
Recent advances in HemN-like radical S-adenosyl-L-methionine enzyme-catalyzed reactions
Wen-Bing
Jin
 ,
Sheng
Wu
,
Yi-Fan
Xu
,
Hua
Yuan
* and
Gong-Li
Tang
*
,
Sheng
Wu
,
Yi-Fan
Xu
,
Hua
Yuan
* and
Gong-Li
Tang
*
State Key Laboratory of Bio-organic and Natural Products Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: huayuan@sioc.ac.cn; gltang@sioc.ac.cn
First published on 10th July 2019
Abstract
Covering: 2012 to 2019
HemN-like radical S-adenosyl-L-methionine (SAM) enzymes have been recently disclosed to catalyze diverse chemically challenging reactions from primary to secondary metabolic pathways. In this highlight, we summarize the reaction examples catalyzed by HemN-like enzymes to date and the enzymatic mechanisms reported. From the recent mechanistic investigations, we reason that there is a shared initiating mechanism wherein a characteristic SAM methylene radical is proposed to abstract a hydrogen atom from an sp3 carbon or add onto an sp2 carbon center although variations occur thereafter from reaction to reaction, as well as providing a brief insight into some future prospects.
 Wen-Bing Jin | Wen-Bing Jin, born in Chongqing, China, obtained his B.S. degree in Polymer Material and Engineering from Sun Yat-Sen University in 2014. He is currently pursuing his PhD studies at the Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Science (CAS), under the supervision of Prof. Gong-Li Tang. His research focuses on the biosynthesis of natural products and the enzymatic reactions and mechanism therein. |
 Sheng Wu | Sheng Wu, born in Jiangsu, China, obtained his B.S. degree in pharmacy from Central South University in 2009. He obtained his PhD degree in 2015 from Shanghai Institute of Organic Chemistry, Chinese Academy of Science, under the supervision of Prof. Gong-Li Tang. His research interests include the mechanisms of enzyme-mediated catalytic reactions, especially metalloproteins-mediated enzymology. |
 Yi-Fan Xu | Yi-Fan Xu, born in Suzhou, Jiangsu, China, obtained his B.S. degree in Kuang Yaming honors school in Nanjing University in 2017. He is currently pursuing his PhD studies at the Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Science (CAS), under the supervision of Prof. Gong-Li Tang. His research focuses on the biosynthesis of natural products and the enzymatic reactions and mechanism therein. |
 Hua Yuan | Hua Yuan was born in Linyi, China and obtained his B.S. degree in Biological Sciences from Weifang University in 2005. He studied for his M.S. degree under Professor Buchang Zhang of Anhui University, investigating erythromycin combinatorial biosynthesis (2008). He then studied for his Ph.D. under Professor Guoping Zhao of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (CAS), investigating rifamycin biosynthesis and regulation (2012). Subsequently, he joined Professor Gong-Li Tang's group in Shanghai Institute of Organic Chemistry, CAS, and has focused on the antibiotic biosynthesis, resistance and regulation. |
 Gong-Li Tang | Gong-Li Tang was born in Longkou, Shandong Province, China. He obtained his B.S. degree in Environmental Sciences from Nankai University in 1994 and Ph.D. degree in Bio-organic Chemistry under Prof. Hai-Bao Chen from Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences in 1999. Subsequently, he joined Prof. Ben Shen's group as a Postdoc. in Department of Chemistry, University of California-Davis (2001) and a research associate in School of Pharmacy, University of Wisconsin-Madison (2003). He joined the faculty team of SIOC from September 2003. He is currently a research professor of State Key Laboratory of Bio-Organic and Natural Products Chemistry. His research interests focus on Natural Products Biosynthesis. |
1. Introduction
In 2001, Sofia et al. firstly defined a radical S-adenosyl-L-methionine (SAM) domain-containing protein superfamily based upon the sensitive bioinformatics analysis of 645 sequences from the NCBI non-redundant protein database.1 These proteins contain a highly conserved cysteine-rich motif, CxxxCxxC, which coordinates a [4Fe–4S] cluster.1 This [4Fe–4S] cluster has a unique Fe that anchors SAM by a classical N/O chelate.2 Once reduced into its +1 form, the resultant [4Fe–4S]+ cluster can trigger the reductive cleavage of SAM to yield a highly reactive 5′-deoxyadenosyl radical (5′-dA·), which will initiate a variety of radical reactions (Scheme 1).2 The diverse reactions catalyzed by radical SAM proteins are found in numerous primary and secondary metabolic pathways, such as glycyl radical enzyme activation,3 rearrangement,4 oxidative decarboxylation,5 C–S bond formation,6 thioether bond formation,7 unusual methylations,8 deoxygenation9 and ring contraction.10 For more comprehensive information of the radical SAM proteins, readers can be directed to several other excellent reviews.11–15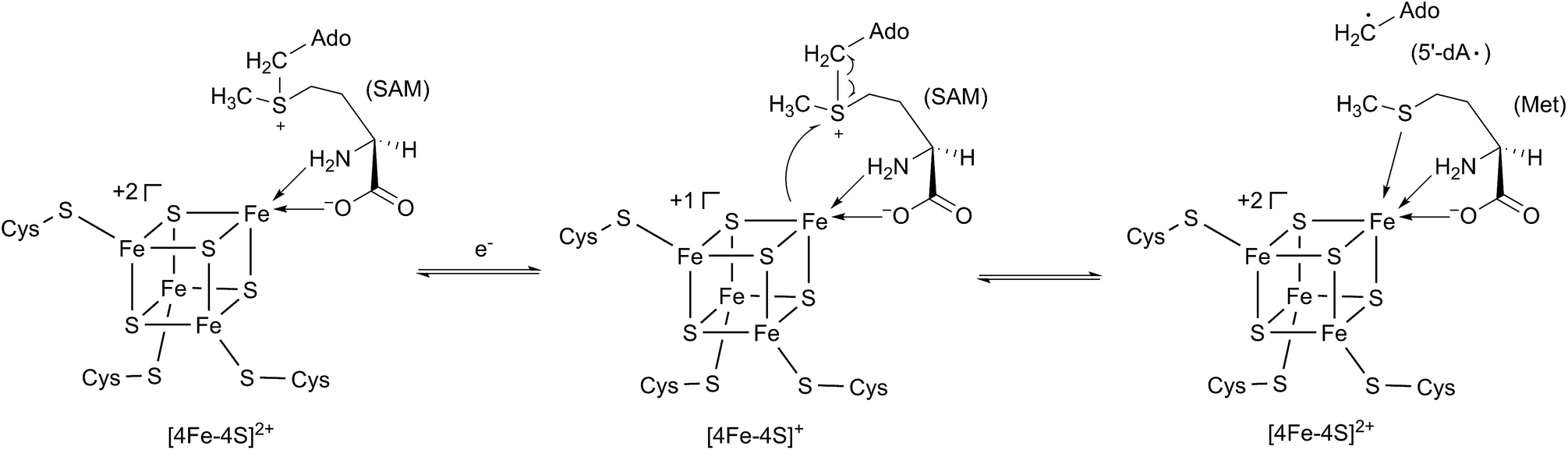 | ||
| Scheme 1 Canonical formation of the 5′-dA· in the radical SAM protein superfamily. | ||
One member of the radical SAM superfamily, HemN, was initially genetically identified as an oxygen-independent coproporphyrinogen III oxidase during the anaerobic biosynthesis of the essential cofactor heme in several bacterial species.16–18 In 2002, the HemN protein from Escherichia coli was overexpressed, anaerobically purified, and characterized to catalyze two consecutive oxidative decarboxylations of the propionate side chains of rings A and B of coproporphyrinogen III to produce protoporphyrinogen IX in vitro (Scheme 2A).19 Structural elucidation of E. coli HemN revealed that HemN harbors two SAM molecules, with SAM1 coordinating to the unique Fe ion of the [4Fe–4S] cluster through its amino nitrogen and carboxylate oxygen and SAM2 located adjacent to SAM1.20 Subsequent investigation of the enzymatic mechanism suggested a radical process wherein 5′-dA· generated from SAM1 abstracts the pro-S-hydrogen atom on the β-position of the propionate side chains and triggers the decarboxylation reaction (this proposed mechanism is revised in 2019, see below),21,22 whereas the role of SAM2 was unsolved (Scheme 2B).
 | ||
| Scheme 2 HemN-catalyzed oxidative decarboxylation in heme biosynthesis. (A) HemN catalyzes two consecutive oxidative decarboxylations of coproporphyrinogen III to from protoporphyrinogen IX.19 (B) Proposed mechanism of the radical-based oxidative decarboxylation,21,22 which is revised in 2019 (see below).69 | ||
During the genetic characterization of Bacillus subtilis hemN, a second hemN-like gene hemZ was identified, which has the same physiological function as hemN.23 Later from 2009 to 2012, a number of hemN-like genes were also found in natural product biosynthetic gene clusters (BGCs). Many of these HemN-like proteins were proposed to be responsible for unusual methylations, such as NosN in nosiheptide biosynthesis,24 NocN in nocathiacin I biosynthesis,25 TpdI, TpdL and TpdU in GE2270 and thiomuracin biosynthesis,26 and Blm-Orf8, Tlm-Orf11 and Zbm-Orf26 in bleomycin family antibiotic biosynthesis;27–29 however, the functions of these enzymes were not confirmed in vitro. Since 2012, there are increasing reports of HemN-like enzymes in regard to enzymatic reactions and mechanistic investigations, and the mechanisms of these enzymes have gradually become clearer. Moreover, a quick analysis of HemN and HemN-like sequences in the InterPro database (IPR034505) in April, 2019 revealed the number of proteins is up to 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 639, which originate from prokaryotes to eukaryotes as well as suggesting evolutionarily important functions in primary and secondary metabolism. Therefore, we believe that it is a good time to summarize the reaction examples catalyzed by HemN-like enzymes and discuss the mechanisms reported in the past 8 years (2012–2019) that illustrate the diversity of radical-based chemical reactions and a shared initiating mechanism found in both primary and secondary metabolic pathways. In this Highlight, we refer to HemN as the prototype HemN and its orthologues that exclusively have the oxygen-independent coproporphyrinogen III oxidase activity, and its other homologous proteins (i.e. paralogues) as HemN-like enzymes.
639, which originate from prokaryotes to eukaryotes as well as suggesting evolutionarily important functions in primary and secondary metabolism. Therefore, we believe that it is a good time to summarize the reaction examples catalyzed by HemN-like enzymes and discuss the mechanisms reported in the past 8 years (2012–2019) that illustrate the diversity of radical-based chemical reactions and a shared initiating mechanism found in both primary and secondary metabolic pathways. In this Highlight, we refer to HemN as the prototype HemN and its orthologues that exclusively have the oxygen-independent coproporphyrinogen III oxidase activity, and its other homologous proteins (i.e. paralogues) as HemN-like enzymes.
2. Cyclopropanation
2.1 YtkT in yatakemycin biosynthesis
In 2012, YtkT became the first member of HemN-like enzymes characterized in vitro, which was essential for the formation of the cyclopropyl moiety in the biosynthesis of the natural product yatakemycin (YTM).30 YTM, isolated from the culture broth of Streptomyces sp. TPA0356, is a novel antitumor antibiotic of the spirocyclopropylcyclohexadienone natural products with potent DNA alkylating activities due to the highly reactive cyclopropyl moiety.31–33Huang et al. first identified the YTM BGC by genome scanning of Streptomyces sp. TPA0356 and then showed that YtkT was necessary for the chemically challenging cyclopropanation reaction in YTM biosynthesis via gene inactivation and complementation experiments.30 The ΔytkT mutant strain abolished the production of YTM but accumulated the biosynthetic intermediate YTM-T lacking the cyclopropyl structure. Subsequently, they purified and reconstructed the YtkT protein to obtain a competent catalytic [4Fe–4S] cluster for enzymatic assays under strictly anaerobic conditions. The biochemical characterization showed that YtkT was unable to transform the substrate YTM-T to YTM, but led to the production of methylated YTM-T (Scheme 3A). The enzymatic mechanism therein was not investigated due to the very low catalytic efficiency, and they proposed the formation of the cyclopropyl moiety of YTM might be assisted by another enzyme(s) encoded by the YTM BGC.
 | ||
| Scheme 3 HemN-like enzyme-catalyzed cyclopropanation reactions. (A) Detected methylation reaction and proposed cyclopropanation reaction of YtkT in YTM biosynthesis.30 (B) Theoretically proposed mechanism of Jaw5-mediated cyclopropanation in jawsamycin biosynthesis.34 (C) Proposed mechanism of the cyclopropanation process catalyzed by C10P and C10Q in CC-1065 biosynthesis.39 The SAM-substrate adduct detected by high resolution mass spectrometry is boxed, indicating the involvement of a SAM methylene radical. | ||
2.2 Jaw5 in jawsamycin biosynthesis
In 2014, Hiratsuka et al. in vivo proved that the HemN-like enzyme Jaw5 was responsible for the construction of the polycyclopropanated backbone in jawsamycin (FR-900848) biosynthesis.34 Jawsamycin, produced by Streptoverticillium fervens HP-891, is a polyketide-nucleoside hybrid with polycyclopropane moieties on a single polyketide chain, which is a potent antifungal agent for a variety of phytopathogenic fungi.35 All the cyclopropane moieties in jawsamycin have the same stereochemistry, and all these methylene groups were confirmed to be derived from L-methionine by isotope feeding experiments.36,37Hiratsuka et al. identified the jawsamycin BGC by its heterologous expression in Streptomyces lividans TK23.34 They showed that in the Δjaw5 mutant strain the production of jawsamycin was abolished and no intermediate was accumulated, and that the complementation of jaw5 into the Δjaw5 mutant recovered the production of jawsamycin. Moreover, when the recombinant S. lividans TK23 strain containing the jaw456 genes (Jaw4 is an iterative polyketide synthase (PKS) containing ketosynthase–acetyltransferase–dehydratase–acyl carrier protein domains, and Jaw6 is a ketoreductase) was fed with the nucleoside precursor, the production of jawsamycin was detected, suggesting that the HemN-like enzyme Jaw5 can collaborate with the iterative PKS system (Jaw4 and Jaw6) to construct the unique polycyclopropanated backbone in a stepwise manner, with the Jaw5-catalyzed cyclopropanation occurring during polyketide extension.
Based on the above in vivo results, they put forward two alternative mechanisms (pathways a and b) for the Jaw5-catalyzed cyclopropanation reaction (Scheme 3B).34 In both mechanisms the role of the second SAM (SAM2) was suggested. In pathway a, the 5′-dA· produced from the reductive cleavage of SAM1 abstracts a hydrogen atom from SAM2 to form a SAM methylene radical, which undergoes addition to the α,β-unsaturated polyketide chain in a radical manner. In pathway b, the SAM methylene radical is first transformed to its SAM ylide by acquiring an electron and then adds to substrate. Subsequently, the cyclopropyl moiety is formed with SAH as the leaving group. This is the first time for the theoretical proposal of a SAM methylene radical involved in the HemN-like enzyme-catalyzed reaction.
Later in 2017, Hiratsuka et al. characterized several cyclopropane deficient jawsamycin analogs from the heterologous host S. lividans TK23 containing the jaw gene cluster, which suggested that a balance between the reaction rates of α,β-unsaturated polyketide chain extension and cyclopropanation may be critical for the construction of the polycyclopropanated backbone, and further confirmed the stepwise mechanism of the cyclopropanation.38
2.3 C10P in CC-1065 biosynthesis
In 2018, the HemN-like enzyme C10P, together with an O-methyltransferase C10Q, was reported to be capable of catalyzing the cyclopropanation reaction in the biosynthesis of the natural product CC-1065.39 As with YTM, CC-1065, produced by Streptomyces zelensis NRRL 11183, is also a potent antitumor antibiotic belonging to the spirocyclopropylcyclohexadienone natural products.40–42Wu et al. first cloned and identified the CC-1065 BGC by degenerate PCR-based screen of YtkT homologues from the S. zelensis NRRL 11183 cosmid library, and then they proposed the biosynthetic pathway of CC-1065 based on gene deletion and intermediate compound characterization.43 Previous isotope feeding experiments indicated that the methylene group of the cyclopropyl moiety in CC-1065 is derived from SAM.44 Just as YtkT in YTM biosynthesis, Jin et al. isolated an equivalent biosynthetic intermediate CC-1065-P from the Δc10P mutant strain, which is also a cyclopropane deficient compound compared to CC-1065.39 Interestingly, this CC-1065-P compound was also accumulated in the Δc10Q mutant strain. Subsequent enzymatic assays confirmed that C10P and C10Q function together to catalyze the cyclopropanation reaction, accompanied by the production of a shunt product Me-CC-1065-P. In addition, they found that 5′-dA and SAH were generated approximately in the stoichiometry of 1:1, which suggests that one of the two SAM molecules is first transformed to the 5′-dA· and then to 5′-dA, and the other SAM is converted to SAH. Fortunately, they detected a key reaction intermediate, SAM-substrate adduct, by high resolution mass spectral analysis of the reaction system, and proposed that this SAM-substrate adduct is likely to be further transformed into the cyclopropane-containing product CC-1065 and the methylated product Me-CC-1065-P, respectively. When CD3-SAM instead of SAM was utilized for mechanistic investigations, the produced 5′-dA showed a mass shift of +1, and the SAM-substrate adduct showed a mass increment of +2. Meanwhile, the produced CC-1065 and Me-CC-1065-P also showed a mass increment of +2, supporting the proposal that both products are derived from the SAM-substrate adduct.
On the basis of these results they proposed that C10P-mediated 5′-dA· generated from SAM1 abstracts a hydrogen atom from the methyl group of SAM2 to yield a SAM methylene radical, which then undergoes radical addition onto an sp2 carbon center of CC-1065-P (Scheme 3C). This is the first time that the existence of the SAM methylene radical is confirmed through the detection of an SAM-substrate adduct. Consistent with this proposal, site-mutagenesis studies of the other enzyme C10Q suggested that the transformation from the SAM-substrate adduct to CC-1065 is catalyzed by C10Q via an intramolecular SN2 reaction with SAH as a leaving group, whereas the shunt product Me-CC-1065-P is formed spontaneously through an intermediate compound containing an exocyclic methylene group, which was also supported by the enzymatic assays in D2O instead of H2O.
Furthermore, they noticed a silent BGC from Shewanella woodyi ATCC 51908 with high homology and synergy with the CC-1065 BGC. Because of the low solubility and purity for the C10P protein, they tried to substitute the equivalent protein Swoo_2002 for C10P and successfully demonstrated that Swoo_2002 and C10Q were also able to catalyze the cyclopropanation of CC-1065-P to produce CC-1065 and Me-CC-1065-P.
3. Ring-opening methylenation
3.1 ChuW in heme degradation
Iron acquisition is essential to all cellular organisms, especially for a pathogenic microorganism during colonization of a mammalian host. For example, several pathogenic bacterial species have evolved a heme utilization operon for iron acquisition.45,46 This operon encodes enzymes for heme uptake, transport and degradation, including an uncharacterized HemN-like enzyme, such as ChuW in Escherichia coli O157:H7 and HutW in Vibrio cholera.In 2016, LaMattina et al. successfully characterized the ChuW-mediated oxygen-independent heme degradation in vitro.47 They first purified and reconstructed ChuW to generate its intact [4Fe–4S] cluster, and then showed that ChuW was able to catalyze heme degradation to form a tetrapyrrole product anaerobilin with the liberation of iron under anaerobic conditions (Scheme 4). When 13CH3-SAM instead of unlabeled SAM was used in the enzymatic assays, the produced anaerobilin showed a mass shift of +1, which indicated that ChuW mediates the transfer of the methyl group from SAM to heme to produce anaerobilin. Furthermore, when CD3-SAM instead of SAM was utilized in the reaction system, the produced anaerobilin showed a mass shift of +2, suggesting that one hydrogen atom of the methyl group from SAM is abstracted during the reaction and the rest two hydrogen atoms are incorporated into the product anaerobilin. Meanwhile, in the CD3-SAM based assays, the isolated product 5′-dA showed a mass increment of +1, implying that the SAM1-derived 5′-dA· abstracts a hydrogen atom from the methyl group of SAM2 to yield a SAM methylene radical, and that the latter radical adds onto the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the substrate heme to trigger the following ring-opening of the porphyrin ring to form anaerobilin with the release of iron (Scheme 4). This proposed process was further substantiated by enzymatic assays in D2O, which revealed that a hydrogen atom from the solvent is incorporated into anaerobilin during the degradation. These isotope labeling experiments actually became the first line of evidence to support the existence of a SAM methylene radical in HemN-like enzyme-catalyzed reactions proposed by Hiratsuka et al. in 2014.34
C double bond of the substrate heme to trigger the following ring-opening of the porphyrin ring to form anaerobilin with the release of iron (Scheme 4). This proposed process was further substantiated by enzymatic assays in D2O, which revealed that a hydrogen atom from the solvent is incorporated into anaerobilin during the degradation. These isotope labeling experiments actually became the first line of evidence to support the existence of a SAM methylene radical in HemN-like enzyme-catalyzed reactions proposed by Hiratsuka et al. in 2014.34
 | ||
| Scheme 4 Proposed mechanism of ChuW-mediated anaerobic heme degradation.47 | ||
4. Methylation
4.1 MenK/MenK2 in methylmenaquinone biosynthesis
In 2017, Hein et al. reported that the HemN-like enzyme MenK can catalyze the methylation reaction in the biosynthesis of methylmenaquinone (MMK) from the substrate menaquinone (MK).48 Isoprenoid quinones are the ubiquitous constituents of bacterial and archaeal membranes, which are the typical redox mediators that connect the oxidoreductases in respiratory electron transport chains.49,50 MK and its methylated form MMK are among the most important isoprenoid quinones, serving as mediators in low-potential electron transport chains involved in anaerobic respiration.51MMK-producing bacteria and archaea harbor either menaquinone (Men) pathway or futalosine (Mqn) pathway.52 Hein et al. demonstrated that the inactivation of the mqnK gene (encoding a HemN-like enzyme) of the Mqn pathway in Wolinella succinogenes abolished the production of 8-MMK6 but accumulated MK6.48 Moreover, when mqnK was complemented back into the ΔmqnK mutant strain, the production of 8-MMK6 was restored, indicating that MqnK is responsible for the methylation of MK6 to yield 8-MMK6. Then they expressed and reconstructed the MqnK homologue MenK of the Men pathway from the human gut bacterium Adlercreutzia equolifaciens, and showed that MenK was capable of catalyzing the methylation of MK6 on the C8 position to give 8-MMK6 (Scheme 5A). Based on these in vivo results, they put forward a mechanism. Once induced by [4Fe–4S]+, SAM1 undergoes reductive cleavage to yield 5′-dA·, and the methyl group of SAM2 is first transferred onto an unknown amino acid residue as a methyl carrier. 5′-dA· then abstracts a hydrogen atom of this methyl group to form a methylene radical linked to the unknown carrier, which is possibly transferred onto MK6 through radical addition.
 | ||
| Scheme 5 HemN-like enzyme-catalyzed methylation reactions. (A) MenK and MenK2-catalyzed methylations and the proposed mechanism in methylmenaquinone biosynthesis.48,53 (B) TbtI-mediated methylation and the proposed mechanism in thiomuracin biosynthesis.54,57 (C) Proposed mechanism of the NosN-catalyzed reaction in the biosynthesis of nosiheptide.64–68 The SAM-substrate adducts detected by mass spectrometry are boxed. | ||
Later in 2018, Hein et al. found that another HemN-like enzyme MenK2 from A. equolifaciens showed the ability to catalyze the methylation of MK6 on the position of C7 to produce 7-MMK6.53 Specifically, when the mqnK inactivated W. succinogenes strain was complemented with the menK2 gene, 7-MMK6 but not 8-MMK6 was produced. Furthermore, when the menK2 gene was introduced into the wild type W. succinogenes strain, 7-MMK6, 8-MMK6, and a dimethylated product 7,8-DMMK6 were produced, suggesting an in vivo cooperation of MqnK and MenK2.53
4.2 TbtI in thiomuracin biosynthesis
In 2017, Mahanta et al. proved that the HemN-like enzyme TbtI is responsible for catalyzing the methylation of a thiazole moiety in thiomuracin biosynthesis.54 Thiomuracin is a ribosomally synthesized and post-translationally modified peptide antibiotic with potent activity against Gram-positive drug-resistant bacteria. In 2015, reconstruction of the thiomuracin biosynthetic pathway in vitro revealed that the precursor peptide (TbtA) first undergoes cyclodehydration of the Cys residues and then dehydrogenation to yield the leader peptide (LP)-linked TbtA hexazole core intermediate, followed by [4 + 2] cycloaddition mediated ring closure and a series of other modifications to give thiomuracin and its analogues.55,56In the report of early 2017, Mahanta et al. demonstrated that after removal of the leader peptide by the endoproteinase GluC, the resultant TbtA hexazole core acts as the substrate of TbtI, which catalyzes the methylation of the second thiazole moiety to generate the methylated TbtA hexazole core (Scheme 5B).54 Later in 2017, Zhang et al. carried out detailed mechanistic investigations of the TbtI-catalyzed methylation reaction.57 In the reaction system, 5′-dA and SAH were found to be produced at a similar rate, implying that the two SAM molecules of the HemN-like enzyme are consumed during one catalytic cycle, with 5′-dA derived from SAM1 through the 5′-dA· intermediate and SAH derived from SAM2. When CD3-SAM instead of SAM was utilized in the enzymatic assays in the H2O solvent, the produced 5′-dA was detected to contain a single deuterium, suggesting that the SAM1-generated 5′-dA· may abstract a hydrogen atom from the methyl group of SAM2 during catalysis, accompanied by the production of a SAM methylene radical. Moreover, the product methylated TbtA hexazole core showed a mass increment of +2 in the assays using CD3-SAM in H2O, indicating that the remaining two deuterium atoms in the SAM methylene radical are transferred onto the substrate probably through radical addition towards the CC double bond on the thiazole ring. Furthermore, when the enzymatic assays were conducted using CD3-SAM in D2O, the majority of products showed a mass increment of +2, with the minority showing a mass shift of +3; however, when the β-deuterated TbtA hexazole core and CD3-SAM were utilized in D2O, the majority of products showed a mass increment of +3, indicating that the hydrogen on the β-position of the thiazole moiety appears to be transferred onto the methyl group of the product, and that solvent exchange occurs in the turnover (Scheme 5B). This is another instance that the existence of a SAM methylene radical in catalysis is supported by isotope labeling studies and that this radical undergoes addition towards the CC double bond of substrates.
4.3 NosN in nosiheptide biosynthesis
Nosiheptide (NOS), produced by Streptomyces actuosus ATCC 25421, is one of the oldest known thiopeptide antibiotics with potent activity against various bacterial pathogens, and consists of a characteristic macrocyclic core and an indolic acid ring system.58,59 In 2009, Yu et al. cloned, sequenced, and characterized the nos gene cluster from S. actuosus ATCC 25421, and they identified an NOS analogue with the methylene group deficient indole side ring-opened structure from the ΔnosN mutant strain. Thus, they proposed that the HemN-like enzyme NosN might first transfer a methyl group onto the indole side ring, which is then hydroxylated to generate a hydroxymethyl group, followed by the formation of the ester linkage.24 Consistent with this proposal, in 2017, Ding et al. showed that NosN recognized a substrate analogue, the N-acetylcysteamine (SNAC) thioester of 3-methyl-2-indolic acid (MIA) (MIA-SNAC), and catalyzed the methylation of MIA-SNAC to produce 3,4-dimethylindolic acid-SNAC (DMIA-SNAC), accompanied by the production of 5′-dA, 5′-methylthioadenosine (MTA) and 5′-thioadenosine (instead of SAH).60 In the reaction system, when CD3-SAM instead of SAM was used, the generated methylated product showed a mass shift of +2, leading to their proposal that the SAM2 molecule of NosN is first converted into MTA to act as the direct methyl donor, which then undergoes hydrogen abstraction by the 5′-dA· generated from SAM1 to form an MTA methylene radical, and that this MTA methylene radical adds onto the substrate to produce the methylated product DMIA-SNAC through the exocyclic methylene-containing intermediate. This proposed mechanism was further supported by the detection of several shunt products with the structure of nucleoside linked to the substrate MIA-SNAC and the mechanistic investigations utilizing an allyl analogue of SAM in the assay.61,62However, in the same year of early 2017, determination of the roles of NosI, NosJ, and NosK led Badding et al. to speculate that NosN catalyzes the production of a methylene group-containing intermediate, followed by the formation of the ester linkage of the side-ring system.63 In accordance with this proposal, in late 2017, LaMattina et al. reported that NosN did not catalyze the methylation reaction but catalyzed the production of a methylene group-containing intermediate.64 In their assays with NosN, they found that the substrate MIA-SNAC was transformed into 4-methylene-3-methyl-indolic acid-SNAC (MMIA-SNAC), which could be derived into 4-(hydroxymethyl)-3-methyl-2-indolic acid (4OHMMIA) when treated with NaOH. In addition, they also noticed that 5′-dA and SAH were produced in nearly 1:1 stoichiometry and that no MTA was detected, indicating that SAM, rather than MTA, serves as the methyl donor in the NosN-mediated reaction. In subsequent CD3-SAM assays, the produced 5′-dA showed a mass shift of +1 and MMIA-SNAC showed a mass increment of +2, which is consistent with the proposal, as in other HemN-like enzyme-catalyzed reactions, that the SAM1-derived 5′-dA· abstracts the hydrogen from the methyl group of SAM2 to generate a SAM methylene radical, which undergoes radical addition towards the CC double bond on the substrate MIA-SNAC, followed by the release of SAH and the formation of MMIA-SNAC (Scheme 5C). Therefore, DMIA-SNAC is actually a shunt product derived from MMIA-SNAC. When a synthesized peptide mimic containing the residues Glu6 and Cys8-linked MIA was used as substrate, NosN catalyzed the methylene transfer reaction, and the formed exocyclic methylene was trapped by Glu6 to generate the ester linkage in the indole side ring system, which was also supported by isotope labeling experiments (Scheme 5C).65 This reaction cascade of the methylene transfer followed by the ester linkage formation is further supported by the two reports by Qiu et al., wherein NosN was showed to be capable of catalyzing the formation of the indole side ring system when the MIA-S-Cys8 peptide intermediate was used as substrate.66,67 More recently, in a reaction system including NosN and a peptide mimic containing the residues Glu6 and Cys8-linked MIA, Wang et al. detected the existence of two SAM-substrate adducts by mass spectrometry, which were likely derived from the proposed aryl radical intermediate generated by the radical addition of a SAM methylene radical towards the substrate (Scheme 5C). These findings verified the proposed mechanism and also supported the existence of a SAM methylene radical.68
5. Oxidative decarboxylation
5.1 Revised mechanism for the HemN-catalyzed reaction in heme biosynthesis
As mentioned above, the prototype protein HemN is well known to catalyze the oxidative decarboxylation of the propionate moieties in coproporphyrinogen III to yield the vinyl groups in protoporphyrinogen IX in the biosynthesis of heme, a ubiquitous cofactor in many fundamental biological processes. The crystal structure of E. coli HemN was determined to contain two SAM molecules, with SAM1 coordinating to the [4Fe–4S] cluster and SAM2 located adjacent to SAM1. Previous mechanistic studies suggested that the hydrogen atom on the β-position of the propionate side chain is abstracted and that the formation of two vinyl groups consumes two SAMs,20–22 leading to the proposed mechanism shown in Scheme 2B, wherein the role of SAM2 remains unclear.However, Ji et al. recently reported the revision of the above mechanism of the HemN-catalyzed oxidative decarboxylation reaction.69 In their assays, when CD3-SAM instead of unlabeled SAM was utilized, the produced 5′-dA showed a mass increment of +1, suggesting that the 5′-dA· does not abstract the hydrogen atom on the β-position of the propionate side chain, but abstracts the hydrogen from the methyl group of SAM2, generating 5′-dA with one deuterium atom and a SAM methylene radical containing the rest two deuterium atoms. This methylene radical is then likely to abstract the β-hydrogen atom of the propionate moieties of substrate and induce the following oxidative decarboxylation to form the vinyl groups with the release of CO2 (Scheme 6). Furthermore, a SAM-product adduct was detected by high resolution mass spectrometry in the enzymatic assays, which was proposed to be a shunt product as a result of the SAM methylene radical adding to the vinyl group of the product. In the CD3-SAM assays, the shunt product was found to show a mass change of +2, which is a further proof of the proposal that the SAM methylene radical also exists in the HemN-catalyzed oxidative decarboxylation. Therefore, the previously proposed mechanism is revised herein, and the role of the second SAM molecule during the HemN-mediated reaction is elucidated.
 | ||
| Scheme 6 Revised enzymatic mechanism of HemN-catalyzed oxidative decarboxylation.69 The SAM-product adduct detected by high resolution mass spectrometry is boxed. | ||
6. Other reactions
6.1 HemW as a heme chaperone in heme transfer
HemW is a HemN-like enzyme widely distributed in prokaryotes and eukaryotes. For example, the Lactococcus lactis HemW protein shares 28% identity with HemN of E. coli. L. lactis is capable of respiratory growth when protoporphyrin IX is supplied, but it is devoid of a heme biosynthetic pathway, indicating that HemW probably do not function as a coproporphyrinogen III oxidase.70,71 In 2012, Abicht et al. investigated the function of L. lactis HemW, and proved that HemW is not responsible for catalyzing the oxidative decarboxylation reaction. Instead, it is able to covalently bind and transfer heme to its membrane acceptor, acting as a heme chaperone.72Later in 2018, Haskamp et al. reported that E. coli HemW was capable of binding heme and catalyzing the transfer of heme to the membrane subunit NarI of the respiratory nitrate reductase NarGHI in vitro,73 wherein HemW was confirmed to harbor a [4Fe–4S] cluster and two SAM molecules and even have the SAM cleavage ability. However, the exact reaction catalyzed by HemW for covalent heme binding remains unknown. Considering the existence of an intact [4Fe–4S] cluster, the binding process may involve a radical-based chemistry. Interestingly, the binding activity of HemW could be enhanced by addition of NADH, making the mechanism of the heme binding and transfer more cryptic. Furthermore, the human HemW homologue RSAD1 from Homo sapiens (showing 30% sequence identity with L. lactis HemW) was also found to bind heme tightly, suggesting the universe significance of the HemW/RSAD1 enzyme family in binding and transferring heme into the cellular proteins.
7. Conclusions and outlook
In 2012, Zhang et al. grouped the identified radical SAM methyltransferases (RSMTs) into three classes (A, B, and C) based on sequence analyses.12 HemN-like enzymes were then classified as class C RSMTs because all the reported members at that time were proposed to catalyze methylation. However, as more diverse chemical reactions are discovered, we suggest that class C RSMTs should be referred to as “HemN-like enzymes” in the future. In this highlight, we summarized all the reaction examples catalyzed by HemN-like enzymes to date and also discussed the recent enzymatic mechanisms reported. Interestingly, from the mechanistic investigations we believe that there is a shared initiating mechanism, that is, once reduced, the [4Fe–4S] cluster triggers the reductive cleavage of SAM1 of HemN and HemN-like enzymes to generate 5′-dA·, which then abstracts a hydrogen from the methyl group of SAM2 to give a key SAM methylene radical, along with a coproduct 5′-dA. The SAM methylene radical can then undergo hydrogen abstraction of an sp3 carbon center or direct radical addition towards an sp2 carbon center, thereby initiating a variety of radical reactions (Scheme 7C). | ||
| Scheme 7 Overall comparison of mechanistic proposals of HemN-like enzymes and class A and class B RSMTs. SAM1 is the SAM molecule that is anchored by the [4Fe–4S] cluster as in all the radical SAM enzymes. For HemN and HemN-like enzymes, SAM2 is the second SAM molecule in the active site adjacent to SAM1, whereas, for classes A and B RSMTs, how SAM2 interacts with the enzyme and how its methyl group is transferred to the enzyme/Cbl are still unknown. The domain structures of different classes of RSMTs are exhibited (radical SAM: the radical SAM core domain; cobalamin BD: the cobalamin binding domain; HemN_C: the C-terminal domain of HemN and HemN-like proteins). | ||
When functioning as a methylase, HemN-like enzymes are distinct from both class A and class B (recently renamed as cobalamin (Cbl)-dependent radical SAM enzymes14) RSMTs although they all consume two SAM molecules within one turnover, with one SAM sequentially generating 5′-dA· and 5′-dA and the other SAM providing the methyl group and producing SAH. For class A RSMTs, they contain two conserved cysteine (Cys) residues with one Cys residue serving as the methyl carrier and the other one as a relay for proton from substrate to assist the methylation (Scheme 7A), while for class B RSMTs, the methyl carrier in Cbl-dependent radical SAM enzymes is Cbl (Scheme 7B). For HemN-like enzymes, because they can simultaneously bind two SAM molecules in the active site, a radical relay occurs, with 5′-dA· triggering the production of the SAM2-based SAM methylene radical, which can directly add to an sp2 carbon center of substrate to form a methylene/methyl group in product.
The structural analysis of E. coli HemN has revealed a clear interaction network of the [4Fe–4S] cluster, SAM1 and SAM2,20 and mechanistic investigations of this HemN enzyme and several other HemN-like enzymes have suggested that they share the same initiating mechanism. However, the structures of complete complexes (substrate included) are required to provide an insight into how the electron and hydrogen are transferred between SAM1, SAM2 and the substrate.
The mechanism of the chaperone HemW-mediated heme transfer is currently not clear,73 however, considering that heme binding and transfer requires an intact [4Fe–4S] cluster and that HemW has the capacity of SAM cleavage, we believe that a radical process induced by the [4Fe–4S] cluster might underlie the covalent binding and transfer. It will be interesting to unravel whether a radical species is involved and what kind of the radical species is, and this related finding will either indicate the same initiating mechanism with HemN and HemN-like enzymes or supply an alternative mechanism.
Sequence analyses of the 48639 HemN and HemN-like proteins from the InterPro database (IPR034505) in April, 2019 revealed that there were 12137 HemN sequences (IPR004558), 3116 HemZ sequences (IPR023995), 1255 ChuW/HutW sequences (IPR026332) and 26356 HemW sequences (IPR004559) that should function in the heme-related primary metabolism from prokaryotes to eukaryotes. The remaining 5775 proteins are proposed to be involved in the secondary metabolic pathways. All the sequences show great diversity as revealed from a sequence similarity network analysis (Fig. 1), which also indicates that more diverse chemical reactions remain to be disclosed. Moreover, although HemN and HemN-like enzymes share the same process of radical initiation and radical transfer to produce the SAM methylene radical species in mechanistic aspect, their functions and capacities are very diverse and may be tailored to a specific substrate or reaction because of the versatility of the SAM methylene radical, thereby implying many possibilities of this class of enzymes.
 | ||
| Fig. 1 Sequence similarity network (SSN) analysis of HemN and HemN-like proteins. The 48639 sequences (IPR034505) in April, 2019 were first clustered to 6433 representative sequences at 60% identity, which were then used to construct SSN. The proteins mentioned in the text include C10P, Jaw5, Swoo_2002 and YtkT involved in cyclopropanation; ChuW and HutW involved in heme degradation; Blm-orf8, MenK, MenK2, MqnK, NocN, NosN, TbtI, TpdI, TpdL, TpdU, Tlm-orf11 and Zbm-orf26 involved in methylation; HemN from E. coli (EcHemN) and HemZ from B. subtilis (BsHemZ) involved in heme biosynthesis; HemW from E. coli (EcHemW), HemW from L. lactis (LlHemW), HemW from P. aeruginosa (PaHemW) and RSAD1 from H. sapiens as heme chaperones. | ||
8. Conflicts of interest
There are no conflicts to declare.9. Acknowledgements
This work was supported in part by grants from the National Natural Science Foundation of China (21632007, 31870034, and 21621002), the Chinese Academy of Sciences (XDB20000000, QYZDJ-SSW-SLH037, and K. C. Wong Education Foundation). We also thank Prof. Wen Liu from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences and Prof. Qi Zhang from Fudan University for their critical reading and helpful suggestions.10. Notes and references
- H. J. Sofia, G. Chen, B. G. Hetzler, J. F. Reyes-Spindola and N. E. Miller, Nucleic Acids Res., 2001, 29, 1097–1106 CrossRef CAS.
- C. J. Walsby, D. Ortillo, W. E. Broderick, J. B. Broderick and B. M. Hoffman, J. Am. Chem. Soc., 2002, 124, 11270–11271 CrossRef CAS.
- T. Selmer, A. J. Pierik and J. Heider, Biol. Chem., 2005, 386, 981–988 CAS.
- Q. Zhang, Y. Li, D. Chen, Y. Yu, L. Duan, B. Shen and W. Liu, Nat. Chem. Biol., 2011, 7, 154–160 CrossRef CAS PubMed.
- N. A. Bruender and V. Bandarian, Biochemistry, 2016, 55, 2813–2816 CrossRef CAS.
- R. Rohac, P. Amara, A. Benjdia, L. Martin, P. Ruffie, A. Favier, O. Berteau, J. M. Mouesca, J. C. Fontecilla-Camps and Y. Nicolet, Nat. Chem., 2016, 8, 491–500 CrossRef CAS.
- A. Benjdia, A. Guillot, B. Lefranc, H. Vaudry, J. Leprince and O. Berteau, Chem. Commun., 2016, 52, 6249–6252 RSC.
- A. Parent, A. Guillot, A. Benjdia, G. Chartier, J. Leprince and O. Berteau, J. Am. Chem. Soc., 2016, 138, 15515–15518 CrossRef CAS.
- H. J. Kim, J. LeVieux, Y. C. Yeh and H. W. Liu, J. Am. Chem. Soc., 2016, 55, 3724–3728 CAS.
- J. Bridwell-Rabb, A. Zhong, H. G. Sun, C. L. Drennan and H. W. Liu, Nature, 2017, 544, 322–326 CrossRef CAS.
- J. L. Vey and C. L. Drennan, Chem. Rev., 2011, 111, 2487–2506 CrossRef CAS.
- Q. Zhang, W. A. van der Donk and W. Liu, Acc. Chem. Res., 2012, 45, 555–564 CrossRef CAS.
- M. R. Bauerle, E. L. Schwalm and S. J. Booker, J. Biol. Chem., 2015, 290, 3995–4002 CrossRef CAS.
- S. C. Wang, Nat. Prod. Rep., 2018, 35, 707–720 RSC.
- J. B. Broderick, B. R. Duffus, K. S. Duschene and E. M. Shepard, Chem. Rev., 2014, 114, 4229–4317 CrossRef CAS.
- S. A. Coomber, R. M. Jones, P. M. Jordan and C. N. Hunter, Mol. Microbiol., 1992, 6, 3159–3169 CrossRef CAS.
- K. Xu and T. Elliott, J. Bacteriol., 1994, 176, 3196–3203 CrossRef CAS.
- B. Troup, C. Hungerer and D. Jahn, J. Bacteriol., 1995, 177, 3326–3331 CrossRef CAS.
- G. Layer, K. Verfurth, E. Mahlitz and D. Jahn, J. Biol. Chem., 2002, 277, 34136–34142 CrossRef CAS.
- G. Layer, J. Moser, D. W. Heinz, D. Jahn and W. D. Schubert, EMBO J., 2003, 22, 6214–6224 CrossRef CAS.
- G. Layer, K. Grage, T. Teschner, V. Schunemann, D. Breckau, A. Masoumi, M. Jahn, P. Heathcote, A. X. Trautwein and D. Jahn, J. Biol. Chem., 2005, 280, 29038–29046 CrossRef CAS.
- G. Layer, A. J. Pierik, M. Trost, S. E. Rigby, H. K. Leech, K. Grage, D. Breckau, I. Astner, L. Jansch, P. Heathcote, M. J. Warren, D. W. Heinz and D. Jahn, J. Biol. Chem., 2006, 281, 15727–15734 CrossRef CAS.
- G. Homuth, A. Rompf, W. Schumann and D. Jahn, J. Bacteriol., 1999, 181, 5922–5929 CAS.
- Y. Yu, L. Duan, Q. Zhang, R. Liao, Y. Ding, H. Pan, E. Wendt-Pienkowski, G. Tang, B. Shen and W. Liu, ACS Chem. Biol., 2009, 4, 855–864 CrossRef CAS.
- Y. Ding, Y. Yu, H. Pan, H. Guo, Y. Li and W. Liu, Mol. BioSyst., 2010, 6, 1180–1185 RSC.
- R. P. Morris, J. A. Leeds, H. U. Naegeli, L. Oberer, K. Memmert, E. Weber, M. J. LaMarche, C. N. Parker, N. Burrer, S. Esterow, A. E. Hein, E. K. Schmitt and P. Krastel, J. Am. Chem. Soc., 2009, 131, 5946–5955 CrossRef CAS.
- L. Du, C. Sanchez, M. Chen, D. J. Edwards and B. Shen, Chem. Biol., 2000, 7, 623–642 CrossRef CAS.
- M. Tao, L. Wang, E. Wendt-Pienkowski, N. P. George, U. Galm, G. Zhang, J. M. Coughlin and B. Shen, Mol. BioSyst., 2007, 3, 60–74 RSC.
- U. Galm, E. Wendt-Pienkowski, L. Wang, N. P. George, T. J. Oh, F. Yi, M. Tao, J. M. Coughlin and B. Shen, Mol. BioSyst., 2009, 5, 77–90 RSC.
- W. Huang, H. Xu, Y. Li, F. Zhang, X. Y. Chen, Q. L. He, Y. Igarashi and G. L. Tang, J. Am. Chem. Soc., 2012, 134, 8831–8840 CrossRef CAS.
- Y. Igarashi, K. Futamata, T. Fujita, A. Sekine, H. Senda, H. Naoki and T. Furumai, J. Antibiot., 2003, 56, 107–113 CrossRef CAS.
- J. P. Parrish, D. B. Kastrinsky, S. E. Wolkenberg, Y. Igarashi and D. L. Boger, J. Am. Chem. Soc., 2003, 125, 10971–10976 CrossRef CAS PubMed.
- M. S. Tichenor, K. S. MacMillan, J. D. Trzupek, T. J. Rayl, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2007, 129, 10858–10869 CrossRef CAS.
- T. Hiratsuka, H. Suzuki, R. Kariya, T. Seo, A. Minami and H. Oikawa, Angew. Chem., Int. Ed., 2014, 53, 5423–5426 CrossRef CAS PubMed.
- M. Yoshida, M. Ezaki, M. Hashimoto, M. Yamashita, N. Shigematsu, M. Okuhara, M. Kohsaka and K. Horikoshi, J. Antibiot., 1990, 43, 748–754 CrossRef CAS.
- W. W. Doubleday, A. G. M. Barrett, K. Kasdorf and G. J. Tustin, J. Org. Chem., 1996, 61, 3280–3288 CrossRef.
- H. Watanabe, T. Tokiwano and H. Oikawa, J. Antibiot., 2006, 59, 607–610 CrossRef CAS.
- T. Hiratsuka, H. Suzuki, A. Minami and H. Oikawa, Org. Biomol. Chem., 2017, 15, 1076–1079 RSC.
- W. B. Jin, S. Wu, X. H. Jian and H. Yuan, Nat. Commun., 2018, 9, 2771 CrossRef PubMed.
- L. J. Hanka, A. Dietz, S. A. Gerpheide, S. L. Kuentzel and D. G. Martin, J. Antibiot., 1978, 31, 1211–1217 CrossRef CAS.
- D. G. Martin, C. G. Chidester, D. J. Duchamp and S. A. Mizsak, J. Antibiot., 1980, 33, 902–903 CrossRef CAS.
- R. M. Garbaccio and D. L. Boger, Acc. Chem. Res., 1999, 32, 1043–1052 CrossRef.
- S. Wu, X. H. Jian and H. Yuan, ACS Chem. Biol., 2017, 12, 1603–1610 CrossRef CAS.
- L. H. Hurley and J. S. Rokem, J. Antibiot., 1983, 36, 383–390 CrossRef CAS PubMed.
- E. E. Wyckoff, D. Duncan, A. G. Torres, M. Mills, K. Maase and S. M. Payne, Mol. Microbiol., 1998, 28, 1139–1152 CrossRef CAS.
- E. E. Wyckoff, M. Schmitt, A. Wilks and S. M. Payne, J. Bacteriol., 2004, 186, 4142–4151 CrossRef CAS.
- J. W. LaMattina, D. B. Nix and W. N. Lanzilotta, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12138–12143 CrossRef CAS.
- S. Hein, O. Klimmek, M. Polly, M. Kern and J. Simon, Mol. Microbiol., 2017, 104, 449–462 CrossRef CAS.
- B. Nowicka and J. Kruk, Biochim. Biophys. Acta, 2010, 1797, 1587–1605 CrossRef CAS.
- T. Dairi, Methods Enzymol., 2012, 515, 107–122 CAS.
- H. D. Juhnke, H. Hiltscher, H. R. Nasiri, H. Schwalbe and C. R. Lancaster, Mol. Microbiol., 2009, 71, 1088–1101 CrossRef CAS.
- X. Y. Zhi, J. C. Yao, S. K. Tang, Y. Huang, H. W. Li and W. J. Li, Genome Biol. Evol., 2014, 6, 149–160 CrossRef PubMed.
- S. Hein, J. von Irmer, M. Gallei, R. Meusinger and J. Simon, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 300–308 CrossRef CAS.
- N. Mahanta, Z. Zhang, G. A. Hudson, W. A. van der Donk and D. A. Mitchell, J. Am. Chem. Soc., 2017, 139, 4310–4313 CrossRef CAS.
- G. A. Hudson, Z. Zhang, J. I. Tietz, D. A. Mitchell and W. A. van der Donk, J. Am. Chem. Soc., 2015, 137, 16012–16015 CrossRef CAS.
- Z. Zhang, G. A. Hudson, N. Mahanta, J. I. Tietz, W. A. van der Donk and D. A. Mitchell, J. Am. Chem. Soc., 2016, 138, 15511–15514 CrossRef CAS.
- Z. Zhang, N. Mahanta, G. A. Hudson, D. A. Mitchell and W. A. van der Donk, J. Am. Chem. Soc., 2017, 139, 18623–18631 CrossRef CAS.
- F. Benazet and J. R. Cartier, Poult. Sci., 1980, 59, 1405–1415 CrossRef CAS.
- F. Benazet, M. Cartier, J. Florent, C. Godard, G. Jung, J. Lunel, D. Mancy, C. Pascal, J. Renaut, P. Tarridec, J. Theilleux, R. Tissier, M. Dubost and L. Ninet, Exper. Suppl., 1980, 36, 414–416 CAS.
- W. Ding, Y. Li, J. Zhao, X. Ji, T. Mo, H. Qianzhu, T. Tu, Z. Deng, Y. Yu, F. Chen and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 3857–3861 CrossRef CAS.
- W. Ding, Y. Wu, X. Ji, H. Qianzhu, F. Chen, Z. Deng, Y. Yu and Q. Zhang, Chem. Commun., 2017, 53, 5235–5238 RSC.
- X. Ji, D. Mandalapu, J. Cheng, W. Ding and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6601–6604 CrossRef CAS.
- E. D. Badding, T. L. Grove, L. K. Gadsby, J. W. LaMattina and A. K. Boal, J. Am. Chem. Soc., 2017, 139, 5896–5905 CrossRef CAS.
- J. W. LaMattina, B. Wang, E. D. Badding, L. K. Gadsby, T. L. Grove and S. J. Booker, J. Am. Chem. Soc., 2017, 139, 17438–17445 CrossRef CAS.
- B. Wang, J. W. LaMattina, E. D. Badding, L. K. Gadsby, T. L. Grove and S. J. Booker, Methods Enzymol., 2018, 606, 241–268 Search PubMed.
- Y. Qiu, Y. Du, F. Zhang, R. Liao, S. Zhou, C. Peng, Y. Guo and W. Liu, J. Am. Chem. Soc., 2017, 139, 18186–18189 CrossRef CAS.
- Y. Qiu, Y. Du, S. Wang, S. Zhou, Y. Guo and W. Liu, Org. Lett., 2019, 21, 1502–1505 CrossRef CAS.
- B. Wang, J. W. LaMattina, S. L. Marshall and S. J. Booker, J. Am. Chem. Soc., 2019, 141, 5788–5797 CrossRef CAS.
- X. Ji, T. Mo, W. Q. Liu, W. Ding, Z. Deng and Q. Zhang, Angew. Chem., Int. Ed., 2019, 58, 6235–6238 CrossRef CAS.
- P. Duwat, S. Sourice, B. Cesselin, G. Lamberet, K. Vido, P. Gaudu, Y. Le Loir, F. Violet, P. Loubiere and A. Gruss, J. Bacteriol., 2001, 183, 4509–4516 CrossRef CAS.
- T. Goto, R. Aoki, K. Minamizaki and Y. Fujita, Plant Cell Physiol., 2010, 51, 650–663 CrossRef CAS.
- H. K. Abicht, J. Martinez, G. Layer, D. Jahn and M. Solioz, Biochem. J., 2012, 442, 335–343 CrossRef CAS.
- V. Haskamp, S. Karrie, T. Mingers, S. Barthels, F. Alberge, A. Magalon, K. Muller, E. Bill, W. Lubitz, K. Kleeberg, P. Schweyen, M. Broring, M. Jahn and D. Jahn, J. Biol. Chem., 2018, 293, 2558–2572 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |