DOI:
10.1039/C9NJ04758A
(Paper)
New J. Chem., 2020,
44, 171-180
Broadband white light emission from one-dimensional zigzag edge-sharing perovskite†
Received
18th September 2019
, Accepted 26th November 2019
First published on 26th November 2019
Abstract
Low dimensional organic–inorganic halide perovskites have gained a great deal of attention due to their augmented performance in optoelectronic applications, such as solid-state lighting and display. Such compounds consist of a layered structure, such as a multiquantum well structure, where the broadband white-light emission has been observed from diverse metal halide structures. Their broadband emission is ascribed to the self-trapped excitons (STE), which are generated from a deformable structure due to the strong electron-phono coupling. The intensity of the broadband emission is improved by lowing the dimensionality to a 1D perovskite due to the enhanced STE. Herein, we report zigzag edge-sharing 1D perovskites, (4-(aminomethyl)pyridinium)PbBr4 (C6H10N22+ = AMP2+), and (AMP)PbCl4, which are intrinsic deformative structures due to a zigzag connected PbBr6 (or PbCl6) octahedra. Through single crystal X-ray diffraction, we have identified the local lattice distortion from the octahedral environmental of Pb2+ in the inorganic sheet. The temperature dependent photoluminescence data indicate that the unusual broadband emission in zigzag edge sharing (AMP)PbBr4 and (AMP)PbCl4 perovskites is attributed to an intrinsic bulk property, in which photogenerated carriers cause the lattice distortion through electron–phonon coupling. When comparing two compounds, (AMP)PbCl4 gives more remarkable efficient white-light emission than (AMP)PbBr4, because the inorganic structure of (AMP)PbCl4 is a much larger distortion than that of (AMP)PbBr4, which results in a high color rendering index of 90.21. These established results allow us to systematically design white-light emitting materials from 1D perovskites by fine-tuning their crystal structures.
1. Introduction
Lead halide perovskite presents promising potential in light emitting diodes (LED) due to a narrow emission bandwidth, a wide emission spectral window in the visible region and band gap, and color tunability via the size control, which is the quantum size effect, and compositional engineering.1–3 Especially, low dimensional perovskites have attracted significant attention, because they consist of alternating organic and inorganic layers with charge carrier confined within the inorganic network, which is similar to a multiquantum well structure.4 It leads to large exciton binding energy that reaches 320 meV,5 which decreases the possibility of exciton dissociation. Recently, broadband white-light emission across the entire visible region has been realized from the corrugated 2D perovskites, which ascribes it to the self-trapping exciton (STE)6 that is generated from the crystal lattice deformation due to strong electron–phonon coupling.7 In previous the work, 2D perovskites, (001)-oriented (N-MPDA)[PbBr4] and (110)-oriented (N-MEDA)[PbBr4],8 self-assembled (C6H11NH3)2PbBr4,9 multilayered (CH3CH2NH3)4Pb3Br10−xClx,10 corrugated α-(DMEN)PbBr4, (DMAPA)PbBr4, and (DMABA)PbBr411 have been reported to show white-light emissions through the STE from structural distortion. However, even though 2D perovskites exhibit efficient white-light emitting material, LED based on 2D perovskites show a low photoluminescence quantum yield (PLQY) due to the thermal quenching of excitons.12,13 To improve the efficiency of 2D perovskites, quasi-2D perovskites have been synthesized with the incorporation of a bulky cation into the corner-sharing octahedral 3D framework, which thereby divides the 3D perovskite architecture into layers. The thickness of each layer in quasi-2D perovskites can be increased by the incorporation of an additional 3D cation, which is related to the high PLQY. Multilayered quasi-2D perovskites,14 which are comprised of quantum-size-tuned grains, function as charge carrier concentrators, ensuring that they increase the radiative recombination intensity within small bandgap grains. As a consequence, the LED device based on these materials exhibits a PLQY of 8.8% and a corresponding radiance of 80 W sr−1 m−2.14 Nevertheless, it requires critical techniques to obtain an accurate quantum well thickness (or n number) and high phase purity of quasi-2D perovskites.15 As an alternative this, lowing the dimensionality to 1D perovskites enhances the STE at any exciton–phonon interactions and increases the PLQY to 20% and 12% for the bulk single crystals and microscale single crystals, respectively.16 In 1D perovskites, the metal halide octahedra are connected by the corner-sharing, edge-sharing, or face-sharing, which form a nanowire surrounded by organic molecules. Their configurations are either linear or zigzag as a bulk assembly of 1D core–shell quantum wires.17 In general, compared to the 3D or 2D perovskites, 1D perovskites have more vibrational degrees of freedom and polarization under photoexcitation, which induce the formation of STE and enhance the broad-band emission.17,18 Because of this, the 1D perovskites provide an ideal principle for designing broadband white-light-emitting materials with a high PLQY. Indeed, 1D perovskites exhibit broad band emissions with large Stokes shifts of approximately 1 eV.19,20 According to this principle, 1D perovskite with STE is the potential candidate as a highly efficient light emitting material. Mitzi et al. first reported 1D perovskites, [NH2C(l)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH2]2(CH3NH3)2Sn21821 and [NH2C(I)NH2]3MI5 (M = Sn, Pb).22 Since then, only a few organic cations17,21,23–29 have been exhibited to form 1D perovskite structures. Thus, the design rule to synthesize the 1D perovskite having a high PLQY and long carrier lifetime is still unknown, in which the connectivity mode of 1D inorganic framework is regarded as design criteria. Recently, edge sharing connectivity in 1D system is anticipated because it shows broadband emission with high PLQY.
NH2]2(CH3NH3)2Sn21821 and [NH2C(I)NH2]3MI5 (M = Sn, Pb).22 Since then, only a few organic cations17,21,23–29 have been exhibited to form 1D perovskite structures. Thus, the design rule to synthesize the 1D perovskite having a high PLQY and long carrier lifetime is still unknown, in which the connectivity mode of 1D inorganic framework is regarded as design criteria. Recently, edge sharing connectivity in 1D system is anticipated because it shows broadband emission with high PLQY.
In this study, we reported 1D perovskites with white-light emission from a combination of 4-(aminomethyl)pyridinium (C6H10N22+ = AMP2+) and PbBr2 (or PbCl2) compounds. The crystal structures of (AMP)PbBr4 and (AMP)PbCl4 consist of a 1D zigzag edge-sharing [PbBr42− (or PbCl42−)]∞ infinite chain, which the AMP2+ cation is positioned between the columns of the inorganic chain and the changing positions every 180 degrees. These unique 1D structures emit pronounced broadband white-light emissions in the entire visible-light spectrum. This broadband white-light emission is directly related to the structural distortion induced by the 1D zigzag structures, because both compounds exhibit a broadband emission up to 10 K with the results of the temperature dependent PL emission. Compared to the (AMP)PbBr4, (AMP)PbCl4 shows white-light emitting properties with a high color rendering index around 90.21, which is attributed to the sufficient carrier confinement due to the strongly deformable lattice. This study confirms that the 1D perovskite systems are favourable for formation of STE to generate an efficient white-light emission and open up a new method for the further development of perovskite emitters based on bulk quantum materials.
2. Experimental section
(1) Starting materials
4-(Aminomethyl)pyridine (C6H8N2, 98%), lead(II) bromide (PbBr2, 98%), lead(II) chloride (PbCl2, 98%), hydrobromic acid (HBr, 48%), hydrochloric acid (HCl, 37%), and hypophosphorous acid (H3PO2, 50 wt% in H2O) were purchased from Sigma-Aldrich, and they were used without further purification.
(2) Synthesis of (AMP)PbBr4 and (AMP)PbCl4 perovskites
For the (AMP)PbBr4 synthesis, 0.367 g of PbBr2 (1 mmol) was dissolved in 5.0 mL HBr solution. The flask was heated in an oil bath to 120 °C, and then stirred vigorously until the solid had completely dissolved. In a separate beaker, 4-(aminomethyl)pyridine (0.216 g, 2 mmol) was neutralized with a 1 mL HBr solution. The 4-(aminomethyl)pyridine solution was slowly added dropwise into the PbBr2 solution while stirring continuously. The heat was removed, and the flask was allowed to air-cool without stirring. A needle-like colorless precipitate crystal appeared during the slow cooling at room temperature. The precipitate crystal was dried with a vacuum oven at 60 °C for 24 hours, and it was used without further purification. The (AMP)PbCl4 perovskite was also prepared in the same manner, and 1 mmol (0.278 g) of PbCl2 and 2 mmol (0.216 g) of 4-(aminomethyl)pyridine were used for this preparation. The crystal shape of (AMP)PbCl4 is similar to the that of (AMP)PbBr4.
(3) Characterization
High resolution powder X-ray diffraction (PXRD) and single crystal X-ray diffraction.
The PXRD was collected at room temperature using an Empyrean PANalytical instrument with Cu Kα radiation (40 kV, 30 mA, λ = 1.54056 Å) and a Ni filter with a graphite monochromator. The perovskite crystal was coated with paratone-N oil, and the diffraction data was measured at 293 K with synchrotron radiation (λ = 0.61000 Å) on a an ADSC Quantum-210 detector at BL2D SMC with a silicon (111) double crystal monochromator (DCM) at the Pohang Accelerator Laboratory in Korea. The PAL BL2D-SMDC program30 was used for data collection. The detector distance was 63 mm, the omega scan (Δω) was 3°, and the exposure time was 1 s per frame. The HKL3000sm (Ver. 703r)31 was used for cell refinement, reduction, and absorption correction. The crystal structure for the perovskite was solved using the direct method with an SHELXT-2014 program32 and refined using full-matrix least-squares calculations with the SHELXL-2014 program package.33
Optical absorption spectroscopy.
The optical diffuse reflectance measurement was performed using an Agilent (Cary-5000) UV-vis NIR spectrometer that operated in the 200–1500 nm region at room temperature. BaSO4 was used as a reference of 100% reflectance for all the measurements. The reflectance versus wavelength data generated was used to estimate the band gap of the material by converting reflectance to absorption data according to the Kubelka–Munk equation:34,35α/S = (1 − R)2(2R)−1, where R is the reflectance and e and α and S are the absorption and scattering coefficients, respectively. As shown in Fig. 8a, the bandgap was determined as the intersection point between the energy axis and the line extrapolated from the linear portion of the absorption edge in an (a/S)2vs. E plot.36,37
Steady state and time-resolved photoluminescence (TRPL).
The static PL measurement was carried out using an Andor iDus DU490A InGaAs detector. A pulse wave 325 nm diode laser was used to photo-excite the samples. A TRPL study was performed using an inverted-type scanning confocal microscope (MicroTime-200, Picoquant, Germany) with a 60× (air) objective. The lifetime measurements were performed at the Korea Basic Science Institute (KBSI), Daegu Center, Korea. Single-mode pulsed diode lasers (375 and 470 nm with a pulse width of ∼30 ps) were used as excitation sources. A dichroic mirror (Z375RDC, AHF), a longpass filter (HQ405lp, AHF), a 75 μm pinhole, a band-pass filter, and an avalanche photodiode detector (PDM series, MPD) were used to collect emissions from the samples when the 375 nm laser irradiated. A dichroic mirror (490 DCXR, AHF), a long-pass filter (HQ500lp, AHF), a 75 μm pinhole, and a single photon avalanche diode (PDM series, MPD) were used to collect emissions when the 470 nm laser irradiated. The time-correlated single-photon counting (TCSPC) technique was used to count the fluorescence photons. The TRPL images, which consisted of 200 × 200 pixels, were recorded using the time-tagged time-resolved (TTTR) data acquisition method. The exponential fittings for the obtained PL decays with a temporal resolution of 16 ps were performed using Symphotime-64 software (Ver. 2.2) with the exponential decay model: 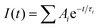 , where I(t) is the time-dependent PL intensity, A is the amplitude, and τ is the PL lifetime.
, where I(t) is the time-dependent PL intensity, A is the amplitude, and τ is the PL lifetime.
Absolute PLQY.
The PLQY was determined using an absolute quantum yield measurement (Hitachi, F-7000 Fluorimeter) with an integrating sphere. The perovskite was excited using a 330 nm laser (CrystaLaser, GCL532-005-L) source. The procedure of data acquisition followed the previous protocol38 in detail.
3. Results and discussion
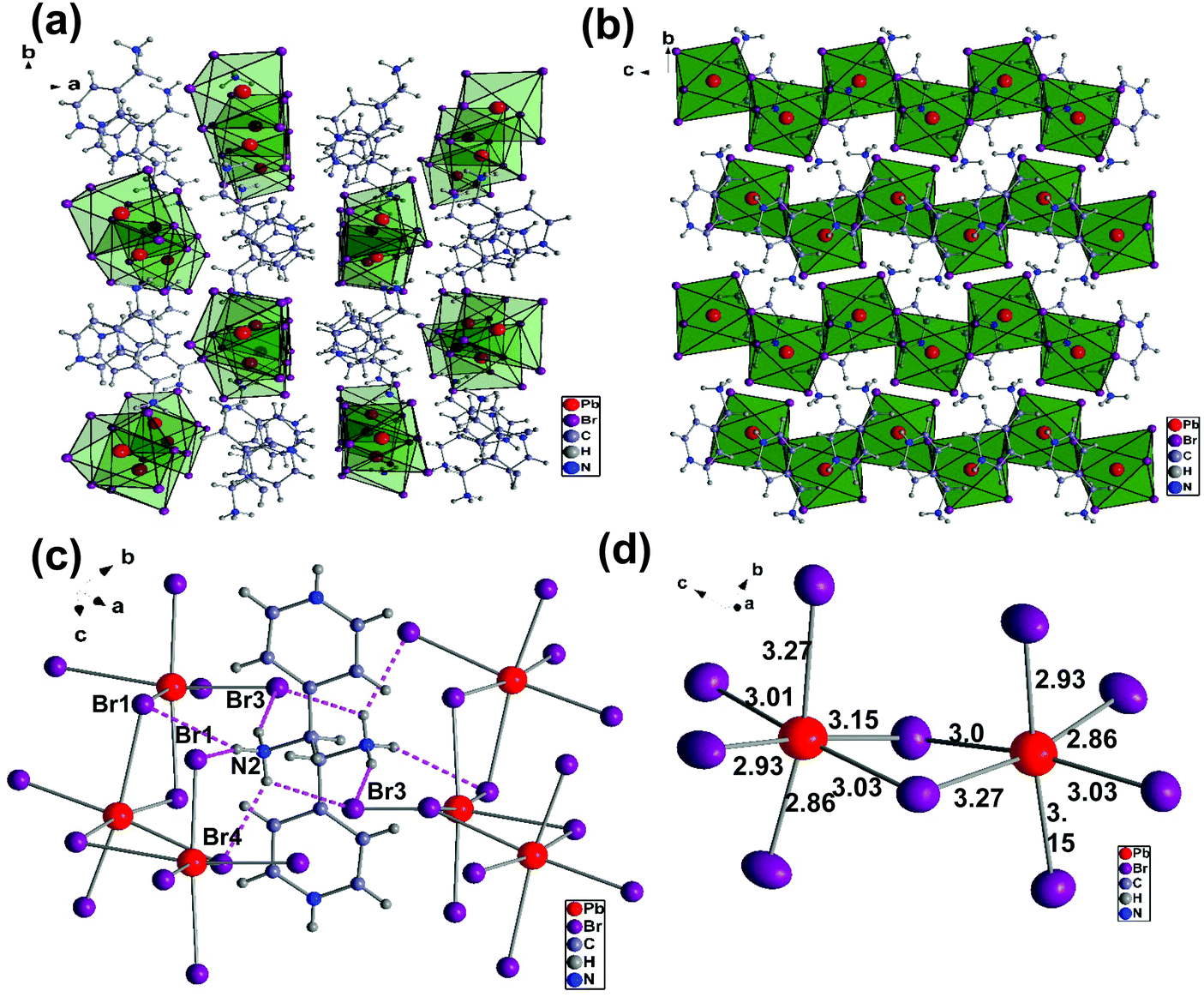
We will first discuss the crystal structures of (AMP)PbBr4 and (AMP)PbCl4 perovskites. These compounds were synthesized by the direct reaction of the cation source and PbBr2 (or PbCl2) in an aqueous HBr (or HCl) solution at 120 °C. The obtained crystals showed a needle-like colorless shape as shown Fig. 1. We selected the crystal from the precipitate in the solution and measured the single crystal X-ray crystallography diffraction. The crystallographic information is presented in Table 1 in detail. The single crystal XRD crystallography measurements revealed that the crystal structure of (AMP)PbBr4 consists of the 1D zigzag edge-sharing octahedral PbBr42− chain, which is connected to the AMP2+ by a hydrogen bond. (AMP)PbBr4 was reported by Li et al.39 They showed the different dimensional lead bromide framework that ranged from 0D and 1D to 2D by varying the substituent position of aminomethyl on a pyridine ring. The combination of 2-(aminomethyl)pyridine and PbBr2 produces 2D (H22-AMP)PbBr4 perovskite, whereas the reaction of 3-(aminomethyl)pyridine and PbBr2 results in 0D (H23-AMP)2PbBr6. When 4-(aminomethyl)pyridine reacted with PbBr2, the 1D zigzag edge-sharing perovskite, (AMP)PbBr4, was obtained. However, they did not report the white-light emission from the (AMP)PbBr4 perovskite. Thus, we report for the first time an approach concerning the white-light emission based on (AMP)PbBr4 perovskite. Fig. 2a presents the 1D crystal structure of (AMP)PbBr4, which crystallized in the monoclinic space group P21/c with unit cell parameters of a = 12.390(3) Å, b = 13.824(3) Å, c = 7.9270(16) Å, α = γ = 90°, and β = 105.47(3)°. The inorganic layer was built from a zigzag edge sharing [PbBr42−]∞ chain, which is a single layer of AMP molecules that are located between consecutive inorganic layers by changing its position by 180° degrees relative to each other. It was well known that molecular packing structure or conformation could influence the emission properties of PL. Fang et al.40 demonstrated the white-light emission by modulation of shape and morphology of dibenzothiophene molecule. It was also observed in supramolecular luminescent materials, in which photoactive molecules are combined with the conformers, changing the crystal structures and stacking of fluorescent molecules, resulting in the improved photophysical properties as well as thermal stability for the potential LED applications.41Fig. 2b gives a closer look at this compound. The packing diagram consists of a 1D [PbBr42−]∞ layer stacked parallel to the ac-plane with AMP cations occupying the interlayer spaces. This structure is similar to the previously reported 1D core–shell quantum wires, which is composed of an insulating organic layer surrounding the core inorganic wire structure for the application of efficient white-light emission.16 Crystal cohesion is achieved on both ends of the AMP molecules by N–H⋯Br hydrogen bonds, which are related to the NH3 polar groups, to the adjacent bromides of the octahedral layers (Fig. 2c). The hydrogen atoms on the N2 take hydrogen bonds with one bridging atom Br1 and tree terminal atoms Br2, Br3, and Br4 with the bond lengths of H–Br that range from 2.52 Å to 3.03 Å. These hydrogen bondings between the AMP molecules and PbBr6 octahedra keep the continuity of the zigzag edge-sharing 1D inorganic chains. Similarly, it was reported that orientational hydrogen bonding regularly arranged the phosphorescence material within the polymer network, resulting in the tunable and efficient white-light phosphorescent emission.42 As the dimensionality decreases in metal halide perovskites, the STEs generated from the local deformation of the crystal lattice due to electron–phonon coupling were enhanced, which is associated with broad band white-light emission. As we can see in Fig. 2d, the structural distortion of (AMP)PbBr4 perovskite was observed in the coordination geometry around the Pb atom, which resulted in one short Pb–Br bond and one long Pb–Br bond from the [Pb–Br–Pb] unit. According to previous reports,10,43,44 the Pb–X octahedrons in a perovskite structure are prone to estimate the structural distortion upon excitation, which generates the broadband emission. The bond distances of Pb–Br to the bridging bromides are the combination of 3.01 Å/3.03 Å, 2.93 Å/3.15 Å, and 3.27 Å/2.86 Å. The vertical Pb–Br bonds have lengths of 2.86 Å/3.27 Å and 3.15 Å/2.93 Å, which are respectively much longer and shorter than the Pb–Br length (2.84 Å) in the 3D perovskite (MAPbBr3).10 The angles of Br–Pb–Br between the neighboring iodides are between 84.04° to 97.72°. The zigzaged connected to the PbBr6-octahedra are tilted at about 90° to each other, which is indicated by the Pb–Br–Pb angles of 94.19° and 91.39°.
 |
| | Fig. 1 Optical microscopy images of (a) (AMP)PbBr4 and (b) (AMP)PbCl4 single crystals. The scale bar is 10 μm. | |
Table 1 Crystal data and structure refinement for (AMP)PbBr4 and (AMP)PbCl4
| Empirical formula |
(AMP)PbBr4 |
(AMP)PbCl4 |
| Formula weight |
636.99 |
459.15 |
| Temperature (K) |
293(2) |
293(2) |
| Wavelength (Å) |
0.700 |
0.700 |
| Crystal system |
Monoclinic |
Orthorhombic |
| Space group |
P21/c |
Pbca
|
| Unit cell dimensions |
a = 12.390(3) Å, α = 90° |
a = 13.428(3) Å, α = 90° |
|
b = 13.824(3) Å, β = 105.47(3)° |
b = 7.6610(15) Å, β = 90° |
|
c = 7.9270(16) Å, γ = 90° |
c = 23.338(5) Å, γ = 90° |
| Volume (Å3) |
1308.5(5) |
2400.8(9) |
|
Z
|
4 |
8 |
| Density (calculated) (Mg m−3) |
3.233 |
2.541 |
| Absorption coefficient (mm−1) |
24.060 |
14.232 |
|
F(000) |
1128 |
1680 |
| Crystal size (mm3) |
0.184 × 0.052 × 0.015 |
0.212 × 0.069 × 0.015 |
| Theta range for data collection |
1.680 to 33.482° |
1.719 to 33.674° |
| Index ranges |
−18 ≤ h ≤ 18, |
−19 ≤ h ≤ 19, |
| −21 ≤ k ≤ 21, |
−9 ≤ k ≤ 9, |
| −12 ≤ l ≤ 12 |
−33 ≤ l ≤ 33 |
| Reflections collected |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 730 730 |
19044 |
| Independent reflections |
4329 [R(int) = 0.1122] |
3832 [R(int) = 0.0569] |
| Completeness to theta = 24.835° |
95.2% |
93.0% |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
4329/0/119 |
3832/0/119 |
| Goodness-of-fit on F2 |
1.293 |
1.130 |
| Final R indices [I > 2 sigma(I)] |
R
1 = 0.0938, wR2 = 0.2789 |
R
1 = 0.0495, wR2 = 0.1503 |
|
R indices (all data) |
R
1 = 0.0975, wR2 = 0.2819 |
R
1 = 0.0503, wR2 = 0.1508 |
| Extinction coefficient |
0.035(4) |
0.0054(4) |
| Largest diff. peak and hole (e Å−3) |
7.025 and −9.939 |
2.615 and −3.713 |
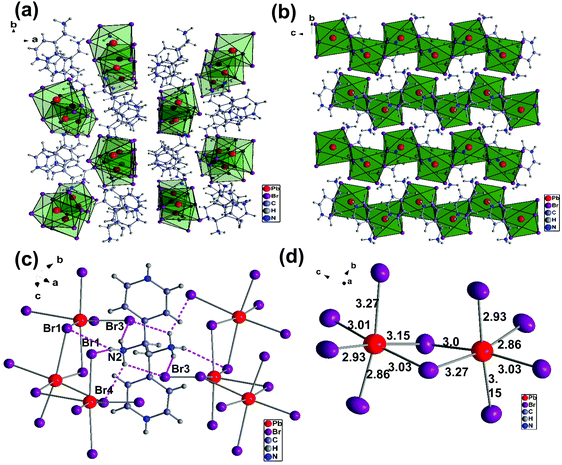 |
| | Fig. 2 (a) Crystal structure of the (AMP)PbBr4 with the monoclinic space group P21/c along the c-axis that shows an infinite zigzag edge sharing [PbBr42−]∞ chain with AMP cations occupying the interlayer spaces. (b) The packing diagram of the 1D [PbBr42−]∞ blocks stacked parallel to the bc-planes. (c) The local hydrogen bridging geometry between the AMP molecules and the zigzag edge sharing octahedral (PbBr6) inorganic layers. The dashed lines denote the N–H⋯Br hydrogen bonds. (d) The Br–Pb–Br angles and the Pb–Br bond lengths in edge sharing PbBr6 octahedral layers. | |
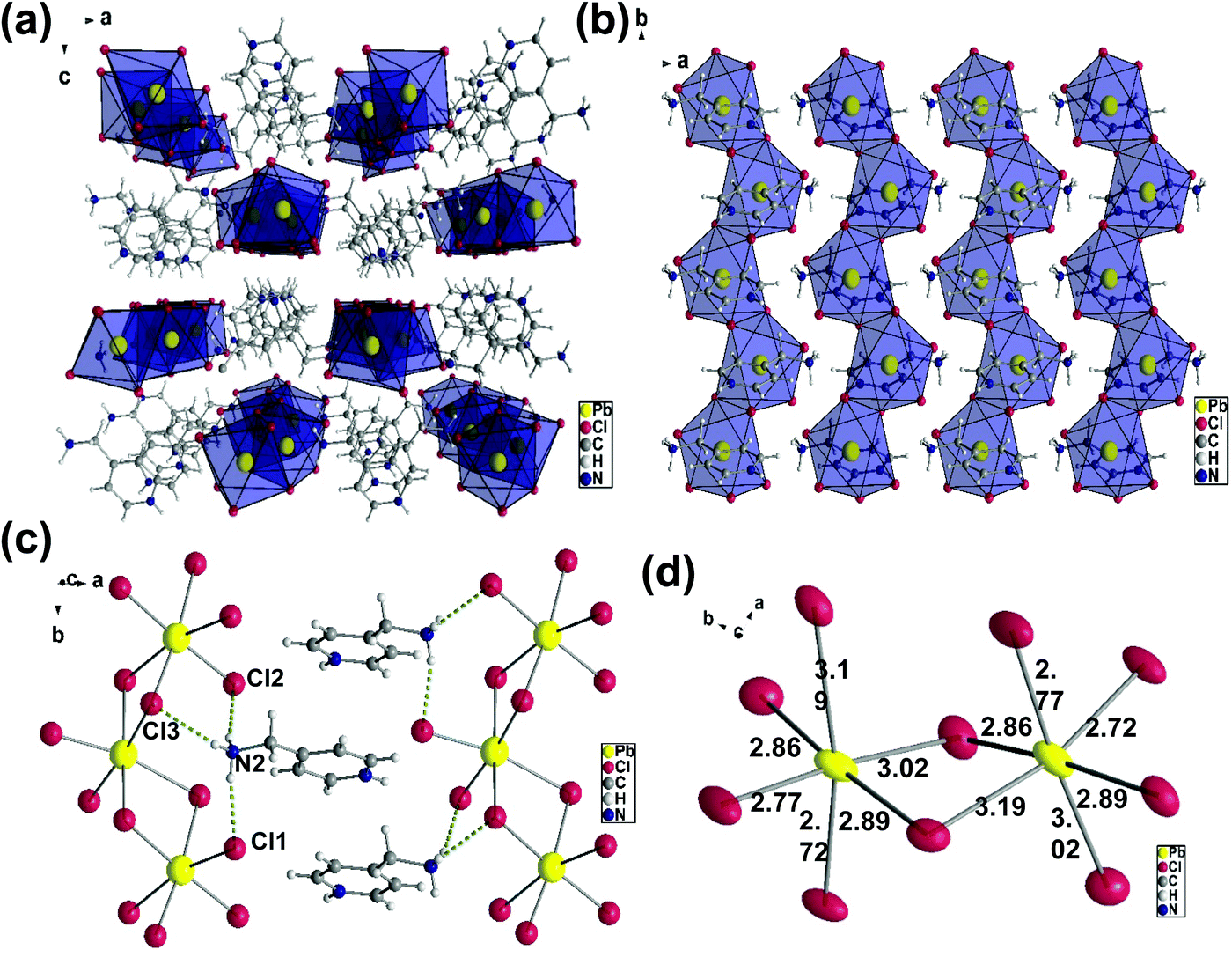
The crystal structure of (AMP)PbCl4, which is shown in Fig. 3a, crystallize in the orthorhombic space group Pbca with unit cell parameters of a = 13.428(3) Å, b = 7.6610(15) Å, and c = 23.338(5) Å. The(AMP)PbCl4 compound consisted of 1D edge-sharing PbI6 octahedra chains propagating along the crystallographic b-axis, which are parallel to the ab-plane (Fig. 3b). The channels in between the 1D infinite chain were filled by the AMP cations the same way as the (AMP)PbBr4. We found that (AMP)PbCl4 exhibited a white-light emission, but the deformation of the inorganic framework is quite different from that of (AMP)PbBr4 perovskite due to the hydrogen bond with the adjacent AMP cations.
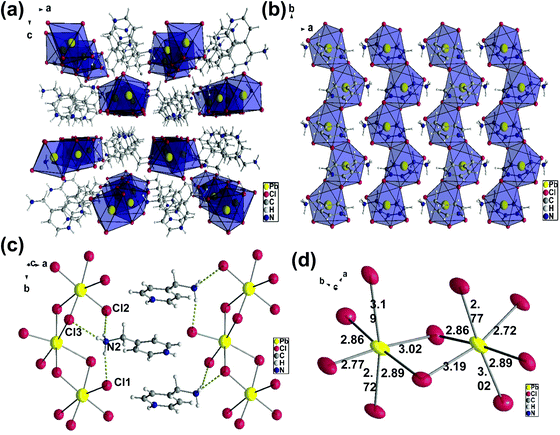 |
| | Fig. 3 (a) Crystal structure of the (AMP)PbCl4 with the orthorhombic space group Pbca along the b-axis. (b) The packing diagram of the 1D [PbCl42−]∞ blocks stacked parallel to the ab-planes with AMP cations. (c) The hydrogen bonds (dashed yellow line) between the [PbCl6]4− octahedra and the AMP cations. (d) The Cl–Pb–Cl angles and the Pb–Cl bond lengths in the edge sharing PbCl6 octahedral layers. | |
As shown in Fig. 3c, there are two hydrogens on the N2 bond to the equatorial Cl1 and Cl2 and the distances are 2.36 Å and 2.37 Å, respectively. The last hydrogen on the N2 is bond to the axial Cl3, and the hydrogen acceptor distance is 2.60 Å. The Pb–Cl bridging distances are the combinations of 3.02 Å/2.77 Å, 2.86 Å/2.89 Å, and 3.19 Å/2.72 Å, while the Pb–Cl axial distances are the combinations of 3.19 Å/2.72 Å and 2.77 Å/3.02 Å (Fig. 3d). The angles of Cl–Pb–Cl between the neighboring iodides are 83.91° to 103.25°, while the bridging Pb–Cl–Pb angles are 94.39° and 90.27°. The distortion in the (AMP)PbCl4 is substantially larger than the (AMP)PbBr4, which confirms the intrinsic deformable lattice.
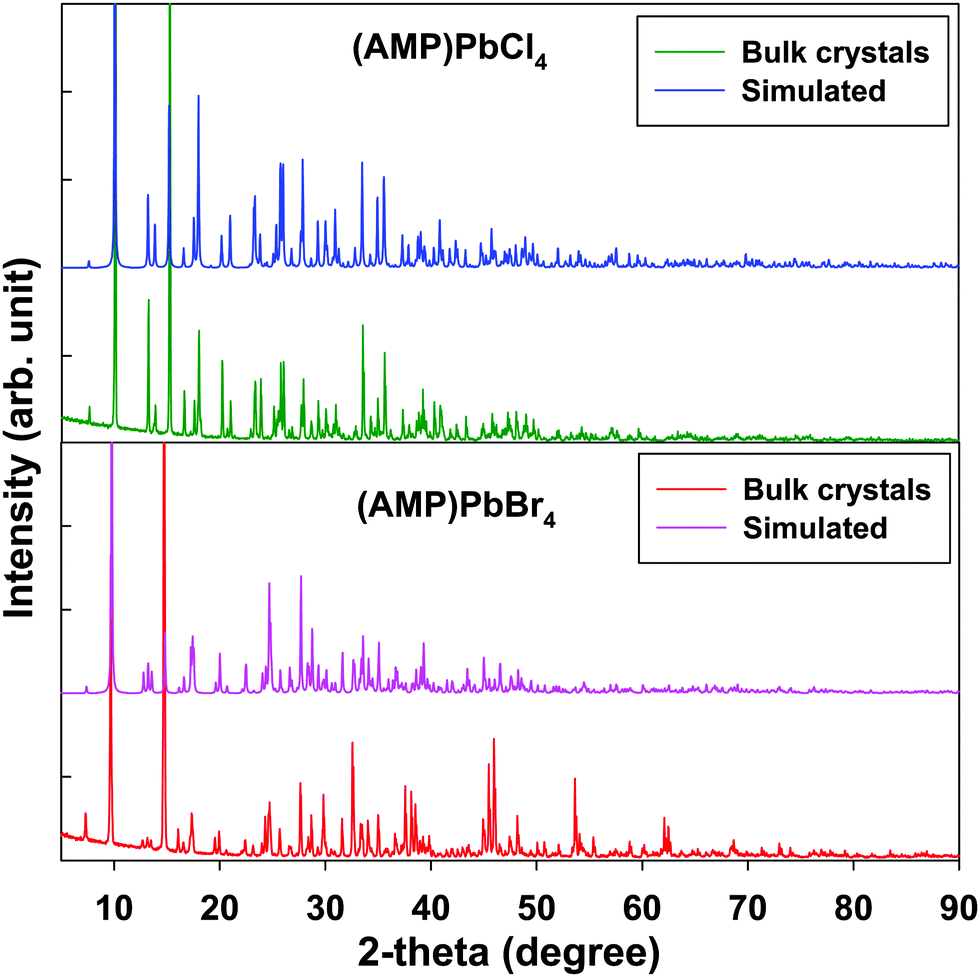
Fig. 4 shows the PXRD patterns of the grinding powder from the single crystals of (AMP)PbBr4 and (AMP)PbCl4 perovskites, which are almost the same features as the simulated patterns from the single crystal X-ray diffraction. This suggests that bulk single crystal synthesized by one-pot syntheis possess no impurities and a uniform crystal structure of the as prepared 1D metal halide perovskite.
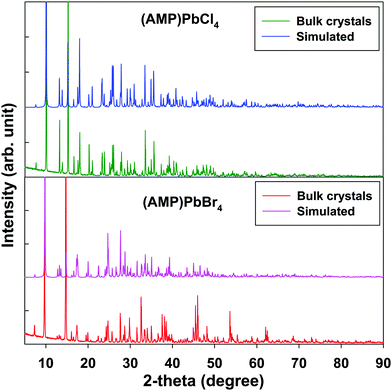 |
| | Fig. 4 Powder X-ray diffraction and the simulated pattern from SCXRD for the (AMP)PbBr4 and the (AMP)PbCl4 perovskites. | |
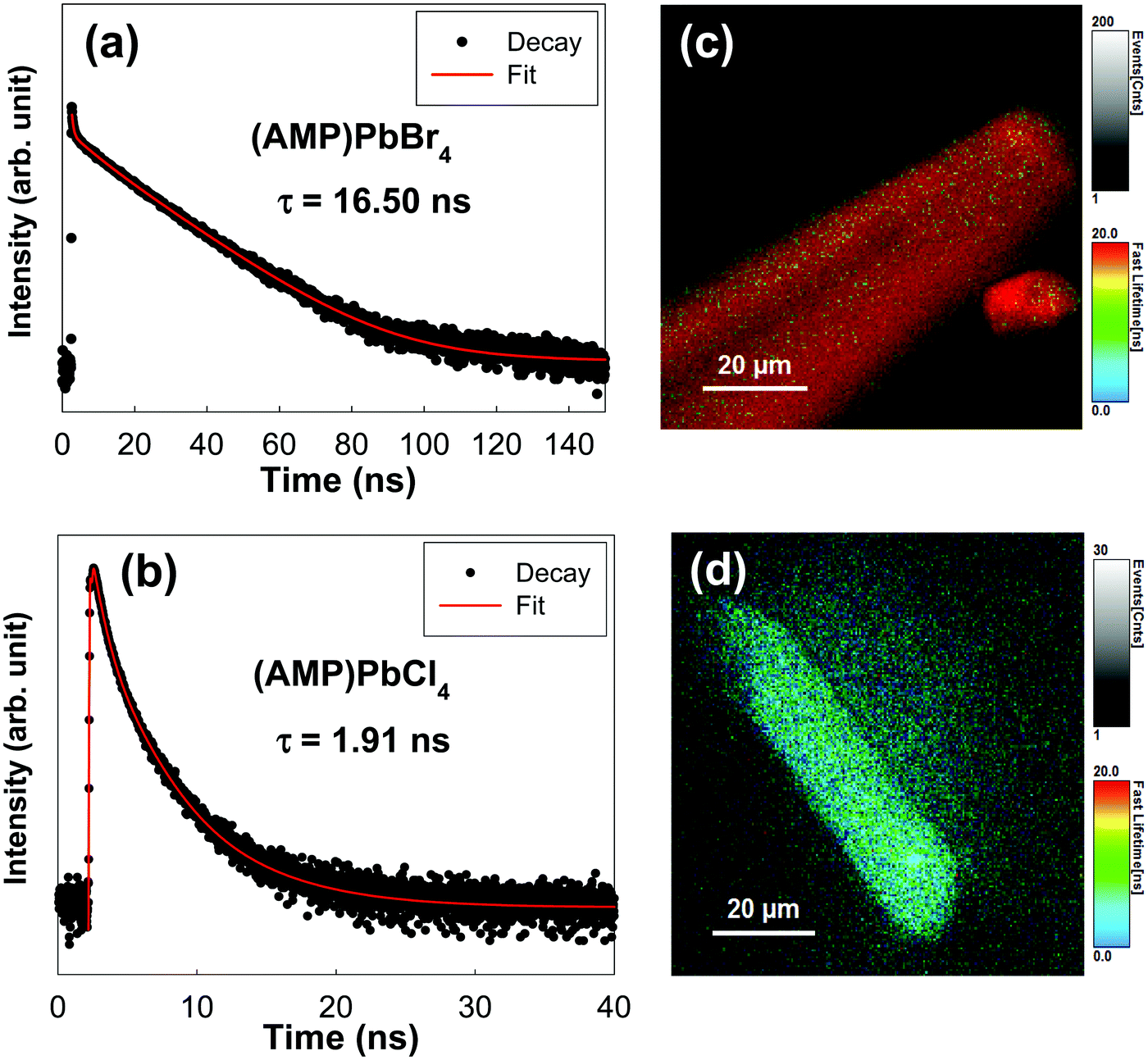
To investigate the kinetics of excitons and the carriers lifetime in both radiative and non-radiative processes in zigzagged 1D perovskites, we conducted the TRPLmeasurement as shown in Fig. 5. The PL decay curves were fitted with a triexponential decay model, and the detailed fitting parameters are listed in Table 2. We assigned τ1 as a fast decay process related to the radiative recombination of excitons, and τ2 and τ3 are a slower decay process corresponding to the trap states formation in the layered perovskites.45,46 For the (AMP)PbBr4 perovskite, a triexponential decay is composed of a 18.00 ns component (52.16%), a 0.43 ns component (36.56%) and a 6.20 ns component (11.28%). In the (AMP)PbCl4 perovskite, it has a 5.50 ns component (2.84%), a 1.90 ns component (25.01%) and a 0.61 ns component (72.15%), which agree well with the triexponential function. As mentioned previously reports,45,47–50 since the characteristics of the trap staps is ascribed to the sub-band-gap states, we only compared the τ1 values, which are related to the band gap edge.16,51,52 In this respect, a remarkable efficient exciton recombination was observed in the (AMP)PbCl4 rather than the (AMP)PbBr4, which in turn indicates that the generation of a free carrier is decreased. In addition, since PL lifetime of (AMP)PbBr4 perovskite consists of 52.16% for τ1 component showing a long lifetime of 18.00 ns, the average lifetime of (AMP)PbBr4 is 16.50 ns, which is much longer than that of the (AMP)PbCl4 of 1.91 ns (Fig. 5a and b). Consistent with these results, the image mapping of PL lifetime also indicates that the PL lifetime of (AMP)PbBr4 perovskite (Fig. 5c) is longer than that of the (AMP)PbCl4 perovskite (Fig. 5d). It was noticed that both perovskites have the uniform PL lifetime, showing fast decay and long lifetime for (AMP)PbCl4 and (AMP)PbBr4 perovskites, respectively.
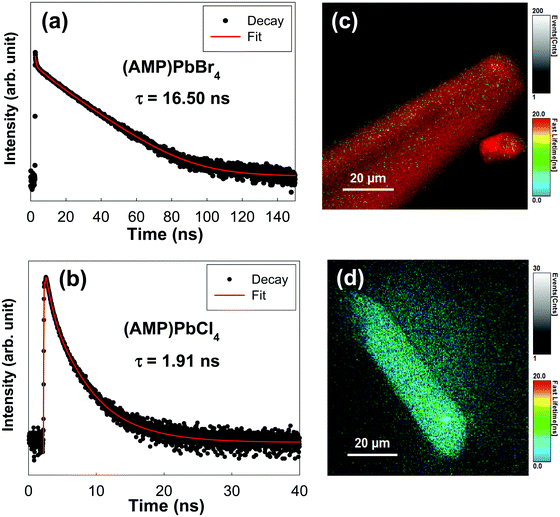 |
| | Fig. 5 Transient photoluminescence decay profile of (a) (AMP)PbBr4 and (b) (AMP)PbCl4 perovskite single crystals. Image mapping of the photoluminescence emission decay of (c) (AMP)PbBr4 and (d) (AMP)PbCl4 perovskite single crystals. | |
Table 2 Photoluminescence lifetime results of (AMP)PbBr4 and (AMP)PbCl4 perovskites from the transient photoluminescence decay analysis
| Perovskites |
A
1/% |
τ
1/ns |
A
2/% |
τ
2/ns |
A
3/% |
τ
3/ns |
τ
avg/ns |
τ
amp/ns |
The emission decay was analyzed using the equation: I(t) = A1exp(−t/τ1) + A2exp(−t/τ2) + A3exp(−t/τ3) + …, where 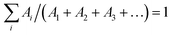 , the average lifetime, 〈τ〉, is defined by , the average lifetime, 〈τ〉, is defined by 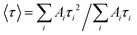 and τamp is the amplitude of average lifetime. and τamp is the amplitude of average lifetime. |
| (AMP)PbBr4 |
52.16 |
18.00 |
36.56 |
0.43 |
11.28 |
6.20 |
16.50 |
10.00 |
| (AMP)PbCl4 |
2.84 |
5.50 |
25.01 |
1.90 |
72.15 |
0.61 |
1.91 |
1.09 |
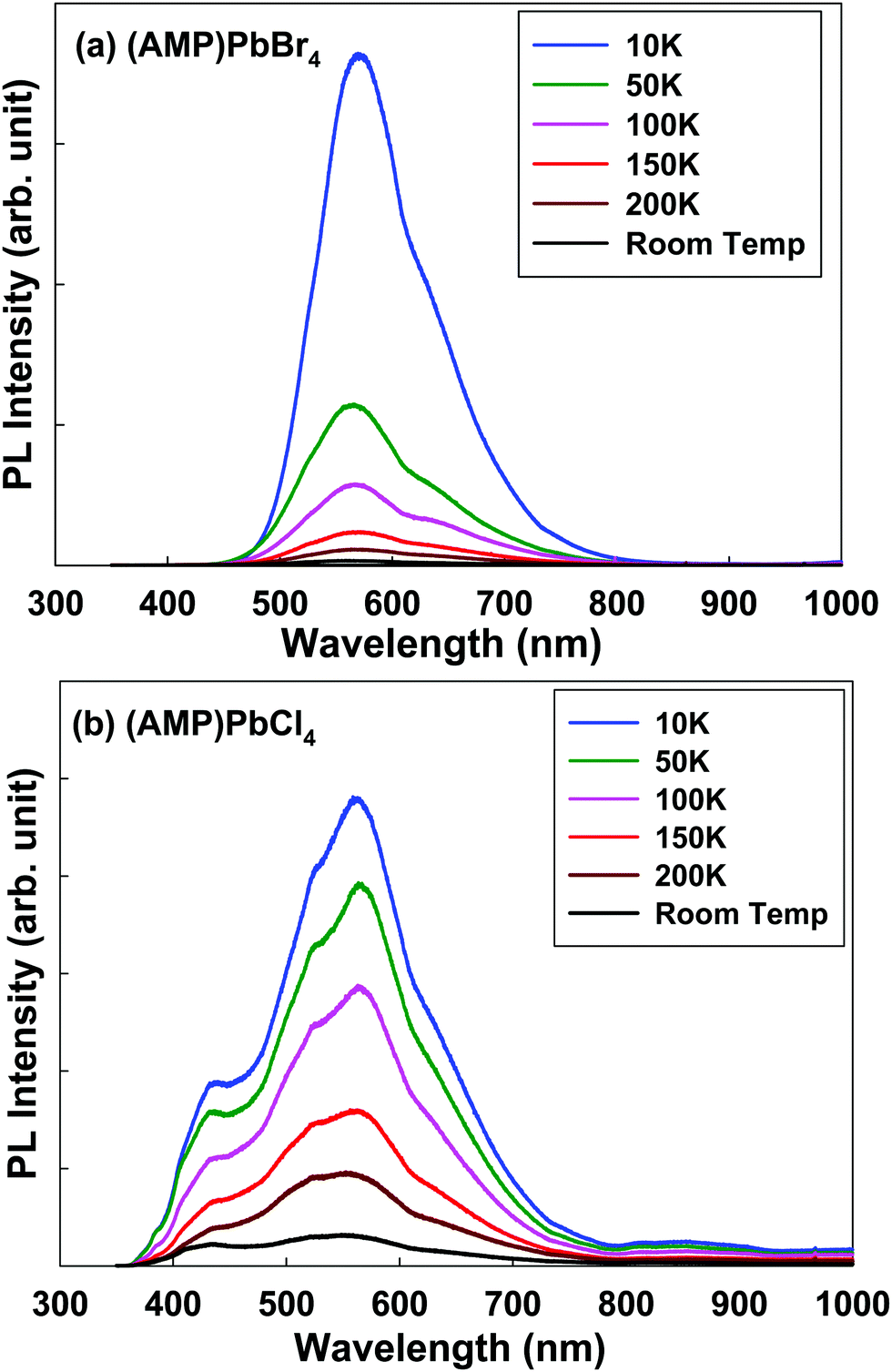
To understand the emission properties of the zigzag edge-sharing perovskite, we conducted the temperature dependent emission properties (Fig. 6). Temperature dependent PL emission for (AMP)PbBr4 perovskite shows the broad peak from 440 nm to 800 nm, centered at ∼570 nm including a shoulder peak at ∼615 nm (Fig. 6a). As the temperature decreases, the broad peak shifts to longer wavelength. Temperature dependent PL measurement of (AMP)PbCl4 shows the unusual evolution of the PL emission from room temperature to 10 K, as shown in Fig. 6b. At room temperature, the PL emission is a broad peak from 350 nm to 800 nm, showing the tree main peaks at 430 nm, 520 nm, 562 nm and one shoulder peak at ∼610 nm. As the temperature decreases, the emission intensity at 430 nm is more pronounced, which is assigned to the free exciton emission at high energy.44 The PL peaks at 520 nm, 562 nm and 610 nm are related to the trap states in the perovskite framework characteristics.46 The trap density is related to the organic/inorganic interface, each consisting of a soft AMP organic layer on a hard inorganic lead chloride. Compared to 3D perovskite, the number of trap states were increased in the low dimensional perovskites. As we can see in Fig. 6b, PL tailing was observed in the low energy region, which comes from the radiative recombination of trap states. As the temperature increases, electron–phonon coupling interaction will be enhanced, which will result in the broadening of the PL emission bandwidth. The electron–phonon coupling induces the structural distortion, which leads to dominant STE and broadening of the PL emission bandwidth.6,53,54 Thus, with the cooling process, the bandwidth of the PL becomes narrower, because the free exciton PL is dominantly occurs at sufficiently low temperature instead of the thermally activated processes. Consistent with this model, the temperature dependent PL measurement of (AMP)PbBr4 and (AMP)PbCl4 showed that the broad peak slightly become narrower and more intense from room temperature to 10 K. However, both compounds still remained with the STE emission, because they showed the broadband emission up to 10 K, which means that their highly distorted structure prevented the STEs from tunneling back to the free exciton states. According to a previous report, the PL of perovskite is related to the combination of the photoexcited electrons in Pb 6p states and a hole in X (I, Br and Cl) outer p.55 The Pb–X bond distance and connectivity of the octahedral PbX6 unit affects exciton energy, which results in different emission energies. The PbX6 octahedral can be connected in the form of a corner, edge, and face sharing, and the distortion of these structures result in the different Pb–X lengths.56 However, (AMP)PbBr4 and (AMP)PbCl4 decrease the full width at half maximum (FWHM) less than the normal perovskite as the temperature decreases, which indicates a weaker phonon–exciton interaction. This result differs from that in a metal–organic framework, in which the guest molecule highly adjust the solid-state photoexcitation and emission processes. Yang et al.57 found the white-light emission and near-infrared phosphorescence due to the host–guest interaction in the Zeolite imidazole framework.
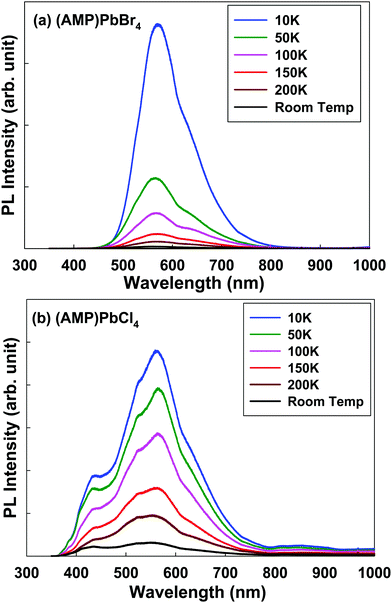 |
| | Fig. 6 Temperature dependent steady-state photoluminescence data for the (a) (AMP)PbBr4 and (b) (AMP)PbCl4 perovskites. The excitation wavelength is 325 nm. | |
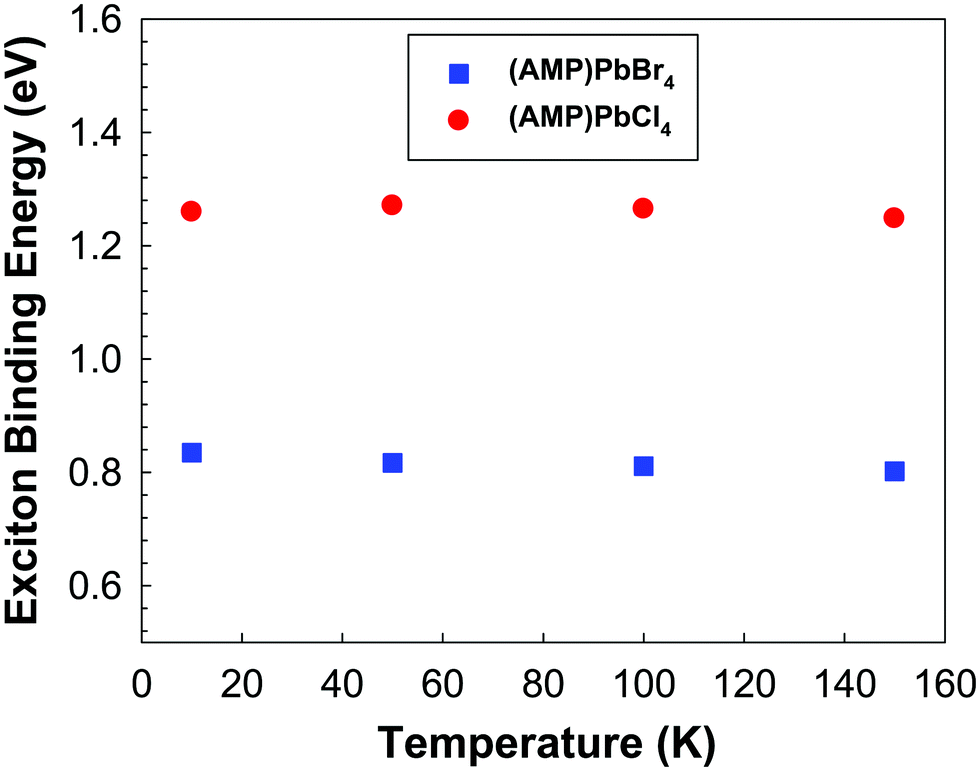
The photoluminescence is essentially the recombination of exciton, which electrically bound electron hole pairs. In order to make them available for electrical conduction, the exciton must be dissociated in the sense the electrons must be transferred to the conduction band and exciton hole must be transferred to the valence band. Since the energy required to get one exciton is optical energy gap, the exciton binding energy is the difference between the absorption edge and the PL emission maximum.58,59 As the temperature increases, the contribution of the excitonic resonance decreases, as a consequence, the exciton binding energy was determined only below 150 K. As shown in Fig. 7, exciton binding energies of (AMP)PbCl4 is much larger than that of (AMP)PbBr4 perovskites. This is attributed to the Cl induced increase in the electron effective mass.60
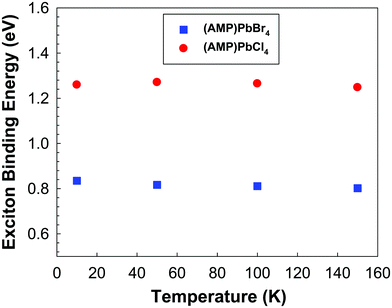 |
| | Fig. 7 The exciton binding energy of the (AMP)PbBr4 and (AMP)PbCl4 perovskites, which was determined from band gap and temperature dependent PL spectra. | |
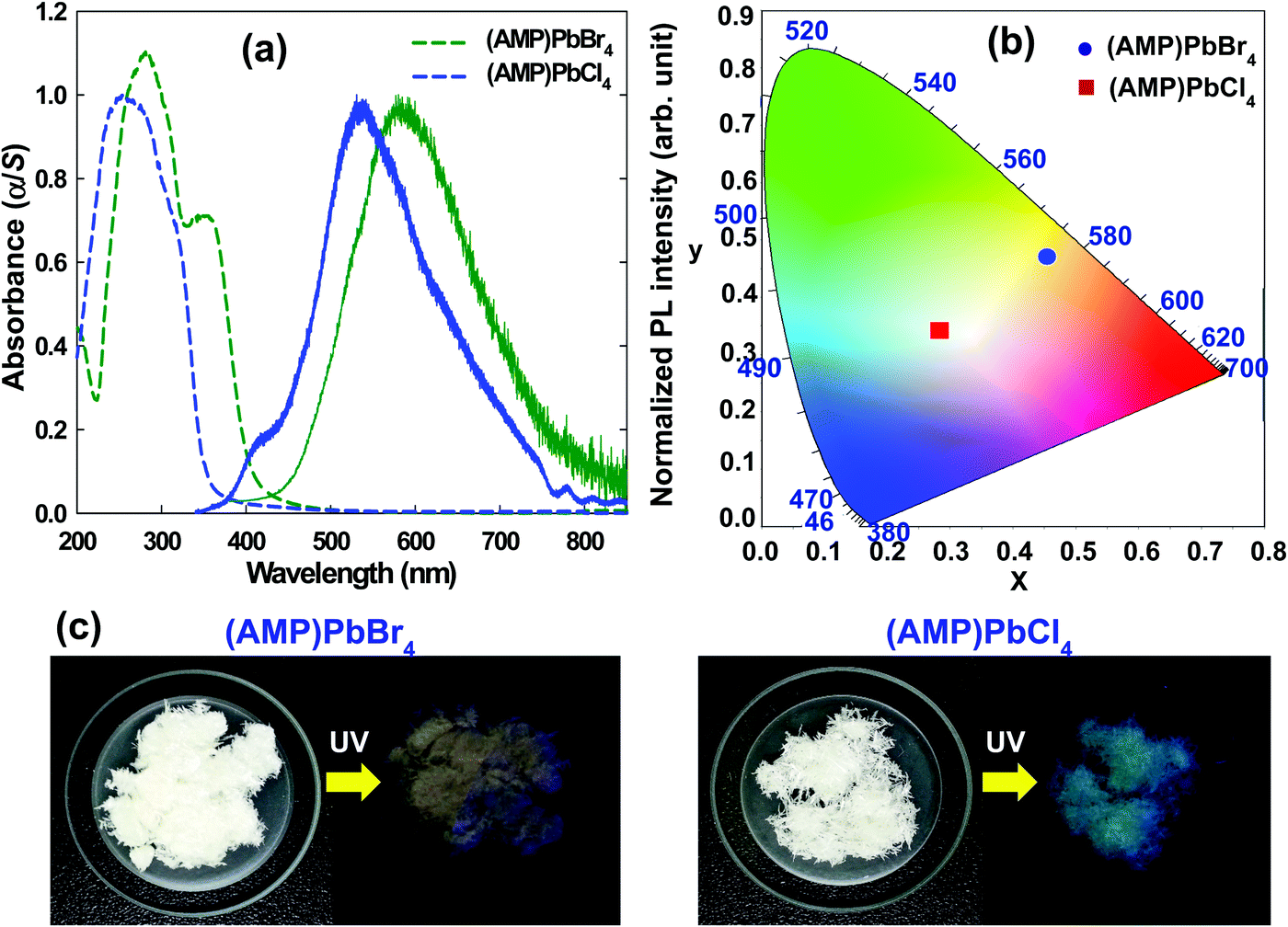
The optical properties of (AMP)PbBr4 and (AMP)PbCl4 were characterized by the UV/vis absorption. As shown in Fig. 8a, an abrupt absorption onset is featured at 412 nm and 358 nm for (AMP)PbBr4 and (AMP)PbCl4, respectively. Upon 325 nm excitation, (AMP)PbBr4 exhibited a broad photoemission covering the entire visible spectrum with a maximum at 581 nm and a large FWHM of 175 nm (0.61 eV).
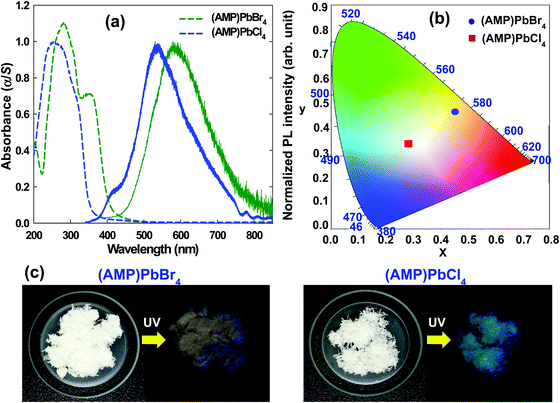 |
| | Fig. 8 (a) A UV-visible absorbance spectra used to determine the band gap for the (AMP)PbBr4 and the (AMP)PbCl4 perovskites (dashed line), and the steady state PL of the (AMP)PbBr4 and the (AMP)PbCl4 perovskites (solid line). (b) CIE color coordination of the (AMP)PbBr4 and the (AMP)PbCl4 perovskites in a 1931 color space chromaticity diagram. (c) The pale orange-light emission from the (AMP)PbBr4 from the powdered polycrystalline sample, and the white-light emission from the single crystal powder of (AMP)PbCl4 under UV light with λex = 365 nm. | |
In contrast to (AMP)PbBr4, (AMP)PbCl4 showed a broad emission with a maximum at 535 nm and an FWHM of 156 nm (0.63 eV) with a high-energy shoulder at 409 nm (Fig. 8a). Based on the previous report,61 the broadband emission and the high-energy shoulder can be attributed to the STEs and free excitons, respectively. The overall light emission of zigzagged 1D perovskites is shown in the Commission Internationale de l'Éclairage (CIE) color coordinate (Fig. 8b). The emission from the (AMP)PbBr4 perovskite has CIE chromaticity coordinates of (0.454, 0.471) with a larger contribution from the red region of the spectrum compared to pure white light, which has chromaticity coordinates of (0.33, 0.33) (Table 3). It gives the emission from (AMP)PbBr4 a correlated color temperature of 3216 K, which corresponds to warm white-light that is almost an ideal value for indoor illumination. The broadband emission also leads to a color-rendering index (CRI) of 74.87. On the other hand, the (AMP)PbCl4 emission was determined to have CIE coordinates of (0.283, 0.342) and a correlated color temperature of 7981 K, which corresponds to cold white light (Table 3). This broad emission leads to a good CRI of 90.21, which is much higher than that of a typical fluorescent light source (approximately 65). The PLQY of (AMP)PbBr4 and (AMP)PbCl4 were measured to be 0.78% and 0.91%, respectively (Table 3). The relatively low PLQYs of the (AMP)PbBr4 and (AMP)PbCl4 perovskites are ascribed to their structural configuration between AMP molecules and inorganic layers. As shown in Fig. 2b, the packing diagram consists of a 1D [PbBr42−]∞ layer stacked parallel to the bc-plane with AMP cations, in which organic cations do not act as spacer between the inorganic layers but coexist with them, forming an ordered laminate structure. The crystal structure of the (AMP)PbCl4 (Fig. 3b) also clearly shows the same feature as that of (AMP)PbBr4. In these structures, it is difficult to expect the quantum confinement effect and quite different from the general 1D perovskite having a high PLQY and long carrier lifetime. Yang et al.62 reported that their manufactured 1D (acridine)Pb2Cl5 perovskite has a high PLQY because the acridine molecules are well restricted and stabilized within the inorganic matrix, leading to the quantum confinement effect. Bo et al.63 synthesized CdCl2-4HP perovskite with PLQY of 63.55% and ultralong carrier lifetime of 103.12 ms due to the 2D confined and ordered laminate structure. Thus, the reason for having low PLQY in the (AMP)PbBr4 and (AMP)PbCl4 perovskites is that the organic molecules are not confined by the inorganic layers, whereas their broadband white-light emissions correlate with intrinsic structural distortion due to the zigzag edge sharing of inorganic layer. The STE generated from a deformable lattice with strong electron–phonon coupling is the main contribution to the broadband emission of (AMP)PbBr4 and (AMP)PbCl4 perovskites. The main difference between the two hybrid lead halide perovskites lies with the intensity of the broadband white-light emission. As shown in Fig. 8c, the (AMP)PbBr4 perovskite emits a pale orange photoluminescence, whereas the (AMP)PbCl4 perovskite emits white-emission under a 365 nm UV light. Thus, to better understand the origin of the white emission from the STE, it is necessary to investigate the structural characteristics, such as perovskite inorganic framework of the corner, edge, and the face sharing of the PbX6 octahedra and the bond length of Pb–X, which affect the crystal lattice distortion to be more deformable under photoexcitation, determining the intensity of the white-light emission.
Table 3 Commission internationale de l'éclairage (CIE) color coordinates (x, y), correlated color temperature (CCT), color purity, color rendering index (CRI), and photoluminescence quantum yield (PLQY) of (AMP)PbBr4 and (AMP)PbCl4 perovskites
| Perovskites |
CIE coordinate (x, y) |
CCT (K) |
Color Purity (%) |
CRI |
PLQY (%) |
|
x
|
y
|
| (AMP)PbBr4 |
0.454 |
0.471 |
3216 |
59.67 |
74.87 |
0.78 |
| (AMP)PbCl4 |
0.283 |
0.342 |
7981 |
91.04 |
90.21 |
0.91 |
4. Conclusions
In this work, the investigation of 1D zigzag edge-sharing perovskite based on (AMP)PbBr4 and (AMP)PbCl4 proved an unusual broadband white-light emission from the intrinsic STE. We have measured the SCXRD for the (AMP)PbBr4 and (AMP)PbCl4 to understand the effect of structural deformability of the lead halide framework by controlling the intensity of broadband white emissions. This measurement revealed that the inorganic lattice of the (AMP)PbCl4 perovskite is more distorted than that of the (AMP)PbBr4, which favored the STE process by lowering the deformation potential and yielding white broadband emissions. A remarkable efficient exciton recombination was observed in (AMP)PbCl4 compared to (AMP)PbBr4 in the TRPL measurement, which was consistent with this result. Specifically, since these compounds are intrinsic deformative structures due to a zigzag connected Pb–Br(Cl) bond, the bandwidth of PL emission is decreased less than the 3D perovskite as the temperature decreases, which suggested a weaker phonon-exciton interaction. This study also indicated an unusual broadband emission in zigzag edge sharing (AMP)PbBr4 and (AMP)PbCl4 perovskites is an intrinsic bulk property. Despite the low PLQY, the high stability of the PL emission in a 1D zigzag edge-sharing perovskite system can underpin a new synthetic design to explore potential white-light solid-state lighting, and it is an important step forward with the practical application of metal halides perovskite in optoelectronics.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1D1A1B04931751). This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2019R1A2C1003108).
References
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, S. Kumar, J. Bär, F. Bertolotti, N. Masciocchi, A. Guagliardi, M. Grotevent, I. Shorubalko, M. I. Bodnarchuk, C.-J. Shih and M. V. Kovalenko, ACS Nano, 2017, 11, 3119–3134 CrossRef CAS PubMed.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk, M. G. Kanatzidis and A. D. Mohite, Nature, 2016, 536, 312 CrossRef CAS PubMed.
- X. Hong, T. Ishihara and A. V. Nurmikko, Phys. Rev. B: Condens. Matter, 1992, 45, 6961–6964 CrossRef CAS.
-
K. S. Song and R. T. Williams, Self-Trapped Excitons, Springer, Berlin, 2nd edn, 1996 Search PubMed.
- T. Hu, M. D. Smith, E. R. Dohner, M.-J. Sher, X. Wu, M. T. Trinh, A. Fisher, J. Corbett, X. Y. Zhu, H. I. Karunadasa and A. M. Lindenberg, J. Phys. Chem. Lett., 2016, 7, 2258–2263 CrossRef CAS.
- E. R. Dohner, E. T. Hoke and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 1718–1721 CrossRef CAS PubMed.
- A. Yangui, D. Garrot, J. S. Lauret, A. Lusson, G. Bouchez, E. Deleporte, S. Pillet, E. E. Bendeif, M. Castro, S. Triki, Y. Abid and K. Boukheddaden, J. Phys. Chem. C, 2015, 119, 23638–23647 CrossRef CAS.
- L. Mao, Y. Wu, C. C. Stoumpos, B. Traore, C. Katan, J. Even, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 11956–11963 CrossRef CAS.
- L. Mao, Y. Wu, C. C. Stoumpos, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 5210–5215 CrossRef CAS.
- L. Dou, A. B. Wong, Y. Yu, M. Lai, N. Kornienko, S. W. Eaton, A. Fu, C. G. Bischak, J. Ma, T. Ding, N. S. Ginsberg, L.-W. Wang, A. P. Alivisatos and P. Yang, Science, 2015, 349, 1518–1521 CrossRef CAS.
- K. Chondroudis and D. B. Mitzi, Chem. Mater., 1999, 11, 3028–3030 CrossRef CAS.
- M. Yuan, L. N. Quan, R. Comin, G. Walters, R. Sabatini, O. Voznyy, S. Hoogland, Y. Zhao, E. M. Beauregard, P. Kanjanaboos, Z. Lu, D. H. Kim and E. H. Sargent, Nat. Nanotechnol., 2016, 11, 872 CrossRef CAS.
- C. C. Stoumpos, D. H. Cao, D. J. Clark, J. Young, J. M. Rondinelli, J. I. Jang, J. T. Hupp and M. G. Kanatzidis, Chem. Mater., 2016, 28, 2852–2867 CrossRef CAS.
- Z. Yuan, C. Zhou, Y. Tian, Y. Shu, J. Messier, J. C. Wang, L. J. van de Burgt, K. Kountouriotis, Y. Xin, E. Holt, K. Schanze, R. Clark, T. Siegrist and B. Ma, Nat. Commun., 2017, 8, 14051 CrossRef CAS PubMed.
- H. Lin, C. Zhou, Y. Tian, T. Siegrist and B. Ma, ACS Energy Lett., 2018, 3, 54–62 CrossRef CAS.
- M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2017, 2, 889–896 CrossRef CAS.
- H. Tanino, W. W. Rühle and K. Takahashi, Phys. Rev. B: Condens. Matter, 1988, 38, 12716–12719 CrossRef CAS PubMed.
- A. Trigui, H. Abid, A. Mlayah and Y. Abid, Synth. Met., 2012, 162, 1731–1736 CrossRef CAS.
- D. B. Mitzi, S. Wang, C. A. Feild, C. A. Chess and A. M. Guloy, Science, 1995, 267, 1473–1476 CrossRef CAS.
- S. Wang, D. B. Mitzi, C. A. Feild and A. Guloy, J. Am. Chem. Soc., 1995, 117, 5297–5302 CrossRef CAS.
- A. C. Véron, A. Linden, N. A. Leclaire, E. Roedern, S. Hu, W. Ren, D. Rentsch and F. A. Nüesch, J. Phys. Chem. Lett., 2018, 9, 2438–2442 CrossRef.
- K. Xiong, W. Liu, S. J. Teat, L. An, H. Wang, T. J. Emge and J. Li, J. Solid State Chem., 2015, 230, 143–148 CrossRef CAS.
- M.-Q. Li, Y.-Q. Hu, L.-Y. Bi, H.-L. Zhang, Y. Wang and Y.-Z. Zheng, Chem. Mater., 2017, 29, 5463–5467 CrossRef CAS.
- W. Zhang, K. Tao, C. Ji, Z. Sun, S. Han, J. Zhang, Z. Wu and J. Luo, Inorg. Chem., 2018, 57, 4239–4243 CrossRef CAS PubMed.
- A. E. Maughan, J. A. Kurzman and J. R. Neilson, Inorg. Chem., 2015, 54, 370–378 CrossRef CAS PubMed.
- G. García-Espejo, D. Rodríguez-Padrón, M. Pérez-Morales, R. Luque, G. de Miguel and L. Camacho, J. Mater. Chem. C, 2018, 6, 7677–7682 RSC.
- G. Wu, C. Zhou, W. Ming, D. Han, S. Chen, D. Yang, T. Besara, J. Neu, T. Siegrist, M.-H. Du, B. Ma and A. Dong, ACS Energy Lett., 2018, 3, 1443–1449 CrossRef CAS.
- J. W. Shin, K. Eom and D. Moon, J. Synchrotron Radiat., 2016, 23, 369–373 CrossRef.
-
Z. Otwinowski and W. Minor, Methods in Enzymology, Academic Press, New York, 1997, vol. 276 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef.
- L. F. Gate, Appl. Opt., 1974, 13, 236–238 CrossRef CAS.
- G. Kortüm, W. Braun and G. Herzog, Angew. Chem., Int. Ed. Engl., 1963, 2, 333–341 CrossRef.
- K. Chondroudis, T. J. McCarthy and M. G. Kanatzidis, Inorg. Chem., 1996, 35, 840–844 CrossRef CAS PubMed.
- T. J. McCarthy and M. G. Kanatzidis, Inorg. Chem., 1995, 34, 1257–1267 CrossRef CAS.
- M.-H. Jung, Inorg. Chem., 2019, 58, 6748–6757 CrossRef CAS PubMed.
- Y. Li, G. Zheng, C. Lin and J. Lin, Solid State Sci., 2007, 9, 855–861 CrossRef CAS.
- X. Fang and D. Yan, Sci. China: Chem., 2018, 61, 397–401 CrossRef CAS.
- D. Yan, H. Yang, Q. Meng, H. Lin and M. Wei, Adv. Funct. Mater., 2014, 24, 587–594 CrossRef CAS.
- R. Gao, X. Fang and D. Yan, J. Mater. Chem. C, 2018, 6, 4444–4449 RSC.
- Z. Zhuang, C. Peng, G. Zhang, H. Yang, J. Yin and H. Fei, Angew. Chem., Int. Ed., 2017, 56, 14411–14416 CrossRef CAS.
- L. Mao, P. Guo, M. Kepenekian, I. Hadar, C. Katan, J. Even, R. D. Schaller, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 13078–13088 CrossRef CAS.
- L. Mao, H. Tsai, W. Nie, L. Ma, J. Im, C. C. Stoumpos, C. D. Malliakas, F. Hao, M. R. Wasielewski, A. D. Mohite and M. G. Kanatzidis, Chem. Mater., 2016, 28, 7781–7792 CrossRef CAS.
- X. Wu, M. T. Trinh, D. Niesner, H. Zhu, Z. Norman, J. S. Owen, O. Yaffe, B. J. Kudisch and X. Y. Zhu, J. Am. Chem. Soc., 2015, 137, 2089–2096 CrossRef CAS PubMed.
- L. Zuo, Q. Chen, N. De Marco, Y.-T. Hsieh, H. Chen, P. Sun, S.-Y. Chang, H. Zhao, S. Dong and Y. Yang, Nano Lett., 2017, 17, 269–275 CrossRef CAS.
- T. Zhao, C.-C. Chueh, Q. Chen, A. Rajagopal and A. K. Y. Jen, ACS Energy Lett., 2016, 1, 757–763 CrossRef CAS.
- P.-W. Liang, C.-Y. Liao, C.-C. Chueh, F. Zuo, S. T. Williams, X.-K. Xin, J. Lin and A. K.-Y. Jen, Adv. Mater., 2014, 26, 3748–3754 CrossRef CAS PubMed.
- B. Yang, J. Chen, F. Hong, X. Mao, K. Zheng, S. Yang, Y. Li, T. Pullerits, W. Deng and K. Han, Angew. Chem., Int. Ed., 2017, 56, 12471–12475 CrossRef CAS PubMed.
- J. Yu, J. Kong, W. Hao, X. Guo, H. He, W. R. Leow, Z. Liu, P. Cai, G. Qian, S. Li, X. Chen and X. Chen, Adv. Mater., 2019, 31, 1806385 Search PubMed.
- R. Zhang, X. Mao, Y. Yang, S. Yang, W. Zhao, T. Wumaier, D. Wei, W. Deng and K. Han, Angew. Chem., Int. Ed., 2019, 58, 2725–2729 CrossRef CAS PubMed.
- J. Lee, E. S. Koteles and M. O. Vassell, Phys. Rev. B: Condens. Matter, 1986, 33, 5512–5516 CrossRef CAS PubMed.
- Z. Y. Xu, Z. D. Lu, X. P. Yang, Z. L. Yuan, B. Z. Zheng, J. Z. Xu, W. K. Ge, Y. Wang, J. Wang and L. L. Chang, Phys. Rev. B: Condens. Matter, 1996, 54, 11528–11531 CrossRef CAS PubMed.
- Y. Wang, T. Gould, J. F. Dobson, H. Zhang, H. Yang, X. Yao and H. Zhao, Phys. Chem. Chem. Phys., 2014, 16, 1424–1429 RSC.
- J. Xing, F. Yan, Y. Zhao, S. Chen, H. Yu, Q. Zhang, R. Zeng, H. V. Demir, X. Sun, A. Huan and Q. Xiong, ACS Nano, 2016, 10, 6623–6630 CrossRef CAS PubMed.
- X. Yang and D. Yan, Chem. Commun., 2017, 53, 1801–1804 RSC.
- A. Biswas, R. Bakthavatsalam, S. R. Shaikh, A. Shinde, A. Lohar, S. Jena, R. G. Gonnade and J. Kundu, Chem. Mater., 2019, 31, 2253–2257 CrossRef CAS.
- Y. Wu, C. Wei, X. Li, Y. Li, S. Qiu, W. Shen, B. Cai, Z. Sun, D. Yang, Z. Deng and H. Zeng, ACS Energy Lett., 2018, 3, 2030–2037 CrossRef CAS.
- N. Ashari-Astani, S. Meloni, A. H. Salavati, G. Palermo, M. Grätzel and U. Rothlisberger, J. Phys. Chem. C, 2017, 121, 23886–23895 CrossRef CAS.
- E. R. Dohner, A. Jaffe, L. R. Bradshaw and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 13154–13157 CrossRef CAS PubMed.
- X. Yang, L.-F. Ma and D. Yan, Chem. Sci., 2019, 10, 4567–4572 RSC.
- B. Zhou and D. Yan, Angew. Chem., Int. Ed., 2019, 58, 15128–15135 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 

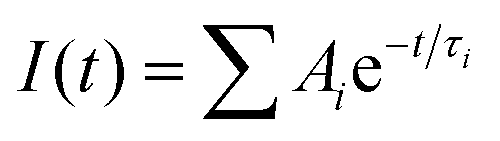 , where I(t) is the time-dependent PL intensity, A is the amplitude, and τ is the PL lifetime.
, where I(t) is the time-dependent PL intensity, A is the amplitude, and τ is the PL lifetime.

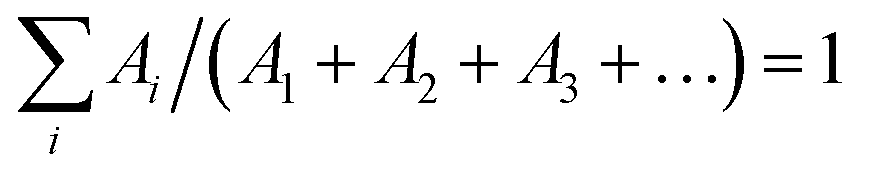 , the average lifetime, 〈τ〉, is defined by
, the average lifetime, 〈τ〉, is defined by 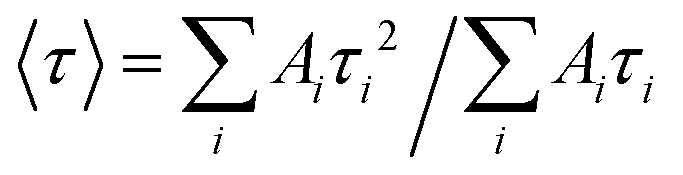 and τamp is the amplitude of average lifetime.
and τamp is the amplitude of average lifetime.