
Methane conversion on cobalt-added liquid-metal indium catalysts†

Abstract
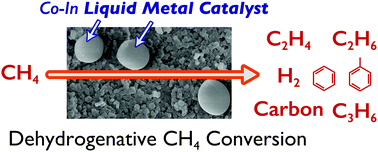
Non-oxidative methane (CH4) conversion was carried out using a silica-supported cobalt-added indium liquid metal catalyst (In–Co/SiO2). In–Co/SiO2 showed different activity from In/SiO2 and Co/SiO2. In–Co/SiO2 promoted the formation of aromatics (e.g., benzene) from CH4. The ethylene conversion reaction implied that Co present in indium liquid metal was effective for the aromatization reaction. We propose bifunctional catalysis by In–Co for CH4 conversion: (1) indium liquid metal activates CH4 molecules to form C2 hydrocarbons by CH4 coupling, and (2) a portion of C2 hydrocarbons is converted to aromatics by Co species present in the liquid metal. This work was compared to reported systems such as supported catalytically active liquid metal solutions (SCALMS).
 Please wait while we load your content...
Please wait while we load your content...

