
Recent advances of metabolic engineering strategies in natural isoprenoid production using cell factories
Meijie
Li
 ac,
Feifei
Hou
ac,
Tong
Wu
ac,
Xinglin
Jiang
e,
Fuli
Li
a,
Haobao
Liu
*bd,
Mo
Xian
*a and
Haibo
Zhang
*a
ac,
Feifei
Hou
ac,
Tong
Wu
ac,
Xinglin
Jiang
e,
Fuli
Li
a,
Haobao
Liu
*bd,
Mo
Xian
*a and
Haibo
Zhang
*a
aKey Laboratory of Biobased Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, No. 135 Songling Road, Qingdao 266101, P. R. China. E-mail: li_mj@qibebt.ac.cn; Houff@qibebt.ac.cn; wutong@qibebt.ac.cn; lifl@qibebt.a.cn; xianmo@qibebt.ac.cn; zhanghb@qibebt.ac.cn; Fax: +86-0532-80662765; Tel: +86-0532-80662768
bMinistry of Agriculture Key Laboratory for Tobacco Biology and Processing, Tobacco Research Institute, Chinese Academy of Agricultural Sciences, No. 11 Keyuanjing 4 Road, Laoshan District, Qingdao, 266101, P. R. China. E-mail: liuhaobao@caas.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
dHainan Cigar Research Institute, Hainan Provincial Branch of China National Tobacco Corporation, No. 22 Hongchenghu Road, Qiongshan District, Haikou, 571100, P. R. China
eThe Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kemitorvet Bygning 220, 2800 Kgs, Lyngby, Denmark. E-mail: xinji@biosustain.dtu.dk
First published on 10th May 2019
Abstract
Covering: up to 2019
As abundant natural products, isoprenoids have many useful industrial applications in the manufacturing of drugs, fragrances, food additives, colorants, rubber and advanced biofuels. The microbial production of isoprenoids has received much attention in recent years. Metabolic engineering approaches and synthetic biology have been utilized to reconstruct and optimize the metabolic pathways for isoprenoid production in cell factories. In this review, the recent advances in isoprenoid production using microbes are summarized, with a focus on MEP and MVA pathway engineering, downstream isoprenoid pathway engineering and microbial host engineering, which mainly includes central carbon pathway engineering. Finally, future perspectives for the improvement of isoprenoid production are discussed.
 Meijie Li | Meijie Li currently is a PhD students under the supervision of Prof. Mo Xian and Prof. Haibo Zhang at Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences (QIBEBT, CAS). Her research is focused on the improvement of isoprene production in microbial cell factories through metabolic engineering and synthetic biology approaches. She has got her master's degree in biological engineering from Institute of Oceanology, CAS in 2015. She expects to graduate with PhD degree in biochemistry and molecular biology in the near future. |
 Haobao Liu | Dr. Haobao Liu is a professor at Tobacco Research Institute, Chinese Academy of Agricultural Sciences (CAAS), the director of Haikou Cigar Research Institute. He is a senior expert on tobacco fermentation, detection, cultivation and nutrition. He made great development in several research field, including cigar fermentation, aroma substance detection, cultivation and modulation, and tobacco potassium nutrition. He obtained his doctor's degree of agriculture at graduate school of the CAAS in 2012. |
 Mo Xian | Dr. Mo Xian is a professor at Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences (QIBEBT, CAS), where he serves as the leader of Key Laboratory of Biobased Materials, CAS. His research interest is focused on biosynthesis of bulk chemicals and materials from renewable resources by biocatalysis and chemical catalysis regarding creating genetic altered microbes, enzymes and chemical catalysts. He obtained his PhD degree at Jilin University in 2001, he work at CNRS in France and MSU in America before he served in QIBEBT in 2007. |
 Haibo Zhang | Dr. Haibo Zhang is currently a professor and the leader of Fine Chemicals group at Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences (QIBEBT, CAS). He is interested in biosynthesis of basic fine chemicals and terpene-based high-density biofuels in engineered microorganisms. His research is focused on several synthetic biology approaches, including biosensor, bacterial microcompartments and phospholipid metabolism regulation technologies. He obtained PhD in microbiology at Shandong University in 2010. He visited Massachusetts Institute of Technology as a visiting scholar in 2016. |
1. Introduction
The family of isoprenoids, which contains more than 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 members, include a large and highly diverse number of natural products in the three domains of life.1 Commonly, with the exception of steroids (C27), isoprenoids can be categorized according to the number of carbons as follows: hemiterpenoids (C5), monoterpenoids (C10), sesquiterpenoids (C15), diterpenoids (C20), triterpenoids (C30) and tetraterpenoids (C40) (Fig. 1). Many isoprenoids have great application value for drugs (e.g., artemisinin and paclitaxel), fragrances (e.g., limonene and santalol), food additives (e.g., lycopene and coenzyme Q10), colorants, rubber manufacturing and particularly advanced biofuels (e.g., farnesene, pinene and isopentenol).2 The metabolic engineering of a microbial host for the production of valuable isoprenoids is a promising strategy that can replace traditional plant extraction and chemical synthesis in industry.3–6
000 members, include a large and highly diverse number of natural products in the three domains of life.1 Commonly, with the exception of steroids (C27), isoprenoids can be categorized according to the number of carbons as follows: hemiterpenoids (C5), monoterpenoids (C10), sesquiterpenoids (C15), diterpenoids (C20), triterpenoids (C30) and tetraterpenoids (C40) (Fig. 1). Many isoprenoids have great application value for drugs (e.g., artemisinin and paclitaxel), fragrances (e.g., limonene and santalol), food additives (e.g., lycopene and coenzyme Q10), colorants, rubber manufacturing and particularly advanced biofuels (e.g., farnesene, pinene and isopentenol).2 The metabolic engineering of a microbial host for the production of valuable isoprenoids is a promising strategy that can replace traditional plant extraction and chemical synthesis in industry.3–6
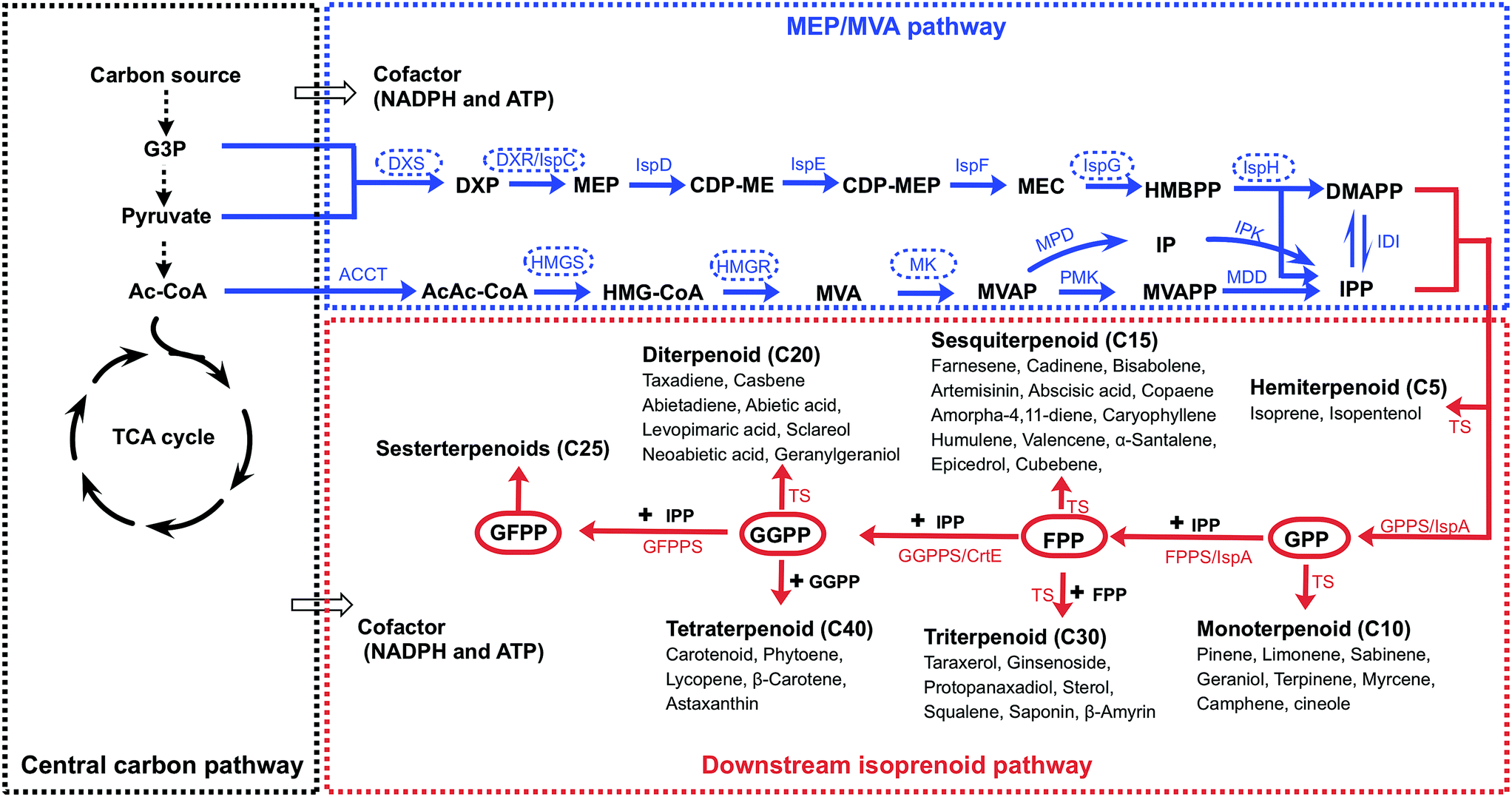 | ||
| Fig. 1 Metabolic pathway of isoprenoid biosynthesis. The metabolic pathway is divided into three parts. The central carbon pathway provides precursors and cofactors for the MVA/MEP pathway and provides cofactors for the downstream isoprenoid pathway. The MVA/MEP pathways produce the common precursors for isoprenoid synthesis, DMAPP and IPP. In the downstream isoprenoid pathway, DMAPP and IPP are condensed to produce GPP, FPP, GGPP and GFPP, which are catalyzed to produce various isoprenoids. The dashed arrows indicate multiple steps. The enzymes with dashed circles indicate the rate-limiting enzymes. | ||
The isoprenoid synthesis pathway can be divided into the following parts: the central carbon pathway, the MVA/MEP pathway and the downstream isoprenoid pathway (Fig. 1).7 The central carbon pathway provides the precursors (acetyl-CoA, G3P and pyruvate) and cofactors (NADPH and ATP) for isoprenoid precursor synthesis from a carbon source (e.g., glucose).8,9 Microbial hosts typically use the central carbon pathway to supply sufficient precursors and cofactors. In the second part, the common precursors, IPP and DMAPP, are produced. Although the MEP or MVA pathway operates in some hosts, metabolic engineering of the MVA or MEP pathway in the microbial host remains necessary for promoting metabolic flux. In the third part, the downstream isoprenoid pathway utilizes the condensation of IPP and DMAPP to produce the common intermediates GPP, FPP, GGPP and GFPP (Fig. 1).10 These diphosphate intermediates are catalyzed by specific enzymes, including terpene synthetase and cytochrome P450, to produce the target isoprenoids. The microbial hosts usually lack the enzymes for specific isoprenoid production, and the heterogeneous expression of specific enzymes is required. Therefore, isoprenoid production from a carbon source is a multigene pathway, and it is necessary to balance the metabolic flux among the three parts.
Numerous studies on the synthesis of isoprenoids in microbial hosts have been published over the past 20 years. Various metabolic engineering strategies and synthetic biology approaches have been developed to improve isoprenoid production in different hosts, including Escherichia coli, Saccharomyces cerevisiae and other appropriate microbes. The production of different types of isoprenoids at different levels has been reported. The titer of taxadiene (C20), a paclitaxel intermediate, was reported to equal 1 g L−1 in 2010,11 and the fermentation titers of antimalarial artemisinin (C20) production reached 25 g L−1 in 2013.12 In 2016, the farnesene (C15) titers after two weeks of fermentation at the 200000 liter scale exceeded 130 g L−1.13 However, research on the biological production of most terpenoids remains at the fundamental stage of identifying the necessary enzyme. In 2017, optimization of the fed-batch cultivation of an engineered strain resulted in the production of 182 mg L−1 betulinic acid and 854 mg L−1 total triterpenoids.14 The metabolic engineering strategies utilized for the high-level production of isoprenoids are instructive for other, less developed isoprenoids. Therefore, reviews summarizing published articles on isoprenoid bioproduction are beneficial to further improvements in their production. Corresponding reviews have been published in the last two years. The metabolic engineering of plants for isoprenoid production was reviewed by Nogueira et al. in 2018.15 Zhang et al. summarized chassis engineering and key enzyme engineering for monoterpene production.16 Metabolic engineering approaches for isoprenoid production in specific hosts, E. coli.1 and yeast,2,17,18 have been reviewed. Metabolic engineering for the production of several specific isoprenoids, carotenoids and isoprenoid-based biofuels, were reviewed.19,20 In another review, several strategies, including an in vitro reconstitution strategy, systematic engineering and the genome mining of novel isoprenoids, were emphasized.21 The isoprenoid pathway has been briefly summarized in a few reviews as an important pathway for hydrocarbon biosynthesis.22,23 These reviews focused on one specific aspect of isoprenoid production, e.g., monoterpene production, specific hosts, isoprenoid-based biofuel production or specific engineering strategies. However, a review focusing on the metabolic engineering strategies for the bioproduction of isoprenoids without any such limitation might be more instructive for different types of isoprenoids. Therefore, without regard to the host and isoprenoid type, this review summarizes most of the developed engineering strategies for isoprenoid production, particularly over the past three years. This review will be instructive for the bioproduction of different isoprenoids in the future, particularly for isoprenoids that have not been successfully synthesized in microbial cell factories or whose production remains at the elementary level.
In this review, the metabolic engineering strategies mainly published in the last three years are divided into three sections according to the three parts of the pathways and engineering targets as follows: MEP and MVA pathway engineering, downstream isoprenoid pathway engineering and microbial host engineering, which mainly includes central carbon pathway engineering. The first section describes the selection between the MEP and MVA pathways, the strategies used for multigene pathway engineering, rate-enzyme engineering and novel pathways. The second section summarizes the approaches for identification of enzymes for target production, enzyme engineering and enzyme modification. In the third section, strategies for microbial host engineering including precursor support, cofactor support, blocking the competitive pathway, cytotoxicity engineering and microbial host evolution, are described. Furthermore, the challenges and perspectives for future isoprenoid bioproduction are discussed.
2. MEP and MVA pathway engineering
The precursors IPP and DMAPP for isoprenoid biosynthesis are produced through the MEP or MVA pathway (Fig. 1). The selection between the MEP and MVA pathways is the first issue to consider, as discussed in Section 2.1. When the MEP or the MVA pathway is selected, it is necessary to perform pathway engineering to improve the metabolic flux. The MEP and MVA pathways are multigene pathways, and the systematic regulation of gene expression is challenging. Multigene pathway engineering strategies are summarized in Section 2.2. The pathways have rate-limiting enzymes, and further engineering to exceed the flux limit is required, as discussed in Section 2.3. However, even though engineering has sometimes been applied for the whole pathway and for the rate-limiting enzymes, the productivity remains low. The exploration of novel pathways might be a powerful alternative, as discussed in Section 2.4. The engineering approaches summarized are shared by different types of isoprenoid production.2.1. Selection between the MEP and MVA pathway for isoprenoid production
Although the MEP and MVA pathways share the same end product, IPP and DMAPP, these pathways differ considerably. In nature, the MVA pathway operates in most eukaryotes, fungi and the cytoplasm of plants, whereas the MEP pathway exists in many bacteria, algae, cyanobacteria and plant chloroplasts (Table 1). Moreover, the energy and cofactor consumption differs between the two pathways. Through the MVA pathway, 1.5 molecules of glucose are consumed, and four molecules of NAD(P)H are produced when one molecule of IPP/DMAPP is produced (Fig. 2, Table 1). However, the MEP pathway requires one molecule of glucose, three molecules of ATP and two molecules of NAD(P)H (Fig. 2, Table 1). Converting the cofactor consumption into glucose consumption, 1.25 molecules of glucose are consumed for one molecule of IPP/DMAPP synthesis (Fig. 2). In general, the MEP pathway shows a higher theoretical mass yield (30.2%), whereas the MVA pathway requires a lower cofactor amount. MEP and MVA pathway selection for isoprenoid production has been performed in different production hosts (Table 1), and different production levels have been obtained.| MEP pathway | MVA pathway | |
|---|---|---|
| a The cofactor consumption is the result of the cofactors produced during the upstream glycolysis pathway, which produces the precursors, and the cofactors required for the MEP or MVA pathway. | ||
| Key enzymes | DXS, DXR, IspG, IspH, IDI | HMGS, HMGR, MK, PMK, IDI |
| Reaction steps | Eight | Seven |
| Precursors | Pyruvate and G3P | Acetyl-CoA |
| Cofactor consumptiona | 2NADPH and 3ATP | 4NAD(P)H |
| Mass yield on glucose | 30.2% | 25.2% |
| Natural distribution | Bacteria, algae, diatoms cyanobacteria and plant chloroplasts | Eukaryotes, fungi, algae, archaea, a few bacteria and plants cytoplasm and mitochondria |
| Engineered microorganisms | Bacteria (including E. coli,31Bacillus subtilis,32Corynebacterium glutamicum,33Methylobacterium extorquens34), Cyanobacteria,35 algae36 | E. coli,37 yeast (including S. cerevisiae),38 fungi (including Schizophyllum commune)39 |
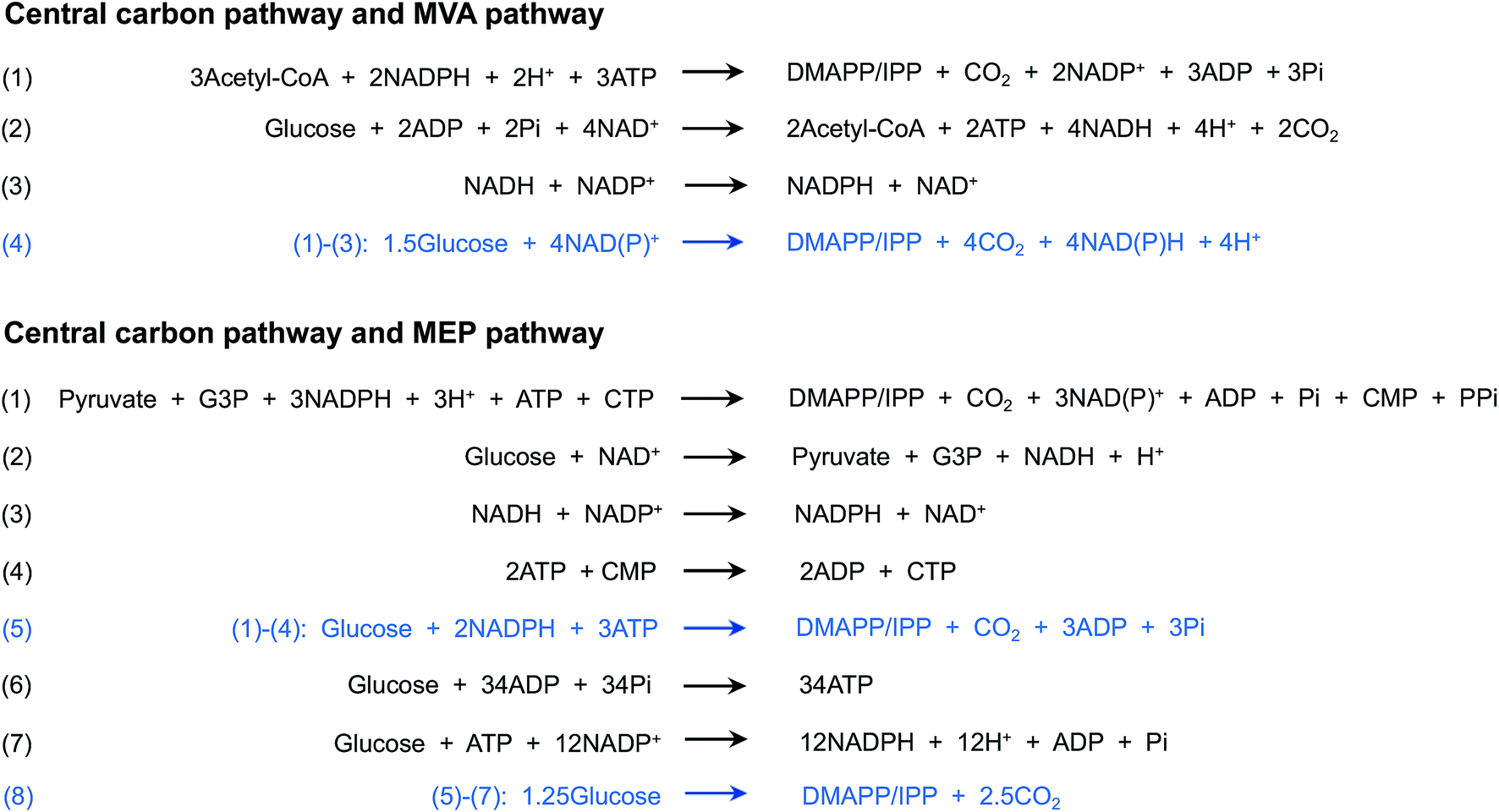 | ||
| Fig. 2 Stoichiometries of the conversion of glucose to IPP/DMAPP through the central carbon pathway and the MVA/MEP pathway. For the synthesis of one IPP/DMAPP molecule, 1.5 glucose molecules are consumed, and four NAD(P)H molecules are produced through the upstream central carbon pathway and MVA pathway. One glucose molecule, two NADPH molecules and one ATP molecule are required for the synthesis of one IPP/DMAPP molecule through the upstream central carbon pathway and MEP pathway. NADPH and ATP are synthesized through the extra central carbon pathway. In summary, 1.25 glucose molecules are needed for the synthesis of one IPP/DMAPP molecule through the central carbon pathway and MEP pathway. | ||
Using E. coli as the chassis, previous studies have modified the native MEP pathway and introduced the heterogeneous MVA pathway. Additionally, utilization of the native MEP pathway usually results in decreased production compared with utilization of heterologous whole MVA pathway overexpression. Using isoprene production in E. coli as an example, overexpression of the restrictive points of the MEP pathway (DXS and DXR) and IspS resulted in the production of 314 mg L−1 isoprene.24 In contrast, the strain with the modified MVA pathway, including the upper mevalonate pathway from Enterococcus faecalis and the lower mevalonate pathway from S. cerevisiae and the same IspS, produced approximately 700 mg L−1 isoprene.25 The large cofactor consumption of the MEP pathway—three molecules of ATP and two molecules of NAD(P)H for one molecule of IPP/DMAPP production (Fig. 2, Table 1)—might lead to the lower isoprenoid production. Moreover, the MEP pathway is regulated in E. coli. Feedback regulation of the flux is achieved through the DXS and IspF enzymes.26,27 The intermediate DXP is also the substrate for the synthesis of thiamine and pyridoxal in E. coli.28 IspG is sensitively inactivated by stress conditions, leading to the accumulation of MEcPP, an anti-stressor.26 The interplay between the MEP pathway and the physiological pathways limits the metabolic flux to IPP/DMAPP formation in E. coli. In another widely utilized host, S. cerevisiae, the native MVA pathway is usually selected for isoprenoid production. In yeast engineered for pentacyclic triterpene production, the rate-limiting enzymes of the MVA pathway, HMG-CoA synthase and HMG-CoA reductase, were overexpressed, and a negative regulator of the MVA pathway encoded by ROX1 was deleted.29 In another species of yeast, Yarrowia lipolytica, key genes involved in the MVA pathway—HMG1, IDI1 and mutant ERG20—were overexpressed for linalool production, and titers of 6.96 ± 0.29 mg L−1 were achieved through shake flask culture.30 To our knowledge, the utilization of the heterogeneous MEP pathway in yeast for isoprenoid production has not been reported.
Utilization of both the MEP and MVA pathways in the same strain has also been performed for isoprenoid production. In E. coli engineered for isoprene, the rate-limiting enzymes of the MEP pathway and the entire MVA pathway were overexpressed by inserting some genes into the chromosome and ligating some genes into the plasmids.40 The strain produced an isoprene titer of 629 mg L−1, which is approximately 19-fold and 32.65-fold higher than those obtained with strains after only MEP and MVA pathway engineering, respectively.40 Interestingly, the E. coli strain in which only the MVA pathway was introduced showed high MEcPP accumulation, an intermediate of the MEP pathway, which indicates an interaction between the MEP and MVA pathways.41 It has been speculated that engineering approaches should be applied to utilize the accumulated MEcPP in the E. coli strain in which the MVA pathway was introduced. Therefore, despite limited research regarding the synergy between the MEP and MVA pathways, we believe that this engineering approach has the potential for higher isoprenoid productivity, particularly in E. coli. More effort should be devoted to examining the interaction between the MEP and MVA pathways and utilizing both pathways for isoprenoid production.
2.2. Strategies used for multigene pathway engineering
Regardless of which pathway is selected, the metabolic pathway for isoprenoid production is a multigene pathway that involves seven steps of the MVA pathway and eight steps of the MEP pathway to produce DMAPP from the precursor (Fig. 1, Table 1). Thus, strategies for multigene pathway engineering are required. With the traditional genetic engineering strategies, several problems appeared. First, gene engineering in recombinant strains often results in metabolic imbalance, which disrupts growth and leads to suboptimal production, particularly when multiple genes are required for the synthesis pathway.11 The screening promoters and RBSs libraries for every gene to obtain the optimized expression pattern is an inefficient strategy. Second, plasmid-based multigene expression systems suffer from genetic instability and cellular burden problems. Third, toxic metabolite accumulation is another serious problem in multigene pathway engineering. Therefore, the optimization of multigene pathways is challenging. In this section, strategies for multigene pathway engineering, including modular engineering, chromosomal integration and dynamic control of the pathway, which would provide effective solutions to these problems, are described (Fig. 3). | ||
| Fig. 3 Strategies for multigene pathway engineering. Modular pathway engineering, chromosomal integration and dynamic control of the pathway are three strategies utilized for multigene pathway engineering. A schematic diagram of an example of the dynamic control of the isoprenoid synthesis pathway is illustrated. PgadE is a promoter that is inhibited by the cytotoxic intermediates IPP and FPP. | ||
Modular pathway engineering, which groups genes into different modules and cooperatively modulates the expression levels of each module, is a powerful strategy for finetuning multigene pathways (Fig. 3). Since the application of modular pathway engineering for paclitaxel production was reported in 2010, it has been performed for the production of various isoprenoids.11 Two modules, an upstream MEP pathway and a downstream terpenoid-forming pathway, were partitioned into the engineered paclitaxel precursor-produced E. coli host.11 The expression levels of the two modules were modulated through promoter screening and gene copy-number screening to obtain the optimal production.11 In the engineered strain for artemisinin production, eight critical genes were classified into four modules.42 Combining the promoter screening for every module and the experimental design-aided systematic pathway optimization (EDASPO) approach, the best promoter combination, which resulted in a three-fold increase in artemisinin production (60 to 201 mg L−1), was identified.42 Metabolic balance between modules can be obtained through promoter library screening (Fig. 3). However, in the same module, although the transcriptional expression of genes is controlled simultaneously by the same promoter, the translational expression and enzyme activity of each gene are difficult to regulate. A multidimensional heuristic process, which screens diverse libraries of three-dimensional elements, including transcription as well as translation and enzyme activity, was developed to generate high astaxanthin-producing strains in which 15 essential genes were partitioned into four modules.37 In this study, RBS library screening was applied to regulate the enzyme expression of the intramodules (Fig. 3). The multidimensional heuristic process works well for not only intermodules but also intramodules. In conclusion, because of the long pathway involved in the synthesis of isoprenoids, the modular engineering approach and improvements of this approach have been applied to regulate the expression of genes belonging to intermodules and intramodules. Nonetheless, modular pathway engineering might lead to the overexploitation of cellular resources (e.g., NADPH and ATP), especially when plasmids are utilized for gene expression. Combining modular pathway engineering with the chromosomal integration method described below might be a solution (Fig. 3). In addition, computer-aided modular engineering, which was developed in recent years, might help balance cell growth and target production.43 In recent years, the different modules of a metabolic pathway were separately expressed in different microbes, such as E. coli and S. cerevisiae, which were designed to be cocultured, and the product of the engineered E. coli strain can diffuse into engineered S. cerevisiae to produce the target isoprenoid.44 Therefore, for multigene pathway engineering, the coupling of modular strategies with other strategies is a promising new frontier for strain optimization.
Due to its easy manipulation and ability to yield high expression, plasmid-based gene overexpression has been extensively utilized for protein and chemical microbial production. Nevertheless, the degree of genetic stability decreases with increases in the plasmid size, an inevitable problem for multigene insertion.45 Moreover, the metabolic burden from plasmid replication and antibiotic expression damage cell growth.46 A chromosomal integration method, which shows stable genetic expression and low metabolic burden to the host cell, is employed in multigene pathway engineering to address these problems (Fig. 3).46 To address the growth defect associated with plasmid engineering for β-carotene production, the Hmg-erg12 operon was integrated into the chromosome at the pflB site and the mvaS-mvaA-mavD1 operon was integrated at the frdB site. After further expression modulation of atoB, mvas and Hmg1, the OD453 of the accumulated carotene was improved to 3.2, which was 1.51-fold higher than that of the control strain, with no damage to cell growth.47 However, chromosomal integration usually suffers from low expression levels. The expression strength of certain genes should be modulated by screening the RBS library and promoter library to obtain the optimized combination.47 Recently, the CRISPR-Cas9-based approach has been applied for the optimization of chromosomally integrated pathways, and the promoters of three modules were regulated.48 Moreover, the gene expression level is also correlated with the integrated chromosomal location, and the expression level of the gfp gene varied by 300-fold depending on its chromosomal location.49,50 In addition, the gene copy number inserted into the chromosome affects gene expression. In an engineered strain for PHB synthesis, 50 copies of the phaCAB operon were inserted into the asnB site and 34.1 wt% of PHB accumulation was obtained; however, no PHB was detected when a single copy of the phaCAB operon was inserted.51 In general, the copy number of the inserted genes, the chromosomal location of the insertion, and expression regulation should be considered to improve the expression levels for chromosomal integration.
Maximizing the flux of the multigene pathway for isoprenoid production usually places tremendous burden on the microbial strain, which is detrimental to cell growth. Moreover, certain intermediates for isoprenoid production are cytotoxic to cell growth. Dynamic control of the pathway might be an effective strategy to solve these problems (Fig. 3).52,53 Coupling the dynamic control of pathways with the modular pathway engineering described above has been performed to maintain the balance between cellular health and target production. The fermentation process is usually divided into the following phases: a growth phase to obtain a high cell density and a production phase in which synthetic pathways for target production are expressed. In carotenoid-producing yeast, a glucose-repressed GAL promoter regulated the carotenoid pathway (modules 1 and 4), and a glucose-activated HXT1 promoter regulated the squalene pathway (module 2), the bypass pathway.53 In the first stage, which is characterized by high glucose concentrations, the genes in module 2 were expressed at a normal level to maintain cell growth. In the second stage, when glucose was consumed and a relatively high cell density was obtained, the genes in module 2 were downregulated and the genes in modules 1 and 4 were overexpressed for carotenoid production.53 Thus, sequential control of the modules was realized. The isoprenoid precursors DMAPP, IPP, GPP and FPP are toxic when they accumulate in E. coli.41,54,55 The dynamic regulation of the expression of the corresponding genes is an effective strategy to avoid toxic intermediate accumulation. The FPP-responsive promoters PgadE and sensor IA44, which were downregulated by IPP and FPP, were used to regulate MVA pathway gene expression, and the corresponding strains produced 69.18 mg L−1 and 58.05 mg L−1 zeaxanthin, respectively, and these titers were 2.1- and 1.76-fold higher than that produced by the control strain, respectively (Fig. 3).52,56 Dynamic control might be successfully utilized for the synthesis of a few isoprenoids; however, it also has limitations. First, not every toxic intermediate naturally has responsive promoters, and not all screened responsive promoters can be successfully applied for improved isoprenoid production. Second, the balance between dynamism and control is difficult to achieve.57 The problems of excessive operator control and excessive dynamism without appropriate control often arise. Open-loop and closed-loop control, coupled with computer-assisted feedback control, are promising strategies for dynamic control.57
For different types of isoprenoid production, the different strategies summarized above have been applied to address the multigene pathway problem. Furthermore, the combination of modular pathway engineering, chromosomal integration methods and dynamic control of the pathway should be considered in future engineering approaches.
2.3. Rate-limiting enzyme engineering
When the multigene pathway, the line, is constructed according to the strategies discussed above, several enzymes of the MEP and MVA pathways might limit the metabolic flux. Therefore, engineering rate-limiting enzymes, the points, is required. In general, DXS, DXR, IspD, IspF, IspG, IspH and IDI in the MEP pathway and HMGR, MK, MPK and IDI in the MVA pathway are rate-limiting enzymes (Fig. 1), as described in a previous review.58 Nonetheless, in different engineered strains, the restrictive points of the metabolic flux are usually different. Several approaches have been applied to identify the target enzyme for optimization. Kinetic flux profiling has been performed to identify bottlenecks in the engineered isoprene-production strain.59 IspG was identified as a bottleneck, and further IspG overexpression led to 30% more isoprene production, approximately 21 mg L−1.59 In the engineered strain for astaxanthin production, in silico flux variability scanning based on enforced objective flux was applied to identify target genes, and 19 potential genes were selected for engineering.31 After the identification of rate-limiting enzymes, various engineering methods have been developed to optimize key enzymes. Enzymes from different species have been screened to find the appropriate enzyme. The downstream MVA pathway genes MK, MPK, MDD and IPP from S. cerevisiae and Streptococcus pneumonia have generally been transformed into E. coli for isoprenoid production.25,60 However, feedback regulation at the transcriptional and posttranslational levels have strongly limited MK expression.61 Then, feedback-resistant MKs from Methanosarcina mazei, Methanosaeta concilii and Methanocella paludicola have been screened and characterized.62,63 The direct evolution of enzymes is also a powerful approach for optimizing the rate-limiting enzyme of the MEP and MVA pathways. The directed evolution of MK and IDI has been performed through the large-scale screening of downstream lycopene production, and enzyme mutants with better enzymatic properties have been identified.64,65 The engineered enzyme with better properties might be applicable to most isoprenoid synthesis. However, research on the direct evolution of the enzymes of the MEP/MVA pathway is far less advanced than that on direct evolution of the enzymes of the downstream isoprenoid pathway. The multiple steps between the enzyme of the MEP/MVA pathway and the target isoprenoid product might be the reason. In conclusion, the identification of rate-limiting enzymes and engineering to release these key points are classic engineering approaches for microbial production. Various techniques for isoprenoid microbial productions have been improved through this strategy.2.4. Novel pathway
The engineering methods described above are performed based on the natural MEP and MVA pathways. In consideration of the metabolic burden resulting from multigene expression and the accumulation of toxic intermediates, novel pathways have been created. Several studies utilizing the novel MVA pathway have been published. In the isoprene production strain, OleTJE and OhyAEM were introduced to produce isoprene from mevalonate through two steps.66 In another study, two novel MVA pathways that circumvent the toxic accumulation of IPP and DMAPP were explored for isoprenol production in E. coli.67 In novel pathway I, MDD directly catalyzes mevalonate to form isoprenol production. In novel pathway II, MVAP is catalyzed by MDD to produce IP, which produces to isoprenol through a reaction catalyzed by phosphatase. However, the low enzyme activity and affinity of OleTJE, OhyAEM and MDD led to low levels of isoprene or isoprenol production. Additional studies on screening enzymes with better activity and higher affinity are necessary. In addition, a novel MEP pathway has been reported, and in this pathway, isoprene is directly produced from HMBPP through a reaction catalyzed by IspH from alkaliphilic Bacillus sp. N16-5 that circumvents IPP and DMAPP accumulation.68 In general, compared with research on the novel MVA pathway, less research on the novel MEP pathway has been reported. The low efficiency of the enzymes in novel pathways remains a limiting factor for novel pathway application, and enzyme screening and engineering are required to improve the metabolic flux.To our knowledge, novel pathway engineering has been mostly restricted to hemiterpenoids, such as isoprene and isopentenol. Hemiterpenoids are simple chemicals that have only five carbons, and no complicated condensation between IPPs and DMAPPs is required. The exploration of a novel pathway for hemiterpenoid production is reasonable and feasible. Moreover, IPP/DMAPP production has been circumvented through the adoption of all the explored novel pathways. However, for the production of other forms of isoprenoids, which have a complicated structure, condensation between IPP and DMAPP molecules is necessary. Thus, exploring a novel pathway that shortens the long pathway without circumventing IPP and DMAPP production would be more useful and could be applied for all types of isoprenoid production. Recently, a two-step pathway for IPP and DMAPP production in which prenol and isoprenol were used as precursors, respectively, was observed in two studies.69,70 Two different kinases catalyzing the first step, i.e., the conversion of prenol and isoprenol to DMAP and IP, respectively, were identified. This novel pathway is decoupled from the central carbon metabolism and has potential for high metabolic flux. The metabolic flux of the novel pathway developed by Stephanopoulos' group is sufficient for almost all the downstream pathways tested.69 The application of this novel pathway for the synthesis of additional isoprenoids is expected in the future. However, further research is required to provide inexpensive and sustainable supplies of the substrate, prenol and isoprenol.
3. Downstream isoprenoid pathway engineering
The engineering approaches involved in the MEP and MVA pathways are usually compatible and shared among different types of isoprenoids. However, more than 50000 different molecules of isoprenoids exist in nature, and more than 55 different types of isoprenoids (Fig. 4) have been successfully produced in microbial cell factories.1 The diversity of the downstream isoprenoid pathway leads to various isoprenoids. Thus, it is necessary to summarize the engineering strategies for downstream isoprenoid production. Usually, target isoprenoids are not naturally produced in engineered microbes, and the identification of enzymes for target isoprenoid production is the first step. The enzymes of the downstream isoprenoid pathway usually show low activity and affinity; thus, enzyme engineering approaches, such as enzyme evolution and enzyme modification, which have been commonly utilized, are discussed in the subsequent section.
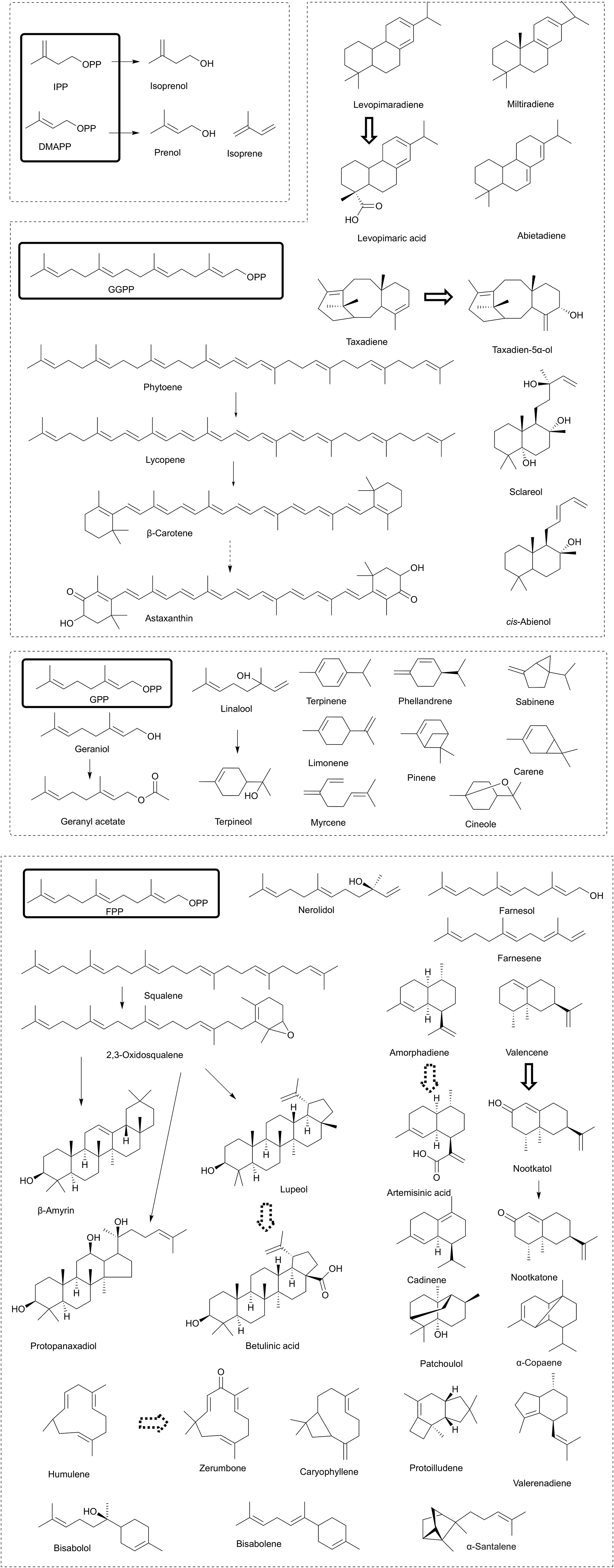 | ||
| Fig. 4 Chemical structures of isoprenoids that have been successfully synthesized in microbial cell factories. The reported biosynthesized isoprenoids in microbes were summarized and classified. The chemicals with similar structures were arranged together. The dashed arrows represent multiple steps. The hollow arrows represent the steps catalyzed by cytochrome P450-dependent monooxygenase. | ||
3.1. Identification of enzymes for target production
The first step for isoprenoid production is the identification of specific genes/enzymes, particularly when the target has not been successfully synthesized in microorganisms. Gene mining is a powerful approach for deciphering secondary metabolic gene clusters within a given system.71 Whole-genome sequencing and gene annotation were performed to characterize the astaxanthin-producing pathway genes of strain Sphingomonas sp. ATCC 55669, which can produce astaxanthin naturally.72 A comparative genomic analysis is usually applied to identify the specific genes among massive genome data, and the cytochrome P450 enzyme that catalyzes artemisinic acid production from amorpha-4,11-diene was first cloned using this method and successfully expressed in yeast.73–75 Transcriptome sequencing is also a powerful method for gene mining. The transcriptome sequencing of hawthorn was performed to identify the specific cytochrome P450 enzyme for the hydroxylation of pentacyclic triterpenoids.76 CYP716, which catalyzes triterpenoid oxidation, was first identified through a comparative analysis of genome and transcriptome data from different related species.77 Therefore, the gene mining approaches, including genome mining and transcriptome mining, have been applied to identify the specific enzymes involved in the biosynthesis of secondary metabolites.75 Genome mining is based on the analysis of genome DNA sequences, which are relatively constant throughout an organism's life.78 Transcriptome mining is based on the analysis of RNA sequences, which are relatively variable and reflect the expression of genes under different factors.78 Nonetheless, both of these gene mining approaches have limits. The genome sequences of certain organisms have not been deciphered, and genome mining is not applicable for all organisms. For transcriptome sequencing, RNA is extremely unstable, and high-quality data are difficult to obtain. In recent years, a variety of genome and transcriptome mining techniques and tools have been developed, and these have yielded improvements in high-throughput sequencing methods.79 Other “omics” technologies, such as proteomics and metabolomics, can also be utilized for gene mining, and a combination of different “omics” technologies should be a powerful strategy for gene mining.80After the genes/enzymes from different organisms are identified, it is necessary to perform enzyme screening to identify the best one for production. For monoterpene geraniol production in S. cerevisiae, GESs from different sources were tested, and GES from Valeriana officinalis was proven to be the most effective.81 The enzymes encoded by CrtW and CrtZ, two necessary genes for the synthesis of astaxanthin from β-carotene, were screened, and the best gene combination that yielded a high astaxanthin production ratio was identified.82 Enzyme screening is a simple and classic engineering strategy for metabolic pathway engineering, and this strategy is commonly utilized at the preliminary stage of research on isoprenoid production. In conclusion, the most suitable genes/enzymes for the production of target isoprenoids are generally identified by first identifying genes/enzymes through gene mining and then comparing the genes/enzymes from different species.
3.2. Enzyme engineering
In the downstream isoprenoid pathway, the identified and screened wild-type enzymes, particularly terpenoid synthetase, usually show low activity, low affinity and poor selectivity. As the end enzyme of the metabolic pathway, the activity of this enzyme is directly related to isoprenoid production. Further enzyme engineering is required. Multiple enzyme engineering approaches, such as rational design, semirational design and directed evolution, have been developed to mutate enzymes to obtain desirable properties.83 Various high-throughput screening methods have been developed.84 In the field of isoprenoid microbial production, the engineering of terpenoid synthetase is more widely performed than the engineering of the rate-limiting enzymes in the MEP and MVA pathways. The abovementioned optimization of enzymes in the MEP and MVA pathways usually shows a relatively lower potential to improve isoprenoid production due to the long distance from the engineered enzyme to the product.Semirational and rational design require in-depth knowledge of the structure and function of an enzyme, particularly the active sites and the binding sites.85 Mutability landscape-guided enzyme engineering was performed for ADS, an enzyme that catalyzes the formation of amorpha-4,11-diene from FPP.86 Based on the 3D model of ADS, 16 putative active sites were selected for site-saturation mutagenesis, and a mutant library with 258 variants was created.86 Rational design can also be applied to obtain the desired enzyme selectivity. The natural TXS shows 92% taxa-4(5)-11(12)-diene production from GGPP.87 To improve paclitaxel production, the higher selectivity of TXS for another isomer, taxa-4(20)-11(12)-diene, was realized through alanine scanning and site-saturation mutagenesis of the six active site residues and six residues in proximity to the active residues.88 Erg20p, a bifunctional enzyme, can catalyze the sequential formation of GPP and FPP.89 Putative residues of Erg20p (F96 and A99), which were essential for FPP synthesis but not GPP synthesis, were identified through comparison of the Erg20p structure with FPP synthase to improve monoterpene production from GPP.89 Rational and semirational design creates a relatively small, high-quality mutant library, which avoids a large amount of work and time required for library screening. Various enzymes in the downstream isoprenoid pathway have been successfully evolved through these engineering strategies.
Nevertheless, for many terpene synthetases, rational design is limited by a lack of basic structure information.90 Directed evolution is a promising strategy for enzyme engineering without prior knowledge of the enzyme structure. Directed evolution consists of two steps: generation and screening of a mutant library. Novel strategies with high efficiency for the generation of mutant libraries have been reported. A Cas9-based approach, CasPER, which can facilitate the directed evolution of large sequences in genomic contexts, was developed and successfully applied for the directed evolution of ERG20 in the yeast genome, and the mutants achieved carotenoid accumulation of approximately 120 mg, which is 5-fold higher than that of the wild type.91 Screening of the mutant library is another determining factor for directed evolution. Color-based high-screening methods have been developed for colorful isoprenoids, such as carotenoids. The directed evolution of β-carotene ketolase was performed based on a color screening system utilizing the red color of canthaxanthin, which was the product. The screened positive mutants with 2.4-fold improved activity were utilized for the microbial production of astaxanthin, and a titer of 47.18 mg L−1 was obtained.92 In the carotenoid synthesis pathway, the CrtYB gene encodes a bifunctional enzyme, CrtYB, which includes CrtB with phytoene synthase function and CrtY with lycopene cyclase function. However, for lycopene production from FPP, CrtB is required, whereas CrtY exerts a negative effect. The directed evolution of CrtYB was performed to inactivate the CrtY function and retain the CrtB function using a color-based screening method.93 Alternatively, for the production of an uncolored isoprenoid, a high-throughput colorimetric assay was developed based on substrate consumption. The target isoprenoid shares the same substrate as the colorful carotenoid, and mutant strains with improved target production have decreased building blocks for carotenoid biosynthesis and thereby show decreased color accumulation. The screening system for the directed evolution of monoterpene synthase, sesquiterpene synthase and diterpene synthase has been explored with the coexpression of the C30 and C40 carotenoid synthesis pathways.94 Geraniol synthase with high enzyme activity was successfully identified in the random mutant library, which was screened based on the color change in the coexpressed carotenoid pathway sharing the same precursor, GPP.95 In addition to the color-based screening system, some specific high-throughput screening methods have been developed. An enzyme-coupled assay was explored for the directed evolution of geraniol synthase. The product geraniol can be converted to geranial along with the production of NADH, which is subsequently utilized by diaphorase to produce fluorescent resorufin from resazurin. The activity of geraniol synthase was then coupled with the fluorescent strength.96 For the directed evolution of enzymes in the isoprenoid synthesis pathway, direct or indirect colorimetric readout based on carotenoid production outperforms other screening strategies, including fluorescence screening, color-byproduct screening, mevalonate-biosensor and growth selection based on intermediate cytotoxicity.84 Although several enzymes in the downstream isoprenoid pathway have been successfully evolved through enzyme engineering, the enzyme activity and selectivity remain limiting factors for the production of most isoprenoids. Further research should focus on the structure–function information of terpene synthetase and high-throughput screening strategies for directed evolution.
3.3. Enzyme modification
Different enzyme engineering strategies, which can change enzyme properties as desired, can greatly improve isoprenoid production. Moreover, the modification of enzyme activity, stability and expression, which are mostly realized by fusion enzymes, has been widely applied for downstream isoprenoid pathways (Fig. 5). Fusion of the sequential enzymes of the metabolic pathway has been performed (Fig. 5a). The proximity of the active site of fusion enzymes facilitates substrate utilization, avoids the diffusion of intermediates and relieves feedback inhibition.97,98 For geraniol production in S. cerevisiae, the fusion protein Erg20p(F96W-N127W)-tVoGES, a bifunctional enzyme with GPP/FPP synthase and geraniol synthase activity, was constructed, and several linkers between the two enzymes were investigated.81 For (S)-linalool production, ERG20 was linked to LIS (Fig. 5a), and different linkers between the enzymes, including short, medium-length and long flexible linkers, have been tested. The results indicated that fusion enzymes with different linkers showed different activities, and the long linker (GGGGS)3 led to the highest (S)-linalool production, 101.55 μg L−1, which was 1.7-fold higher than that obtained with the strain with two independent enzymes.99 Notably, the ligation point of the two enzymes affects the catalytic efficiency. From the structural models of fusion enzymes, it has been speculated that the proximity of two active sites is different in fusion enzymes with different ligation points.59 The active sites of the two enzymes in the fusion enzyme IDI-IspS are closer than those in IspS-IDI; thus, IDI-IspS enzyme fusion showed 2-fold higher isoprene production than that of IspS-IDI.59 In addition to the fusion of sequential enzymes of the metabolic pathway, fusion tags are ligated to enzymes, which can stabilize protein or improve protein expression (Fig. 5b). Several tags, including six-arginine tags, six-aspartic acid tags and one protein-stabilizing tag, TrxA, were fused to the N terminus of ADS. The use of six-arginine-tagged ADS resulted in improved ADS protein expression and a 2.5-fold increase in amorphadiene production to 5 mg L−1.100 In another study, the fusion tag OmpF, which guides the localization of the enzyme to the membrane, and TrxA were attached to the N and C termini of trCrBKT, respectively, which encodes β-carotene ketolase. The fusion tags led to the production of 12.9 mg L−1 astaxanthin, and this level was 2.08-fold higher than that obtained with the strain without tags.31 Traditionally, the tags are fused to enzymes through gene cloning, which is laborious when several candidate tags need to be cloned. A Cas9-based toolkit, which includes 10 fusion tags, 37 promoters and 23 Cas9-sgRNA plasmids, was developed.101 Enzyme expression and solubility were optimized through this toolkit, and a 25-fold improvement in taxadiene production to 20 mg L−1 was obtained.101 In other studies, the fusion of heterogeneously expressed enzymes with the native highly expressed enzymes was applied to improve isoprenoid production (Fig. 5c). Several studies have demonstrated that the fusion of enzymes with cpcB, a highly expressed native β-subunit of phycocyanin in cyanobacteria, helps improve enzyme expression. For isoprene production in cyanobacteria, the fusion enzyme cpcB-IspS was constructed and linkers with different lengths were examined, leading to a 300-fold increase in IspS expression.102 In another study, SQS was attached to the same cpcB, resulting in the production of 11.98 mg per L per OD730 squalene, which corresponded to a 3.1-fold increase.103 Enzyme expression, stability and catalytic ability are usually considered to be optimized when a fusion enzyme is used. Occasionally, the enzyme structure and function are influenced when it is attached to another enzyme, and linker engineering is needed to obtain the optimal fusion enzyme structure. In conclusion, enzyme modification through enzyme fusion has been proven to be a powerful strategy for enzyme optimization in the downstream isoprenoid pathway.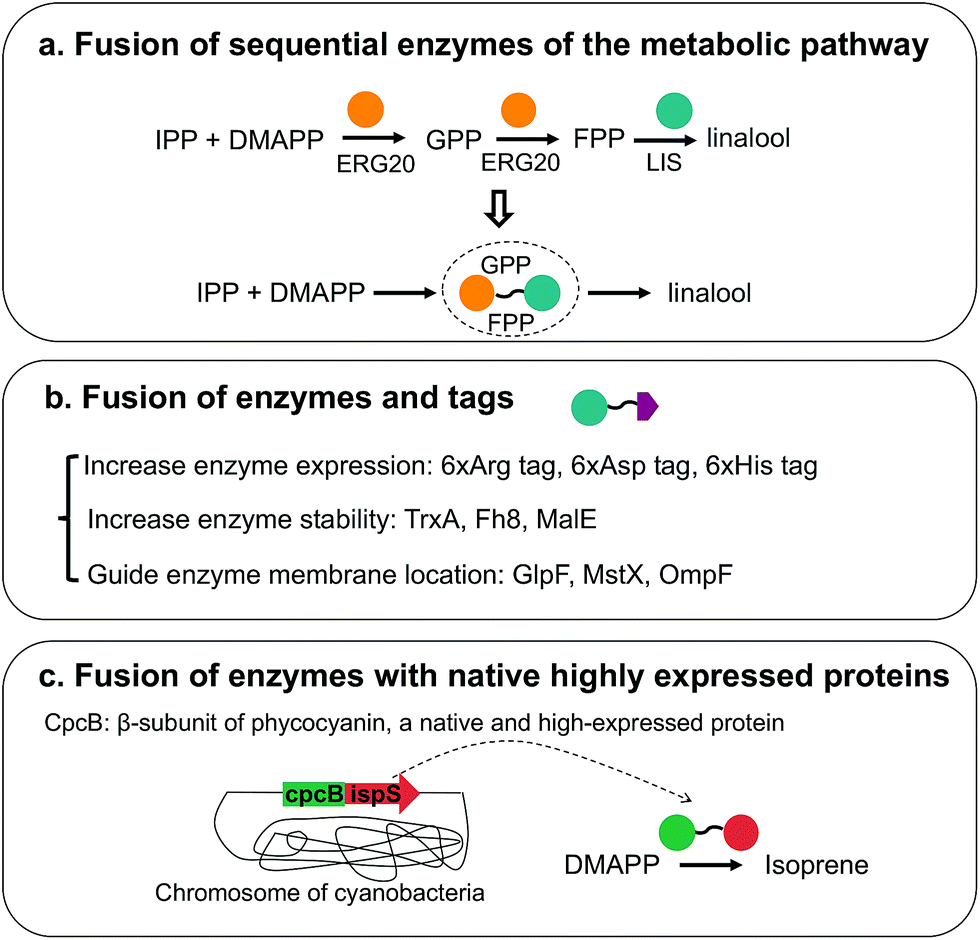 | ||
| Fig. 5 Enzyme modification through fusion enzymes utilized for the downstream isoprenoid pathway. (a) Fusion of sequential enzymes of the metabolic pathway. The schematic diagram illustrates the fusion enzyme of ERG20 and LIS utilized for linalool production. (b) Fusion of enzyme and tags. The expression improving, stability-improving and membrane-located tags are summarized. (c) Fusion of enzymes with native highly expressed proteins. The fusion of rate-limiting enzymes with CpcB, a fusion strategy primarily performed in cyanobacteria, is illustrated. | ||
4. Microbial host engineering
In addition to the MEP and MVA pathway engineering and the downstream isoprenoid pathway engineering, engineering strategies targeting the central carbon pathway, including precursor support and cofactor support (Fig. 6a and b), have been widely developed. The microbial host is engineered when the central carbon pathway is modified, which is different from the engineering strategies summarized above. In addition to precursor support (4.1) and cofactor support (4.2), the microbial host can affect the synthetic pathway in other aspects. First, the native competitive pathway might consume the carbon flux or inhibit the metabolic pathway for target production. Second, the intermediates and products of the pathway might be toxic to microbial cells. Therefore, blocking the competitive pathway (4.3) and cytotoxicity engineering (4.4) are discussed (Fig. 6c and d). In some cases, the strain still has low productivity when the abovementioned strategies have been applied or when engineering strategies are infeasible, and microbial host evolution through a random mutagenesis strategy and systemic analysis should then be applied, as summarized in the last section (4.5, Fig. 6e).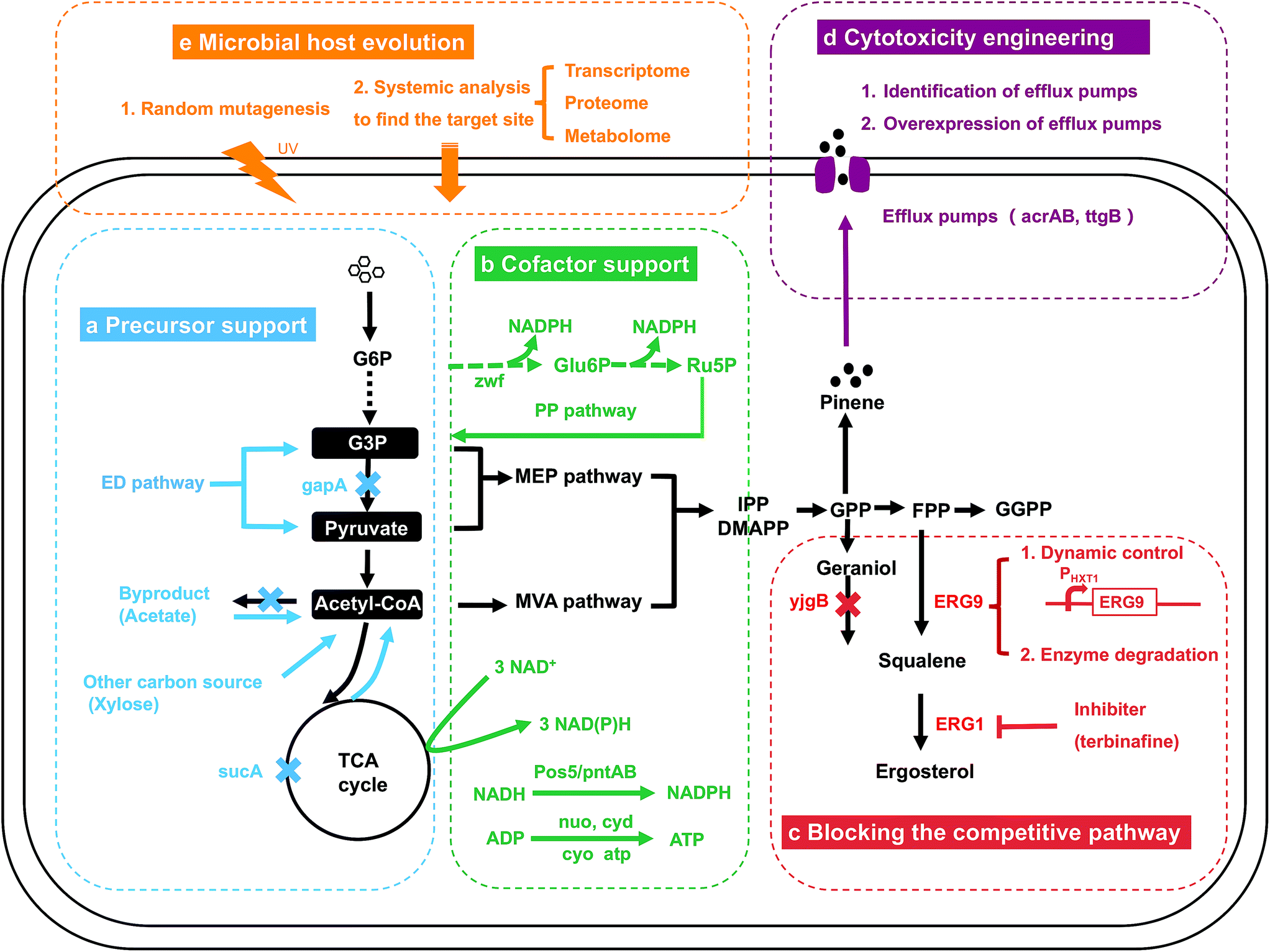 | ||
| Fig. 6 Engineering strategies targeting microbial hosts. Several strategies targeting microbial hosts for isoprenoid biosynthesis, including precursor support (a), cofactor support (b), blocking the competitive pathway (c), cytotoxicity engineering (d) and microbial host evolution (e), are illustrated. | ||
4.1. Precursor support
The common precursors for isoprenoid production, G3P, pyruvate and acetyl-CoA, are produced through the native glycolysis pathway in microbial hosts (Fig. 1). Engineering of the host for sufficient precursor support is very important for isoprenoid production (Fig. 6a). When the MVA pathway is engineered for isoprenoid production, engineering strategies should be applied to accumulate more acetyl-CoA. The deletion or downregulation of genes related to byproduct formation is a powerful approach. In the E. coli AceCo strain, which showed reduced the formation of the byproducts derived from acetyl-CoA (Fig. 6a), such as lactate, acetate and ethanol, isoprene production increased to 1832 mg L−1 in flask culture.104 Deletion of the sucA gene which consumes acetyl-CoA in the TCA cycle, led to a 1.15-fold increase in the mevalonate yield, 0.502 g g−1 of glucose (Fig. 6a).9 Redirecting the carbon flux from the byproduct to acetyl-CoA is also a strategy to address the byproduct problem. In yeast, the overexpression of ADH2 enables the redirection of the carbon flux from ethanol to acetyl-CoA, whereas the overexpression of ALD6 and acsSEL641P allows acetate to be pulled to acetyl-CoA, which ultimately increases the metabolic flux to santalene production (Fig. 6a).105 The introduction of a novel precursor synthesis pathway/carbon source has also been reported. Meadows et al. and Lian et al. introduced the heterologous acetyl-CoA synthetic pathway into S. cerevisiae to increase the acetyl-CoA levels.13,106 The dual utilization of cytoplasmic and mitochondrial acetyl-CoA has also been applied in S. cerevisiae, which increased the production of isoprene to 246 mg L−1, which is approximately 2.1-fold and 1.6-fold greater than that obtained with strains generated by mitochondrial or cytoplasmic engineering alone.107 In yeast, the utilization of xylose as a carbon source resulted in slower xylose consumption, less ethanol and more acetate accumulation, and the production of 150 mg L−1 squalene, which was 8-fold higher than the level obtained with glucose (Fig. 6a).108 Not only isoprenoids but also several chemicals with broad application, including polyketides and lipids, are derived from acetyl-CoA. The engineering approaches summarized above in this section for isoprenoid production are suitable for other acetyl-CoA-derived chemicals.When the MEP pathway is utilized for isoprenoid production, an imbalanced supply of G3P and pyruvate is the major bottleneck because G3P is converted to pyruvate by several steps in the glycolysis pathway (Fig. 1). Equivalent supplies of G3P and pyruvate are necessary. The downregulation of gapA led to a decreased flux from G3P to pyruvate, achieving a balance between precursors.109 Another glycolysis pathway, the ED pathway, which produces equal amounts of G3P and pyruvate, has been utilized for isoprenoid production.110–112 Improvements in isoprenoid production, including carotenoid, isoprene and isopentenol, were obtained. Along with the traditional pathway, a novel pathway was explored to synthesize DXP of the MEP pathway from ribulose 5-phosphate through a reaction catalyzed to by YajO, a putative xylose reductase, and mutant RibB.113 This novel pathway circumvents the first rate-limiting step, which catalyzes the conversion of G3P and pyruvate to DXP. This novel pathway is not dependent on the central carbon system and is promising for high metabolic flux. In conclusion, for the MEP pathway, precursor engineering usually focuses on the equivalent supply of G3P and pyruvate. Precursors of the MVA and MEP pathway are intermediates of the central carbon pathway, and it is necessary to direct sufficient flux to the isoprenoid pathway without affecting normal cell growth.
In recent years, novel microbial hosts have been successfully engineered to produce isoprenoids. Mevalonate and isoprene production was successfully achieved in engineered Clostridium ljungdahlii, which is capable of metabolizing syngas (H2, CO2 or CO) through the Wood–Ljungdahl pathway to produce acetyl-CoA.114 The precursor engineering strategies summarized above are more meaningful for these novel microbial hosts.
4.2. Cofactor support
In addition to sufficient precursor support for the MEP and MVA pathways, cofactor support, e.g., NADPH and ATP, is necessary. The manipulation of cofactor availability should be exploited to improve the reaction flux for isoprenoid production (Fig. 6b). As summarized in the previous section, the MEP pathway consumes ATP and NADPH to produce one molecule of DMAPP from glucose (Fig. 2, Table 1). Multiple strategies have been explored for NADPH engineering. In E. coli, three major sources, namely, the PP pathway, TCA cycle and NADH, which is converted to NADPH by PntAB, are dedicated to NADPH production.115 Engineering based on the PP pathway has been explored. The overexpression of Zwf1, an enzyme of the PP pathway, resulted in an 18.8% increase in the β-carotene titer (0.32 mg L−1) obtained with engineered S. cerevisiae (Fig. 6b).116 In engineered E. coli, overexpression of the zwf gene led to only a 5% increase in the β-carotene yield, and overexpression of the tktA and talB genes, the other two PP pathway genes, led to 16% and 17% increases, respectively, relative to the yield of the control strain (18.4 mg g−1).8 The expression of three genes of the TCA modules, sucAB, gltA and sdhABCD, improved NAD(P)H supply, and 1.39-fold, 1.35-fold and 1.25-fold increases in the β-carotene yield were detected, respectively, based on the same control strain (Fig. 6b).8 The reducing agent NADPH is primarily required in cellular anabolism, but NADH is the main redox product in cellular catabolism, e.g., the glycolysis pathway. An NADH kinase (Pos5), which catalyzes the conversion of NADH to NADPH, was overexpressed in engineered S. cerevisiae and resulted in a 1.5-fold increase in protoilludene production to 479.5 mg L−1 (Fig. 6b).117 In E. coli, the proton-translocating transhydrogenase PntAB exerted the same effect, and PntAB was overexpressed for isoprenoid production (Fig. 6b).115 Deletion of the competitive pathway that consumes NADPH can aid the accumulation of NADPH. Deletion of YjgB, an NADPH-dependent aldehyde reductase, was applied, and the target protoilludene yield was improved to 512.7 mg L−1, which corresponded to a 1.1-fold increase.117 Similarly, deletion of GDH1, which consumes NADPH in the ammonium assimilation pathway, has also been explored in a few studies, and a cubebol production yield of 6 mg L−1, which corresponded to a 1.9-fold increase, was obtained.118,119 However, deletion of GDH1 also caused a decrease in cell growth.118,119 In addition to NADPH, a sufficient supply of ATP is essential for isoprenoid production. Modulation of the atp gene operons in the ATP synthesis module (Fig. 6b) led to a 1.21-fold increase in the β-carotene yield to 22.3 mg L−1.8 In general, when the MEP pathway is utilized for isoprenoid production, cofactor engineering is a powerful strategy for improvement, and a sufficient NADPH supply is the highlight of cofactor engineering.However, very few cofactor engineering strategies were performed for the MVA pathway. The MVA pathway consumes two molecules of NADPH for one molecule of DMAPP production, and the upstream glycolysis pathway produces six molecules of NADH (Fig. 2, Table 1). Overexpression of Pos5, an enzyme that converts NADH to NADPH, is the major strategy utilized for cofactor engineering of the strain utilizing the MVA pathway.116 In conclusion, less cofactor is required in the MVA pathway, and cofactor engineering might not significantly promote production.
4.3. Blocking the competitive pathway
In microorganisms, the isoprenoid synthetic pathway is related to many physiological pathways for cell growth, leading to many bypass and competitive pathways for target isoprenoid production. Redirecting the carbon flux from the bypass pathway and the competitive pathway is necessary for improved isoprenoid production. Deletion of the competing enzyme is usually performed. Specifically, deletion of yjgB in E. coli and OYE2 and ATF1 in S. cerevisiae, which encode enzymes for geraniol degradation, redirected more flux to geraniol production (Fig. 6c).120,121 Deletion of CrtR, which represses crt operon expression, led to a 15–30 fold increase in decaprenoxanthin accumulation, up to 4.07 mg g−1 CDW.122 However, when the bypass pathway is indispensable for cell growth, complete knockout is lethal.123 Downregulation strategies at the transcriptional level, including selecting a weaker promoter or performing RNA interference, have been applied. For sesquiterpenoid production from FPP, squalene production from FPP catalyzed by SQS (ERG9) is considered a bypass pathway (Fig. 6c). Ergosterol, produced from squalene, is an essential cell membrane component (Fig. 6c). Engineering strategies should be applied to achieve a balance between production of the target sesquiterpenoid and the production of squalene. Downregulation of ERG9 was achieved by replacing the native promoter with PBTS1, a weak promoter.53 Dynamic control of the bypass pathway has also been explored. In the engineered zerumbone-producing S. cerevisiae, the native ERG9 promoter was replaced by the PHXT1 promoter to couple its expression with the concentration of glucose in the medium (Fig. 6c).124,125 Protein destabilization approaches have also been applied for flux-competing enzymes, such as ERG9 and ERG20. A PEST peptide tag-activated endoplasmic reticulum-associated protein degradation mechanism was established to control ERG9 expression at the posttranslational level, and the production of sesquiterpene (nerolidol) was improved by 1.85-fold to 100 mg L−1.126 In another study, ERG20 degradation was mediated by an N-degron-dependent protein degradation strategy, and this engineering strategy has been successfully applied for several monoterpenes.127 In contrast to the dynamic control of flux-limiting enzymes, a protein degradation strategy does not require any addition of inducers, repressors or specific conditions. The addition of an inhibitor is also a feasible strategy for flux-competing enzymes. ERG1 catalyzes ergosterol synthesis from squalene, which is a necessary process for cell growth. For squalene accumulation, terbinafine, an ERG1 inhibitor, was added to maintain the content of ergosterol at a low level and thereby control the loss of carbon from squalene.128 In conclusion, the strategies summarized above, including deletion, downregulation or dynamic control of the expression of flux-competing enzymes and utilization of enzyme inhibitors, have been successfully applied for target isoprenoid production.4.4. Cytotoxicity engineering
Most isoprenoids, particularly monoterpenes, are highly toxic to microorganisms (Table 2).129 Product cytotoxicity is a difficult problem to solve for isoprenoid production in microorganisms. Several strategies have been explored to address this problem (Fig. 6d). Exports of toxic compounds by efflux pumps have been applied to several isoprenoid products (Fig. 6d). The published toxic dose and efflux pumps for certain isoprenoids are summarized in Table 2. Pinene shows high toxicity to E. coli (Table 2).130 Efflux pumps, such as AcrAB from E. coli and TtgB from Pseudomonas putida KT2440, which can enhance pinene tolerance, were overexpressed in the pinene-producing strain, and this overexpression improved pinene production to 8 mg L−1 and 7.5 mg L−1, which corresponded to approximately 1.5-fold and 1.4-fold increases, respectively.131 Pleiotropic resistant pumps that have a wide spectrum of substrates with antimicrobial activities, such as amorphadiene, were screened for isoprenoids tolerance.132 The best combination of efflux pumps, TolC and TolC and AcrB, was identified to improve the yield of amorphadiene to 404.83 mg L−1, which was 63% greater than that of the control.132 However, when the efflux pump information is unclear, efforts should be made to identify efflux pumps. A library containing 43 efflux pumps was constructed and a competitive growth assay was utilized to screen the library and identify the matched pumps for seven advanced biofuels.130 In addition, genomic library approaches have been applied. The genomic DNA from Marinobacter aquaeolei VT8, which survives in hydrocarbon-rich environments, was digested into various fragments, inserted into plasmids and transformed into E. coli. The E. coli library was cultivated with the selector pinene and screened by OD600, and the yceI gene improved pinene tolerance.133 In general, chemicals with similar structures share the same efflux pumps and the same tolerance mechanisms. The strategies successfully applied for one isoprenoid component might be feasible for another isoprenoid component.134 Although many isoprenoids have been shown to be toxic, relatively few studies on cytotoxicity engineering has been reported. With improvements in the isoprenoid production level, more research should be focused on cytotoxicity. In addition to the final product of the metabolic pathway, the cellular accumulation of toxic intermediates is observed when the heterogeneous pathway is overexpressed. Unlike the efflux pump engineering strategy for the target isoprenoid, circumventing the synthesis of toxic intermediates is the major solution. The intermediates of the MVA pathway, IPP and DMAPP, were ultimately proven to impair cell growth and influence product synthesis.41,135 As the novel pathway noted above, the IPP/DMAPP-bypass pathway, which directly produces isopentenol from mevalonate or mevalonate monophosphate, has been established to avoid the accumulation of IPP and DMAPP.67 Alternatively, a cell-free system that mixes the required enzymes together in a reaction tube can circumvent the toxicity of products and intermediates; moreover, other limiting factors can be resolved, such as the comparative pathway in the host and the isolation cost of the target product. A cell-free system with 27 enzymes was designed for monoterpene production, and existing titers (>15 g L−1) were obtained.136 In general, the cytotoxicity of certain isoprenoids and intermediates to the microbial host has been proved in many studies, and cytotoxicity engineering is expected to promote production in the future.| Isoprenoid | Toxic dosea | Efflux pumps | References |
|---|---|---|---|
| a The lowest dose that completely inhibited the growth of E. coli. b The isoprenoid that cannot inhibit E. coli growth completely and the dose higher than the toxic dose have no further effect on cell growth. c The pumps that improve pinene tolerance but not its production are indicated. | |||
| Geraniol | 0.05% | ArcB | 130 |
| Pineneb | 0.5%, 0.2% | TtgB from Pseudomonas putida KT2440, AcrBDFac from Alcanivorax borkumensis, MexFc from P. putida, YceI from Marinobacter aquaeolei, AcrB, AcrAB | 130, 131 and 133 |
| Terpinolene | 0.6% | YceI from M. aquaeolei | 133 |
| Limonene | 0.025% | ArcB, AcrBDFa from Alcanivorax borkumensis, | 130 |
| Farnesyl hexanoateb | 1% | MexF from P. putida | 129 and 130 |
| 2-Methyl-1-butanol | 0.1% | 129 | |
| 3-Methyl-1-butanol | 0.1% | 129 | |
| Geranyl acetateb | 0.5% | AcrBDFa from A. borkumensis | 129 and 130 |
| Amorphadiene | — | AcrB, TolC | 132 and 137 |
| Kaurene | — | MexB from P. aeruginosa, AcrB, TolC | 132 |
| Canthaxanthin | — | MsbA | 138 |
| β-Carotene | — | MsbA | 138 |
| Sabinene | 0.5 g L−1 | 139 | |
4.5. Microbial host evolution
The engineering methods noted above target specific areas, including the bypass pathway, precursors, cofactors and cytotoxicity. The evolution of microbial hosts through random mutagenesis and systemic analysis is usually applied with no specific engineering target. Several studies on the random mutagenesis of the astaxanthin-hyperproducing Haematococcus pluvialis have been published. A three-stage mutagenesis breeding strategy, including UV irradiation, ethyl methane sulfonate and diphenylamine, was performed on H. pluvialis, and the study yielded a mutant with an astaxanthin production yield of 47.21 mg g−1 dry cells, which corresponded to a 1.7-fold increase compared with the wild type.140 In another study, random UV mutagenesis was performed on wild-type H. pluvialis, and a rapid screening method, which involved a combination of an azide-based colorimetric assay with oil-based extraction, was utilized.141 However, similar to the directed evolution of enzymes, this strategy requires considerable work, and a high-throughput screening method should be feasible. Based on the whole cell, the identification of target genes for overexpression or knockout might be an alternative method (Fig. 6e). Systemic analysis, including transcriptome, proteome and metabolome analyses, has been mostly applied for the identification of target genes (Fig. 6e). Through an in silico analysis of the S. cerevisiae metabolic network, genes whose knockout can potentially increase IPP accumulation were identified, and the yield of amorphadiene produced by the relevant mutant strain reached 54.55 mg L−1, which was 12-fold greater than that obtained with the wild-type strain.142 In another study, a computational algorithm was built to identify the overexpression target for improving isoprenoid production.143 The random mutagenesis strategy and systemic analysis are two strategies applied without considering the product, host and pathway. In general, these two strategies are time-consuming, and substantial work is required to screen the positive mutants and genes. However, the identified mutants and genes for production improvement are usually creative and original.5. Conclusions and perspectives
As an alternative to the largely energy-consuming and environmentally unfriendly traditional approaches, the microbial production of isoprenoids is expected to be successfully applied in industry, not just in the laboratory. The microbial production of isoprenoids has been explored in the past twenty years. Several reviews on isoprenoid production have been published, and the present review summarizes the metabolic engineering approaches that have been applied to target the MEP and MVA pathways, downstream isoprenoid pathway and microbial host, particularly those developed in the past three years.Although increasingly higher productivity levels, novel isoprenoid production and novel engineering strategies have been reported, few engineered strains have been widely applied in industry. However, the successful production of the antimalarial agent artemisinin provides strong encouragement for additional efforts to optimize isoprenoid production. More attention should be paid to the following prospects in future research. First, in prokaryotic hosts, the utilization of both the MEP and MVA pathways for isoprenoid production should be engineered. Communication between the native MEP and heterogeneous MVA pathways has been confirmed, but the mechanisms involved remain unclear. Second, the cytotoxicity of the pathway intermediates is a serious problem that limits isoprenoid production. The toxicity of IPP and DMAPP has been reported in several articles, but no engineering approaches to address this problem have been published. The dynamic control of IPP and DMAPP expression might be a solution. Compartmentalization of the partial MEP or MVA pathway can also resolve this problem through the accumulation of intermediate concentrations in specific regions, which can improve production. Third, with the increasingly higher levels of isoprenoid production being reported, the cellular tolerance of some end-products, e.g., pinene,130 sabinene139 and certain biofuels, limits further improvement. An engineering approach aiming to improve the cellular tolerance of a specific isoprenoid without an additional metabolic burden is needed. Fourth, in the downstream isoprenoid pathway, terpene synthase from plants is expressed in microbes, and the low-level catalytic efficiency of terpene synthetase limits isoprenoid production. The lack of structural information on terpene synthetase and the high-throughput screening method lead to difficulty in terpene synthase engineering. Fifth, the isoprenoid synthesis pathway is a multigene pathway, and gene expression regulation with highly efficient tools, such as the Cas9-based toolkit, would solve the low-efficiency problem of the traditional gene manipulation tools.101 Finally, nonnatural isoprenoid synthesis in microbial hosts might be a promising research aspect. The synthetic pathway of a nonnatural C11 isoprenoid was successfully established in yeast.144 Of course, the breakthroughs for improvements in isoprenoid production include but are not limited to these research directions.
6. Abbreviations
| (G3P) | Glyceraldehyde-3-phosphate |
| (Ac-CoA) | Acetyl-CoA |
| (DXP) | 1-Deoxy-D-xylulose-5-phosphate |
| (MEP) | Methylerythritol phosphate |
| (CDP-ME) | 4-Diphosphocytidyl-2C-methyl-D-erythritol |
| (CDP-MEP) | 4-Diphosphocytidyl-2C-methyl-D-erythritol-2-phosphate |
| (MEC) | 2C-Methyl-D-erythritol-2,4-cyclo-diphosphate |
| (HMBPP) | 4-Hydroxy-3-methyl-2-(E)-butenyl-4-diphosphate |
| (IPP) | Isopentenyl diphosphate |
| (DMAPP) | Dimethylallyl diphosphate |
| (AcAc-CoA) | Acetoacetyl-CoA |
| (HMG-CoA) | 3-Hydroxy-3-methylglutaryl-CoA |
| (MVA) | Mevalonate |
| (MVAP) | Mevalonate-5-phosphate |
| (MVAPP) | Mevalonate-5-pyrophosphate |
| (IP) | Isopentenyl phosphate |
| (GPP) | Geranyl diphosphate |
| (FPP) | Farnesyl diphosphate |
| (GGPP) | Geranylgeranyl diphosphate |
| (GFPP) | Geranylfarnesyl diphosphate |
| (DXS) | DXP synthase |
| (DXR/IspC) | DXP reductoisomerase |
| (IspD) | CDP-ME cytidylyltransferase |
| (IspE) | CDP-ME kinase |
| (IspF) | MEC synthase |
| (IspG) | HMBPP synthase |
| (IspH) | HMBPP reductase |
| (IDI) | Isopentenyl-diphosphate isomerase |
| (ACCT) | Acetoacetyl-CoA thiolase |
| (HMGS) | HMG-CoA synthase |
| (HMGR) | HMG-CoA reductase |
| (MK) | Mevalonate kinase |
| (PMK) | MVAP kinase |
| (MDD) | MVAPP decarboxylase |
| (MPD) | MVAP decarboxylase |
| (IPK) | IP kinase |
| (TS) | Terpene synthetase |
| (GPPS/IspA) | GPP synthetase |
| (FPPS/IspA) | FPP synthetase |
| (GGPPS/CrtE) | GGPP synthetase |
| (GFPPS) | GFPP synthetase |
| (OleTJE) | Fatty acid decarboxylase |
| (OhyAEM) | Oleate hydratase |
| (GES) | Geraniol synthase |
| (ADS) | Amorpha-4,11-diene synthase |
| (TXS) | Taxadiene synthase |
| (ED pathway) | Entner–Doudoroff pathway |
| (PP pathway) | Pentose phosphate pathway |
| (TCA cycle) | Tricarboxylic acid cycle |
| (PntAB) | Transhydrogenase |
| (Zwf1) | Glucose-6-phosphate dehydrogenase |
| (ERG20) | GPP/FPP synthase |
| (LIS) | (S)-Linalool synthase |
| (PHB) | Polyhydroxybutyrate |
| (SQS) | Squalene synthase |
7. Conflicts of interest
There are no conflicts of interest to declare.8. Acknowledgements
This work was supported by Hainan's Key Project of Research and Development Plan (No. ZDYF2017155), Youth Innovation Promotion Association CAS (No. 2017252) and Taishan Scholars Climbing Program of Shandong (No. TSPD20150210).9. Notes and reference
- C. Wang, B. Zada, G. Wei and S.-W. Kim, Bioresour. Technol., 2017, 241, 430–438 CrossRef CAS PubMed.
- C. E. Vickers, T. C. Williams, B. Peng and J. Cherry, Curr. Opin. Chem. Biol., 2017, 40, 47–56 CrossRef CAS PubMed.
- J. Yang, Q. Nie, M. Ren, H. Feng, X. Jiang, Y. Zheng, M. Liu, H. Zhang and M. Xian, Biotechnol. Biofuels, 2013, 6, 60 CrossRef CAS PubMed.
- Y. Li and G. Wang, J. Appl. Microbiol., 2016, 121, 932–940 CrossRef CAS PubMed.
- J. Kirby and J. D. Keasling, Nat. Prod. Rep., 2008, 25, 656–661 RSC.
- M. C. Chang and J. D. Keasling, Nat. Chem. Biol., 2006, 2, 674–681 CrossRef CAS PubMed.
- P. M. Dewick, Nat. Prod. Rep., 1999, 16, 97–130 RSC.
- J. Zhao, Q. Li, T. Sun, X. Zhu, H. Xu, J. Tang, X. Zhang and Y. Ma, Metab. Eng., 2013, 17, 42–50 CrossRef CAS PubMed.
- J. Wang, S. Niyompanich, Y. S. Tai, J. Wang, W. Bai, P. Mahida, T. Gao and K. Zhang, Appl. Environ. Microbiol., 2016, 82, 7176–7184 CrossRef CAS PubMed.
- J. S. Dickschat, Nat. Prod. Rep., 2011, 28, 1917–1936 RSC.
- P. K. Ajikumar, W. H. Xiao, K. E. Tyo, Y. Wang, F. Simeon, E. Leonard, O. Mucha, T. H. Phon, B. Pfeifer and G. Stephanopoulos, Science, 2010, 330, 70–74 CrossRef CAS PubMed.
- C. J. Paddon, P. J. Westfall, D. J. Pitera, K. Benjamin, K. Fisher, D. McPhee, M. D. Leavell, A. Tai, A. Main, D. Eng, D. R. Polichuk, K. H. Teoh, D. W. Reed, T. Treynor, J. Lenihan, H. Jiang, M. Fleck, S. Bajad, G. Dang, D. Dengrove, D. Diola, G. Dorin, K. W. Ellens, S. Fickes, J. Galazzo, S. P. Gaucher, T. Geistlinger, R. Henry, M. Hepp, T. Horning, T. Iqbal, L. Kizer, B. Lieu, D. Melis, N. Moss, R. Regentin, S. Secrest, H. Tsuruta, R. Vazquez, L. F. Westblade, L. Xu, M. Yu, Y. Zhang, L. Zhao, J. Lievense, P. S. Covello, J. D. Keasling, K. K. Reiling, N. S. Renninger and J. D. Newman, Nature, 2013, 496, 528 CrossRef CAS PubMed.
- A. L. Meadows, K. M. Hawkins, Y. Tsegaye, E. Antipov, Y. Kim, L. Raetz, R. H. Dahl, A. Tai, T. Mahatdejkul-Meadows, L. Xu, L. Zhao, M. S. Dasika, A. Murarka, J. Lenihan, D. Eng, J. S. Leng, C.-L. Liu, J. W. Wenger, H. Jiang, L. Chao, P. Westfall, J. Lai, S. Ganesan, P. Jackson, R. Mans, D. Platt, C. D. Reeves, P. R. Saija, G. Wichmann, V. F. Holmes, K. Benjamin, P. W. Hill, T. S. Gardner and A. E. Tsong, Nature, 2016, 537, 694–697 CrossRef CAS PubMed.
- E. Czarnotta, M. Dianat, M. Korf, F. Granica, J. Merz, J. Maury, S. A. Baallal Jacobsen, J. Förster, B. E. Ebert and L. M. Blank, Biotechnol. Bioeng., 2017, 114, 2528–2538 CrossRef CAS.
- M. Nogueira, E. M. Enfissi, J. Almeida and P. D. Fraser, Curr. Opin. Biotechnol., 2018, 49, 80–87 CrossRef CAS PubMed.
- L. Zhang, W.-H. Xiao, Y. Wang, M.-D. Yao, G.-Z. Jiang, B.-X. Zeng, R.-S. Zhang and Y.-J. Yuan, Biotechnol. Adv., 2017, 35, 1022–1031 CrossRef CAS PubMed.
- K. Paramasivan and S. Mutturi, Crit. Rev. Biotechnol., 2017, 37, 974–989 CrossRef CAS PubMed.
- P. Liao, A. Hemmerlin, T. J. Bach and M.-L. Chye, Biotechnol. Adv., 2016, 34, 697–713 CrossRef CAS PubMed.
- F. X. Niu, Q. Lu, Y. F. Bu and J. Z. Liu, Synth. Syst. Biotechnol., 2017, 2, 167–175 CrossRef PubMed.
- H. R. Beller, T. S. Lee and L. Katz, Nat. Prod. Rep., 2015, 32, 1508–1526 RSC.
- G. Bian, Z. Deng and T. Liu, Curr. Opin. Biotechnol., 2017, 48, 234–241 CrossRef CAS.
- Y. J. Zhou, E. J. Kerkhoven and J. Nielsen, Nat. Energy, 2018, 3, 925–935 CrossRef CAS.
- S. Y. Lee, H. U. Kim, T. U. Chae, J. S. Cho, J. W. Kim, J. H. Shin, D. I. Kim, Y.-S. Ko, W. D. Jang and Y.-S. Jang, Nat. Catal., 2019, 2, 18–33 CrossRef CAS.
- Y. Zhao, J. Yang, B. Qin, Y. Li, Y. Sun, S. Su and M. Xian, Appl. Microbiol. Biotechnol., 2011, 90, 1915–1922 CrossRef CAS PubMed.
- J. Yang, M. Xian, S. Su, G. Zhao, Q. Nie, X. Jiang, Y. Zheng and W. Liu, PLoS One, 2012, 7, e33509 CrossRef CAS PubMed.
- W. Chang, H. Song, H. Liu and P. Liu, Curr. Opin. Chem. Biol., 2013, 17, 571–579 CrossRef CAS PubMed.
- A. Banerjee, Y. Wu, R. Banerjee, Y. Li, H. Yan and T. D. Sharkey, J. Biol. Chem., 2013, 288, 16926–16936 CrossRef CAS PubMed.
- J. H. Julliard and R. Douce, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 2042–2045 CrossRef CAS PubMed.
- J. N. Broker, B. Muller, N. van Deenen, D. Prufer and C. Schulze Gronover, Appl. Microbiol. Biotechnol., 2018, 102, 6923–6934 CrossRef PubMed.
- X. Cao, L. J. Wei, J. Y. Lin and Q. Hua, Bioresour. Technol., 2017, 245, 1641–1644 CrossRef CAS PubMed.
- S. Y. Park, R. M. Binkley, W. J. Kim, M. H. Lee and S. Y. Lee, Metab. Eng., 2018, 49, 105–115 CrossRef CAS.
- D. Xue, I. I. Abdallah, I. E. de Haan, M. J. Sibbald and W. J. Quax, Appl. Microbiol. Biotechnol., 2015, 99, 5907–5915 CrossRef CAS PubMed.
- N. A. Henke, J. Frohwitter, P. Peters-Wendisch and V. F. Wendisch, in Microbial carotenoids, ed. C. Barreiro and J. Barredo, Humana Press, New York, 2018, ch. 8, pp. 127–141 Search PubMed.
- F. Sonntag, C. Kroner, P. Lubuta, R. Peyraud, A. Horst, M. Buchhaupt and J. Schrader, Metab. Eng., 2015, 32, 82–94 CrossRef CAS PubMed.
- E. Englund, K. Shabestary, E. P. Hudson and P. Lindberg, Metab. Eng., 2018, 49, 164–177 CrossRef CAS.
- K. J. Lauersen, J. Wichmann, T. Baier, S. C. Kampranis, I. Pateraki, B. L. Moller and O. Kruse, Metab. Eng., 2018, 49, 116–127 CrossRef CAS PubMed.
- C. Zhang, V. Y. Seow, X. Chen and H.-P. Too, Nat. Commun., 2018, 9, 1858 CrossRef.
- P. Zhou, W. Xie, Z. Yao, Y. Zhu, L. Ye and H. Yu, Biotechnol. Bioeng., 2018, 115, 1321–1330 CrossRef CAS.
- K. Scholtmeijer, K. Cankar, J. Beekwilder, H. A. Wosten, L. G. Lugones and D. Bosch, Appl. Microbiol. Biotechnol., 2014, 98, 5059–5068 CrossRef CAS PubMed.
- C. Yang, X. Gao, Y. Jiang, B. Sun, F. Gao and S. Yang, Metab. Eng., 2016, 37, 79–91 CrossRef CAS PubMed.
- K. W. George, M. G. Thompson, J. Kim, E. E. K. Baidoo, G. Wang, V. T. Benites, C. J. Petzold, L. J. G. Chan, S. Yilmaz, P. Turhanen, P. D. Adams, J. D. Keasling and T. S. Lee, Metab. Eng., 2018, 47, 60–72 CrossRef CAS PubMed.
- C. Zhang, R. Zou, X. Chen, G. Stephanopoulos and H. P. Too, Appl. Microbiol. Biotechnol., 2015, 99, 3825–3837 CrossRef CAS.
- H. Lu, J. C. Villada and P. K. H. Lee, Trends Biotechnol., 2019, 37, 152–166 CrossRef CAS PubMed.
- K. Zhou, K. Qiao, S. Edgar and G. Stephanopoulos, Nat. Biotechnol., 2015, 33, 377–383 CrossRef CAS.
- K. Friehs, in New trends and developments in biochemical engineering, ed. S. Thomas, Springer, Berlin, Heidelberg, Berlin, Heidelberg, 2003, pp. 47–82 Search PubMed.
- B. Ou, C. Garcia, Y. Wang, W. Zhang and G. Zhu, Biotechnol. Bioeng., 2018, 115, 2467–2478 CrossRef CAS.
- L. Ye, C. Zhang, C. Bi, Q. Li and X. Zhang, Microb. Cell Fact., 2016, 15, 202 CrossRef.
- J. Alonso-Gutierrez, D. Koma, Q. Hu, Y. Yang, L. J. G. Chan, C. J. Petzold, P. D. Adams, C. E. Vickers, L. K. Nielsen, J. D. Keasling and T. S. Lee, Biotechnol. Bioeng., 2018, 115, 1000–1013 CrossRef CAS.
- J. A. Bryant, L. E. Sellars, S. J. Busby and D. J. Lee, Nucleic Acids Res., 2014, 42, 11383–11392 CrossRef CAS.
- V. Gerganova, M. Berger, E. Zaldastanishvili, P. Sobetzko, C. Lafon, M. Mourez, A. Travers and G. Muskhelishvili, Nucleic Acids Res., 2015, 43, 8215–8226 CrossRef CAS.
- J. Yin, H. Wang, X. Fu, X. Gao, Q. Wu and G. Chen, Appl. Microbiol. Biotechnol., 2015, 99, 5523–5534 CrossRef CAS PubMed.
- H. J. Shen, B. Y. Cheng, Y. M. Zhang, L. Tang, Z. Li, Y. F. Bu, X. R. Li, G. Q. Tian and J. Z. Liu, Metab. Eng., 2016, 38, 180–190 CrossRef CAS PubMed.
- W. Xie, L. Ye, X. Lv, H. Xu and H. Yu, Metab. Eng., 2015, 28, 8–18 CrossRef CAS.
- K. W. George, A. Chen, A. Jain, T. S. Batth, E. E. K. Baidoo, G. Wang, P. D. Adams, C. J. Petzold, J. D. Keasling and T. S. Lee, Biotechnol. Bioeng., 2014, 111, 1648–1658 CrossRef CAS PubMed.
- V. J. J. Martin, D. J. Pitera, S. T. Withers, J. D. Newman and J. D. Keasling, Nat. Biotechnol., 2003, 21, 796–802 CrossRef CAS PubMed.
- R. H. Dahl, F. Zhang, J. Alonso-Gutierrez, E. Baidoo, T. S. Batth, A. M. Redding-Johanson, C. J. Petzold, A. Mukhopadhyay, T. S. Lee, P. D. Adams and J. D. Keasling, Nat. Biotechnol., 2013, 31, 1039–1046 CrossRef CAS PubMed.
- M. A. Lalwani, E. M. Zhao and J. L. Avalos, Curr. Opin. Biotechnol., 2018, 52, 56–65 CrossRef CAS PubMed.
- M. Li, R. Nian, M. Xian and H. Zhang, Appl. Microbiol. Biotechnol., 2018, 102, 7725–7738 CrossRef CAS PubMed.
- X. Gao, F. Gao, D. Liu, H. Zhang, X. Nie and C. Yang, Energy Environ. Sci., 2016, 9, 1400–1411 RSC.
- A. Zurbriggen, H. Kirst and A. Melis, BioEnergy Res., 2012, 5, 814–828 CrossRef CAS.
- D. D. Hinson, K. L. Chambliss, M. J. Toth, R. D. Tanaka and K. M. Gibson, J. Lipid Res., 1997, 38, 2216–2223 CAS.
- Y. A. Primak, M. Du, M. C. Miller, D. H. Wells, A. T. Nielsen, W. Weyler and Z. Q. Beck, Appl. Environ. Microbiol., 2011, 77, 7772–7778 CrossRef CAS PubMed.
- E. Kazieva, Y. Yamamoto, Y. Tajima, K. Yokoyama, J. Katashkina and Y. Nishio, Microbiology, 2017, 163, 1283–1291 CrossRef CAS PubMed.
- H. Chen, M. Li, C. Liu, H. Zhang, M. Xian and H. Liu, Microb. Cell Fact., 2018, 17, 65 CrossRef.
- H. Chen, C. Liu, M. Li, H. Zhang, M. Xian and H. Liu, RSC Adv., 2018, 8, 15021–15028 RSC.
- J. Yang, Q. Nie, H. Liu, M. Xian and H. Liu, BMC Biotechnol., 2016, 16, 5 CrossRef PubMed.
- A. Kang, K. W. George, G. Wang, E. Baidoo, J. D. Keasling and T. S. Lee, Metab. Eng., 2015, 34, 25–35 CrossRef PubMed.
- D. Ge, Y. Xue and Y. Ma, Microb. Cell Fact., 2016, 15, 79 CrossRef PubMed.
- A. O. Chatzivasileiou, V. Ward, S. M. Edgar and G. Stephanopoulos, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 506–511 CrossRef CAS PubMed.
- S. Lund, R. Hall and G. J. Williams, ACS Synth. Biol., 2019, 8, 232–238 CrossRef CAS.
- B. O. Bachmann, S. G. Van Lanen and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2014, 41, 175–184 CrossRef CAS PubMed.
- T. Ma, Y. Zhou, X. Li, F. Zhu, Y. Cheng, Y. Liu, Z. Deng and T. Liu, Biotechnol. J., 2016, 11, 228–237 CrossRef CAS PubMed.
- D. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Y. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Nature, 2006, 440, 940–943 CrossRef CAS.
- M. C. Chang, R. A. Eachus, W. Trieu, D. K. Ro and J. D. Keasling, Nat. Chem. Biol., 2007, 3, 274–277 CrossRef CAS.
- N. Ziemert, M. Alanjary and T. Weber, Nat. Prod. Rep., 2016, 33, 988–1005 RSC.
- Z. Dai, Y. Liu, Z. Sun, D. Wang, G. Qu, X. Ma, F. Fan, L. Zhang, S. Li and X. Zhang, Metab. Eng., 2018, 51, 70–78 CrossRef PubMed.
- K. Miettinen, J. Pollier, D. Buyst, P. Arendt, R. Csuk, S. Sommerwerk, T. Moses, J. Mertens, P. D. Sonawane, L. Pauwels, A. Aharoni, J. Martins, D. R. Nelson and A. Goossens, Nat. Commun., 2017, 8, 14153 CrossRef CAS PubMed.
- D. C. Chambers, A. M. Carew, S. W. Lukowski and J. E. Powell, Respirology, 2019, 24, 29–36 CrossRef.
- X. Song and Q. Lin, Rheumatol. Int., 2017, 37, 1257–1265 CrossRef CAS.
- H. Machado, R. N. Tuttle and P. R. Jensen, Curr. Opin. Microbiol., 2017, 39, 136–142 CrossRef CAS.
- J. Zhao, X. Bao, C. Li, Y. Shen and J. Hou, Appl. Microbiol. Biotechnol., 2016, 100, 4561–4571 CrossRef CAS.
- Q. Lu, Y. F. Bu and J. Z. Liu, Mar. Drugs, 2017, 15, 296 CrossRef.
- R. A. Chica, N. Doucet and J. N. Pelletier, Curr. Opin. Biotechnol., 2005, 16, 378–384 CrossRef PubMed.
- A. Emmerstorfer-Augustin, S. Moser and H. Pichler, J. Biotechnol., 2016, 235, 112–120 CrossRef CAS PubMed.
- S. Lutz, Curr. Opin. Biotechnol., 2010, 21, 734–743 CrossRef CAS PubMed.
- I. I. Abdallah, R. van Merkerk, E. Klumpenaar and W. J. Quax, Sci. Rep., 2018, 8, 9961 CrossRef.
- A. E. Koepp, M. Hezari, J. Zajicek, B. S. Vogel, R. E. LaFever, N. G. Lewis and R. Croteau, J. Biol. Chem., 1995, 270, 8686–8690 CrossRef CAS PubMed.
- S. Edgar, F. S. Li, K. Qiao, J. K. Weng and G. Stephanopoulos, ACS Synth. Biol., 2017, 6, 201–205 CrossRef CAS.
- C. Ignea, M. Pontini, M. E. Maffei, A. M. Makris and S. C. Kampranis, ACS Synth. Biol., 2014, 3, 298–306 CrossRef CAS.
- Y. Gao, R. B. Honzatko and R. J. Peters, Nat. Prod. Rep., 2012, 29, 1153–1175 RSC.
- T. Jakociunas, L. E. Pedersen, A. V. Lis, M. K. Jensen and J. D. Keasling, Metab. Eng., 2018, 48, 288–296 CrossRef CAS.
- P. Zhou, W. Xie, A. Li, F. Wang, Z. Yao, Q. Bian, Y. Zhu, H. Yu and L. Ye, Enzyme Microb. Technol., 2017, 100, 28–36 CrossRef CAS.
- W. Xie, X. Lv, L. Ye, P. Zhou and H. Yu, Metab. Eng., 2015, 30, 69–78 CrossRef CAS.
- M. Furubayashi, M. Ikezumi, J. Kajiwara, M. Iwasaki, A. Fujii, L. Li, K. Saito and D. Umeno, PLoS One, 2014, 9, e93317 CrossRef PubMed.
- M. Tashiro, A. Fujii, S. Kawai-Noma, K. Saito and D. Umeno, J. Gen. Appl. Microbiol., 2017, 63, 287–295 CrossRef CAS PubMed.
- J. Lin, H. Ekas, K. Markham and H. S. Alper, Biochem. Eng. J., 2018, 139, 95–100 CrossRef CAS.
- R. J. Conrado, J. D. Varner and M. P. DeLisa, Curr. Opin. Biotechnol., 2008, 19, 492–499 CrossRef CAS PubMed.
- P. A. Srere, Annu. Rev. Biochem., 1987, 56, 89–124 CrossRef CAS.
- Y. Deng, M. Sun, S. Xu and J. Zhou, J. Appl. Microbiol., 2016, 121, 187–195 CrossRef CAS.
- K. Zhou, R. Zou, C. Zhang, G. Stephanopoulos and H.-P. Too, Biotechnol. Bioeng., 2013, 110, 2556–2561 CrossRef CAS PubMed.
- A. Reider Apel, L. d'Espaux, M. Wehrs, D. Sachs, R. A. Li, G. J. Tong, M. Garber, O. Nnadi, W. Zhuang, N. J. Hillson, J. D. Keasling and A. Mukhopadhyay, Nucleic Acids Res., 2017, 45, 496–508 CrossRef.
- J. E. Chaves, P. Rueda-Romero, H. Kirst and A. Melis, ACS Synth. Biol., 2017, 6, 2281–2292 CrossRef CAS PubMed.
- S. Y. Choi, J. Y. Wang, H. S. Kwak, S. M. Lee, Y. Um, Y. Kim, S. J. Sim, J. I. Choi and H. M. Woo, ACS Synth. Biol., 2017, 6, 1289–1295 CrossRef CAS PubMed.
- J. H. Kim, C. Wang, H. J. Jang, M. S. Cha, J. E. Park, S. Y. Jo, E. S. Choi and S. W. Kim, Microb. Cell Fact., 2016, 15, 214 CrossRef.
- Y. Chen, L. Daviet, M. Schalk, V. Siewers and J. Nielsen, Metab. Eng., 2013, 15, 48–54 CrossRef CAS.
- J. Lian, T. Si, N. U. Nair and H. Zhao, Metab. Eng., 2014, 24, 139–149 CrossRef CAS PubMed.
- X. Lv, F. Wang, P. Zhou, L. Ye, W. Xie, H. Xu and H. Yu, Nat. Commun., 2016, 7, 12851 CrossRef CAS.
- S. Kwak, S. R. Kim, H. Xu, G. Zhang, S. Lane, H. Kim and Y. Jin, Biotechnol. Bioeng., 2017, 114, 2581–2591 CrossRef CAS PubMed.
- J. Jung, J. H. Lim, S. Y. Kim, D. Im, J. Y. Seok, S. V. Lee, M. Oh and G. Y. Jung, Metab. Eng., 2016, 38, 401–408 CrossRef CAS PubMed.
- H. Liu, Y. Sun, K. R. Ramos, G. M. Nisola, K. N. Valdehuesa, W. K. Lee, S. J. Park and W. J. Chung, PLoS One, 2013, 8, e83290 CrossRef PubMed.
- H. Liu, Y. Wang, Q. Tang, W. Kong, W. J. Chung and T. Lu, Microb. Cell Fact., 2014, 13, 135 CrossRef PubMed.
- C. Li, L. Q. Ying, S. S. Zhang, N. Chen, W. F. Liu and Y. Tao, Microb. Cell Fact., 2015, 14, 117 CrossRef PubMed.
- J. Kirby, M. Nishimoto, R. W. N. Chow, E. E. K. Baidoo, G. Wang, J. Martin, W. Schackwitz, R. Chan, J. L. Fortman and J. D. Keasling, Appl. Environ. Microbiol., 2015, 81, 130–138 CrossRef PubMed.
- B. A. Diner, J. Fan, M. C. Scotcher, D. H. Wells and G. M. Whited, Appl. Environ. Microbiol., 2018, 84, e01723-17 CrossRef PubMed.
- U. Sauer, F. Canonaco, S. Heri, A. Perrenoud and E. Fischer, J. Biol. Chem., 2004, 279, 6613–6619 CrossRef CAS PubMed.
- X. Zhao, F. Shi and W. Zhan, Lett. Appl. Microbiol., 2015, 61, 354–360 CrossRef CAS PubMed.
- J. Zhou, L. Yang, C. Wang, E. Choi and S. Kim, J. Biotechnol., 2017, 248, 1–8 CrossRef CAS PubMed.
- G. Scalcinati, S. Partow, V. Siewers, M. Schalk, L. Daviet and J. Nielsen, Microb. Cell Fact., 2012, 11, 117 CrossRef CAS PubMed.
- M. A. Asadollahi, J. Maury, K. R. Patil, M. Schalk, A. Clark and J. Nielsen, Metab. Eng., 2009, 11, 328–334 CrossRef CAS PubMed.
- J. Zhou, C. Wang, S. H. Yoon, H. J. Jang, E. S. Choi and S. W. Kim, J. Biotechnol., 2014, 169, 42–50 CrossRef CAS PubMed.
- J. Zhao, C. Li, Y. Zhang, Y. Shen, J. Hou and X. Bao, Microb. Cell Fact., 2017, 16, 17 CrossRef PubMed.
- N. A. Henke, S. A. E. Heider, S. Hannibal, V. F. Wendisch and P. Peters-Wendisch, Front. Microbiol., 2017, 8, 633 CrossRef PubMed.
- M. S. Anderson, J. G. Yarger, C. L. Burck and C. D. Poulter, J. Biol. Chem., 1989, 264, 19176–19184 CAS.
- C. Zhang, J. Liu, F. Zhao, C. Lu, G. Zhao and W. Lu, Metab. Eng., 2018, 49, 28–35 CrossRef CAS PubMed.
- G. Scalcinati, C. Knuf, S. Partow, Y. Chen, J. Maury, M. Schalk, L. Daviet, J. Nielsen and V. Siewers, Metab. Eng., 2012, 14, 91–103 CrossRef CAS PubMed.
- B. Peng, M. R. Plan, P. Chrysanthopoulos, M. P. Hodson, L. K. Nielsen and C. E. Vickers, Metab. Eng., 2017, 39, 209–219 CrossRef CAS PubMed.
- B. Peng, L. K. Nielsen, S. C. Kampranis and C. E. Vickers, Metab. Eng., 2018, 47, 83–93 CrossRef CAS PubMed.
- J. Y. Han, S. H. Seo, J. M. Song, H. Lee and E. S. Choi, J. Ind. Microbiol. Biotechnol., 2018, 45, 239–251 CrossRef CAS PubMed.
- M. J. Dunlop, Biotechnol. Biofuels, 2011, 4, 32 CrossRef CAS PubMed.
- M. J. Dunlop, Z. Y. Dossani, H. L. Szmidt, H. C. Chu, T. S. Lee, J. D. Keasling, M. Z. Hadi and A. Mukhopadhyay, Mol. Syst. Biol., 2011, 7, 487 CrossRef PubMed.
- F. Niu, X. He, Y. Wu and J. Liu, Front. Microbiol., 2018, 9, 1623 CrossRef PubMed.
- J. Wang, Z. Xiong, S. Li and Y. Wang, Appl. Microbiol. Biotechnol., 2013, 97, 8057–8067 CrossRef CAS PubMed.
- T. A. Tomko and M. J. Dunlop, Biotechnol. Biofuels, 2015, 8, 165 CrossRef PubMed.
- S. Atsumi, T. Wu, I. M. P. Machado, W. Huang, P. Chen, M. Pellegrini and J. C. Liao, Mol. Syst. Biol., 2010, 6, 449 CrossRef PubMed.
- A. M. Miguez, M. P. McNerney and M. P. Styczynski, Front. Microbiol., 2018, 9, 760 CrossRef PubMed.
- T. P. Korman, P. H. Opgenorth and J. U. Bowie, Nat. Commun., 2017, 8, 15526 CrossRef CAS PubMed.
- C. Zhang, X. Chen, G. Stephanopoulos and H. P. Too, Biotechnol. Bioeng., 2016, 113, 1755–1763 CrossRef CAS PubMed.
- R. Doshi, T. Nguyen and G. Chang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7642–7647 CrossRef CAS PubMed.
- H. Zhang, Q. Liu, Y. Cao, X. Feng, Y. Zheng, H. Zou, H. Liu, J. Yang and M. Xian, Microb. Cell Fact., 2014, 13, 20 CrossRef PubMed.
- N. Wang, B. Guan, Q. Kong, H. Sun, Z. Geng and L. Duan, J. Biotechnol., 2016, 236, 71–77 CrossRef CAS PubMed.
- M. E. Hong, H. I. Choi, H. S. Kwak, S.-W. Hwang, Y. J. Sung, W. S. Chang and S. J. Sim, Bioresour. Technol., 2018, 267, 175–181 CrossRef CAS PubMed.
- A. Galli, Z. Sun, H. Meng, J. Li, J. Wang, Q. Li, Y. Wang and Y. Zhang, PLoS One, 2014, 9, e112615 CrossRef PubMed.
- B. A. Boghigian, J. Armando, D. Salas and B. A. Pfeifer, Appl. Microbiol. Biotechnol., 2012, 93, 2063–2073 CrossRef CAS PubMed.
- C. Ignea, M. Pontini, M. S. Motawia, M. E. Maffei, A. M. Makris and S. C. Kampranis, Nat. Chem. Biol., 2018, 14, 1090–1098 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |