
Improved efficiency of single-component active layer photovoltaics by optimizing conjugated diblock copolymers†
 *a
and
*a
and
Abstract
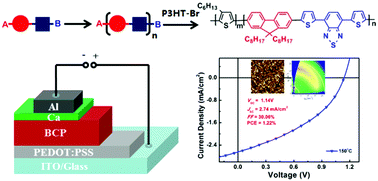
The synthesis of single-component, all-conjugated D–A diblock copolymers that exhibit high photovoltaic activity remains a significant challenge. To date, the synthesis of all-conjugated block copolymers typically produces a mixture of donor and acceptor homopolymers, diblock copolymers, ternary block copolymers or multi-block copolymers. In this work, we report the use of an A–B type monomer to synthesise a diblock copolymer. Importantly, by avoiding AA + BB polymerization, we have eliminated the production of ternary block and multi-block copolymers. In addition, the donor and acceptor blocks had orthogonal solubility in order to increase the purity of the block copolymer and give significant nanoscale phase separation. Two single-component diblock copolymers (P3HT-b-TBTF8 and P3HT-b-F8TBT) were synthesized, of which different molecular weights were obtained using Soxhlet extraction and preparative GPC. Both copolymers exhibited good thermal stability and effective self-aggregation. Thermal annealing was found to reduce the self-aggregation time and enhance the crystallization of the P3HT block, which effectively reduced the self-aggregation of the polymers. The power conversion efficiency of a single component active layer based P3HT-b-F8TBT photovoltaic device reached 1.22%. The higher efficiency of this device was thought to be caused by the linkage between the donor and acceptor blocks, whose P3HT and PF8TBT chains are separated by the wider bandgap F8 unit. Moreover, the unique molecular structure and self-assembly properties of these diblock copolymers will allow them to be used as model materials for further studies on charge and energy transfer in photovoltaic devices.
 Please wait while we load your content...
Please wait while we load your content...

