. However, in CHO cells and human keratinocytes, melatonin was not able to inhibit RORα or RORγ transactivation. In addition, relatively low docking scores in in silico modeling have been obtained for RORα or RORγ, suggesting low affinity of melatonin for these receptors.119 In summary, the ability of melatonin to influence ROR transcriptional activity seems to be cell-type specific and at least in human breast cancer cells a link to the calmodulin antagonism of melatonin is suggested. However, a causal relation could not be established. Moreover, it is likely that melatonin modulates ROR expression via its influence on circadian rhythm.178,1794.2 Selected indirect modulators of RORs
A summary of studies on selected indirect ROR modulators is available in Table 2. The mechanism of action of most indirect ROR-modulators has not been elucidated, though hypotheses have been proposed. For instance, saikosaponin A has been suggested to influence RORγt protein expression via inhibition of NF-κB activation and hence IL-6 expression, a known activator of STAT3 signaling.134–137,180 Similarly, anti-inflammatory mechanisms might explain the effects of compound sophorae decoction181 and ginger extract182 on ROR. Other proposed mechanisms include the STAT5-dependent modulation of IFN-γ, or PPARγ expression, ultimately decreasing RORγt levels (e.g. primisterin,183 arctigenin,184 astragalus polysaccharide185) and the downregulation of the transcription factor HIF-1α via mTOR, leading to a decrease in RORC transactivation as suggested for rapamycin186,187 (also reviewed in ref. 188).| Substance class | Examples | Effect on RORs | Study results |
|---|---|---|---|
| Steroids and terpenoids | Dioscin, pristimerin, 3β-acetyloxy-oleanolic acid, saikosaponin A | • Decrease of RORγt mRNA189,190 and protein180,183 levels | • Amelioration of inflammatory diseases in rodents180,183,189,190 |
| • Inhibition of RORγt transcriptional activity190 | • Decrease of proinflammatory cytokines (e.g. IL-17)180,183,189,190 | ||
| • Decrease in Th17 cell differentiation183,189,190 | |||
| Polyketides | Bavachalcone, poncirin/ponciretin, quercetin, baicalein | • Increase of RORα transcriptional activity191 and Rora/RORA expression191,192 | • Increased expression of RORα target genes (e.g. BMAL1, Fgf21)191,192 |
| • Decreased expression of RORγt193,194 | • Decrease of proinflammatory cytokines (e.g. IL-17)193,194 | ||
| • Decrease in Th17 cell differentiation193,194 | |||
| • Protection from liver damage in rodents194 | |||
| Cardiac glycosides | Uscharin, calcein, calotropin, digoxigenin, dihydroouabain, strophanthidine | • Decrease of RORγt transcriptional activity157 | • Decrease in Th17 cell differentiation157 |
| • Decrease of RORγt protein levels157 | • Increased expression of RORγ(t) target genes (e.g. G6PC, IL-17)156 | ||
| • Increase of RORγ transcriptional activity156 | • Favorable scores when docked into RORγ-LBD156,157 | ||
| • Direct interaction with RORγt suggested by docking156,157 | |||
| Other substance classes | Arctigenin (lignan), epigallocatechin-3-gallate, astragalus polysaccharide & astragaloside IV (saponin), oxymatrine (quinolizidine alkaloid), rapamycin (macrolide), α-mangostin (xanthone) | • Decrease of RORγt mRNA184,186,195–198 and protein185,198 levels | • Amelioration of inflammatory diseases184,185,195,196,198 and emphysema186 in rodents |
| • Decrease of RORγt transcriptional activity199 | • Decrease of proinflammatory cytokines (e.g. IL-17)184,185,195,198,199 | ||
| • Decrease in Th17 (ref. 184 and 199) and Tc17(ref. 186) cell differentiation | |||
| Extracts | Grape seed proanthocyanidin extract, ginger extract, compound sophorae decoction | • Increase of cyclic RORA expression200 | • Amelioration of inflammatory diseases in rodents181 |
| • Increase of RORα transcriptional activity in a Gal4 system, thus direct interaction suggested200 | • Decrease of proinflammatory cytokines (e.g. IL-17)181 | ||
| • Decrease of RORγt mRNA levels181,182 | • Decrease in Th17 cell differentiation181 |
It must be stated that the extent of RORs' influence on the outcomes of these studies was not explicitly investigated in most cases. It can be assumed (and is often pointed out by the authors themselves) that other mechanisms are involved as well.
5 Conclusion and outlook
Many studies discussed the question as to whether the transcriptional activity of ROR is ligand dependent. Interestingly, one study showed that apo-RORα (expressed in E. coli and therefore deemed ligand free) is active,103 but in most reports, RORs were active only in the presence of sterol ligands (examples: ref. 1, 8, 104 and 155). To explain this discrepancy, the concept of “silent ligands” was brought up,18 which, however, also excludes RORs in an apo-state. Additionally, expression of RORs in E. coli do not necessarily yield empty LBDs.144 It is likely that intermediates and metabolites of the cholesterol metabolism act as endogenous ligands for RORs.1–3 Furthermore, it was shown that 500 nM of an endogenous ligand are sufficient to occupy 80% of available RORγ. Calculations revealed that a further increase in occupancy would require very high concentration of ligands, which might explain the moderate effect of exogenous ligands in reporter assays containing serum.1 When discussing natural products interacting with RORs, one of the most prominent and heavily investigated ligands is the cardiac glycoside digoxin. Despite its relatively high IC50 value,155 its benefits were examined in numerous preclinical studies in rodents. The cardenolide was deemed both, inverse agonist6,155 and agonist171 of RORγ(t) in different studies. Variations in experimental settings and the possibility of observing concentration-dependent effects were proposed as explanations,171 but further validation is needed. As inhibitors of the Na+/K+-ATPase, cardiac glycosides like digoxin are used to treat heart failure, among other conditions, due to their positive inotropic effects. However, the cardenolide is used with caution and only when strictly indicated due to its narrow therapeutic window.201 Digoxin dose-dependently decreased cell viability in 10 human tumor cell lines with a mean IC50 of 80 nM (ref. 168) and it is recommended to aim for serum concentrations not exceeding 0.8 ng ml−1 (approx. 1.0 nM) when treating patients.202 However, digoxin's inhibition of RORγ occurs at the micromolar concentration range6,155 and even its possible agonistic effect occurs at much higher concentrations.171 In a study on digoxin's use in atherosclerosis therapy in mice, the authors measured plasma levels after the last injection and ascertained that they were “at or below the therapeutic range for humans”,166 at least in the low dose group. But even if digoxin could be used for its effects on RORγ(t) in humans, pharmacokinetic issues arising for instance from kidney impairments203 would then have to be taken into account to prevent poisoning.In general, RORs are challenging targets, since their apparent therapeutic potential is accompanied by various difficulties that still need to be overcome. RORs were first discovered in the mid-90s, the crystal structures were solved in the early 2000s and yet, to this day, there are no drugs on the market that have RORs as their target. As RORs are connected to several prominent biological systems, from circadian rhythm to metabolism and cancer, caution and a targeted approach is vital. The question to be answered is how natural products can be of use in this regard. Most of the compounds described in this study are either (i) toxic in the concentrations needed (e.g. digoxin), (ii) too ineffective (e.g. betulinaldehyde), (iii) not bioavailable enough (e.g. uvaol), and/or (iv) lack selectivity (e.g. ursolic acid). On the other hand, natural products have provided important mechanistical insights and may serve as templates for improved synthetic substances, as it was often the case in the past. Such substances would ideally be selective for one ROR protein, with certain exceptions (see chapter 2.2), and active in lower nanomolar concentrations while having no to little off-target activity and good safety. A good example for such an approach is the chemical conversion of digoxin to 20,22-dihydrodigoxin-21,23-diol which did not exhibit cytotoxic effects on human cells even at 40 μM while still inhibiting RORγ with an IC50 of 12 μM (in vitro competition assay).155 In the last couple of years, progress has been made in this regard204,205 (some further examples reviewed in ref. 206).
6 Conflicts of interest
There are no conflicts of interest to declare.7 Acknowledgements
This work was supported by the Vienna Anniversary Foundation for Higher Education (H-268438/2018). Special thanks to Andrea Szabo for drawing the figures.8 References
- F. R. Santori, P. Huang, S. A. Van De Pavert, E. F. Douglass, D. J. Leaver, B. A. Haubrich, R. Keber, G. Lorbek, T. Konijn, B. N. Rosales, D. Rozman, S. Horvat, A. Rahier, R. E. Mebius, F. Rastinejad, W. D. Nes and D. R. Littman, Cell Metab., 2015, 21, 286–298 CrossRef CAS.
- X. Hu, Y. Wang, L. Y. Hao, X. Liu, C. A. Lesch, B. M. Sanchez, J. M. Wendling, R. W. Morgan, T. D. Aicher, L. L. Carter, P. L. Toogood and G. D. Glick, Nat. Chem. Biol., 2015, 11, 141–147 CrossRef CAS.
- A. M. Jetten and D. N. Cook, Annu. Rev. Pharmacol. Toxicol., 2020, 60, 371–390 CrossRef CAS.
- A. Wärnmark, E. Treuter, A. P. H. Wright and J. Å. Gustafsson, Mol. Endocrinol., 2003, 17, 1901–1909 CrossRef.
- Y. Zhang, X. Y. Luo, D. H. Wu and Y. Xu, Acta Pharmacol. Sin., 2015, 36, 71–87 CrossRef CAS.
- S. Fujita-Sato, S. Ito, T. Isobe, T. Ohyama, K. Wakabayashi, K. Morishita, O. Ando and F. Isono, J. Biol. Chem., 2011, 286, 31409–31417 CrossRef CAS.
- A. Aranda and A. Pascual, Physiol. Rev., 2001, 81, 1269–1304 CrossRef CAS.
- J. A. Kallen, J. M. Schlaeppi, F. Bitsch, S. Geisse, M. Geiser, I. Delhon and B. Fournier, Structure, 2002, 10, 1697–1707 CrossRef CAS.
- J. Kallen, A. Izaac, C. Be, L. Arista, D. Orain, K. Kaupmann, C. Guntermann, K. Hoegenauer and S. Hintermann, ChemMedChem, 2017, 12, 1014–1021 CrossRef CAS.
- X. Li, M. Anderson, D. Collin, I. Muegge, J. Wan, D. Brennan, S. Kugler, D. Terenzio, C. Kennedy, S. Lin, M. E. Labadia, B. Cook, R. Hughes and N. A. Farrow, J. Biol. Chem., 2017, 292, 11618–11630 CrossRef CAS.
- P. J. Watson, L. Fairall and J. W. R. Schwabe, Mol. Cell. Endocrinol., 2012, 348, 440–449 CrossRef CAS.
- C. A. Snyder, M. L. Goodson, A. C. Schroeder and M. L. Privalsky, Mol. Cell. Endocrinol., 2015, 413, 228–235 CrossRef CAS.
- M. L. Privalsky, Annu. Rev. Physiol., 2004, 66, 315–360 CrossRef CAS.
- D. P. Marciano, M. R. Chang, C. A. Corzo, D. Goswami, V. Q. Lam, B. D. Pascal and P. R. Griffin, Cell Metab., 2014, 19, 193–208 CrossRef CAS.
- Y. W. He, M. L. Deftos, E. W. Ojala and M. J. Bevan, Immunity, 1998, 9, 797–806 CrossRef CAS.
- D. N. Cook, H. S. Kang and A. M. Jetten, Nucl. Recept. Res., 2015, 2, 101185, DOI:10.11131/2015/101185.
- D. J. Kojetin and T. P. Burris, Nat. Rev. Drug Discovery, 2014, 13, 197–216 CrossRef CAS.
- L. A. Solt, P. R. Griffin and T. P. Burris, Curr. Opin. Lipidol., 2010, 21, 204–211 CrossRef CAS.
- N. Sun, H. Guo and Y. Wang, Expert Opin. Ther. Pat., 2019, 29, 663–674 CrossRef.
- V. Amirkia and M. Heinrich, Front. Pharmacol., 2015, 6, 1–8 Search PubMed.
- J. A. Beutler, Curr. Protoc. Pharmacol., 2019, 86, e67 Search PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS.
- A. M. Jetten, S. Kurebayashi and E. Ueda, Prog. Nucleic Acid Res. Mol. Biol., 2001, 69, 205–247 CAS.
- A. M. Jetten, Nucl. Recept. Signaling, 2009, 7(1), e003, DOI:10.1621/nrs.07003.
- L. A. Solt and T. P. Burris, Trends Endocrinol. Metab., 2012, 23, 619–627 CrossRef CAS.
- R. L. Sidman, P. W. Lane and M. M. Dickie, Science, 1962, 137, 610–612 CrossRef CAS.
- K. Herrup and R. J. Mullen, Brain Res., 1979, 172, 1–12 CrossRef CAS.
- B. A. Hamilton, W. N. Frankel, A. W. Kerrebrock, T. L. Hawkins, W. FitzHugh, K. Kusumi, L. B. Russell, K. L. Mueller, V. Van Berkel, B. W. Birren, L. Kruglyak and E. S. Lander, Nature, 1996, 379, 736–739 CrossRef CAS.
- M. Steinmayr, E. André, F. Conquet, L. Rondi-Reig, N. Delhaye-Bouchaud, N. Auclair, H. Daniel, F. Crépel, J. Mariani, C. Sotelo and M. Becker-André, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3960–3965 CrossRef CAS.
- C. I. Jarvis, B. Staels, B. Brugg, Y. Lemaigre-Dubreuil, A. Tedgui and J. Mariani, Mol. Cell. Endocrinol., 2002, 186, 1–5 CrossRef CAS.
- M. Akashi and T. Takumi, Nat. Struct. Mol. Biol., 2005, 12, 441–448 CrossRef CAS.
- N. Schaeren-Wiemers, E. André, J. P. Kapfhammer and M. Becker-André, European Journal of Neuroscience, 1997, 9, 2687–2701 CrossRef CAS.
- E. André, F. Conquet, M. Steinmayr, S. C. Stratton, V. Porciatti and M. Becker-André, EMBO J., 1998, 17, 3867–3877 CrossRef.
- Z. Sun, D. Unutmaz, Y. R. Zou, M. J. Sunshine, A. Pierani, S. Brenner-Morton, R. E. Mebius and D. R. Littman, Science, 2000, 288, 2369–2373 CrossRef CAS.
- S. Kurebayashi, E. Ueda, M. Sakaue, D. D. Patel, A. Medvedev, F. Zhang and A. M. Jetten, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10132–10137 CrossRef CAS.
- T. K. Sato, S. Panda, L. J. Miraglia, T. M. Reyes, R. D. Rudic, P. McNamara, K. A. Naik, G. A. Fitzgerald, S. A. Kay and J. B. Hogenesch, Neuron, 2004, 43, 527–537 CrossRef CAS.
- C. L. Partch, C. B. Green and J. S. Takahashi, Trends Cell Biol., 2014, 24, 90–99 CrossRef CAS.
- J. P. DeBruyne, D. R. Weaver and S. M. Reppert, Nat. Neurosci., 2007, 10, 543–545 CrossRef CAS.
- Y. Takeda, H. S. Kang, M. Angers and A. M. Jetten, Nucleic Acids Res., 2011, 39, 4769–4782 CrossRef CAS.
- Y. Takeda, R. Jothi, V. Birault and A. M. Jetten, Nucleic Acids Res., 2012, 40, 8519–8535 CrossRef CAS.
- V. Mongrain, X. Ruan, H. Dardente, E. E. Fortier and N. Cermakian, Genes Cells, 2008, 13, 1197–1210 CrossRef CAS.
- Y. Takeda, H. S. Kang, J. Freudenberg, L. M. DeGraff, R. Jothi and A. M. Jetten, PLoS Genet., 2014, 10(5), e1004331, DOI:10.1371/journal.pgen.1004331.
- Y. Takeda, H. S. Kang, F. B. Lih, H. Jiang, W. S. Blaner and A. M. Jetten, Nucleic Acids Res., 2014, 42, 10448–10459 CrossRef CAS.
- S. K. Hong, M. Angers, Y. B. Ju, X. Wu, J. M. Gimble, T. Wada, W. Xie, J. B. Collins, S. F. Grissom and A. M. Jetten, Physiol. Genomics, 2007, 31, 281–294 CrossRef.
- J. Fan, Z. Lv, G. Yang, T. ting Liao, J. Xu, F. Wu, Q. Huang, M. Guo, G. Hu, M. Zhou, L. Duan, S. Liu and Y. Jin, Front. Immunol., 2018, 9, 1187, DOI:10.3389/fimmu.2018.01187.
- G. Xiong, C. Wang, B. M. Evers, B. P. Zhou and R. Xu, Cancer Res., 2012, 72, 1728–1739 CrossRef CAS.
- H. Kim, J. M. Lee, G. Lee, J. Bhin, S. K. Oh, K. Kim, K. E. Pyo, J. S. Lee, H. Y. Yim, K. Il Kim, D. Hwang, J. Chung and S. H. Baek, Mol. Cell, 2011, 44, 797–810 CrossRef CAS.
- Y. Wang, L. A. Solt, D. J. Kojetin and T. P. Burris, PLoS One, 2012, 7(4), e34921, DOI:10.1371/journal.pone.0034921.
- X. Pan, J. Zhao, W. N. Zhang, H. Y. Li, R. Mu, T. Zhou, H. Y. Zhang, W. L. Gong, M. Yu, J. H. Man, P. J. Zhang, A. N. Li and X. M. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3788–3793 CrossRef CAS.
- J. S. Tzartos, M. A. Friese, M. J. Craner, J. Palace, J. Newcombe, M. M. Esiri and L. Fugger, Am. J. Pathol., 2008, 172, 146–155 CrossRef CAS.
- E. Lubberts, M. I. Koenders, B. Oppers-Walgreen, L. Van Den Bersselaar, C. J. J. Coenen-De Roo, L. A. B. Joosten and W. B. Van Den Berg, Arthritis Rheuma, 2004, 50, 650–659 CrossRef CAS.
- J. Yang, Y. Chu, X. Yang, D. Gao, L. Zhu, X. Yang, L. Wan and M. Li, Arthritis Rheuma, 2009, 60, 1472–1483 CrossRef.
- S. Kagami, H. L. Rizzo, J. J. Lee, Y. Koguchi and A. Blauvelt, J. Invest. Dermatol., 2010, 130, 1373–1383 CrossRef CAS.
- L. Novelli, M. S. Chimenti, A. Chiricozzi and R. Perricone, Autoimmun. Rev., 2014, 13, 64–69 CrossRef CAS.
- S. Molet, Q. Hamid, F. Davoine, E. Nutku, R. Taha, N. Pagé, R. Olivenstein, J. Elias and J. Chakir, J. Allergy Clin. Immunol., 2001, 108, 430–438 CrossRef CAS.
- I. Agache, C. Ciobanu, C. Agache and M. Anghel, Respir. Med., 2010, 104, 1131–1137 CrossRef.
- W. Zou and N. P. Restifo, Nat. Rev. Immunol., 2010, 10, 248–256 CrossRef CAS.
- C. M. Wilke, I. Kryczek, S. Wei, E. Zhao, K. Wu, G. Wang and W. Zou, Carcinogenesis, 2011, 32, 643–649 CrossRef CAS.
- R. P. Singh, S. Hasan, S. Sharma, S. Nagra, D. T. Yamaguchi, D. T. W. Wong, B. H. Hahn and A. Hossain, Autoimmun. Rev., 2014, 13, 1174–1181 CrossRef CAS.
- I. I. Ivanov, B. S. McKenzie, L. Zhou, C. E. Tadokoro, A. Lepelley, J. J. Lafaille, D. J. Cua and D. R. Littman, Cell, 2006, 126, 1121–1133 CrossRef CAS.
- K. Oboki, T. Ohno, H. Saito and S. Nakae, Allergol. Int., 2008, 57, 121–134 CrossRef CAS.
- G. L. Stritesky, N. Yeh and M. H. Kaplan, J. Immunol., 2008, 181, 5948–5955 CrossRef CAS.
- R. Nurieva, X. O. Yang, G. Martinez, Y. Zhang, A. D. Panopoulos, L. Ma, K. Schluns, Q. Tian, S. S. Watowich, A. M. Jetten and C. Dong, Nature, 2007, 448, 480–483 CrossRef CAS.
- C. Dong, Nat. Rev. Immunol., 2008, 8, 337–348 CrossRef CAS.
- X. O. Yang, B. P. Pappu, R. Nurieva, A. Akimzhanov, H. S. Kang, Y. Chung, L. Ma, B. Shah, A. D. Panopoulos, K. S. Schluns, S. S. Watowich, Q. Tian, A. M. Jetten and C. Dong, Immunity, 2008, 28, 29–39 CrossRef CAS.
- W. J. Chen, W. Jin, N. Hardegen, K. J. Lei, L. Li, N. Marinos, G. McGrady and S. M. Wahl, J. Exp. Med., 2003, 198, 1875–1886 CrossRef CAS.
- L. Zhou, J. E. Lopes, M. M. W. Chong, I. I. Ivanov, R. Min, G. D. Victora, Y. Shen, J. Du, Y. P. Rubtsov, A. Y. Rudensky, S. F. Ziegler and D. R. Littman, Nature, 2008, 453, 236–240 CrossRef CAS.
- P. R. Mangan, L. E. Harrington, D. B. O'Quinn, W. S. Helms, D. C. Bullard, C. O. Elson, R. D. Hatton, S. M. Wahl, T. R. Schoeb and C. T. Weaver, Nature, 2006, 441, 231–234 CrossRef CAS.
- T. Korn, E. Bettelli, W. Gao, A. Awasthi, A. Jäger, T. B. Strom, M. Oukka and V. K. Kuchroo, Nature, 2007, 448, 484–487 CrossRef CAS.
- L. Zhou, I. I. Ivanov, R. Spolski, R. Min, K. Shenderov, T. Egawa, D. E. Levy, W. J. Leonard and D. R. Littman, Nat. Immunol., 2007, 8, 967–974 CrossRef CAS.
- I. I. Ivanov, L. Zhou and D. R. Littman, Semin. Immunol., 2007, 19, 409–417 CrossRef CAS.
- E. Raspé, H. Duez, P. Gervois, C. Fiévet, J. C. Fruchart, S. Besnard, J. Mariani, A. Tedgui and B. Staels, J. Biol. Chem., 2001, 276, 2865–2871 CrossRef.
- P. Lau, R. L. Fitzsimmons, S. Raichur, S. C. M. Wang, A. Lechtken and G. E. O. Muscat, J. Biol. Chem., 2008, 283, 18411–18421 CrossRef CAS.
- P. Lau, R. L. Fitzsimmons, M. A. Pearen, M. J. Watt and G. E. O. Muscat, Diabetologia, 2011, 54, 1169–1180 CrossRef CAS.
- N. Kumar, D. J. Kojetin, L. A. Solt, K. G. Kumar, P. Nuhant, D. R. Duckett, M. D. Cameron, A. A. Butler, W. R. Roush, P. R. Griffin and T. P. Burris, ACS Chem. Biol., 2011, 6, 218–222 CrossRef CAS.
- P. Madsen and N. Westergaard, Expert Opin. Ther. Pat., 2001, 11, 1429–1441 CrossRef CAS.
- P. Delerive, D. Monté, G. Dubois, F. Trottein, J. Fruchart-Najib, J. Mariani, J. C. Fruchart and B. Staels, EMBO Rep., 2001, 2, 42–48 CrossRef CAS.
- A. Mamontova, S. Séguret-Macé, B. Esposito, C. Chaniale, M. Bouly, N. Delhaye-Bouchaud, G. Luc, B. Staels, N. Duverger, J. Mariani and A. Tedgui, Circulation, 1998, 98, 2738–2743 CrossRef CAS.
- T. Meyer, M. Kneissel, J. Mariani and B. Fournier, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9197–9202 CrossRef CAS.
- M. M. Roforth, G. Liu, S. Khosla and D. G. Monroe, J. Bone Miner. Res., 2012, 27, 891–901 CrossRef CAS.
- C. Gege, T. Schlüter and T. Hoffmann, Bioorg. Med. Chem. Lett., 2014, 24, 5265–5267 CrossRef CAS.
- R. Patouret, C. Doebelin, R. D. Garcia-Ordonez, M. Ra Chang, C. Ruiz, M. D. Cameron, P. R. Griffin and T. M. Kamenecka, Bioorg. Med. Chem. Lett., 2018, 28, 1178–1181 CrossRef CAS.
- M. R. Chang, V. Dharmarajan, C. Doebelin, R. D. Garcia-Ordonez, S. J. Novick, D. S. Kuruvilla, T. M. Kamenecka and P. R. Griffin, ACS Chem. Biol., 2016, 11, 1012–1018 CrossRef CAS.
- X. Hu, X. Liu, J. Moisan, Y. Wang, C. A. Lesch, C. Spooner, R. W. Morgan, E. M. Zawidzka, D. Mertz, D. Bousley, K. Majchrzak, I. Kryczek, C. Taylor, C. Van Huis, D. Skalitzky, A. Hurd, T. D. Aicher, P. L. Toogood, G. D. Glick, C. M. Paulos, W. Zou and L. L. Carter, Oncoimmunology, 2016, 5(12), e1254854, DOI:10.1080/2162402x.2016.1254854.
- D. Mahalingam, J. S. Wang, E. P. Hamilton, J. Sarantopoulos, J. Nemunaitis, G. Weems, L. Carter, X. Hu, M. Schreeder and H. J. Wilkins, Clin. Cancer Res., 2019, 25, 3508–3516 CrossRef CAS.
- D. I. Gabrilovich, Cancer Immunol. Res., 2017, 5, 3–8 CrossRef CAS.
- T. Condamine, I. Ramachandran, J. I. Youn and D. I. Gabrilovich, Annu. Rev. Med., 2015, 66, 97–110 CrossRef CAS.
- L. Strauss, S. Sangaletti, F. M. Consonni, G. Szebeni, S. Morlacchi, M. G. Totaro, C. Porta, A. Anselmo, S. Tartari, A. Doni, F. Zitelli, C. Tripodo, M. P. Colombo and A. Sica, Cancer Cell, 2015, 28, 253–269 CrossRef CAS.
- M. Takaishi, M. Ishizaki, K. Suzuki, T. Isobe, T. Shimozato and S. Sano, J. Dermatol. Sci., 2017, 85, 12–19 CrossRef CAS.
- J. Wang, J. X. Zou, X. Xue, D. Cai, Y. Zhang, Z. Duan, Q. Xiang, J. C. Yang, M. C. Louie, A. D. Borowsky, A. C. Gao, C. P. Evans, K. S. Lam, J. Xu, H. J. Kung, R. M. Evans, Y. Xu and H. W. Chen, Nat. Med., 2016, 22, 488–496 CrossRef CAS.
- D. Cai, J. Wang, B. Gao, J. Li, F. Wu, J. X. Zou, J. Xu, Y. Jiang, H. Zou, Z. Huang, A. D. Borowsky, R. J. Bold, P. N. Lara, J. J. Li, X. Chen, K. S. Lam, K. F. To, H. J. Kung, O. Fiehn, R. Zhao, R. M. Evans and H. W. Chen, Nat. Commun., 2019, 10(1), 4621, DOI:10.1038/s41467-019-12529-3.
- C. S. Constantinescu, N. Farooqi, K. O'Brien and B. Gran, Br. J. Pharmacol., 2011, 164, 1079–1106 CrossRef CAS.
- J. Dai, P. T. Ram, L. Yuan, L. L. Spriggs and S. M. Hill, Mol. Cell. Endocrinol., 2001, 176, 111–120 CrossRef CAS.
- M. Becker-André, I. Wiesenberg, N. Schaeren-Wiemers, E. André, M. Missbach, J. H. Saurat and C. Carlberg, J. Biol. Chem., 1994, 269, 28531–28534 CrossRef.
- F. Bitsch, R. Aichholz, J. Kallen, S. Geisse, B. Fournier and J. M. Schlaeppi, Anal. Biochem., 2003, 323, 139–149 CrossRef CAS.
- J. P. Renaud and D. Moras, Cell. Mol. Life Sci., 2000, 57, 1748–1769 CrossRef CAS.
- O. René, B. P. Fauber, G. D. L. Boenig, B. Burton, C. Eidenschenk, C. Everett, A. Gobbi, S. G. Hymowitz, A. R. Johnson, J. R. Kiefer, M. Liimatta, P. Lockey, M. Norman, W. Ouyang, H. A. Wallweber and H. Wong, ACS Med. Chem. Lett., 2015, 6, 276–281 CrossRef.
- C. Doebelin, R. Patouret, R. D. Garcia-Ordonez, M. R. Chang, V. Dharmarajan, D. S. Kuruvilla, S. J. Novick, L. Lin, M. D. Cameron, P. R. Griffin and T. M. Kamenecka, ChemMedChem, 2016, 11, 2607–2620 CrossRef CAS.
- L. Jin, D. Martynowski, S. Zheng, T. Wada, W. Xie and Y. Li, Mol. Endocrinol., 2010, 24, 923–929 CrossRef CAS.
- P. Soroosh, J. Wu, X. Xue, J. Song, S. W. Sutton, M. Sablad, J. Yu, M. I. Nelen, X. Liu, G. Castro, R. Luna, S. Crawford, H. Banie, R. A. Dandridge, X. Deng, A. Bittner, C. Kuei, M. Tootoonchi, N. Rozenkrants, K. Herman, J. Gao, X. V. Yang, K. Sachen, K. Ngo, W. P. Fung-Leung, S. Nguyen, A. De Leon-Tabaldo, J. Blevitt, Y. Zhang, M. D. Cummings, T. Rao, N. S. Mani, C. Liu, M. McKinnon, M. E. Milla, A. M. Fourie and S. Sun, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12163–12168 CrossRef CAS.
- C. Stehlin-Gaon, D. Willmann, D. Zeyer, S. Sanglier, A. Van Dorsselaer, J. P. Renaud, D. Moras and R. Schüle, Nat. Struct. Biol., 2003, 10, 820–825 CrossRef CAS.
- Y. Wang, N. Kumar, L. A. Solt, T. I. Richardson, L. M. Helvering, C. Crumbley, R. D. Garcia-Ordonez, K. R. Stayrook, X. Zhang, S. Novick, M. J. Chalmers, P. R. Griffin and T. P. Burris, J. Biol. Chem., 2010, 285, 5013–5025 CrossRef CAS.
- J. Kallen, J. M. Schlaeppi, F. Bitsch, I. Delhon and B. Fournier, J. Biol. Chem., 2004, 279, 14033–14038 CrossRef CAS.
- O. Hanyu, H. Nakae, T. Miida, Y. Higashi, H. Fuda, M. Endo, A. Kohjitani, H. Sone and C. A. Strott, Biochem. Biophys. Res. Commun., 2012, 428, 99–104 CrossRef CAS.
- X. Zhang, J. Jin, X. Peng, V. S. Ramgolam and S. Markovic-Plese, J. Immunol., 2008, 180(10), 6988–6996, DOI:10.4049/jimmunol.180.10.6988.
- S. I. Kagami, T. Owada, H. Kanari, Y. Saito, A. Suto, K. Ikeda, K. Hirose, N. Watanabe, I. Iwamoto and H. Nakajima, Int. Immunol., 2009, 21(6), 679–689, DOI:10.1093/intimm/dxp037.
- Z. K. Tuong, P. Lau, J. C. Yeo, M. A. Pearen, A. A. Wall, A. C. Stanley, J. L. Stow and G. E. O. Muscat, Endocrinology, 2013, 154(1), 140–149, DOI:10.1210/en.2012-1889.
- Z. K. Tuong, P. Lau, X. Du, N. D. Condon, J. M. Goode, T. G. Oh, J. C. Yeo, G. E. O. Muscat and J. L. Stow, PLoS One, 2016, 11, 1–16 CrossRef.
- J. Y. L. Chiang, F1000Research, 2017, 6, 2029, DOI:10.12688/f1000research.12449.1.
- A. R. Chopra, J. F. Louet, P. Saha, J. An, F. DeMayo, J. Xu, B. York, S. Karpen, M. Finegold, D. Moore, L. Chan, C. B. Newgard and B. W. O'Malley, Science, 2008, 322, 1395–1399 CrossRef CAS.
- Y. Wang, N. Kumar, C. Crumbley, P. R. Griffin and T. P. Burris, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2010, 1801, 917–923 CrossRef CAS.
- D. Lütjohann, O. Breuer, G. Ahlborg, I. Nennesmo, Å. Sidén, U. Diczfalusy and I. Björkhem, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9799–9804 CrossRef.
- J. Wong, C. M. Quinn, G. Guillemin and A. J. Brown, J. Neurochem., 2007, 103(5), 1764–1773, DOI:10.1111/j.1471-4159.2007.04913.x.
- T. A. Spencer, A. K. Gayen, S. Phirwa, J. A. Nelson, F. R. Taylor, A. A. Kandutsch and S. K. Erickson, J. Biol. Chem., 1985, 260(25), 13391–13394 CrossRef CAS.
- J. M. Lehmann, S. A. Kliewer, L. B. Moore, T. A. Smith-Oliver, B. B. Oliver, J. L. Su, S. S. Sundseth, D. A. Winegar, D. E. Blanchard, T. A. Spencer and T. M. Willson, J. Biol. Chem., 1997, 272, 3137–3140 CrossRef CAS.
- S. Hirayama, S. Nakagawa, S. Soda, Y. Kamimura, E. Nishioka, T. Ueno, Y. Fukushima, K. ichi Higuchi, M. Inoue, U. Seino, H. Ohmura, S. Yamato and T. Miida, Atherosclerosis, 2013, 230(1), 48–51, DOI:10.1016/j.atherosclerosis.2013.06.016.
- A. J. Brown and W. Jessup, Atherosclerosis, 1999, 142(1), 1–28 CrossRef CAS.
- A. T. Slominski, T. K. Kim, Y. Takeda, Z. Janjetovic, A. A. Brozyna, C. Skobowiat, J. Wang, A. Postlethwaite, W. Li, R. C. Tuckey and A. M. Jetten, FASEB J., 2014, 28, 2775–2789 CrossRef CAS.
- A. T. Slominski, T. K. Kim, J. V. Hobrath, A. S. W. Oak, E. K. Y. Tang, E. W. Tieu, W. Li, R. C. Tuckey and A. M. Jetten, J. Steroid Biochem. Mol. Biol., 2017, 173, 42–56 CrossRef CAS.
- S. Helleboid, C. Haug, K. Lamottke, Y. Zhou, J. Wei, S. Daix, L. Cambula, G. Rigou, D. W. Hum and R. Walczak, J. Biomol. Screening, 2014, 19, 399–406 CrossRef CAS.
- Y. Ikeda, A. Murakami and H. Ohigashi, Mol. Nutr. Food Res., 2008, 52, 26–42 CrossRef CAS.
- T. Xu, X. Wang, B. Zhong, R. I. Nurieva, S. Ding and C. Dong, J. Biol. Chem., 2011, 286, 22707–22710 CrossRef CAS.
- S. Y. Baek, J. Lee, D. G. Lee, M. K. Park, J. Lee, S. K. Kwok, M. La Cho and S. H. Park, Acta Pharmacol. Sin., 2014, 35, 1177–1187 CrossRef CAS.
- K. Kim, E. A. Shin, J. H. Jung, J. E. Park, D. S. Kim, B. S. Shim and S. H. Kim, Int. J. Mol. Sci., 2018, 20(1), 114, DOI:10.3390/ijms20010114.
- M. Zhai, J. Guo, H. Ma, W. Shi, D. Jou, D. Yan, T. Liu, J. Tao, J. Duan, Y. Wang, S. Li, J. Lv, C. Li, J. Lin, C. Zhang and L. Lin, Atherosclerosis, 2018, 271, 128–135, DOI:10.1016/j.atherosclerosis.2018.02.022.
- M. K. Shanmugam, T. H. Ong, A. P. Kumar, C. K. Lun, P. C. Ho, P. T. H. Wong, K. M. Hui and G. Sethi, PLoS One, 2012, 7(3), e32476, DOI:10.1371/journal.pone.0032476.
- S. Prasad, V. R. Yadav, B. Sung, S. Reuter, R. Kannappan, A. Deorukhkar, P. Diagaradjane, C. Wei, V. Baladandayuthapani, S. Krishnan, S. Guha and B. B. Aggarwal, Clin. Cancer Res., 2012, 18(18), 4942–4953, DOI:10.1158/1078-0432.ccr-11-2805.
- S. Prasad, V. R. Yadav, B. Sung, S. C. Gupta, A. K. Tyagi and B. B. Aggarwal, Oncotarget, 2016, 7(11), 13182–13196, DOI:10.18632/oncotarget.7537.
- X. Ma, Y. Zhang, Z. Wang, Y. Shen, M. Zhang, Q. Nie, Y. Hou and G. Bai, Mol. Nutr. Food Res., 2017, 61(12), 1700332, DOI:10.1002/mnfr.201700332.
- T. Liu, H. Ma, W. Shi, J. Duan, Y. Wang, C. Zhang, C. Li, J. Lin, S. Li, J. Lv and L. Lin, Int. J. Oncol., 2017, 51(2), 555–562, DOI:10.3892/ijo.2017.4035.
- T. J. Harris, J. F. Grosso, H.-R. Yen, H. Xin, M. Kortylewski, E. Albesiano, E. L. Hipkiss, D. Getnet, M. V. Goldberg, C. H. Maris, F. Housseau, H. Yu, D. M. Pardoll and C. G. Drake, J. Immunol., 2007, 179(7), 4313–4317, DOI:10.4049/jimmunol.179.7.4313.
- S. Shishodia, S. Majumdar, S. Banerjee and B. B. Aggarwal, Cancer Res., 2003, 63(15), 4375–4383 CAS.
- N. Wang, H. Liang and K. Zen, Front. Immunol., 2014, 5, 614 Search PubMed.
- R. Catlett-Falcone, T. H. Landowski, M. M. Oshiro, J. Turkson, A. Levitzki, R. Savino, G. Ciliberto, L. Moscinski, J. L. Fernández-Luna, G. Nuñez, W. S. Dalton and R. Jove, Immunity, 1999, 10(1), 105–115, DOI:10.1016/S1074-7613(00)80011-4.
- Z. Zhong, Z. Wen and J. E. Darnell, Science, 1994, 264(5155), 95–98, DOI:10.1126/science.8140422.
- T. Liu, L. Zhang, D. Joo and S. C. Sun, Signal Transduction Targeted Ther., 2017, 2, 17023, DOI:10.1038/sigtrans.2017.23.
- M. Noguchi, A. Nomura, K. Murase, S. Doi, K. Yamaguchi, K. Hirata, M. Shiozaki, S. Hirashima, M. Kotoku, T. Yamaguchi, Y. Katsuda, R. Steensma, X. Li, H. Tao, B. Tse, M. Fenn, R. Babine, E. Bradley, P. Crowe, S. Thacher, T. Adachi and M. Kamada, Genes Cells, 2017, 22, 535–551 CrossRef CAS.
- F. Yang, R. Zhang, D. Ni, X. Luo, S. Chen, C. Luo and W. Xiao, Fitoterapia, 2019, 137, 9–10 Search PubMed.
- Ł. Woźniak, S. Skąpska and K. Marszałek, Molecules, 2015, 20, 20614–20641 CrossRef.
- X. Zhou, H. Chen, F. Wei, Q. Zhao, Q. Su, Y. Lei, M. Yin, X. Tian, Z. Liu, B. Yu, C. Bai, X. He and Z. Huang, Immunol. Invest., 2020, 49(6), 632–647, DOI:10.1080/08820139.2019.1698599.
- S. M. Fu, C. Dai, Z. Zhao and F. Gaskin, F1000Research, 2015, 4, 939 Search PubMed.
- S. Thacher, X. Li, R. Babine and B. Tse, US Pat., US8389739, 2013.
- C. Stehlin, J. M. Wurtz, A. Steinmetz, E. Greiner, R. Schüle, D. Moras and J. P. Renaud, EMBO J., 2001, 20, 5822–5831 CrossRef CAS.
- C. L. Zhao, M. S. Sarwar, J. H. Ye, C. F. Ku, W. F. Li, G. Y. Luo, J. J. Zhang, J. Xu, Z. F. Huang, S. W. Tsang, L. T. Pan and H. J. Zhang, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 635–640 CrossRef CAS.
- N. Li, X. Wu, W. Zhuang, L. Xia, Y. Chen, R. Zhao, M. Yi, Q. Wan, L. Du and Y. Zhou, Mol. Nutr. Food Res., 2020, 64(4), e1900751 CrossRef.
- H. Kojima, Y. Takeda, R. Muromoto, M. Takahashi, T. Hirao, S. Takeuchi, A. M. Jetten and T. Matsuda, Toxicology, 2015, 329, 32–39, DOI:10.1016/j.tox.2015.01.007.
- M. Takahashi, R. Muromoto, H. Kojima, S. Takeuchi, Y. Kitai, J. Ichi Kashiwakura and T. Matsuda, Biochem. Biophys. Res. Commun., 2017, 489, 503–508 CrossRef CAS.
- A. T. Dias, S. B. R. de Castro, C. C. de Souza Alves, M. G. Evangelista, L. C. da Silva, D. R. L. Reis, M. A. Machado, M. A. Juliano and A. P. Ferreira, Inflammation Res., 2018, 67(7), 597–608, DOI:10.1007/s00011-018-1146-7.
- M. N. Mansoori, A. M. Tyagi, P. Shukla, K. Srivastava, K. Dev, R. Chillara, R. Maurya and D. Singh, Menopause, 2016, 23(5), 565–576, DOI:10.1097/gme.0000000000000646.
- F. Zhang, Z. Wang, M. Li, Y. Lan, Y. Chen and C. Wang, Int. Immunopharmacol., 2015, 28(1), 546–553, DOI:10.1016/j.intimp.2015.07.023.
- B. He, K. Nohara, N. Park, Y. S. Park, B. Guillory, Z. Zhao, J. M. Garcia, N. Koike, C. C. Lee, J. S. Takahashi, S. H. Yoo and Z. Chen, Cell Metab., 2016, 23, 610–621 CrossRef CAS.
- K. Nohara, V. Mallampalli, T. Nemkov, M. Wirianto, J. Yang, Y. Ye, Y. Sun, L. Han, K. A. Esser, E. Mileykovskaya, A. D’Alessandro, C. B. Green, J. S. Takahashi, W. Dowhan, S. H. Yoo and Z. Chen, Nat. Commun., 2019, 10(1), 3923, DOI:10.1038/s41467-019-11926-y.
- K. Nohara, T. Nemkov, A. D’Alessandro, S. H. Yoo and Z. Chen, Int. J. Mol. Sci., 2019, 20(17), 4281, DOI:10.3390/ijms20174281.
- J. R. Huh, M. W. L. Leung, P. Huang, D. A. Ryan, M. R. Krout, R. R. V. Malapaka, J. Chow, N. Manel, M. Ciofani, S. V. Kim, A. Cuesta, F. R. Santori, J. J. Lafaille, H. E. Xu, D. Y. Gin, F. Rastinejad and D. R. Littman, Nature, 2011, 472, 486–490 CrossRef CAS.
- K. Karaś, A. Sałkowska, A. Walczak-Drzewiecka, K. Ryba, J. Dastych, R. A. Bachorz and M. Ratajewski, Toxicol. Lett., 2018, 295, 314–324 CrossRef.
- J. Liu, L. P. Bai, F. Yang, X. Yao, K. Lei, C. W. Kei Lam, Q. Wu, Y. Zhuang, R. Xiao, K. Liao, H. Kuok, T. Li and L. Liu, Mol. Pharm., 2019, 16, 798–807 CrossRef CAS.
- V. Gorbacheva, R. Fan, X. Li and A. Valujskikh, Am. J. Pathol., 2010, 177, 1265–1273 CrossRef CAS.
- S. Itoh, N. Kimura, R. C. Axtell, J. B. Velotta, Y. Gong, X. Wang, N. Kajiwara, A. Nambu, E. Shimura, H. Adachi, Y. Iwakura, H. Saito, K. Okumura, K. Sudo, L. Steinman, R. C. Robbins, S. Nakae and M. P. Fischbein, Circulation, 2011, 124(11 Suppl.), S187–S196, DOI:10.1161/circulationaha.110.014852.
- M. A. Antonysamy, W. C. Fanslow, F. Fu, W. Li, S. Qian, A. B. Troutt and A. W. Thomson, J. Immunol., 1999, 162, 577–584 CAS.
- J. Wu, C. Zhou, W. Chen, A. Xie, J. Li, S. Wang, P. Ye, W. Wang and J. Xia, Transplantation, 2013, 95, 434–441 CrossRef CAS.
- Z. Wei, Y. Wang, K. Zhang, Y. Liao, P. Ye, J. Wu, Y. Wang, F. Li, Y. Yao, Y. Zhou and J. Liu, Arterioscler., Thromb., Vasc. Biol., 2014, 34, 2429–2438 CrossRef CAS.
- A. K. Sharma, G. Lu, A. Jester, W. F. Johnston, Y. Zhao, V. A. Hajzus, M. Reza Saadatzadeh, G. Su, C. M. Bhamidipati, G. S. Mehta, I. L. Kron, V. E. Laubach, M. P. Murphy, G. Ailawadi and G. R. Upchurch, Circulation, 2012, 126(11 Suppl. 1), S38–S45, DOI:10.1161/circulationaha.111.083451.
- A. Wanhainen, F. Verzini, I. Van Herzeele, E. Allaire, M. Bown, T. Cohnert, F. Dick, J. van Herwaarden, C. Karkos, M. Koelemay, T. Kölbel, I. Loftus, K. Mani, G. Melissano, J. Powell, Z. Szeberin, G. J. de Borst, N. Chakfe, S. Debus, R. Hinchliffe, S. Kakkos, I. Koncar, P. Kolh, J. S. Lindholt, M. de Vega, F. Vermassen, M. Björck, S. Cheng, R. Dalman, L. Davidovic, K. Donas, J. Earnshaw, H. H. Eckstein, J. Golledge, S. Haulon, T. Mastracci, R. Naylor, J. B. Ricco and H. Verhagen, Eur. J. Vasc. Endovasc. Surg., 2019, 57, 8–93 CrossRef.
- J. Lee, S. Baek, J. Lee, J. Lee, D. G. Lee, M. K. Park, M. La Cho, S. H. Park and S. K. Kwok, Int. Immunopharmacol., 2015, 26, 103–111 CrossRef CAS.
- H. Shi, X. Mao, Y. Zhong, Y. Liu, X. Zhao, K. Yu, R. Zhu, Y. Wei, J. Zhu, H. Sun, Y. Mao and Q. Zeng, Br. J. Pharmacol., 2016, 173, 1517–1528 CrossRef CAS.
- S. Tani, R. Takano, S. Tamura, S. Oishi, M. Iwaizumi, Y. Hamaya, K. Takagaki, T. Nagata, S. Seto, T. Horii, I. Kosugi, T. Iwashita, S. Osawa, T. Furuta, H. Miyajima and K. Sugimoto, Inflammatory Bowel Dis., 2017, 23, 728–738 CrossRef.
- S. Johansson, P. Lindholm, J. Gullbo, R. Larsson, L. Bohlin and P. Claeson, Anticancer Drugs, 2001, 12, 475–483 CrossRef CAS.
- E. M. Price and J. B. Lingrel, Biochemistry, 1988, 27, 8400–8408 CrossRef CAS.
- R. Thomas, P. Gray and J. Andrews, Adv. Drug Res., 1990, 19, 311–562 CAS.
- K. Karaś, A. Sałkowska, M. Sobalska-Kwapis, A. Walczak-Drzewiecka, D. Strapagiel, J. Dastych, R. A. Bachorz and M. Ratajewski, Front. Pharmacol., 2019, 9, 1460, DOI:10.3389/fphar.2018.01460.
- N. Kumar, L. a. Solt, J. J. Conkright, Y. Wang, M. a. Istrate, S. a. Busby, R. D. Garcia-ordonez, T. P. Burris and P. R. Griffin, Mol. Pharmacol., 2010, 77, 228–236 CrossRef CAS.
- I. Wiesenberg, M. Missbach, J. pierre Kahlen, M. Schräder and C. Carlberg, Nucleic Acids Res., 1995, 23, 327–333 CrossRef CAS.
- C. D. Kane and A. R. Means, EMBO J., 2000, 19(4), 691–701, DOI:10.1093/emboj/19.4.691.
- G. Benítez-King, L. Huerto-Delgadillo and F. Antón-Tay, Life Sci., 1993, 53(3), 201–207, DOI:10.1016/0024-3205(93)90670-x.
- L. Brydon, F. Roka, L. Petit, P. De Coppet, M. Tissot, P. Barrett, P. J. Morgan, C. Nanoff, A. D. Strosberg and R. Jockers, Mol. Endocrinol., 1999, 13(12), 2025–2038, DOI:10.1210/mend.13.12.0390.
- J. Vanecek and D. C. Klein, Endocrinology, 1992, 130(2), 701–707, DOI:10.1210/endo.130.2.1733718.
- N. Zisapel, Br. J. Pharmacol., 2018, 175, 3190–3199 CrossRef CAS.
- M. C. Beker, B. Caglayan, A. B. Caglayan, T. Kelestemur, E. Yalcin, A. Caglayan, U. Kilic, A. T. Baykal, R. J. Reiter and E. Kilic, Sci. Rep., 2019, 9(1), 19082, DOI:10.1038/s41598-019-55663-0.
- C. H. Piao, C. H. Song, E. J. Lee and O. H. Chai, Chem.-Biol. Interact., 2020, 315, 1–8 CrossRef.
- M. Xu, X. Y. Duan, Q. Y. Chen, H. Fan, Z. Chao Hong, S. J. Deng, Z. Nan, H. Wu, Y. L. Dong, Y. J. Liu and C. Z. Zhou, Biomed. Pharmacother., 2019, 109, 2396–2408 CrossRef CAS.
- M. Kardan, A. Rafiei, J. Ghaffari, R. Valadan, Z. Morsaljahan and S. T. Haj-ghorbani, Allergol. Immunopathol., 2019, 47, 378–385 CrossRef CAS.
- L. Tong, S. M. Nanjundaiah, S. H. Venkatesha, B. Astry, H. Yu and K. D. Moudgil, Clin. Immunol., 2014, 155, 220–230 CrossRef CAS.
- W. Li, Z. Zhang, K. Zhang, Z. Xue, Y. Li, Z. Zhang, L. Zhang, C. Gu, Q. Zhang, J. Hao, Y. Da, Z. Yao, Y. Kong and R. Zhang, Mol. Neurobiol., 2016, 53, 5356–5366 CrossRef CAS.
- H. M. Zhao, Y. Wang, X. Y. Huang, M. F. Huang, R. Xu, H. Y. Yue, B. G. Zhou, H. Y. Huang, Q. M. Sun and D. Y. Liu, World J. Gastroenterol., 2016, 22, 3175–3185 CrossRef CAS.
- H. Zhang, X. Zhou, X. Chen, Y. Lin, S. Qiu, Y. Zhao, Q. Tang, Y. Liang and X. Zhong, Inflammation Res., 2019, 68, 957–968 CrossRef CAS.
- E. V. Dang, J. Barbi, H. Y. Yang, D. Jinasena, H. Yu, Y. Zheng, Z. Bordman, J. Fu, Y. Kim, H. R. Yen, W. Luo, K. Zeller, L. Shimoda, S. L. Topalian, G. L. Semenza, C. V. Dang, D. M. Pardoll and F. Pan, Cell, 2011, 146, 772–784 CrossRef CAS.
- W. Ren, J. Yin, J. Duan, G. Liu, B. Tan, G. Yang, G. Wu, F. W. Bazer, Y. Peng and Y. Yin, Eur. J. Immunol., 2016, 46, 291–299 CrossRef CAS.
- Y. J. Cao, Y. Xu, B. Liu, X. Zheng, J. Wu, Y. Zhang, X. S. Li, Y. Qi, Y. M. Sun, W. B. Wen, L. Hou and C. P. Wan, Am. J. Chin. Med., 2019, 47, 423–437 CrossRef CAS.
- X. Zhou, H. Chen, F. Wei, Q. Zhao, Q. Su, J. Liang, M. Yin, X. Tian, Z. Liu, B. Yu, C. Bai, X. He and Z. Huang, J. Immunol. Res., 2019, 2431617, DOI:10.1155/2019/2431617.
- Y. Dang, S. Ling, J. Ma, R. Ni and J. W. Xu, J. Evidence-Based Complementary Altern. Med., 2015, 920431, DOI:10.1155/2015/920431.
- T. Hirai, K. Nomura, R. Ikai, K. ichi Nakashima and M. Inoue, Biomed. Pharmacother., 2019, 109, 503–510 CrossRef CAS.
- G. D. Kang and D. H. Kim, J. Ethnopharmacol., 2016, 189, 175–185 CrossRef CAS.
- C. B. Wei, K. Tao, R. Jiang, L. Di Zhou, Q. H. Zhang and C. S. Yuan, Int. Immunopharmacol., 2017, 53, 73–82 CrossRef CAS.
- S. J. Yu, R. Jiang, Y. Z. Mazzu, C. B. Wei, Z. L. Sun, Y. Z. Zhang, L. Di Zhou and Q. H. Zhang, Am. J. Chin. Med., 2016, 44, 1221–1236 CrossRef CAS.
- K. Li, Y. Chen, R. Jiang, D. Chen, H. Wang, W. Xiong, D. Li, Z. Liu, X. Li, J. Li and K. Yuan, Mol. Med. Rep., 2017, 16, 1207–1215 CrossRef CAS.
- Z. Yan, Z. Xiaoyu, S. Zhixin, Q. Di, D. Xinyu, X. Jing, H. Jing, D. Wang, Z. Xi, Z. Chunrong and W. Daoxin, Sci. Rep., 2016, 6, 1–11 CrossRef.
- A. Ma, Y. Yang, Q. Wang, Y. Wang, J. Wen and Y. Zhang, Mol. Med. Rep., 2017, 15, 3615–3622 CrossRef CAS.
- X. Zhou, H. Chen, F. Wei, Q. Zhao, Q. Su, Y. Lei, M. Yin, X. Tian, Z. Liu, B. Yu, C. Bai, X. He and Z. Huang, Int. J. Rheum. Dis., 2020, 23, 74–83 CrossRef CAS.
- A. Ribas-Latre, J. M. Del Bas, L. Baselga-Escudero, E. Casanova, A. Arola-Arnal, M. J. Salvadó, C. Bladé and L. Arola, J. Funct. Foods, 2015, 13, 336–344 CrossRef CAS.
- P. Ponikowski, A. A. Voors, S. D. Anker, H. Bueno, J. G. F. Cleland, A. J. S. Coats, V. Falk, J. R. González-Juanatey, V. P. Harjola, E. A. Jankowska, M. Jessup, C. Linde, P. Nihoyannopoulos, J. T. Parissis, B. Pieske, J. P. Riley, G. M. C. Rosano, L. M. Ruilope, F. Ruschitzka, F. H. Rutten, P. Van Der Meer, H. S. Sisakian, E. Isayev, A. Kurlianskaya, W. Mullens, M. Tokmakova, P. Agathangelou, V. Melenovsky, H. Wiggers, M. Hassanein, T. Uuetoa, J. Lommi, E. S. Kostovska, Y. Juilliere, A. Aladashvili, A. Luchner, C. Chrysohoou, N. Nyolczas, G. Thorgeirsson, J. M. Weinstein, A. Di Lenarda, N. Aidargaliyeva, G. Bajraktari, M. Beishenkulov, G. Kamzola, T. Abdel-Massih, J. Celutkiene, S. Noppe, A. Cassar, E. Vataman, S. AbirKhalil, P. van Pol, R. Mo, E. Straburzynska-Migaj, C. Fonseca, O. Chioncel, E. Shlyakhto, M. Zavatta, P. Otasevic, E. Goncalvesova, M. Lainscak, B. D. Molina, M. Schaufelberger, T. Suter, M. B. Yılmaz, L. Voronkov and C. Davies, Eur. Heart J., 2016, 37, 2129–2200 CrossRef.
- Z. D. Goldberger and A. L. Goldberger, Am. J. Cardiol., 2012, 109, 1818–1821 CrossRef CAS.
- E. Iisalo, Clin. Pharmacokinet., 1977, 2, 1–16 CrossRef CAS.
- M. Dal Prà, D. Carta, G. Szabadkai, M. Suman, Y. Frión-Herrera, N. Paccagnella, G. Castellani, S. De Martin and M. G. Ferlin, Bioorg. Med. Chem., 2018, 26, 1686–1704 CrossRef.
- H. Toyama, M. Nakamura, M. Nakamura, Y. Matsumoto, M. Nakagomi and Y. Hashimoto, Bioorg. Med. Chem., 2014, 22, 1948–1959 CrossRef CAS.
- B. P. Fauber and S. Magnuson, J. Med. Chem., 2014, 57, 5871–5892 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |