
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyketide β-branching: diversity, mechanism and selectivity
P. D.
Walker†
 a,
A. N. M.
Weir†
b,
C. L.
Willis
*b and
M. P.
Crump
*b
a,
A. N. M.
Weir†
b,
C. L.
Willis
*b and
M. P.
Crump
*b
aInstitute of Metabolism and Systems Research, College of Medical and Dental Sciences, University of Birmingham, Birmingham, B15 2TT, UK
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: matt.crump@bristol.ac.uk; Chris.willis@bristol.ac.uk
First published on 15th October 2020
Abstract
Covering: 2008 to August 2020
Polyketides are a family of natural products constructed from simple building blocks to generate a diverse range of often complex chemical structures with biological activities of both pharmaceutical and agrochemical importance. Their biosynthesis is controlled by polyketide synthases (PKSs) which catalyse the condensation of thioesters to assemble a functionalised linear carbon chain. Alkyl-branches may be installed at the nucleophilic α- or electrophilic β-carbon of the growing chain. Polyketide β-branching is a fascinating biosynthetic modification that allows for the conversion of a β-ketone into a β-alkyl group or functionalised side-chain. The overall transformation is catalysed by a multi-protein 3-hydroxy-3-methylglutaryl synthase (HMGS) cassette and is reminiscent of the mevalonate pathway in terpene biosynthesis. The first step most commonly involves the aldol addition of acetate to the electrophilic carbon of the β-ketothioester catalysed by a 3-hydroxy-3-methylglutaryl synthase (HMGS). Subsequent dehydration and decarboxylation selectively generates either α,β- or β,γ-unsaturated β-alkyl branches which may be further modified. This review covers 2008 to August 2020 and summarises the diversity of β-branch incorporation and the mechanistic details of each catalytic step. This is extended to discussion of polyketides containing multiple β-branches and the selectivity exerted by the PKS to ensure β-branching fidelity. Finally, the application of HMGS in data mining, additional β-branching mechanisms and current knowledge of the role of β-branches in this important class of biologically active natural products is discussed.
 P. D. Walker | Paul Walker obtained a MChem from the University of Oxford in 2012, undertaking his Master's research in the lab of Professor Stuart Conway. He moved to the University of Bristol and completed his PhD in 2019 under the supervision of Professor Matt Crump and Professor Chris Willis as part of the Bristol Chemical Synthesis Centre for Doctoral Training. His work focussed on the in vitro characterisation of polyketide biosynthetic pathways, including the kalimantacin β-branching pathway. Paul is currently a post-doctoral research fellow in the lab of Dr Dan Tennant at the University of Birmingham investigating altered metabolism in brain tumours. |
 A. N. M. Weir | Angus Weir graduated from the University of Bristol in 2017 with an MSci in Chemistry with Industrial Experience having spent one year on placement at Evotec. He is currently in the final year of his PhD studies with Professors Chris Willis and Matt Crump in Bristol having also spent an internship with Dr Joleen Masschelein at KU Leuven in Belgium. His interdisciplinary research centres on natural product biosynthesis with a focus on polyketide derived antibiotics. |
 C. L. Willis | Chris Willis is Professor of Organic Chemistry and Head of Organic and Biological Chemistry at the University of Bristol. Her research focuses on natural product biosynthesis including the application of total synthesis, isotopic labelling, pathway engineering and mechanistic studies to produce biocatalysts and new bioactive molecules. She was the recipient of the Natural Product Chemistry Award of the Royal Society of Chemistry in 2020. |
 M. P. Crump | Matt Crump is Professor of NMR and Structural Biology at the University of Bristol. He was appointed as a lecturer in NMR at the School of Biological Sciences, University of Southampton (1999–2002) before taking up his current post in biological NMR at the University of Bristol (2003–2020). His major focus is on structural and mechanistic aspects of natural product biosynthesis that began with reporting the first polyketide protein NMR structure in 1997 and continues with NMR and X-ray crystallography of natural product enzymes and their interactions with intermediates. |
1. Introduction
Polyketides are natural products produced by bacteria, fungi and plants. In their natural environments, these secondary metabolites provide a competitive advantage for the producing species and have been harnessed in the modern world for their pharmacological and agrochemical properties as herbicides, insecticides, antibiotics, antifungal, immunosuppressive and anticancer agents. The range of biological activity exhibited by these natural products is partly due to the vast array of complex chemical structures that are assembled, primarily, from simple acyl building blocks. The diversity of polyketide products is achieved by a combinatorial array of sequential condensations and post-condensation molecular processing catalysed by polyketide synthases (PKSs) and subsequent tailoring enzymes.1–3Polyketide biosynthesis is initiated by the priming of the PKS with a starter unit, most commonly containing alkyl groups (e.g. acetate, propionate), aromatic rings (e.g. benzoate) or amino acid-derived.4 Chain extension is catalysed by a ketosynthase (KS) domain through a decarboxylative Claisen condensation with malonyl-derived building blocks to form a β-ketothioester (Fig. 1A). The β-ketone may be processed sequentially by ketoreductase (KR), dehydratase (DH) and enoylreductase (ER) domains to produce hydroxyl, alkenyl or alkyl moieties respectively. Exquisite control of stereochemistry is seen when substituted extender units are utilised and during reductive processing of the β-ketone.5,6 Cleavage of the growing chain from the PKS may be achieved in a variety of ways, e.g. through hydrolysis of the thioester bond, cyclisation, transesterification, peptide bond formation or macrolactonisation.7–9 Through these canonical steps of polyketide biosynthesis, an extremely diverse range of chemical scaffolds may be generated. Furthermore, cascade ring closures,10 dimerisation,11 small and medium-sized ring formation,12 oxidoreductions (OX)13 and glycosylation14,15 are among many additional modifications that may expand the complexity of the chemical structure created.16–19
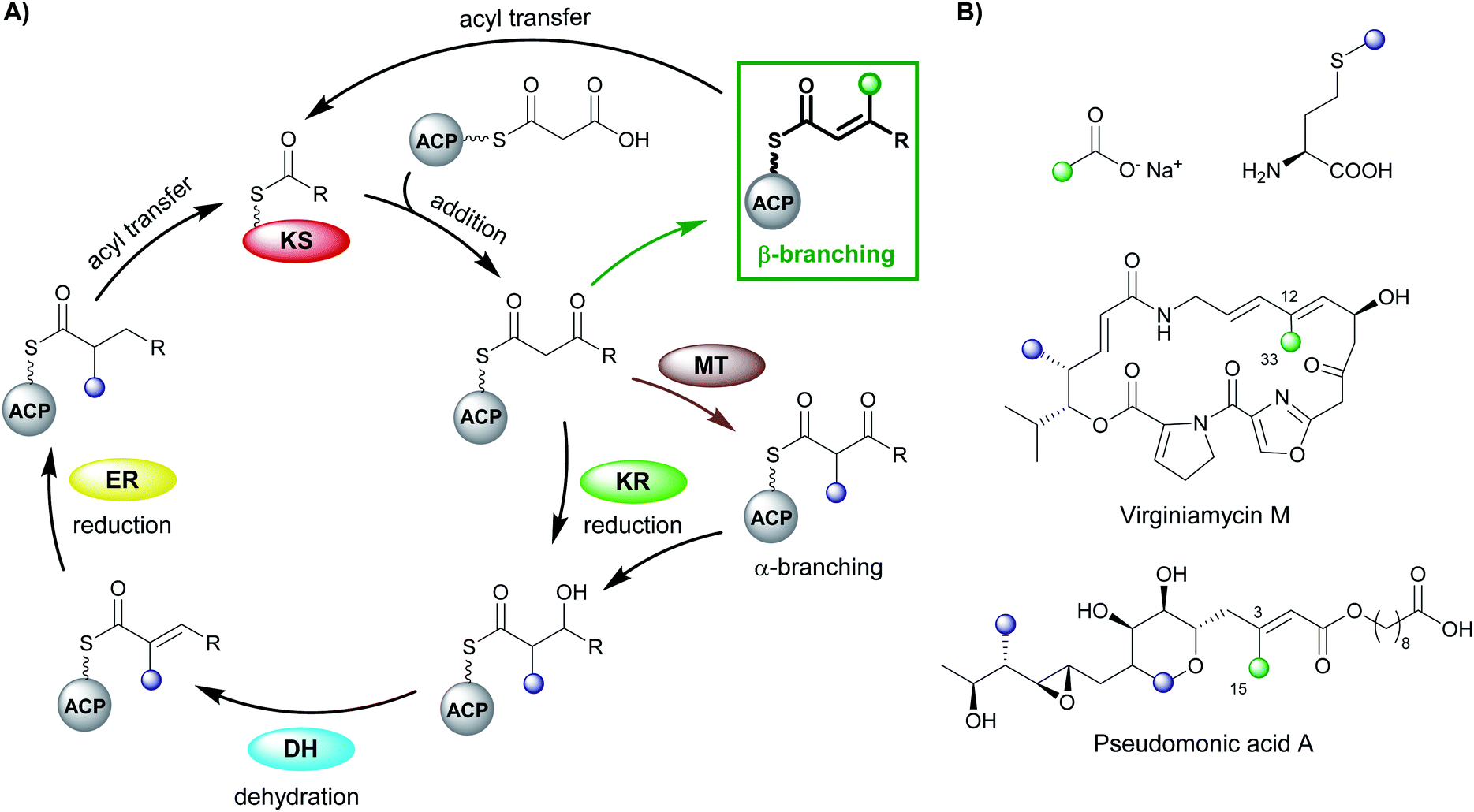 | ||
| Fig. 1 (A) Polyketide biosynthesis by a polyketide synthase. A Claisen-like condensation of malonyl-ACP with the extending polyketide chain catalysed by a ketosynthase (KS) gives a β-ketothioester that may be processed by ketoreductase (KR), dehydratase (DH) and enoylreductase (ER) domains. Chain branching (when malonyl-CoA extender units are used) may occur through α-methylation derived from S-adenosyl methionine (SAM) or β-branching catalysed by a 3-hydroxy-3-methylglutaryl (HMGS) cassette. (B) Stable isotope precursors fed to Streptomyces virginiae and Pseudomonas fluorescens highlight the source of α- and β-branching in the polyketide structure in virginiamycin M and pseudomonic acid A respectively. | ||
PKS assemblies are organised in a number of different ways. Type II PKSs contain a collection of discrete enzymes that orchestrate an iterative series of polyketide extensions followed by a series of processing steps. Conversely, type I PKSs are comprised of covalently linked domains that collectively form modules, which are capable of catalysing chain extension and processing of the growing polyketide chain in an assembly-line manner. The two phylogenetically-distinct classes of type I PKS are identified by the presence of an in-cis or in-trans acyltransferase (AT) domain that is responsible for the loading of extender units.20 The cis-AT PKS contain AT domains within each module and generally follow a co-linearity logic whereby modular domain architecture can be correlated to the structure of the final polyketide. trans-AT PKSs rely on a small number of free-standing AT domains to load all of the ACPs within the PKS.
trans-AT systems can be extremely complex and often do not conform to co-linearity rules and show many other unusual features including extensive involvement of other trans-acting enzymes during polyketide assembly. trans-ATs, like cis-AT PKSs, can incorporate α-carbon branches into the product backbone by the use of building blocks that possess α-substituents (e.g. methylmalonyl). However, trans-AT systems can also achieve this by including a C-methyltransferase domain (MT) in a module that utilises S-adenosylmethionine (SAM) as a source of methyl groups.21–24 Other unusual features of trans-AT PKS include extensive involvement of trans-acting enzymes during polyketide biosynthesis and in particular the introduction of β-branches. Polyketide β-branching is less common than α-branching and requires the addition of a carbon nucleophile to the electrophilic β-carbon and subsequent processing which elaborates the chemical structure of the branch. Numerous examples have now been discovered including different chemical functionality, single or multiple β-branch incorporations in a single molecule and their discovery outside of trans-AT systems. This review will focus on the diversity, selectivity and mechanism of polyketide β-branching.
1.1 β-Branching
Early studies of polyketide β-branching were undertaken on Streptomyces virginiae and Pseudomonas fluorescens that produce the virginiamycins and pseudomonic acids respectively (Fig. 1B). A study by Feline et al. in 1977 speculated that 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) might be the precursor for the C-3 methyl group observed in pseudomonic acid A.25 During the 1980s, feeding studies of isotopically labelled precursors with cultures of S. virginiae revealed the origin of the β-branching carbon (C-33) in virginiamycin M as the methyl group of an acetate precursor.26,27 The authors proposed that the β-branch may arise from either: (i) an aldol addition of acetate to the polyketide chain, or (ii) an aldol addition of two polyketide chains. Differential enrichment between C-33 and the rest of the polyketide chain ruled out the condensation of two polyketide chains and a novel mechanism, akin to the biosynthesis of mevalonic acid in the formation of isoprenes was alluded to.In 2003, following the sequencing of the gene cluster responsible for the production of pseudomonic acid A from Pseudomonas fluorescens, it was predicted that an HMG intermediate or analogue would be formed prior to the loss of water and carbon dioxide in the formation of the C-3 β-branch.28 MupH, a 3-hydroxy-3-methylglutaryl synthase (HMGS), was identified but it wasn't until 2006 that Calderone et al. reported the cassette of proteins responsible for the β-branch incorporation in the biosynthesis of bacillaene.29 This cassette has been called a 3-hydroxy-3-methylglutaryl synthase (HMGS or HCS) cassette as the key catalytic step is carried out by an HMGS domain. Since these early reports, many natural products possessing β-branches have been isolated, mostly commonly from trans-AT PKSs,1,2 but also cis-AT and type II PKSs as well.30,31
The mechanism for β-branch formation in the biosynthesis of bacillaene was elucidated in vitro by Calderone et al. in 2006 (Fig. 2A).29 β-Branching arises through the interaction of the HMGS cassette with a β-ketothioester biosynthetic intermediate bound to a modular acceptor ACP (ACPA). The cassette contains a free-standing donor ACP (ACPD), a KS lacking the active site cysteine required for condensation (KS0) and a 3-hydroxy-3-methylglutaryl synthase (HMGS) that together form HMG-ACPA. Dehydration to give methylglutaconyl-ACP (MG-ACP) is achieved by an enoyl-CoA hydratase (ECH) domain (termed ECH1) that belong to the crotonase superfamily (CS) of enzymes which gives rise to the carboxylated β-branched products. The decarboxylation of MG-ACPA is catalysed by the second ECH domain in the cassette (ECH2) and gives rise to the second and third class of β-branch products: α,β-unsaturated β-branches or β,γ-unsaturated β-branches. Throughout this review the simplest α,β-unsaturated β-methyl branch will be referred to as an endo-β-methyl whilst the simplest β,γ-unsaturated β-methylene branch will be referred to as an exo-β-methylene.
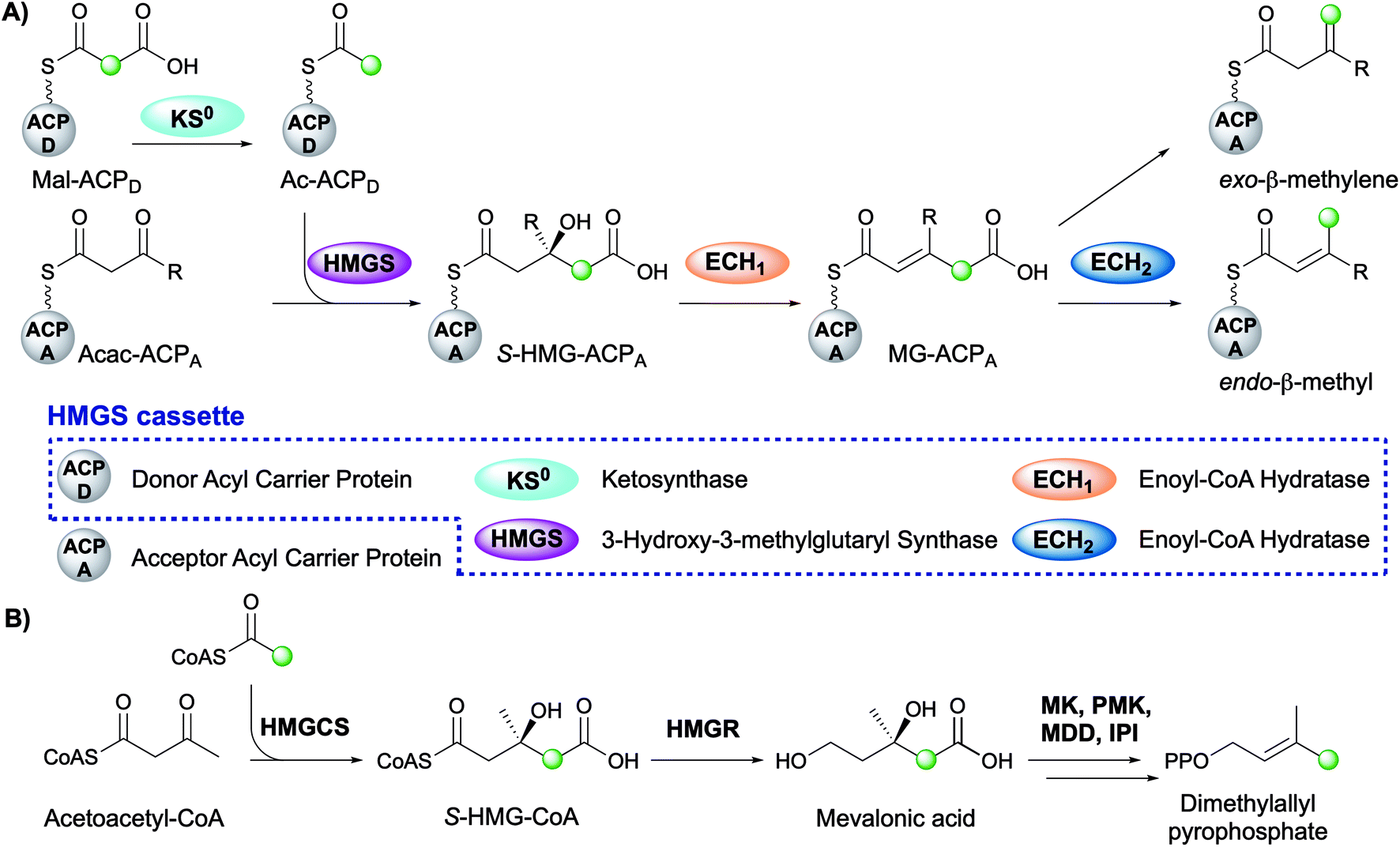 | ||
| Fig. 2 (A) β-Branching pathway by an HMGS cassette. The green dot indicates the position of the first carbon atom of the β-branch, most commonly originating from C-2 of acetate. The HMGS cassette consists of five proteins which are shown in the box and the colour coding for each domain is used throughout this review. (B) The mevalonate pathway that produces dimethylallyl pyrophosphate. | ||
Comparisons may be drawn between polyketide β-branching and mevalonate-dependent isoprenoid biosynthesis. The mevalonate pathway uses a 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS) to form S-HMG-CoA (Fig. 2B). Whilst phylogenetically similar, this pathway diverges from the β-branching pathway through subsequent reductive cleavage by 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR) of the thioester bond and further processing through mevalonate kinase (MK), phosphomevalonate kinase (PMK), mevalonate-5-pyrophosphate decarboxylase (MDD) and isopentenyl-PP isomerase (IPI) to form dimethylallyl pyrophosphate, a key intermediate in terpene biosynthesis (Fig. 2B).32 The similarities of the pathways were reviewed in detail by Calderone in 2008.33
2. Diversity of branches
Molecular complexity may be introduced at each stage of the multi-enzyme β-branching pathway as is apparent from the different groups which may be installed by an HMGS cassette and associated tailoring enzymes (Fig. 3). In addition to the canonical pathway that produces an endo-β-methyl branch (1), PKSs may accept modified substrates (methylmalonyl (4, 7, 12) or halogenated intermediates (8)), contain domains with alternate selectivity (ECH2 (6)), lack catalytic domains (10, 11) or contain those that encode tailoring reactions (oxidation (9), reduction (3), ring formation (5) and methyl ether formation (2)). With a large number of enzymatic partners required for the synthesis of complex β-branches, high fidelity of the pathway is required. This is achieved through a mixture of protein–protein interactions (ACP/HMGS interaction), use of in-cis vs. in-trans domains (ECH2, ER, Ox) and exclusion of specific domains (ECH2). The details of the β-branching pathways analysed in this review, including the parent organism, the type of β-branch incorporation and the biosynthetic machinery that installs the β-branch, are shown in Table 1 (see end of review).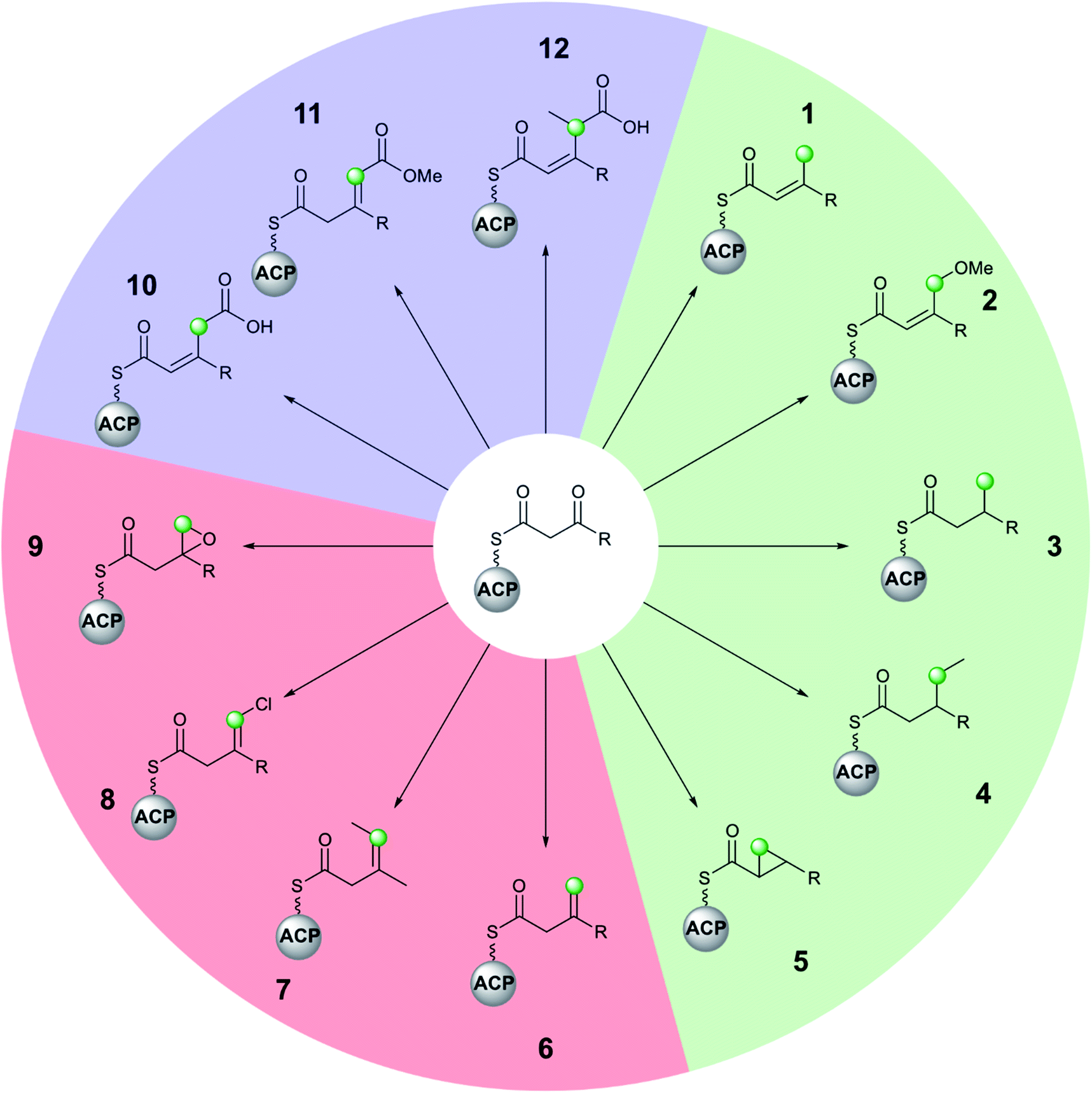 | ||
| Fig. 3 The diversity of polyketide β-branching by an HMGS cassette is shown by the different moieties that may be installed by homologous cassettes. The green dots indicate the position of the first carbon atom of the β-branch. β-Branches 1–5 (green) arise from decarboxylation and C-4 protonation to form an α,β-unsaturated branch prior to subsequent modification. β-Branches 6–9 (red) arise from C-2 protonation by ECH2 domains to give a β,γ-unsaturated branch that may contain additional functionality or undergo further modification. The carboxylated β-branches 10–12 (blue) do not undergo decarboxylation by the HMGS cassette. Although stereochemistry may have been determined in select examples above (e.g.5, Section 5.3), this is not shown to maintain generality. | ||
3. Techniques for probing β-branching pathways
The challenges associated with reconstituting PKS pathways in vitro has been documented by Clay Brown and co-workers.34 The substantial number of interacting in-cis and in-trans domains required for the in vitro reconstitution of β-branching pathways provides a challenge for exploring these pathways. Experiments rely on the expression and purification of multiple proteins and priming them with appropriate chemical intermediates. The proteins may be single or tandem domains and may also require excision from a modular PKS. Furthermore, consideration must be given to the synthesis of intermediates or model substrates with or without isotopic labels, purification of microgram quantities of reaction products and the use of multiple analytical techniques (LC/MS, protein MS and NMR).A common feature of PKS pathways is the tethering of reaction intermediates to an ACP that delivers substrates to catalytic domains.35 A phosphopantetheine (Ppant) arm is transferred from CoA to the apo-ACP, resulting in the active holo-form of the carrier protein bearing a terminal thiol that can shuttle intermediates via thioester linkages. A vital tool for the dissection of ACP-bound biosynthetic intermediates is the Ppant ejection assay developed during analysis of these pathways (Fig. 4A).36,37 An abundant ion of X-charge state (green) is isolated and subjected to collision-induced dissociation (CID) resulting in the ejection of a 1+ ion derived from the phosphopantetheine arm (red) and an ACP fragment corresponding to the X − 1 charge state (blue). Importantly, the thioester bond is not cleaved which allows for the determination of the thioester-bound substrate with higher resolution compared to the deconvolution of the multiple charge states of an ACP-bound intermediate.
 | ||
| Fig. 4 (A) Mass spectrum of acetoacetyl-ACP showing the m/z envelope and the resulting m/z from collision-induced dissociation (CID) of the ACP precursor (green) into a Ppant ejection ion (orange) and the resulting ACP peak (purple). (B) The structure of coenzyme A and common structural mimics used to monitor enzymatic reactions. (C) Chemoenzymatic synthesis of functionalised coenzyme A from commercially available D-pantothenic acid and subsequent ACP loading. | ||
Chemical synthesis and biotransformations play a key role in the preparation of mimics of biosynthetic intermediates which may be used for functional studies and identification of enzymatic products. Studies have taken advantage of acyl-SNAC or pantetheine intermediates as these smaller molecules may be synthesised from readily available starting materials (Fig. 4B).38,39 Whilst these mimics only contain fragments of a Ppant arm often required for recognition/interaction with enzymatic partners, they may be directly detected by LCMS methods allowing for simpler assay setup. Acyl pantetheines may also be converted into their respective CoA analogues in vitro by incubation with enzymes from the CoA biosynthetic pathway (CoaA/D/E) (Fig. 4C).40 This method has allowed the synthesis of more complex biosynthetic intermediates, including those incorporating isotopic labels, which complement the few commercially available CoA derivatives. The Ppant arm bearing any chosen chemistry may be subsequently transferred to an appropriate apo-ACP using a Ppant transferase enzyme (PPTase).41,42 The derivatised ACP more closely mimics the substrate delivery seen in the native PKS systems and ensures the correct protein–protein interactions can be formed.
Structural biology has enabled the accurate dissection of β-branching pathways through protein crystallography and structural NMR studies. The advancement in NMR technology has also allowed for the design of more complex assays utilising isotopic labels and identification of trace intermediates in β-branching assays.43
4. Mechanism of β-branching
4.1 Malonyl-loading and decarboxylation
The first step in canonical polyketide β-branching is the transfer of a malonyl group from CoA to the donor ACPD catalysed by an acyl transferase (AT) which may load multiple ACPs within the gene cluster (Fig. 5).44 Subsequently, a KS0 domain lacking the cysteine required for condensation, decarboxylates malonyl-ACPD to form Ac-ACPD. An acyltransferase/decarboxylase (AT/DC) di-domain which catalyses both the transfer of the acyl chain and the subsequent decarboxylation is utilised in the loading of methylmalonyl-CoA to form propionyl-ACPD.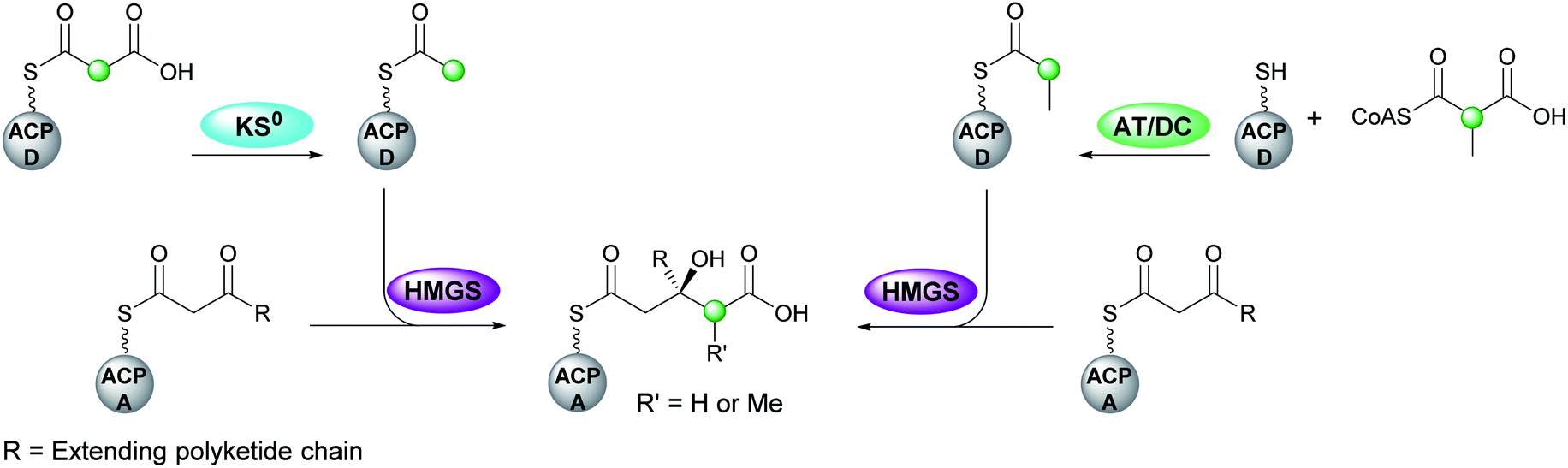 | ||
| Fig. 5 Acetyl-ACPD is formed from the decarboxylation of malonyl-ACPD by a KS0 domain. Propionyl-ACPD is generated by a separate enzymatic pathway that utilises a bi-functional AT/DC domain that catalyses acyl transfer and decarboxylation. The next step is the aldol addition of the acyl-ACPD to the β-ketothioester-ACPA, where R is the extending polyketide chain. | ||
4.2 Aldol addition by HMGS
The aldol addition is complex and requires the interaction of two distinct ACPs that each deliver a different substrate to the HMGS. Ac-ACPD associates with the HMGS and transfers the acetyl group to an active site cysteine residue of the HMGS. Following deprotonation of the acetyl moiety by an adjacent histidine residue, the resulting enolate attacks the ketone of the ACPA-bound β-ketothioester, which is the growing polyketide chain, to give the cross-linked intermediate. Hydrolysis of the HMGS thioester bond results in the formation of HMG-ACPA which dissociates from the HMGS active site (HMG-ACPA is used for brevity although strictly HMG-like as part of a more complex polyketide). Consistent with the formation of S-HMG-CoA in mevalonate-dependent isoprene biosynthesis, S-HMG-ACPA is the product of the condensation.45–47A key challenge for the selectivity of the HMGS is the delivery of the correct substrates at the appropriate time. A similar demand arises in the homologous reaction catalysed by the 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS) of primary metabolism (Fig. 2B) as well as KS domains in polyketide biosynthesis. HMGCS discriminates between the acyl groups of the two substrates (acetyl-CoA and acetoacetyl-CoA) as both access the active site through the same entrance. KS domains achieve the correct substrate delivery of ACP-tethered substrates through different channels to the active site, often arising from the local PKS architecture or use of docking domains. By homology, a single channel to access the active site in HMGSs, requires interaction in-trans with both ACP-tethered substrates. Phylogenetic analysis shows that ACPA and ACPD clade separately from each other and both have been structurally and functionally characterised.48 Taking ACPD and the acetyl transfer step first, this must involve a specific interaction of acetyl-ACPD with HMGS. The binding constants for apo- and holo-CurB (ACPD) to CurD (HMGS) have been measured at 1.1 μM and 0.5 μM, respectively, representing a two-fold increase in binding affinity through interaction of the Ppant arm with the HMGS. The strength of interaction is several fold higher than most ACP-synthase interactions and is in excess of 100-fold higher affinity than the equivalent HMGS/ACPA interaction. Strict specificity has been demonstrated and ACPD could not be substituted by ACPA.
The 2.1 Å X-ray structure of the HMGS from the curacin pathway revealed a catalytic triad comprised of Glu82, Cys114 and His250 (Fig. 6A) buried deeply within the enzyme at the end of a binding cavity.46 This was homologous to the HMGCS that generates HMG-CoA by direct aldol condensation of acetyl- and acetoacetyl-CoA substrates. However, near the entrance to this cavity, the HMGS presents a longer disordered loop of fourteen residues containing a conserved insertion comprised of residues 155–164 (highlighted as a broken chain in Fig. 6A). Lining the remainder of the cavity entrance, negatively charged residues Asp222 and Glu225 in HMGS helix8, positively charged residue Arg266 on helix9 along with Arg38 and Asp214 form a distinct pattern that define the electrostatic potential of the ACP binding site.
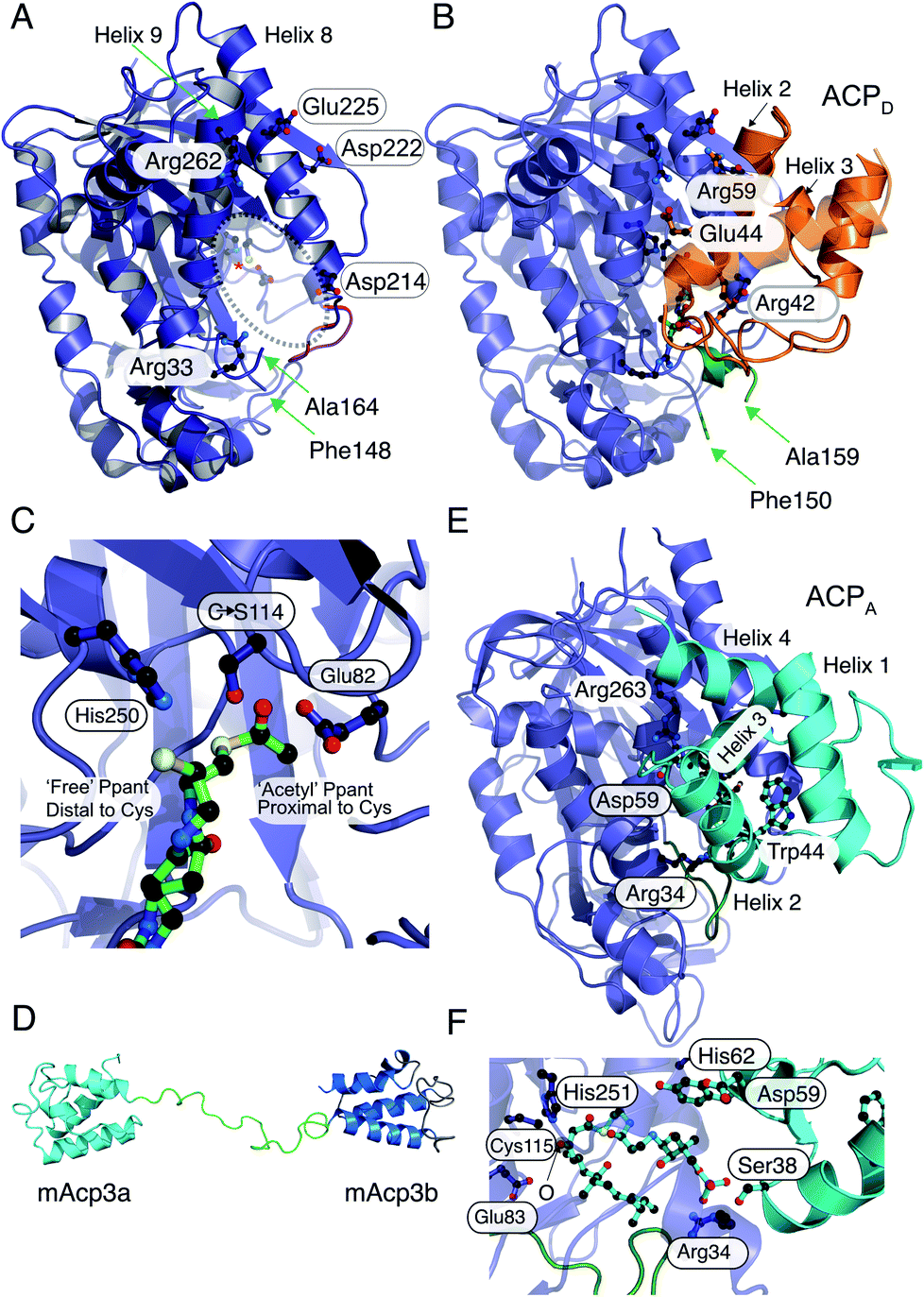 | ||
| Fig. 6 X-ray crystallography, NMR solution structures and modelling of ACPD, ACPA and HMGS interactions. (A) X-ray crystal structure of the curacin HMGS in free form (PDB:5KP5). The catalytic triad (Glu82, Cys114 and His250) are circled and highlighted with an asterisk. Residues identified as important for ACPD binding and/or function are labelled. Green arrows from Phe148 to Ala164 mark the ends of a disordered loop unique to the ACP binding HMGSs and this was not visible in the X-ray structure. (B) X-ray crystal structure of the complex between acetyl-/holo-ACPD (gold) and HMGS (blue) (PDB:5KP8). Key ACPD residues involved in the interaction with HMGS are shown and the ordering of the disordered loop (green) upon complexation with ACPD is apparent. (C) Close-up of the active site of the acetyl-/holo-ACPD HMGS (blue) complex showing the positioning of the acetyl group proximal to the active site cysteine residue and the ‘blocked’ terminal thiol of the holo-ACPD. (D) NMR solution structure of the ACPA didomain (mAcp3a and mAcp3b) from the mupirocin biosynthetic pathway. The two ACP domains are connected by a flexible linker shown in green. (E) Model for mAcp3a docked to the mupirocin HMGS. The conserved tryptophan residue (Trp44) is shown packed adjacent to helix 3 that packs at the interface with the HMGS. The disordered loop is shown in green. (F) Close-up of the β-ketothioester polyketide intermediate (pre β-branching) bound to the HMGS. The β-keto group is labelled with an O. | ||
Through co-crystallisation of ACPD (CurB) and HMGS (CurD) from the curacin pathway, the key interactions between ACPD and HMGS have been demonstrated. The unusual position of helix 3 in ACPD gives rise to a cleft that is complementary to a hydrophobic ridge located on helix α8 of the HMGS (Fig. 6B). Additionally, the surface presented to the HMGS contains a positive/neutral/negative charge distribution which is different to PKS ACPs and FAS ACPs in general which are positively and negatively charged on the surfaces that interact with cognate proteins. A Cys114 to Ser114 mutant of HMGS was utilised in co-crystallisation studies with acetyl-ACPD. The resulting 1.9 Å structure revealed the positioning of the Ppant chain and the acetyl group buried deeply in the HMGS binding pocket and positioned proximal to the active site Cys (Fig. 6C). The Ppant arm made numerous electrostatic interactions with HMGS, i.e. Arg38 helping to account for the 2-fold increase in affinity of holo- versus apo-ACPD (0.5 μM and 1.1 μM respectively). By comparison to other ACP partner interactions, both apo and holo are considered to be tight binding compared to the ranges typically observed (>2 μM).35 Despite the Ser substitution, hydrolysis of the acetyl group led to the production of bound holo-ACPD in the same sample. Solving the holo-ACPD bound structure revealed the positioning of the Ppant arm and free thiol, now distal to the active site Cys and essentially occluded from the acetyl binding pocket.
The second chemical step in the branching mechanism involves the aldol addition of the acetyl group to the polyketide substrate covalently linked to ACPA. To prevent aberrant β-branching, the HMGS cassette must only interact with the correct modular ACPA. Non-branching PKS modules usually contain a single ACP and the molecular recognition features controlling the sequence of protein–protein interactions have been extensively studied.49,50 ACPA may be distinguished from their non-branching counterparts, however, by the presence of a conserved tryptophan positioned six residues after the conserved serine bearing the Ppant arm and forming part of a GxDSxxxxxW signature motif.51 The binding constant for the bryostatin HMGS (BryR) to a cognate ACPA (BryM3) was determined by SPR to be 177 μM, 180 μM and 200 μM for apo-, holo- and acetyl-ACPA respectively. This demonstrates the importance of protein–protein interactions in ACP and HMGS recognition rather than purely substrate control.
β-Branching modules may also contain between 1 to 3 copies of ACPA. Tandem ACPs in the mupirocin gene cluster have been shown in vivo to increase the flux of metabolites through the pathway at a rate-limiting, in-trans biosynthetic step.52 This view was reinforced by an NMR structure of the di-domain ACPA from mupirocin that showed two, independent domains that were predicted to function in parallel.51,53 Studies on the curacin biosynthetic pathway demonstrated in vitro an increased efficiency with tandem ACPs, however, some synergistic interaction of the triplet domain was also observed.54 Wang et al. genetically engineered module 6 of the 6-deoxyerythronolide B (DEBS) cis-AT PKS by inserting two additional ACPs to give a triplet ACP. This module was chosen as it has been shown to accept non-native substrates as SNAC thioesters, and catalyse in vitro chain extension with methylmalonyl-CoA and hydrolysis of the products by the C-terminal TE. By measuring the total polyketide product formation in vitro, a 2.5-fold increase in production was demonstrated for the triplet ACP compared to the native single ACP. This further suggests that increasing ACP concentration results in higher flux and polyketide production by a modular PKS.55 Shen and co-workers have also demonstrated that the number of ACPs in polyunsaturated fatty acid synthesis strongly influenced product titre with increasing numbers of deactivated ACPs giving a proportionate reduction in yield.56
The NMR structure of the didomain ACPAs, mupA3a and mupA3b from the mupirocin biosynthetic pathway from P. fluorescens revealed that both ACPs possessed similar folds connected by a flexible linker (Fig. 6D). The burial of the conserved tryptophan side chain creates a hydrophobic core that presents helix II and III in a distinctive orientation for interaction with the HMGS and was shown to be essential for ACP protein folding and production of pseudomonic acid A. Simultaneous or sequential binding of the ACPD and ACPA has not been determined but combined docking and bioinformatics have been used to model the complex between mupA3a and the β-branching cassette HMGS (without a priori knowledge of the X-ray structures). MupA3a docked in the same place as ACPD and the phosphate of the phosphopantetheine engaged with the homologous arginine residue, Arg34, at the entrance to the HMGS cavity. However, mupA3a packs quite differently against HMGS, with helix III running perpendicularly to helices 8 and 9 of HMGS (Fig. 6E). The extended polyketide substrate attached to ACPA (Fig. 6F) is forced to double back on itself to be accommodated in the cleft and in doing so interacts with the disordered loop, moving it to a new location distinct from the ACPD complex.
4.3 Dehydration by ECH1
Following the formation of HMG-ACPA, the subsequent dehydration appears to be ubiquitous across all β-branching pathways to form methylglutaconyl-ACPA (MG-ACPA), a transient intermediate that is difficult to observe due to the equilibrium favouring the reverse reaction to form HMG-ACPA (Fig. 7). This phenomenon was first reported by Gu et al.30 who described the coupling of ECH1/ECH2 steps in the formation of an endo-β-methyl. As a result, the precise structure of MG-ACP is hard to assign. However, the α,β-unsaturation (relative to the thioester) may often be inferred in the decarboxylated β-branches, due to the mechanism of CO2 loss. For the carboxylated β-branched polyketides, the ECH1-catalysed dehydration is the final step along their β-branch biosynthetic pathway, resulting in both α,β- and β,γ-unsaturated polyketides that retain a carboxyl group.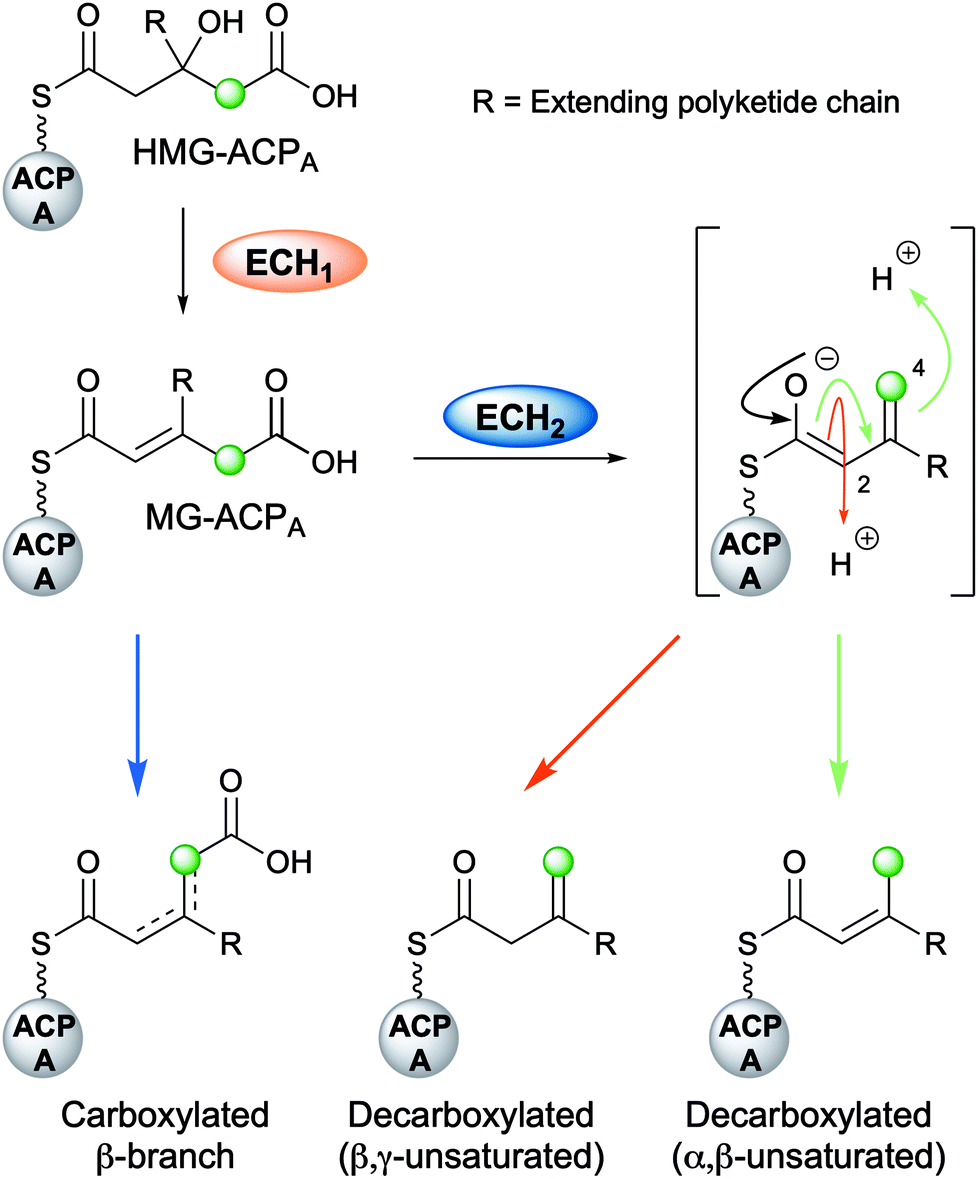 | ||
| Fig. 7 Role of ECH domains. HMG-ACPA may be differentially processed to produce the three distinct families of β-branches: carboxylated β-branches, decarboxylated with α,β-unsaturation or decarboxylated with β,γ-unsaturation. The decarboxylated products arise from a vinylogous enolate intermediate which can be selectively protonated at either C-2 or C-4. | ||
Despite the transient nature of MG-ACP, the stereochemistry of the double bond was determined by Walker et al.57 The newly incorporated acetate is positioned cis to the alkene proton and the growing polyketide chain trans to the alkene proton. This is the opposite stereochemistry to that observed in the formation of an endo-β-methyl branch which has the growing polyketide chain cis to the alkene proton and the β-methyl branch trans. This implies a bond rotation must take place during the decarboxylation step and the absence of this rotation would result in a kink in the polyketide chain.
Recently, the first crystal structure of a β-branching ECH1 domain, pksH, was published by Nair et al. (Fig. 8).58 PksH catalyses dehydration in the bacillaene β-branching pathway and is a member of the crotonase superfamily of enzymes. It retains the classical CS-fold, forms a trimer in solid and solution state and the backbone residues Gly67 and Gly114 were predicted to form the oxyanion hole that stabilises the enolate intermediate. Through inspection of the active site, generation of point mutants and functional studies it was proposed that Glu137 deprotonates the HMG intermediate whilst Asp68 reprotonates following the loss of water (Fig. 8C). The system was difficult to study as only trace amounts of product were identified following incubation with high concentrations of proteins (5 mg mL−1) with long reaction times (>72 h) at 37 °C confirming the equilibrium of the reaction favouring HMG intermediates. Therefore, coupled reactions with PksI (ECH2) were used as a readout of catalytic competency of point mutants.
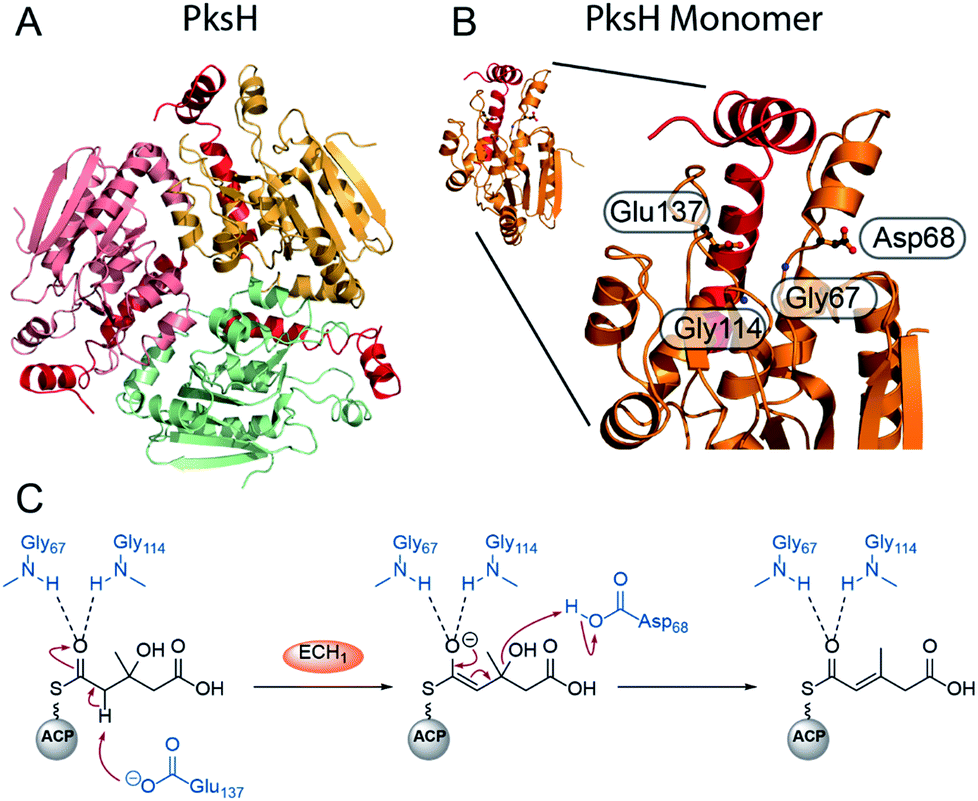 | ||
| Fig. 8 Structure and mechanism of ECH1 domain PksH. (A) Trimeric structure of PksH (PDB: 3HP0) with each subunit coloured differently and the C-terminal helices that form the self-association fold are shown in red. (B) A single monomer and expanded active site showing annotated residues thought to form the OAH and catalytic residues for deprotonation and re-protonation steps. (C) The proposed mechanism of dehydration by PksH uses catalytic residues Glu137 and Asp68 whilst Gly67 and Gly114 form the oxyanion hole. | ||
4.4 Decarboxylation by ECH2
Following the loss of water from HMG-ACP, the final step for the majority of β-branching mechanisms is the ECH2-catalysed decarboxylation (Fig. 7). This step is important for creating diversity in β-branching through regioselective protonation which may occur at either C-2 or C-4 of the reaction intermediate resulting in the formation of endo-β-methyl and exo-β-methylene branches. Downstream processing may subsequently occur on either of the unsaturated moieties to further diversify the β-branch architecture.The crystal structure of the N-terminal ECH2 domain of CurF from the curacin gene cluster revealed the excised domain to have the overall fold of a crotonase enzyme and to possess characteristic catalytic residues (Fig. 9A and B).59 Deprotonation of the carboxylic acid could be achieved by an active site histidine (His240) and following decarboxylation, the enolate could be stabilised within an oxyanion hole formed by the backbone NHs of alanine (Ala78) and glycine (Gly118) residues (Fig. 9E). Finally, lysine (Lys86) was proposed to be positioned appropriately for protonation of the intermediate. This was the first reported crystal structure of a crotonase enzyme that accepts ACP-bound substrates and the preference of CurF ECH2 for ACP-bound intermediates over CoA-bound intermediates was also highlighted.
 | ||
| Fig. 9 Structure and mechanism of ECH2 domains CurF and PksI. (A) Trimeric structure of CurF (PDB: 2Q2X) with each subunit coloured differently and the C-terminal helices that form the self-association fold are shown in red. (B) Closeup of the CurF active site with OAH and catalytic residues annotated. (C) PksI active site (PDB: 4Q1G) in the closed form. Residues 76–84 were not visible in this form. (D) PksI active site in the open form where the additional residues Lys80 and Phe81 are now visible. (E) Mechanism of decarboxylation by ECH2 domains from CurF (Gly118, Ala78 and His240) and PksI (Gly66, Gly108, His230). | ||
The trans-acting ECH2 domain (PksI) from the bacillaene β-branching pathway was characterised by Nair et al. (Fig. 9C and D).58 Expression, crystallisation and functional studies of wild-type and mutant enzymes showed the characteristic CS-fold, catalytic His230 and oxyanion hole-forming residues (Gly66 and Gly108). Two distinct states of the wild-type enzyme were crystallised in the presence and absence of glycerol. The unliganded structure occupies a closed conformation with Phe136 and Phe81 orientated towards the active site and His230 pointed away. Upon ligand binding, an open-conformation is adopted whereby His230 points towards the centre of the active site and forms a hydrogen bond with the glycerol hydroxyl group. Phe81 and Phe136 rotate out of the active site, with Phe136 forming a stacking interaction with Phe234 and His235. No active site lysine for re-protonation was identified and two further candidates (Lys80 and Lys232) were ruled out by point mutations. It was concluded that His230 likely catalyses the deprotonation and subsequent re-protonation steps (Fig. 9E).
4.5 Full-length and truncated modular ECH domains
Most β-branches are incorporated through the interaction of a trans-acting HMGS cassette with the modular ACPAs. However, in the incorporation of exo-β-methylene branches the trans-acting ECH2 domain is often absent and the HMGS cassette is instead supplemented by an in-cis ECH2 located proximal to the ACPAs (denoted in pink in subsequent figures). Additionally, an upstream ECH domain is often annotated in these pathways and it may be assumed this is an extra ECH1 domain. However, closer inspection reveals that this domain is truncated, shows low sequence homology to characterised ECH1 domains and is therefore assumed to be non-functional. This feature will be highlighted in the discussions below of the relevant pathways.5. Polyketides containing a single β-branch
The details of individual mechanistic steps have been rationalised following extensive in vitro reconstitution of β-branching cassettes from many pathways. Selected pathways are discussed in more detail below beginning with natural products with a single β-branch followed by examples with multiple branches. The types of β-branches incorporated into each compound are numbered according to Fig. 3.5.1 endo-β-Methyl (1) – mupirocin and bacillaene
A single endo-β-methyl branch is the most common β-branching moiety. Pseudomonic acid A, the major component of the antibiotic mixture mupirocin, contains a single endo-β-methyl branch and is one of the earliest gene clusters to be identified to contain an HMGS cassette (Fig. 10).28,60–62 The initially reported incorporation of a cleaved acetate group was confirmed through a combination of isotope labelling and gene cluster comparison.25,63 The mechanism of such β-branching was elucidated in the bacillaene biosynthetic pathway in vitro by Calderone et al. using a combination of biochemical assays and mass spectrometry.29 Both of these pathways utilise tandem ACPAs.51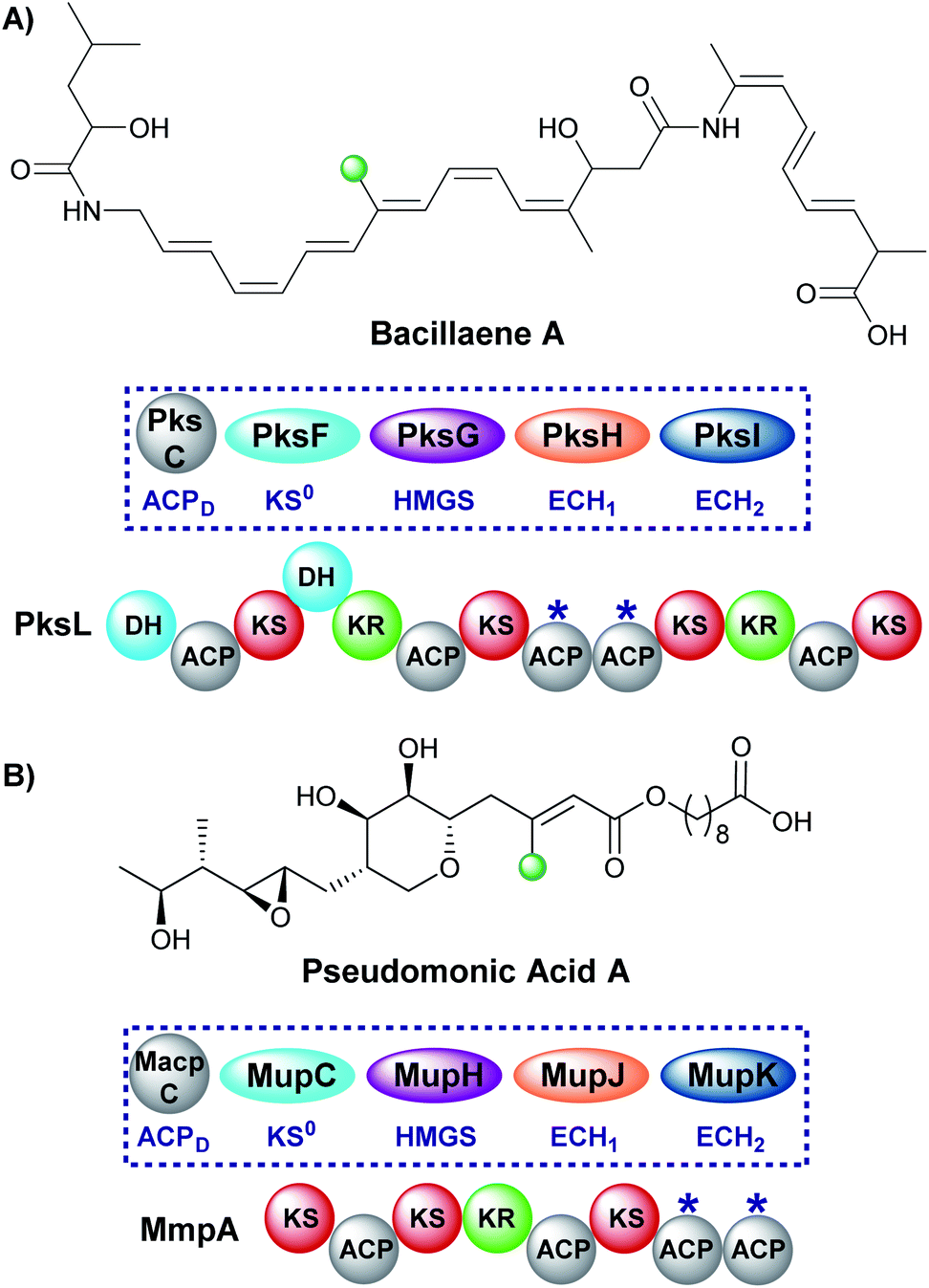 | ||
| Fig. 10 Bacillaene (A) and pseudomonic acid A (B) both contain an endo-β-methyl branch. The respective HMGS cassettes and modular architecture of the respective polyketide synthases are shown, whilst ACPAs are labelled *. | ||
5.2 Saturated β-methyl (3) – patellazole and cylindrocyclophane
During the creation of a saturated β-methyl branch the intermediate α,β-unsaturated thioester may be reduced by an ER to give the β-methyl group. In patellazole C biosynthesis, a saturated β-methyl is incorporated by an unusual HMGS cassette lacking a trans-acting ACPD (Fig. 11A).64 The modular PKS PtzD contains a di-domain ACPA but also a candidate for ACPD located at the C-terminus of the polypeptide. This ACP showed homology to donor ACPs providing a rare example of an in-cis ACPD. PtzQ is a trans-acting ER domain that is hypothesised to carry out the reduction of the endo-β-methyl to produce the saturated β-methyl branch.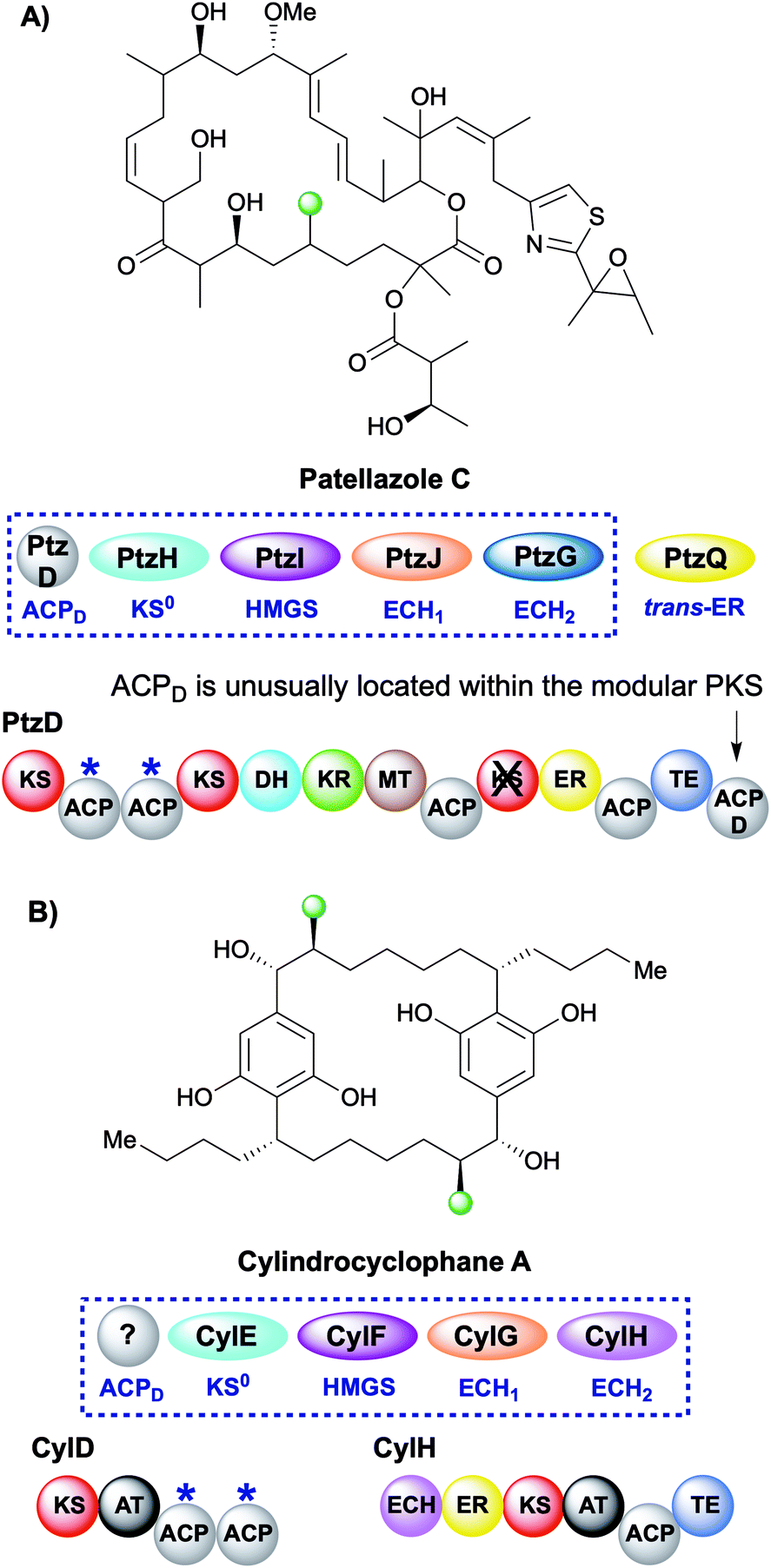 | ||
| Fig. 11 Patellazole C and cylindrocyclophane A contain one and two saturated β-methyl branches respectively. The modular architecture of the polyketide synthase where branching occurs is highlighted. (A) In the case of patellazole C a trans-acting ER reduces the endo-β-methyl branch formed by a trans-acting ECH2 PtzG. (B) In cylindrocyclophane A biosynthesis, both the ECH2 (pink) and ER (yellow) are cis-acting and located on CylH whilst the ACPAs reside on CylD. | ||
The multi-faceted biosynthesis of cylindrocyclophane combines a fatty acid starter unit, polyketide chain extension and β-branching via a type I cis-AT PKS and a dedicated type III PKS that aromatises the intermediate prior to a final dimerisation to yield the product (Fig. 11B).65,66 A saturated β-methyl branch is introduced into each monomer by a combination of in-cis (ECH2, ER) and in-trans (KS0, HMGS, ECH1) domains, whilst no ACPD was identified. The type I PKS architecture is unusual as the ECH2 and ER domains are part of a split module and reside on a different ORF (CylH) from the acceptor ACPAs (CylD). This modular architecture is also observed in the apratoxin biosynthetic pathway whereby an in-cis ECH2 and ER domains are located on a different protein to the ACPAs, whilst a split module of ACPAs and in-cis ER (with trans-acting ECH2) is found in the biosynthesis of macrobrevin,67,68 leptolyngbyalide (Section 6.4) and myxovirescin (Section 6.8).
5.3 Cyclopropane (5) – curacin and vinyl chloride (8) – jamaicamide
Curacin and jamaicamide, isolated from Lyngbya majuscula, are produced by cis-AT PKS pathways that share common enzymatic machinery to produce different products (Fig. 12).69,70 The HMGS cassette contains free-standing ACPD, KS0, HMGS and ECH1, however, the ECH2 domains are located at the N-terminus of a type I PKS (CurF) that would associate downstream of the ACPAs in CurA. The compounds are unusual in that the β-branching intermediate HMG-ACP in both pathways is chlorinated at C-4 to give γ-Cl-HMG-ACP. In vitro reconstitution of the pathway utilised the non-chlorinated derivative, indicating that the halogen was not essential for HMGS cassette recognition or activity.45,46 Whilst the chlorine atom is retained in the final vinyl chloride β-branch in jamaicamide, it is lost in the biosynthesis of the cyclopropane ring of curacin.30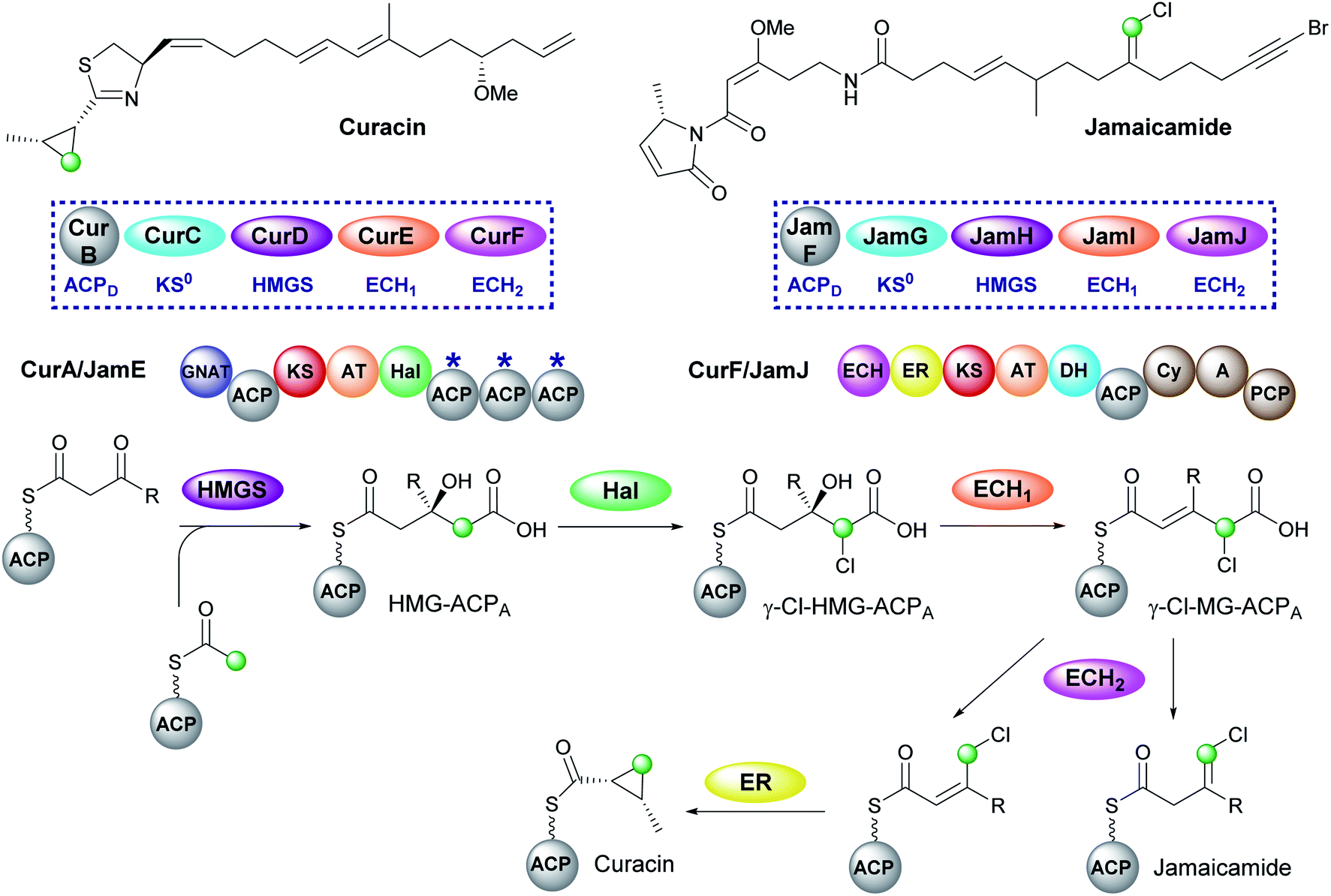 | ||
| Fig. 12 Curacin and jamaicamide contain a cyclopropane and vinyl chloride moiety respectively. The two pathways share homologous HMGS cassettes and modular architecture consisting of a split module whereby the ACPAs (CurA/JamE) and cis-acting ECH2 and ER (CurF/JamJ) reside on different proteins. γ-Cl-HMG-ACPA is made from HMG-ACPA by a halogenase domain (Hal) located in CurA/JamE. | ||
Chlorination is catalysed by a Fe2+/α-ketoglutarate (αKG) dependent halogenase domain located immediately upstream of the triplet of ACPAs in the type I PKS CurA.71,72 In these halogenases a Cl− ion typically replaces a Glu or Asp residue in the coordination shell of the iron and reacts with a substrate radical in a homologous fashion to the hydroxyl group of Fe2+/αKG dependent hydroxylases. Using a combination of crystallographic studies and in vitro assays, the cofactor dependency (Cl−, O2, Fe2+ and α-ketoglutarate), conformational switching between inactive and active states and the structure/function of the halogenase was demonstrated in the conversion of HMG-ACP to γ-Cl-HMG-ACP. From this common intermediate, both pathways continue with the ECH1-catalysed dehydration to give γ-Cl-MG-ACP. The metabolic coupling of CurE (ECH1) and the modular ECH domain of CurF (ECH2) was required to furnish an endo-β-methyl branch in curacin indicating the unfavourable position of the equilibrium in the dehydration reaction.45 The key divergence between curacin and jamaicamide β-branching incorporation occurs at the ECH2-catalysed decarboxylation step. In the curacin biosynthetic pathway, protonation at C-4 generates a chlorinated endo-β-methyl branch. From this intermediate, the cyclopropane ring is formed by a modified ER domain downstream of the ECH2 domain which results in the loss of chloride ion.73 Conversely, protonation at C-2 by the ECH2 domain of JamJ in jamaicamide biosynthesis results in the production of the vinyl chloride moiety.30 These differences arise despite the two ECH2 domains sharing 61% identity. Subsequently, although the downstream ER in JamJ is catalytically active, the chlorinated exo-β-methylene intermediate is not a compatible substrate. The identity of the two β-branches was confirmed by hydrolysis of ACP-bound products as amides, GC/MS analysis and comparison to synthetic standards.
5.4 exo-β-Methylene (6) – pederin-like compounds
The pederin-like compounds (pederin,74–77 labrenzin,78 diaphorin,79 mycalamide,80–82 onnamide A,76,83,84 psymberin,85–87 nosperin88 and cusperin A89) exhibit a range of biological activities and have been isolated from numerous species including rove beetles and marine sponges. With the exception of psymberin, which contains a shortened carbon chain, they all share a common structural feature: a tetrahydropyran ring with an exo-β-methylene branch. In all cases, the exo-β-methylene is incorporated by an HMGS cassette lacking a trans-acting ECH2 but containing a cis-acting ECH2 domain immediately upstream of the ACPAs which vary in number between 1–3 ACPs (Fig. 13). Most pathways are annotated as containing two modular ECH domains, however, closer inspection reveals that the N-terminal domain is truncated, and assumed to be non-functional, or absent. García et al. presented an alignment of the truncated N-terminal ECH domains, and little sequence homology was observed.78 The second domain is always a full-length domain of 248 residues which shows homology to characterised ECH2 domains. With only one ECH2 domain in the gene cluster, it is assumed the exo-β-methylene arises from decarboxylation and selective protonation at C-2 to give the β,γ-alkene. | ||
| Fig. 13 The pederin-like compounds share a common β-branching pathway to install an exo-β-methylene branch. The HMGS cassettes lack an in-trans ECH2 domain but ECH2 domains are located prior to the β-branching ACPs in the modular PKS. A truncated (#) N-terminal ECH domain may be present or absent altogether (−). The onnamide gene cluster lacks ACPD, KS0 and ECH1 domains, but this is likely due to a lack of genomic data encoding these genes rather than complete absence. The number of ACPAs vary for each pathway as annotated and are labelled *. | ||
5.5 exo-β-Propylene (7) – fogacin
In 2019, Sato et al. reported the first β-alkylation in a type II PKS following the isolation of the minor metabolite fogacin C from Actinoplanes missouriensis (Fig. 14).31 The core polyketide backbone of fogacin C is typical of aromatic polyketides produced by actinomycetes, whereby basic chain assembly is catalysed by a minimal PKS comprised of a KS, a chain length (or initiation) factor (CLF) and an ACP. However, the structure contains a novel tertiary alkene β-branch arising from the incorporation of a methylmalonyl building block.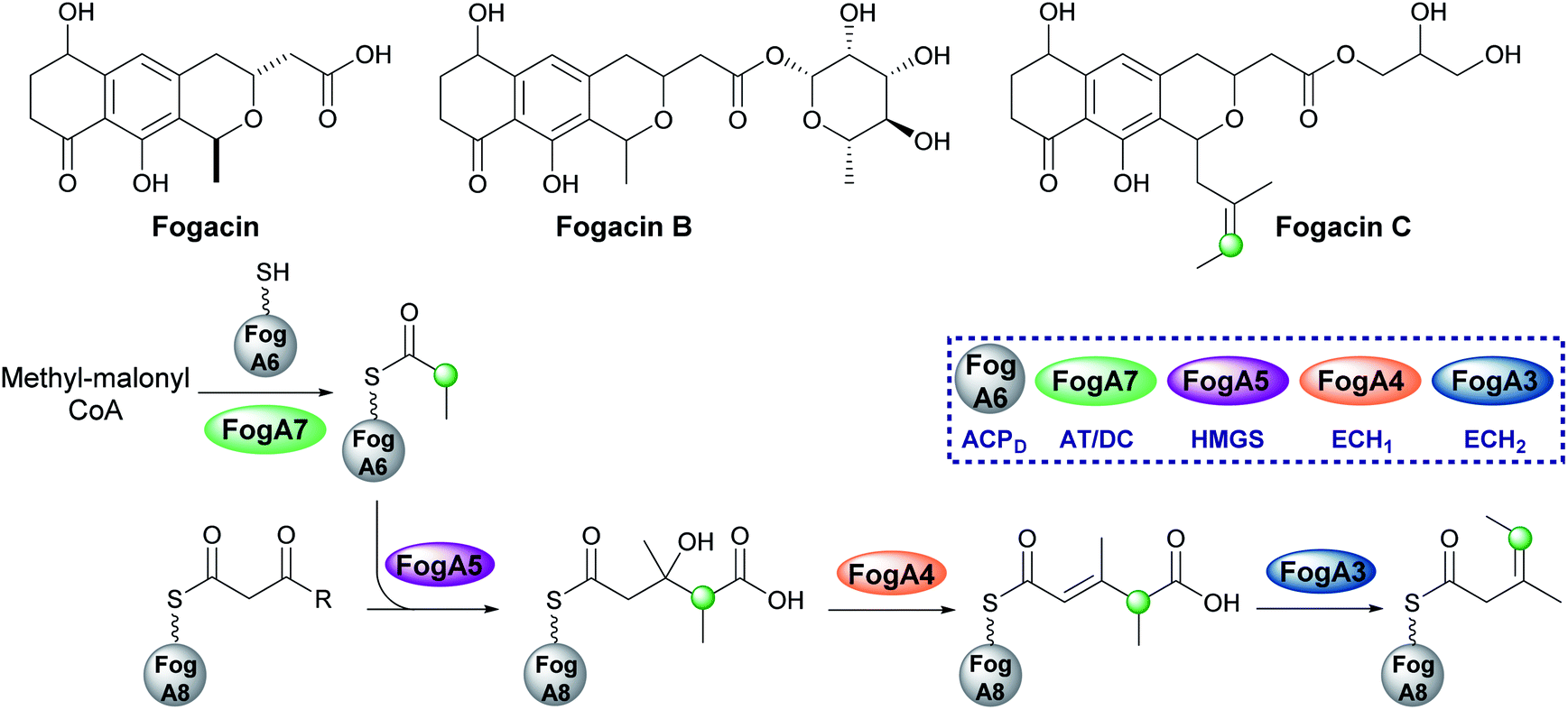 | ||
| Fig. 14 Actinoplanes missouriensis 431 produces fogacin derivatives, however, fogacin C is the only one to contain a β-branch and is currently the only known β-branched secondary metabolite from a type II PKS system. The HMGS cassette contains an AT/DC didomain (Fog A7) to form propionyl-ACPD which produces an exo-β-ethyl branch. | ||
Analogous to β-branching in the biosynthesis of leinamycin (see Section 5.8), whereby methylmalonyl-CoA is used as a starter unit, no AT and KS genes required for acyl-ACPD synthesis were found in the HMGS cassette. Instead, FogA7, a domain with homology to LnmK (45%) was proposed to catalyse acyl-transfer and decarboxylation in forming propionyl-ACPFogA6 for β-alkylation (Fig. 14). As this is a type II system, the acceptor ACPFogA8 is free standing rather than modular and thought to bear an acetoacetyl group following a single Claisen condensation step catalysed by a specific KS, FogA2. β-Branching is then catalysed in a normal fashion by the combined action of the HMGS (FogA5), ECH1 (FogA4) and ECH2 (FogA3) that generates methyl-3-pentenoyl-ACPFogA8. Methyl-3-pentenoyl-ACPFogA8 essentially comprises an unusual acyl starter unit for the remainder of the biosynthetic pathway, requiring a dedicated priming KASIII-like KS, analogous to DpsC, prior to ‘classical’ aromatic polyketide chain extension catalysed by the minimal PKS, FogA9 (CLF), FogA10 (KS) and FogA11 (ACP).90 Interestingly, FogA11 (ACP) may be shared with a second KS/CLF (FogN/FogO) in the same biosynthetic cluster that synthesises both of the major metabolites fogacin and fogacin B in a parallel pathway, both of which lack the trisubstituted exo-alkene and share common biosynthetic intermediates with the classic aromatic polyketide actinorhodin.
5.6 Epoxide (9) – spliceostatin
Spliceostatins are produced by hybrid PKS-NRPS pathways and display promising anticancer activity (Fig. 15).91,92 They act as inhibitors of the eukaryotic spliceosome and modulate splicing activity by binding to a subunit of the large ribonucleoprotein complex responsible for mRNA processing. The gene cluster from Burkholderia sp. FERM BP-3421 was identified and the biosynthesis of spliceostatin A determined.93 An exo-β-methylene is installed by a trans-acting HMGS cassette which lacks a free-standing ECH2 domain. At the point of β-branching a truncated and full-length ECH resides upstream of three ACPAs in the type I PKS Fr9GH (a single protein). The presence of a single ECH2 within the gene cluster suggests the exo-β-methylene branch is derived from decarboxylation and C-2 reprotonation.93–95 Through in vivo knock-out studies, the flavin-dependent monooxygenase (Ox) located at the C-terminus of Fr9GH, downstream of the acceptor ACPAs, was shown to epoxidise the exo-β-methylene to give spliceostatin A. Derivatives have been isolated from numerous bacterial species that contain differential functionality around the six-membered oxygen heterocycle, including spliceostatin B that lacks the epoxide group.96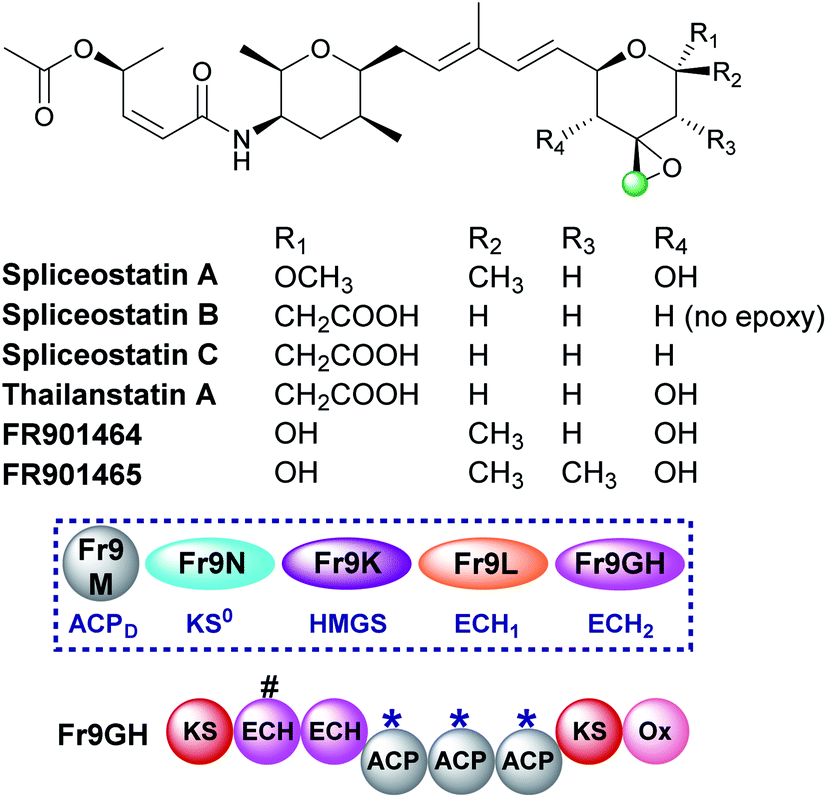 | ||
| Fig. 15 The structure of the spliceostatins which contain a tetrahydropyran ring with varying oxygenation patterns. The HMGS cassette and modular architecture (Fr9GH is a single ORF) for spliceostatin A isolated from Burkholderia sp. FERM BP-3421 is shown. A modular ECH2 domain in Fr9GH (with a truncated upstream modular ECH labelled #) forms an exo-β-methylene branch prior to epoxidation by a cis-acting flavin-dependant oxygenase (Ox). This functionality is found in all compounds with the exception of spliceostatin B which retains the exo-β-methylene. | ||
5.7 Methylacrylate (11) – bryostatin
Bryostatin 1 is a macrolactone isolated from a bacterial symbiont of the marine bryozoan Bugula neritina and is a potent protein kinase C agonist which has been investigated for the treatment of cancer, HIV/AIDS and Alzheimer's disease.97–99 In the biosynthesis of bryostatin 1, two methyl acrylate esters are introduced by a β-branching cassette that lacks an ECH2 domain (either trans-acting or modular) (Fig. 16A).100 Sherman and co-workers chemoenzymatically synthesised γ-d2-HMG-CoA and used HPLC/MS analysis to show that surprisingly the ECH1 (BryT) catalysed an α,β-dehydration despite the final product containing a β,γ-double bond and the absence of other enzymatic components that subsequently isomerase the product (Fig. 16B).101 The lack of an ECH2 domain leads to retention of the carboxylate group and subsequent esterification catalysed by BryA yielded the 3-methylglutaconyl methyl ester. BryA was shown to methylate MG-ACP but did not methylate HMG-ACP or acyl-CoA thioesters used as substrate mimics. The recognition of the α,β-dehydration intermediate by BryA suggested this regioisomer had not been produced aberrantly by the use of non-native substrates in the in vitro assays. This led to the hypothesis that isomerisation then produces the β/γ-unsaturated product prior to tetrahydropyran ring formation by an unknown mechanism. A possible candidate for the isomerisation is an ER-like domain located in module 4 of BryB, immediately downstream of the ACPAs, but this has not been determined experimentally.34,83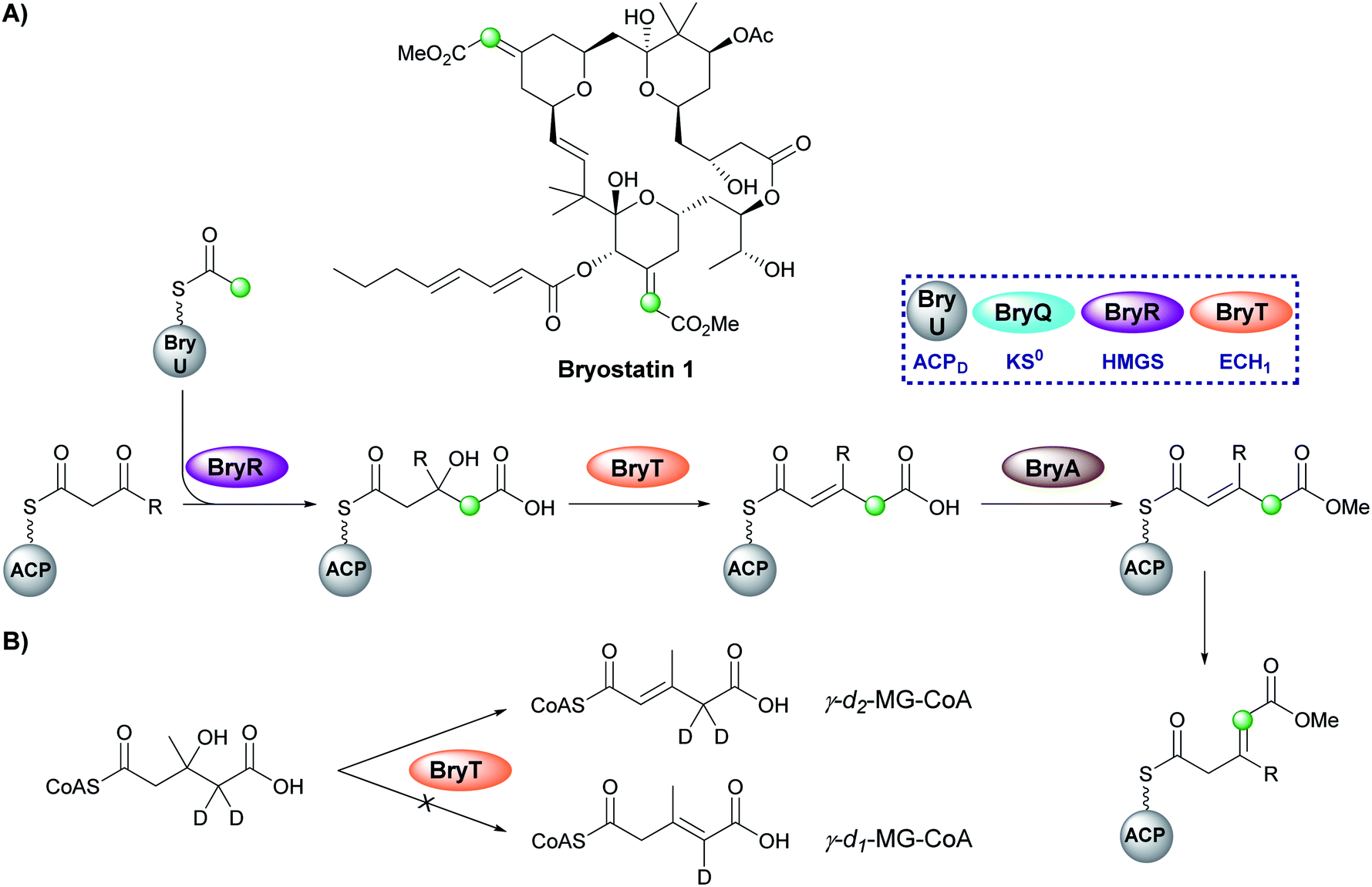 | ||
| Fig. 16 (A) Bryostatin 1 contains two methyl acrylate ester β-branches installed by an HMGS cassette lacking an ECH2. After dehydration by BryT, esterification (BryA) gives a methyl ester and subsequent double bond isomerism by an as yet unknown domain forms the acrylate branch. (B) Dehydration by BryT was investigated using deuterium labelling which showed retention of both deuterium atoms in the product (γ-d2-MG-ACP) in accord with α,β-unsaturation. | ||
5.8 1,3-Dioxo-1,2-dithiolane (12) – leinamycin
Leinamycin possesses a very unusual 1,3-dioxo-1,2-dithiolane moiety that is introduced via a modified HMGS cassette-mediated β-branching pathway.44,102 The pathway diverges from the canonical steps by the lack of KS0 in the gene cluster and the selection of methylmalonyl-CoA to provide the substrate for the HMGS (Fig. 17).103 LnmK, a domain of previously unassigned function and with no sequence homology to known proteins, was proposed to act as an acyltransferase/decarboxylase (AT/DC) to replace the absent KS0.104 The bifunctional domain was purified, crystallised and probed in functional studies to demonstrate transfer of a methylmalonyl group from CoA to ACPD (LnmL) prior to decarboxylation to produce propionyl-ACPD.105 In contrast to the AT/KS0 model for substrate loading, no active site serine or cysteine residues were identified in the active site, however, through site direct mutagenesis, Tyr62 was proposed to act as the nucleophile for acyl transfer of methylmalonate whereas residues for decarboxylation could not be clearly determined. Homologous domains have since been identified in the production of linear and β-branched starter units in the type II PKS that produce lomaiviticin and fogacin C respectively (see Section 5.5).31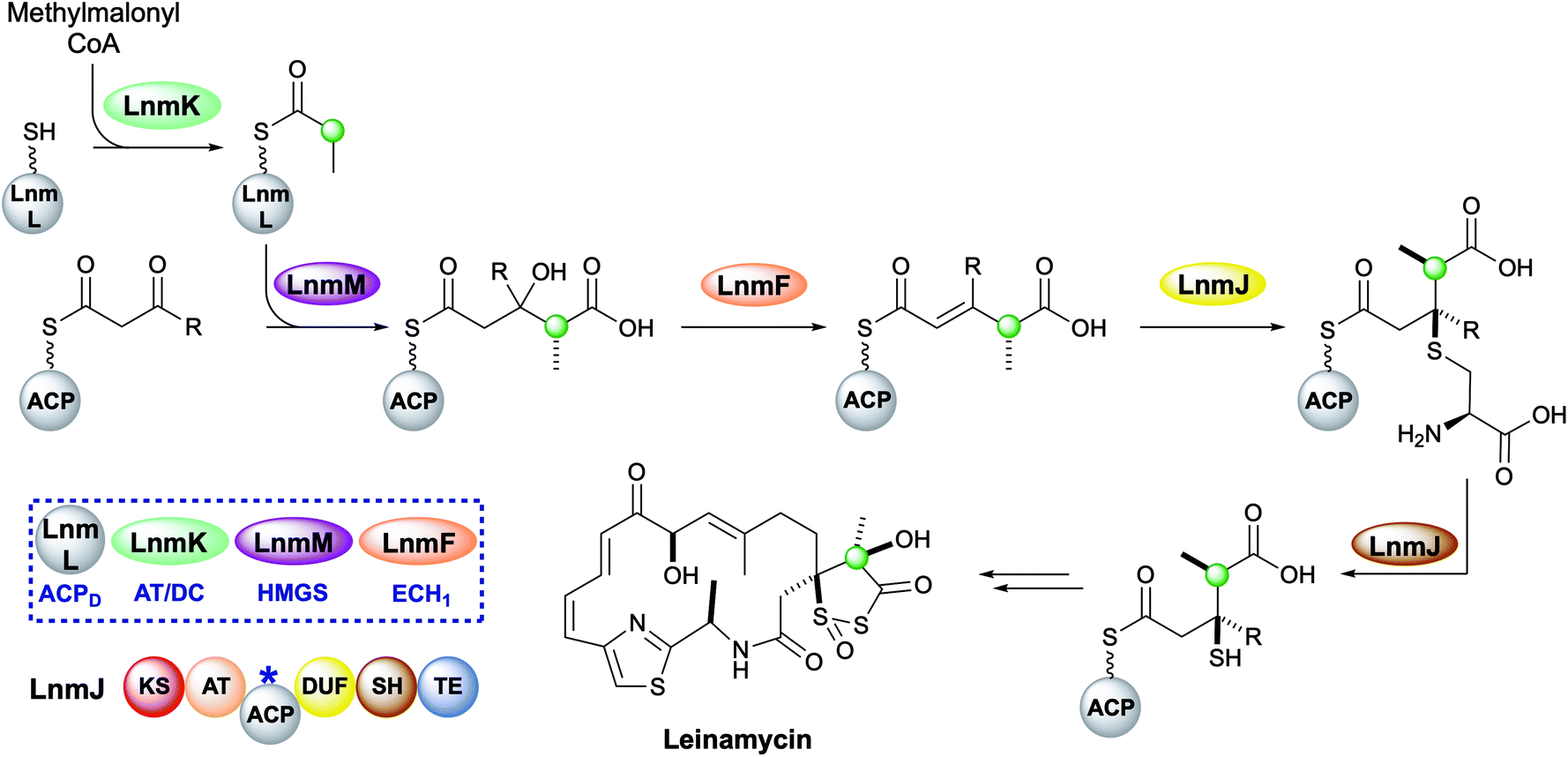 | ||
| Fig. 17 Leinamycin contains a 1,3-dioxo-1,2-dithiolane moiety introduced by a trans-acting HMGS cassette. | ||
The HMGS-catalysed conversion of propionyl-ACPD and β-ketothioester-ACPA produces a γ-methyl substituted HMG-ACPA that then undergoes LnmF-catalysed dehydration. The gene cluster lacks an ECH2 domain but instead alternate processing takes place on the newly-installed β-branch. Module 8 of the type I PKS contains a domain of unknown function (DUF) and a cysteine lyase (SH) that catalyse the Michael addition of a cysteine thiol to the α,β-unsaturated thioester and subsequent C–S bond cleavage generates a β-thiol moiety.106 Further processing to produce the 5-membered heterocycle and oxidation throughout the molecule takes place, however, the identity and sequence of the catalytic steps have not been proven.44,102,107–109
6. Polyketides containing multiple β-branches
In some cases, multiple β-branches may be incorporated into the natural product requiring the ACPs, HMGS cassette and tailoring enzymes to selectively install each β-branch with high fidelity. This may be achieved by (i) physically locating the correct catalytic domains in-cis where required, (ii) providing highly selective in-trans domains or (iii) deploying multiple cassettes with different selectivity's within the biosynthetic pathway.6.1 Etnangien (1) and gladiolin (1 and 3)
The related compounds etnangien and gladiolin share many structural features but differ in their β-methyl incorporation pattern. Etnangien contains two endo-β-methyl branches (C-4 and C-18) incorporated by a five-enzyme HMGS cassette that performs the same functionalisation of the β-ketothioester at each of the ACPAs in EtnD and EtnE respectively (Fig. 18A).110 In gladiolin biosynthesis, the gene cluster encodes an expanded trans-acting HMGS cassette that contains two sets of ECH1/ECH2 domains giving a total of 7 enzymes (Fig. 18B). This cassette incorporates an endo-β-methyl (C-25) and saturated β-methyl branch (C-35).111 An ER in module 1 of GbnD1 is co-localised at the point of saturated β-methyl incorporation to carry out the reduction, of presumably an endo-β-methyl group at C-35. This common functionality brought into question the requirement for two sets of ECH1/ECH2 domains, when, as for etnangien, the successive action of one set would suffice. It has been speculated that the modular ER domain may require additional molecular recognition features of the module 1 ACPA that therefore require the unified action of the ER with a specific pair of ECH1/ECH2 domains.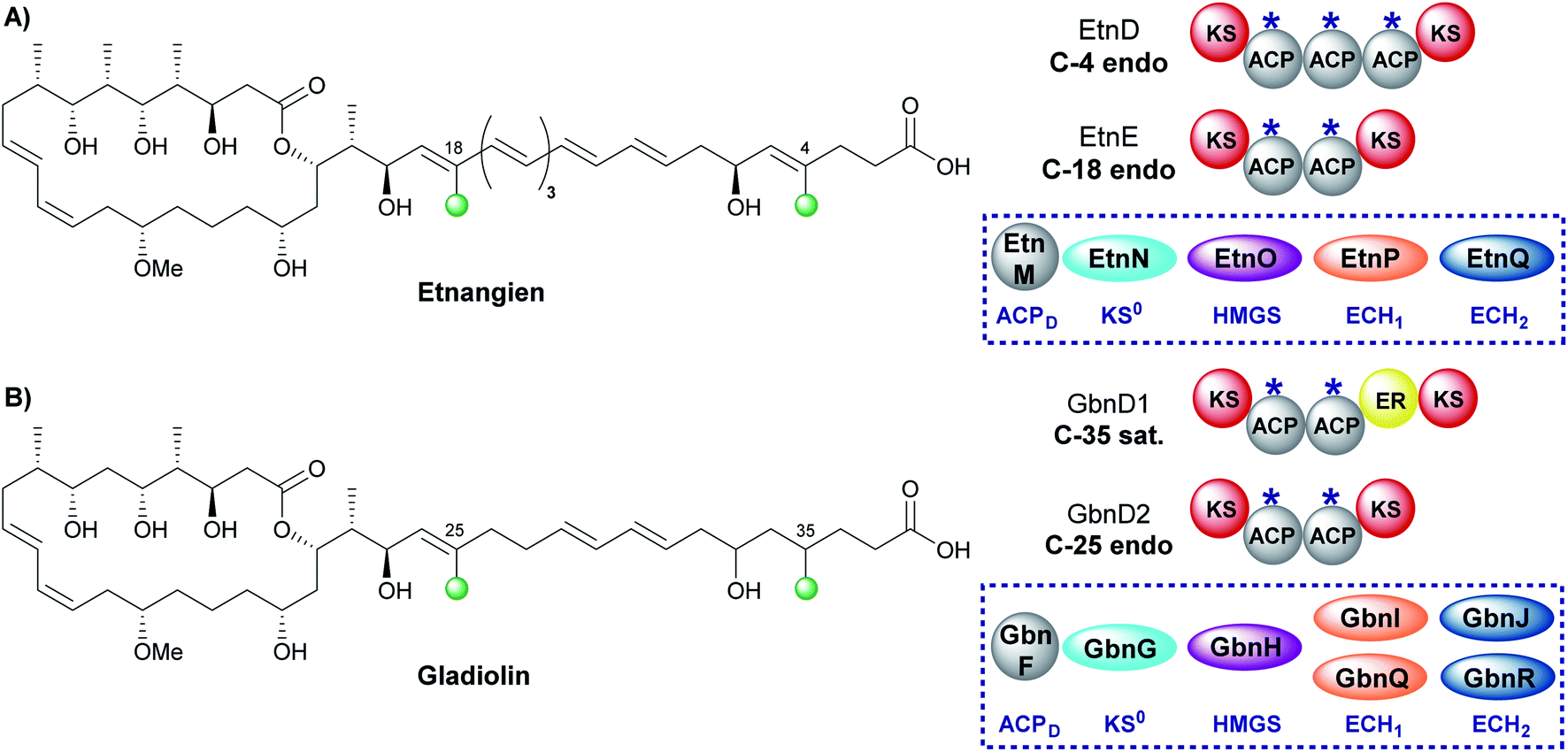 | ||
| Fig. 18 (A) Etnangien contains two endo-β-methyl branches (C-4 and C-18) that are installed by a single HMGS cassette. (B) Gladiolin contains an endo-β-methyl (C-25) and sat.-β-methyl branch (C-35) and is installed by an HMGS cassette that contains two pairs of ECH1/ECH2 domains and an in-cis ER located in module 1 of GbnD1 to reduce the α,β-unsaturated β-branch. | ||
6.2 Kalimantacin (1, 3 and 6)
The kalimantacins are potent antibiotics isolated from a number of Pseudomonas species and Alcaligenes species (Fig. 19).112–114 The kalimantacin gene cluster contains a single trans-acting HMGS cassette that installs a total of four β-branches with three different structures.114,115 This is one of the most complex β-branching patterns observed in polyketides and of particular interest is the consecutive installation of an exo-β-methylene, saturated β-methyl and endo-β-methyl via ACP4, 5 and 6 of the type I PKS Bat3 respectively. A previously unidentified modular ECH (mECH) domain located downstream of the acceptor ACP4 in Bat3 was proposed to install the exo-β-methylene. Due to the wide range of catalytic activities displayed by CS enzymes, it was postulated that the mECH might act as either a decarboxylase that selectively reprotonates to form an exo-β-methylene or an isomerase to convert a BatE-installed endo-β-methyl to an exo-β-methylene.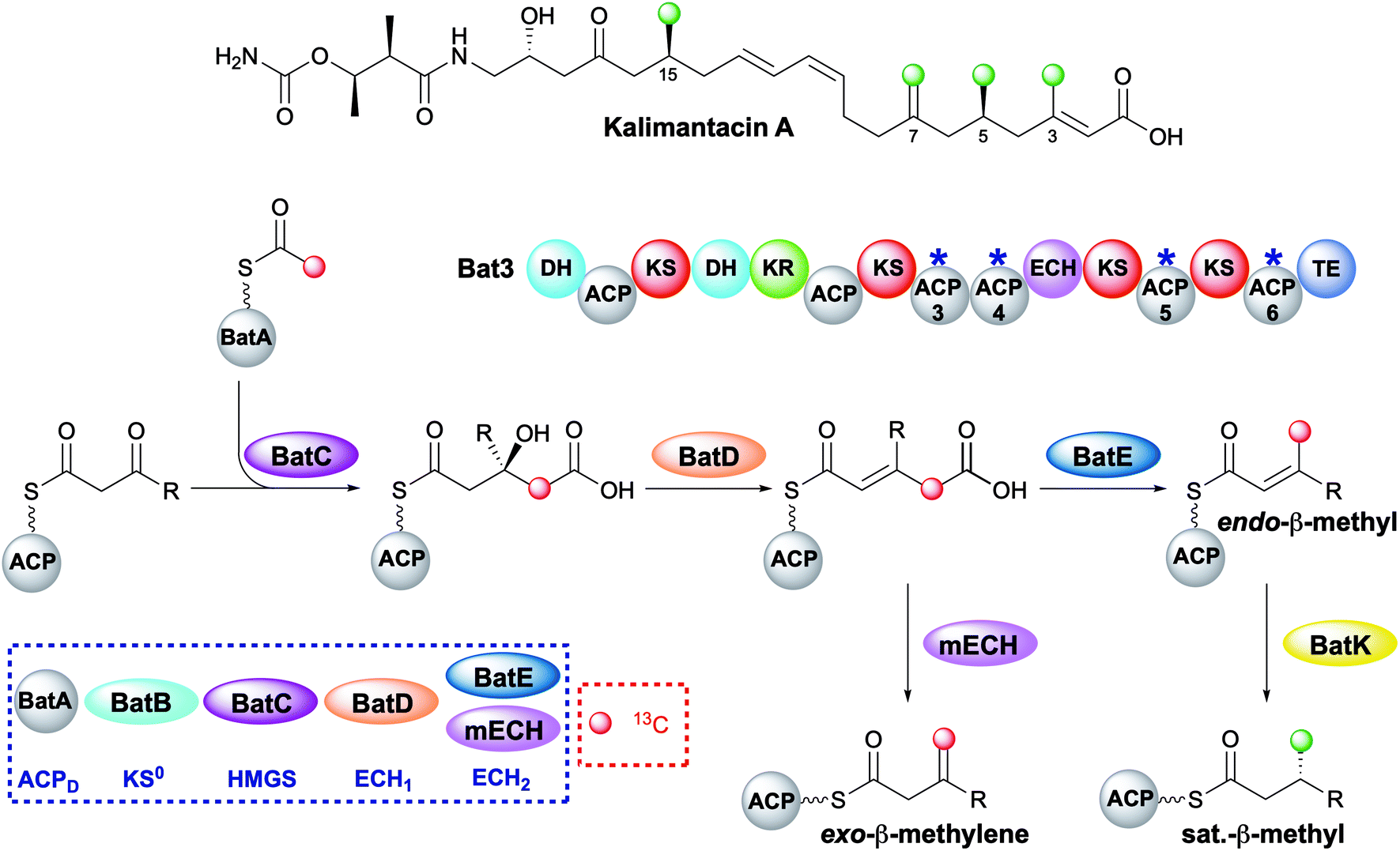 | ||
| Fig. 19 Kalimantacin A contains four β-branches with three different structures; two saturated β-methyl branches (C-5 and C-15), an endo-β-methyl branch (C-3) and an exo-β-methyl (C-7). The HMGS cassette contains a trans-acting ECH2 (BatE) and a modular ECH2 (mECH). Using [2-13C]-Ac-ACPD, the pathway was monitored by NMR and the red dots indicate the carbon atoms that were enhanced due to [13C] incorporation. | ||
Through protein expression and chemical synthesis of 13C labelled intermediates, pathways for the selective formation of each β-branch type were reconstituted in vitro.57 Incubating [2-13C]-acetyl-ACPD with Acac-ACP4 and HMGS (BatC) resulted in the formation of [γ-13C]-HMG-ACP4 as determined by monitoring of the 13C signal by 1D 13C-observe and 2D homonuclear NMR experiments as well as the Ppant ejection assay. Addition of ECH1 (BatD) and ECH2 (BatE) to this multi-protein in vitro assay resulted in loss of the [γ-13C]-HMG-ACP4 signal and the generation of a 13C methyl signal at 23.51 ppm and a Ppant ejection ion consistent with the formation of an [γ-13C]-endo-β-methyl moiety, an unnatural β-branch for this ACP. Preparation of an active mECH for in vitro assays required the expression and purification of an ACP4-mECH di-domain (4M) where the two domains retained the native interdomain linker region. Subsequent in vitro reconstitution with [13C]-Ac-ACPD, BatC and BatD, however, demonstrated the decarboxylase activity of the mECH which unequivocally yielded an [γ-13C]-exo-β-methyl in the absence of BatE. Further, in a competition reaction containing 4M and BatE, a mixture of both [γ-13C]-endo-β-methyl and [γ-13C]-exo-β-methylene branches were formed. Therefore, surprisingly, ACP4 does not appear to be specific for the mECH (cf. gladiolin where this tailoring of the ACPA is implied). This means that BatE cannot be excluded in the simplified in vitro model but in the native PKS steric occlusion of BatE or faster in-cis reaction of MG-ACP4 with mECH must occur to prevent aberrant β-branching. Finally, the trans-acting ER BatK was shown to be ACP selective and would only reduce the endo-β-methyl branch when attached to ACP5 but not ACP6. Interestingly, an endo-β-methyl branch could be reduced at ACP4 indicating the promiscuity of this ACP that normally produces an exo-β-methylene.
6.3 Phormidolide (1 and 6)
Phormidolide is a complex molecule isolated from a Leptolyngbya species and contains five β-methyl branches: two exo-β-methylenes and three endo-β-methyls (Fig. 20).116 The gene cluster encodes a type I trans-AT PKS and a five-enzyme trans-acting HMGS cassette which installs the endo-β-methyl branches. The pathway is initiated by a 1,3-disphosphoglycerate unit which primes PhmE utilising the FkbH domain, which is known to integrate glycolytic intermediates into PKS pathways, and is subsequently dehydrated by the DH domain and methylated by the O-MT to give glyceryl-ACP. Chain extension then takes place prior to exo-β-methylene incorporation. A pair of modular ECH domains are annotated within PhmE and PhmI at the point of exo-β-methylene incorporation. In both ORFs, the upstream ECH domain is truncated and assumed non-functional whilst the downstream of each pair of these modular ECH domains shows sequence similarity to characterised ECH2 (decarboxylase) domains but their biochemical role has not been shown experimentally. Following insights into the decarboxylase activity of the modular ECH domain in exo-β-methylene formation in kalimantacin biosynthesis, it is highly likely that selective re-protonation gives the exo-β-methylene moieties in phormidolide. An unusual feature of the modular PKS is the apparent separation of one pair of ACPAs (both contain the conserved tryptophan HMGS recognition motifs)51 across two ORFs at the C-terminal of PhmH and N-terminal of PhmI. Tandem ACPA domains are commonly encountered at the point of β-branching, however, how the split across separate multi-modular proteins affects the relationship and catalytic activity of these two domains remains to be experimentally determined. It should be noted that an ambiguity in labelling of ECH1 and ECH2 domains was identified and analysis of the sequences deposited in genbank suggest that PhmO is in fact the ECH1 domain and PhmN is the ECH2 domain. Using chemical synthesis, computation and NMR analysis, the stereochemistry of phormidolide has also been reassigned by Ndukwe et al.117–119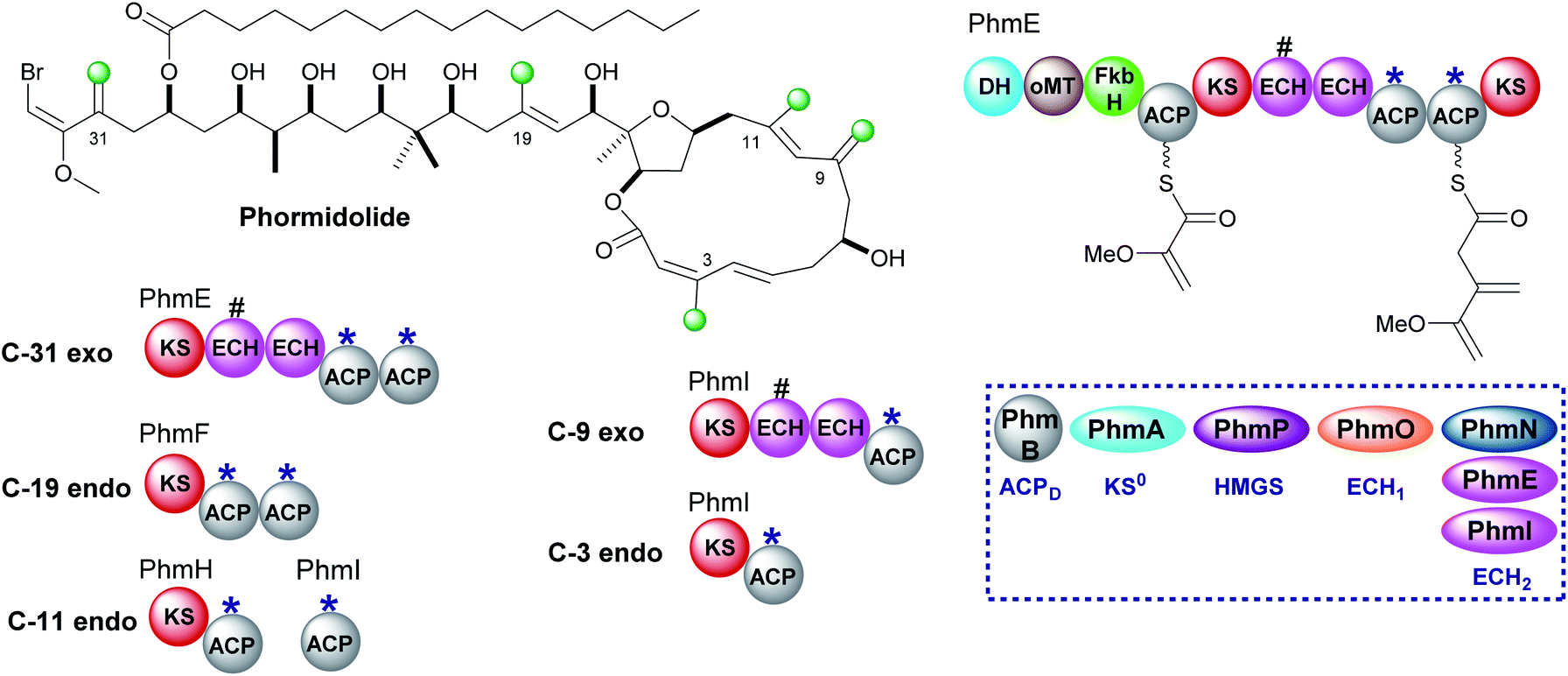 | ||
| Fig. 20 Phormidolide contains five β-branches, three endo-β-methyl branches (C-3, C-11 and C-19) and two exo-β-methylenes (C-9 and C-31). The HMGS cassette contains three ECH2 domains, one trans-acting (PhmN) and two modular cis-acting (PhmE and PhmI) located at the sites of exo-β-methylene formation. A truncated (#) ECH domain is located upstream of the ACPAs. The modular architecture of PhmE which loads a glycerate unit to initiate biosynthesis prior to exo-β-methylene incorporation is shown. | ||
6.4 Leptolyngbyalide (1, 3 and 6)
Through mining a collection of cyanobacteria, a 91 kb trans-AT PKS cluster that produces leptolyngbyalide A was identified.119 This compound is structurally related to phormidolide but contains a different β-branching pattern. There are again five β-branches in total: two endo-β-methyl, one exo-β-methylene and two saturated β-methyl branches (Fig. 21). At the point of exo-β-methylene incorporation (C-29) an in-cis ECH domain likely acts as a decarboxylase to give β,γ-unsaturation in preference to the trans-acting ECH2. An endo-β-methyl is introduced at C-17 and an in-cis ER domain gives the saturated β-methyl at C-9. The ER is part of a split module whereby the two ACPAs are again split across two polypeptide chains (LeptP and LeptQ).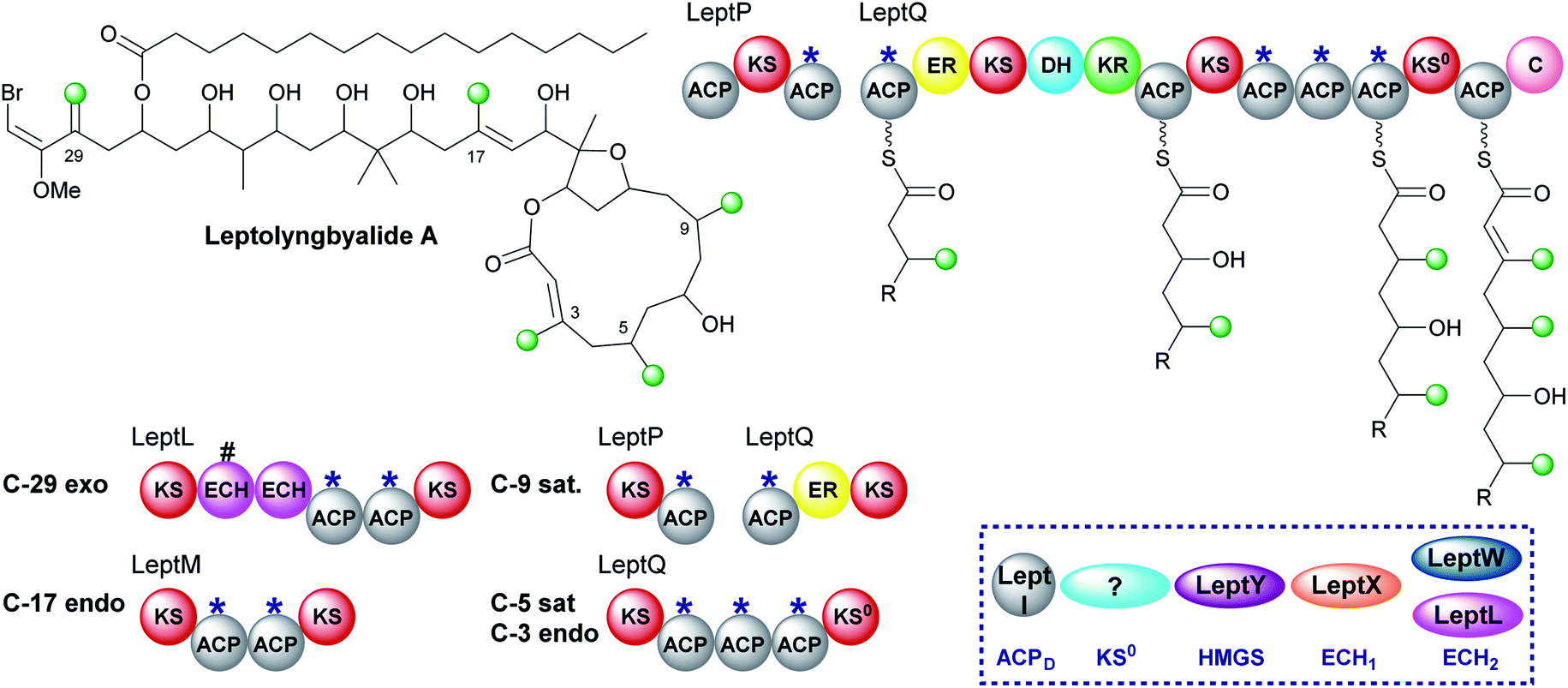 | ||
| Fig. 21 Leptolyngbyalide A contains five β-branches with three different branch structures; two saturated β-methyl branches (C-5 and C-9), two endo-β-methyl branches (C-3 and C-17) and one exo-β-methylene (C-29). The HMGS cassette contains a trans-acting ECH2 (LeptW) and an in-cis ECH2 (LeptL) adjacent to a truncated (#) ECH at the point of exo-β-methylene incorporation. The modular architecture and proposed biosynthetic intermediates of LeptQ suggests a saturated β-methyl and endo-β-methyl are incorporated by the triplet of ACPAs. | ||
The final steps of the pathway are uncertain as there is one less module in the LeptQ than expected for the creation of a 14-membered macrolactone ring. It is predicted that an iterative use of the KS and triplet of ACPAs is used to install the final two β-branches as the final ACP lacks the Trp-flag of ACPAs. However, this would require the selective installation of a saturated β-methyl at C-5 followed by an endo-β-methyl at C-3 possibly whilst tethered to the triplet ACPs with no obvious mechanism for the selective incorporation of each branch. Additionally, the PKS lacks an in-cis ER located within the module so the enzyme responsible for reduction is also uncertain. Deciphering the mechanism of consecutive β-branch installation in LeptQ would provide greater knowledge into the constraints required for correct β-branching.
6.5 Oocydin, haterumalides and biselides (1 and 6)
The haterumalides, and the structurally related biselides, are a series of chlorinated natural products that have shown anti-tumour activity against breast, lung and colon cancers (Fig. 22A).120–127 They have been isolated from a range of organisms, including protobacteria, sponges/porifera and tunicates. All compounds share a macrolactone core but vary in their patterns of β-branching, oxygenation and esterification partners. The gene cluster for oocydin A (also known as haterumalide NA) from the genome of the rhizosphere bacterium Serratia plymuthica A153 was identified and a biosynthetic pathway proposed.127 The gene cluster contains a 5 enzyme trans-acting HMGS cassette as well as two modular ECH domains in OocL, the first of which is truncated and the second a full-length ECH2 domain. The originally proposed biosynthesis suggests the DH-oMT-FbkH tri-domain normally used to load glycerate units is inactive and instead a malonyl starter unit is utilised (Fig. 22B). Incorporation of an endo-β-methyl takes place within module 1 of the PKS and, due to the lack of ketone in the final product, it is assumed that module 3 is skipped. Mutation of the trans-acting ECH2 (OocC) resulted in lack of product formation and the authors therefore suggested that the trans-acting HMGS is responsible for endo-β-methyl branching rather than the modular ECH domains.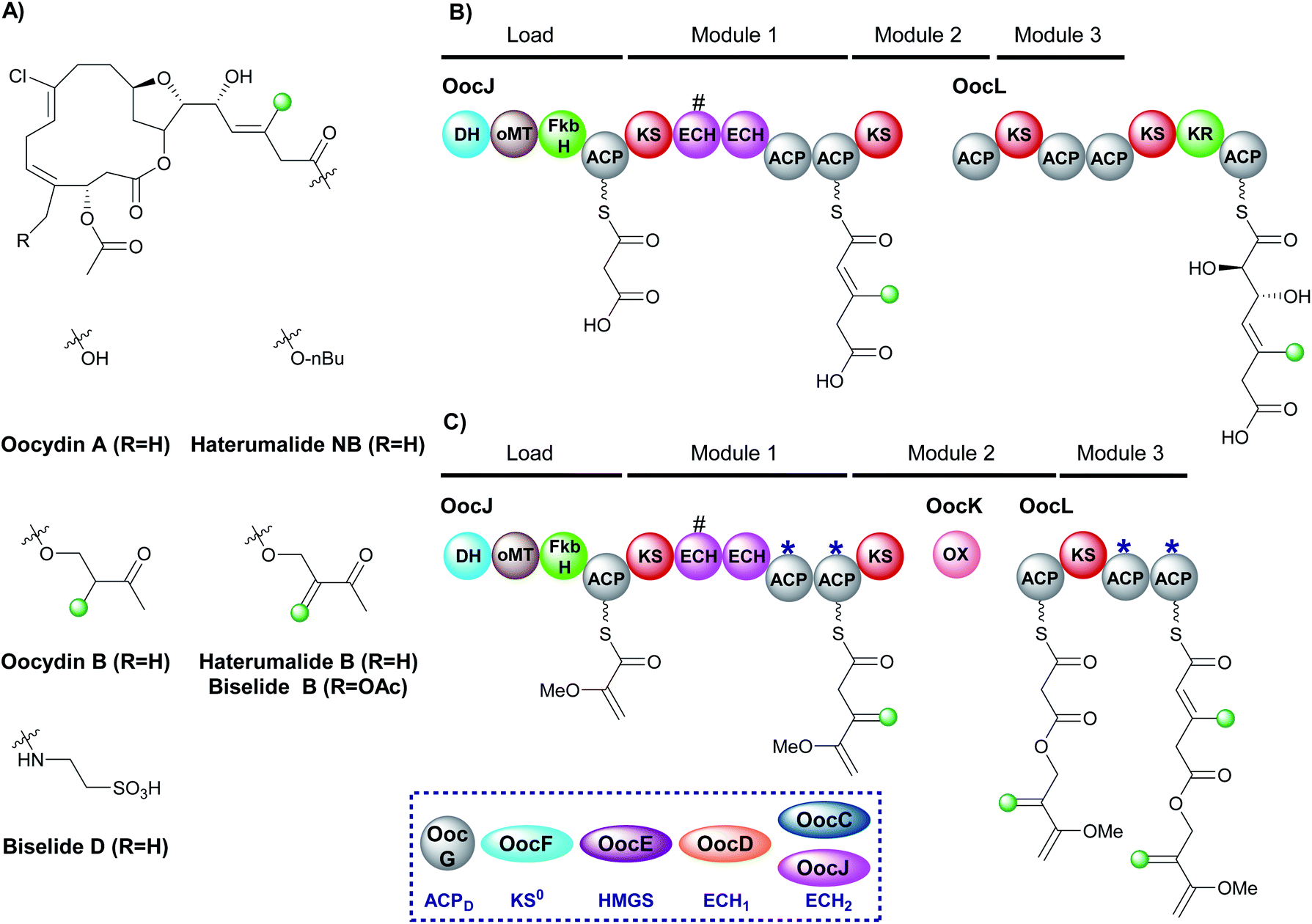 | ||
| Fig. 22 β-Branching in the haterumalide and biselide natural products. (A) Structures of the natural products. (B) The original proposal for the biosynthesis of oocydin A (haterumalide NA). (C) Updated proposal based on the oxygen insertion of OocK, a Baeyer–Villiger monooxygenase, suggests an exo-β-methylene is introduced in module 1 of OocJ and an endo-β-methyl in module 3. The HMGS cassette utilises a modular ECH2 domain with a truncated (#) upstream ECH at the point of exo-β-methylene incorporation. | ||
During the investigation of OocK, a flavin-dependent monooxygenase also defined as a Baeyer–Villiger monooxygenase (BVMO), Meoded et al. revealed the domain inserts oxygen atoms into β-ketoacyl functionality to yield esters.128 The authors re-examined the early stages of biosynthesis on the type I PKS OocJ and noted the identical set of domains are found at the start of PhmE in the biosynthesis of phormidolide (Fig. 22C). Whilst the structural features predicted to be incorporated are lacking in oocydin A, examination of other members of the haterumalide and biselide families showed they possess similar chemical structure to phormidolide. By analogy, it was therefore proposed that a glycerate unit may prime the PKS followed by exo-β-methylene incorporation facilitated by the modular ECH2 domain to introduce the β,γ-unsaturation. Chain extension and oxygen insertion by OocK yields the ester moiety which then undergoes endo-β-methyl incorporation at module 3 (previously thought to be silent) using the trans-acting ECH2 to produce the α,β-unsaturation. The functional groups introduced in this way are not always transferred through to the final product architecture, however, it is suggested that additional enzymatic transformations may take place in different producing organisms to give rise to the diversity of β-branched compounds in the haterumalide and biselide family.
The proposed β-branching mechanistic steps show homology to phormidolide and kalimantacin that introduce both an endo-β-methyl and an exo-β-methylene into the same compound (Sections 6.2 and 6.3). Selectivity is achieved by in-cis modular ECH2 domain activity in preference to the trans-acting ECH2 to produce the exo-β-methylene, whilst the endo-β-methyl arises from trans-acting ECH2 reactivity. Further credence is added to the newly proposed biosynthesis as the two pairs of ACPs in OocJ (exo-β-methylene) and OocL (endo-β-methyl) both contain the tryptophan-flag indicating they are ACPAs and so both the β-branching events are feasible in these pathways.
6.6 Ripostatin (1 and 10)
Ripostatin, an antibiotic with a novel binding mode to bacterial RNA polymerase, is produced by Sorangium cellulosum So ce377 by a mixed cis/trans-AT PKS. In the biosynthetic pathway, selective incorporation of two different β-branches into the final polyketide structure must be achieved by a single HMGS cassette (Fig. 23).129,130 The endo-β-methyl forms via the canonical pathway whilst tethered to the di-domain of ACPAs in RipD. However, the carboxymethyl β-branch lacks the final ECH2-catalysed decarboxylation and occurs while tethered to the single ACPA in RipE, immediately prior to off-loading by the terminal thioesterase. As a result, a degree of control is required during assembly to prevent decarboxylation either by excluding the ECH2 domain or a faster in-cis reaction. The isotopic labelling pattern for the polyketide terminus shows the incorporation of an intact acetate into both the linear chain and carboxylmethyl β-branch. As a result, this information alone does not define the type of β-branch incorporated into the final structure. Considering the stereochemistry of previously characterised dehydration reactions, the ECH1-catalysed reaction likely gives the α,β-alkene. Macrolactonisation with the acrylate moiety gives rise to ripostatin A with a carboxymethyl β-branch but must occur in a manner to prevent decarboxylation by the ECH2 domain.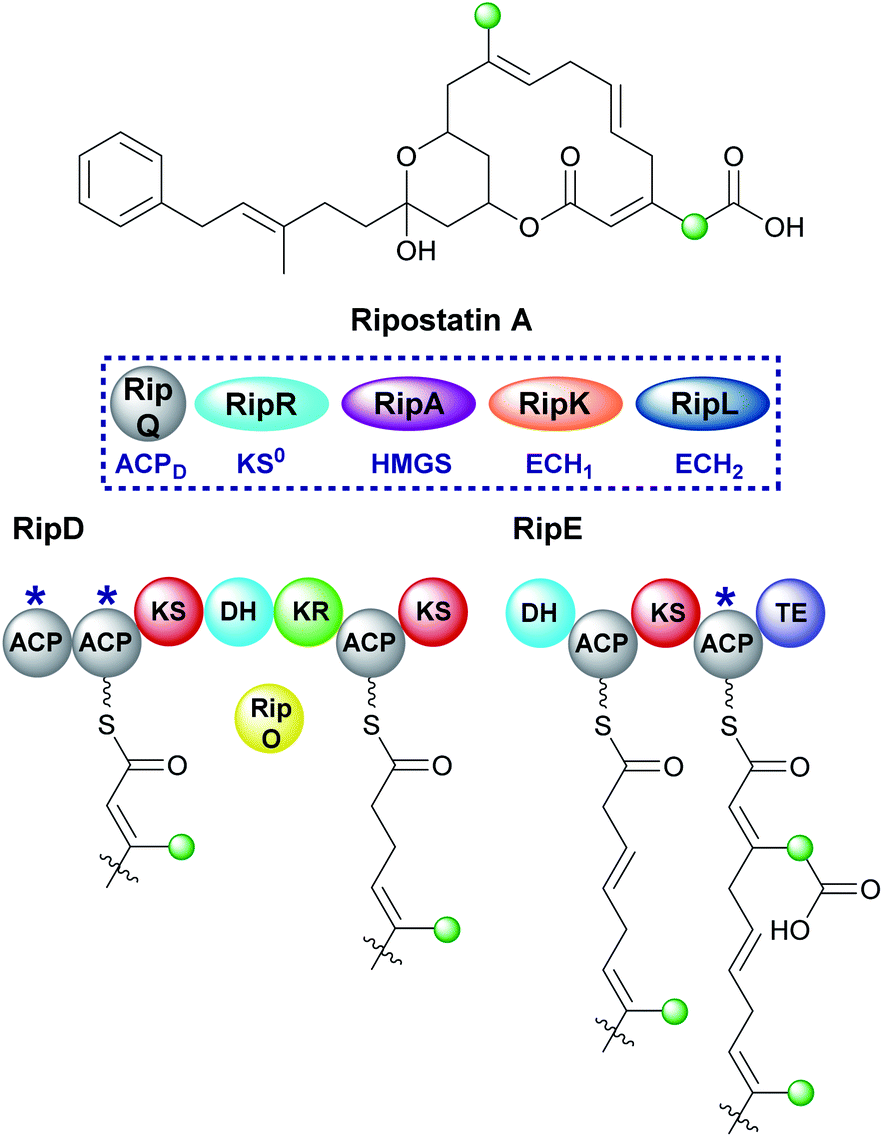 | ||
| Fig. 23 Ripostatin contains an endo-β-methyl branch and a carboxymethyl-β-branch installed by a trans-acting HMGS. An endo-β-methyl is installed in RipD and the carboxylated β-branch in the final step before release from the PKS by the terminal TE domain. | ||
6.7 Bongkrekic acid (1 and 10)
Bongkrekic acid, produced by Burkholderia gladioli, is an acute respiratory toxin targeting the mitochondrial ADP/ATP transporter required for oxidative phosphorylation. Its biosynthesis is an interesting example of the divergence of a biosynthetic pathway from a common intermediate to introduce two different β-branches within one biosynthetic assembly line: an endo-β-methyl and a carboxymethyl β-branch (Fig. 24). Isotopic feeding studies with the bongkrekic acid producer B. gladioli strain revealed a cleaved acetate as part of the endo-β-methyl branch incorporated at an early stage via the single ACPA in BonA. It also demonstrated intact acetate incorporation into the late-stage, carboxymethyl β-branch at the β-branching di-domain ACPs in BonD. A five-enzyme HMGS cassette was found in the gene cluster with no obvious mechanism for the selective installation of either β-branch.131,132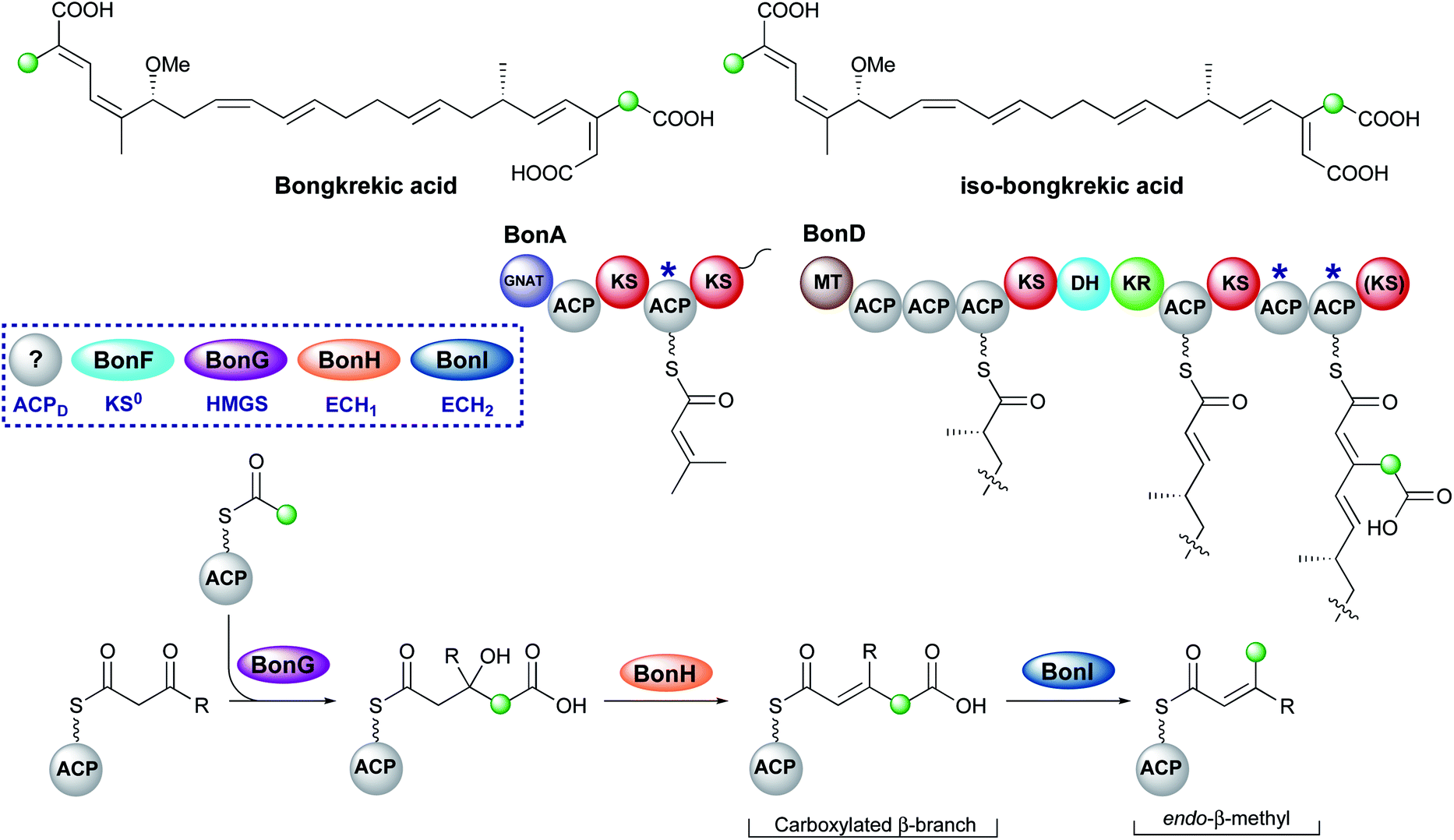 | ||
| Fig. 24 Bongkrekic acid and iso-bongkrekic acid contain two β-branches; an endo-β-branch and a carboxymethyl branch. The installation of an endo-β-methyl occurs on BonA whilst the carboxylated β-branch is formed in the final step catalysed by the type I PKS (BonD) which lacks an off-loading domain. | ||
The original publication annotated a β,γ-unsaturated β-branch arising from ECH1-catalysed dehydration.131 The β,γ-alkene is not a substrate for ECH2 domains and therefore this alkene regiochemistry might hint at a mechanism for selective β-branch incorporation. However, parallels have been drawn with the biosynthesis of bryostatin in which a β,γ-unsaturated acrylate moiety is introduced but the ECH1 domain (BryT) in bryostatin was shown to selectively produce an α,β-alkene rather than β,γ-alkene as found in the product (Section 5.7).
The acetate labelling pattern of bongkrekic acid is also consistent with the formation of α,β-unsaturation from the dehydration reaction that simply does not undergo decarboxylation and results in a carboxymethyl β-branch, analogous to ripostatin biosynthesis (Section 6.6). Isotopic feeding studies would not be able to differentiate between these two outcomes due to the incorporation of an intact acetate moiety into each position. However, the ubiquitous α,β-unsaturation arising from ECH1-catalysed dehydration either observed or reasonably assumed for all other pathways suggests this is a more likely result. No isomerase has been reported in the gene cluster to interconvert the double bond isomers, which suggests the outcome of dehydration is maintained in the final product unlike the biosynthesis of bryostatin.
Studies on the ECH1-catalysed dehydration in kalimantacin biosynthesis revealed the thioester is orientated cis to the growing polyketide chain in the MG-ACP intermediate arising from dehydration whilst the α-proton and β-branched group are cis-orientated (Section 6.2). The alkene geometry in bongkrekic acid is consistent with this stereochemical outcome further suggesting an α,β-dehydration occurs. Migration of the double bond between the two carboxylic acids could therefore result in the formation of iso-bongkrekic acid which contains the opposite double bond isomer.
The lack of off-loading domain from the modular PKS led to a currently unproven and unique hypothesis of concurrent β-branching and off-loading. If α,β-dehydration is the correct ECH1 product, then either a steric or kinetic mechanism may exist to occlude the ECH2 domain from accessing MG-ACP or to hydrolyse the final product from the PKS prior to aberrant catalysis by ECH2. This would be similar to the selectivity seen in the kalimantacin β-branching pathway that demonstrated the preferential in-cis production of an exo-β-methylene branch rather than an in-trans endo-β-methyl branch.
6.8 Myxovirescin (2 and 4)
One of the most complex β-branching cassettes is found in the gene cluster of myxovirescin (Fig. 25).133,134 The natural product contains two different β-branches: an endo-β-methoxymethyl at C-12 arising from hydroxylation (TaH) and O-methylation (TaQ) of an endo-β-methyl branch, and an unusual saturated β-ethyl branch at C-16 formed from the reduction (TaO) of an endo-β-ethyl branch.134 There are two pairs of ACPD/HMGS domains in the gene cluster, however, only single copies of KS0, ECH1 and ECH2. In vivo feeding studies with labelled precursors established the origin of the C-12 branch to be acetate and the C-16 branch to be methylmalonyl/propionate.135 An AT (TaV) and KS0 (TaK) are responsible for the formation of Ac-ACPD from malonyl-CoA, whilst it is predicted an AT/DC domain (TaD) loads the propionyl-ACPD from methylmalonyl-CoA in a similar way to leinamycin (Section 5.8).104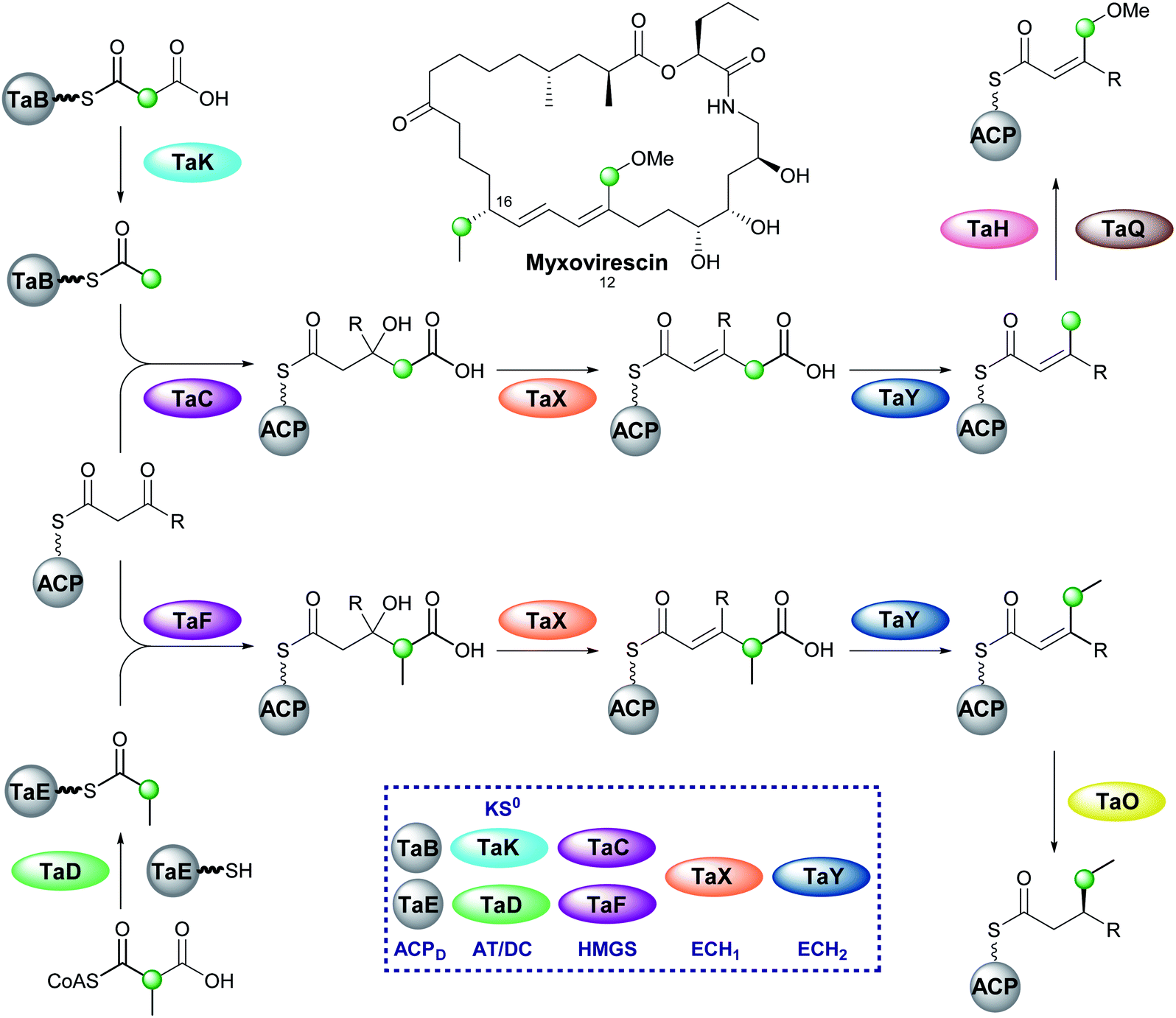 | ||
| Fig. 25 Incorporation of endo-β-methoxymethyl and sat-β-ethyl branches in the biosynthesis of myxovirescin. The gene cluster encodes a KS0 (TaK) and AT/DC (TaD) to form acetyl-ACPD and propionyl-ACPD respectively. Two ACP/HMGS pairs then select and react either acetyl-ACPD (TaB/TaC) or propionyl-ACPD (TaE/TaF) with the modular β-ketothioester. Dehydration and decarboxylation are carried out by the same ECH1/ECH2 pair (TaX/TaY) prior to oxidation (TaH) and O-methylation (TaQ) to produce the endo-β-methoxymethyl at C-12 or reduction (TaO) to produce the sat.-β-ethyl group at C-16. | ||
In vitro reconstitution of the HMGS cassette demonstrated two translationally coupled ACPD/HMGS pairs whereby TaB/TaC selectively introduce the C-12 ethyl branch and TaE/TaF the C-16 branch,136 which was supported by in vivo gene deletions.137 Furthermore, a ΔTaF mutant produced a compound with endo-β-methyl branches at C-12 and C-16, proving TaF could complement for TaC.133 However, the opposite complementation was not observed, and these results were further supported in vitro whereby TaC may accept substrates bound to either of the ACPAs, whilst TaF only functioned with its cognate ACPA. Overall, two distinct sets of enzymatic machinery are utilised in the formation of different β-branches, with the shared steps occurring only once a selective HMGS-catalysed condensation has taken place.
6.9 Guangnanmycin and weishanmycin (6 and 10)
Mining of bacterial genomes using the DUF-SH di-domain from the leinamycin gene cluster led to the identification of new clusters than encode structurally-related compounds.72 Guangnanmycin and weishanmycin are two of the compounds identified through this screen and both possess unusual β-branching cassettes and mechanisms of incorporation (Fig. 26). Both compounds contain an exo-β-methylene and a carboxymethyl β-branch, two HMGS homologues, a trans-acting ECH1 domain but no trans-acting ECH2. The only ECH2 present is a cis-acting ECH2 domain located at the point of exo-β-methylene incorporation. By locating the ECH2 domain within the type I PKS high fidelity of the pathway is achieved by preventing aberrant decarboxylation at the point of carboxymethyl β-branch installation. This is different to the selective installation of carboxymethyl β-branches in ripostatin and bongkrekic acid which contain trans-acting ECH2 domains and therefore differential installation of carboxylated and decarboxylated β-branches (Sections 6.6 and 6.7). Interestingly, the clusters both contain two HMGS domains which accept Ac-ACPD as the starter unit for β-branching. GnmU and WsmS clade with HMGS domains that use propionyl-ACPD as the β-branching group, however, neither utilise this alternate substrate. Considering the rest of the HMGS cassette is shared the reason for these two rather unusual homologues in the pathway is not completely clear.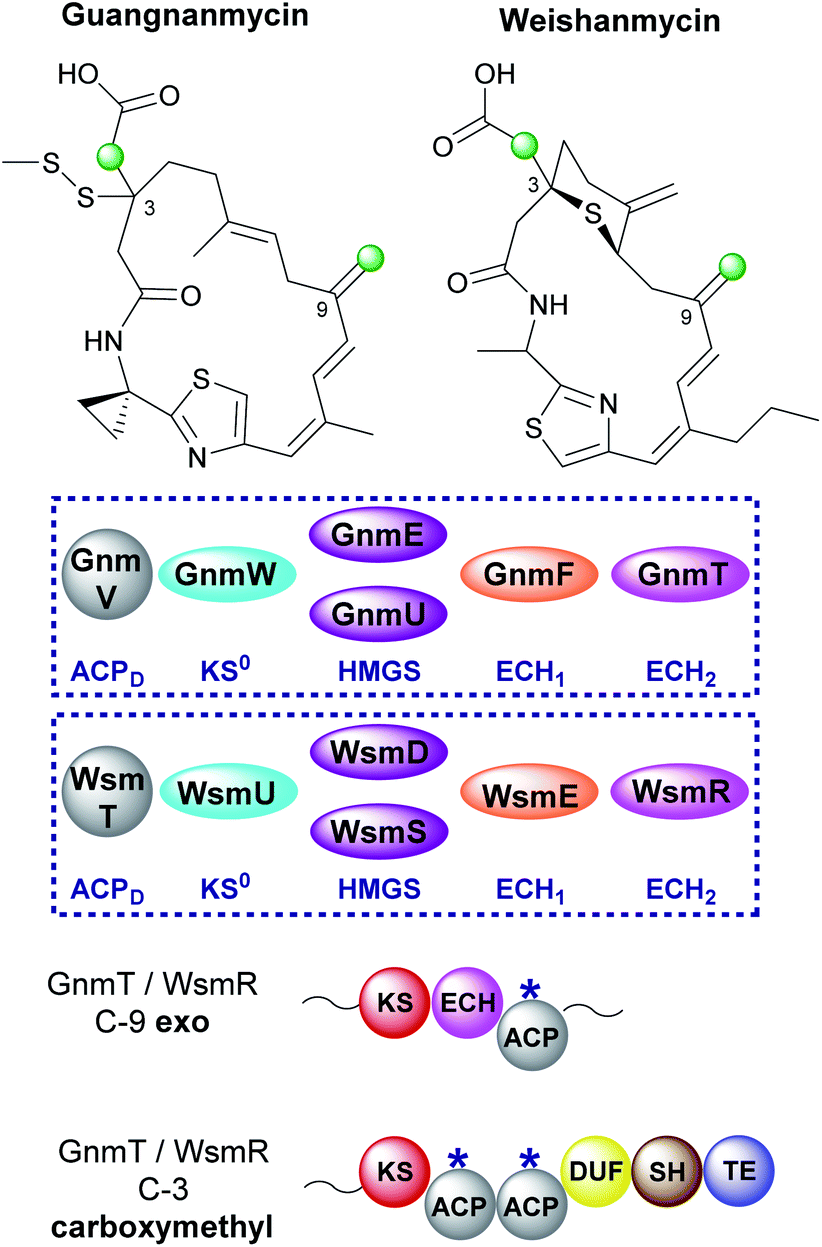 | ||
| Fig. 26 Guangnanmycin and weishanmycin both contain an exo-β-methylene and a carboxylated β-branch. The HMGS cassette only contains an in-cis ECH2 domain at the point of exo-β-methylene installation which prevents aberrant decarboxylation at C-3. There are two HMGS domains in the gene cluster but it is currently unknown whether they are both active. | ||
Further analysis of the DUF and SH domains from 29 leinamycin-family pathways (including guangnanmycin and weishanmycin) revealed these domains cluster according to the β-branching pattern at C-3. One clade contains domains that react at the site of β-branches derived from propionyl-ACPD whilst the other reacts at β-branches that derive from Ac-ACPD. The DUF-SH di-domains within GnmT and WsmR clade with those that utilise Ac-ACPD which highlights the unusual requirement for HMGS homologues.
6.10 Largimycin (6 and 12)
The largimycins were discovered from genome mining of Streptomyces strains where several BGCs were discovered but were poorly expressed.138 One such gene cluster, showing similarity to the leinamycin cluster was activated and a range of natural products termed the largimycins were isolated (Fig. 27). This class was differentiated from the leinamycins by the presence of an oxazole heterocycle instead of a thiazole moiety, cyclisation through an oxime ester to form a 19-membered macrolactone ring, the presence of an epoxide and one or more S-conjugated CysNAc sidechains in addition to a number of derivatisations of C-17 that together form the family largimycin A1–A4. Similar to the leinamycin biosynthetic pathway, the C-3 propionyl β-branch and subsequent derivatives were again proposed to be formed from methylmalonyl-CoA. The β-branching cassette is composed of a specialised propionyl-accepting ACPD (LrgL), HMGS (LrgM1) and a dual function AT/DC (LrgK) that is homolgous to LnmK and replaces the separate AT and KS0 domains. Interestingly, a second β-branching cassette (LrgA, LrgD, LrgM2) included an in-cis ECH2 (LrgJ) which suggests that an exo-β-methylene group should be present at C-9 as observed in the guangnanmycin/weishanmycins. However, largimycin A1–A4 all contain a C-9 ketone suggesting the β-branching cassette was skipped or inactive. Inactivation of LrgO, a putative NAD(P)/FAD-dependent oxidoreductase thought to be responsible for oxime formation, resulted in the production of largimycin O1, a compound containing a C-9 exo-β-methylene branch. It was suggested that the exo-β-methylene is the natural precursor before processing enzymes oxidatively cleave the alkene to form the ketone.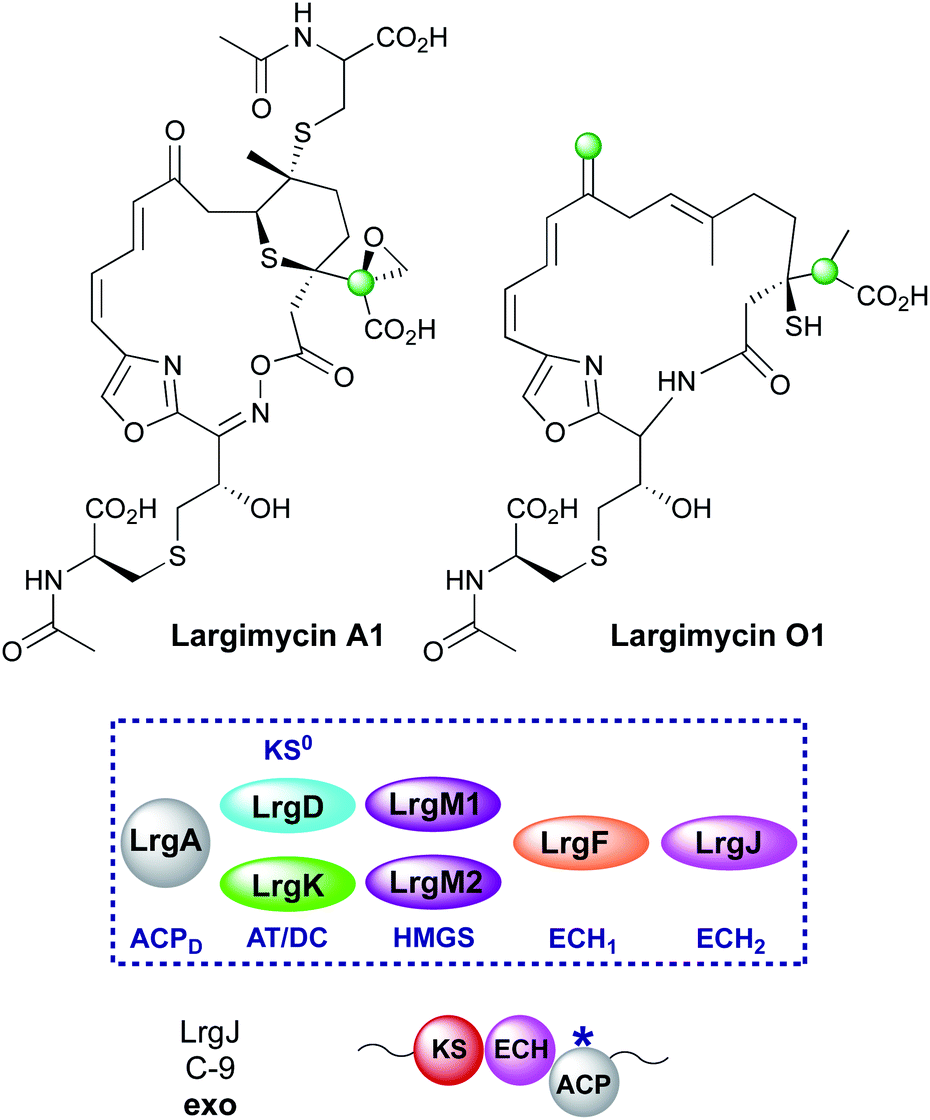 | ||
| Fig. 27 The structures of largimycin A1 and O1. The gene cluster encodes an acetyl-accepting ACPD, KS0 and HMGS for the introduction of an exo-β-methylene and a propionyl-accepting ACPD, AT/DC and HMGS to install the 1-carboxyethyl β-branch. | ||
7. Bioinformatics
The diverse and often unique chemistry encoded within components of trans-AT assemblies has been exploited in the genome mining and bioinformatic analysis of Nature's vast array of biosynthetic pathways and natural products. To date several different approaches have been adopted but these fundamentally rest on the same principle; the probes are anticipated to target trans-AT pathways. The choice of probe might uncover a wide range of trans-AT pathways or alternatively select for a highly specific chemistry that allows the expansion, for example, of known homologs around a scaffold of potential therapeutic value. Both approaches utilise or reveal patterns in the fundamental β-branching machinery and chemistry of the natural products.7.1 HMGSs as probes for trans-AT pathways
These key features have been demonstrated by Shen and co-workers by combining knowledge of published pathways, gene cluster analysis and bioinformatics for superior predictions of new or orphan clusters. The clading of HMGSs and their associated donor ACPs was initially uncovered during the identification of leinamycin-type biosynthetic pathways using a highly specific probe.72 In leinamycin biosynthesis the DUF-SH domain incorporates sulfur into the polyketide backbone and this unusual sequence was used to search for biosynthetic pathways incorporating a similar domain that may encode for leinamycin analogues with potentially potent anti-cancer properties. This discovery based genome mining chemistry approach led to the annotation of 28 lnm type gene clusters and the discovery of the guangnanmycins and weishanmycins within this new family of leinamycin natural products. Variations in β-branching were a prominent feature of these homologs and phylogenetic analysis from the 28 biosynthetic pathways showed that the HMGSs responsible for the β-branching at C-3 clustered into two subgroups depending on their use of either acetyl-ACPD or propionyl-ACPD as substrates. HMGSs responsible for β-branching at C-9 where acetyl-ACPD was used exclusively also claded as a separate group. This highlighted the power of the HMGS sequences in bioinformatic analysis of these biosynthetic pathways.More generally, the presence of an HMGS cassette in a gene cluster provides evidence that a form of β-branching is incorporated into the biosynthetic product. Whilst a diverse range of β-branches may be introduced by changing the effective enzymatic composition of these cassettes, the HMGS remains common to them all. Applying HMGS DNA sequences as experimental probes also benefits from strongly conserved regions of encoded primary sequence and relatively low numbers of occurrence per genome compared to more abundant PKS domains (KS, KR, DH). Huang and co-workers adopted this approach to apply both in silico genome mining of bacterial strains as well as experimental surveying of soil samples using HMGS DNA probes.139 From the in silico analysis, a total of 251 HMGSs derived from β-branching pathways could be grouped as a distinct cluster. Associated acetyl and propionyl ACPDs also formed separate clades and the study has now provided a far more general confirmation that a substrate specific HMGS clades with the appropriate ACPD. Donor ACPs also claded separately to non-β-branching trans-AT PKS ACPs, suggesting their co-evolutionary relationship with trans-AT PKS assembly lines.
7.2 KS domains
Initial bioinformatic results from Nguyen et al. showed phylogenetic analysis from trans-AT systems whereby KS domains did not clade by individual pathways but by the accepted substrate that is to be processed downstream.140 Research by Jenner et al. showed that alongside this analysis KSs exert a level of selective pressure over which substrates are accepted or rejected to ensure fidelity within polyketide biosynthesis.141 A recent paper by Helfrich et al. not only broadened the specificity of KS clades to a myriad of specific condensation products but also showed the predictive powers available to the modern chemist.119 Bioinformatics has allowed for the development of predictive tools and models to mine organisms for previously unstudied biosynthetic gene clusters. Furthermore, Piel and co-workers have created a tool that allows for the prediction of chemical structure from genomic information for trans-AT PKSs.1198. Other forms of branching at the β-position
8.1 Michael addition
β-Alkylation of polyketides may also be achieved through Michael addition to an α,β-unsaturated thioester, a mechanistically different pathway from that used by HMGS cassettes.142 In the biosynthesis of rhizoxin, a two-carbon unit is introduced at the β-position and then cyclisation occurs to generate a δ-lactone (Fig. 28).142 An unprecedented modular architecture (KS-B-ACP) was observed with a newly defined branching domain (B). In vitro reconstitution of the module, X-ray crystallography, protein cross-linking and stable isotope studies were used to elucidate the mechanism of branching. An ACP-bound malonyl group is decarboxylated and attacks the β-position of a KS-bound α,β-unsaturated thioester (Fig. 28A). The 5-hydroxy group subsequently attacks the KS-thioester to liberate the δ-lactone product in which the malonyl-derived thioester becomes the point for chain extension.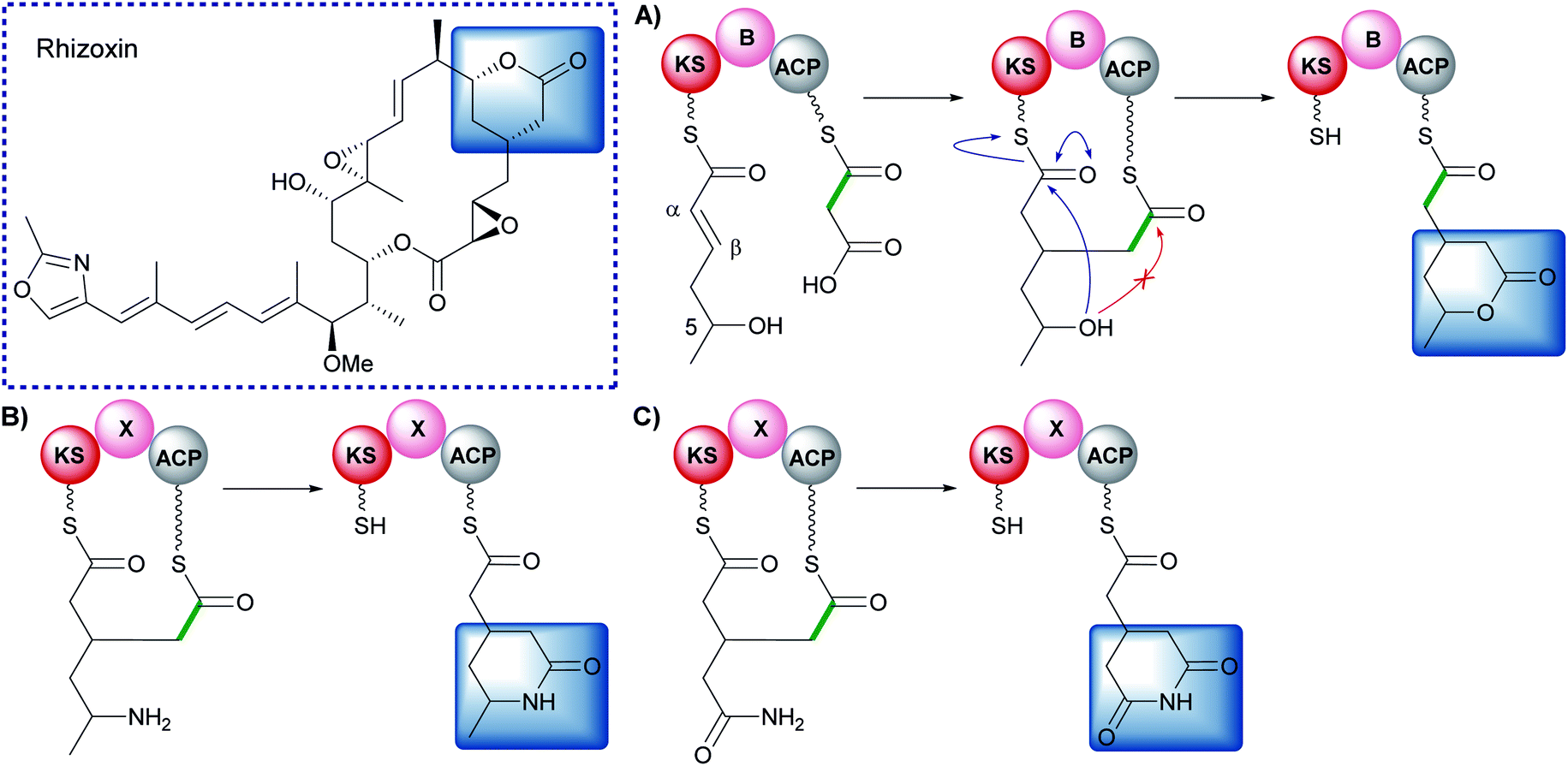 | ||
| Fig. 28 Bacterial polyketide rhizoxin contains an unusual two-carbon β-branch. (A) The mechanism of δ-lactone formation via Michael addition onto the unsaturated α,β-thioester followed by cyclisation. (B) The amino variant in the synthesis of lactams. (C) The formation of pharmacologically-important glutarimide moiety by attack of a carboxamide. | ||
Investigation into the scope of the reaction revealed amine and carboxamide nucleophiles could be used to synthesise lactam and glutarimide analogues (Fig. 28B and C).143 Genetic analysis of the glutarimide-containing polyketides revealed a homolog to the B-domain that was labelled an X-domain. A range of chimeras were constructed and used to demonstrate complementation between B/X-domains and the structural rather than catalytic role they play in the formation of β-branched polyketides.144 For more detailed examples see the review by Miyanaga.145
8.2 Pseudo α/β-branching in marine dinoflagellates
The biosynthesis of polyketides from marine dinoflagellates presents an intriguing array of structural diversity arising from α/β-branching. In 2014 Wright et al. stated that all isolated compounds isolated from these sources contain at least one β-alkylation and the majority of these arise from cleaved acetate.146Amphidinolide B is a macrolactone and is a member of a large family of similar compounds that possesses a range of β-branches off the core ring (Fig. 29).147,148 Feeding studies with isotopically labelled precursors revealed that whilst the β-branches arose from C-2 of acetate, the labelling of the carbon backbone was inconsistent with sequential acetate incorporation. As these branches originate from acetate rather than SAM (as for α-alkylation) it has been suggested that β-branching is under the control of an HMGS cassette. It is currently proposed that of the six branches, four have been installed by β-branching and two are installed by a new mechanism called pseudo α-alkylation. Pseudo α-alkylation occurs through in-chain carbon deletion, a phenomena rarely seen outside dinoflagellate PKS.146,149–151
 | ||
| Fig. 29 Structure of amphidinolide B with coloured labels representing each type of branching. Blue: presumed methyl from β-alkylation. Red: methyl from in-chain carbon deletion. Green: methyls from pseudo α-alkylation, after in-chain carbon deletion a carbon in the β-position can be alkylated but would appear as an α-carbon from acetate feeding studies. | ||
Due to large and complex genomic DNA, the synthases responsible for these complex molecules are not easily identified so currently labelling studies provide the only evidence of β-branch incorporation by an HMGS cassette; vicinal carbon-13 labelling and carbon-13 and deuterium labelling have shown consistent incorporation patterns for exclusive acetate incorporation in the polyketide chain and branching products, ruling out proposals of succinate or α-ketoglutarate precursors.152,153
9. The role of β-branching
β-Branching equips biosynthetic pathways with a powerful mechanism by which polyketide chemistry can be diversified and may be of particular importance for classes of polyketides characterised by their extended linear or macrocyclic nature. However, the true role of β-branching remains to be elucidated and the many examples presented here are yet to be mapped to a specific structure–activity relationships. There are however several examples where β-branched products have been co-crystallised with biological targets, providing valuable insights into their structure–function relationships.9.1 Specific functionalisation
Perhaps the most obvious observation is that a branch may provide a unique chemical handle, imbuing the natural products with additional chemistry, polarity or charge. In its simplest form this may amount to selectively injecting patches of tailored hydrophobicity to replace a carbonyl group but with the introduction of more tailored chemistry, other interactions are made possible. The crystal structure of the inner mitochondrial matrix ADP/ATP transporter from the fungus Thermothelomyces thermophile complexed with bongkrekic acid has recently been reported (Fig. 30).154 The structure captured the “matrix-facing” conformation complementing the previous reported “cytoplasm-facing” conformation. Bongkrekic acid competitively inhibits the native nucleotide substrates by binding in a central cavity and forming a large number of electrostatic interactions and hydrogen bonds. These interactions include the carboxymethyl β-branch and distal carboxylate which is adjacent to an endo-β-methyl branch. The two ends of the molecule are brought together in a horseshoe shape with the di-acid end mimicking the phosphate binding of ATP whilst the unsaturated carbon backbone partially mimics the adenine ring and also excludes ATP. The β-branches appear to play an important role in the tight-binding of bongkrekic acid to the transporter, particularly the non-decarboxylated branch. | ||
| Fig. 30 Bongkrekic acid binding to the mitochondrial ADP/ATP transporter from Thermothelomyces thermophila. (A) Structure of ADP/ATP transporter (pdb: 6GCI) with bongkrekic acid shown in green. (B) Expanded view of the active site showing the key electrostatic, hydrogen bonding interactions and the horseshoe shape formed by the backbone. | ||
9.2 Molecular conformation
β-Branches may also modulate the conformational and energetic landscape sampled by a molecule. Perhaps most intriguing is the role of multiple consecutive β-branches in the same molecule. Recently the crystal structures of kalimantacins A and B bound to Staphylococcus aureus enoyl-acyl carrier protein reductase (saFabI) were reported (Fig. 31).155 In both structures, kalimantacin lies across the entirety of the bound NADPH co-factor, blocking entry of the enoyl-acyl carrier protein substrate. The consecutive arrangement of sp2 and sp3 β-carbons form a hindered hydrophobic portion of the molecule that interacts with a hydrophobic patch on FabI. As well as key hydrophobic and hydrogen-bonding interactions to the enzyme, kalimantacin deploys its two hydrophilic ‘ends’ to form a number of defined interactions with the NADPH nicotinamide ring and the ribose group at one extreme and the adenine ring at the other. Interestingly the consecutive arrangement of sp2 and sp3 β-carbons imparts a specific curvature to the molecule that may mimic the bent conformation of the natural bound fatty acid. This pose places the carboxylate group close to the site of the carbonyl of the thioester that would be reduced in the complex of bound acyl-ACP. Despite the switch in double-bond geometry, kalimantacin B appears to maintain these key interactions but molecular dynamics simulations revealed this complex was less stable in line with reduced activity. | ||
| Fig. 31 Kalimantacin A and B binding to saFabI. (A) Structure of FabI (PDB: 6TBB) bound to kalimantacin A (green backbone) and NADPH (cyan). (B) Expanded view of kalimantacin A in the binding pocket showing key hydrophobic and hydrogen-bonding interactions. The E/Z geometry of the diene is highlighted and the sp2/sp3 centres of the β-branches are annotated. (C) Expanded view of FabI (PDB: 6TBC) bound to kalimantacin B and NADPH highlighting the altered geometry due to the presence of the E/E double bond geometry. | ||
10. Conclusion
An important structural feature of many biologically active polyketide-derived natural products is the presence of one of more β-branches. Carbon–carbon bond formation at the electrophilic β-carbon of the growing polyketide chain is catalysed by a multi-protein HMGS cassette which is responsible for loading, aldol addition, dehydration and (commonly) decarboxylation to install structurally different types of β-branches. Whilst the canonical steps are shared by many pathways, exquisite selectivity is achieved by adding or omitting domains or employing domains with altered selectivity. The PKS must use a variety of methods (protein–protein interactions, in-cis tethered domains and in-cis vs. in-trans reactions) to ensure the correct fidelity at the point of β-branch installation. Whilst the role of β-branches is not well understood, influencing shape and functionality available for interaction with biological targets are two possible benefits.| Pathway | B | N | AT | MiBiG/DOI | ACPD | KS0 | HMGS | ECH1 | ECH2 | ACPA | ER | Producing organism | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Pathway: the β-branched polyketide biosynthetic pathway. B: type of branch observed in each compound (numbering refers to β-branch type in Fig. 3). N: total number of β-branches present in the compound. AT: acyl transferase architecture of type I PKS system (type II PKS systems are labelled type II). MIBiG/DOI: the reference number for the MIBiG database, where this is not possible, the DOI for the biosynthetic pathway reference has been used in its place.176 ACPD: donor acyl carrier protein (ACP). KS0: ketosynthase domain (* no KS0 is present but an AT/DC di-domain responsible for formation of propionyl-ACPD). HMGS: the 3-hydroxy-3-methylglutaryl synthase domain. ECH1: the enoyl-CoA hydratase domain catalysing dehydration. ECH2: the enoyl-CoA hydratase domain catalysing decarboxylation (*cis-acting domains with their respective modular location described where known). ACPA: acceptor acyl carrier protein (ACP) with the multiplicity given in brackets (ǂ ACPAs that are split across two ORFs). ER: enoylreductase (*trans-acting ERs). Producing organism: the species of organism from which the biosynthetic gene cluster was isolated and characterised. A — indicates a protein function that is not present and a ? indicates a protein that is expected to be present but is currently unknown. | |||||||||||||
| Bacillaene | 1 | 1 | trans | BGC0001089 | AcpK | BaeF | BaeF | BaeH | BaeI | BaeL-M6 (2), PksL-M6 (2) | — | Bacillus velenzensis FZB42 | 156–158 |
| PksF | PksG | PksH | PksI | ||||||||||
| Crocacin | 1 | 1 | cis | BGC0000974 | CroH | CroD | CroE | CroF | CroG | CroC-M3 | — | Chondromyces crocatus | 159 |
| Elansolid | 1 | 1 | trans | BGC0000178 | ElaE | ElaF | ElaL | ElaM | ElaN | ElaK-M5 (2) | — | Chitinophaga sancti | 160 |
| Myxopyronin | 1 | 1 | trans | DOI: 10.1002/cbic.201300289 | MxnC | MxnD | MxnE | MxnF | MxnG | MxnK-M3 (2) | — | Myxococcus fulvus Mx f50 | 161 |
| Pristinamycin | 1 | 1 | trans | DOI: 10.1111/j.1751–7915.2010.00213.x | SnaG | SnaH | SnaI | SnaJ | SnaK | SnaE2-M5 (3) | — | Streptomyces pristinaespiralis | 162 |
| Pseudomonic acid | 1 | 1 | trans | BGC0000182 | MacpC | MupG | MupH | MupJ | MupK | MmpA-M6 (2) | — | Pseudomonas fluorescens | 28 and 163 |
| Pyxipyrrolone | 1 | 1 | trans | BGC0001751 | PyxK | PyxL | PyxM | PyxN | PyxO | PyxD-M7 (1) | — | Pyxidicoccus sp. MCy9557 | 164 |
| SIA7248 | 1 | 1 | trans | DOI: 10.1002/cbic.201300068 | SiaL | SiaA | SiaJ | SiaC | SiaK | SiaF-M8 (3) | — | Streptomyces sp. A7248 | 165 |
| Thiomarinol | 1 | 1 | trans | BGC0001115 | TacpC | TmlG | TmlH | TmlJ | TmpE | TmpA-M6 (3) | — | Pseudoalteromonas sp. SANK 73390 | 166 |
| Virginiamycin | 1 | 1 | trans | BGC0001116 | ? | VirB | VirC | VirD | VirE | VirA-M5 (2) | — | Streptomyces virginiae | 27 and 167 |
| Calyculin | 1 | 2 | trans | BGC0000967 | CalX | CalW | CalT | CalS | CalR | CalG-M23 (2), CalH-M25 (2) | — | Discodermia calyx symbiont | 168 |
| CalH-M25* | Candidatus Entotheonella sp. | ||||||||||||
| Corallopyronin | 1 | 2 | trans | BGC0001091 | CorC | CorD | CorE | CorF | CorG | CorK-M3′ (2), CorL-M5′ (1) | — | Corallococcus coralloides | 169 |
| Etnangien | 1 | 2 | trans | BGC0000179 | EtnM | EtnN | EtnO | EtnP | EtnQ | EtnD-M1 (3), EtnE-M6 (2) | — | Sorangium cellulosum so ce56 | 110 |
| Pulvomycin | 1 | 2 | trans | DOI: 10.1021/acs.orglett.0c01249 | PulM | PulN | PulJ | PulK | PulL | PulG-M12 (2), PulG-M14 (2) | — | Streptomyces sp. HRS33 | 170 |
| Thailandamide | 1 | 2 | trans | BGC0000186 | ThaI | ThaJ | ThaK | ThaL | ThaM | ThaH-M5 (2), ThaO-M10 (2) | — | Burkholderia thailandensis E264 | 171 and 172 |
| Apratoxin | 3 | 1 | cis | DOI: 10.1371/journal.pone.0018565 | AprC | AprD | AprE | AprF | AprG-M* | AprB (2) | AprG-M ER | Lyngbya bouillonii | 173 |
| Macrobrevin | 3 | 1 | trans | BGC0001470 | BreH | BreI | BreJ | BreK | BreL | BreG (2) | BreM-M12 ER | Brevibacillus sp. Leaf182 | 67 and 68 |
| Patellazole | 3 | 1 | trans | DOI: 10.1073/pnas.1213820109 | PtzD | PtzH | PtzI | PtzJ | PtzG | PtzD-M13 (4) (2 predicted inactive) | PtzQ* | Lissoclinum patella Endolissoclinum faulkneri symbiont | 64 |
| Cylindrocyclophane | 3 | 2 | cis | BGC0001566 | ? | CylE | CylF | CylG | CylH-M2* | CylD-M1 (2) | CylH-M2 ER | Cylindrospermum licheniforme UTEX B 2014 | 65 and 66 |
| BGC0001064 | |||||||||||||
| Curacin | 5 | 1 | cis | BGC0000976 | CurB | CurC | CurD | CurE | CurF* | CurA (3) | — | Moorea producens 3L, Lyngbya majuscula | 69 |
| BGC0001165 | |||||||||||||
| Cusperin | 6 | 1 | trans | BGC0001564 | CusG | CusH | CusI | CusJ | CusA-M2* | CusA-M2 (2) | — | Cuspidothrix issatschenkoi CHARLIE-1 | 89 |
| Diaphorin | 6 | 1 | trans | DOI: 10.1016/j.cub.2013.06.027 | DipR | DipF | DipE | DipC | DipP* | DipP (3) | — | Diaphorina citri symbiont candidatus Profftella armatura | 79 |
| Difficidin | 6 | 1 | trans | BGC0000176 | DifC | ? | DifN | ? | DifO* | DifL-M13 (2) | — | Bacillus velenzensis FZB42 | 156 |
| Labrenzin | 6 | 1 | trans | DOI: 10.3389/fmicb.2019.02561 | Lab2 | Lab12 | Lab3 | Lab11 | Lab4* | Lab4 (2) | — | Labrenzia sp. PHM005 | 78 |
| Mycalamide | 6 | 1 | trans | BGC0002055 | MycN | MycM | MycP | MycL | MycI-M2* | MycI-M2 (3) | — | Mycale hentscheli | 81 and 82 |
| Nosperin | 6 | 1 | trans | BGC0001071 | NspH | NspG | NspI | NspJ | NspA-M2* | NspA-M2 (2) | — | Nostoc sp. ‘Peltigera membranacea cyanobiont’ | 88 |
| Onnamide | 6 | 1 | trans | BGC0001105 | ? | ? | OnnA | ? | OnnB* | OnnB (3) | — | Bacterial symbiont of Theonella swinhoei | 76 |
| Pederin | 6 | 1 | trans | BGC0001108 | PedN | PedM | PedP | PedL | PedI* | PedI (1) | — | Bacterial symbiont of Paedarus fuscipes | 75–77 |
| Psymberin | 6 | 1 | trans | BGC0001110 | PsyL | PsyM | PsyI | PsyJ | PsyA* | PsyA (1) | — | Psammonicia aff. Bulbosa | 87 |
| Ircinia ramosa | |||||||||||||
| Fogacin C | 7 | 1 | Type II | BGC0002021 | FogA6 | FogA7* | FogA5 | FogA4 | FogA3 | FogA8 | — | Actinoplanes missouriensis | 31 |
| Jamaicamide | 8 | 1 | cis | BGC0001001 | JamF | JamG | JamH | JamI | JamJ* | JamE (3) | — | Lyngbya majuscula | 70 |
| Malyngamide | 8 | 1 | cis | BGC0001971 | MgcL | MgcM | MgcN | MgcP | MgcQ-M1* | MgcK (2) | — | Okeania hirsuta | 174 |
| Spliceostatin | 9 | 1 | trans | BGC0001113 | Fr9M | Fr9N | Fr9K | Fr9L | Fr9GH-M8* | Fr9GH-M8 (3) | — | Burkholderia sp. FERM BP-3421, Pseudomonas sp. 2663 | 93 |
| BGC0000995 | |||||||||||||
| Thailanstatin | 9 | 1 | trans | BGC0001114 | TstM | TstN | TstK | TstL | TstGH-M8* | TstGH-M8 (3) | — | Burkholderia thailandensis | 95 |
| Bryostatin | 11 | 2 | trans | BGC0000174 | BryU | BryQ | BryR | BryT | — | BryA-M3 (1), BryB-M7 (1) | — | Candidatus Endobugula sertula | 48 and 100 |
| BGC0000205 | |||||||||||||
| Leinamycin | 12 | 1 | trans | BGC0001101 | LnmL | LnmK* | LnmM | LnmF | — | LnmJ-M8 (1) | — | Streptomyces atroolivaceus | 44, 103 and 104 |
| Aurantinin | 1, 3 | 2 | trans | BGC0001520 | ? | BG616_04390 | BG616_04395 | BG616_0440 | BG616_0445 | ? | ? | Bacillus subtillis, Bacillus aurantin | 175 |
| Gladiolin | 1, 3 | 2 | trans | DOI: 10.1021/jacs.7b03382 | GbnF | GbnG | GbnH | GbnI | GbnJ | GbnD1-M1 (2), GbnD2-M6 (2) | GbnD1-M1 ER | Burkholderia gladioli | 111 |
| GbnQ | GbnR | ||||||||||||
| Pateamine | 1, 3 | 3 | trans | BGC0002057 | PamE | PamF | PamG | PamH | PamI | PamA-M2 (2)ǂ, PamC-M8 (1), PamD-M12 (2) | PamC-M8 ER | Kiritimatiellaeota bacterium | 81 and 82 |
| Myxovirescin | 2, 4 | 2 | trans | BGC0001025 | TaB | TaK | TaC | TaX | TaY | Ta1-M5 (1) | TaO-M8 ER | Myxococcus xanthus DK 1622 | 134 |
| TaE | TaD* | TaF | TaO-M8 (1) | ||||||||||
| Oocydin | 1, 6 | 2 | trans | BGC0001031 | OocG | OocF | OocE | OocD | OocC | OocJ-M1 (2) | — | Serratia marcescens, | 127 |
| BGC0001032 | OocJ* | OocL-M4 (2) | Serratia plymuthica | ||||||||||
Bongrekic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Acid Acid |
1, 10 | 2 | trans | BGC0000173 | ? | BonF | BonG | BonH | BonI | BonA-M1 (1), BonD-M11 (2) | — | Burkholderia gladioli | 131 and 132 |
| Ripostatin | 1, 10 | 2 | Both | BGC0001761 | RipQ | RipR | RipA | RipL | RipK | RipD-M6 (2), RipE-M9 (1) | — | Sorangium cellulosum | 130 |
| Guangnanmycin | 6, 10 | 2 | trans | BGC0001611 | GnmV | GnmW | GnmE | GnmF | GnmT-M5* | GnmT-M5 (1), GnmT-M8 (2) | — | Streptomyces sp. CB01883 | 72 |
| GnmU | |||||||||||||
| Weishanmycin | 6, 10 | 2 | trans | BGC0001823 | WsmT | WsmU | WsmD | WsmE | WsmR-M5* | WsmR-M5 (1), WsmR-M8 (2) | — | Streptomyces sp. CB02120-2 | 72 |
| WsmS | |||||||||||||
| Largimycin | 6, 12 | 2 | trans | DOI: 10.1021/acschembio.0c00160 | LrgA | LrgD | LrgM2 | LrgF | LrgJ-M5* | LrgJ-M5 (1), LrgJ-M8 (2) | — | Streptomyces argillaceus sp. ATCC 12956 | 138 |
| LrgL | LrgK* | LrgM1 | |||||||||||
| Kalimantacin | 1, 3, 6 | 4 | trans | BGC0001099 | BatA | BatB | BatC | BatD | BatE | Bat2-M6 (3), Bat3-M11 (2), Bat3-M12 (1), Bat3-M13 (1) | BatK* | Pseudomonas fluorescens | 114 |
| Bat3-mECH* | |||||||||||||
| Leptolyngbyalide | 1, 3, 6 | 5 | trans | BGC0001837 | LeptI | ? | LeptY | LeptX | LeptW | LeptL-M2 (2), LeptM-M8 (2), LeptQ-M13 (2)ǂ, LeptQ-M14 (3), LeptQ-M15 (1) | LeptQ-13M ER | Leptolyngbya sp. PCC 7375 | 119 |
| LeptL-M2* | |||||||||||||
| Phormidolide | 1, 6 | 5 | trans | BGC0001350 | PhmB | PhmA | PhmP | PhmO | PhmN | PhmE-M2 (2), PhmF-M8 (2), PhmH-M13 (2)ǂ, PhmI-M14 (1), PhmI-M17 (1) | — | Leptolyngbya sp. ISBN3-Nov-94-8 | 116 |
| PhmE-M2* | |||||||||||||
| PhmI-M14* | |||||||||||||
11. Conflicts of interest
There are no conflicts to declare.12. Acknowledgements
We thank the EPSRC Bristol Chemical Synthesis Centre for Doctoral Training (EP/L015366/1) and CRUK (C42109/A26982) for funding for P Walker and the doctoral training grant for A Weir. We also thank the BBSRC for support (BB/R007853/1) and BBSRC/EPSRC for funding through the Bristol Centre for Synthetic Biology (BB/L01386X/1).13. References
- J. Piel, Nat. Prod. Rep., 2010, 27, 996–1047 RSC.
- E. J. N. Helfrich and J. Piel, Nat. Prod. Rep., 2016, 33, 231–316 RSC.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS.
- L. Ray and B. S. Moore, Nat. Prod. Rep., 2016, 33, 150–161 RSC.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2016, 33, 141–149 RSC.
- K. J. Weissman, Beilstein J. Org. Chem., 2017, 13, 348–371 CrossRef CAS.
- J. W. Giraldes, D. L. Akey, J. D. Kittendorf, D. H. Sherman, J. L. Smith and R. A. Fecik, Nat. Chem. Biol., 2006, 2, 531–536 CrossRef CAS.
- A. Miyanaga, F. Kudo and T. Eguchi, Nat. Prod. Rep., 2018, 35, 1185–1209 RSC.
- H. Nakamura, J. X. Wang and E. P. Balskus, Chem. Sci., 2015, 6, 3816–3822 RSC.
- A. R. Gallimore, C. B. W. Stark, A. Bhatt, B. M. Harvey, Y. Demydchuk, V. Bolanos-Garcia, D. J. Fowler, J. Staunton, P. F. Leadlay and J. B. Spencer, Chem. Biol., 2006, 13, 453–460 CrossRef CAS.
- C. P. Gorst-Allman, B. A. M. Rudd, C.-J. Chang and H. G. Floss, J. Org. Chem., 1981, 46, 455–456 CrossRef CAS.
- F. Hemmerling and F. Hahn, Beilstein J. Org. Chem., 2016, 12, 1512–1550 CrossRef CAS.
- P. Wang, X. Gao and Y. Tang, Curr. Opin. Chem. Biol., 2012, 16, 362–369 CrossRef CAS.
- J. Thorson, T. Hosted Jr, J. Jiang, J. Biggins and J. Ahlert, Curr. Org. Chem., 2001, 5, 139–167 CrossRef CAS.
- C. Walsh, C. L. Freel Meyers and H. C. Losey, J. Med. Chem., 2003, 46, 3425–3436 CrossRef CAS.
- S. Sundaram and C. Hertweck, Curr. Opin. Chem. Biol., 2016, 31, 82–94 CrossRef CAS.
- C. T. Walsh, Acc. Chem. Res., 2008, 41, 4–10 CrossRef CAS.
- C. Olano, C. Méndez and J. A. Salas, Nat. Prod. Rep., 2010, 27, 571–616 RSC.
- A. Nivina, K. P. Yuet, J. Hsu and C. Khosla, Chem. Rev., 2019, 119, 12524–12547 CrossRef CAS.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- D. K. Liscombe, G. V. Louie and J. P. Noel, Nat. Prod. Rep., 2012, 29, 1238–1250 RSC.
- M. A. Skiba, A. P. Sikkema, W. D. Fiers, W. H. Gerwick, D. H. Sherman, C. C. Aldrich and J. L. Smith, ACS Chem. Biol., 2016, 11, 3319–3327 CrossRef CAS.
- M. Z. Ansari, J. Sharma, R. S. Gokhale and D. Mohanty, BMC Bioinf., 2008, 9, 454 CrossRef.
- Y. A. Chan, A. M. Podevels, B. M. Kevany and M. G. Thomas, Nat. Prod. Rep., 2009, 26, 90–114 RSC.
- T. C. Feline, R. B. Jones, G. Mellows and L. Phillips, J. Chem. Soc. Perkin Trans. I, 1977, 309 RSC.
- D. G. I. Kingston, M. X. Kolpak, J. W. LeFevre and I. Borup-Grochtmann, J. Am. Chem. Soc., 1983, 105, 5106–5110 CrossRef CAS.
- D. G. I. Kingston and M. X. Kolpak, J. Am. Chem. Soc., 1980, 102, 5964–5966 CrossRef CAS.
- A. K. El-Sayed, J. Hothersall, S. M. Cooper, E. Stephens, T. J. Simpson and C. M. Thomas, Chem. Biol., 2003, 10, 419–430 CrossRef CAS.
- C. T. Calderone, W. E. Kowtoniuk, N. L. Kelleher, C. T. Walsh and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8977–8982 CrossRef CAS.
- L. Gu, B. Wang, A. Kulkarni, T. W. Geders, R. V. Grindberg, L. Gerwick, K. Hkansson, P. Wipf, J. L. Smith, W. H. Gerwick and D. H. Sherman, Nature, 2009, 459, 731–735 CrossRef CAS.
- K. Sato, Y. Katsuyama, K. Yokota, T. Awakawa, T. Tezuka and Y. Ohnishi, ChemBioChem, 2019, 20, 1039–1050 CrossRef CAS.
- E. J. N. Helfrich, G. M. Lin, C. A. Voigt and J. Clardy, Beilstein J. Org. Chem., 2019, 15, 2889–2906 CrossRef CAS.
- C. T. Calderone, Nat. Prod. Rep., 2008, 25, 845–853 RSC.
- M. A. Skiba, F. P. Maloney, Q. Dan, A. E. Fraley, C. C. Aldrich, J. L. Smith and W. C. Brown, in Methods in Enzymology, Academic Press Inc., 2018, vol. 604, pp. 45–88 Search PubMed.
- J. Crosby and M. P. Crump, Nat. Prod. Rep., 2012, 29, 1111–1137 RSC.
- P. C. Dorrestein, S. B. Bumpus, C. T. Calderone, S. Garneau-Tsodikova, Z. D. Aron, P. D. Straight, R. Kolter, C. T. Walsh and N. L. Kelleher, Biochemistry, 2006, 45, 12756–12766 CrossRef CAS.
- D. Meluzzi, W. H. Zheng, M. Hensler, V. Nizet and P. C. Dorrestein, Bioorg. Med. Chem. Lett., 2008, 18, 3107–3111 CrossRef CAS.
- P. K. Mishra and D. G. Drueckhammer, Chem. Rev., 2000, 100, 3283–3309 CrossRef CAS.
- J. Franke and C. Hertweck, Cell Chem. Biol., 2016, 23, 1179–1192 CrossRef CAS.
- V. Agarwal, S. Diethelm, L. Ray, N. Garg, T. Awakawa, P. C. Dorrestein and B. S. Moore, Org. Lett., 2015, 17, 4452–4455 CrossRef CAS.
- R. J. Cox, T. S. Hitchman, K. J. Byrom, I. S. C. Findlow, J. A. Tanner, J. Crosby and T. J. Simpson, FEBS Lett., 1997, 405, 267–272 CrossRef CAS.
- J. Beld, E. C. Sonnenschein, C. R. Vickery, J. P. Noel and M. D. Burkart, Nat. Prod. Rep., 2014, 31, 61–108 RSC.
- M. Cummings, R. Breitling and E. Takano, FEMS Microbiol. Lett., 2014, 351, 116–125 CrossRef CAS.
- Y. Q. Cheng, G. L. Tang and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3149–3154 CrossRef CAS.
- L. Gu, J. Jia, H. Liu, K. Håkansson, W. H. Gerwick and D. H. Sherman, J. Am. Chem. Soc., 2006, 128, 9014–9015 CrossRef.
- F. P. Maloney, L. Gerwick, W. H. Gerwick, D. H. Sherman and J. L. Smith, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10316–10321 CrossRef CAS.
- H. M. Miziorko, Arch. Biochem. Biophys., 2011, 505, 131–143 CrossRef CAS.
- T. J. Buchholz, C. M. Rath, N. B. Lopanik, N. P. Gardner, K. Håkansson and D. H. Sherman, Chem. Biol., 2010, 17, 1092–1100 CrossRef CAS.
- S. Dutta, J. R. Whicher, D. A. Hansen, W. A. Hale, J. A. Chemler, G. R. Congdon, A. R. H. Narayan, K. Håkansson, D. H. Sherman, J. L. Smith and G. Skiniotis, Nature, 2014, 510, 512–517 CrossRef CAS.
- J. R. Whicher, S. Dutta, D. A. Hansen, W. A. Hale, J. A. Chemler, A. M. Dosey, A. R. H. Narayan, K. Håkansson, D. H. Sherman, J. L. Smith and G. Skiniotis, Nature, 2014, 510, 560–564 CrossRef CAS.
- A. S. Haines, X. Dong, Z. Song, R. Farmer, C. Williams, J. Hothersall, E. Płoskoń, P. Wattana-Amorn, E. R. Stephens, E. Yamada, R. Gurney, Y. Takebayashi, J. Masschelein, R. J. Cox, R. Lavigne, C. L. Willis, T. J. Simpson, J. Crosby, P. J. Winn, C. M. Thomas and M. P. Crump, Nat. Chem. Biol., 2013, 9, 685–692 CrossRef CAS.
- A. S. Rahman, J. Hothersall, J. Crosby, T. J. Simpson and C. M. Thomas, J. Biol. Chem., 2005, 280, 6399–6408 CrossRef CAS.
- D. J. Daley, Adv. Appl. Probab., 1976, 8, 395–415 CrossRef.
- L. Gu, E. B. Eisman, S. Dutta, T. M. Franzmann, S. Walter, W. H. Gerwick, G. Skiniotis and D. H. Sherman, Angew. Chem., Int. Ed., 2011, 50, 2795–2798 CrossRef CAS.
- Z. Wang, S. R. Bagde, G. Zavala, T. Matsui, X. Chen and C. Y. Kim, ACS Chem. Biol., 2018, 13, 3072–3077 CrossRef CAS.
- H. Jiang, R. Zirkle, J. G. Metz, L. Braun, L. Richter, S. G. Van Lanen and B. Shen, J. Am. Chem. Soc., 2008, 130, 6336–6337 CrossRef CAS.
- P. D. Walker, C. Williams, A. N. M. Weir, L. Wang, J. Crosby, P. R. Race, T. J. Simpson, C. L. Willis and M. P. Crump, Angew. Chem., Int. Ed., 2019, 58, 12446–12450 CrossRef CAS.
- A. V. Nair, A. Robson, T. D. Ackrill, M. Till, M. J. Byrne, C. R. Back, K. Tiwari, J. A. Davies, C. L. Willis and P. R. Race, Sci. Rep., 2020, 10, 15323 CrossRef CAS.
- T. W. Geders, L. Gu, J. C. Mowers, H. Liu, W. H. Gerwick, K. Håkansson, D. H. Sherman and J. L. Smith, J. Biol. Chem., 2007, 282, 35954–35963 CrossRef CAS.
- J. Hothersall, J. Wu, A. S. Rahman, J. A. Shields, J. Haddock, N. Johnson, S. M. Cooper, E. R. Stephens, R. J. Cox, J. Crosby, C. L. Willis, T. J. Simpson and C. M. Thomas, J. Biol. Chem., 2007, 282, 15451–15461 CrossRef CAS.
- J. Wu, S. M. Cooper, R. J. Cox, J. Crosby, M. P. Crump, J. Hothersall, T. J. Simpson, C. M. Thomas and C. L. Willis, Chem. Commun., 2007, 2040–2042 RSC.
- J. A. Shields, A. S. Rahman, C. J. Arthur, J. Crosby, J. Hothersall, T. J. Simpson and C. M. Thomas, ChemBioChem, 2010, 11, 248–255 CrossRef CAS.
- F. M. Martin and T. J. Simpson, J. Chem. Soc. Perkin Trans. I, 1989, 207–209 RSC.
- J. C. Kwan, M. S. Donia, A. W. Han, E. Hirose, M. G. Haygood and E. W. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20655–20660 CrossRef CAS.
- H. Nakamura, H. A. Hamer, G. Sirasani and E. P. Balskus, J. Am. Chem. Soc., 2012, 134, 18518–18521 CrossRef CAS.
- H. Nakamura, E. E. Schultz and E. P. Balskus, Nat. Chem. Biol., 2017, 13, 916–921 CrossRef CAS.
- E. J. N. Helfrich, C. M. Vogel, R. Ueoka, M. Schäfer, F. Ryffel, D. B. Müller, S. Probst, M. Kreuzer, J. Piel and J. A. Vorholt, Nat. Microbiol., 2018, 3, 909–919 CrossRef CAS.
- Y. Bai, D. B. Müller, G. Srinivas, R. Garrido-Oter, E. Potthoff, M. Rott, N. Dombrowski, P. C. Münch, S. Spaepen, M. Remus-Emsermann, B. Hüttel, A. C. McHardy, J. A. Vorholt and P. Schulze-Lefert, Nature, 2015, 528, 364–369 CrossRef CAS.
- Z. Chang, N. Sitachitta, J. V. Rossi, M. A. Roberts, P. M. Flatt, J. Jia, D. H. Sherman and W. H. Gerwick, J. Nat. Prod., 2004, 67, 1356–1367 CrossRef CAS.
- D. J. Edwards, B. L. Marquez, L. M. Nogle, K. McPhail, D. E. Goeger, M. A. Roberts and W. H. Gerwick, Chem. Biol., 2004, 11, 817–833 CrossRef CAS.
- D. Khare, B. Wang, L. Gu, J. Razelun, D. H. Sherman, W. H. Gerwick, K. Håkansson and J. L. Smith, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14099–14104 CrossRef CAS.
- G. Pan, Z. Xu, Z. Guo, Hindra, M. Ma, D. Yang, H. Zhou, Y. Gansemans, X. Zhu, Y. Huang, L. X. Zhao, Y. Jiang, J. Cheng, F. Van Nieuwerburgh, J. W. Suh, Y. Duan and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E11131–E11140 CrossRef CAS.
- D. Khare, W. A. Hale, A. Tripathi, L. Gu, D. H. Sherman, W. H. Gerwick, K. Håkansson and J. L. Smith, Structure, 2015, 23, 2213–2223 CrossRef CAS.
- C. Cardani, D. Ghiringhelli, R. Mondelli and A. Quilico, Tetrahedron Lett., 1965, 6, 2537–2545 CrossRef.
- J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14002–14007 CrossRef CAS.
- J. Piel, D. Hui, G. Wen, D. Butzke, M. Platzer, N. Fusetani and S. Matsunaga, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16222–16227 CrossRef CAS.
- J. Piel, G. Wen, M. Platzer and D. Hui, ChemBioChem, 2004, 5, 93–98 CrossRef CAS.
- D. Kačar, C. Schleissner, L. M. Cañedo, P. Rodríguez, F. de la Calle, B. Galán and J. L. García, Front. Microbiol., 2019, 10, 2561 CrossRef.
- A. Nakabachi, R. Ueoka, K. Oshima, R. Teta, A. Mangoni, M. Gurgui, N. J. Oldham, G. Van Echten-Deckert, K. Okamura, K. Yamamoto, H. Inoue, M. Ohkuma, Y. Hongoh, S. Y. Miyagishima, M. Hattori, J. Piel and T. Fukatsu, Curr. Biol., 2013, 23, 1478–1484 CrossRef CAS.
- L. K. Pannell, N. B. Perry, J. W. Blunt and M. H. Munro, J. Am. Chem. Soc., 1988, 110, 4850–4851 CrossRef.
- M. A. Storey, S. K. Andreassend, J. Bracegirdle, A. Brown, R. A. Keyzers, D. F. Ackerley, P. T. Northcote and J. G. Owen, mBio, 2020, 11, e02997–19 CrossRef.
- M. Rust, E. J. N. Helfrich, M. F. Freeman, P. Nanudorn, C. M. Field, C. Rückert, T. Kündig, M. J. Page, V. L. Webb, J. Kalinowski, S. Sunagawa and J. Piel, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 9508–9518 CrossRef CAS.
- S. Sakemi, T. Ichiba, S. Kohmoto, G. Saucy and T. Higa, J. Am. Chem. Soc., 1988, 110, 4851–4853 CrossRef CAS.
- J. Piel, D. Butzke, N. Fusetani, D. Hui, M. Platzer, G. Wen and S. Matsunaga, J. Nat. Prod., 2005, 68, 472–479 CrossRef CAS.
- G. R. Pettit, J. P. Xu, J. C. Chapuis, R. K. Pettit, L. P. Tackett, D. L. Doubek, J. N. A. Hooper and J. M. Schmidt, J. Med. Chem., 2004, 47, 1149–1152 CrossRef CAS.
- R. H. Cichewicz, F. A. Valeriote and P. Crews, Org. Lett., 2004, 6, 1951–1954 CrossRef CAS.
- K. M. Fisch, C. Gurgui, N. Heycke, S. A. Van Der Sar, S. A. Anderson, V. L. Webb, S. Taudien, M. Platzer, B. K. Rubio, S. J. Robinson, P. Crews and J. Piel, Nat. Chem. Biol., 2009, 5, 494–501 CrossRef CAS.
- A. Kampa, A. N. Gagunashvili, T. A. M. Gulder, B. I. Morinaka, C. Daolio, M. Godejohann, V. P. W. Miao, J. Piel and Ó. S. Andrésson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3129–E3137 CrossRef CAS.
- A. Kust, J. Mareš, J. Jokela, P. Urajová, J. Hájek, K. Saurav, K. Voráčová, D. P. Fewer, E. Haapaniemi, P. Permi, K. Řeháková, K. Sivonen and P. Hrouzek, ACS Chem. Biol., 2018, 13, 1123–1129 CrossRef CAS.
- D. R. Jackson, G. Shakya, A. B. Patel, L. Y. Mohammed, K. Vasilakis, P. Wattana-Amorn, T. R. Valentic, J. C. Milligan, M. P. Crump, J. Crosby and S. C. Tsai, ACS Chem. Biol., 2018, 13, 141–151 CrossRef CAS.
- D. Kaida, H. Motoyoshi, E. Tashiro, T. Nojima, M. Hagiwara, K. Ishigami, H. Watanabe, T. Kitahara, T. Yoshida, H. Nakajima, T. Tani, S. Horinouchi and M. Yoshida, Nat. Chem. Biol., 2007, 3, 576–583 CrossRef CAS.
- R. J. Van Alphen, E. A. C. Wiemer, H. Burger and F. A. L. M. Eskens, Br. J. Cancer, 2009, 100, 228–232 CrossRef CAS.
- A. S. Eustáquio, J. E. Janso, A. S. Ratnayake, C. J. O'Donnell and F. E. Koehn, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3376–E3385 CrossRef.
- F. Zhang, H. Y. He, M. C. Tang, Y. M. Tang, Q. Zhou and G. L. Tang, J. Am. Chem. Soc., 2011, 133, 2452–2462 CrossRef CAS.
- X. Liu, S. Biswas, M. G. Berg, C. M. Antapli, F. Xie, Q. Wang, M. C. Tang, G. L. Tang, L. Zhang, G. Dreyfuss and Y. Q. Cheng, J. Nat. Prod., 2013, 76, 685–693 CrossRef CAS.
- X. Liu, S. Biswas, G. L. Tang and Y. Q. Cheng, J. Antibiot. (Tokyo), 2013, 66, 555–558 CrossRef CAS.
- T. J. Nelson, M. K. Sun, C. Lim, A. Sen, T. Khan, F. V. Chirila and D. L. Alkon, J. Alzheim. Dis., 2017, 58, 521–535 CAS.
- M. D. Marsden, B. A. Loy, X. Wu, C. M. Ramirez, A. J. Schrier, D. Murray, A. Shimizu, S. M. Ryckbosch, K. E. Near, T. W. Chun, P. A. Wender and J. A. Zack, PLoS Pathog., 2017, 13, e1006575 CrossRef.
- P. Kollár, J. Rajchard, Z. Balounová and J. Pazourek, Pharm. Biol., 2014, 52, 237–242 CrossRef.
- S. Sudek, N. B. Lopanik, L. E. Waggoner, M. Hildebrand, C. Anderson, H. Liu, A. Patel, D. H. Sherman and M. G. Haygood, J. Nat. Prod., 2007, 70, 67–74 CrossRef CAS.
- S. T. Slocum, A. N. Lowell, A. N. Tripathi, V. V. Shende, J. L. Smith and D. H. Sherman, in Methods in Enzymology, Academic Press Inc., 2018, vol. 604, pp. 207–236 Search PubMed.
- Y. Q. Cheng, G. L. Tang and B. Shen, J. Bacteriol., 2002, 184, 7013–7024 CrossRef CAS.
- Y. Huang, S. X. Huang, J. Ju, G. Tang, T. Liu and B. Shen, Org. Lett., 2011, 13, 498–501 CrossRef CAS.
- T. Liu, Y. Huang and B. Shen, J. Am. Chem. Soc., 2009, 131, 6900–6901 CrossRef CAS.
- J. R. Lohman, C. A. Bingman, G. N. Phillips and B. Shen, Biochemistry, 2013, 52, 902–911 CrossRef CAS.
- M. Ma, J. R. Lohman, T. Liu and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10359–10364 CrossRef CAS.
- G. L. Tang, Y. Q. Cheng and B. Shen, J. Biol. Chem., 2007, 282, 20273–20282 CrossRef CAS.
- G. L. Tang, Y. Q. Cheng and B. Shen, Chem. Biol., 2004, 11, 33–45 CrossRef CAS.
- T. Kwong, M. Ma, G. Pan, Hindra, D. Yang, C. Yang, J. R. Lohman, J. D. Rudolf, J. L. Cleveland and B. Shen, Biochemistry, 2018, 57, 5005–5013 CrossRef CAS.
- D. Menche, F. Arikan, O. Perlova, N. Horstmann, W. Ahlbrecht, S. C. Wenzel, R. Jansen, H. Irschik and R. Müller, J. Am. Chem. Soc., 2008, 130, 14234–14243 CrossRef CAS.
- L. Song, M. Jenner, J. Masschelein, C. Jones, M. J. Bull, S. R. Harris, R. C. Hartkoorn, A. Vocat, I. Romero-Canelon, P. Coupland, G. Webster, M. Dunn, R. Weiser, C. Paisey, S. T. Cole, J. Parkhill, E. Mahenthiralingam and G. L. Challis, J. Am. Chem. Soc., 2017, 139, 7974–7981 CrossRef CAS.
- T. Tokunaga, K. Kamigiri, M. Orita, T. Nishikawa, M. Shimizu and H. Kaniwa, J. Antibiot. (Tokyo), 1996, 49, 140–144 CrossRef CAS.
- K. Kamigiri, Y. Suzuki, M. Shibazaki, M. Morioka, K.-I. Suzuki, T. Tokunaga, B. Setiawan and R. M. Rantiatmodjo, J. Antibiot. (Tokyo), 1996, 49, 136–139 CrossRef CAS.
- W. Mattheus, L. J. Gao, P. Herdewijn, B. Landuyt, J. Verhaegen, J. Masschelein, G. Volckaert and R. Lavigne, Chem. Biol., 2010, 17, 149–159 CrossRef CAS.
- I. R. G. Thistlethwaite, F. M. Bull, C. Cui, P. D. Walker, S. S. Gao, L. Wang, Z. Song, J. Masschelein, R. Lavigne, M. P. Crump, P. R. Race, T. J. Simpson and C. L. Willis, Chem. Sci., 2017, 8, 6196–6201 RSC.
- M. J. Bertin, A. Vulpanovici, E. A. Monroe, A. Korobeynikov, D. H. Sherman, L. Gerwick and W. H. Gerwick, ChemBioChem, 2016, 17, 164–173 CrossRef CAS.
- I. E. Ndukwe, X. Wang, N. Y. S. Lam, K. Ermanis, K. L. Alexander, G. E. Martin, G. Muir, I. Paterson, R. Britton, J. M. Goodman, J. Piel, W. H. Gerwick, R. T. Williamson, M. J. Bertin, G. E. Martin, G. Muir, I. Paterson, R. Britton, J. M. Goodman, E. J. N. Helfrich, J. Piel, W. H. Gerwick and R. T. Williamson, Chem. Commun., 2020, 7565–7568 RSC.
- N. Y. S. Lam, G. Muir, V. R. Challa, R. Britton and I. Paterson, Chem. Commun., 2019, 55, 9717–9720 RSC.
- E. J. N. Helfrich, R. Ueoka, A. Dolev, M. Rust, R. A. Meoded, A. Bhushan, G. Califano, R. Costa, M. Gugger, C. Steinbeck, P. Moreno and J. Piel, Nat. Chem. Biol., 2019, 15, 813–821 CrossRef CAS.
- K. Ueda and Y. Hu, Tetrahedron Lett., 1999, 40, 6305–6308 CrossRef CAS.
- N. Takada, H. Sato, K. Suenaga, H. Arimoto, K. Yamada, K. Ueda and D. Uemura, Tetrahedron Lett., 1999, 40, 6309–6312 CrossRef CAS.
- T. Teruya, H. Shimogawa, K. Suenaga and H. Kigoshi, Chem. Lett., 2004, 33, 1184–1185 CrossRef CAS.
- T. Teruya, K. Suenaga, S. Maruyama, M. Kurotaki and H. Kigoshi, Tetrahedron, 2005, 61, 6561–6567 CrossRef CAS.
- M. A. Matilla, F. J. Leeper and G. P. C. Salmond, Environ. Microbiol., 2015, 17, 2993–3008 CrossRef CAS.
- B. Sato, H. Nakajima, T. Fujita, S. Takase, S. Yoshimura, T. Kinoshita and H. Terano, J. Antibiot. (Tokyo), 2005, 58, 634–639 CrossRef CAS.
- H. Kigoshi and I. Hayakawa, Chem. Rec., 2007, 7, 254–264 CrossRef CAS.
- M. A. Matilla, H. Stöckmann, F. J. Leeper and G. P. C. Salmond, J. Biol. Chem., 2012, 287, 39125–39138 CrossRef CAS.
- R. A. Meoded, R. Ueoka, E. J. N. Helfrich, K. Jensen, N. Magnus, B. Piechulla and J. Piel, Angew. Chem., Int. Ed., 2018, 57, 11644–11648 CrossRef CAS.
- H. Augustiniak, G. Höfle, H. Irschik and H. Reichenbach, Liebigs Ann., 1996, 1996, 1657–1663 CrossRef.
- C. Fu, D. Auerbach, Y. Li, U. Scheid, E. Luxenburger, R. Garcia, H. Irschik and R. Müller, Angew. Chem., Int. Ed., 2017, 56, 2192–2197 CrossRef CAS.
- N. Moebius, C. Ross, K. Scherlach, B. Rohm, M. Roth and C. Hertweck, Chem. Biol., 2012, 19, 1164–1174 CrossRef CAS.
- B. Rohm, K. Scherlach and C. Hertweck, Org. Biomol. Chem., 2010, 8, 1520–1522 RSC.
- V. Simunovic and R. Müller, ChemBioChem, 2007, 8, 497–500 CrossRef CAS.
- V. Simunovic, J. Zapp, S. Rachid, D. Krug, P. Meiser and R. Müller, ChemBioChem, 2006, 7, 1206–1220 CrossRef CAS.
- W. Trowitzsch-Kienast, K. Schober, V. Wray, K. Gerth, H. Reichenbach and G. Hefle, Liebigs Ann. Chem., 1989, 345–355 CrossRef CAS.
- C. T. Calderone, D. F. Iwig, P. C. Dorrestein, N. L. Kelleher and C. T. Walsh, Chem. Biol., 2007, 14, 835–846 CrossRef CAS.
- V. Simunovic and R. Müller, ChemBioChem, 2007, 8, 1273–1280 CrossRef CAS.
- A. Becerril, I. Pérez-Victoria, S. Ye, A. F. Branã, J. Martín, F. Reyes, J. A. Salas and C. Méndez, ACS Chem. Biol., 2020, 15, 1541–1553 CrossRef CAS.
- H. Liang, L. Jiang, Q. Jiang, J. Shi, J. Xiang, X. Yan, X. Zhu, L. Zhao, B. Shen, Y. Duan and Y. Huang, Environ. Microbiol., 2019, 21, 4270–4282 CrossRef CAS.
- T. A. Nguyen, K. Ishida, H. Jenke-Kodama, E. Dittmann, C. Gurgui, T. Hochmuth, S. Taudien, M. Platzer, C. Hertweck and J. Piel, Nat. Biotechnol., 2008, 26, 225–233 CrossRef CAS.
- M. Jenner, S. Frank, A. Kampa, C. Kohlhaas, P. Pöplau, G. S. Briggs, J. Piel and N. J. Oldham, Angew. Chem., Int. Ed., 2013, 52, 1143–1147 CrossRef CAS.
- T. Bretschneider, J. B. Heim, D. Heine, R. Winkler, B. Busch, B. Kusebauch, T. Stehle, G. Zocher and C. Hertweck, Nature, 2013, 502, 124–128 CrossRef CAS.
- D. Heine, T. Bretschneider, S. Sundaram and C. Hertweck, Angew. Chem., Int. Ed., 2014, 53, 11645–11649 CrossRef CAS.
- S. Sundaram, D. Heine and C. Hertweck, Nat. Chem. Biol., 2015, 11, 949–951 CrossRef CAS.
- A. Miyanaga, Nat. Prod. Rep., 2019, 36, 531–547 RSC.
- R. M. Van Wagoner, M. Satake and J. L. C. Wright, Nat. Prod. Rep., 2014, 31, 1101–1137 RSC.
- M. Ishibashi, Y. Ohizumi, M. Hamashima, H. Nakamura, Y. Hirata, T. Sasaki and J. Kobayashi, J. Chem. Soc., Chem. Commun., 1987, 1127–1129 RSC.
- J. Kobayashi and M. Tsuda, Nat. Prod. Rep., 2004, 21, 77–93 RSC.
- X. Wan, G. Yao, Y. Liu, J. Chen and H. Jiang, Mar. Drugs, 2019, 17, 594 CrossRef CAS.
- L. Xiang, J. A. Kalaitzis and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15609–15614 CrossRef CAS.
- J. H. Wisecaver and J. D. Hackett, Annu. Rev. Microbiol., 2011, 65, 369–387 CrossRef CAS.
- M. Tsuda, T. Kubota, Y. Sakuma and J. Kobayashi, Chem. Pharm. Bull. (Tokyo), 2001, 49, 1366–1367 CrossRef CAS.
- J. L. C. Wright, T. Hu, J. L. McLachlan, J. Needham and J. A. Walter, J. Am. Chem. Soc., 1996, 118, 8757–8758 CrossRef CAS.
- J. J. Ruprecht, M. S. King, T. Zögg, A. A. Aleksandrova, E. Pardon, P. G. Crichton, J. Steyaert and E. R. S. Kunji, Cell, 2019, 176, 435–447 CrossRef CAS.
- C. D. Fage, T. Lathouwers, M. Vanmeert, L.-J. Gao, K. Vrancken, E.-M. Lammens, A. N. M. Weir, R. Degroote, H. Cuppens, S. Kosol, T. J. Simpson, M. P. Crump, C. L. Willis, P. Herdewijn, E. Lescrinier, R. Lavigne, J. Anné and J. Masschelein, Angew. Chem., Int. Ed., 2020, 59, 10549–10556 CrossRef CAS.
- X. H. Chen, J. Vater, J. Piel, P. Franke, R. Scholz, K. Schneider, A. Koumoutsi, G. Hitzeroth, N. Grammel, A. W. Strittmatter, G. Gottschalk, R. D. Süssmuth and R. Borriss, J. Bacteriol., 2006, 188, 4024–4036 CrossRef CAS.
- R. A. Butcher, F. C. Schroeder, M. A. Fischbach, P. D. Straight, R. Kolter, C. T. Walsh and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1506–1509 CrossRef CAS.
- J. Moldenhauer, X. H. Chen, R. Borriss and J. Piel, Angew. Chem., Int. Ed., 2007, 46, 8195–8197 CrossRef CAS.
- S. Müller, S. Rachid, T. Hoffmann, F. Surup, C. Volz, N. Zaburannyi and R. Müller, Chem. Biol., 2014, 21, 855–865 CrossRef.
- R. Dehn, Y. Katsuyama, A. Weber, K. Gerth, R. Jansen, H. Steinmetz, G. Höfle, R. Müller and A. Kirschning, Angew. Chem., Int. Ed., 2011, 50, 3882–3887 CrossRef CAS.
- H. Sucipto, S. C. Wenzel and R. Müller, ChemBioChem, 2013, 14, 1581–1589 CrossRef CAS.
- Y. Mast, T. Weber, M. Gölz, R. Ort-Winklbauer, A. Gondran, W. Wohlleben and E. Schinko, Microb. Biotechnol., 2011, 4, 192–206 CrossRef CAS.
- R. Gurney and C. M. Thomas, Appl. Microbiol. Biotechnol., 2011, 90, 11–21 CrossRef CAS.
- L. Kjaerulff, R. Raju, F. Panter, U. Scheid, R. Garcia, J. Herrmann and R. Müller, Angew. Chem., Int. Ed., 2017, 56, 9614–9618 CrossRef CAS.
- Y. Zou, H. Yin, D. Kong, Z. Deng and S. Lin, ChemBioChem, 2013, 14, 679–683 CrossRef CAS.
- D. Fukuda, A. S. Haines, Z. Song, A. C. Murphy, J. Hothersall, E. R. Stephens, R. Gurney, R. J. Cox, J. Crosby, C. L. Willis, T. J. Simpson and C. M. Thomas, PLoS One, 2011, 6, e18031 CrossRef CAS.
- N. Pulsawat, S. Kitani and T. Nihira, Gene, 2007, 393, 31–42 CrossRef CAS.
- T. Wakimoto, Y. Egami, Y. Nakashima, Y. Wakimoto, T. Mori, T. Awakawa, T. Ito, H. Kenmoku, Y. Asakawa, J. Piel and I. Abe, Nat. Chem. Biol., 2014, 10, 648–655 CrossRef CAS.
- Ö. Erol, T. F. Schäberle, A. Schmitz, S. Rachid, C. Gurgui, M. El Omari, F. Lohr, S. Kehraus, J. Piel, R. Müller and G. M. Königd, ChemBioChem, 2010, 11, 1253–1265 CrossRef.
- K. Moon, J. Cui, E. Kim, E. S. Riandi, S. H. Park, W. S. Byun, Y. Kal, J. Y. Park, S. Hwang, D. Shin, J. Sun, K. B. Oh, S. Cha, J. Shin, S. K. Lee, Y. J. Yoon and D. C. Oh, Org. Lett., 2020, 22, 5358–5362 CrossRef CAS.
- K. Ishida, T. Lincke, S. Behnken and C. Hertweck, J. Am. Chem. Soc., 2010, 132, 13966–13968 CrossRef CAS.
- K. Ishida, T. Lincke and C. Hertweck, Angew. Chem., Int. Ed., 2012, 51, 5470–5474 CrossRef CAS.
- R. V. Grindberg, T. Ishoey, D. Brinza, E. Esquenazi, R. C. Coates, W. Liu, L. Gerwick, P. C. Dorrestein, P. Pevzner, R. Lasken and W. H. Gerwick, PLoS One, 2011, 6, e18565 CrossRef CAS.
- N. A. Moss, T. Leão, M. R. Rankin, T. M. McCullough, P. Qu, A. Korobeynikov, J. L. Smith, L. Gerwick and W. H. Gerwick, ACS Chem. Biol., 2018, 13, 3385–3395 CrossRef CAS.
- J. Yang, X. Zhu, M. Cao, C. Wang, C. Zhang, Z. Lu and F. Lu, J. Agric. Food Chem., 2016, 64, 8811–8820 CrossRef CAS.
- M. H. Medema, R. Kottmann, P. Yilmaz, M. Cummings, J. B. Biggins, K. Blin, I. De Bruijn, Y. H. Chooi, J. Claesen, R. C. Coates, P. Cruz-Morales, S. Duddela, S. Düsterhus, D. J. Edwards, D. P. Fewer, N. Garg, C. Geiger, J. P. Gomez-Escribano, A. Greule, M. Hadjithomas, A. S. Haines, E. J. N. Helfrich, M. L. Hillwig, K. Ishida, A. C. Jones, C. S. Jones, K. Jungmann, C. Kegler, H. U. Kim, P. Kötter, D. Krug, J. Masschelein, A. V. Melnik, S. M. Mantovani, E. A. Monroe, M. Moore, N. Moss, H. W. Nützmann, G. Pan, A. Pati, D. Petras, F. J. Reen, F. Rosconi, Z. Rui, Z. Tian, N. J. Tobias, Y. Tsunematsu, P. Wiemann, E. Wyckoff, X. Yan, G. Yim, F. Yu, Y. Xie, B. Aigle, A. K. Apel, C. J. Balibar, E. P. Balskus, F. Barona-Gómez, A. Bechthold, H. B. Bode, R. Borriss, S. F. Brady, A. A. Brakhage, P. Caffrey, Y. Q. Cheng, J. Clardy, R. J. Cox, R. De Mot, S. Donadio, M. S. Donia, W. A. Van Der Donk, P. C. Dorrestein, S. Doyle, A. J. M. Driessen, M. Ehling-Schulz, K. D. Entian, M. A. Fischbach, L. Gerwick, W. H. Gerwick, H. Gross, B. Gust, C. Hertweck, M. Höfte, S. E. Jensen, J. Ju, L. Katz, L. Kaysser, J. L. Klassen, N. P. Keller, J. Kormanec, O. P. Kuipers, T. Kuzuyama, N. C. Kyrpides, H. J. Kwon, S. Lautru, R. Lavigne, C. Y. Lee, B. Linquan, X. Liu, W. Liu, A. Luzhetskyy, T. Mahmud, Y. Mast, C. Méndez, M. Metsä-Ketelä, J. Micklefield, D. A. Mitchell, B. S. Moore, L. M. Moreira, R. Müller, B. A. Neilan, M. Nett, J. Nielsen, F. O'Gara, H. Oikawa, A. Osbourn, M. S. Osburne, B. Ostash, S. M. Payne, J. L. Pernodet, M. Petricek, J. Piel, O. Ploux, J. M. Raaijmakers, J. A. Salas, E. K. Schmitt, B. Scott, R. F. Seipke, B. Shen, D. H. Sherman, K. Sivonen, M. J. Smanski, M. Sosio, E. Stegmann, R. D. Süssmuth, K. Tahlan, C. M. Thomas, Y. Tang, A. W. Truman, M. Viaud, J. D. Walton, C. T. Walsh, T. Weber, G. P. Van Wezel, B. Wilkinson, J. M. Willey, W. Wohlleben, G. D. Wright, N. Ziemert, C. Zhang, S. B. Zotchev, R. Breitling, E. Takano and F. O. Glöckner, Nat. Chem. Biol., 2015, 11, 625–631 CrossRef CAS.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |