
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of repeat unit charge density on the physical and electrochemical properties of novel heterocationic poly(ionic liquid)s†
Merlin
Cotessat
a,
Dimitri
Flachard
b,
Daniil
Nosov
 a,
Elena I.
Lozinskaya
c,
Denis O.
Ponkratov
c,
Daniel F.
Schmidt
a,
Eric
Drockenmuller
*a and
Alexander S.
Shaplov
*a
a,
Elena I.
Lozinskaya
c,
Denis O.
Ponkratov
c,
Daniel F.
Schmidt
a,
Eric
Drockenmuller
*a and
Alexander S.
Shaplov
*a
aLuxembourg Institute of Science and Technology (LIST), 5 avenue des Hauts-Fourneaux, L-4362 Esch-sur-Alzette, Luxembourg. E-mail: alexander.shaplov@list.lu
bUniv. Lyon, Université Lyon 1, CNRS, Ingénierie des Matériaux Polymères, UMR 5223, F-69003, Lyon, France. E-mail: eric.drockenmuller@univ-lyon1.fr
cA. N. Nesmeyanov Institute of Organoelement Compounds Russian Academy of Sciences (INEOS RAS), Vavilov str. 28, 119991, Moscow, Russia
First published on 11th December 2020
Abstract
We report the synthesis and structure/property correlations of a series of eight poly(ionic liquid)s (PILs) obtained from sequential AA + BB polyaddition by copper(I)-catalyzed azide–alkyne cycloaddition and the subsequent N-alkylation reaction. The different repeat units contain one to four ion pairs, with one to four bis(trifluorosulfonyl)imide (TFSI) anions and one or two types of ammonium, imidazolium or 1,2,3-triazolium counter-cations. Their physical, ion conducting and electrochemical properties are discussed based on the repeat unit charge density and structure of the cationic moieties. The comparison of several pairs of polyelectrolytes revealed that ionic conductivity is dependent on (1) the ratio between the number of charge carriers per monomer unit and the number of surrounding atoms/groups that can solvate the ions and (2) the nature of the cation. The highest conductivity (1.8 × 10−5 S cm−1 at 25 °C) was reached when PILs contain two 1,2,3-triazolium cations that are separated by an oxyethylene spacer. The incorporation of an additional type of cation (either imidazolium or ammonium with 1,2,3-triazolium) in one PIL allows its cathodic limit to be increased up to −0.4 V vs. Li+/Li (70 °C) and the oxidation instability of 1,2,3-triazolium cations to be overcome.
Introduction
In the last decade, intensive efforts focused on flexible, lightweight and safe energy storage devices have been stimulated by the increased demand of next-generation portable and flexible electronics, wearable devices and electric cars. Both flexible solid-state supercapacitors (SCs)1,2 and solid-state Li batteries3–6 have attracted increased interest given the convincing advantages of transitioning from volatile, flammable and toxic liquid electrolytes to lightweight, non-volatile polymer electrolytes. In comparison with the existing technologies, solid-state SCs and Li batteries are smaller in size, light weight, and easy to handle, and offer a broader range of operating temperatures, and greater safety and reliability.1,3,4,7,8 The development of safe, efficient solid-state SCs and lithium batteries requires solid polymer electrolytes (SPEs) with high ionic conductivity (>10−4 S cm−1 at 25 °C) as well as good electrochemical and thermal stabilities.3,9Among promising candidates for SPEs, a new class of polyelectrolytes, namely polymeric analogs of ionic liquids, a.k.a. poly(ionic liquid)s (PILs), has recently gained significant attention.10–17 PILs can be considered as the macromolecular analogs of ionic liquids (ILs), in the best cases combining the advantages of polymers (viscoelasticity, processability, film-forming properties, etc.) and ILs (high thermal and electrochemical stabilities, enhanced ionic conductivity, etc.).10,11,15 The use of PILs in SPEs instead of common neutral polymers offers several advantages. Firstly, PILs being polyelectrolytes, impart additional charges to the system. Secondly, when ILs are added to PILs, the optimal homogeneous distribution of charges can be reached at the molecular level due to strong electrostatic interactions, thus minimizing phase separation, IL leakage and providing the possibility of increasing the IL content up to 70–90 wt%.9,18
Nowadays a broad variety of PILs are prepared using countless combinations of cations (ammonium, pyridinium, imidazolium, phosphonium, 1,2,3- and 1,2,4-triazoliums, etc.) and anions (halides, perfluorinated sulfonimides, etc.).10,19–22 An analysis of the published data allows the identification of the main factors influencing the bulk conductivity of PILs: the structure of the cation and anion, the length and type of the spacer between the polymer backbone and chemically grafted ion, the glass-transition temperature and the polymerization degree.3,10–17,23,24 It should be noted that all of the aforementioned factors are interrelated and that it is their combination that governs the ionic conductivity of PILs. The structures of the most highly conductive PILs synthesized to date25–31 are listed in Scheme 1; the maximum bulk ionic conductivity reported in a neat, well-purified linear PIL is 6.7 × 10−5 S cm−1 at 30 °C under anhydrous conditions.31
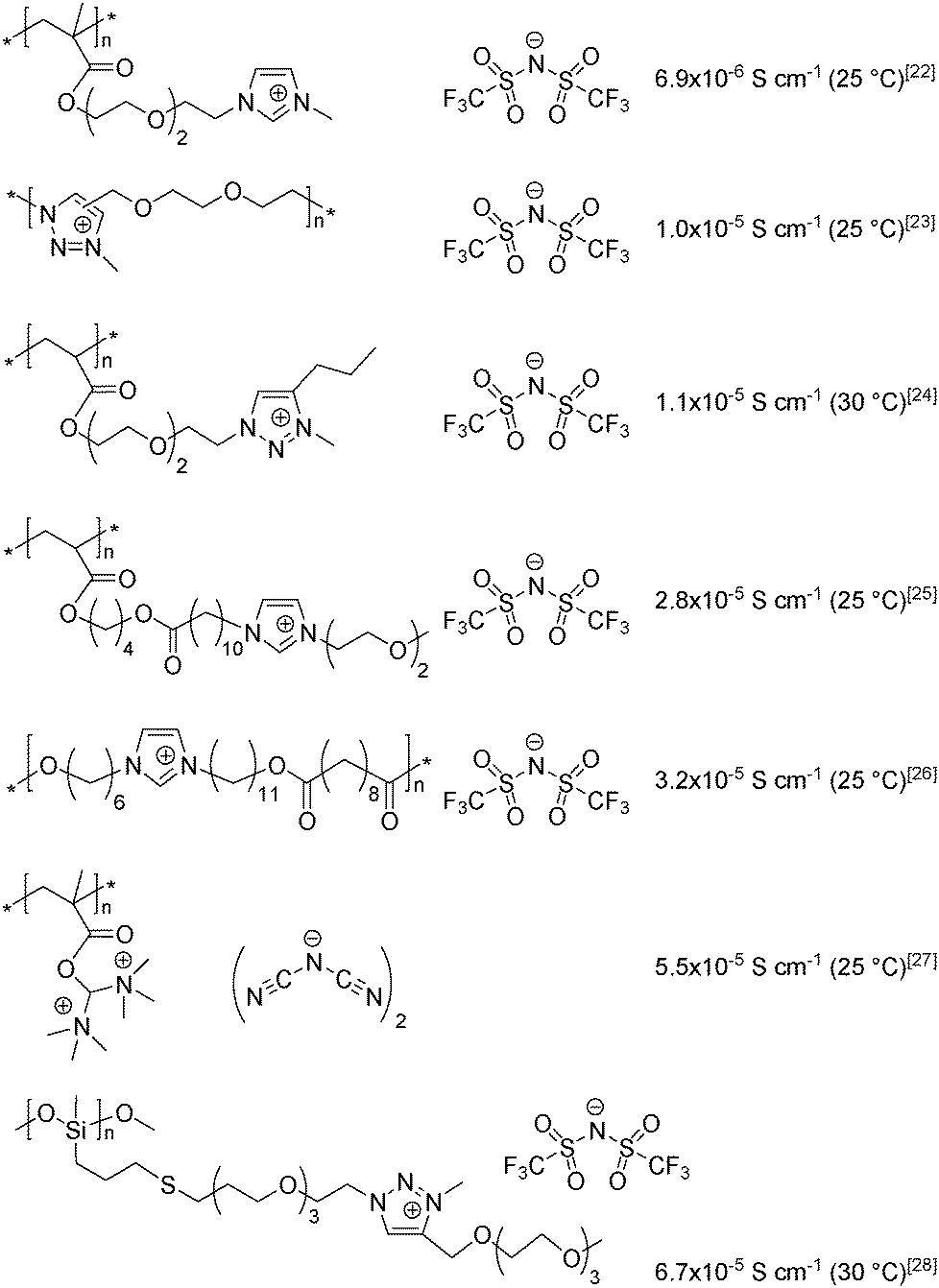 | ||
| Scheme 1 An overview of the most highly conductive PILs reported to date. | ||
Although attempts to enhance the ionic conductivity of PILs have been made by varying the anion and cation structures, the length of the spacer and the placement of the ions in the polymer backbone vs. in the side chain, the influence of repeat unit charge density on the bulk conductivity of PILs has been scarcely addressed.29,32–39 As seen in Scheme 2, the maximum number of ion pairs per monomer unit reported to date is three,32 and the structure/conductivity relationships remain sometimes contradictory. For instance, comparable hydroxide conductivities were reported for imidazolium-based PILs having cationic or dicationic repeat units and hydroxide counter-anions.33,34 Conversely, Fan et al.35 and Drockenmuller et al.36 observed a ca. 2 fold increase in ionic conductivity for PILs carrying one and two pendent imidazolium or 1,2,3-triazolium cations in each repeating unit (Scheme 2). In total contradiction, Gibson et al. observed a fourfold decrease in ionic conductivity for polyesters when the number of main-chain imidazolium cations was increased from one to two.29 Finally, while extremely low ionic conductivity was reported for poly(1-vinyl-3-ethylimidazolium) bis(trifluoromethylsulfonyl)-imide,37,38 the ionic conductivity of the only example of heterodicationic PIL reported so far (i.e. poly(N,N,N-trimethyl)-N-(1-vinylimidazolium-3-ethyl)-ammonium bis(trifluoromethyl-sulfonyl)imide39) has not been reported.
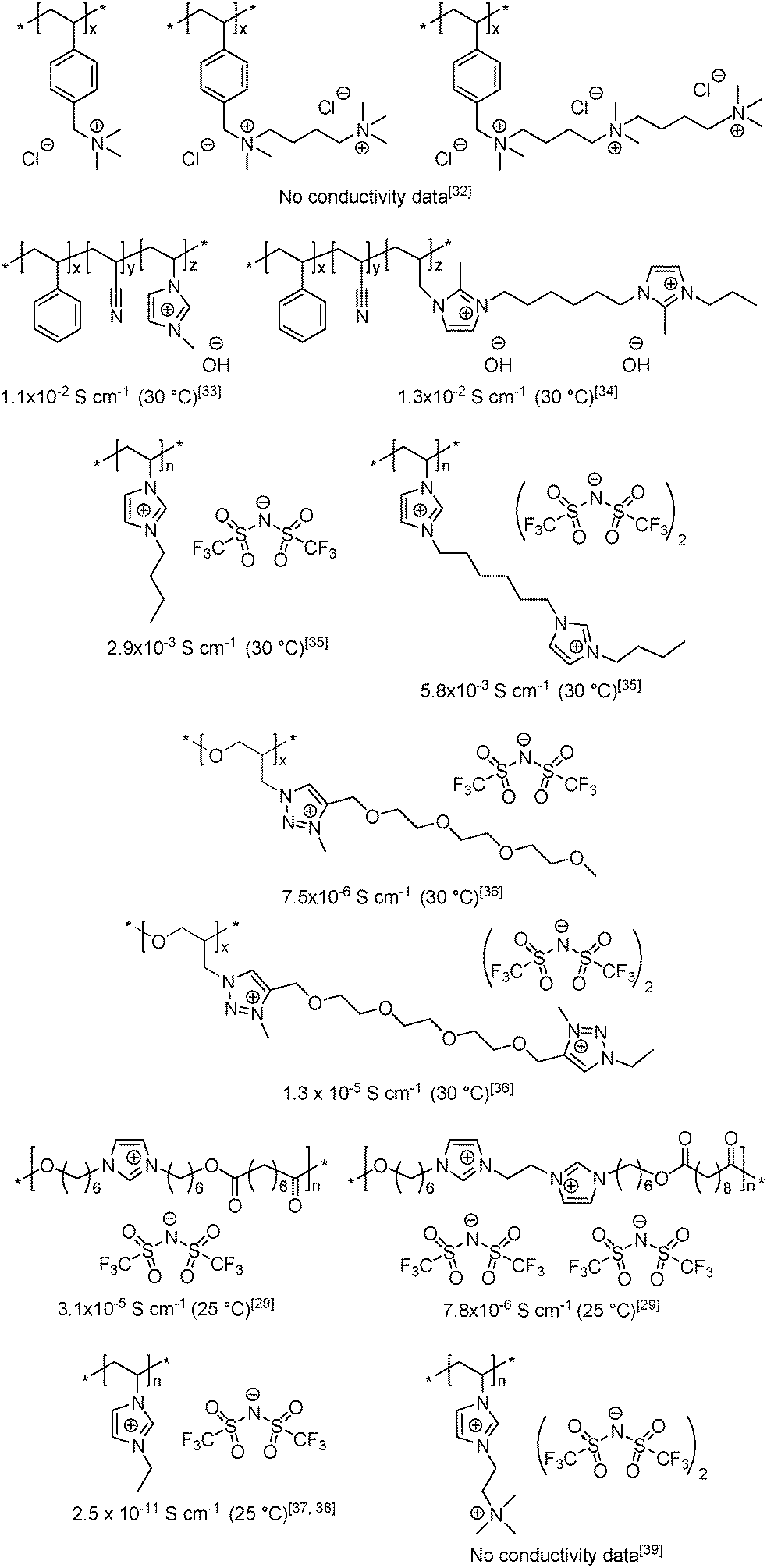 | ||
| Scheme 2 Examples of the repeat unit charge density influence on the ionic conductivity of PILs. | ||
To fill this gap in fundamental knowledge concerning the influence of repeat unit charge density on the bulk ionic conductivity of SPEs, in this work we report the synthesis and comparison of eight PILs obtained from sequential AA + BB polyaddition by copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) using ammonium or imidazolium dialkynes with α,ω-diazido-tetra(ethylene glycol) and subsequent N-alkylation of the 1,2,3-triazole groups. This versatile and general approach afforded the preparation of a series of PILs carrying TFSI counter-anions and differing by the chemical nature and number of cations in the repeat unit (Scheme 4). Their physical and electrochemical properties are discussed based on the repeat unit charge density as well as on the combination and chemical nature of the cations. These novel PILs exhibit conductivity up to 1.6 × 10−5 S cm−1 at 25 °C as well as electrochemical stability up to 3.6 V VS. Li+/Li.
Results and discussion
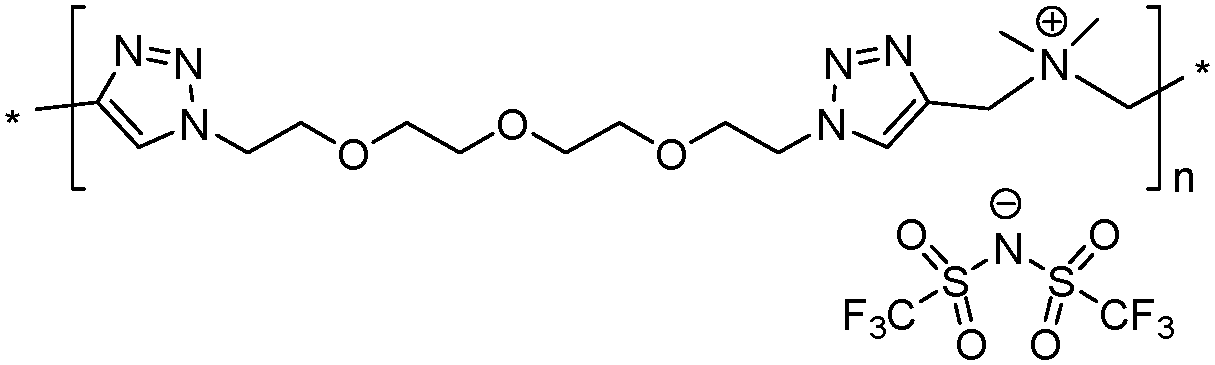
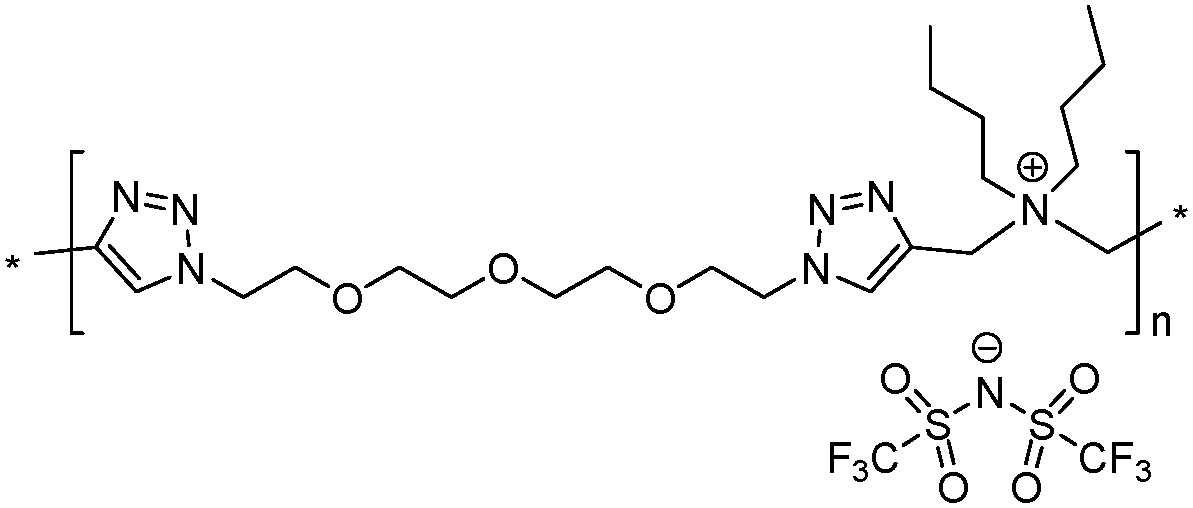
The analysis of Schemes 1 and 2 led us to the conclusion that the synthesis of (meth)acrylate or siloxane monomers with multiple ion pairs would require advanced multistep synthetic procedures. In contrast, synthesis of 1,2,3-triazolium-based PILs (TPILs) through AA + BB CuAAC polyaddition and subsequent quantitative N-alkylation of poly(1,2,3-triazole) intermediates allows unprecedented opportunities for macromolecular design taking advantage of the appealing attributes of the “click chemistry” philosophy.14 Such an approach has been previously applied to the synthesis of aliphatic, aromatic, and ethylene glycol-based TPILs.14,40,41 Thus, to fulfill the purpose of this study the synthetic platform towards TPILs was selected as the most favorable approach for the synthesis and investigation of a library of monocationic and heterodicationic PILs with tunable repeat unit charge density (Scheme 4 and Table 1). 1,11-Diazido-3,6,9-trioxaundecane (6) was chosen as the diazide monomer due to its easy accessibility and the presence of oxyethylene units that on one hand impart flexibility to the polymer chain and on another improve the solubility of ionic species, facilitate their dissociation, and promote an increase in the ionic conductivity of the resulting PILs.10 A TFSI ion was selected as an anion providing PILs both with high ionic conductivity (Scheme 1) and electrochemical stability.6| Polymer | M n (kDa) | Đ | T g (°C) | T onset (°C) | σ DC at 25 °C (S cm−1) | Electro-chemical windowd (V) | Charge density (mmol g−1) | |
|---|---|---|---|---|---|---|---|---|
| Structure | No. | |||||||
| a Obtained by GPC in 0.1 M LiTFSI in DMF at 50 °C with polystyrene standard calibration. b Obtained by DSC. c Obtained by TGA. d Obtained by cyclic voltammetry at 70 °C (stainless steel as a working electrode and Li foil as counter and reference electrodes, scan rate 0.5 mV s−1). e Not determined due to the very low conductivity. f For comparison. | ||||||||
|
7 | 50.5 | 2.79 | 13 | 235 | 9.8 × 10−10 | —e | 1.55 |
|
8 | 62.8 | 1.94 | 8 | 220 | 2.8 × 10−9 | 4.0 | 1.37 |
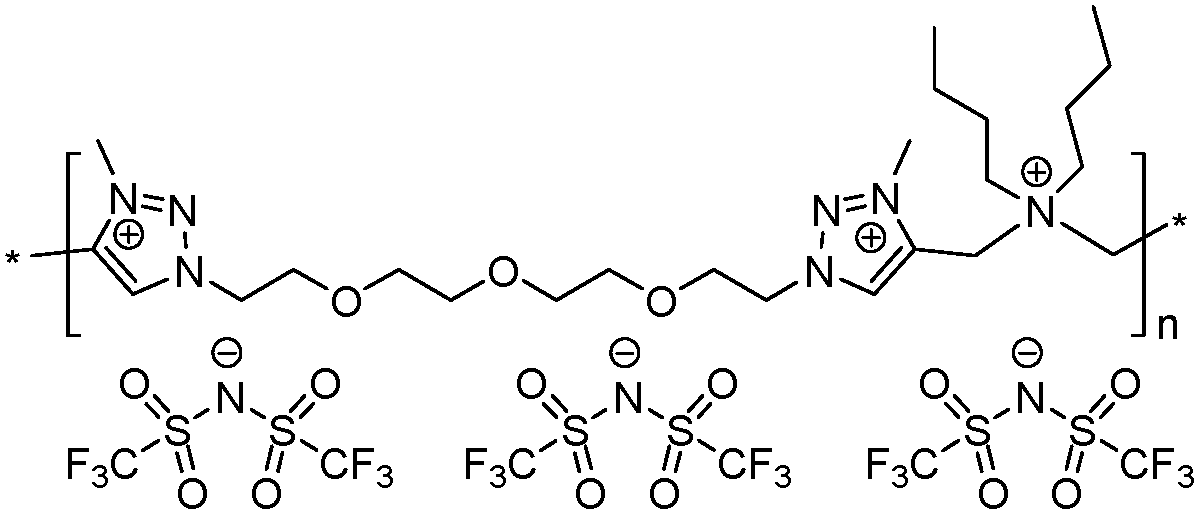
|
12 | 19.9 | 1.06 | 15 | 175 | 4.9 × 10−10 | 3.3 | 2.27 |
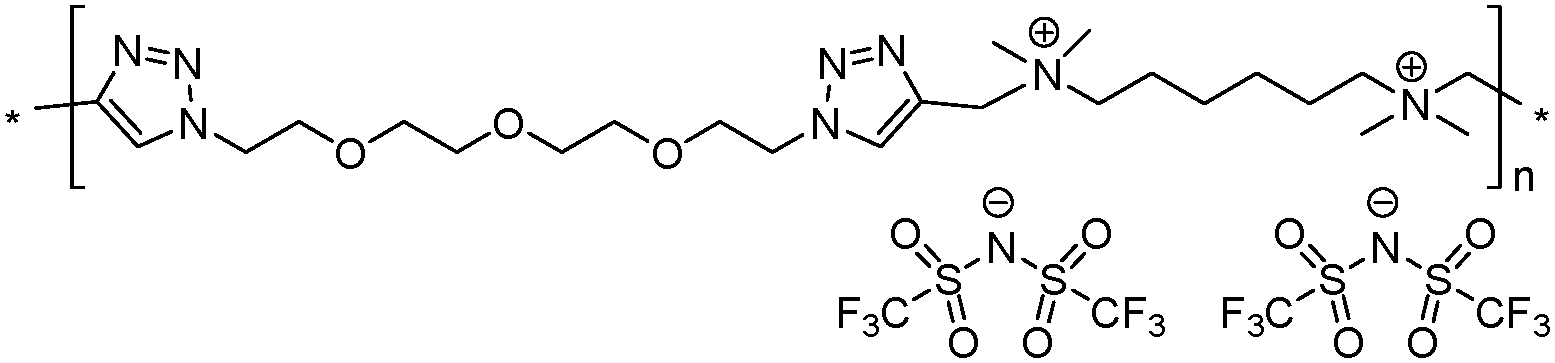
|
9 | 57.4 | 1.77 | 3 | 270 | 5.8 × 10−8 | 4.3 | 1.90 |
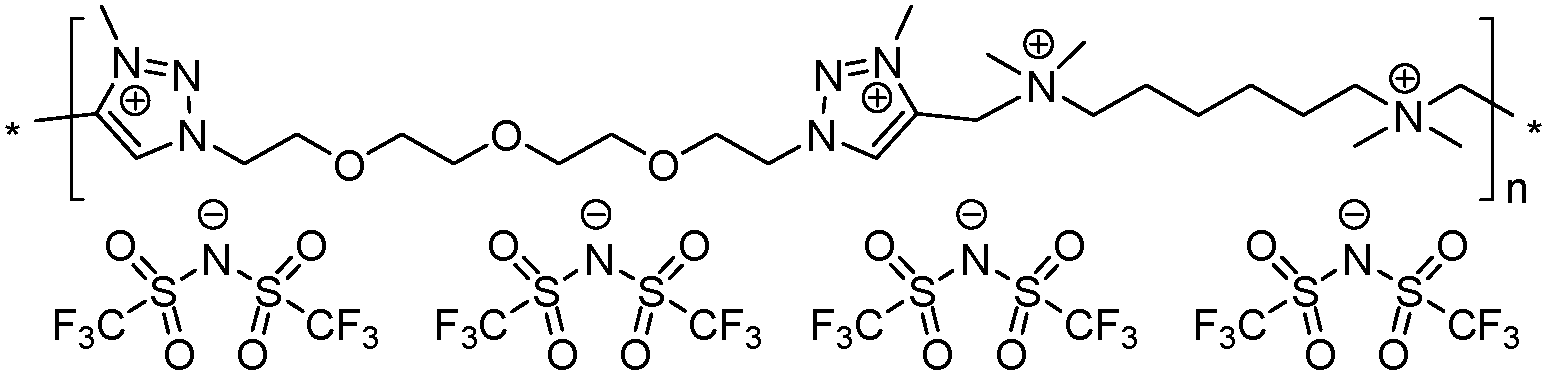
|
13 | 56.2 | 1.22 | 8 | 230 | 8.3 × 10−9 | 3.3 | 2.43 |
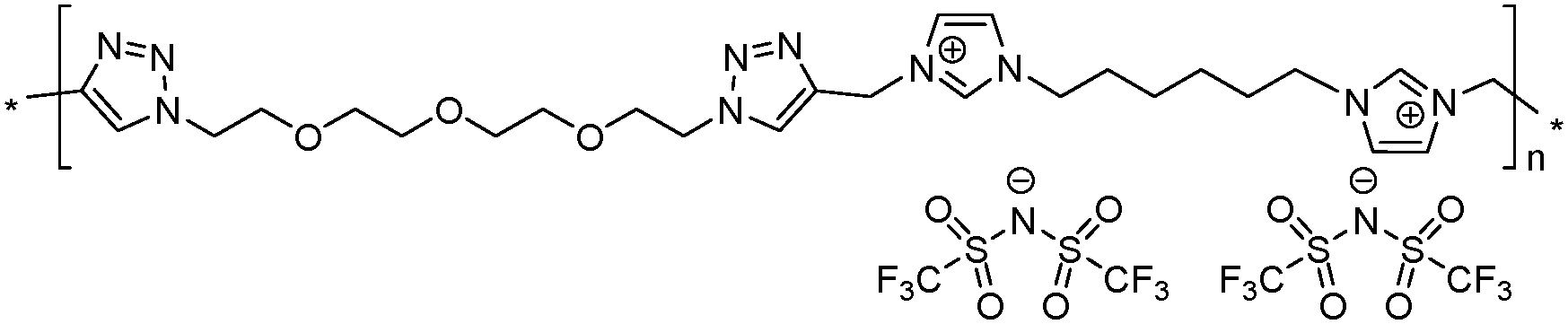
|
10 | 119.0 | 1.82 | −11 | 290 | 6.0 × 10−7 | 3.9 | 1.82 |
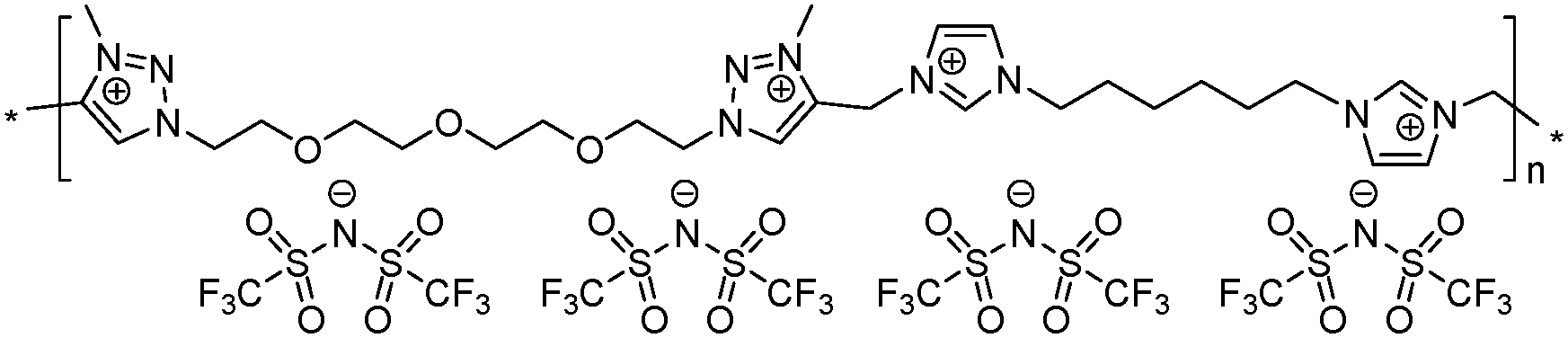
|
14 | 121.9 | 1.48 | −1 | 265 | 4.2 × 10−7 | 3.1 | 2.37 |
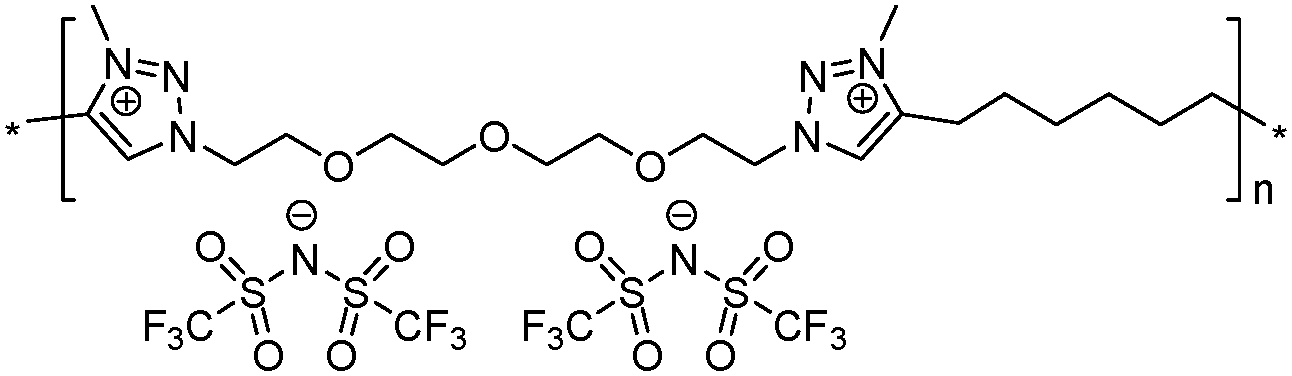
|
15 | 71.7 | 2.59 | −32 | 280 | 1.8 × 10−5 | 4.8 | 2.06 |
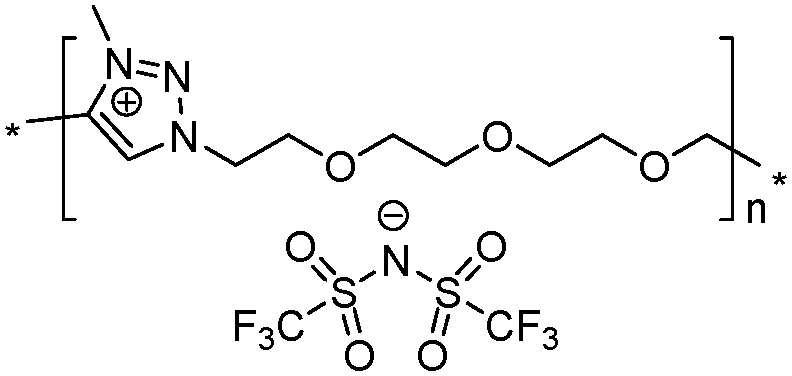
|
16 | — | — | −3543 | — | 9.0 × 10−6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43 43 |
— | 1.97 |
Synthesis of dialkyne monomers
The first step of our investigation started with the design of dialkyne monomers. As first candidates N,N-dimethyl-N,N-dipropargylammonium bis(trifluoromethylsulfonyl)imide (1) and N,N-dibutyl-N,N-dipropargylammonium bis(trifluoromethylsulfonyl)imide (2) each having one ammonium cation were prepared as reported previously.42Dipropargyl monomer 3 having two ammonium cations and a flexible n-hexyl spacer in between was designed in order to increase the number of cations and to enable sufficient ion motion (Scheme 3). The synthetic strategy for the preparation of 3 included N-alkylation of N,N,N′,N′-tetramethyl-1,6-hexanediamine by propargyl bromide and an ion exchange reaction with excess LiTFSI. As a result, monomer 3 was obtained as a white crystalline solid showing a relatively low melting point (Tm = 68–70 °C, Fig. S15 in the ESI†). Monomer 4 was further developed to contain two imidazolium cations and by analogy with 3 has the same linear n-hexyl spacer (Scheme 3). For monomer 4 the reaction pathway contained three steps: (1) synthesis of N-propargyl imidazole through N-alkylation of imidazole with propargyl tosylate, (2) N-alkylation of N-propargyl imidazole by 1,6-dibromohexane and (3) ion exchange reaction with an excess of LiTFSI. After isolation and purification monomer 4 was obtained as a slightly viscous yellow oil. DSC analysis confirmed the glassy behavior of 4 with a single glass transition temperature (Tg = –56 °C) and the absence of any crystallization or melting processes (Fig. S16, ESI†).
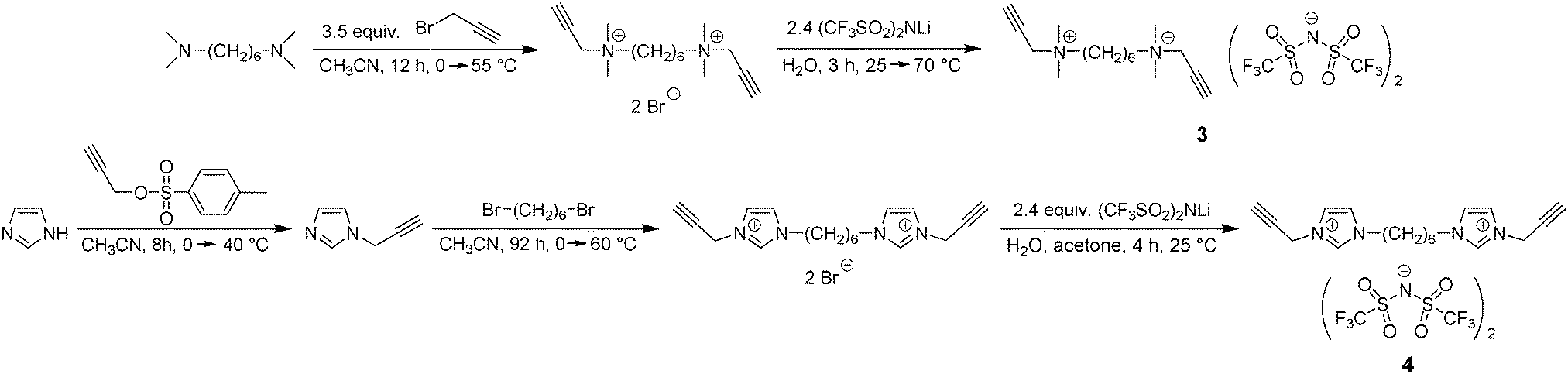 | ||
| Scheme 3 Synthesis of dicationic dipropargyl monomers 3 and 4. | ||
The structure and purity of monomers 3 and 4 were proved by 1H, 13C and 19F NMR spectroscopy, FTIR spectroscopy and elemental analysis (Fig. S1–S6, ESI†). The FTIR spectra of monomers 3 and 4 show absorption bands at ∼3050, 2990–2960, 2930 and 2880–2850 cm−1 that were assigned to CH2 stretching (Fig. S3 and S6, ESI†). The absorption bands at 3310–3250 (νCH), 2135 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and ∼615 (δCH) cm−1 were identified as characteristic bands of alkyne groups. The characteristic bands of the TFSI anion were observed at ∼1350–1340 (asymmetric SO), ∼1200–1190 (CF), ∼1150–1140 (symmetric SO) and ∼1060–1050 (CF) cm−1. Finally, 19F NMR spectra of 3 and 4 contain a single signal at −78.7 ppm assigned to trifluoromethyl groups (Fig. S2 and S5, ESI†).
C) and ∼615 (δCH) cm−1 were identified as characteristic bands of alkyne groups. The characteristic bands of the TFSI anion were observed at ∼1350–1340 (asymmetric SO), ∼1200–1190 (CF), ∼1150–1140 (symmetric SO) and ∼1060–1050 (CF) cm−1. Finally, 19F NMR spectra of 3 and 4 contain a single signal at −78.7 ppm assigned to trifluoromethyl groups (Fig. S2 and S5, ESI†).
Synthesis of PILs
Both ionic dialkynes 1–4 and neutral dialkyne 5 were first polymerized with 1,11-diazido-3,6,9-trioxaundecane (6) by AA + BB CuAAC polyaddition in N,N-dimethylformamide (DMF) (Scheme 4). In order to obtain high molar mass PILs, the previously optimized conditions involving CuIP(OEt)3 as a catalyst and a monomer concentration of 20 wt% were used.26,44,45 The change from CH2Cl2 to more polar DMF was governed by the solubility of the formed polymers. The application of the abovementioned reaction conditions to the polyaddition of dialkynes 1–4 with diazide 6 resulted in the preparation of PILs having one (Scheme 4, polymers 7 and 8) or two (Scheme 4, polymers 9 and 10) ion pairs per repeat unit.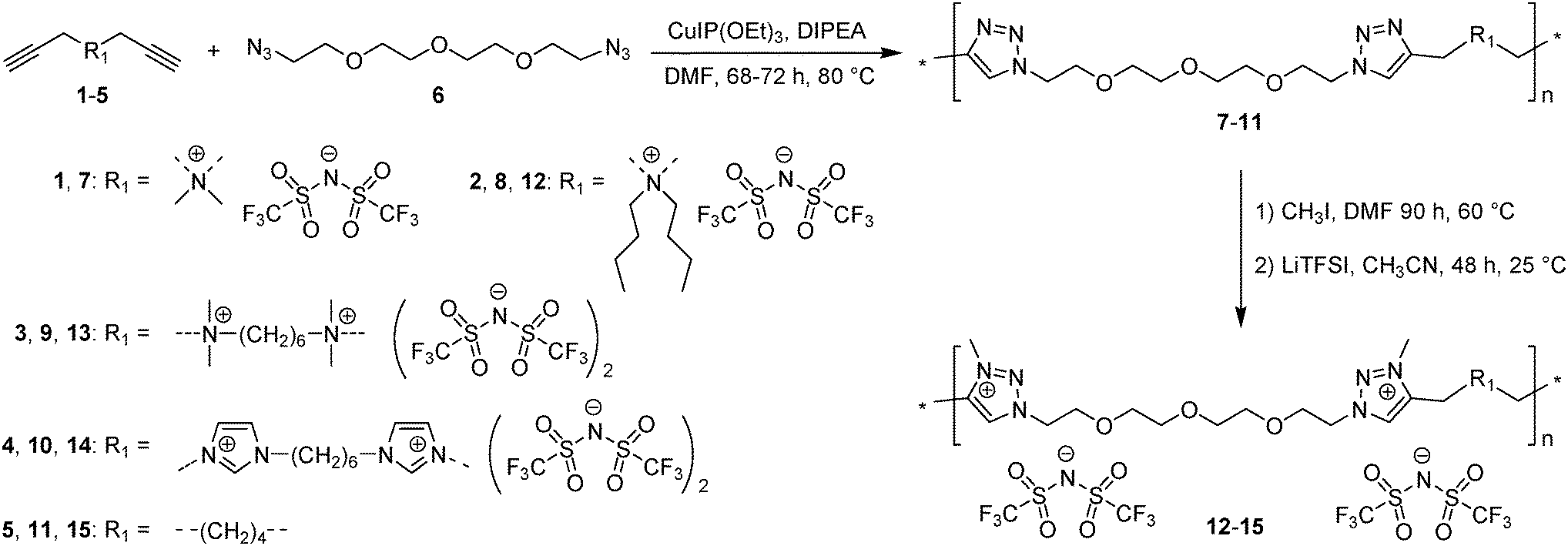 | ||
| Scheme 4 Synthetic pathway for the preparation of cationic PILs 7-10 and 15 and heterocationic PILs 12–14. | ||
The molar mass of the obtained polymers was estimated by size exclusion chromatography (SEC). To suppress the insufficiently charged screening or the so-called “polyelectrolyte effect” during SEC studies, an electrolyte, namely LiTFSI, was added to the polymer solution in DMF. It was revealed that in all cases CuAAC polyaddition resulted in the formation of high molar mass polymers, with number average molar masses (Mn) ranging from 50 to 120 kg mol−1 for PILs 8–11 (Table 1). PIL 10, based on bisimidazolium monomer 4, showed the highest Mn value most probably due to its better solubility (Table 1, polymer 10).
To increase the repeat unit charge density poly(1,2,3-triazole)s 8–11 were further subjected to N-alkylation reaction using iodomethane and ion exchange reaction with LiTFSI to obtain the corresponding 1,2,3-triazolium-containing analogues 12–15 (Scheme 4). PIL 7 was not considered further due to its limited solubility that prevented the quantitative N-alkylation of 1,2,3-triazole groups. It should be mentioned that the previously developed single step method involving N-alkylation of poly(1,2,3-triazole)s 8–11 by N-methyl bis(trifluoromethylsulfonyl)imide26 could not be performed quantitatively even after 72 h at 90 °C. Probably this can be explained by steric hindrance neighboring the 1,2,3-triazole rings due to the near proximity of an ammonium or imidazolium cation and a bulky TFSI anion. Optimization of the reaction duration and the amount of CH3I allowed achieving quantitative N-alkylation of the 1,2,3-triazole groups as confirmed by 1H NMR. Further ion exchange reaction with excess LiTFSI in aqueous medium led to the preparation of polymers 12–15 having from two to four ion pairs per repeat unit.
PILs 8–10 and 12–15 are amorphous and readily soluble in acetone, acetonitrile and polar aprotic solvents (i.e. DMF, DMSO, DMAc and NMP). Their structure, composition and purity were supported by 1H, 13C and 19F NMR and FTIR spectroscopy (see e.g. Fig. S7–S14, ESI†) as well as by elemental analysis. 1H NMR corroborated the quantitative N-alkylation of the 1,2,3-triazole groups in 12–15 by: (1) the appearance of the N-3 methyl signal at 4.34 ppm; (2) the shift from 4.55 to 4.83 ppm of the N-1 methylene signal; and (3) the shift from 8.22 to 8.97 ppm for the signal of the 1,2,3-triazole and 1,2,3-triazolium protons, respectively. The quantitative formation of 1,2,3-triazolium rings in 12–15 was also supported by elemental analysis. Finally, FTIR spectra of 7–10 and 12–15 showed the presence of aliphatic C–H groups (2930 and 2880 cm−1) and the bands related to the TFSI anion (1346, 1328, 1175, 1131 and 1050 cm−1).
Thermal properties of PILs
Thermal properties of PILs 7–10 and 12–15 were assessed via differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). DSC analysis revealed several interconnections between Tg and the chemical structure of PILs (e.g. Fig. S17 and S18 (ESI†) for polymers 10 and 14). Firstly, the increase in the length of side chains in the ammonium cations affords a decrease in Tg (Table 1, polymers 7 and 8). This tendency fully correlates with the melting point variation with the carbon number in the side alkyl chain for 1-methyl-3-alkyl imidazolium ILs46 and with the size of the side chain in 1,2,3-triazolium PILs.43 The separation of the ammonium cations by a flexible aliphatic spacer increases the flexibility of the polymer backbone and thus reduces Tg (Table 1, polymers 7 and 9). The transition from ammonium to imidazolium cations results in the lowering of PIL heat resistance (Table 1, polymers 9 and 10, 13 and 14), which was found to be in full agreement with data reported for (meth)acrylate-based PILs.47,48 The correlation between repeat unit charge density and Tg can be summarized as follows:| Tg7 (1 charge) > Tg13 (4 charges) > Tg9 (2 charges) |
| Tg12 (3 charges) > Tg8 (1 charge) |
Thus, it can be concluded that with the exception of polymer 7, which does not contain a flexible spacer between ammonium and 1,2,3-triazole groups, the increase in repeat unit charge density induces a rise in Tg. This reduction of polymer chain mobility is most likely due to electrostatic interactions.
The thermal stability of PILs in air was found to be strongly dependent on the cation structure (Table 1 and Fig. S19, ESI†). The overall evolution of the thermal stability of PILs (Tonset values) according to the cation's nature can be summarized as follows:
| Tonset10 (imidazolium cations, 290 °C) > Tonset15 (1,2,3-triazolium cations, 280 °C) > Tonset9 (ammonium cations, 270 °C) |
As generally observed for ILs49 and methacrylate PILs,47 the imidazolium PIL 10 demonstrates higher thermal stability than PILs 7, 8 and 9 having ammonium cations (Table 1). The transition to 1,2,3-triazolium cations in polymer 15 results in the lowering of the Tonset value in comparison with imidazolium analog 10. The thermal stability of PILs decreased following the orders below with respect to the repeat unit charge density:
| Tonset9 (2 charges, 270 °C) > Tonset7 (1 charge, 235 °C) ≈ Tonset13 (4 charges, 230 °C) |
| Tonset8 (1 charge, 220 °C) > Tonset12 (3 charge, 175 °C) |
| Tonset10 (2 charges, 290 °C) > Tonset14 (4 charges, 265 °C) |
Analyzing the thermal stability data mentioned above, it is possible to summarize that the increase in the repeat unit charge density of PILs affords a decrease in their decomposition temperatures. This is mainly due to the limited thermal stability of the 1,2,3-triazolium cation towards de-N-alkylation.50
Electrochemical properties of PILs
Ionic conductivities of polymer electrolytes were investigated as a function of temperature by electrochemical impedance spectroscopy (Table 1 and Fig. 1). Depending on the chemical nature of the cations and the repeat unit charge density the σ values determined at 25 °C under anhydrous conditions for synthesized PILs varied from 9.8 × 10−10 to 1.8 × 10−5 S cm−1. The influence of the cation chemical structure on the ionic conductivity of monocationic PILs can be ranked as follows:| σ15 (1,2,3-triazolium, 1.6 × 10−5 S cm−1) ≫ σ10 (imidazolium, 6.0 × 10−7 S cm−1) > σ9 (ammonium, 5.8 × 10−8 S cm−1) |
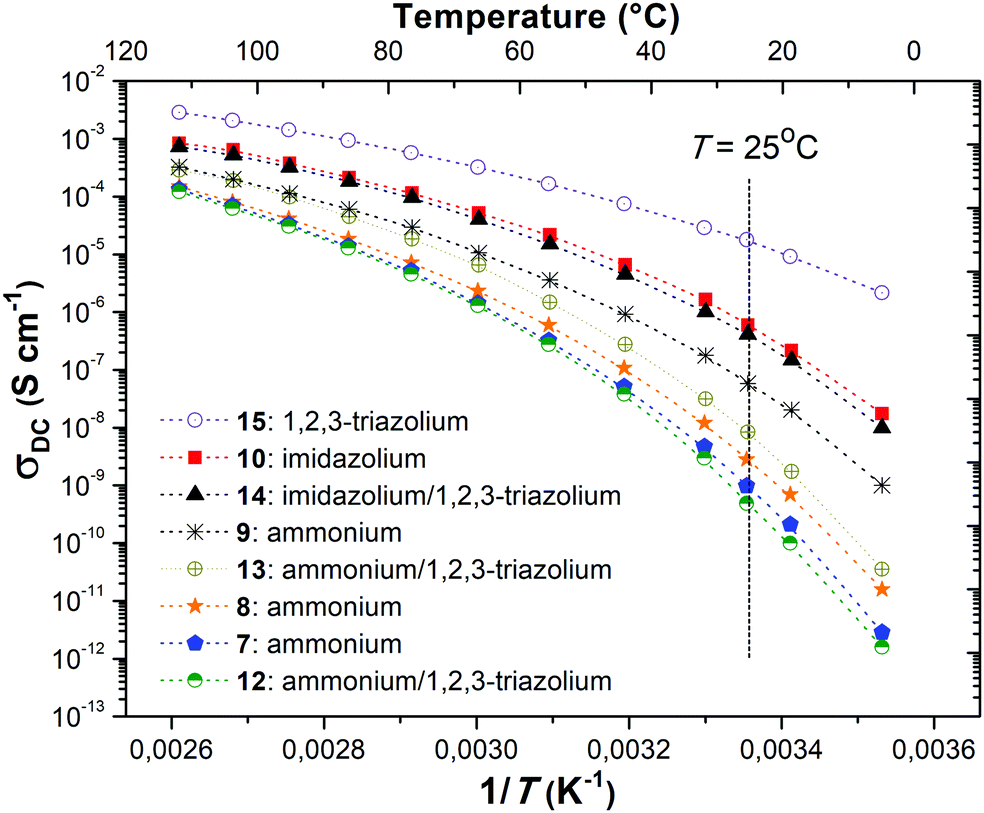 | ||
| Fig. 1 Evolution of ionic conductivity (σDC) with inverse temperature for PILs 7–10 and 12–15. | ||
This order fully correlates with the previously reported influence of cation structures on the conductivity of ILs.51,52 The insertion of side “dangling” chains decreases Tg and improves ionic conductivity as it can be concluded from the comparison of PILs 7 and 8 (Table 1). Such an increase in σ can be explained by the decrease in packing density of the corresponding polyelectrolyte.10 The separation of cations by an aliphatic spacer in 9 increases the solvation of ions and thus influences their dissociation and improves ionic conductivity (Table 1, polymers 7 and 9). Finally, the influence of repeat unit charge density on the ionic conductivity of PILs can be ranked in the following decreasing orders:
| σ9 (2 charges, 5.8 × 10−8 S cm−1) > σ13 (4 charges, 8.3 × 10−9 S cm−1) > σ7 (1 charge, 9.8 × 10−10 S cm−1) |
| σ10 (2 charges, 6.0 × 10−7 S cm−1) > σ14 (4 charges, 4.2 × 10−7 S cm−1) |
| σ15 (2 charges, 1.8 × 10−5 S cm−1) > σ16 (1 charge, 9.6 × 10−6 S cm−1) |
Taking these data into account it is possible to conclude that in the present work the repeat unit charge density vs. conductivity dependence passes through the maximum at PILs carrying two ion pairs. There are several possible explanations of the observed phenomenon. Possibly, the initial increase in the number of cations leads to the increase in the repeat unit charge density or in the concentration of free anions and results in the conductivity magnification. However, at some point in a solid polymer, only the surrounding atoms/groups can solvate the ions, the ratio between the number of charges and the solvating groups becomes too low for efficient dissociation and the conductivity starts to decrease. Following this assumption, the dilution of ions by incorporation of an aliphatic spacer leads to an immediate rise in ionic conductivity (Table 1, polymers 7 and 9). Another explanation can be connected with the highly documented influence of the PIL structure/composition on Tg.10 As the increase in repeat unit charge density generally induces a rise of Tg (Table 1 and Fig. S20, ESI†) and as ionic conductivity has an inverse relationship with the latter,10 it becomes obvious that the increase in the number of charges will reduce the ionic conductivity of PILs. In accordance with this assumption the transfer from monocationic or dicationic PILs (Table 1, polymers 8, 9, and 10) to tricationic or tetracationic ones (Table 1, polymers 12, 13, and 14) is accompanied by an increase in Tg and as a result in a decrease in σ. Finally, it seems obvious that 1,2,3-triazolium being the most ion conducting cation studied herein and in the literature,31 its presence systematically enhances σ, except for PIL 12, compared to the corresponding poly(1,2,3-triazole) analogue 8.
Additionally, the charge density of PILs was calculated as the number of charges per gram of polymeric material (Table 1 and Fig. S21, ESI†) and its influence on the ionic conductivity of PILs was estimated by the following decreasing orders:
| σ9 (1.90 mmol g−1, 5.8 × 10−8 S cm−1) > σ13 (2.43 mmol g−1, 8.3 × 10−9 S cm−1) > σ7 (1.55 mmol g−1, 9.8 × 10−10 S cm−1) |
| σ10 (1.82 mmol g−1, 6.0 × 10−7 S cm−1) > σ14 (2.37 mmol g−1, 4.2 × 10−7 S cm−1) |
| σ15 (2.06 mmol g−1, 1.8 × 10−5 S cm−1) > σ16 (1.97 mmol g−1, 9.6 × 10−6 S cm−1) |
Similarly to repeat unit charge density, the charge density vs. conductivity dependence passes through the maximum at an average value of 1.92 mmol g−1. It can also be concluded that the ionic conductivity of PILs is mainly ruled by segmental motion and Tg of polyelectrolytes rather than by their charge density.
The conductivity of PILs as a function of inverse temperature between 10 and 110 °C is shown in Fig. 1. Independently of the repeat unit charge density the ionic conductivity of all studied PILs increased with increasing temperature and reached a level ranging from ca. 1 × 10−4 to 3 × 10−3 S cm−1 at 110 °C. The temperature dependence of conductivity did not follow linear Arrhenius behavior indicating that anion diffusion results from various mechanisms: hopping of TFSI anions along a positively charged polymer backbone and local segmental motion of polymer chains.
Besides ionic conductivity another important property that should be considered for practical applications of PILs in e.g. the field of energy is their electrochemical stability. The electrochemical stability towards oxidative processes at the interface with the cathode is particularly important for battery applications.3,53 The electrochemical stability limits of PILs were assessed via cyclic voltammetry. As reasonable for electrochemical applications the ionic conductivity (>10−5 S cm−1) for PILs studied in this work was achieved only at 70 °C (Fig. 1); the latter was chosen as the experimental temperature for ESW studies. It should be noted that the selected temperature exceeds the Tg of all PILs and therefore during measurements they were in a rubber-like elastic state. Fig. 2 shows the anodic and cathodic scans of most ionic conductive polymers 9, 10, 13, 14 and 15 at 70 °C. For all PILs two reversible redox peaks at −0.45 V and between −0.05 and 0.30 V vs. Li+/Li were observed that are associated with the reversible lithium plating and stripping at the copper electrode surface. Generally plating/stripping depends on the number of Li+ ions that are able to reach the surface of the Cu electrode. In its turn, this process is primarily affected by the lithium ion transport54 and secondarily by the overall conductivity, which instead considers the movement of all charges (anions and cations). In the studied systems the intensity of the peaks was directly dependent on the ionic transport in polymers. The higher is the overall conductivity of PIL and, respectively, the ion movement, the more intense were the lithium plating and stripping processes.
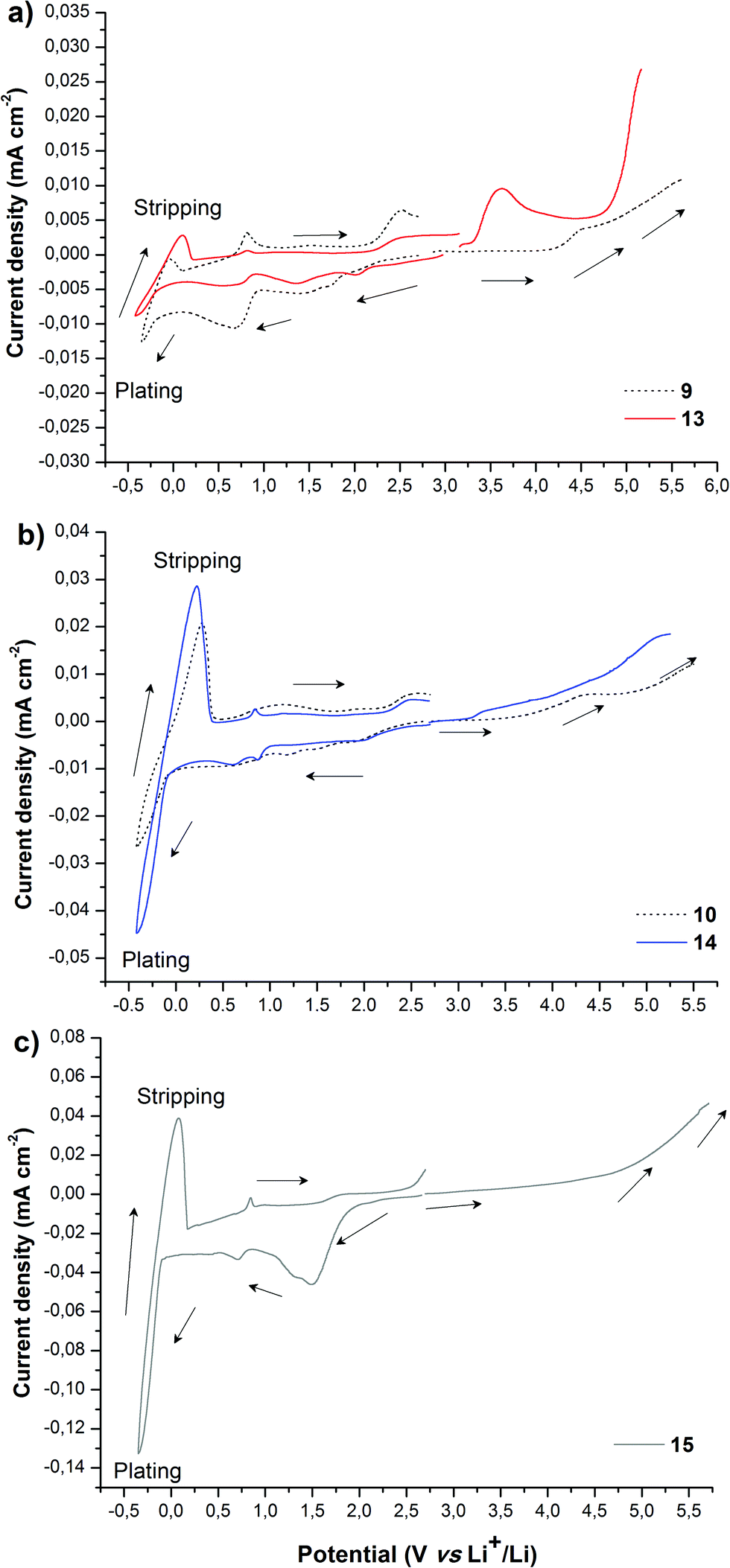 | ||
| Fig. 2 Electrochemical stability windows obtained by CV for (a) PILs 9 and 13, (b) PILs 10 and 14 and (c) PIL 15 at 70 °C (stainless steel as a working electrode and Li foil as counter and reference electrodes, scan rate 0.5 mV s−1). | ||
Fig. 2 suggests that the cathodic limit potential is mainly determined by the reduction of the respective cations. The irreversible oxidation peak at 1.5 V vs. Li+/Li found for polymer 15 (Fig. 2c) can be attributed to the partial degradation of the 1,2,3-triazolium cations as was observed previously.26 The increased cathodic stability up to −0.4 V vs. Li+/Li and observation for Li stripping and platting in comparison with previously reported 1,2,3-triazolium-based PILs26 may on one hand arise from the presence of the oxyethylene chain that could contribute to preventing the full reduction of the 1,2,3-triazolium cation. A similar influence of CH2CH2O groups on the cathodic limit of 1,2,3-triazolate anions was recently reported.55 On another hand, the cathodic stability of 1,2,3-triazolium-based PILs can be improved by the presence of LiTFSI as was noted formerly for pyrrolidinium ILs and PILs.6,56 It was previously observed that 1,2,3-triazolium cations exhibit less cathodic stability than quaternary ammonium and imidazolium cations.57 Following these observations for ILs, imidazolium-based polymer 10 shows better cathodic stability in comparison with 15 and reveals no oxidation peaks until Li plating at −0.41 V (Fig. 2b and c). An ammonium-based polymer 9 demonstrates one additional marked oxidation peak at ca. 1.5 V (Fig. 2a). Although in PIL 9 the ammonium cation is separated from the 1,2,3-triazole ring by a methylene group, these peaks can probably be attributed to the cleavage of the ring by Dimroth rearrangement.58 Interestingly, the presence of ammonium and imidazolium cations in heterocationic PILs 13 and 14 fully suppresses the reduction of 1,2,3-triazolium cations, thus increasing the electrochemical stability of PILs (Fig. 2a and b). Additionally, the couple of reversible peaks at 0.7 and 0.9 V versus Li+/Li may be attributed to a redox process involving oxide species at the electrode surface. The other couple of small peaks observable in the potential range between 1.5 and 2.7 V is most likely associated with the multistep decomposition of some impurity traces in PILs such as solvents. Indeed, these peaks disappear in the subsequent voltammetric cycles, which clearly states for the non-reproducibility of the processes and their insignificance for the overall performance of the system.
The anodic limit potential of cationic PILs is commonly determined by the oxidation of the anion. As all studied PILs contain the same TFSI anion, their anodic stability was expected to be above 4.0 V vs. Li+/Li.6,39 Indeed, polymers 9 and 15 demonstrate electrochemical stability windows up to 4.3 and 4.8 V, respectively (Fig. 2a and c). In accordance with what was observed previously for ILs,51 imidazolium-based polymer 10 shows a slightly reduced anodic limit equal to 3.9 V vs. Li+/Li (Fig. 2b). Unexpectedly, some irreversible reduction peaks at 3.6 and 3.3 V were found for polymers 13 and 14 having mixed cations (Fig. 2a and b). The authors were not able to find reasonable explanation for this phenomenon as usually the mixtures of Li salts either in EC-EMC solution59 or in polymer electrolyte60 show an improved stability in comparison with systems containing single salts. Overall, except for 13 and 15, the breakdown voltage accounting for the full degradation of polyelectrolyte was not clearly observed. This can be attributed to the diffusion resistance and slow process kinetics in polymer electrolytes.61,62
Experimental
Iodomethane (>99%, Aldrich), deca-1,9-diyne (5, 99%, Aldrich), 1,11-diazido-3,6,9-trioxaundecane (6, 99%, Aldrich), N,N,N′,N′-tetramethyl-1,6-hexanediamine (99%, Aldrich), propargyl bromide (>97.0%, TCI chemicals), diisopropylethylamine (DIPEA, 99%, Aldrich), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%, Aldrich), copper(I) iodide triethylphosphite (CuIP(OEt)3, 97%, Aldrich), lithium bis(trifluoromethylsulfonyl)imide (LiTFSI, 99+%, Aldrich), acetonitrile (CH3CN, HPLC grade 99%, Acros), dimethyl formamide (DMF, 99.8%, anhydrous, Acros), lithium metal foil (Chemetall Foote Corporation) and activated decolorizing charcoal (Acros) were used without further purification.Imidazole (>99%, Aldrich) was purified by vacuum sublimation at 80 °C/10−2 mmHg. 1,6-Dibromohexane (96%, Aldrich) was distilled under vacuum. Propargyl tosylate was prepared by the reaction of tosyl chloride with an excess of propargyl alcohol in accordance with the well-established procedure.63
Ionic liquid monomers (ILMs) N,N-dimethyl-N,N-dipropargylammonium bis(trifluoromethylsulfonyl)imide (1) and N,N-dibutyl-N,N-dipropargylammonium bis(trifluoromethylsulfonyl)imide (2) were synthesized as described previously.42
Synthesis of N,N,N′,N′-tetramethyl-N,N′-dipropargyl-hexane-1,6-diammonium bis(trifluoromethylsulfonyl)imide (3)
Monomer 3 was synthesized in two steps: (1) N-alkylation of N,N,N′,N′-tetramethyl-1,6-hexanediamine by propargyl bromide and (2) ion exchange reaction with an excess of LiTFSI.A solution of propargyl bromide (9.13 g, 76.8 mmol) in anhydrous CH3CN (10 mL) was added dropwise to a solution of N,N,N′,N′-tetramethyl-1,6-hexanediamine (3.78 g, 21.9 mmol) in anhydrous CH3CN (20 mL) maintained at 0–5 °C under an inert atmosphere. After stirring for 5 minutes the start of the precipitation of a beige powder was observed. Stirring was continued for 1 h at 0–5 °C, then for 1 h at 25 °C and finally for 10 h at 55 °C. The reaction mixture was centrifuged for 5 min at 5000 rpm, the mother liquor was decanted and a fresh portion of CH3CN was added to the precipitate. After intense shaking the suspension was again centrifuged and this procedure was repeated three times. N,N,N′,N′-Tetramethyl-N,N′-dipropargyl-hexane-1,6-diammonium bromide was isolated by filtration as a white-beige powder and dried for 8 h at 70 °C/1 mmHg (hereinafter with a special flask filled with P2O5 and introduced into the vacuum line). Yield: 8.64 g (96%); m.p. = 204–206 °C.
Lithium bis(trifluoromethylsulfonyl)imide (3.53 g, 12.2 mmol) was dissolved in H2O (8 mL) and added dropwise to a solution of N,N,N′,N′-tetramethyl-N,N′-dipropargyl-hexane-1,6-diammonium bromide (2.10 g, 5.12 mmol) in H2O (20 mL) at room temperature. The mixture was stirred for 2 h at 25 °C and the precipitation of a yellow powder was observed. The temperature was raised to 70 °C and stirring was continued for 1 h. After cooling down to room temperature the upper aqueous layer was decanted and the residual powder was redissolved in 80 mL of dichloromethane (CH2Cl2). The CH2Cl2 solution was washed with water (3 × 30 mL) and dried over anhydrous MgSO4. The magnesium sulfate was filtered off, the decolorizing charcoal was added to the solution and the suspension was stirred at room temperature for another 2 h. The charcoal was filtered off and CH2Cl2 was stripped off under reduced pressure. Bispropargyl monomer 3 was obtained as a white crystalline powder, which was dried at 50 °C/1 mmHg for 4 h. Yield: 3.45 g (83%); m.p. = 68–70 °C (68 °C by DSC); cryst.p. = 14 °C (DSC); Tg = −45 °C (DSC); 1H NMR (300 MHz, DMSO-d6, δ, ppm): 4.35 (d, 4H, JHH = 3.0 Hz, CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 4.02 (t, 2H, JHH = 3.0 Hz, CCH), 3.33 (m, 4H, NCH2CH2), 3.07 (s, 12H, NCH3), 1.71 (m, 4H, NCH2CH2), 1.33 (m, 4H, NCH2CH2CH2); 13C NMR (75.5 MHz, DMSO-d6, δ, ppm): 125.8–113.0 (q, 1JCF = 322 Hz, CF3), 82.8, 72.2, 62.9, 53.2, 49.8, 25.1, 21.6; 19F NMR (282 MHz, DMSO-d6, δ, ppm): −78.7 (s, CF3); IR (KBr pellet): 3305 (m, νCH), 3251 (m, νCH), 3049 (w, νCH), 2996 (w, νCH), 2966 (m, νCH), 2626 (w, νCH), 2867 (w, νCH), 2135 (w, νCH), 1484 (m), 1423 (w), 1348 (vs, νasSO), 1196 (vs, νCF), 1141 (s, νsSO), 1054 (vs, νCF), 969 (w), 890 (m), 796 (m), 766 (w), 741 (m), 688 (w), 654 (w), 615 (vs, δCH), 570 (s), 514 (s) cm−1; anal. calcd for C20H30F12N4O8S4 (810.7), %: C 29.63; H 3.73; N 6.91; found, %: C 29.48; H 3.66; N 6.90.
CH), 4.02 (t, 2H, JHH = 3.0 Hz, CCH), 3.33 (m, 4H, NCH2CH2), 3.07 (s, 12H, NCH3), 1.71 (m, 4H, NCH2CH2), 1.33 (m, 4H, NCH2CH2CH2); 13C NMR (75.5 MHz, DMSO-d6, δ, ppm): 125.8–113.0 (q, 1JCF = 322 Hz, CF3), 82.8, 72.2, 62.9, 53.2, 49.8, 25.1, 21.6; 19F NMR (282 MHz, DMSO-d6, δ, ppm): −78.7 (s, CF3); IR (KBr pellet): 3305 (m, νCH), 3251 (m, νCH), 3049 (w, νCH), 2996 (w, νCH), 2966 (m, νCH), 2626 (w, νCH), 2867 (w, νCH), 2135 (w, νCH), 1484 (m), 1423 (w), 1348 (vs, νasSO), 1196 (vs, νCF), 1141 (s, νsSO), 1054 (vs, νCF), 969 (w), 890 (m), 796 (m), 766 (w), 741 (m), 688 (w), 654 (w), 615 (vs, δCH), 570 (s), 514 (s) cm−1; anal. calcd for C20H30F12N4O8S4 (810.7), %: C 29.63; H 3.73; N 6.91; found, %: C 29.48; H 3.66; N 6.90.
Synthesis of 1,1′-(hexane-1,6-diyl)bis(3-propargyl-imidazolium) bis(trifluoromethylsulfonyl)imide (4)
Monomer 4 was prepared via a three step reaction: (1) synthesis of N-propargyl imidazole, (2) N-alkylation of N-propargyl imidazole by 1,6-dibromohexane and (3) ion exchange reaction with an excess of LiTFSI.A solution of propargyl tosylate (48.08 g, 228.7 mmol) in anhydrous CH3CN (100 mL) was added dropwise to a solution of imidazole (34.2 g, 503 mmol) in anhydrous CH3CN (140 mL) at 0–5 °C under an inert atmosphere. Stirring was continued for 1 h at 0–5 °C and then for 2 h at 40 °C. CH3CN was evaporated under reduced pressure at room temperature and the residual oil was distilled under vacuum (bp = 62 °C/0.08 mmHg). The obtained colorless oil still contained 10 mol% of imidazole (1H NMR). To remove impurity 1.5 mL of 37 wt% HCl was added to the oil with vigorous stirring at room temperature. Stirring was continued for 15 minutes and CH2Cl2 (100 mL) was added to the solution. The organic layer was separated, washed with H2O (3 × 5 mL) and dried over anhydrous MgSO4. The magnesium sulfate was filtered off and CH2Cl2 was evaporated under reduced pressure. N-Propargyl imidazole was obtained as a yellow oil, which was dried at 25 °C/1 mmHg for 4 h. Yield: 9.78 g (40%); 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.70 (s, 1H, NCHN), 7.22 (s, 1H, NCHCHN), 6.95 (s, 1H, NCHCHN), 4.93 (m, 2H, CH2CCH–), 3.52 (m, 2H, CH2CCH); 13C NMR (100 MHz, DMSO-d6, δ, ppm): 137.1 (C2im), 128.9 (C4im), 119.5 (C5im), 78.7 (CH2CCH), 76.2 (CH2CCH–), 35.6 (CH2N); IR (KBr pellet): 3290 (vs, νCH), 3191 (s, νCH imidazole), 3112 (vs, νCHνCH imidazole), 2961 (m, νCH), 2927 (m, νCH), 2121 (s, νCH), 1670 (w), 1637 (w), 1594 (m), 1505 (vs), 1435 (s), 1392 (s), 1348 (s), 1282 (vs), 1231 (vs), 1107 (s), 1075 (vs), 1029 (s), 944 (m), 907 (s), 820 (vs), 736 (vs, δCH imidazole), 660 (vs), 616 (vs, δCH), 458 (w) cm−1.
A solution of 1,6-dibromohexane (4.50 g, 18.5 mmol) in anhydrous CH3CN (25 mL) was added dropwise to a solution of N-propargyl imidazole (4.29 g, 40.6 mmol) in anhydrous CH3CN (25 mL) maintained at 0–5 °C under an inert atmosphere. The stirring was continued for 1 h at 0–5 °C, 2 h at 25 °C, 66 h at 45 °C and 23 h at 60 °C. The precipitation of the light-brown oil was observed and the emulsion was placed in the fridge (5 °C) for 48 h, whereupon the crystallization occurred. The light-brown crystals were filtered, washed with Et2O, then thrice with CH2Cl2, filtered again and smashed into powder with a mortar. 1,1′-(Hexane-1,6-diyl)bis(3-propargyl-imidazolium) bromide was crystallized from a CH3CN/H2O mixture (150/11 mL) as light-yellow crystals that were dried at 60 °C/1 mmHg for 12 h. Yield: 6.20 g (73%); m.p. = 143–144 °C (139 °C by DSC).
Lithium bis(trifluoromethylsulfonyl)imide (11.30 g, 39.3 mmol) was dissolved in H2O (25 mL) and added dropwise to a solution of 1,1′-(hexane-1,6-diyl)bis(3-propargyl-imidazolium) bromide (5.98 g, 13.1 mmol) in H2O (30 mL) at room temperature. The mixture was stirred for 2 h at 25 °C and the precipitation of a yellow powder was observed. The yellow powder was filtered and dissolved in acetone (60 mL). A solution of LiTFSI (6.50 g, 22.6 mmol) in acetone (15 mL) was added dropwise under stirring to the obtained solution. The reaction continued overnight at room temperature, whereupon the solvent was evaporated and the residual orange oil was extensively washed with H2O (5 × 20 mL). The oil was dissolved in CH3CN and dried over anhydrous MgSO4. The magnesium sulfate was filtered off, the decolorizing charcoal was added to the solution and the suspension was stirred at room temperature for another 2 h. The charcoal was filtered off and CH3CN was evaporated under reduced pressure. Monomer 4 was obtained as a slightly viscous yellow oil, which was dried at 55 °C/1 mmHg for 12 h. Yield: 7.09 g (63%); Tg = −56 °C (DSC); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 9.26 (s, 2H, NCHN), 7.81 (s, 4H, NCHCHN), 5.18 (m, 4H, CH2CCH–), 4.19 (t, 4H, JHH = 7.2 Hz, CH2N), 3.81 (m, 2H, CH2CCH–), 1.80 (t, 4H, JHH = 7.2 Hz, NCH2CH2), 1.28 (m, 4H, NCH2CH2CH2); 13C NMR (151 MHz, DMSO-d6, δ, ppm): 136.1 (C2im), 122.8 (C4im), 122.4 (C5im), 122.7–116.3 (q, 1JCF = 322 Hz, CF3), 78.9 (CH2CCH–), 75.9 (CH2CCH–), 48.9 (NCH2), 38.6 (CH2CCH), 29.1 (NCH2CH2), 24.9 (NCH2CH2CH2); 19F NMR (565 MHz, DMSO-d6, δ, ppm): −78.8 (s, CF3); IR (KBr pellet): 3277 (m, νCH), 3151 (s, νCH imidazolium), 2949 (m, νCH), 2868 (w, νCH), 2138 (w, νCH), 1564 (s), 1475 (w), 1444 (w), 1350 (vs, νasSO), 1330 (vs), 1193 (vs, νCF), 1137 (vs, νsSO), 1055 (vs, νCF), 955 (w), 842 (m), 790 (s), 740 (s, δCH imidazolium), 654 (m), 614 (vs, δCH), 570 (vs), 513 (s), 406 (m) cm−1; anal. calcd for C22H24F12N6O8S4 (856.7), %: C 30.84; H 2.82; F 26.61; found, %: C 31.18; H 2.80; F 26.69.
CuAAC polyaddition of bisalkynes with 1,11-diazido-3,6,9-trioxaundecane (6). Synthesis of poly(1,2,3-triazole)s 7–11
Polymers 7–11 were synthesized via AA + BB CuAAC polyaddition and a typical procedure is detailed below by the example of poly(1,2,3-triazole) 10 preparation.Dialkyne 4 (1.01 g, 1.18 mmol), diazide 6 (0.29 g, 1.18 mmol) and N,N-diisopropylethylamine (0.31 g, 2.36 mmol) were dissolved in anhydrous DMF (6 mL) under an inert atmosphere at room temperature. Afterwards CuIP(OEt)3 (42.2 mg, 0.118 mmol) was added in one portion to the transparent yellow solution and stirring was continued in the dark for 68 h at 80 °C. The resulting brown solution was precipitated into excess of diethyl ether and the brown oil was centrifuged (10000 rpm, 5 min). The oil was dissolved in CH3CN (5 mL) and precipitated twice into an excess of H2O containing 2–3 drops of PMEDTA and then thrice in H2O. Poly(1,2,3-triazole) 10 was recovered as a dark brown gummy material and was dried at 80 °C/1 mmHg for 12 h. Yield: 0.82 g (63%); Tg = −12 °C (DSC); Mn = 119.3 kDa, Mw/Mn = 1.82 (GPC); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 9.26 (s, 2H, imNCHN), 8.22 (s, 2H, CCHNtriazole), 7.78 (s, 2H, imNCHCHN), 7.76 (s, 2H, imNCHCHN), 5.53 (s, 4H, imNCH2triazole), 4.55 (t, 4H, JHH = 4.8 Hz, NCH2CH2O), 4.17 (t, 4H, JHH = 7.8 Hz, imNCH2CH2), 3.82 (t, 4H, JHH = 5.4 Hz, NCH2CH2O), 3.52 (m, 4H, NCH2CH2OCH2CH2O), 3.46 (m, 4H, NCH2CH2OCH2CH2O), 1.79 (m, 4H, imNCH2CH2), 1.28 (m, 4H, imNCH2CH2CH2); 13C NMR (151 MHz, DMSO-d6, δ, ppm): 140.2 (CCHNtriazole), 136.1 (C2im), 124.9 (CCHNtriazole), 122.6 (C4im and C5im), 123.7–116.3 (q, 1JCF = 322 Hz, CF3), 69.6, 69.5 (NCH2CH2OCH2CH2OCH2CH2), 68.6 (NCH2CH2O), 49.6 (NCH2CH2O), 48.9 (imNCH2CH2), 43.8 (imNCH2triazole), 29.1 (imNCH2CH2), 24.9 (imNCH2CH2CH2); 19F NMR (565 MHz, DMSO-d6, δ, ppm): −78.8 (s, CF3); IR (ATR mode): 3152 (m, νCH imidazolium), 3113 (w, νCH triazole), 3096 (w, νCH triazole), 2934 (w, νCH), 2916 (w, νCH), 2868 (m, νCH), 1697 (w), 1598 (w), 1563 (m), 1515 (w), 1449 (m), 1348 (s, νasSO), 1329 (s), 1180 (vs, νCF), 1132 (vs, νsSO), 1051 (vs, νCF), 833 (m), 740 (s, δCH imidazolium), 652 (m), 613 (vs, δCH), 599 (s), 570 (vs), 531 (w), 512 (vs) cm−1; Anal. calcd for C31H44F12N12O11S4 (1117.0), %: C 33.33; H 3.97; N 15.05; found, %: C 33.19; H 3.96; N 14.94.
Synthesis of poly(1,2,3-triazolium)s 12–15 by N-alkylation and ion exchange reactions
Polymers 12–15 were synthesized in two steps: N-alkylation of the 1,2,3-triazole groups of polymers 8–11 by an excess of CH3I and subsequent ion exchange reaction with LiTFSI. A typical procedure is given below by the example of PIL 14 preparation.Iodomethane (0.93 g, 6.54 mmol) was added to a solution of poly(1,2,3-triazole) 10 (0.72 g, 0.654 mmol) in anhydrous DMF (10 mL) at room temperature under an inert atmosphere. Stirring was continued at 60 °C for 90 h, whereupon additional CH3I (0.50 g, 3.52 mmol) was added and the solution was further stirred at 80 °C for 72 h. The crude reaction mixture was diluted with DMF and precipitated two times in acetone to yield a dark brown viscous oil which was separated by centrifugation (10000 rpm, 10 min).
The obtained oil was redissolved in H2O (20 mL) and an aqueous solution of LiTFSI (0.69 g, 2.4 mmol in 5 mL of H2O) was added dropwise under vigorous stirring at room temperature. The mixture was stirred for 12 h at 40 °C and the precipitation of a viscous brown oil was observed. The aqueous layer was decanted and the oil was washed with H2O (3 × 20 mL), dissolved in acetone (6 mL) and precipitated again into an excess of H2O. Polymer 14 was recovered as a brown gummy solid after centrifugation (10000 rpm, 10 min) and drying at 80 °C/1 mmHg for 12 h. Yield: 0.67 g (69%); Tg = −1 °C (DSC); Mn = 121.9 kDa, Mw/Mn = 1.48 (GPC); 1H NMR (600 MHz, DMSO-d6, δ, ppm): 9.30 (s, 2H, imNCHN), 8.97 (s, 2H, CCHNtriazolium), 7.85 (m, 4H, imNCHCHN), 5.84 (s, 4H, imNCH2triazolium), 4.83 (t, 4H, JHH = 4.8 Hz, NCH2CH2O), 4.34 (s, 6H, triazolium NCH3), 4.18 (t, 4H, JHH = 7.8 Hz, imNCH2CH2), 3.92 (t, 4H, JHH = 5.4 Hz, NCH2CH2O), 3.58 (m, 4H, NCH2CH2OCH2CH2O), 3.51 (m, 4H, NCH2CH2OCH2CH2O), 1.82 (m, 4H, imNCH2CH2), 1.35 (m, 4H, imNCH2CH2CH2); 13C NMR (151 MHz, DMSO-d6, δ, ppm): 137.2 (C2im), 131.4 (CCHNtriazolium), 122.9 (C4im), 122.7 (C5im), 122.6–116.3 (q, 1JCF = 322 Hz, CF3), 69.5 (NCH2CH2OCH2CH2OCH2CH2), 67.4 (NCH2CH2), 53.4 (NCH2CH2O), 49.1 (imNCH2CH2), 40.3 (imNCH2triazolium), 38.4 (NCH3), 29.2 (imNCH2CH2), 25.1 (imNCH2CH2CH2); 19F NMR (565 MHz, DMSO-d6, δ, ppm): −78.8 (s, CF3); IR (ATR mode): 3151 (w, νCH imidazolium), 3119 (w, νCH triazolium), 3089 (w, νCH triazolium), 2929 (w, νCH), 2875 (m, νCH), 1561 (m), 1457 (m), 1346 (s, νasSO), 1328 (s), 1175 (vs, νCF), 1131 (vs, νsSO), 1050 (vs, νCF), 935 (w), 843 (m), 791 (s), 741 (s, δCH imidazolium), 692 (m, δCH triazolium), 652 (s), 611 (vs, δCH), 598 (s), 569 (vs), 505 (vs) cm−1; anal. calcd for C37H50F24N14O19S8 (1707.4), %: C 26.03; H 2.95; N 11.49; found, %: C 26.07; H 2.91; N 11.51.
Methods
NMR spectra were recorded on AMX-400, Avance II 500 MHz and Avance III HD 600 MHz spectrometers (Bruker) at 25 °C in the indicated deuterated solvents and are listed in ppm. The signal corresponding to the residual protons or carbons of the deuterated solvent was used as an internal standard for 1H and 13C NMR, respectively. C6F6 was utilized as an external standard for 19F NMR. IR spectra were acquired on a Nicolet Magna-750 Fourier IR-spectrometer using KBr pellets or on a Bruker Tensor 27 Fourier IR-spectrometer using ATR technology (128 scans, resolution is 2 cm−1) and Spectragryph optical spectroscopy software.64A 1200 Infinity gel permeation chromatograph (GPC, Agilent Technologies) was used to determine Mn, Mw and Mw/Mn of PILs. The chromatograph was equipped with an integrated IR detector, a PL PolarGel-M column and a PL PolarGel-M guard column (Agilent Technologies). The eluent was a 0.1 M solution of LiTFSI in DMF and the flow rate was 1.0 mL min−1 at 50 °C. Polystyrene standards (EasiVial PS-M, Agilent Technologies, Mp = 162–500 × 103 g mol−1) were used to perform calibration.
For Differential Scanning Calorimetry (DSC) measurements all samples (ionic monomers and PILs) were hermetically sealed in aluminum pans inside an argon-filled glove-box (MBRAUN MB-Labstar, H2O and O2 content < 0.5 ppm). DSC experiments were performed on a DSC3 + STARe System differential calorimeter (Mettler Toledo). For PILs DSC measurements were conducted with a heating rate of 10 °C min−1 in the range of -80 to 150 °C, while for ionic monomers a very low rate of 3 °C min−1 was applied as recommended for highly viscous ionic liquids.65 Glass transition temperatures (Tg) were determined during the second heating cycle.
Thermal gravimetric analysis (TGA) was carried out in air on a TGA2 STARe System (Mettler Toledo) applying a heating rate of 5 °C min−1.
Ionic conductivity (σDC) was determined by electrochemical impedance spectroscopy (EIS or complex alternating-current (AC) impedance spectroscopy) with a VSP potentiostat/galvanostat (Bio-Logic Science Instruments). To avoid any influence of moisture/humidity on the ionic conductivity of polymer electrolytes, the latter were preliminary dried at 60 °C/1 mmHg for 12 h in a B-585 oven (Buchi Glass Drying Oven, Switzerland) filled with P2O5 and were transferred under vacuum inside an argon-filled glovebox. Polymers were sandwiched between two stainless steel electrodes. The distance between the electrodes was kept equal to 250 μm using a Teflon spacer ring with an inner area of 0.5 cm2. Symmetrical stainless steel/PIL/stainless steel assembly was clamped into the 2032 coin cell and afterwards was taken out from the glovebox. EIS experiments were carried out by applying a 10 mV perturbation in the frequency range from 10−2 to 2 × 105 Hz at the open circuit potential (OCV). The ohmic resistance (RΩ) of the sample, obtained from the Nyquist plot at the low frequency end of the semicircle, was used to calculate the ionic conductivity using the following equation:
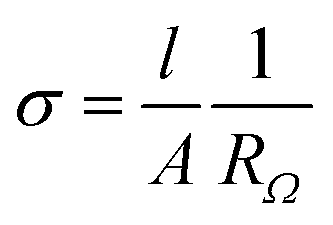
Cyclic voltammetry (CV) and VSP potentiostat/galvanostat (Bio-Logic Science Instruments, France) were used to determine the electrochemical stability window of solid polymer electrolytes at 70 °C. Prior to measurement PILs (0.2 g) were dissolved in anhydrous CH3CN (2 mL) inside the glovebox and 10 wt% of LiTFSI (0.02 g) were added under stirring until the formation of a clear solution. The solvent was evaporated under reduced pressure in a B-585 oven (Buchi Glass Drying Oven, Switzerland) and gummy samples were further dried there at 60 °C/1 mmHg for 12 h. To avoid moisture or oxygen contamination samples were afterwards transferred under vacuum back to an argon-filled glovebox, where they were used for the two-electrode 2032 coin cell assembly. The two-electrode cells were compiled by sandwiching samples between the working electrode and a lithium metal foil, which served as the reference and the counter electrode, simultaneously. Stainless steel and copper disks were used as working electrodes during anodic and cathodic stability measurements, respectively. To evaluate anodic limits, potential sweeps were carried out between OCV and 5.5 V vs. Li+/Li at a constant rate of 0.5 mV s−1. To determine cathodic limits, potential sweeps were performed between OCV and −0.5 V vs. Li+/Li at the same constant rate.
The charge density of PILs as the number of charges per gram of material was calculated using the following equation:
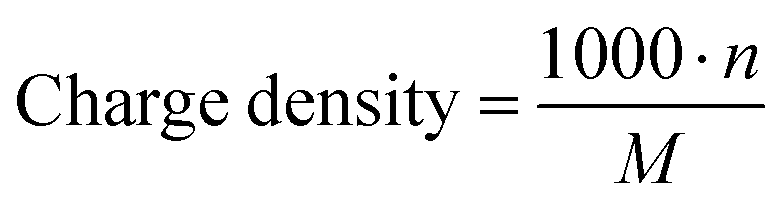
Conclusions
The aim of this paper was to obtain polyelectrolytes with enhanced ionic conductivity and electrochemical stability as well as to fill the gap in fundamental knowledge concerning the influence of repeat unit charge density on the structure/property relationships of PILs. Within this scope a series of TFSI-based cationic PILs was obtained from (1) CuAAC polyaddition using cationic ILMs and (2) subsequent N-alkylation of poly(1,2,3-triazole) intermediates. The synthesized PILs differed by the number of charge carriers per monomer unit (from 1 to 4 ion pairs) and by the nature of cation combinations (1,2,3-triazolium, imidazolium or ammonium as well as 1,2,3-triazolium/imidazolium or 1,2,3-triazolium/ammonium associations).Within the studied PILs thermal stability, heat resistance (glass transition temperature), ionic conductivity and electrochemical stability were all found to be significantly affected by repeat unit charge density. Generally, the higher the repeat unit charge density of PILs the higher their Tg and the lower their ionic conductivity and thermal stability. The observed dependences passed through a maximum when PILs had two cations per monomer unit that were separated by alkyl or oxyethylene chains. For almost all combinations, 1,2,3-triazolium-containing PILs showed the highest ionic conductivity. A good cathodic stability on the Li metal electrode was seen when PILs with a mixed type of cation were tested. It is worth noting that the presence of ammonium and imidazolium cations in heterocationic PILs was able to hamper the reduction of 1,2,3-triazolium cations and to increase the cathodic limit down to −0.4 V vs. Li+/Li at 70 °C. The highest ionic bulk conductivity (1.8 × 10−5 S cm−1) and anodic stability (4.8 V vs. Li+/Li at 70 °C) were demonstrated by PIL 15 containing solely 1,2,3-triazolium cations. Such PILs can be potential candidates for the next generation of safe and flexible polymer-based electrochemical devices requiring high conductivity in the solid state, for example supercapacitors, smart windows or actuators. However, when an application demands high electrochemical stability, it is preferred to include more stable cations such as ammonium or imidazolium in the polymer backbone. Therefore, the future application area of PILs with mixed type cations can include Li batteries.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Merlin Cotessat is grateful to the Luxembourg Institute of Science and Technology (LIST) for the sponsorship of his internship and work in the Materials and Research Technology Department (MRT). This work was supported by the Luxembourg National Research Fund (FNR) and Agency Nationale de la Recherche (ANR) through ANR-FNR project DISAFECAP (Agreement number INTER/ANR/19/13358226). Elemental analysis and IR spectroscopy were performed with the financial support from the Ministry of Science and Higher Education of the Russian Federation using the equipment of Center for molecular composition studies of INEOS RAS. This manuscript is in honor of the 50 year anniversary of the French Polymer Group (Groupe Français des Polymères – GFP).Notes and references
- X. Lu, M. Yu, G. Wang, Y. Tong and Y. Li, Energy Environ. Sci., 2014, 7, 2160 RSC.
- W. K. Chee, H. N. Lim, Z. Zainal, N. M. Huang, I. Harrison and Y. Andou, J. Phys. Chem. C, 2016, 120, 4153–4172 CrossRef CAS.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- L. Yue, J. Ma, J. Zhang, J. Zhao, S. Dong, Z. Liu, G. Cui and L. Chen, Energy Storage Mater., 2016, 5, 139–164 CrossRef.
- M. J. Park, I. Choi, J. Hong and O. Kim, J. Appl. Polym. Sci., 2013, 129, 2363–2376 CrossRef CAS.
- A.-L. Pont, R. Marcilla, I. De Meatza, H. Grande and D. Mecerreyes, J. Power Sources, 2009, 188, 558–563 CrossRef CAS.
- A. Balducci, J. Power Sources, 2016, 326, 534–540 CrossRef CAS.
- G. Ayalneh Tiruye, D. Munoz-Torrero, J. Palma, M. Anderson and R. Marcilla, J. Power Sources, 2015, 279, 472–480 CrossRef CAS.
- I. Osada, H. de Vries, B. Scrosati and S. Passerini, Angew. Chem., Int. Ed., 2016, 55, 500–513 CrossRef CAS.
- A. S. Shaplov, R. Marcilla and D. Mecerreyes, Electrochim. Acta, 2015, 175, 18–34 CrossRef CAS.
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- M. M. Obadia and E. Drockenmuller, Chem. Commun., 2016, 52, 2433–2450 RSC.
- M. D. Green and T. E. Long, Polym. Rev., 2009, 49, 291–314 CrossRef CAS.
- E. B. Anderson and T. E. Long, Polymer, 2010, 51, 2447–2454 CrossRef CAS.
- W. Qian, J. Texter and F. Yan, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC.
- G. B. Appetecchi, G.-T. Kim, M. Montanino, M. Carewska, R. Marcilla, D. Mecerreyes and I. D. Meatza, J. Power Sources, 2010, 195, 3668–3675 CrossRef CAS.
- A. S. Shaplov, D. O. Ponkratov and Y. S. Vygodskii, Polym. Sci., Ser. B, 2016, 58, 73–142 CrossRef CAS.
- W. Zhang, Z. Kochovski, B. V. K. J. Schmidt, M. Antonietti and J. Yuan, Polymer, 2016, 107, 509–516 CrossRef CAS.
- W. Zhang and J. Yuan, Macromol. Rapid Commun., 2016, 1124–1129 CrossRef CAS.
- M. Yahia, S. Mei, A. P. Mathew and J. Yuan, ACS Macro Lett., 2019, 8, 1372–1377 CrossRef CAS.
- H. Ohno, Macromol. Symp., 2007, 249–250, 551–556 CrossRef.
- B. Qiu, B. Lin and F. Yan, Polym. Int., 2013, 62, 335–337 CrossRef CAS.
- A. S. Shaplov, D. O. Ponkratov, P.-H. Aubert, E. I. Lozinskaya, C. Plesse, F. Vidal and Y. S. Vygodskii, Chem. Commun., 2014, 50, 3191–3193 RSC.
- G. Colliat-Dangus, M. M. Obadia, Y. S. Vygodskii, A. Serghei, A. S. Shaplov and E. Drockenmuller, Polym. Chem., 2015, 6, 4299–4308 RSC.
- E. Drockenmuller, J. Bernard, A. Serghei, B. Zhang and R. Sood, Polym. Chem., 2015, 6, 3521–3528 RSC.
- M. Lee, U. H. Choi, R. H. Colby and H. W. Gibson, Chem. Mater., 2010, 22, 5814–5822 CrossRef CAS.
- M. Lee, U. H. Choi, D. Salas-de la Cruz, A. Mittal, K. I. Winey, R. H. Colby and H. W. Gibson, Adv. Funct. Mater., 2011, 21, 708–717 CrossRef CAS.
- A. S. Shaplov, E. I. Lozinskaya, R. Losada, C. Wandrey, A. T. Zdvizhkov, A. A. Korlyukov, K. A. Lyssenko, I. A. Malyshkina and Y. S. Vygodskii, Polym. Adv. Technol., 2011, 22, 448–457 CrossRef CAS.
- A. Jourdain, A. Serghei and E. Drockenmuller, ACS Macro Lett., 2016, 5, 1283–1286 CrossRef CAS.
- S. Yi, W. Leon, D. Vezenov and S. L. Regen, ACS Macro Lett., 2016, 5, 915–918 CrossRef CAS.
- B. Qiu, B. Lin, L. Qiu and F. Yan, J. Mater. Chem., 2012, 22, 1040–1045 RSC.
- B. Qiu, B. Lin, Z. Si, L. Qiu, F. Chu, J. Zhao and F. Yan, J. Power Sources, 2012, 217, 329–335 CrossRef CAS.
- X. Chen, J. Zhao, J. Zhang, L. Qiu, D. Xu, H. Zhang, X. Han, B. Sun, G. Fu, Y. Zhang and F. Yan, J. Mater. Chem., 2012, 22, 18018–18024 RSC.
- M. M. Obadia, A. Jourdain, A. Serghei, T. Ikeda and E. Drockenmuller, Polym. Chem., 2017, 8, 910–917 RSC.
- Y. S. Vygodskii, O. A. Mel’nik, E. I. Lozinskaya, A. S. Shaplov, I. A. Malyshkina, N. D. Gavrilova, K. A. Lyssenko, M. Y. Antipin, D. G. Golovanov, A. A. Korlyukov, N. Ignat’ev and U. Welz-Biermann, Polym. Adv. Technol., 2007, 18, 50–63 CrossRef CAS.
- R. Marcilla, F. Alcaide, H. Sardon, J. A. Pomposo, C. Pozo-Gonzalo and D. Mecerreyes, Electrochem. Commun., 2006, 8, 482–488 CrossRef CAS.
- K. Yin, Z. Zhang, X. Li, L. Yang, K. Tachibana and S. Hirano, J. Mater. Chem. A, 2014, 3, 170–178 RSC.
- J. M. C. Puguan and H. Kim, J. Ind. Eng. Chem., 2019, 74, 1–6 CrossRef CAS.
- D. Flachard, A. Serghei, M. Fumagalli and E. Drockenmuller, Polym. Int., 2019, 68, 1591–1598 CrossRef CAS.
- Y. S. Vygodskii, A. S. Shaplov, E. I. Lozinskaya, P. S. Vlasov, I. A. Malyshkina, N. D. Gavrilova, P. Santhosh Kumar and M. R. Buchmeiser, Macromolecules, 2008, 41, 1919–1928 CrossRef CAS.
- B. P. Mudraboyina, M. M. Obadia, I. Allaoua, R. Sood, A. Serghei and E. Drockenmuller, Chem. Mater., 2014, 26, 1720–1726 CrossRef CAS.
- P. Dimitrov-Raytchev, S. Beghdadi, A. Serghei and E. Drockenmuller, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 34–38 CrossRef CAS.
- S. Binauld, E. Fleury and E. Drockenmuller, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2470–2476 CrossRef CAS.
- S. Zhang, N. Sun, X. He, X. Lu and X. Zhang, J. Phys. Chem. Ref. Data, 2006, 35, 1475 CrossRef CAS.
- A. S. Shaplov, E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, F. Vidal, P.-H. Aubert, O. V. Okatova, G. M. Pavlov, L. I. Komarova, C. Wandrey and Y. S. Vygodskii, Electrochim. Acta, 2011, 57, 74–90 CrossRef CAS.
- W. Ogihara, S. Washiro, H. Nakajima and H. Ohno, Electrochim. Acta, 2006, 51, 2614–2619 CrossRef CAS.
- Y. Cao and T. Mu, Ind. Eng. Chem. Res., 2014, 53, 8651–8664 CrossRef CAS.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Adv. Funct. Mater., 2017, 27, 1703258 CrossRef.
- H. Matsumoto, H. Sakaebe and K. Tatsumi, J. Power Sources, 2005, 146, 45–50 CrossRef CAS.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS.
- G. G. Eshetu, D. Mecerreyes, M. Forsyth, H. Zhang and M. Armand, Mol. Syst. Des. Eng., 2019, 4, 294–309 RSC.
- G. M. A. Girard, M. Hilder, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D. R. MacFarlane and P. C. Howlett, J. Phys. Chem. C, 2017, 121, 21087–21095 CrossRef CAS.
- D. Flachard, J. Rolland, M. M. Obadia, A. Serghei, R. Bouchet and E. Drockenmuller, Chem. Commun., 2018, 54, 9035–9038 RSC.
- J.-H. Shin, W. A. Henderson and S. Passerini, Electrochem. Solid-State Lett., 2005, 8, A125 CrossRef CAS.
- S. Sanghi, E. Willett, C. Versek, M. Tuominen and E. B. Coughlin, RSC Adv., 2012, 2, 848–853 RSC.
- E. S. H. El Ashry, S. Nadeem, M. R. Shah and Y. E. Kilany, Advances in Heterocyclic Chemistry, Elsevier, 2010, vol. 101, pp. 161–228 Search PubMed.
- X. Chen, W. Xu, M. H. Engelhard, J. Zheng, Y. Zhang, F. Ding, J. Qian and J.-G. Zhang, J. Mater. Chem. A, 2014, 2, 2346 RSC.
- S. Li, Y.-M. Chen, W. Liang, Y. Shao, K. Liu, Z. Nikolov and Y. Zhu, Joule, 2018, 2, 1838–1856 CrossRef CAS.
- Y. V. Oza, D. R. MacFarlane, M. Forsyth and L. A. O’Dell, Electrochim. Acta, 2015, 175, 80–86 CrossRef CAS.
- M. Martinez-Ibañez, E. Sanchez-Diez, L. Qiao, Y. Zhang, X. Judez, A. Santiago, I. Aldalur, J. Carrasco, H. Zhu, M. Forsyth, M. Armand and H. Zhang, Adv. Funct. Mater., 2020, 2000455 CrossRef.
- M. N. Soltani Rad, S. Behrouz, J. Mohammadtaghi-Nezhad, E. Zarenezhad and M. Agholi, Appl. Organomet. Chem., 2019, 33, e4799 CrossRef.
- F. Menges, Spectragryph – optical spectroscopy software, Version 1.2.9, 2019, http://www.effemm2.de/spectragryph/ Search PubMed.
- E. Gómez, N. Calvar and Á. Domínguez, in Ionic Liquids - Current State of the Art, ed. S. Handy, InTech, London, 2015, pp. 199–228 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C and 19F NMR and FT-IR spectra, and DSC and TGA traces of ionic monomers and PILs. See DOI: 10.1039/d0nj04143b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |