
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of ammonium ions by electrochemical oxidation of urea with a boron-doped diamond electrode†
Norihiro
Suzuki‡
 *a,
Akihiro
Okazaki‡
b,
Kai
Takagi
b,
Izumi
Serizawa
b,
Genji
Okada
c,
Chiaki
Terashima
a,
Ken-ichi
Katsumata
ad,
Takeshi
Kondo
c,
Makoto
Yuasa
c and
Akira
Fujishima
a
*a,
Akihiro
Okazaki‡
b,
Kai
Takagi
b,
Izumi
Serizawa
b,
Genji
Okada
c,
Chiaki
Terashima
a,
Ken-ichi
Katsumata
ad,
Takeshi
Kondo
c,
Makoto
Yuasa
c and
Akira
Fujishima
a
aPhotocatalysis International Research Center, Research Institute for Science and Technology, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan. E-mail: suzuki.norihiro@rs.tus.ac.jp
bORC Manufacturing Co., Ltd, 4896 Tamagawa, Chino, Nagano 391-0011, Japan
cFaculty of Science and Technology, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan
dFaculty of Industrial Science and Technology, Tokyo University of Science, 6-3-1 Niijyuku, Katsushika, Tokyo 125-8585, Japan
First published on 16th September 2020
Abstract
Ammonium ions were formed by electrochemical oxidation of urea with a boron-doped diamond (BDD) electrode. Almost complete decomposition of urea was achieved. When the BDD electrode was used together with a mesoporous titanium dioxide photocatalyst, the amount of ammonium ions produced increased.
Nitrogen (N) is one of the three major nutrients, along with phosphorus and potassium, that are indispensable for plant growth. However, because molecular nitrogen (N2) is chemically inert, plants cannot directly uptake N2 from air. Therefore, N-containing ions such as ammonium (NH4+) and nitrate ions (NO3−) are needed in fertiliser. In addition, numerous N-containing inorganic and organic compounds have been industrially produced that support our society. Reactive N sources are needed for life on Earth and ammonia (NH3) is the most important N source.
NH3 has been attracting great interest as a hydrogen carrier. NH3 easily liquefies under moderate conditions such as 0.857 MPa at 20 °C or ambient pressure at −33 °C. In addition, the volumetric hydrogen storage density of NH3 is very high—three times that of molecular hydrogen (H2) at 70 MPa.1 Furthermore, the risk of ignition and explosion of NH3 is lower than that of H2. These features mean that NH3 is a promising carrier for the realisation of a hydrogen society.
NH3 is commonly fabricated by the Haber–Bosch method, in which H2 and N2 are directly reacted with a metal catalyst. Although this process is well established, because the reaction is conducted at high temperature (400–600 °C) and high pressure (20–40 MPa), it requires a huge amount of energy. Therefore, development of an alternative method to produce NH3 is important.
As an alternative process to fabricate NH3, we focused on oxidation of urea in urine. This process is of interest from the perspective of making a useful product (i.e., NH3) from a waste product (i.e., urine). To realise cost-effective and environmentally benign process, using common equipment at atmospheric pressure and room temperature is desired. However, photolysis and photocatalytic reaction is not suitable because urea is hard to decompose by them.
In this study, we adopted electrochemical treatment and use boron-doped diamond (BDD) as an electrode to oxidise urea. BDD has several unique properties, such as a wide potential window, low background current and high physical/chemical stability. The wide potential window of BDD enables water oxidation at high voltage, leading to the production of reactive oxygen species (ROS).2 Previous studies have revealed that BDD is a suitable electrode for electrochemical oxidation of urea. Hernández et al.3 compared the performance of Pt, Ti–Ru oxide, BDD and antimony-doped tin oxide electrodes in urea oxidation and found that only BDD could decompose urea completely. Li and co-workers used BDD and Ti/IrO2 electrodes to oxidise urea.4 They reported that toxic N2H4 formed when urea was oxidised with Ti/IrO2, whereas this by-product did not form during electrochemical oxidation with BDD.
Here, we report synthesis of NH4+ from simplified artificial urine by electrochemical oxidation of urea with a BDD electrode. By positioning the BDD electrode and counter electrode in different cells, oxidation and (undesired) reduction are completely separated. In addition, the effect of introducing a mesoporous TiO2 photocatalyst during the oxidation process by BDD is explored.
Experiments were conducted with an H-type cell consisting of two glass cells, as described in our previous study.5 One half cell contained the BDD working electrode and the other contained a Pt counter electrode. When the TiO2 photocatalyst was used together with the BDD electrode, a Kr–Br excimer lamp producing 207 nm UV light (ORC Manufacturing Co., Ltd (Machida, Tokyo, Japan)) and mesoporous TiO2/BDD photocatalyst were included in the system. The fabrication process of the photocatalyst was described in our previous paper.6 The half cells were connected with a Nafion® NRE-212 membrane (Sigma-Aldrich (St. Louis, MO, US)). A schematic of the experimental setup is shown in Fig. 1.
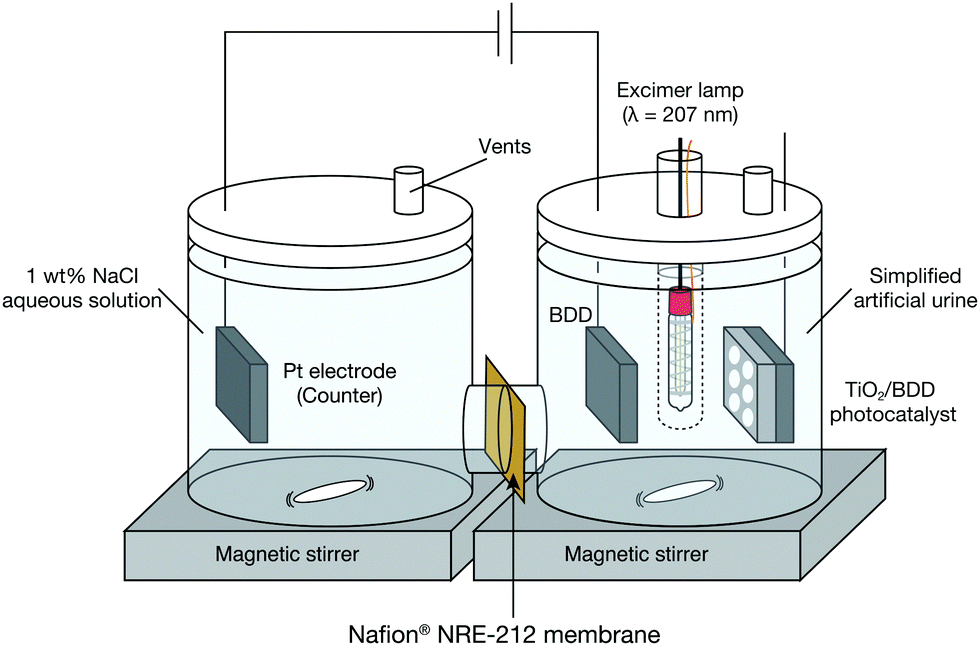 | ||
| Fig. 1 Schematic of the experimental setup. When only electrochemical treatment was conducted, the excimer lamp and TiO2/BDD photocatalyst were removed beforehand. | ||
Simplified artificial urine consists of urea and NaCl (main sub component) in which the concentration of both matches with actual urine (i.e., Urea solution (2 wt%) with 1 wt% NaCl aqueous solution) was prepared and used as the test material. The simplified artificial urea (50 mL) was poured into the glass half cell with the BDD electrode, which is hereafter denoted it as the reaction cell. The other half cell (i.e., the Pt electrode side), which is hereafter denoted as the counter cell, was filled with 1 wt% NaCl aqueous solution (50 mL). The BDD electrode was positioned horizontally. A constant current of 75 mA was applied to the BDD electrode to produce ROS by water electrolysis. When the mesoporous TiO2/BDD photocatalyst was used, 2.0 mW cm−2 of 207 nm UV light from an excimer lamp was irradiated onto the TiO2/BDD photocatalyst through a quartz sample tube. The glass cell was capped with a silicone rubber cap and Teflon tape to prevent evaporation during the test. Aliquots of the urea solution (1 mL) for further chemical analysis were collected several times during electrochemical treatment by opening the cap. The experiment was conducted at room temperature. During the test, the urea solution was constantly stirred. The concentrations of urea and formed ions were estimated from high-performance liquid chromatography (HPLC) and ion chromatography measurements, respectively.
First, electrochemical decomposition of urea using only the BDD electrode was examined. Fig. 2 shows the time dependence of urea concentration during the electrochemical treatment. The urea concentration decreased monotonically with lengthening treatment time and almost all the urea in the solution was decomposed after 18 h. This result is consistent with a previous study that showed complete removal of urea was achieved by electrochemical oxidation using a BDD electrode.3 Nitrogen containing ions (i.e., NH4+, NO2− and NO3−) derived from the decomposition of urea increased with processing time until 12 h and then saturated. The detailed explanation on each ions is described below.
 | ||
| Fig. 2 Time dependence of urea concentration during the electrochemical treatment estimated from HPLC measurements. C and C0 are the remaining and initial urea concentrations, respectively. Sum of concentration of nitrogen containing ions measured with ion chromatography is also included. | ||
Fig. 3 depicts the time dependence of ion concentrations during the electrochemical treatment. It was found that NH4+, nitrite ions (NO2−) and NO3− formed in the reaction cell (Fig. 3(a)), confirming that urea was mineralised during electrochemical treatment. Previous studies reported that the oxidation of urea was associated with hydroxyl radicals (OH˙) electrogenerated at the BDD surface during the electrolysis at high potentials in the oxygen evolution region3 and OH˙ were involved in the generation of NH4+, NO2− and NO3− by oxidation of intermediate species.7
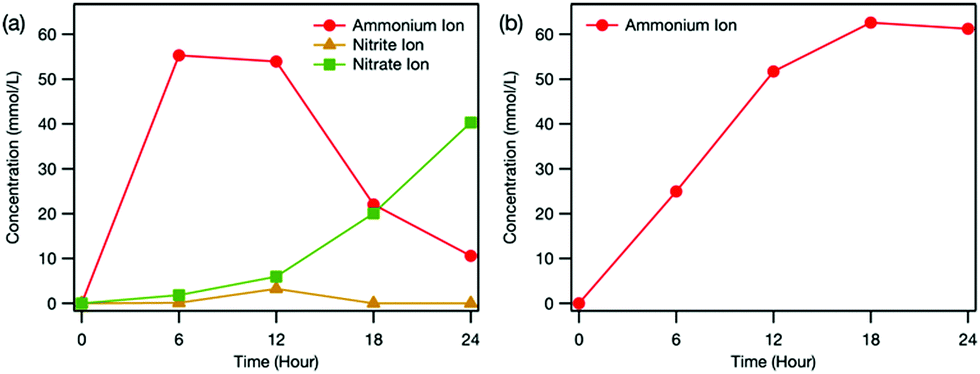 | ||
| Fig. 3 Time dependence of ion concentration in (a) the reaction cell and (b) the counter cell during the electrochemical treatment estimated from ion chromatography measurements. Note that nitrite and nitrate ions were not detected in the counter cell. | ||
Park et al.8 proposed a mechanism for the reaction between urea and OH˙ (Scheme S1, ESI†). First, OH˙ attacks an amino group (–NH2) to form a nitroso group (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). Then, OH˙ attacks the –NO group to form NH2CONO2. Hydrolysis of NH2CONO2 releases NO2− and its further oxidation by OH˙ forms NO3−.9 The resulting carbamic acid, which mainly exists as a zwitterion,10 can be oxidised in two different ways: one leads to the formation of NH4+ and the other produces NO3−via NO2− oxidation.
O). Then, OH˙ attacks the –NO group to form NH2CONO2. Hydrolysis of NH2CONO2 releases NO2− and its further oxidation by OH˙ forms NO3−.9 The resulting carbamic acid, which mainly exists as a zwitterion,10 can be oxidised in two different ways: one leads to the formation of NH4+ and the other produces NO3−via NO2− oxidation.
We found that during electrochemical treatment, the NO2− concentration in the reaction cell first increased and then decreased until it reached zero, whereas the NO3− concentration kept increasing over time (Fig. 3(a)). These results revealed the formation of NO2− as an intermediate and its further oxidation to NO3−, which is consistent with the proposed mineralisation mechanism of urea described above. Compared with the concentrations of NO2− and NO3−, more NH4+ existed in the reaction cell (Fig. 3(a)), clarifying that the formation of NH4+ from carbamic acid is strongly favoured over the production of NO3−. With lengthening electrochemical treatment time, the concentration of NH4+ in the reaction cell first rapidly increased and then gradually decreased (Fig. 3(a)), whereas that in the counter cell increased monotonically (Fig. 3(b)). Because Pt electrode was negatively biased, the NH4+ formed in the reaction cell was electrically guided to the counter cell, where it accumulated.
Next, a mesoporous TiO2/BDD photocatalyst and Kr–Br excimer lamp (λ = 207 nm) were introduced into the reaction cell and both photocatalytic and electrochemical treatments were conducted at the same time. The time dependence of urea concentration with and without photocatalysis is shown in Fig. S1 (ESI†). The measured urea concentration was almost identical regardless of the presence or absence of photocatalyst, indicating that the initial oxidation of urea is predominantly induced by electrochemical reaction with BDD. However, the time dependence of nitrogen containing ions differed when photocatalyst was also used, implying that photocatalytic reaction had an influence on mineralization process.
The photocatalyst clearly affected the distribution of formed ions (Fig. 4). First, NO2− was not detected in the reaction cell when the photocatalyst was used (Fig. 4(a)). This means that the photocatalyst promoted the oxidation of the intermediate NO2− to NO3−. In our previous study, we demonstrated that oxidation ability was enhanced by using electrochemical treatment together with photocatalysis because electrochemically formed H2O2 was reduced by electrons in the conduction band of TiO2 to form OH˙.5 The observed promotion of NO2− oxidation suggests that the photocatalyst also enhanced OH˙ production in this study. Although the concentration of NO3− formed decreased (Fig. 4(a)), the NH4+ accumulated in the counter cell increased upon using the photocatalyst (Fig. 4(b)). Therefore, the photocatalyst promoted the oxidation of carbamic acid to NH4+ over that to NO3−. Although the exact reason for the enhanced selectivity using the photocatalyst is still unclear, because one electron as well as one OH˙ is required for NH4+ production (Scheme S1, ESI†), the electrons in the conduction band of the photocatalyst may play an important role.
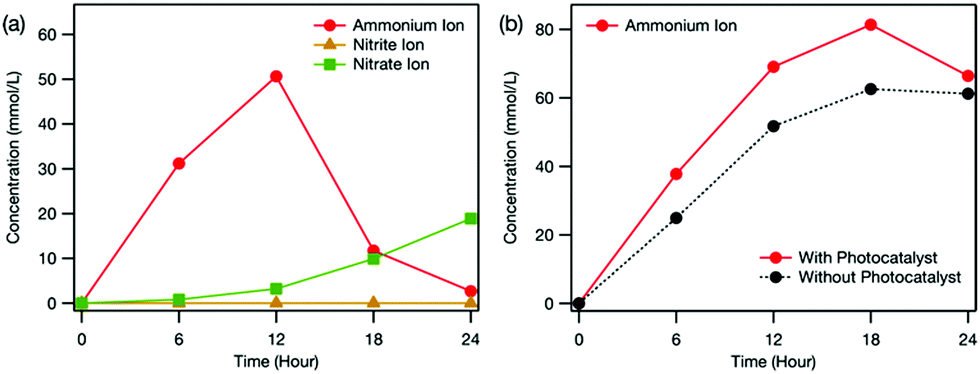 | ||
| Fig. 4 Time dependence of ion concentration in (a) the reaction cell and (b) the counter cell during the electrochemical and photocatalytic treatment estimated from ion chromatography measurements. The dotted line in (b) indicates the ion concentration when only the electrochemical treatment was conducted (i.e., Fig. 3(b)). Nitrite and nitrate ions were not detected in the counter cell. | ||
In conclusion, we obtained valuable NH4+ from a simplified artificial urine by electrochemical treatment. Because this process can be conducted at ambient pressure and room temperature, it shows potential as an energy-efficient NH3 fabrication process. It is hoped that this technique can be applied to actual urine, which contains various inorganic/organic components; initial studies are now underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was partly supported by the Joint Usage/Research Program of the Photocatalysis International Research Center, Research Institute for Science and Technology. We thank Natasha Lundin, PhD, from Edanz Group (https://en-author-services.edanzgroup.com/) for editing a draft of this manuscript.References
- J. Andersson and S. Grönkvist, Int. J. Hydrogen Energy, 2019, 44, 11901–11919 CrossRef CAS.
- Y. Einaga, J. Appl. Electrochem., 2010, 40, 1807–1816 CrossRef CAS; Y. Einaga, Bull. Chem. Soc. Jpn., 2018, 91, 1752–1762 CrossRef.
- M. Cataldo Hernández, N. Russo, M. Panizza, P. Spinelli and D. Fino, Diamond Relat. Mater., 2014, 44, 109–116 CrossRef.
- H. Li, Q. Yu, B. Yang, Z. Li and L. Lei, J. Electroanal. Chem., 2015, 738, 14–19 CrossRef CAS.
- N. Suzuki, A. Okazaki, H. Kuriyama, I. Serizawa, Y. Hirami, A. Hara, Y. Hirano, Y. Nakabayashi, N. Roy, C. Terashima, K. Nakata, K. Katsumata, T. Kondo, M. Yuasa and A. Fujishima, RSC Adv., 2020, 10, 1793–1798 RSC.
- N. Suzuki, A. Okazaki, H. Kuriyama, I. Serizawa, A. Hara, Y. Hirano, Y. Nakabayashi, N. Roy, C. Terashima, K. Nakata, K. Katsumata, T. Kondo, M. Yuasa and A. Fujishima, Molecules, 2018, 23, 3095 CrossRef.
- E. Pelizzetti, P. Calza, G. Mariella, V. Maurino, C. Minero and H. Hidaka, Chem. Commun., 2004, 1504–1505 RSC; P. Calza, E. Pelizzetti and C. Minero, J. Appl. Electrochem., 2005, 35, 665–673 CrossRef CAS.
- S. Park, J. T. Lee and J. Kim, Environ. Sci. Pollut. Res., 2019, 26, 1044–1053 CrossRef CAS.
- A. Zafra, J. Garcia, A. Milis and X. Domènech, J. Mol. Catal., 1991, 70, 343–349 CrossRef CAS.
- S. L. Johnson and D. L. Morrison, J. Am. Chem. Soc., 1972, 94, 1323–1334 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, the time dependence of urea concentration with and without photocatalytic reaction (Fig. S1), and assumed urea oxidation mechanism (Scheme S1). See DOI: 10.1039/d0nj03347b |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |