
Extracting value: mechanistic insights into the formation of natural product artifacts – case studies in marine natural products
Robert J.
Capon
 *
*
Division of Chemistry and Structural Biology, Institute for Molecular Bioscience, The University of Queensland, St Lucia, QLD 4072, Australia. E-mail: r.capon@uq.edu.au
First published on 2nd May 2019
Abstract
Covering: up to the end of 2018
Natural selection relentlessly drives the evolution of natural products, favoring those that enhance the survival of species. During this evolutionary journey, some natural products acquire heightened chemical reactivity towards environmental stimuli, most likely benefiting host species through more agile ecological responses, leading to superior survival outcomes (i.e., more potent and faster acting toxins and chemical defences, infection control, and intra/inter species chemical communication). Although knowledge of natural products informs our understanding of the living world, inspiring new medicines, agrochemicals and scientific tools, the study of chemically reactive natural products can be particularly informative, albeit quite challenging. For example, such natural products are often prone to facile transformations during handling, yielding new “un-natural” products commonly referred to as artifacts. These transformations can be induced by many stimuli, including modest changes in pH or temperature, or exposure to light or air (i.e., oxygen), or even common organic solvents or chromatography media. Sadly, in the race to explore and report on new natural products, knowledge of the relationship between chemically reactive natural products and their artifacts is not always recognised, documented or valued. This review seeks to recalibrate and encourage a greater awareness of the relationship between natural products and artifacts, by examining case studies in the field of marine natural products chemistry (i.e., natural products and associated artifacts from marine invertebrates and algae, and marine-derived microbes). While every effort has been made to provide a comprehensive coverage up to early 2019, any omissions are inadvertent not deliberate. Structured around the types of chemical transformations that can deliver artifacts, and the functional groups involved, the review concludes with observations on how to regulate, detect, rationalise and even exploit artifacts, going forward.
 Robert J. Capon | Rob Capon is a Professorial Research Fellow and Group Leader at The University of Queensland Institute for Molecular Bioscience, where he leads a research group specializing in the detection, isolation and structure elucidation of natural products from Australian marine and microbial biodiversity. Rob and his team have successfully discovered and documented thousands of metabolites spanning all biosynthetic classes, many featuring rare or new scaffolds, annotated with diverse functional groups and rich in stereo complexity. Knowledge of the chemical and biological properties of these metabolites informs both basic and applied science outcomes, including new molecular tools to better understand living systems, and new bioactive motifs to inspire innovative solutions in human and animal health, as well as crop and environmental protection. |
1. Introduction
Modern natural products science is a multidisciplinary endeavour that explores, learns from, and seeks to apply knowledge of chemical diversity found in Nature (i.e., secondary metabolites from plants, animals, fungi and bacteria). Natural products chemists often target molecules that feature rare and even unique carbon skeletons, annotated with a wide array of functional groups, and replete with stereo-complexity. Knowledge of the chemistry, ecology and pharmacology of natural products holds the promise of new medicines, agrochemicals, materials and research tools. That natural products are primed to deliver on this promise is informed by billions of years of evolution, which has positioned natural products as an accessible, high value pool of chemical diversity, pre-optimised to interact with the natural world. The last 100 years have shown just how successful natural products can be in advancing science, healthcare, agriculture and commerce, to the betterment of society. That natural products exhibit such exquisitely potent and selective chemical and biological properties stems largely from their having evolved to deliver a rapid and highly selective ecological (survival) benefit, in response to specific environmental stimuli, often while exposed to a storm of other stimuli. This pre-programmed reactivity is two-edged, as it also renders some natural products chemically unstable, and prone to facile conversion into artifacts. In the context of this review, artifacts are defined as the products obtained following the non-enzymatic transformation of a natural product during extraction, fractionation, analysis, handling and/or storage. These chemical transformations can be simple and unremarkable, such as the esterification of a carboxylic acid, or solvolysis of an acetal. On the other hand, they can be remarkably elegant, providing valuable insights into fundamental organic chemistry, biosynthetic pathways and biological mechanisms of action, and even inspire biomimetic syntheses and new chemical reactions. Notwithstanding their capacity to reveal valuable knowledge, published accounts of natural products artifacts are limited, and for the most part down played. For example, on far too many occasions artifacts are misidentified as natural products, or simply ignored. This is regrettable, as the under-reporting and under-valuing of artifacts represents a lost opportunity, and compromises our ability to fully understand the properties and potential of natural products. This review seeks to recalibrate and encourage a greater awareness and appreciation of natural product artifacts, by documenting their diversity and occurrence in the field of marine natural products chemistry. The review, loosely structured around the types of chemical transformations that can deliver artifacts, and the functional groups involved in these transformations, concludes with some observations on how to detect, assess, value and exploit artifacts going forward.2. Discussion
2.1. Solvolysis (alcohols)
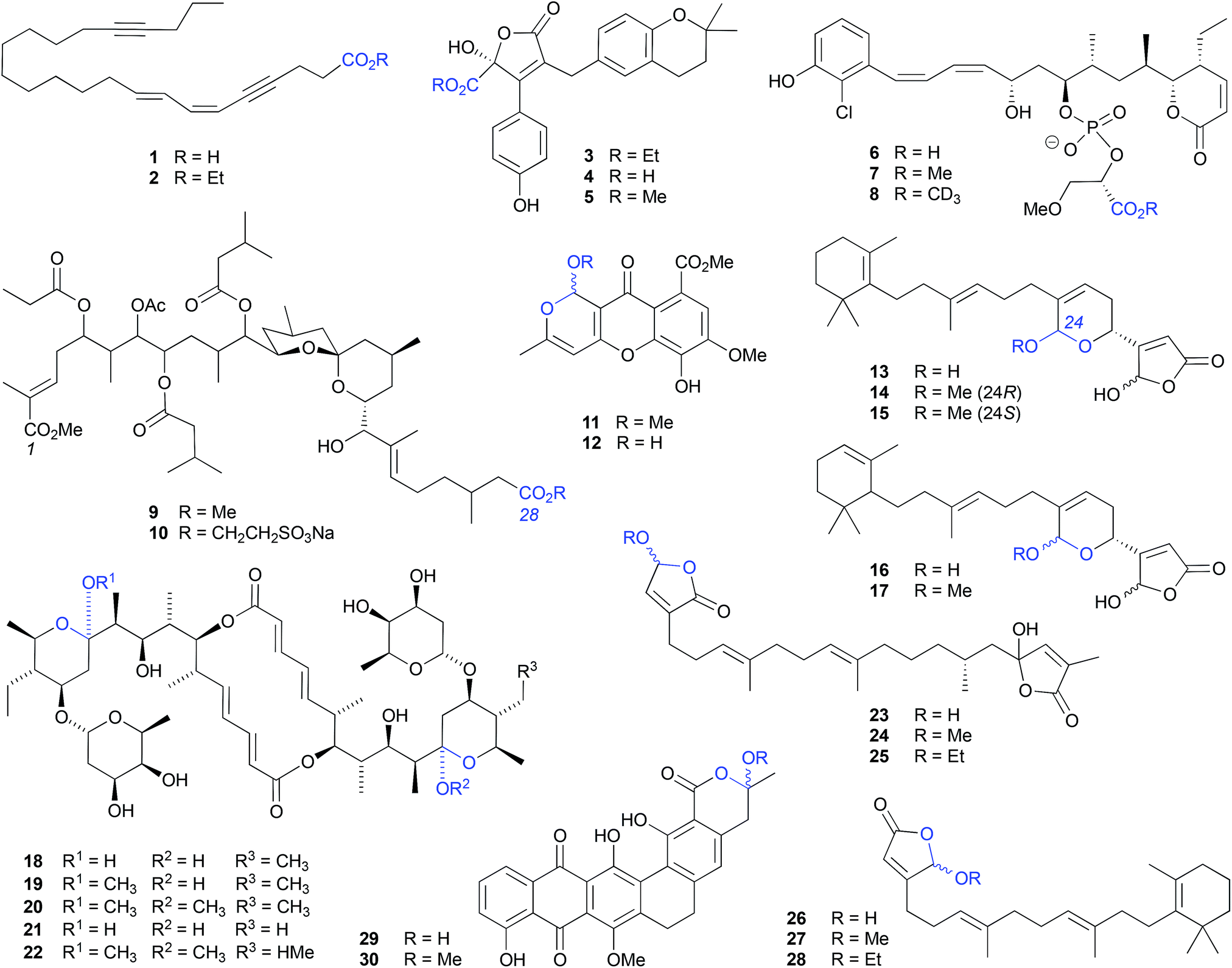 | ||
| Fig. 1 Examples of the solvolysis of carboxylic acids (1–10) and cyclic acetals (11–30). | ||
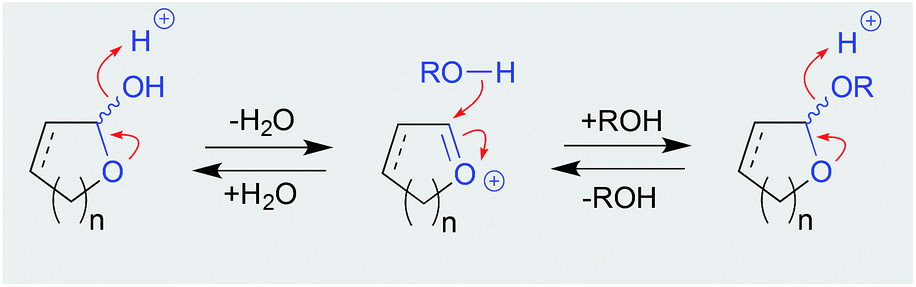 | ||
| Scheme 1 Mechanism for acetal solvolysis (equilibrium). | ||
Although a well-documented transformation, particularly in carbohydrate chemistry, O-alkyl cyclic acetal artifacts are regularly mischaracterised as natural products. For example, first designated a natural product from the marine algae-derived fungus, Chaetomium sp. Gö 100/2, the O-methyl cyclic acetal chaetocyclinone A (11) is more likely a solvolysis adduct of the co-metabolite chaetocyclinone A (12), produced during silica (MeOH) chromatography.6 Similarly, silica (MeOH/CH2Cl2) fractionation of a West Atlantic marine sponge, Luffariella sp., likely induced the known cyclic acetal manoalide (13) to undergo solvolysis to the resolved O-methyl artifacts 14 and 15,7 while silica (MeOH/CHCl3) chromatography of the Okinawan marine sponge Luffariella sp. (SS-996) likely caused the undetected precursor 16 to undergo solvolysis to the artifact luffariolide H (17).8
Notwithstanding that most natural product O-alkyl acetal artifacts are epimeric mixtures, asymmetric induction by adjacent chiral centres can influence configurational outcomes. For example, when exposed to MeOH the natural product elaiophylin (18), isolated from a Chinese marine sediment-derived Streptomyces sp. 7-145, undergoes stereospecific acetal alkylation to 11-O-methylelaiophylin (19) and 11,11′-O-dimethylelaiophylin (20), while the co-metabolite efomycin G (21) undergoes transformation to 11,11′-O-dimethyl-14′deethyl-14′-methylelaiophylin (22).9
The γ-hydroxybutenolide moiety can also be highly susceptible to solvolysis, proceeding through an oxonium intermediate to a mixture of O-alkyl-γ-hydroxybutenolide epimers. For example, the sesterterpene cacolide C (23) isolated from the southern Australian marine sponge Cacospongia sp. (CMB-03404), underwent solvolysis to the artifact cacolides D (24) and E (25).10 Similar transformations are likely to have been in effect during the MeOH/EtOH fractionation of the Indonesian marine sponge Acanthodendrilla sp., causing the sponge natural product luffariellolide (26) to undergo solvolysis to the artifacts 27 and 28.11
Likewise, MeOH fractionation of a marine sediment-derived Streptosporangium sp. CGMCC 4.7309 yielded the natural product hexarin F (29), together with the solvolysis artifact hexarin G (30).12
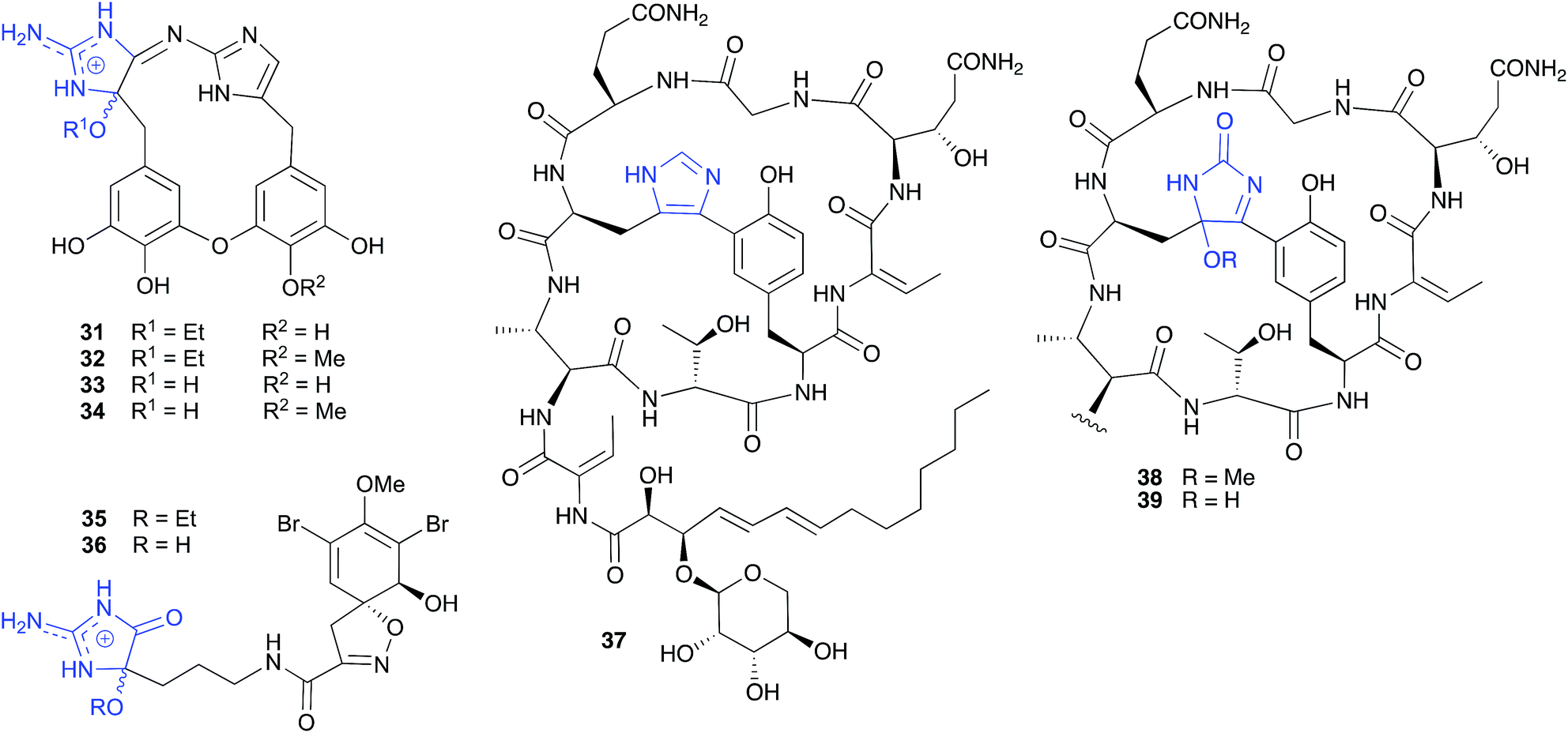 | ||
| Fig. 2 Examples of the solvolysis of cyclic aminols (31–38). | ||
Similarly, and notwithstanding the elaborate multi-step published hypothesis, the ethanolysis artifact 35 reported from a Western Australian marine sponge, Pseudoceratina cf. verrucosa, is most likely an ethanolysis artefact of an undetected cyclic aminol 36.14
 | ||
| Fig. 3 Examples of the solvolysis of cyclic aminols (40–45). | ||
The cyclic peptide bearing an imidazole bridge, aciculatin B (37), isolated from the Philippines marine sponge Aciculites orientalis (NCI-013), was reported to generate an oxidation artifact, aciculitamide A (38), by a mechanism invoking a “[4 + 2] cycloaddition of singlet oxygen across the imidazole ring, followed by cleavage of the peroxide bond and replacement of the hydroxyl group by methoxyl”.17 Although this singlet oxygen transformation could be achieved in the lab, it involved treatment with Rose Bengal and 6 h 400 W UV irradiation at −70 °C, following by quenching and a further 1 h reaction with a solution of 5% diisopropylethylamine in MeOH. As these conditions are at odds with those encountered during isolation and handling, perhaps a simpler and more plausible explanation is that 38 is a methanolysis artifact of the undetected, and highly reactive, hydroxylated analogue 39. Interestingly, the appearance of the artifact 38 in the crude sponge extract was reported to coincide with a loss of biological activity, raising the possibility that the putative precursor 39 may be the source of promising biological properties.
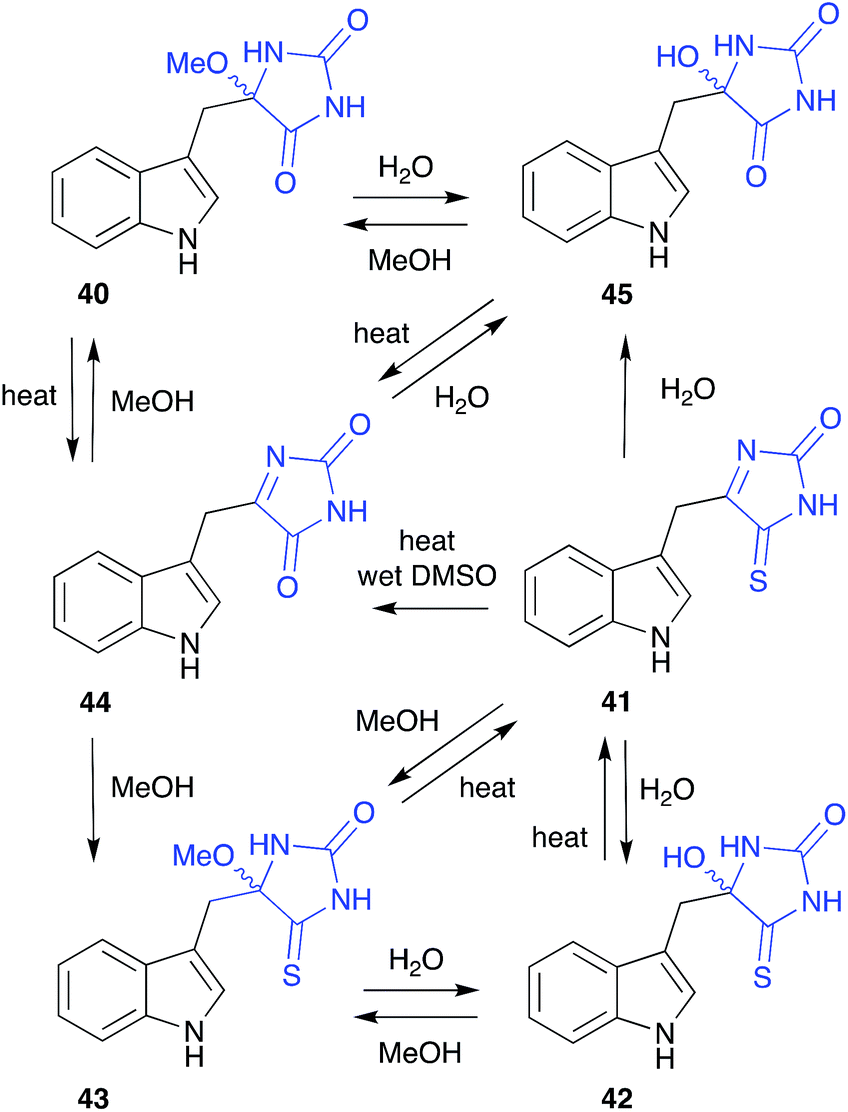
The cyclic O-alkyl aminol polyandrocarpamide D (40), isolated by MeOH chromatography of the Philippines colonial ascidian Polyandrocarpa sp., yielded a complex artifact provenance,15 only fully appreciated after careful analysis of the metabolites of the Coral Sea marine sponge Zyzza massalis.16 This latter study determined that even cyclic aminols can be artifacts, by documenting a network of transformations originating from the thioxo natural product 41. These included; (i) the thermally reversible, facile conversion of 41 to the H2O and MeOH adducts 42 and 43, respectively; (ii) hydrolysis of 41 to the oxo artifact 44 and (iii) the thermally reversible, facile conversion of 44 to the H2O and MeOH adducts 45 and 40.
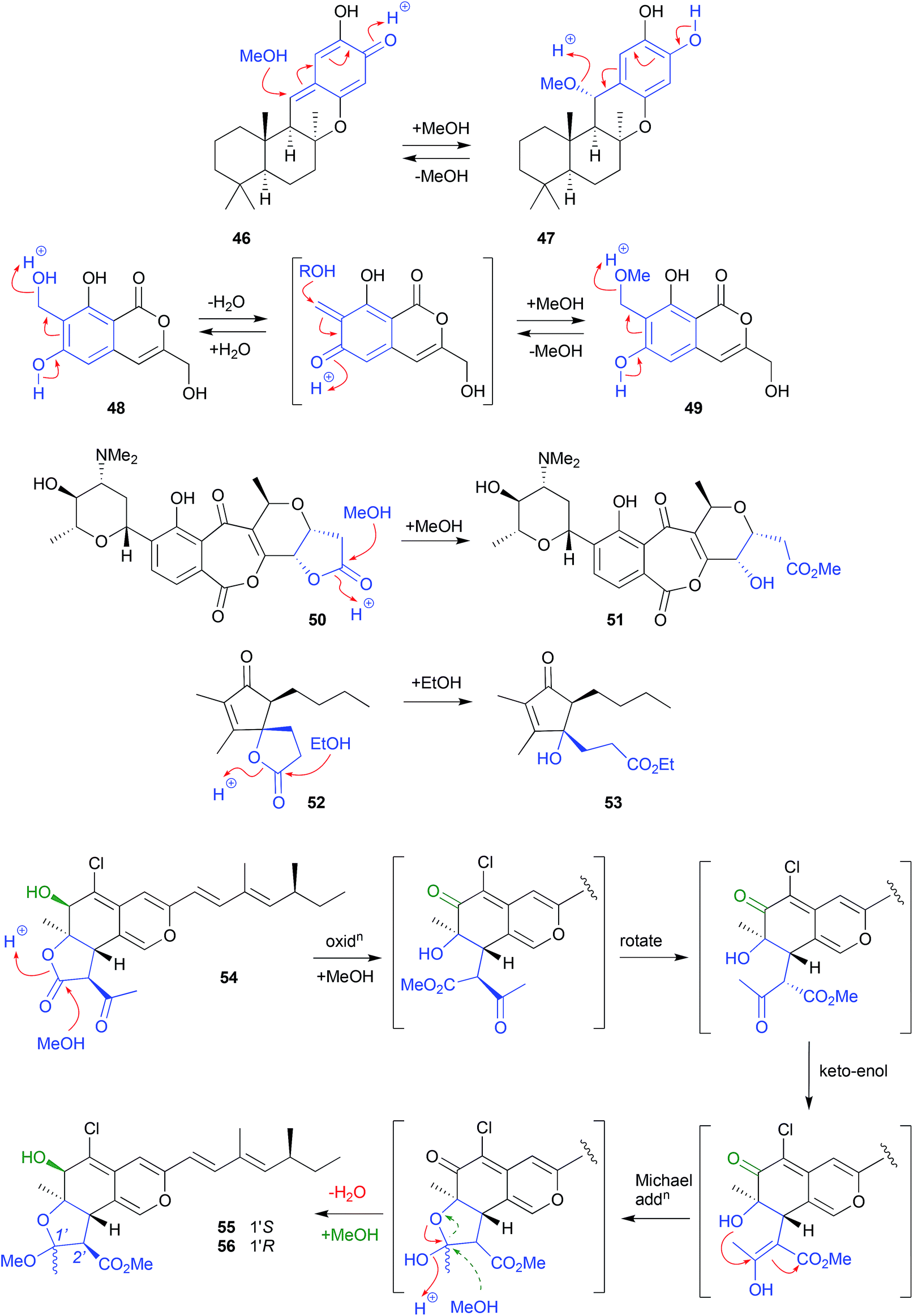 | ||
| Fig. 4 Examples of the solvolysis of quinone-methides (46–49) and lactones (50–56). | ||
By contrast, isochromophilone A (54), recovered from the mangrove derived fungus Diaporthe sp. SCSIO 41011, underwent solvolytic ring opening to a methyl ester intermediate, that subsequently recyclized to a cyclic acetal, before a second solvolysis yielded two isomeric artifacts, isochromophilones C (55) and D (56).24 The apparent inversion of configuration about C-2′ could be explained by an keto–enol tautomerisation of the ring opened methanolysis intermediate, followed by stereospecific intramolecular Michael addition, and methanolysis, to yield only the 2′R diasteromers. Alternatively, the Michael addition may yield both the 2′R and 2′S isomers, with the latter being either unstable and/or not isolated.
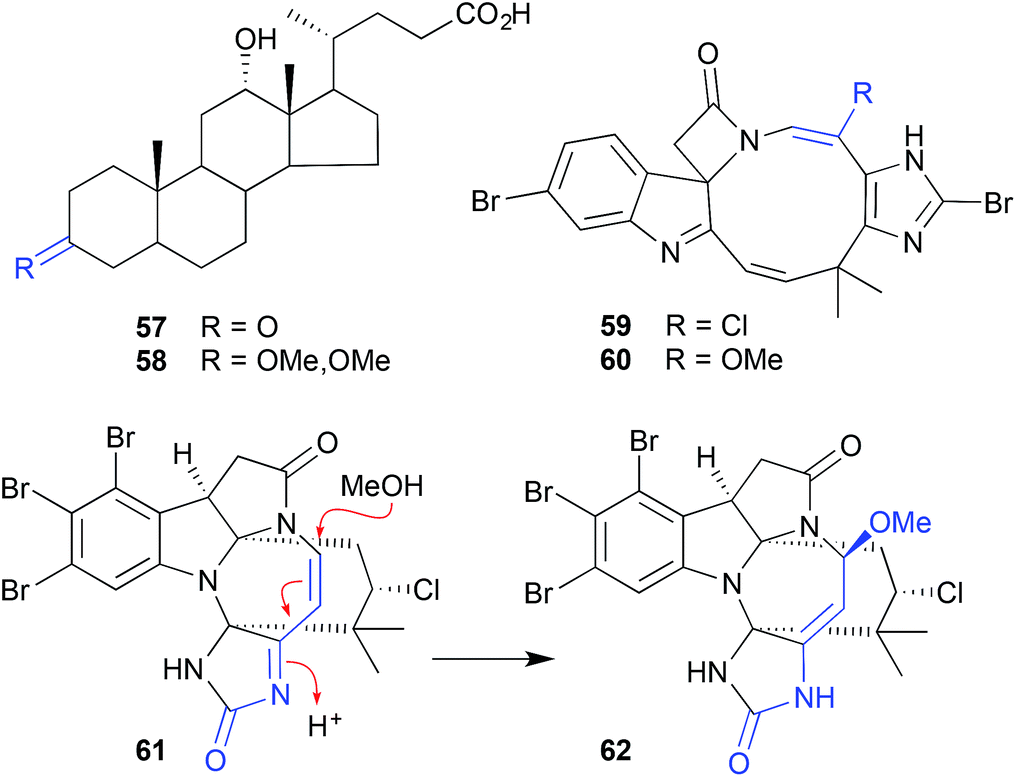 | ||
| Fig. 5 Examples of miscellaneous solvolysis (57–62). | ||
2.2. Solvolysis (acetone) (Fig. 6)
Acetone can be quite reactive, particularly under low or high pH conditions, and for this reason is generally used with caution in natural products research. That said, it is a useful lab solvent, and can make unanticipated appearances. For example, although not used in the extraction and chromatographic fractionation of the Chinese mangrove plant Bruguiera gymnorrhiza, acetone was nevertheless used for transferring residues from distillation flasks. Even this brief exposure transformed the known natural product (+)-lyoniresinol (63) into the artifact bruguierol D (64).28 On the other hand, the use of acetone was opportune when used to fractionate the Texas seawater-derived Streptomyces spinoverrucosus SNB-032, where it successfully trapped and revealed the presence of the cryptic iminoquinone 65, as the isopropylidence artifact 66.29 Subsequent LC-MS analysis detect the presence of 65 in the crude SNB-032 extract.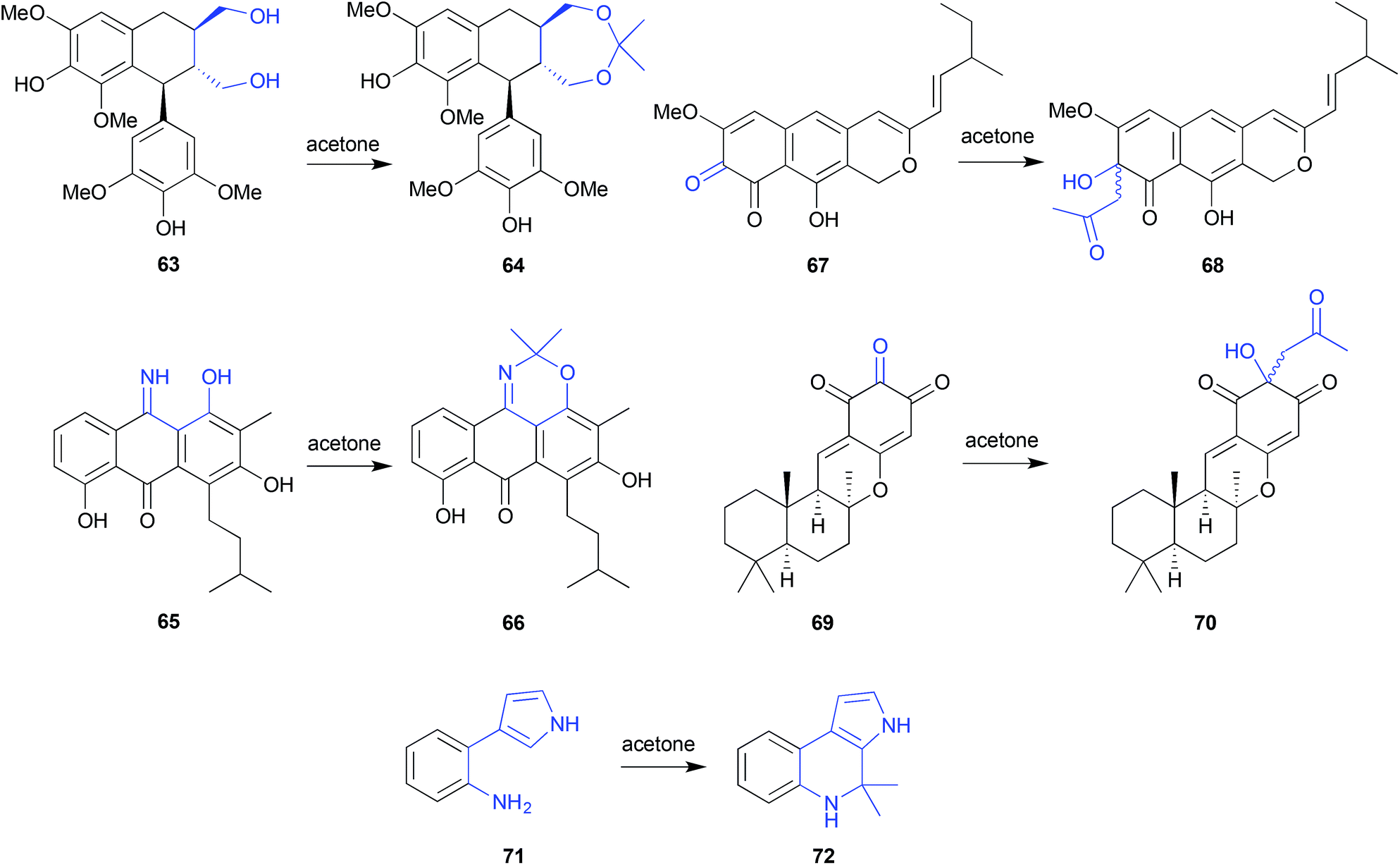 | ||
| Fig. 6 Examples of solvolysis with acetone (63–72). | ||
Acetone extraction of the marine-derived ascomycete, Leptosphaeria oraemis, saw the cryptic o-quinone leptosphaerodione (67) transformed to the epimeric mixture 68,30 while acetone extraction of an Okinawan marine sponge, Dysidea sp., saw the natural trione 69 transformed to the epimeric mixture 70.31 In both these cases, the identity of the acetone Aldol condensation products 68 and 70 was confirmed by the retro-Aldol transformation, and the associated natural products 67 and 69 were subsequently detected in the crude extracts. In yet another example, silica (acetone) chromatography of a marine gliding bacterium, Rapidithrix thailandica, initiated a Pictet–Spengler transformation of the natural product 71 to the artifact 72.32
2.3. Solvolysis (dichloromethane) (Fig. 7)
Generally, CH2Cl2 is considered a benign solvent, notwithstanding its capacity to become acidic through the release of HCl, and tendency to react with tertiary amines to yield chloromethyl quaternary ammonium salts. An example of the latter, CH2Cl2 extraction of an Indonesian marine sponge Acanthostrongylospora ingens initiated transformation of the tertiary amine natural product halicyclamine B (73) to the quaternary ammonium artifact chlorohalicyclamine B (74).33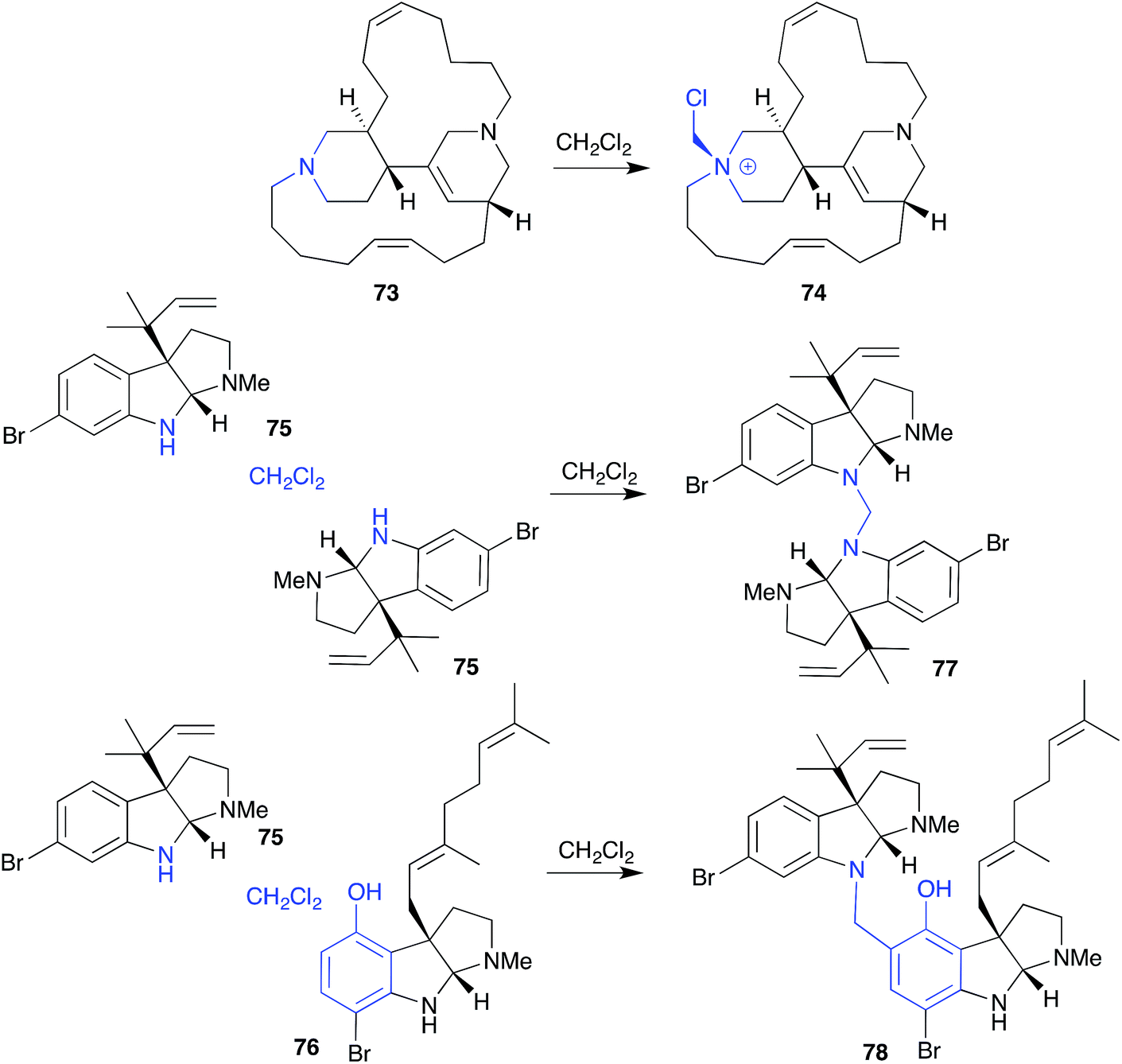 | ||
| Fig. 7 Examples of solvolysis with dichloromethane (73–78). | ||
A variation on this theme, exposure to CH2Cl2 caused dihydroflustramine C (75) and flustramine I (76), isolated from the Canadian bryozoan Flustra foliacea, to form the symmetric and asymmetric dimers, flustramine O (77) and P (78), respectively.34
2.4. Solvolysis (hydrolysis) (Fig. 8)
Whereas simple solvent extraction of an Australian Great Barrier Reef sponge Thorecta sp. yielded the imine aplysinopsin (79), more aggressive soxhlet extraction generated the hydrolysis artifact 80.39 Similarly, on overnight storage in aqueous EtOH, the imine secobatzelline A (81) isolated from a deep-water Caribbean sponge Batzella sp. underwent hydrolysis to the artifact secobatzelline B (82),40 while merely lyophilizing the Australian marine sponge Zyzzya fuliginosa caused the natural makaluvamines H (83) and C (84) to undergo hydrolysis to the artifact damirones A (85) and B (86).41 A deep water Swedish marine sponge, Geodia baretti, yielded the imino cytidine 87, which was believed to partially transform during handling to the hydrolysis artifact 88.42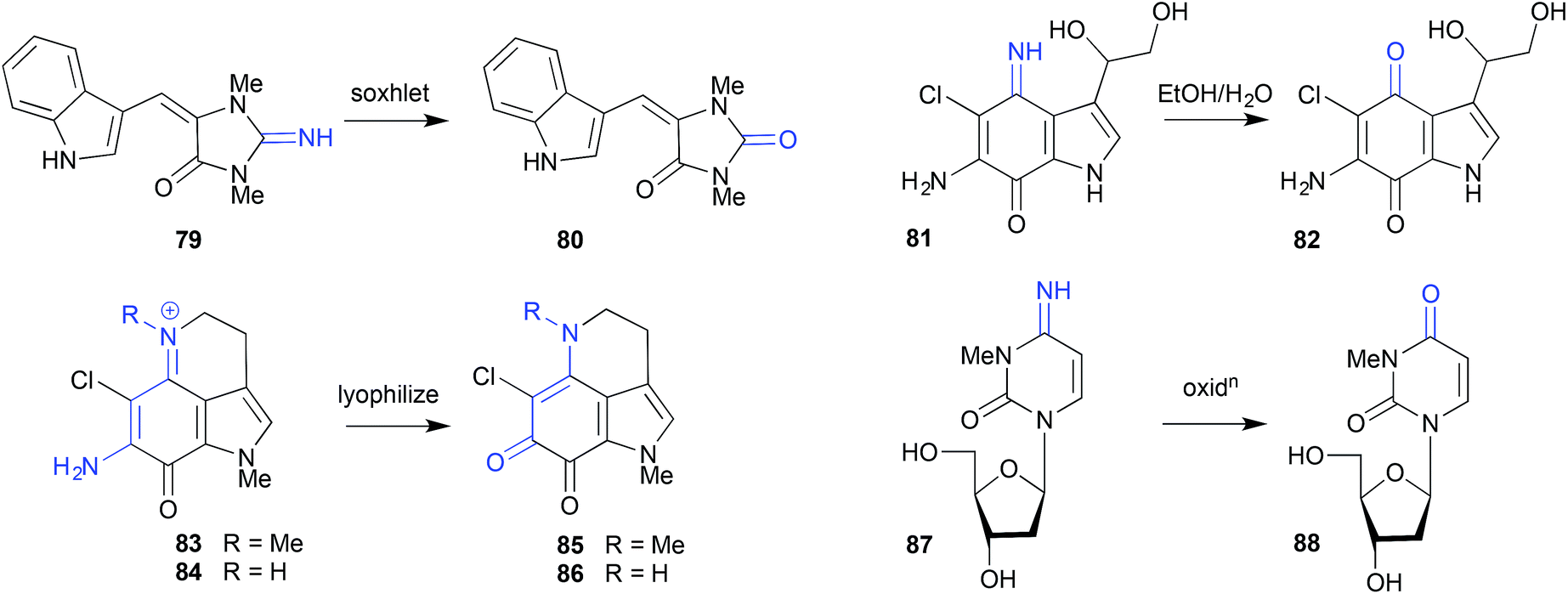 | ||
| Fig. 8 Examples of solvolytic hydrolysis (79–88). | ||
2.5. Oxidation
 | ||
| Fig. 9 Examples of oxidation of dihydroquinones (89–95) and benzylic alcohols (96–97). | ||
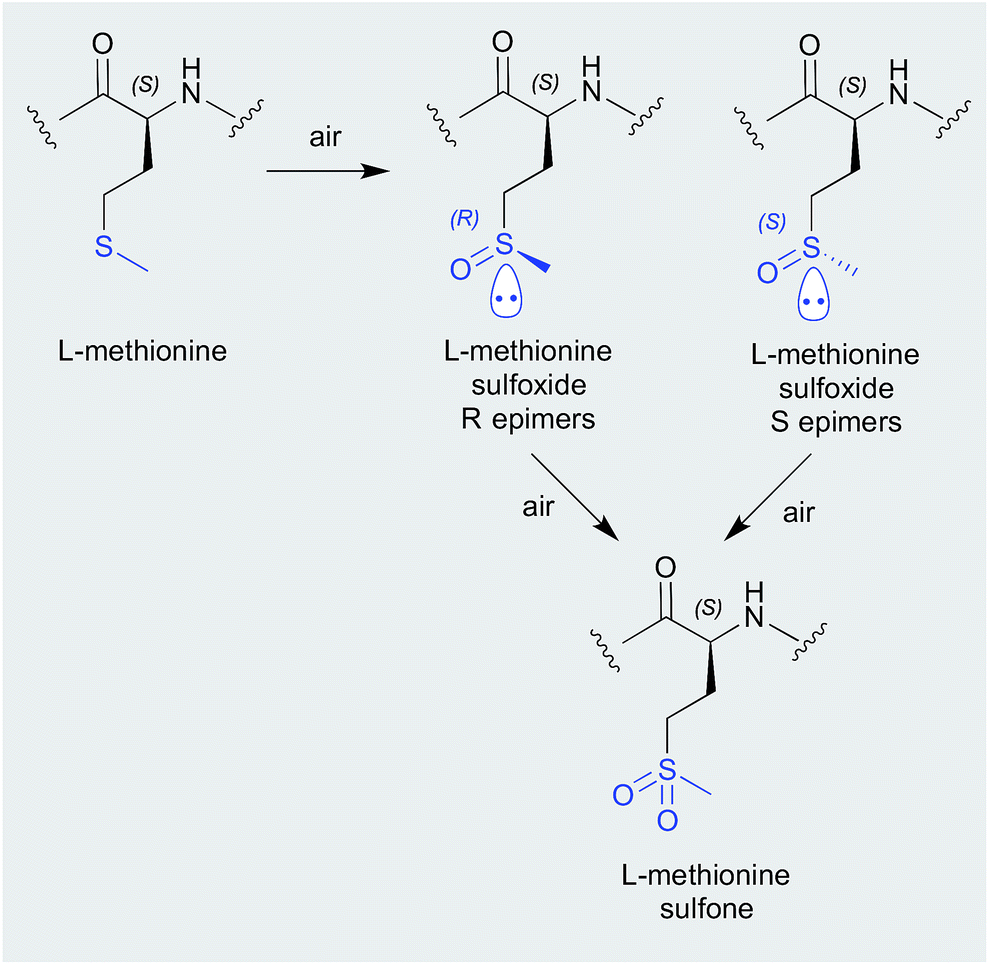 | ||
| Scheme 2 Air oxidation of methionine. | ||
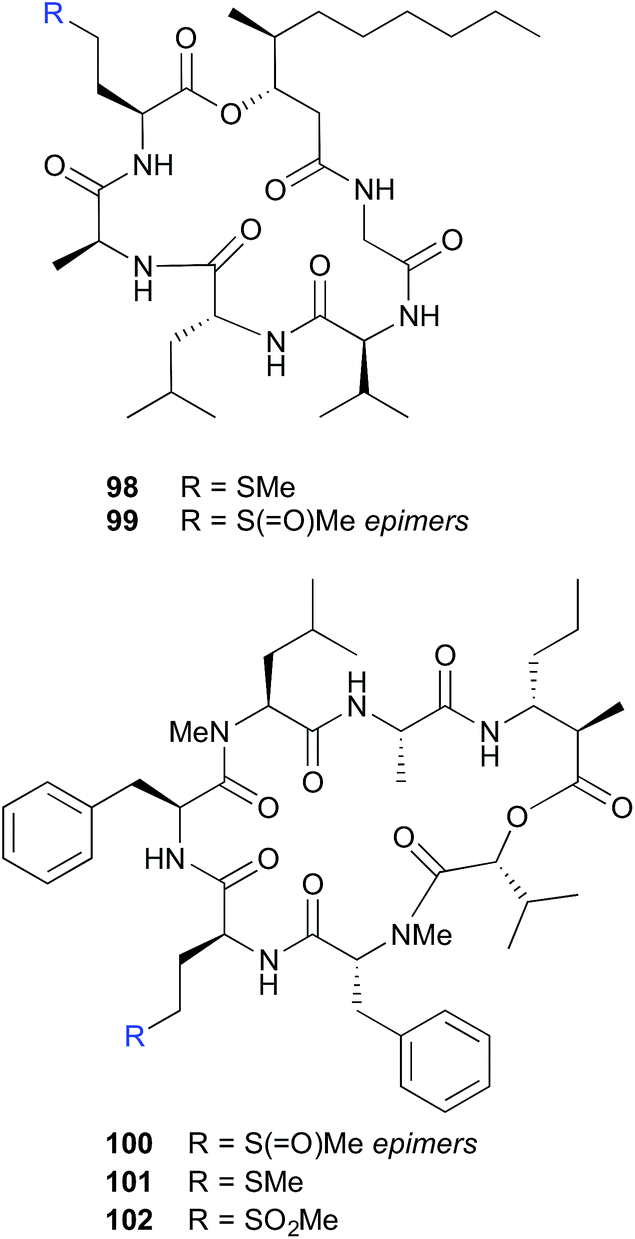 | ||
| Fig. 10 Air oxidation of methionine peptides (98–102). | ||
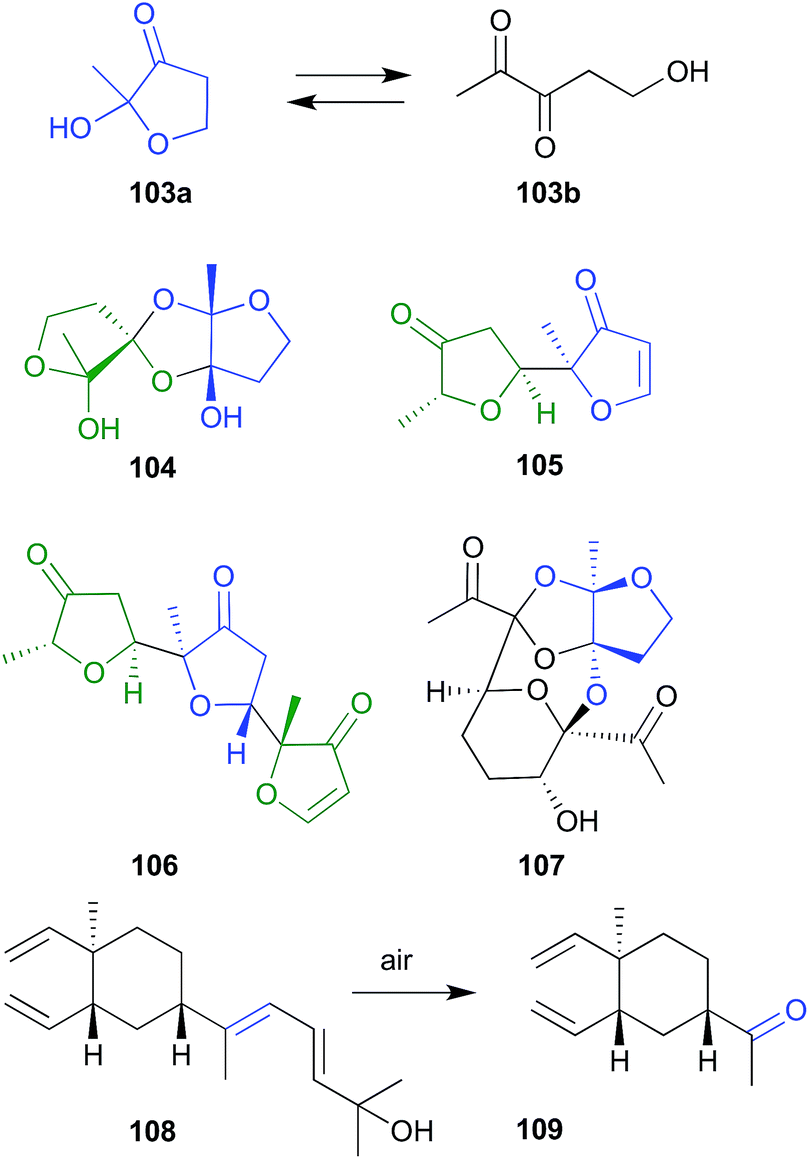 | ||
| Fig. 11 Miscellaneous oxidations (103–109). | ||
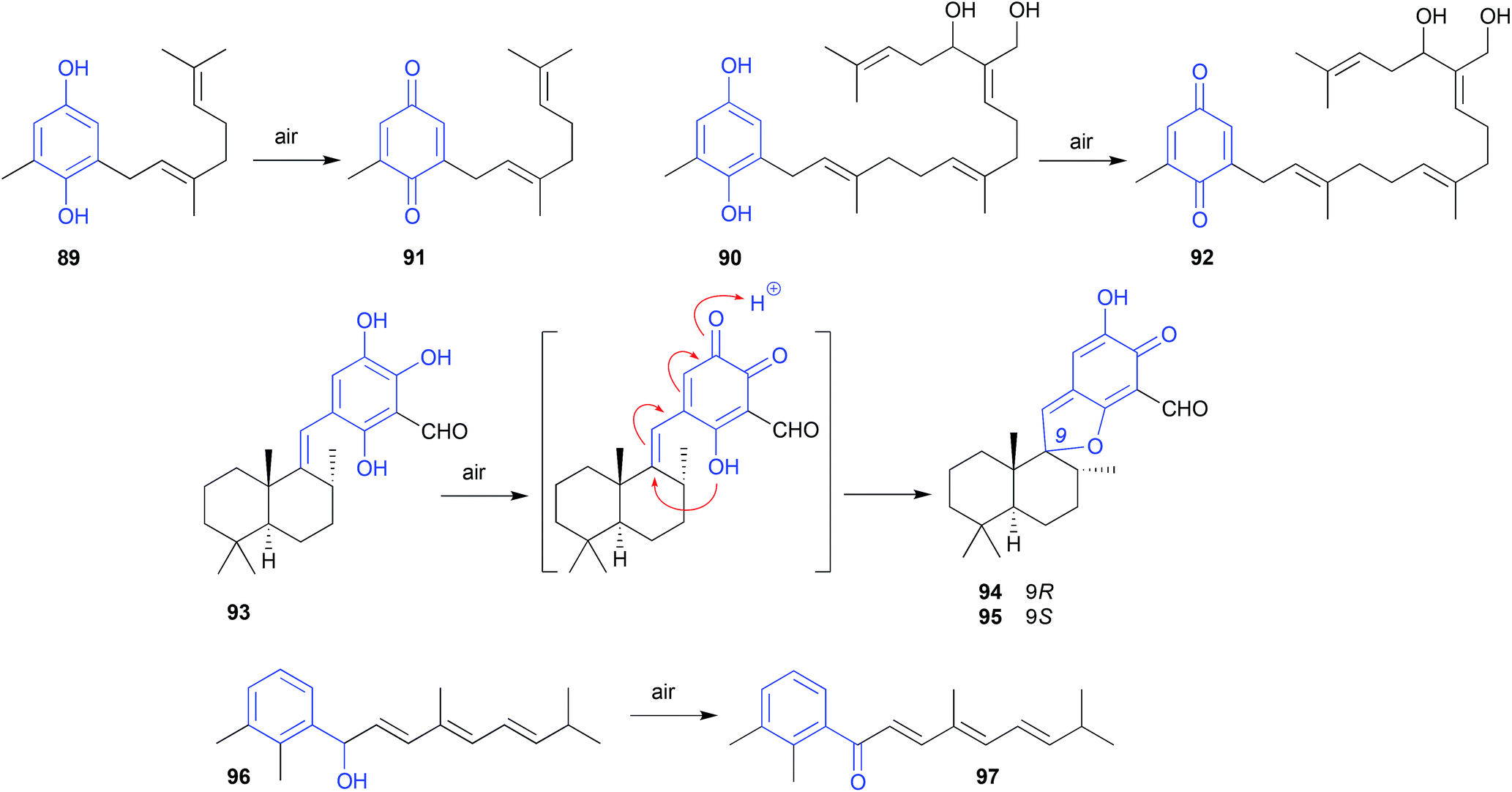
During sequential chromatography on TLC grade silica gel, and 5% AgNO3-silica gel, the natural product fuscol (108), produced by the West Indies gorgonian Eunicea fusca, ungoes oxidative cleavage of the side chain to return the degraded artifact 109.54
2.6. pH (acid)
There are many routes through which natural products can encounter acidic conditions during extraction, isolation, handling and storage. Natural products can, for example, incorporate functional groups that make them auto-acidic (i.e., phenols, carboxylic acids), or could occur naturally with acidic co-metabolites. Certain solvents can become acidic over time (i.e., CHCl3 and DMSO), or can be deliberately augmented with acidic modifiers (i.e., the addition of traces of TFA to facilitate chromatography). Some chromatographic media are notoriously acidic (i.e., silica gel), while even laboratory glassware incorporates acidic sites.As a great many marine natural product artifacts are generated by either accidental or deliberate exposure to acid, modern natural products researchers aspiring to best practice typically make the effort to perform all operations under neutral conditions – however this is not always possible.
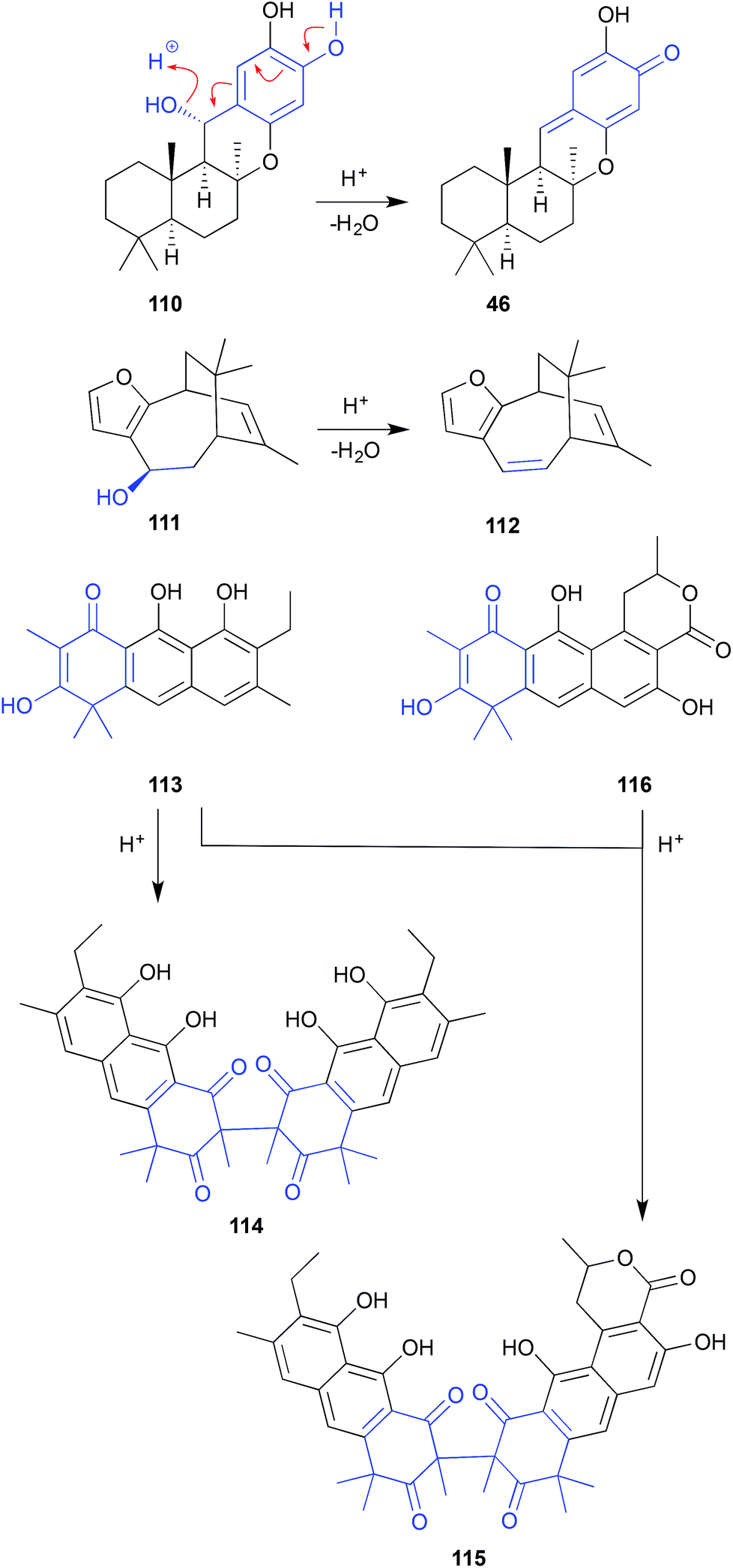 | ||
| Fig. 12 pH (acid) mediated NMR analysis artifacts (110–116). | ||
Similarly, after 3 days exposure to CDCl3 in an NMR tube, euryspongin A (111) from the Okinawan sponge Eurospongia sp., underwent dehydration to dehydroeuryspongin A (112).57
The MeOH extract of a British Columbia hydroid Garveia annulata yielded an array of oxidized anthracenes, with NMR (CDCl3) analysis of garveatin B (113) generating the symmetric dimer artifact 114. Although isolated from the crude extract, the closely related asymmetric dimer 115 was viewed as an artefact, generated by dimerization of 113 with the co-metabolite garvin B (116).58
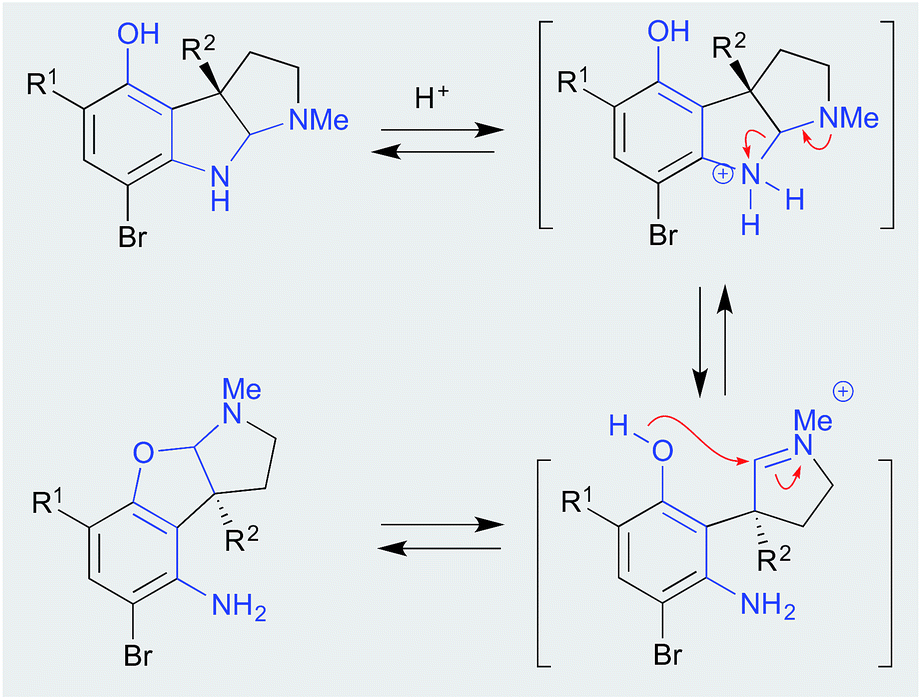 | ||
| Scheme 3 pH (acid) mediated flustramine rearrangement. | ||
On storage at −30 °C in DMSO the alkaloid trachycladindoles E (120) and F (121), isolated from the southern Australian sponge, Trachycladus laevispirulifer, undergo quantitative conversion to the deformylated trachycladindoles A (122) and C (123). Despite this reactivity, as 120–123 were present in the crude sponge extract, all are deemed natural products.59 The oxoglyantrypine epimers 124 and 125 isolated from a mangrove-derived fungus Cladosporium sp. PJX-41 undergo interconversion when stored in DMSO at 4 °C for 3 months.60
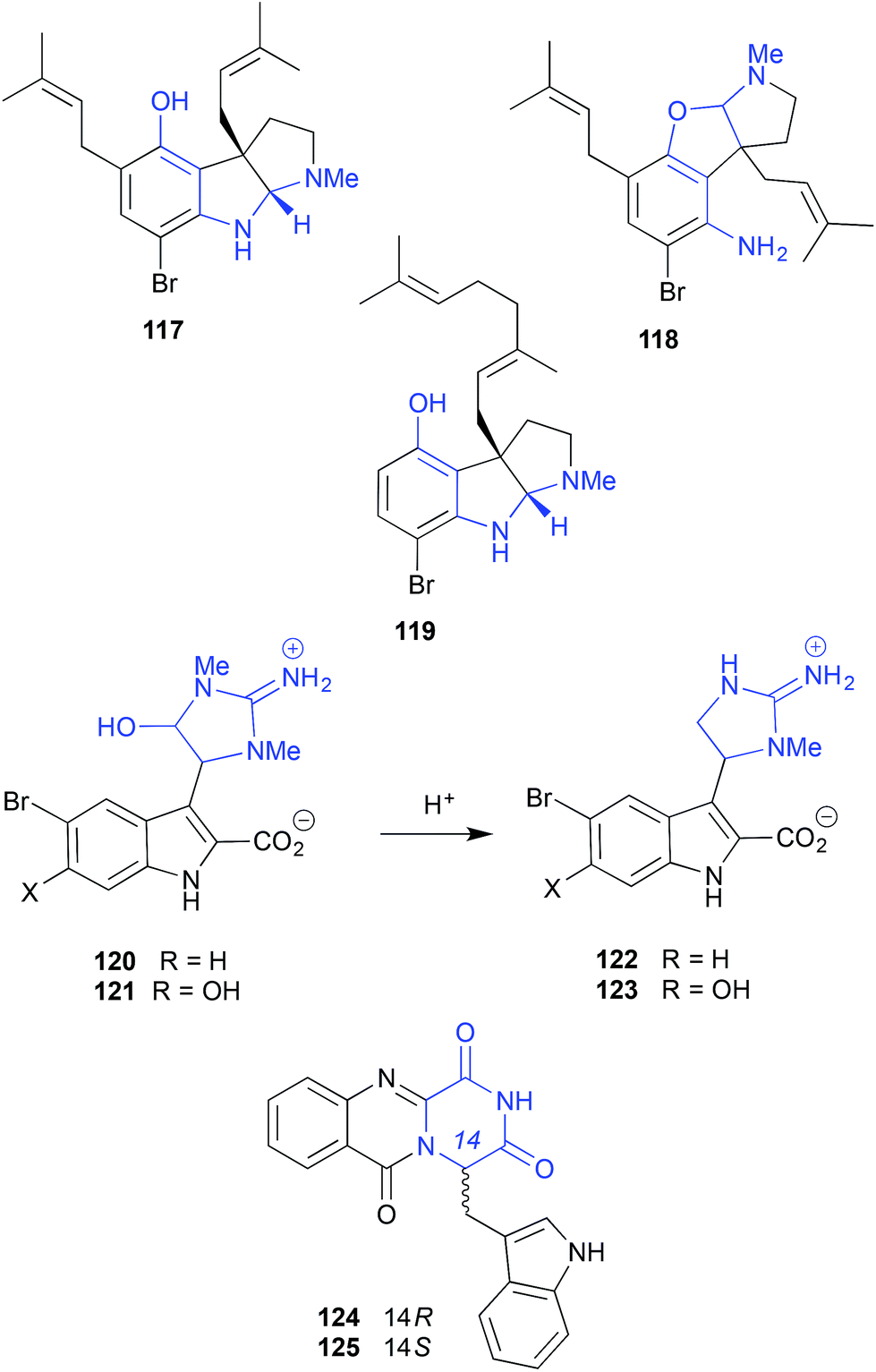 | ||
| Fig. 13 pH (acid) mediated storage artifacts (117–125). | ||
The sesquiterpene calenzanol (126) isolated from a sample of red seaweed Laurencia microcladia collected off Elba Island, proved highly acid (and heat) labile, transforming to the indene-type artifact 127, and the blue guaiazulenium-type ion 128.61
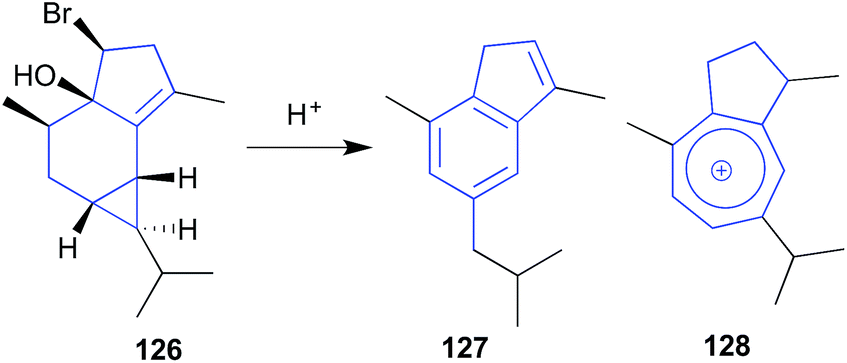 | ||
| Fig. 14 pH (acid) mediated storage artifacts (126–128). | ||
Shornephine A (131), a rare diketomorpholine produced by the Australian marine-derived Aspergillus sp. CMB-M081F, proved to be highly susceptible to auto acid-mediated rearrangement in hydrolytic solvents. For example, whereas extraction with MeOH initiated a cascade of transformations that led to the quantitative transformation of 131 to seco-shornephine A (132) (Scheme 4), extraction with MeCN yielded only 131.63 Knowledge of the chemical reactivity of natural and synthetic diketomorpholines informed structure–activity relationship (SAR) investigations that documented P-glycoprotein (P-gp) inhibitory properties, and permitted the design of simplified analogues that impeded drug efflux in P-gp over-expressing, multidrug resistant carcinoma cells.
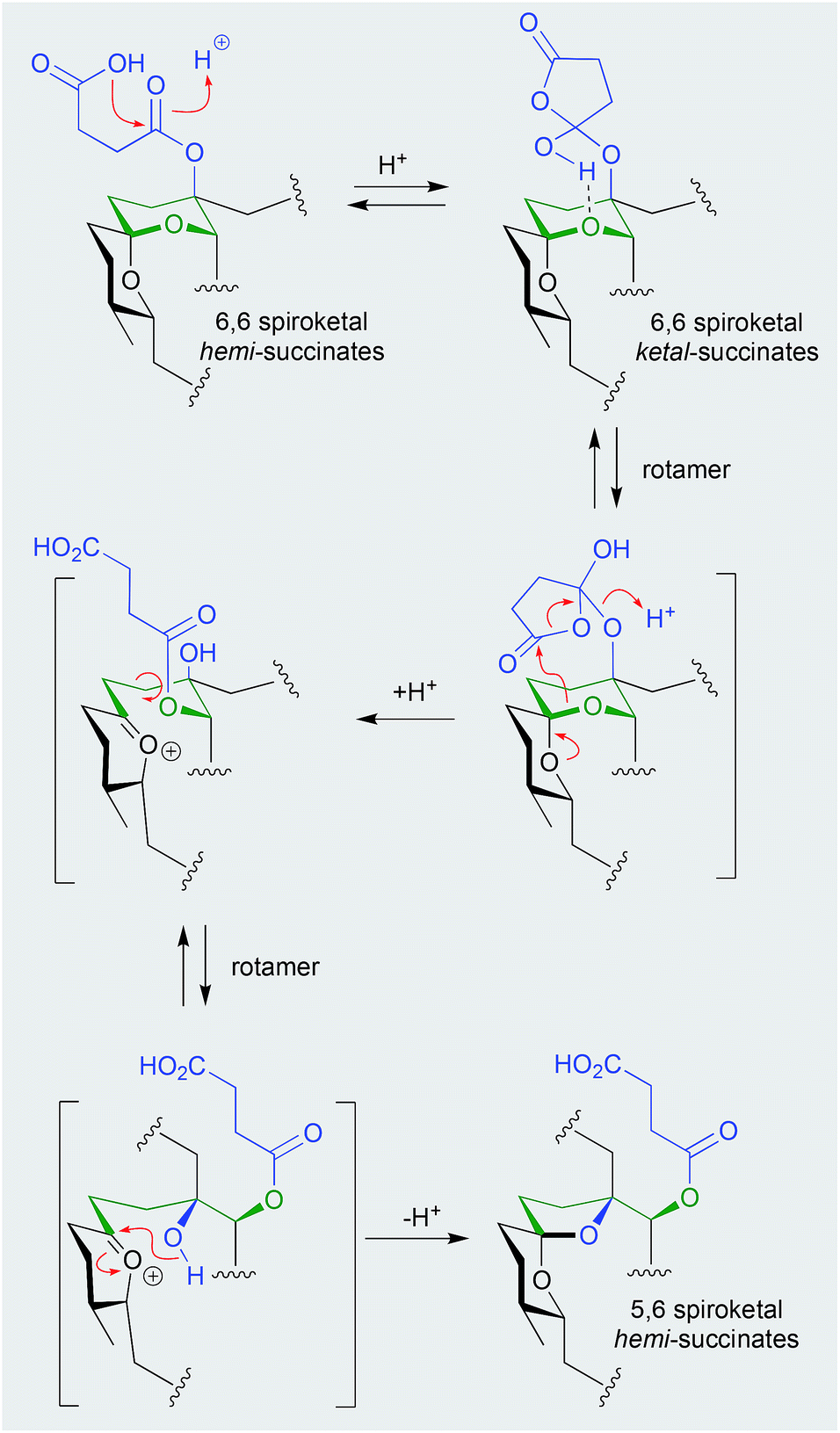 | ||
| Scheme 4 pH (acid) mediated rearrangement of 6,6-spiriketal reveromycins to 5,6-spiroketal reveromycins. | ||
Careful extraction and fractionation of an Australian marine brown alga Zonaria spiralis yielded the highly acid labile acylphloroglucinol hemiketal spiralisone B (133), and related hemiketals.64 This study also returned known chromene natural products such as 134, first reported several decades ago following silica (CHCl3) chromatography of a Pacific brown alga, Zonaria tournefortii.65 That the phenolic hemiketal 133 undergoes facile dehydration to yield the chromene 134, both raised questions about the natural product status of 134 and related chromene algal metabolites, and informed an effective biomimetic synthesis.
As phenols with o-isoprenyl substituents are prone to acid-mediated cyclization during isolation and handling, either with or without added oxidation of the isoprenyl side chain (Scheme 5), it is prudent to carefully restrict exposure to acid during isolation, to avoid artifacts. Unfortunately, the natural products literature has many accounts where the impact of acid has not been constrained.
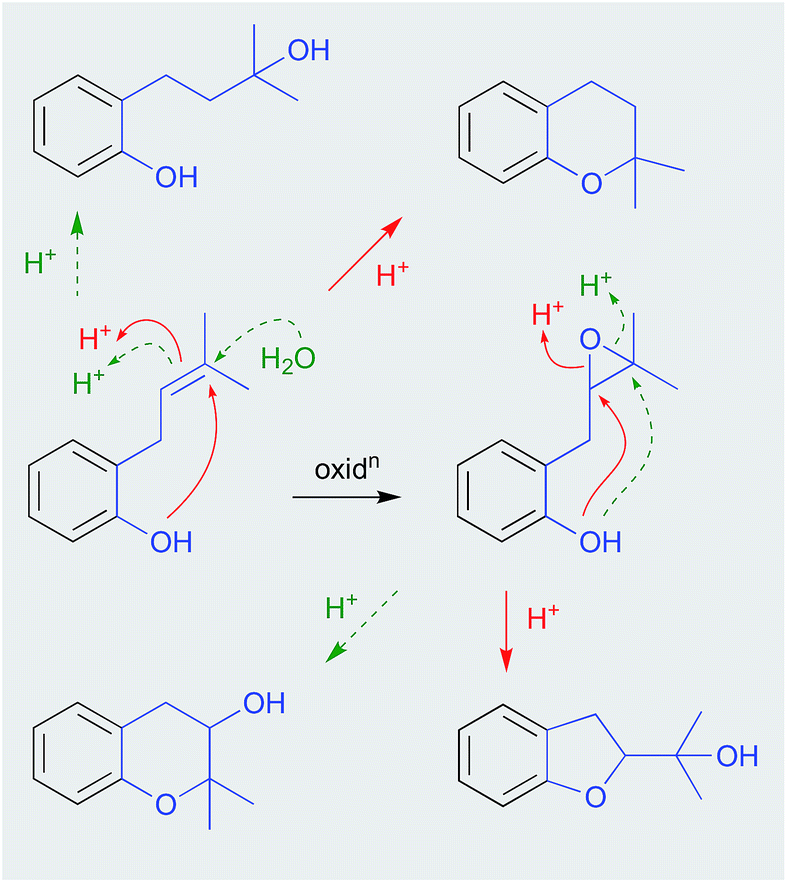 | ||
| Scheme 5 pH (acid) mediated phenol rearrangements. | ||
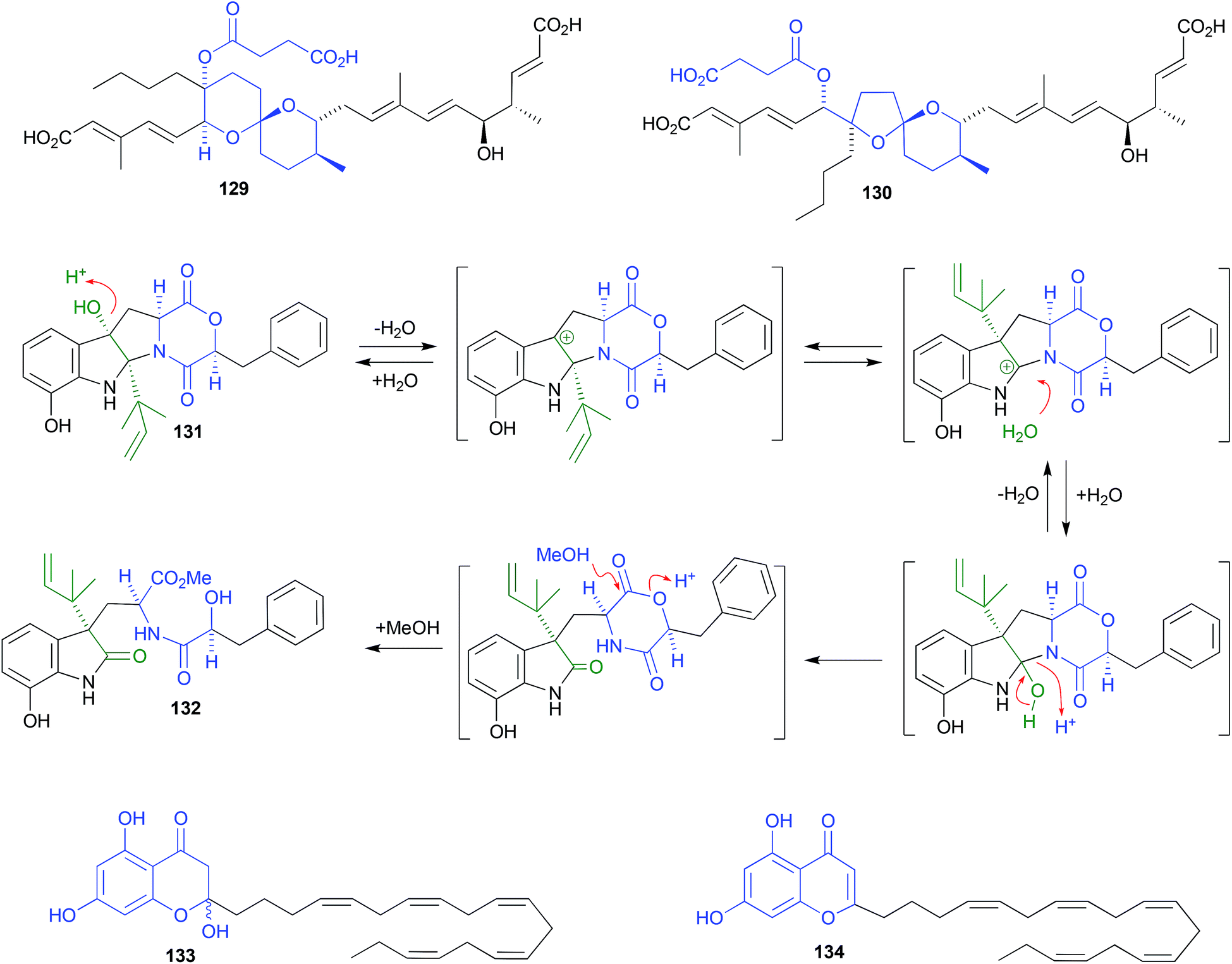 | ||
| Fig. 15 pH (acid) mediated isolation artifacts (129–134). | ||
Consider for example accounts of the isolation of butenolides from three separate marine-derived isolates of the fungus Aspergillus terreus, all of which involve silica gel chromatography with chlorinated solvents (i.e., acidic conditions). In a 2009 example, an Indian soft coral-derived A. terreus yielded the well-known fungal natural product butyrolactone I (135), together with two new natural products, aspernolides A (136) and B (137).66 In a 2011 example, a Chinese marine sediment-derived isolate of A. terreus PT06-2 yielded twelve known natural products, including butyrolactone I (135), aspernolide A (136) and butyrolactone III (138), together with three new natural products, which included aspernolide B (137), renamed as terralactone A.67 In a 2018 example, a marine sponge-derived A. terreus yielded nine known natural products, including 135, 136, butyrolactone IV (139) and V (140), together with three new natural products, the resolved (+) and (−) epimers of asperteretal D (141a/141b), and asperteretal E (142).68 Notwithstanding a subsequent corrigendum to this latter study,69 given the acidic isolation conditions used in all three studies, the putative natural products (+) and (−) asperteretal D (141a/141b) are almost certainly methanolysis artifacts of asperteretal E (142). Furthermore, given that none of these studies carry out analyses to validate the presence of isolated compounds in the unprocessed crude extract, it's quite likely that 136–137 are artifacts of 135, while 139–140 are artifacts of 138, which itself could be an oxidation artefact of 135. Extending this assessment to many other published accounts of “natural product” butenolides from terrestrial strains of A. terreus reveals even more confusion, with many other likely artifacts mis-characterised as natural products.
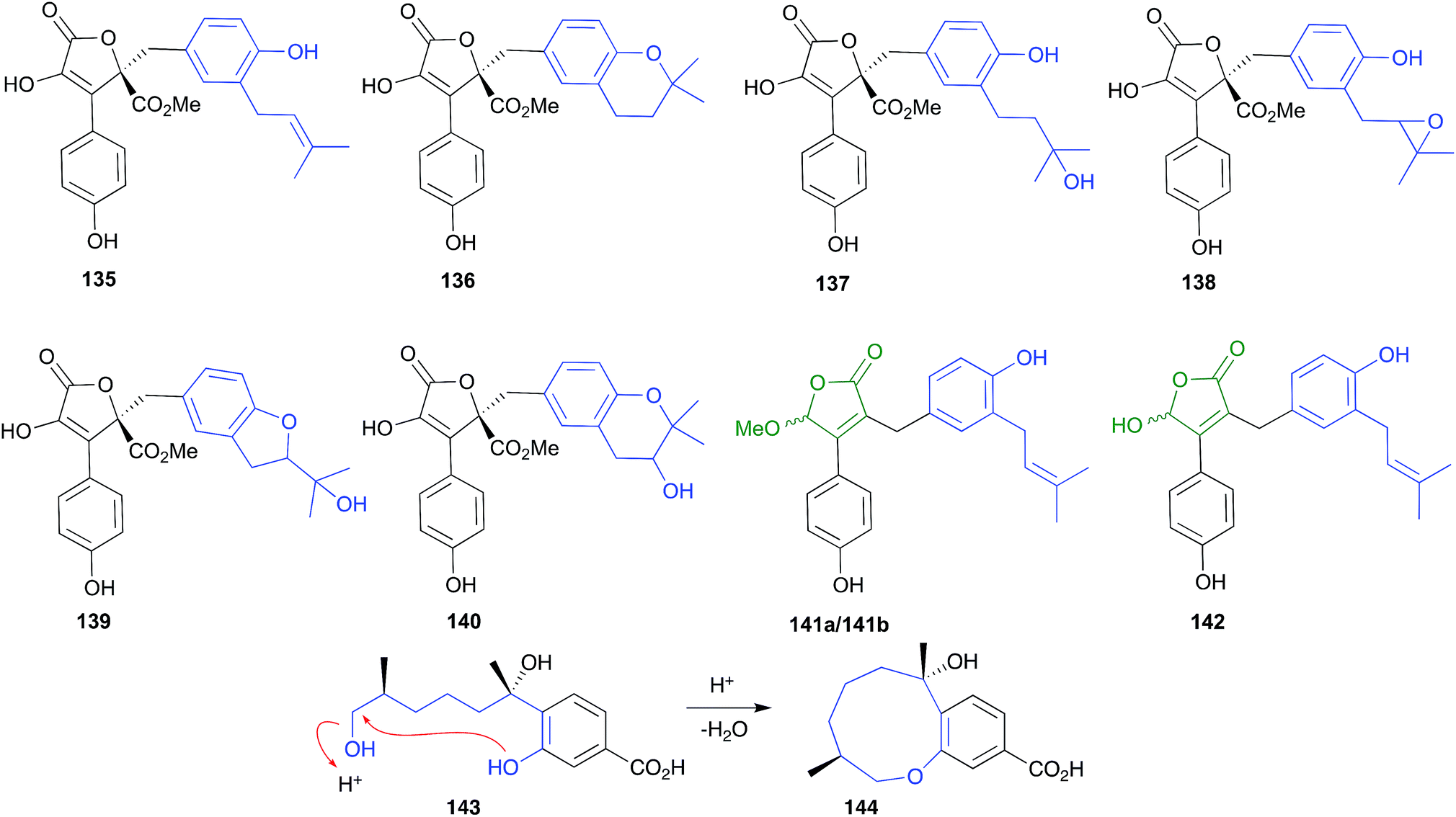 | ||
| Fig. 16 pH (acid) mediated isolation artifacts (135–144). | ||
The bisabolene sesquiterpene, (+)-12-hydroxysydonic acid (143), produced by the Palauan red alga-derived fungus, Trichoderma sp. TPU199, yielded the acid-mediated dehydration cyclic artifact 144 when subjected to acidic (0.05% TFA) HPLC fractionation.70
An Australian coastal marine sediment-derived fungus, Phomopsis sp. CMB-M0042F, yielded the two known cytochalasins J (145) and H (146). Both these metabolites were acid labile and, during acidic (0.01% TFA) HPLC fractionation, underwent stereo and regiospecific intramolecular cycloadditions, with 145 yielding the artifacts 147–149, and 146 yielding the artifacts 150–151.71
Increasing the TFA concentration greatly enhanced the yields of these transformations, and all artifacts were isolated and fully characterised. Of note, the fused scaffolds in 148 and 150 are quite rare, with the only known natural product examples being phomopchalasins A (152) and B (153), both recovered along with cytochalasin J (145) after repeated silica (CHCl3) chromatography of an endophytic fungus, Phomopsis sp. shj2.72 Based on these latter observations, 152 and 153 might be viewed as acid-mediated isolation artifacts of 145.
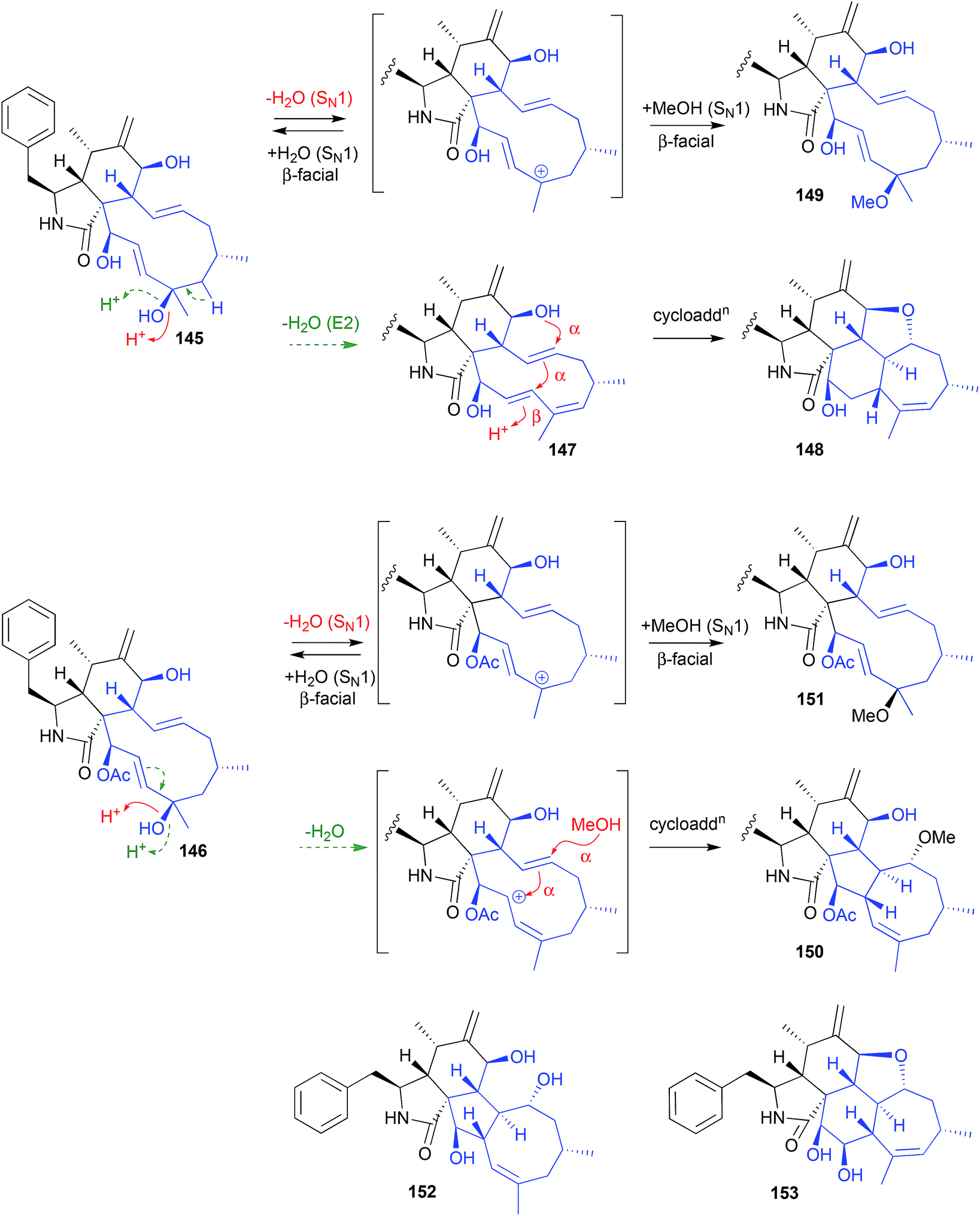 | ||
| Fig. 17 pH (acid) mediated isolation artifacts (145–153). | ||
HPLC (0.1% TFA) fractionation of an Okinawan marine sponge, Pseudoceratina sp. (SS-214), yielded nine known natural products including purealidin J (154), and three new natural products, including ceratinadin C (155).73 To solve the structure and configuration of the putative natural product 155, it was prepared semi-synthetically by treating a sample of 154 with TFA at room temperature for 1 h. This acid-mediated demethylation and subsequent enol–keto tautomerisation suggests 155 may be an artifact, and mirrors that of an earlier report where the sponge natural product (+)-aeroplysinin-1 (156) was subjected to (albeit more forcing) TFA-mediated conversion into the epimeric dibromoketones 157 and 158.74
Cyclobromoveratrylene (159) was isolated as a putative methylated natural product from the red alga Halopytis pinastroides, following an isolation process that was brutal by modern standards. This process included (i) refluxing the EtOH extract for 30 min in the presence of 1 N HCl, (ii) partitioning into Et2O, (iii) concentration and exhaustive methylation with diazomethane, (iv) saponification by 3 h reflux in the presence of 0.5 N KOH, followed by (v) alumina (benzene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et2O) chromatography.75 That any algal natural product could survive such treatment seems remarkable! Indeed, a subsequent account of the synthesis of 159 revealed that on standing in TFA at room temperature for 3 days, 5-bromoveratryl alcohol (160) undergoes cyclic oligomerisation to yield 159 in good yield, supporting the proposition that 159 is an artifact.76
:Et2O) chromatography.75 That any algal natural product could survive such treatment seems remarkable! Indeed, a subsequent account of the synthesis of 159 revealed that on standing in TFA at room temperature for 3 days, 5-bromoveratryl alcohol (160) undergoes cyclic oligomerisation to yield 159 in good yield, supporting the proposition that 159 is an artifact.76
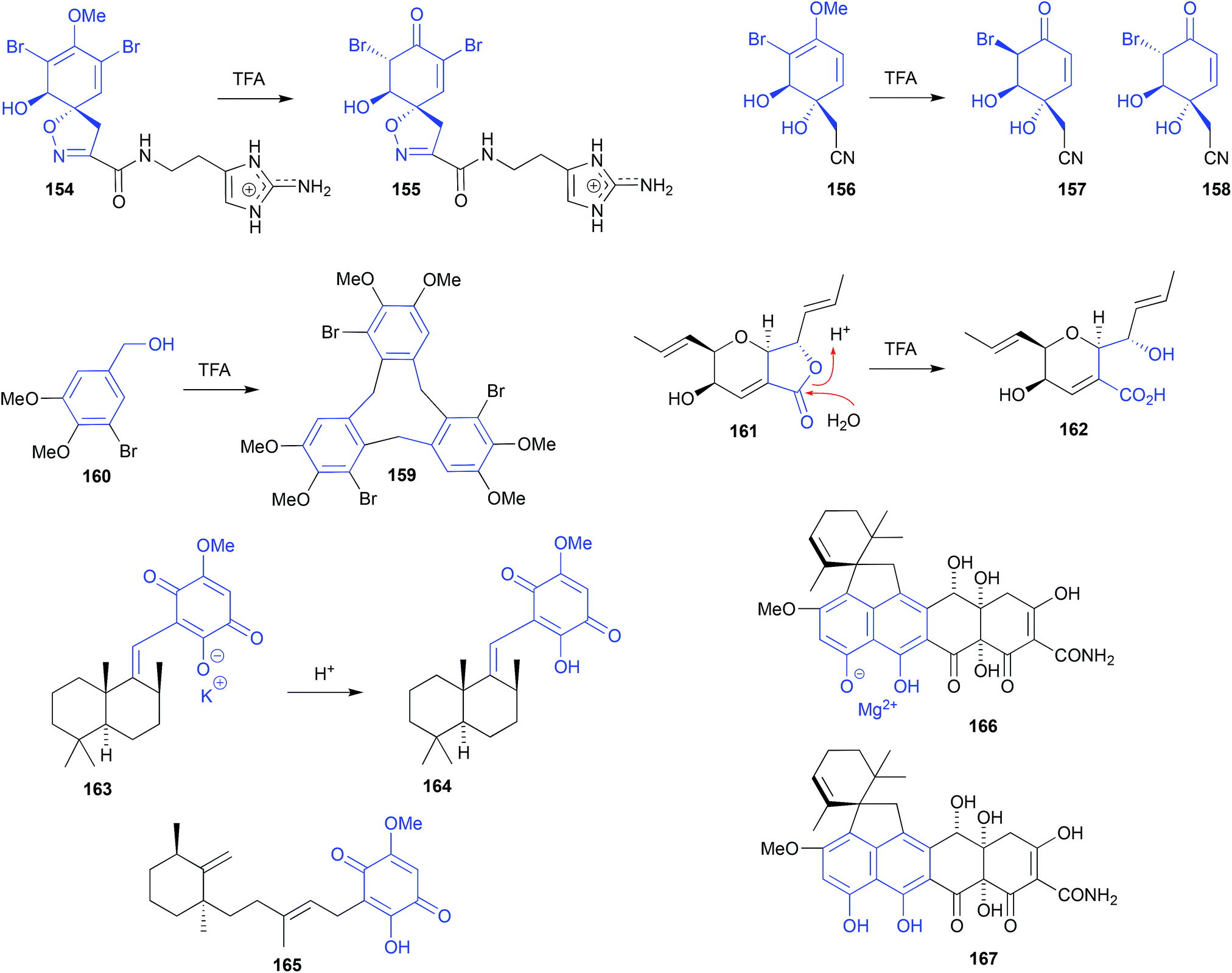 | ||
| Fig. 18 pH (acid) mediated isolation artifacts (154–167). | ||
Silica gel, size exclusion and reversed HPLC fractionation of the Vietnamese mangrove-derived endophytic fungus Acremonium strictum, yielded seven known natural products including waol A (161), and five new polyketides including waol acid (162).77 Despite using acidified HPLC (0.1% TFA) conditions, the new carboxylic acid 162 was designated as a natural product, and the known lactone 161 was redefined as a dehydration artifact? This explanation seems implausible, as it's more likely the lactone 161 is the natural product, and putative new natural product 162 is the acid-mediated hydrolysis artifact.
Sephadex LH-20 (MeOH) fractionation of a purple-coloured southern Australian marine sponge, Spongia sp., recovered the purple pigment as the first naturally occurring sesquiterpene-quinone salt, spongiaquinone potassium salt (163).78 In contrast, silica chromatography of the extract, or Sephadex LH-20 chromatography of the acidified extract, yielded the corresponding yellow-coloured free acid, spongiaquinone (164). The discovery of a natural product salt (i.e., 163), let alone a potassium salt, was unexpected, and while it did not exclude the possibility that the yellow pigment 164 was also a natural product, it did draw attention to the need to carefully correlate the colour of a crude extract, with the colour of the isolated pigments. For example, although silica chromatography of the purple-coloured Okinawan marine sponge Hippospongia cf. metachromia yielded the orange pigment metachromin A (165), the acidic isolation strategy almost certainly precluded discovery of any purple-coloured natural salts.79
Isolation of the known tetracycline-like mycotoxin viridicatumtoxin from an Australian marine mollusc-derived fungus, Paecilomyces sp. CMB-MF010, together with several related known and new metabolites, revealed the excellent acid stability and antibiotic properties of this structure class (compared to commercial tetracycline antibiotics), while simultaneously exposing an unexpected acid/salt relationship.80,81 For example, viridicatumtoxin A recovered from Paecilomyces sp. CMB-MF010 proved to be the magnesium salt 166 ([α]D, −10.0 EtOH), which on exposure to acid during isolation was converted to the free acid 167 ([α]D +31.8, EtOH). Although both 166 and 167 had near identical NMR, UV-vis and MS data, they had distinctly different [α]D values, and could be quantitatively interconverted by pH adjustments.
Significantly, a commercial sample of viridicatumtoxin A, marketed as the free acid, was determined to be the (−)-salt 166,80 while the (+) free acid 167 isolated from Penicillium brasilianum FKI-3368, was mischaracterised as enantio-viridicatumtoxin A.82 Although the distinction between a salt (i.e., (−)-166) and an acid (i.e., (+)-167) may be viewed as falling outside the remit of natural product vs. artifact, the practical implications of failing to make such a distinction can, nevertheless, lead to incorrect structure assignments.
2.7. pH (basic) (Fig. 19)
A Palauan sponge, Agelas sp., yielded quaternary diterpenyl 9-methyladenine natural product salts, exemplified by ageline A (168). Significantly, during silica (6:3:1, CHCl3:MeOH:NH4OH) chromatography 168 underwent quantitative hydrolysis to equilibrating mixtures of isomeric formamide artifacts, 169a and 169b.83 This artifact transformation greatly assisted the identification of 168, and informed an efficient biomimetic synthesis of the natural product hymeniacidin (170a and 170b), an equilibrating mixture of formamide rotamers isolated from a southern Australian marine sponge, Hymeniacidion sp.84 In that synthesis, caffeine (171) was methylated to the salt 172, before being treated with NH4OH to deliver a high yield of 170a/170b.
 | ||
| Fig. 19 pH (base) mediated artifacts (168–176). | ||
When exposed to broth media at pH 10, chlorizidine A (173) isolated from a marine-derived Streptomyces sp. CNH-287 undergoes ring opening and decarboxylation to yield the artifact chlorizidine B (174).85 Knowledge of this transformation permitted assembly of an analogue library, for example, by treating 173 with MeOH/K2CO3 to yield 175, and benzylamine to yield 176.
2.8. Diels–Alder (Fig. 20)
Although dicitrinin A (177) was reported as a natural product from co-culture of the marine algal-derived fungi, Aspergillus sydowii EN-534 and Penicillium citrinum EN-535,86 and Penicillium citrinum IFM 53298,87 it had also been identified as an artifact of the well-known natural product and co-metabolite, citrinin (178). For example, heating of a MeOH solution of 178 to 50 °C, over several days, yielded as the major product the heterocyclic Diels–Alder dimer and artifact 177, together with dicitrinins B–D.88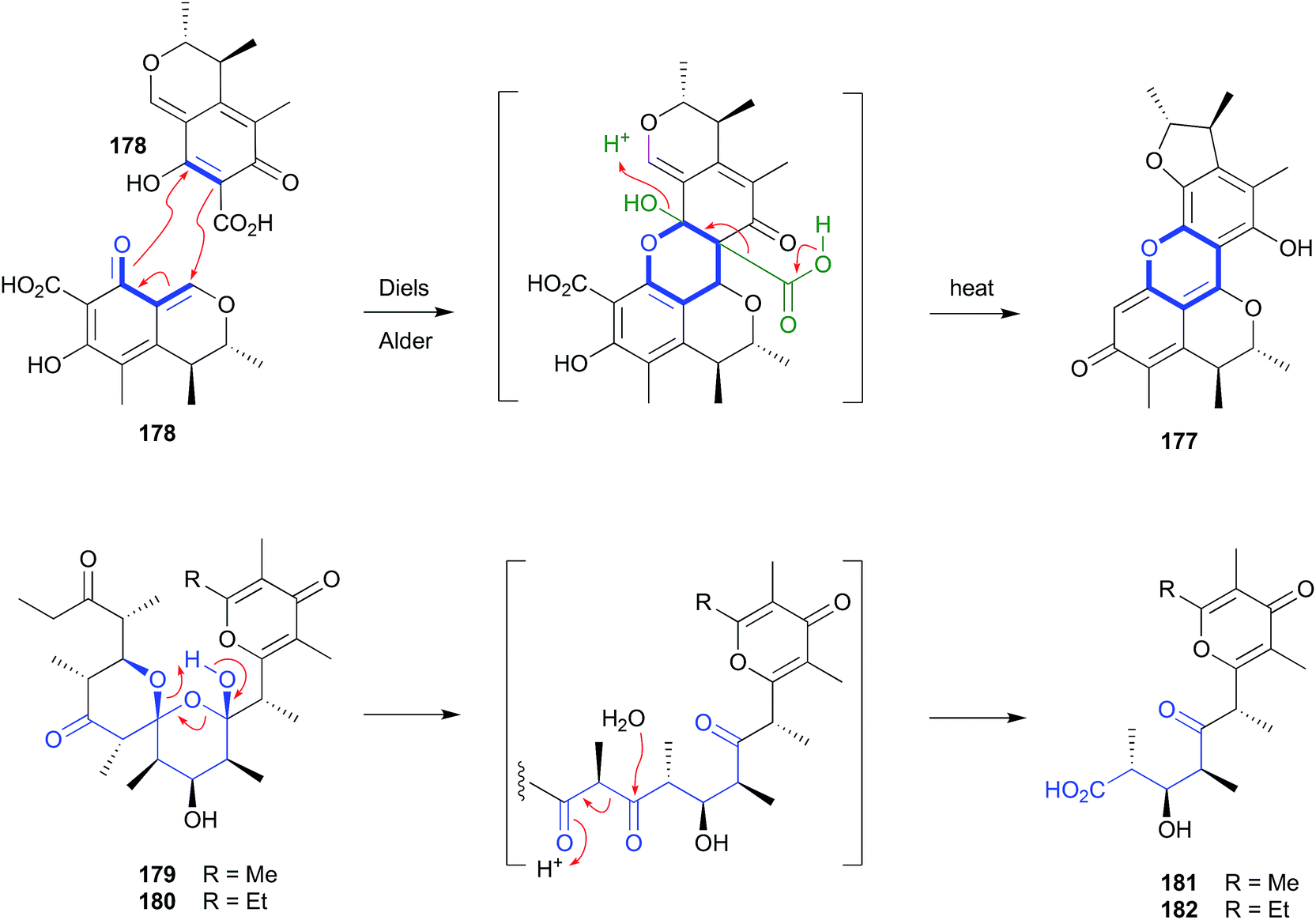 | ||
| Fig. 20 Artifacts derived from Diels–Alder (177–178) and retro Claisen condensation (179–182) reactions. | ||
2.9. Retro-Claisen condensation (Fig. 20)
Silica chromatography of a selection of Australian and Hawaiian molluscs, Siphonaria spp., yielded an array of polyketides, including dihydrosiphonarin A (179) and B (180), and the associated retro-Claisen condensation artifacts 181–182.962.10. Michael addition (Fig. 21)
The polyketide abyssomicin C (183), isolated from a Japanese Sea deep sea (−289 m) sediment-derived Verrucosispora sp AB 18-032, was reported as an inhibitor of p-aminobenzoic acid biosynthesis, with promising antibiotic properties.89,90 Subsequent re-investigation yielded an even more potent antibiotic isomer, atrop-abyssomicin C (184), which transformed to 183 on exposure to acidic HPLC solvents.91 The same transformation had been observed during the acquisition of NMR data on synthetic 184 in unstabilized (i.e., acidic) CDCl3.92 These observations suggest atrop-abyssomicin C (184) is a natural product, while abyssomicin C (183) may be a handling artifact. The re-isolation of 183 together with the new analogue abyssomicin J (185) from a South China Sea deep-sea (−2733 m) sediment-derived Verrucosispora sp. MS100128, was supportive of this hypothesis.93 For example, where prior studies had determined that only Michael acceptor abyssomicins such as 183 and 184 exhibit antibiotic properties, the observation that 185 was equipotent was anomalous. This anomaly prompted speculation that 185 was biosynthetically derived from a quadruple Michael addition dimerization of 183 (or 184), which in turn prompted a successful biomimetic synthesis in which treatment of 183 with 0.1 M Na2S resulted in near quantitative yields of 185. Importantly, when added to bacteria (i.e., mycobacteria) analytical studies determined that 185 underwent an in situ (intracellular) oxidation of the thioether to a sulfone, followed by a quadrupule reverse Michael addition to deliver two equivalents of ONLY atrop-abyssomicin C (184). The kinetic atropisomer 184 is a far more potent Michael acceptor and antibiotic than the thermodynamically more stable atropisomer 183. Given this, it is entirely possible that the antibacterial properties attributed to 183 reflect partial in situ equilibration to 184.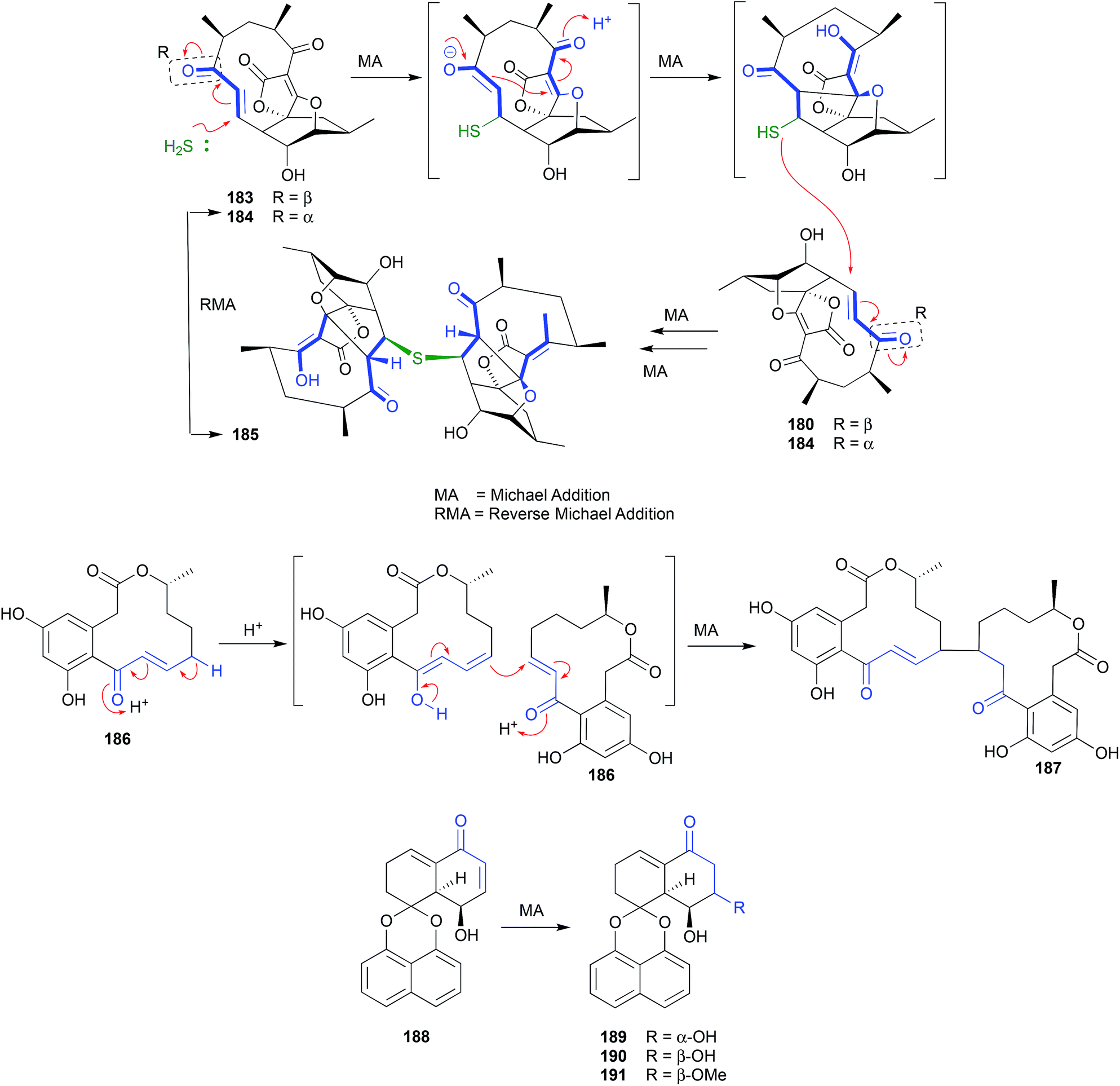 | ||
| Fig. 21 Artifacts derived from Michael addition reactions (183–191). | ||
A fungus, Curvularia sp., isolated from a red alga collected off Guam yielded a selection of curvularin polyketides, including the new metabolite (+)-(10E,15R)-10,11-dehydrocurvularin (186), which underwent a Michael addition dimerization under the extraction and handling conditions to the artifact 187.94 A Thai mangrove-derived fungus, Rhytidhysteron sp., yielded an array of five new spirobisnaphthalenes including the Michael acceptor rhytidenone F (188), and the handling Michael addition adducts rhytidenones C-E (189–191).95 The dimerization and trimerization of prolinimine A (192) to yield prolinimines C (194) and D (195) is also a nice example of artifact formation through Michael addition (see Section 2.11. Schiff bases, below).
2.11. Schiff bases (Fig. 22)
Analysis of the EtOAc extract of a rice cultivation of Trichoderma sp. CMB-F563, a fungus recovered from the gastrointestinal tract of a fish-market purchased fresh mullet, detected the presence of two unprecedented Schiff bases, prolinimines A–B (192–193).97 As the presence of phenolic co-metabolites rendered the crude extract slightly acidic, it was determined that during handling 192 underwent quantitative conversion to the artifact dimer and trimer, prolinimines C (194) and D (195), respectively. This observation informed an effective biomimetic synthesis of 192–195, from the putative biosynthetic precursors N-amino-L-proline methyl ester (196) and furans 197–198. During subsequent investigations into the biosynthetic origins of prolinimines, analytical studies on M1 broth cultivations detected the precursor amino acid 196 in extracts of partitioned (i.e., centrifuged) mycelia, but failed to detect it in the partitioned broth. Likewise, no prolinimines could be detected in extracts of partitioned mycelia, or broth. Instead, the known thermolysis (i.e., autoclave) artifact 197 was detected in the partitioned broth, but not the partitioned mycelia. Subsequent studies determined that solvent extraction of M1 broth cultivations initiated the release of 196 from the mycelia, enabling its rapid Schiff's base condensation with 197 in the media, to form the prolinimines. These observations suggest that 196 is a natural product, and all the prolinimines 192–185 are artifacts.98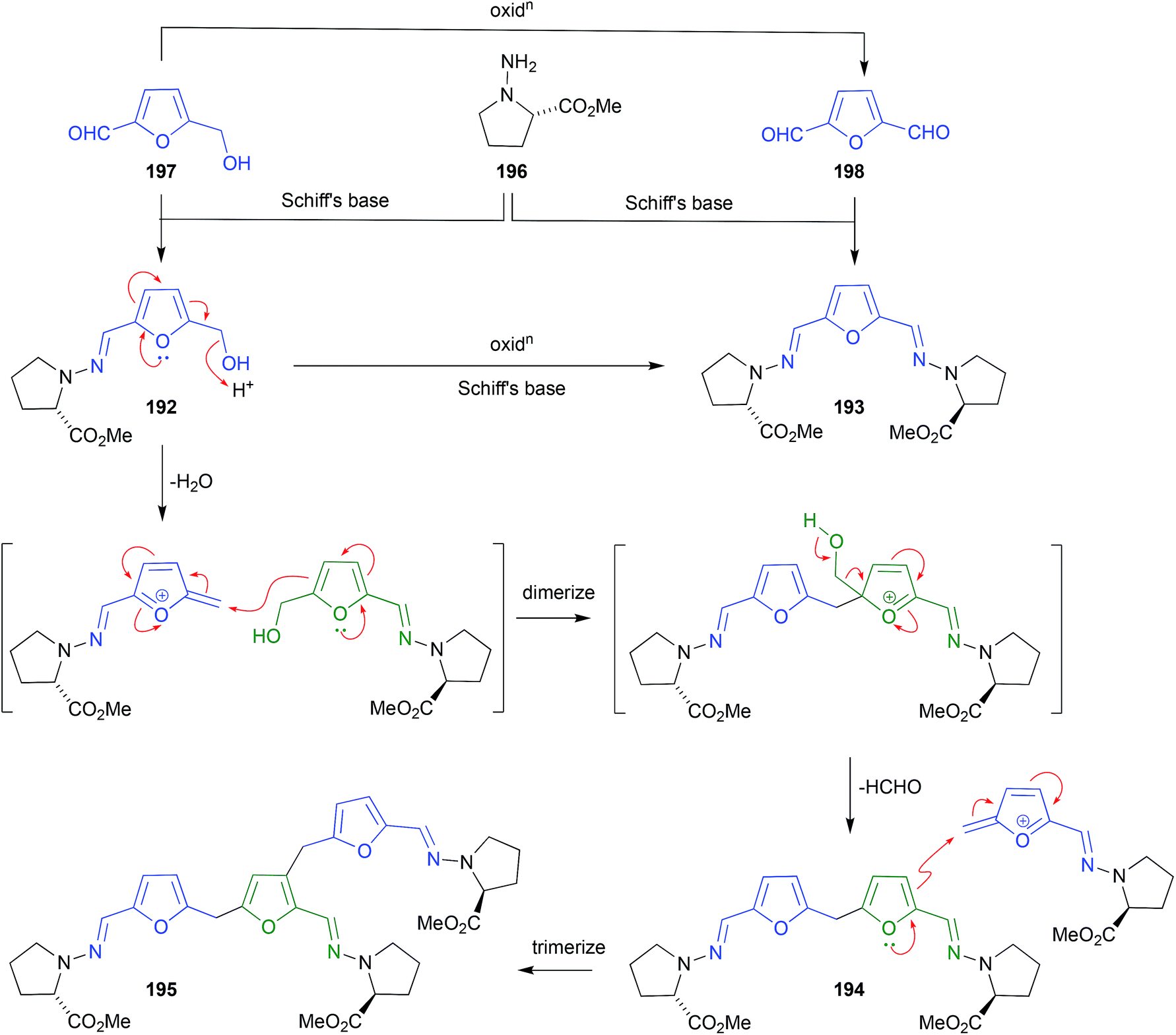 | ||
| Fig. 22 Artifacts derived from Schiff bases (192–198). | ||
2.12. Benzylic rearrangement (Scheme 6, Fig. 23)
The EtOH extract of two southern Australian marine sponges, Ircinia spp., yielded an array of sesterterpene lactams, including ircinialactam A (199) and D (200),99 whereas the MeOH extract of a Korean sponge, Sarcotragus sp., returned a comparable array of sesterterpenes, including (7E,12E,18R,20Z)-variabilin (201) and the related dimethyl ester 202.100 Although 200 and 202 were not reported as artifacts of 199 and 201, respectively, this does nevertheless seem plausible. For example, the generation of artifacts could occur through oxidation of the tetronic acid moiety to a highly reactive intermediate, that undergoes solvolysis and ring opening followed by a benzylic rearrangement (Scheme 6). Supportive of this hypothesis, two southern Australian sponges, Psammocinia spp., yielded just such an oxidized tetronic acid moiety in the natural products (−)-oxoircinianin (203) and (−)-oxoircinianin lactam A (204), which are presumably stable to solvolysis due to the absence of Δ20,21.101 | ||
| Scheme 6 Benzylic acid rearrangement mechanism. | ||
2.13. Heat (Fig. 23)
A Western Australian inter-tidal collection of the marine red alga, Laurencia filiformis, yielded the sesquiterpene 6β-hydroxyaplysistatin (205).102 Observations made during acquisition of a melting point suggested that 205 was thermally labile. This prompted isolation and identification,102 and total synthesis103 of the thermolysis artifact 206.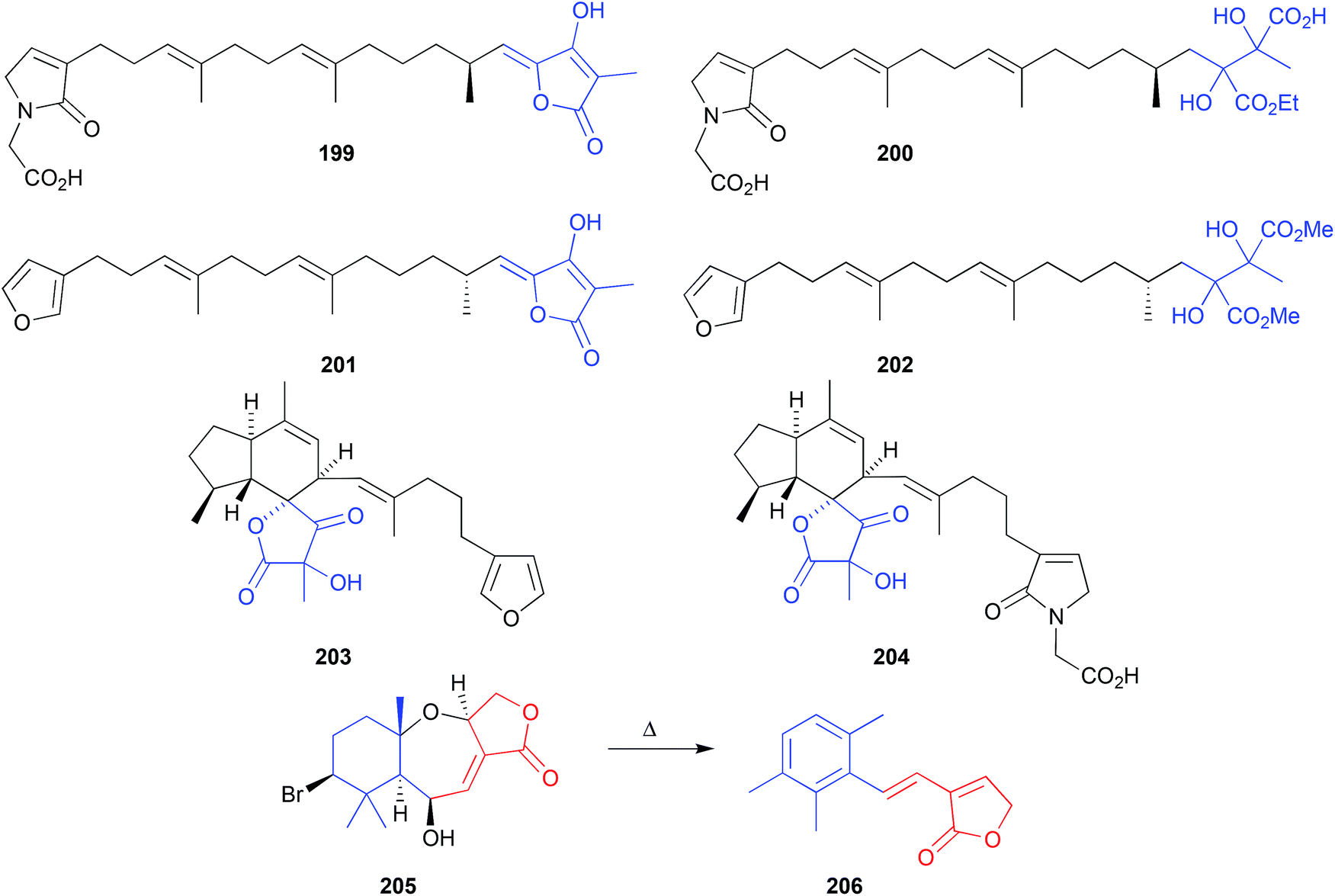 | ||
| Fig. 23 Artifacts derived from benzylic (199–204) and thermal (205–206) rearrangements. | ||
2.14. Light
 | ||
| Fig. 24 Artifacts derived from light mediated isomerisation (207–216), cycloaddition (217–221) and rearrangement (222–224). | ||
2.15. Electrophiles (Fig. 25)
The putative natural product ammosamides A–C (225–227) were reported from a deep-water (−1681 m) marine sediment-derived Streptomyces sp. CNR-698,114,115 while ammosamide D (228) was reported from Streptomyces variabilis SNA-020,116 both collected off the Bahamas Islands. Subsequent studies established that the potent electrophile ammosamide C (227) was the likely precursor to all ammosamides. For example, 5 d exposure of 227 at pH 10 in A1 media yielded B (226), while a PBS solution containing an excess of cysteine yielded A (225) and B (226), and prolonged exposure of the latter to air and sunlight yielded D (228).117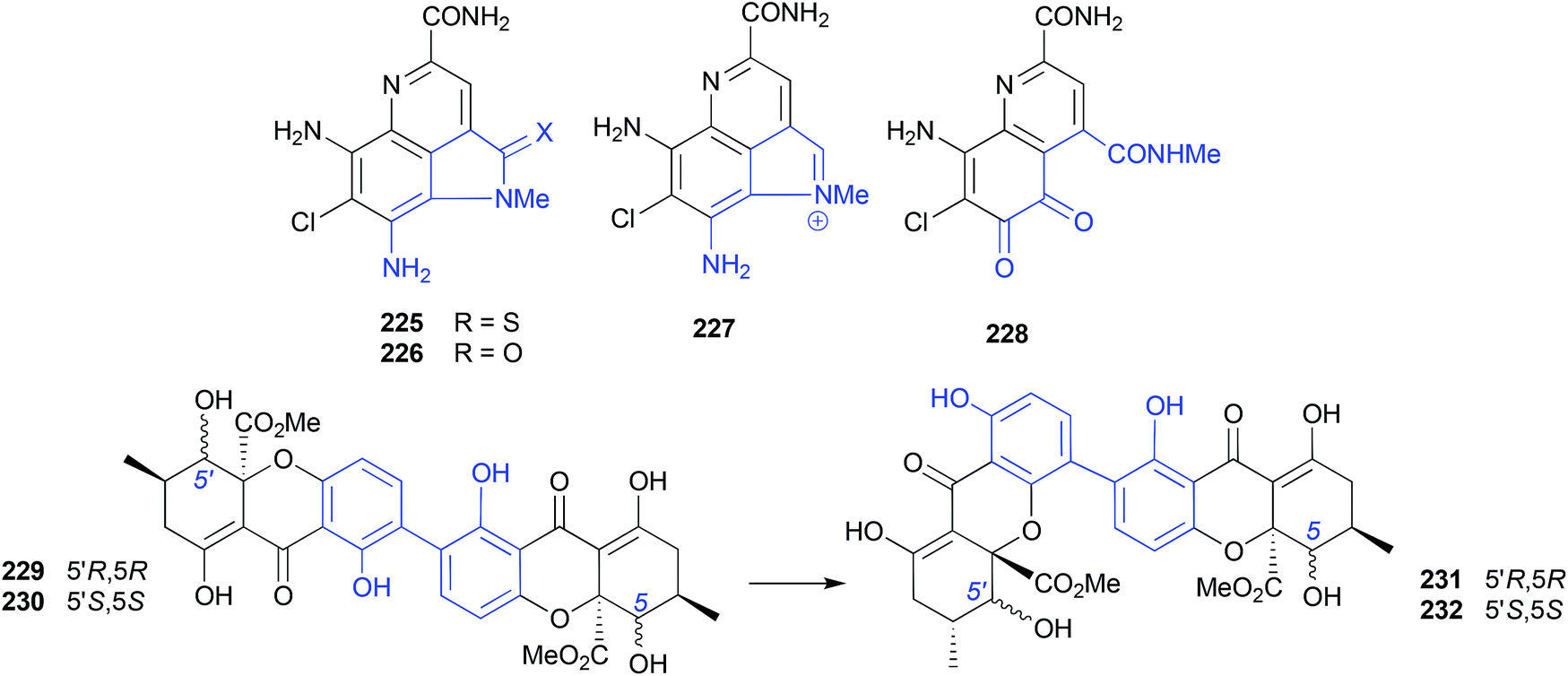 | ||
| Fig. 25 Artifacts derived from electrophiles (225–228) and rearrangements (229–232). | ||
2.16. Rearrangements (Fig. 25 and 26)
The mangrove leaf litter-derived fungus, Setophoma terrestris MSX45109, yielded and array of polyketides, including the known fungal natural products, secalonic acids A (229) and E (230). When stored at room temperature in polar organic solvents (i.e., acetone or MeCN) 229 and 230 rearrange to the isomers 231 and 232, respectively.118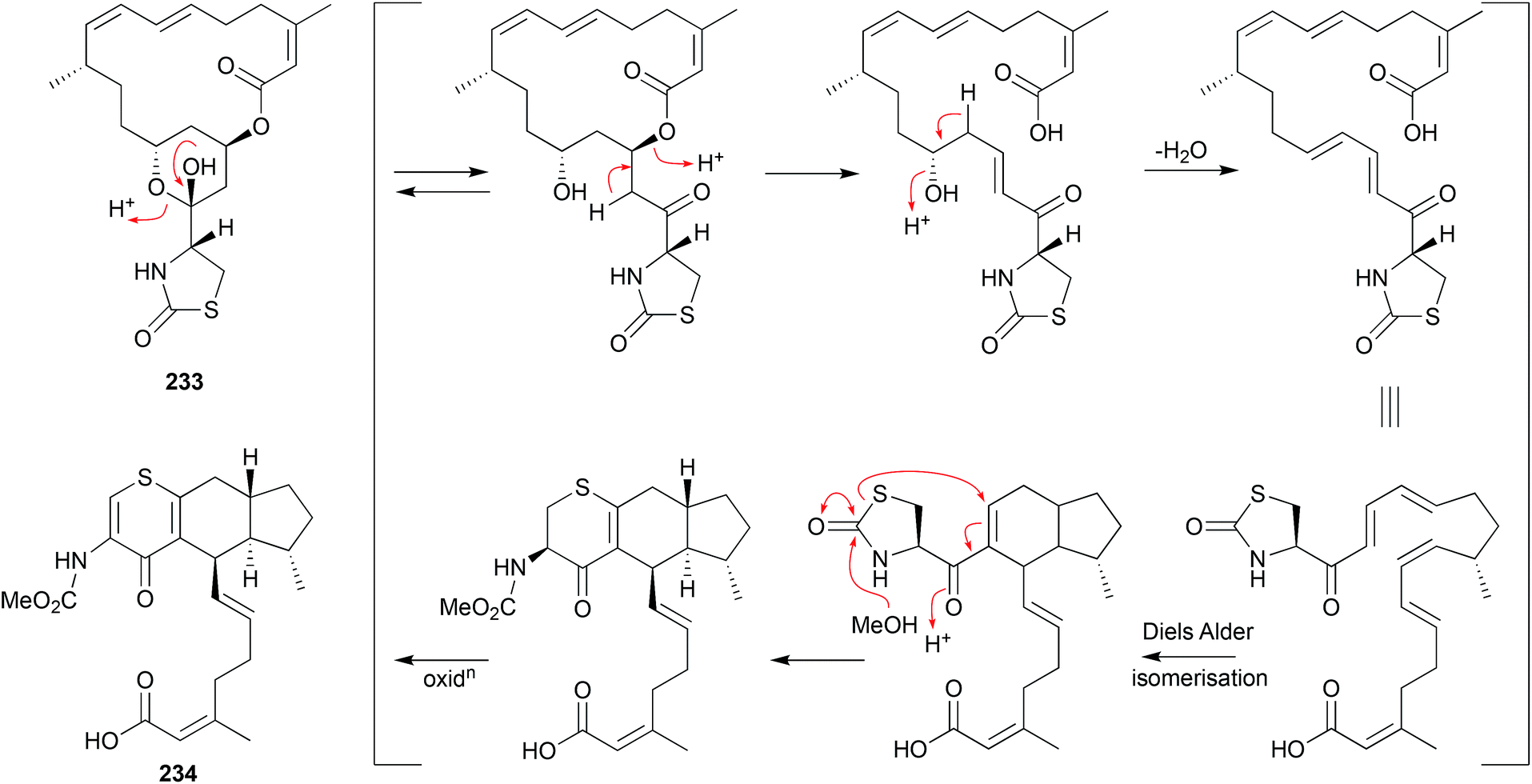 | ||
| Fig. 26 Artifacts derived from rearrangements on prolonged storage (233–234). | ||
As a classic illustration of both the dangers and opportunities encountered during the storage of natural products, long term storage of the sponge natural product latrunculin A (233) in MeOH led to transformation into the unprecedented artifact CTP-431 (234). A plausible explanation for the transformation has been proposed.119
3. Conclusions
Although this review summarizes multiple published accounts of marine natural product artifacts, spanning simple esterifications to intricate rearrangements (i.e., 1 to 2versus233 to 234), the scientific literature clearly under reports the occurrence and significance of artifacts. As a result, artifacts are regularly mischaracterised as natural products, or simply ignored. The following observations are made in the hope they inform and encourage researchers to persist in the detection, characterisation and reporting of natural product artifacts.3.1. Regulating artifacts
Notwithstanding that the study of artifacts can disclose valuable knowledge, they can also impede the investigation of scarce, highly reactive natural products. To control this adverse impact, it's important to be aware of the conditions that may initiate the formation of artifacts (i.e., as summarized in this review). Being forewarned and alert to risk factors makes it easier to either avoid, or at least minimize such conditions. For example, in many instances artifact production can be controlled by limiting exposure to high or low pH, direct sunlight and/or excessive heat. In the case of the latter this could be achieved by simply setting rotary evaporator water baths, and other sample drying technologies, to <45 °C. Similarly, natural product stability can be enhanced by sample storage in the dark in freezers, rather than on the bench in the light at room temperature. The use of nitrogen rather than air when drying down samples can also reduce the risk of oxidation artifacts. On the other hand, knowledge of conditions that promote artifact formation can be used to generate new analogues, to further explore chemical reactivity, and support structure–activity relationship investigations.3.2. Detecting artifacts
Irrespective of the care taken to limit (or promote) artifact formation, it is always prudent to preserve a portion of unprocessed biodiversity crude extracts for comparative analysis. For example, if chemical analysis of the archived crude extract does not detect the putative natural product, then its most likely, but not always, an artifact (or a contaminant). Equally important, careful analysis of all solvent partitions and chromatographic fractions throughout the isolation and handling process, with comparison back to the archived crude extract, will detect the appearance of artifacts, and draw attention to conditions that coincided with their formation (i.e., silica chromatography, and/or exposure to certain solvents).Importantly, it is possible for a compound to be both an artifact produced during fractionation and handling, and a natural product detected in the archived crude extract. While this latter observation may appear a contradiction, it merely acknowledges that nature can take advantage of the same facile non-enzymatic transformations that drive artifact formation during fractionation and handling. As an even more intriguing caveat, just because a compound is detected in the crude extract does not always mean it's a natural product, as the extraction process itself can provide the transforming conditions that generate an artifact. Examples of this phenomena are evident in the facile solvolysis of the diketomorphilino shornephine A,63 and solvent extraction-mediated Schiff's base formation of the prolinimines.97,98
3.3. Rationalising artifacts
Having isolated and identified one or more new or known natural products, and related artifacts, it is always helpful to speculate on plausible biosynthetic origins for the natural products, and the chemical mechanisms behind the transformations that drive artifact formation. A useful strategy for probing the latter is to use analytical methods (i.e., UPLC-DAD, UPLC-MS and UPLC-MS/MS) to monitor the fate of pure natural products over time, when exposed to conditions known or suspected to generate artifacts. Such studies greatly enhance knowledge of the properties of both the precursor natural products and associated artifacts, and provide a rational explanation for how both were created.3.4. Exploiting artifacts
The chemical mechanisms by which natural products transform to artifacts can provide valuable insights into biological mechanisms of action, as evident in the antitubercular Michael adduct and Michael acceptor abyssomicins.93 Likewise, knowledge of natural product to artifact chemical reactivity can inspire the synthesis of new chemical diversity to advance structure–activity relationship investigations, as observed for the electrophilic ammosamides.117 It can also inspire new synthetic routes to novel scaffolds, such as the synchronous cycloadditions at play within the heronamides110,112 and cytochalasins.71 As noted on various occasions throughout this review, an appreciation of artifacts from one source, can highlight instances where artifacts have been mis-assigned as natural products in another source, such as the didemnaketals4,5 and carriebowmides.44,45 Artifacts can also provide a glimpse of highly reactive natural products that do not survive extraction and handling, as noted for the leucettazoles13 and possibly the aciculitamides.17In short, a commitment to detect, identify and document the chemical and biological properties of artifacts, provides a valuable new window into natural product chemical space. This can only enhance future prospects for exciting new discoveries, and improved molecular tools and products.
4. Conflicts of interest
There are no conflicts to declare.5. Acknowledgements
The support of The University of Queensland, and the UQ Institute for Molecular Bioscience, and past and present members of my research group, is gratefully acknowledged.6. References
- R. A. Barrow and R. J. Capon, Aust. J. Chem., 1994, 47, 1901–1918 CrossRef CAS.
- C. Wang, L. Guo, J. Hao, L. Wang and W. Zhu, J. Nat. Prod., 2016, 79, 2977–2981 CrossRef CAS PubMed.
- H. Zhang, M. M. Conte and R. J. Capon, Angew. Chem., Int. Ed., 2010, 49, 9904–9906 CrossRef CAS PubMed.
- B. C. M. Potts, D. J. Faulkner, J. A. Chan, G. C. Simolike, P. Offen, M. E. Hemling and T. A. Francis, J. Am. Chem. Soc., 1991, 113, 6321–6322 CrossRef CAS.
- A. Pika and J. Faulkner, Nat. Prod. Lett., 1995, 7, 291–296 CrossRef.
- S. Lösgen, O. Schlörke, K. Meindl, R. Herbst-Irmer and A. Zeeck, Eur. J. Org. Chem., 2007, 2191–2196 CrossRef.
- G.-X. Zhou and T. F. Molinski, J. Asian Nat. Prod. Res., 2006, 8, 15–20 CrossRef CAS PubMed.
- M. Tsuda, T. Endo, Y. Mikami, J. Fromont and J. Kobayashi, J. Nat. Prod., 2002, 65, 1507–1508 CrossRef CAS PubMed.
- C. Wu, Y. Tan, M. Gan, Y. Wang, Y. Guan, X. Hu, H. Zhou, X. Shang, X. You, Z. Yang and C. Xiao, J. Nat. Prod., 2013, 76, 2153–2157 CrossRef CAS PubMed.
- S. Khushi, L. Nahar, A. A. Salim and R. J. Capon, Mar. Drugs, 2018, 16, 456 CrossRef CAS PubMed.
- E. Elkhayat, R. Edrada, R. Ebel, V. Wray, R. van Soest, S. Wiryowidagdo, M. H. Mohamed, W. E. G. Müller and P. Proksch, J. Nat. Prod., 2004, 67, 1809–1817 CrossRef CAS PubMed.
- C. Gao, Z. Guo, X. Lu, H. Chen, L. Liu, Z. Yu and Y. Chen, J. Nat. Prod., 2018, 81, 2069–2074 CrossRef CAS PubMed.
- P. Prasad, A. A. Salim, S. Khushi, Z. G. Khalil and M. Quezada, Mar. Drugs, 2019, 17(2), 106 CrossRef CAS PubMed.
- K. Ragini, J. Fromont, A. M. Piggott and P. Karuso, J. Nat. Prod., 2017, 80, 215–219 CrossRef CAS PubMed.
- N. Lindquist and W. Fenical, Tetrahedron Lett., 1990, 31, 2521–2524 CrossRef CAS.
- I. Mancini, G. Guella, C. Debitus, D. Dubet and F. Pietra, Helv. Chim. Acta, 1994, 77, 1886–1894 CrossRef CAS.
- C. A. Bewley, H. He, D. H. Williams and D. J. Faulkner, J. Am. Chem. Soc., 1996, 118, 4314–4321 CrossRef CAS.
- M. L. Bourguet-Kondracki, F. Lacombe and M. Guyot, J. Nat. Prod., 1999, 62, 1304–1305 CrossRef CAS PubMed.
- S. Urban and R. J. Capon, J. Nat. Prod., 1996, 59, 900–901 CrossRef CAS.
- M. T. Hamann, P. J. Scheuer and M. Kelly-Borges, J. Org. Chem., 1993, 58, 6565–6569 CrossRef CAS.
- C. F. Pérez Hemphill, G. Daletos, Z. Liu, W. Lin and P. Proksch, Tetrahedron Lett., 2016, 57, 2078–2083 CrossRef.
- Y.-J. Jiang, D.-S. Zhang, H.-J. Zhang, J.-Q. Li, W.-J. Ding, C.-D. Xu and Z.-J. Ma, J. Nat. Prod., 2018, 81, 2120–2124 CrossRef CAS PubMed.
- W. Yuan, S. Cheng, W. Fu, M. Zhao, X. Li, Y. Cai, J. Dong, K. Huang, K. R. Gustafson and P. Yan, J. Nat. Prod., 2016, 79, 1124–1131 CrossRef CAS PubMed.
- X. Luo, X. Lin, H. Tao, J. Wang, J. Li, B. Yang, X. Zhou and Y. Liu, J. Nat. Prod., 2018, 81, 934–941 CrossRef CAS PubMed.
- H. Li, P. B. Shinde, H. J. Lee, E. S. Yoo, C.-O. Lee, J. Hong, S.-H. Choi and J. H. Jung, Arch. Pharmacal Res., 2009, 32, 857–862 CrossRef CAS PubMed.
- U. Anthoni, L. Chevolot, C. Larsen, P. H. Nielsen and C. Christophersen, J. Org. Chem., 1987, 52, 4709–4712 CrossRef CAS.
- K. Ø. Hansen, J. Isaksson, A. Bayer, J. A. Johansen, J. H. Andersen and E. Hansen, J. Nat. Prod., 2017, 80, 3276–3283 CrossRef CAS.
- X. S. Huang, I. Sattler, H.-Z. Fu, S. Grabley and W.-H. Lin, J. Asian Nat. Prod. Res., 2007, 9, 327–331 CrossRef PubMed.
- Y. Hu, E. D. Martinez and J. B. MacMillan, J. Nat. Prod., 2012, 75, 1759–1764 CrossRef CAS PubMed.
- A. Guerriero, M. D'Ambrosio, V. Cuomo and F. Pietra, Helv. Chim. Acta, 1991, 74, 1445–1450 CrossRef CAS.
- K. Ueda, T. Ueta, E. Richard, O. Siwu, M. Kita and D. Uemura, Chem. Lett., 2005, 34, 1530–1531 CrossRef CAS.
- Y. Sangnoi, O. Sakulkeo, S. Yuenyongsawad, A. Kanjana-opas, K. Ingkaninan, A. Plubrukarn and K. Suwanborirux, Mar. Drugs, 2008, 6, 578–586 CrossRef CAS.
- G. Esposito, M.-L. Bourguet-Kondracki, L. H. Mai, A. Longeon and R. van Soest, J. Nat. Prod., 2016, 79, 2953–2960 CrossRef CAS PubMed.
- S. J. Rochfort, S. Moore, C. Craft, N. H. Martin, R. M. Van Wagoner and J. L. C. Wright, J. Nat. Prod., 2009, 72, 1773–1781 CrossRef CAS PubMed.
- R. J. Capon, E. L. Ghisalberti and P. R. Jefferies, Phytochemicals, 1981, 20, 2598–2600 CrossRef CAS.
- P. Reddy and S. Urban, Phytochemicals, 2009, 70, 250–255 CrossRef CAS PubMed.
- A. W. Markwell-Heys and J. H. George, Org. Biomol. Chem., 2016, 14, 5546–5549 RSC.
- R. J. Capon, M. Miller and F. Rooney, J. Nat. Prod., 2000, 63, 821–824 CrossRef CAS.
- R. Kazlauskas, P. T. Murphy, R. J. Quinn and R. J. Wells, Tetrahedron Lett., 1977, 18, 61–64 CrossRef.
- S. P. Gunasekera, P. J. McCarthy, S. A. Pomponi and A. E. Wright, J. Nat. Prod., 1999, 62, 1208–1211 CrossRef CAS PubMed.
- N. K. Utkina, A. V. Gerasimenko and D. Y. Popov, Russ. Chem. Bull., 2003, 52, 258–260 CrossRef CAS.
- G. Lidgren, L. Bohlin and C. Christophersen, J. Nat. Prod., 1988, 51, 1277–1280 CrossRef CAS.
- L.-J. Ding, W. Yuan, X.-J. Liao, B. N. Han, S.-P. Wang, Z.-Y. Li, S.-H. Xu, W. Zhang and H.-W. Lin, J. Nat. Prod., 2016, 79, 2045–2052 CrossRef CAS PubMed.
- S. P. Gunasekera, R. Ritson-Williams and V. J. Paul, J. Nat. Prod., 2008, 71, 2060–2063 CrossRef CAS PubMed.
- E. Mevers, T. Byrum and W. H. Gerwick, J. Nat. Prod., 2013, 76, 1810–1814 CrossRef CAS.
- L. M. Nogle, R. T. Williamson and W. H. Gerwick, J. Nat. Prod., 2001, 64, 716–719 CrossRef CAS PubMed.
- G. G. Harrigan, H. Luesch, W. Y. Yoshida, R. E. Moore, D. G. Nagle and V. J. Paul, J. Nat. Prod., 1999, 62, 655–658 CrossRef CAS PubMed.
- S. Matthew, C. Ross, V. J. Paul and H. Luesch, Tetrahedron, 2008, 64, 4081–4089 CrossRef CAS.
- M. W. Bernart, W. H. Gerwick, E. E. Corcoran, A. Y. Lee and J. Clardy, Phytochemicals, 1992, 31, 1273–1276 CrossRef CAS.
- H. Wiedenfeld, F. Knoch and M. Koch, Arch. Pharm., 1985, 318, 289–293 CrossRef CAS.
- A. S. Martin, J. Rovirosa, O. Muñoz, M. H. M. Chen, R. D. Guneratne and J. Clardy, Tetrahedron Lett., 1983, 24, 4063–4066 CrossRef.
- A. San-Martín, J. Rovirosa, C. Xu, H. S. M. Lu and J. Clardy, Tetrahedron Lett., 1987, 28, 6013–6014 CrossRef.
- M. Bittner, F. Gonzalez, H. Valdebenito, M. Silva, V. J. Paul, W. Fenical, M. H. M. Chen and J. Clardy, Tetrahedron Lett., 1987, 28, 4031–4032 CrossRef CAS.
- Y. Gopichand and F. J. Schmitz, Tetrahedron Lett., 1978, 19, 3641–3644 CrossRef.
- K. Hagiwara, J. E. Garcia Hernandez, M. K. Harper, A. Carroll, C. A. Motti, J. Awaya, H. Y. Nguyen and A. D. Wright, J. Nat. Prod., 2015, 78, 325–329 CrossRef CAS PubMed.
- B. N. Ravi, H. P. Perzanowski, R. A. Ross, T. R. Erdman and P. J. Scheuer, Pure Appl. Chem., 1979, 51, 1893–1900 CAS.
- H. Yamazaki, O. Takahashi, S.-I. Kanno, T. Nakazawa, S. Takahashi, K. Ukai, D. A. Sumilat, M. Ishikawa and M. Namikoshi, Bioorg. Med. Chem., 2015, 23, 797–802 CrossRef CAS PubMed.
- E. Fahy and R. J. Andersen, Can. J. Chem., 1987, 65, 376–383 CrossRef CAS.
- R. J. Capon, C. Peng and C. Dooms, Org. Biomol. Chem., 2008, 6, 2765–2771 RSC.
- J. Peng, T. Lin, W. Wang, Z. Xin, T. Zhu, Q. Gu and D. Li, J. Nat. Prod., 2013, 76, 1133–1140 CrossRef CAS PubMed.
- G. Guella, D. Skropeta, I. Mancini and F. Pietra, Chem.–Eur. J., 2003, 9, 5770–5777 CrossRef CAS PubMed.
- L. Fremlin, M. Farrugia, A. M. Piggott, Z. Khalil, E. Lacey and R. J. Capon, Org. Biomol. Chem., 2011, 9, 1201–1211 RSC.
- Z. G. Khalil, X.-C. Huang, R. Raju, A. M. Piggott and R. J. Capon, J. Org. Chem., 2014, 79, 8700–8705 CrossRef CAS PubMed.
- H. Zhang, X. Xiao, M. M. Conte, Z. Khalil and R. J. Capon, Org. Biomol. Chem., 2012, 10, 9671–9676 RSC.
- C. Tringali and M. Piattelli, Tetrahedron Lett., 1982, 23, 1509–1512 CrossRef CAS.
- R. R. Parvatkar, C. D'Souza, A. Tripathi and C. G. Naik, Phytochemicals, 2009, 70, 128–132 CrossRef CAS PubMed.
- Y. Wang, J. Zheng, P. Liu, W. Wang and W. Zhu, Mar. Drugs, 2011, 9, 1368–1378 CrossRef CAS PubMed.
- Y. Sun, J. Liu, L. Li, C. Gong, S. Wang, F. Yang, H. Hua and H. Lin, Bioorg. Med. Chem. Lett., 2017, 28, 315–318 CrossRef PubMed.
- H. Hua and H. Lin, Bioorg. Med. Chem. Lett., 2018, 28, 3281–3282 CrossRef PubMed.
- H. Yamazaki, H. Rotinsulu, O. Takahashi, R. Kirikoshi and M. Namikoshi, Tetrahedron Lett., 2016, 57, 5764–5767 CrossRef CAS.
- Z. Shang, R. Raju, A. A. Salim, Z. G. Khalil and R. J. Capon, J. Org. Chem., 2017, 82, 9704–9709 CrossRef CAS PubMed.
- B.-C. Yan, W.-G. Wang, D.-B. Hu, X. Sun, L.-M. Kong, X.-N. Li, X. Du, S.-H. Luo, Y. Liu, Y. Li, H.-D. Sun and J.-X. Pu, Org. Lett., 2016, 18, 1108–1111 CrossRef CAS PubMed.
- Y. Kon, T. Kubota, A. Shibazaki, T. Gonoi and J. Kobayashi, Bioorg. Med. Chem. Lett., 2010, 20, 4569–4572 CrossRef CAS PubMed.
- R. J. Capon and J. K. MacLeod, Aust. J. Chem., 1987, 40, 341–346 CrossRef CAS.
- G. Combaut, J.-M. Chantraine, J. Teste and K.-W. Glombitza, Phytochemicals, 1978, 17, 1791–1792 CrossRef CAS.
- E. Al-Farhan, O. M. Falana, P. M. Keehn and R. Stevenson, Tetrahedron Lett., 1992, 33, 5885–5886 CrossRef CAS.
- L. Hammerschmidt, A. Debbab, T. D. Ngoc, V. Wray, C. P. Hemphil, W. Lin, H. Broetz-Oesterhelt, M. U. Kassack, P. Proksch and A. H. Aly, Tetrahedron Lett., 2014, 55, 3463–3468 CrossRef CAS.
- R. J. Capon, D. R. Groves, S. Urban and R. G. Watson, Aust. J. Chem., 1993, 46, 1245–1253 CrossRef CAS.
- M. Ishibashi, Y. Ohizumi, J. F. Cheng, H. Nakamura, Y. Hirata, T. Sasaki and J. Kobayashi, J. Org. Chem., 1988, 53, 2855–2858 CrossRef CAS.
- Z. Shang, A. A. Salim, Z. Khalil, M. Quezada, P. V. Bernhardt and R. J. Capon, J. Org. Chem., 2015, 80, 12501–12508 CrossRef CAS PubMed.
- Z. Shang, A. A. Salim, Z. Khalil, P. V. Bernhardt and R. J. Capon, J. Org. Chem., 2016, 81, 6186–6194 CrossRef CAS PubMed.
- J. Inokoshi, Y. Nakamura, Z. Hongbin, R. Uchida, K.-I. Nonaka, R. Masuma and H. Tomoda, J. Antibiot., 2013, 66, 37–41 CrossRef CAS PubMed.
- R. J. Capon and D. J. Faulkner, J. Am. Chem. Soc., 1984, 106, 1819–1822 CrossRef CAS.
- R. J. Capon, C. Skene, D. Vuong, E. Lacey, K. Heiland and T. Friedel, J. Nat. Prod., 2002, 65, 368–370 CrossRef CAS PubMed.
- X. Alvarez-Mico, P. R. Jensen, W. Fenical and C. C. Hughes, Org. Lett., 2013, 15, 988–991 CrossRef CAS PubMed.
- S.-Q. Yang, X.-M. Li, X. Li, H.-L. Li, L.-H. Meng and B.-G. Wang, Phytochem. Lett., 2018, 25, 191–195 CrossRef CAS.
- D. Wakana, T. Hosoe, T. Itabashi, K. Okada, G. M. De Campos Takaki, T. Yaguchi, K. Fukushima and K. I. Kawai, J. Nat. Med., 2006, 60, 279–284 CrossRef CAS.
- B. R. Clark, R. J. Capon, E. Lacey, S. Tennant and J. H. Gill, Org. Biomol. Chem., 2006, 4, 1520–1528 RSC.
- B. Bister, D. Bischoff, M. Ströbele, J. Riedlinger, A. Reicke, F. Wolter, A. T. Bull, H. Zähner, H.-P. Fiedler and R. D. Süssmuth, Angew. Chem., Int. Ed., 2004, 43, 2574–2576 CrossRef CAS.
- J. Riedlinger, A. Reicke, H. Zähner, B. Krismer, A. T. Bull, L. A. Maldonado, A. C. Ward, M. Goodfellow, B. Bister, D. Bischoff, R. D. Süssmuth and H.-P. Fiedler, J. Antibiot., 2004, 57, 271–279 CrossRef CAS.
- S. Keller, G. Nicholson, C. Drahl, E. Sorensen, H.-P. Fiedler and R. D. Süssmuth, J. Antibiot., 2007, 60, 391–394 CrossRef CAS PubMed.
- K. C. Nicolaou and S. T. Harrison, J. Am. Chem. Soc., 2007, 129, 429–440 CrossRef CAS PubMed.
- Q. Wang, F. Song, X. Xiao, P. Huang, L. Li, A. Monte, W. M. Abdel-Mageed, J. Wang, H. Guo, W. He, F. Xie, H. Dai, M. Liu, C. Chen, H. Xu, M. Liu, A. M. Piggott, X. Liu, R. J. Capon and L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 1231–1234 CrossRef CAS PubMed.
- H. Greve, P. J. Schupp, E. Eguereva, S. Kehraus, G. Kelter, A. Maier, H.-H. Fiebig and G. M. König, Eur. J. Org. Chem., 2008, 2008, 5085–5092 CrossRef PubMed.
- K. Pudhom and T. Teerawatananond, J. Nat. Prod., 2014, 77, 1962–1966 CrossRef CAS.
- J. E. Hochlowski, D. J. Faulkner, J. C. Coll, J. E. Biskupiak, C. M. Ireland, Z. Qi-tai, H. Cun-heng and J. Clardy, J. Am. Chem. Soc., 1984, 106, 6748–6750 CrossRef.
- O. G. Mohamed, Z. G. Khalil and R. J. Capon, Org. Lett., 2018, 20, 377–380 CrossRef CAS PubMed.
- O. G. M. Mohamed, Adventures in Australian Microbial Biodiscovery, PhD thesis, 2018.
- W. Balansa, R. Islam, F. Fontaine, A. M. Piggott, H. Zhang, T. I. Webb, D. F. Gilbert, J. W. Lynch and R. J. Capon, Bioorg. Med. Chem., 2010, 18, 2912–2919 CrossRef CAS.
- N. Wang, J. Song, K. H. Jang, H.-S. Lee, X. Li, K.-B. Oh and J. Shin, J. Nat. Prod., 2008, 71, 551–557 CrossRef CAS.
- W. Balansa, R. Islam, F. Fontaine, A. M. Piggott, H. Zhang, X. Xiao, T. I. Webb, D. F. Gilbert, J. W. Lynch and R. J. Capon, Org. Biomol. Chem., 2013, 11, 4695–4701 RSC.
- R. Capon, E. L. Ghisalberti, P. R. Jefferies, B. W. Skelton and A. White, Tetrahedron, 1981, 37, 1613–1621 CrossRef CAS.
- R. J. Capon, E. L. Ghisalberti and P. R. Jefferies, J. Chem. Res., Synop., 1987, 118–119 CAS.
- J. Zheng, H. Zhu, K. Hong, Y. Wang, P. Liu, X. Wang, X. Peng and W. Zhu, Org. Lett., 2009, 11, 5262–5265 CrossRef CAS PubMed.
- H. C. Kwon, C. A. Kauffman, P. R. Jensen and W. Fenical, J. Org. Chem., 2009, 74, 675–684 CrossRef CAS.
- P. Fu, P. Liu, X. Li, Y. Wang, S. Wang, K. Hong and W. Zhu, Org. Lett., 2011, 13, 5948–5951 CrossRef CAS.
- H. Gao, W. Guo, Q. Wang, L. Zhang, M. Zhu, T. Zhu, Q. Gu, W. Wang and D. Li, Bioorg. Med. Chem. Lett., 2013, 23, 1776–1778 CrossRef CAS.
- H. Shigemori, M.-A. Bae, K. Yazawa, T. Sasaki and J. Kobayashi, J. Org. Chem., 1992, 57, 4317–4320 CrossRef CAS.
- W. J. Moree, O. J. McConnell, D. D. Nguyen, L. M. Sanchez, Y.-L. Yang, X. Zhao, W.-T. Liu, P. D. Boudreau, J. Srinivasan, L. Atencio, J. Ballesteros, R. G. Gavilán, D. Torres-Mendoza, H. M. Guzmán, W. H. Gerwick, M. Gutiérrez and P. C. Dorrestein, ACS Chem. Biol., 2014, 9, 2300–2308 CrossRef CAS PubMed.
- R. Raju, A. M. Piggott, M. M. Conte and R. J. Capon, Org. Biomol. Chem., 2010, 8, 4682–4689 RSC.
- N. Kanoh, S. Itoh, K. Fujita, K. Sakanishi, R. Sugiyama, Y. Terajima, Y. Iwabuchi, S. Nishimura and H. Kakeya, Chem.–Eur. J., 2016, 22, 8586–8595 CrossRef CAS.
- T. J. Booth, S. Alt, R. J. Capon and B. Wilkinson, Chem. Commun., 2016, 52, 6383–6386 RSC.
- S. Cai, Y. Luan, X. Kong, T. Zhu, Q. Gu and D. Li, Org. Lett., 2013, 15, 2168–2171 CrossRef CAS.
- C. C. Hughes, J. B. MacMillan, S. P. Gaudêncio, P. R. Jensen and W. Fenical, Angew. Chem., Int. Ed., 2009, 48, 725–727 CrossRef CAS PubMed.
- C. C. Hughes and W. Fenical, J. Am. Chem. Soc., 2010, 132, 2528–2529 CrossRef CAS.
- E. Pan, M. Jamison, M. Yousufuddin and J. B. MacMillan, Org. Lett., 2012, 14, 2390–2393 CrossRef CAS.
- D. Reimer and C. C. Hughes, J. Nat. Prod., 2017, 80, 126–133 CrossRef CAS.
- T. El-Elimat, M. Figueroa, H. A. Raja, T. N. Graf, S. M. Swanson, J. O. Falkinham, M. C. Wani, C. J. Pearce and N. H. Oberlies, Eur. J. Org. Chem., 2015, 2015, 109–121 CrossRef CAS.
- A. D. Przeslak, M. Inman, W. Lewis and C. J. Moody, J. Org. Chem., 2018, 83, 10595–10601 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |