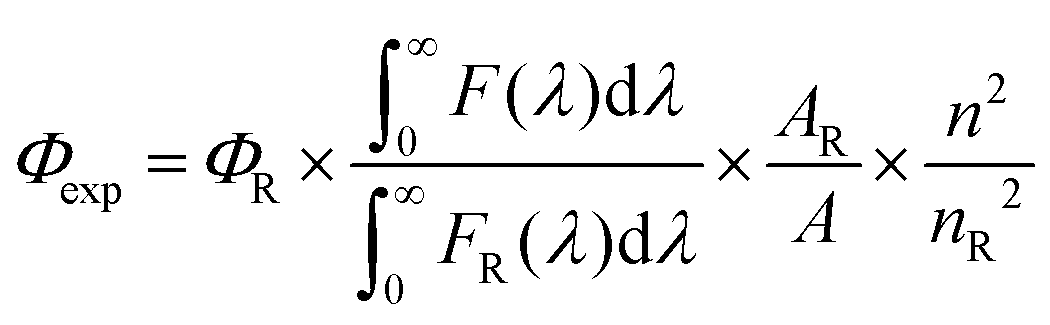DOI:
10.1039/C9NJ04612G
(Paper)
New J. Chem., 2020,
44, 29-37
Thieno[1,3,2]oxazaborinine-containing aza-BODIPYs with near infrared absorption bands: synthesis, photophysical properties, and device applications†
Received
7th September 2019
, Accepted 29th October 2019
First published on 30th October 2019
Abstract
As part of an ongoing study of near infrared (NIR) absorbing dyes applicable to optoelectronics, thieno[1,3,2]oxazaborinine-containing aza-BODIPYs were synthesized for the first time. 3,5-Di(thiophene)-substituted N2O2-type 1 showed a NIR absorption band centered at 780 nm, with a molar extinction coefficient (εmax) of 5.51 × 104 M−1 cm−1 in CH2Cl2; however, it was easily hydrolyzed in solution. In comparison, 3-thiophene-containing N2O-type analogues 2 and 3 had greater stability, and NIR absorption bands at 768 nm (εmax = 8.62 × 104 M−1 cm−1) and 779 nm (εmax = 6.32 × 104 M−1 cm−1), respectively, in CH2Cl2. Notably, these dyes had longer absorption bands than those of all of the structurally constraining aza-BODIPYs reported thus far. CV measurements and theoretical calculations indicated that this shift was the result of a higher-lying HOMO energy level generated by incorporation of thiophene into the aza-BODIPY core. As a potential device application, the 3-loaded film was prepared on indium tin oxide (ITO). The film showed a NIR absorption band at 827 nm, with a wide spectral range, and a λonset value of 897 nm; thus, its applicability as a NIR photodetector was evaluated through fabrication of a single-component device. The ITO/3 (85 nm)/Al (100 nm) device produced a photocurrent of 9.57 × 10−7 A cm−2 at a bias potential of 1 V upon illumination at 850 nm and a fluence of 130 μW cm−2. This demonstrated the potential of thieno[1,3,2]oxazaborinine-containing aza-BODIPY materials in optoelectronic applications such as NIR photodetectors.
Introduction
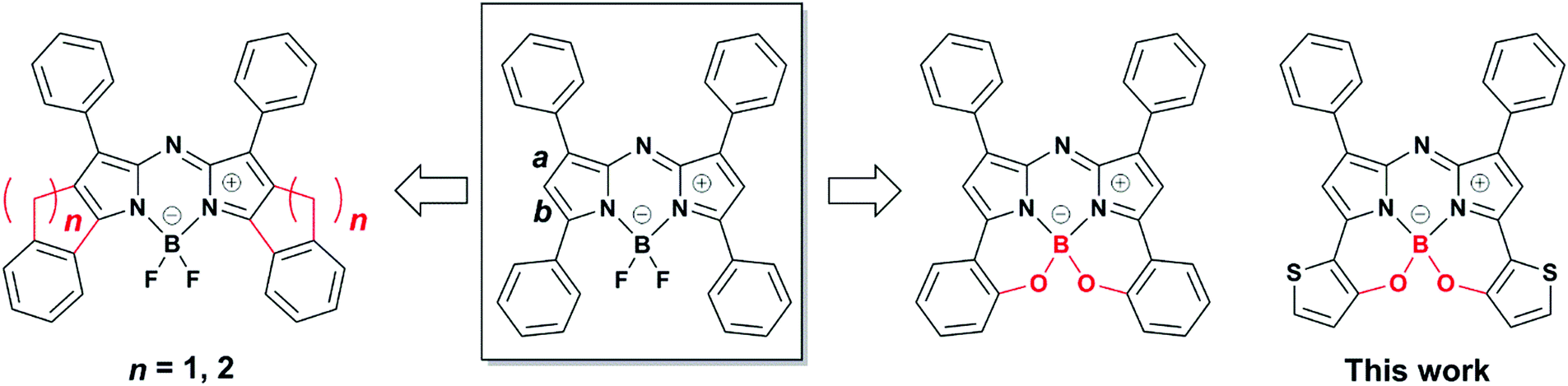
Aza-boron-dipyrromethene complexes (aza-BODIPYs)1 have been recognized as promising candidates for near infrared (NIR) absorbing dyes.2–5 Replacement of the C atom at the meso position of BODIPY6 with a N atom leads to a significant, ≈100 nm, bathochromic shift in the absorbance and fluorescence spectra of the dye. This feature is advantageous for numerous applications in optoelectronics,7 photovoltaics,8–10 probes in biology11 such as in vivo imaging,12 photodynamic therapy13 and chemosensing.14,15 Various synthetic strategies for dye core modification have enabled extensions of π-conjugation to produce [a]-benzene-fused,16,17 [b]-thiophene-fused18 and phenanthrene-fused19,20 aza-BODIPYs with a NIR region. Additionally, a number of NIR dyes have been produced through the introduction of electron donor or acceptor groups at the 1,7 and/or 3,5-positions,21–23 with a few examples of dyes synthesized from electron-donating groups introduced at the 2,6-positions.24 Specifically, substitution of thiophene at the 3,5-positions leads to effective spectral perturbation in the form of a redshift,25–27 which is the result of a high-lying HOMO energy level.27 Notably, a conjugated polymer composed of 1,3,5,7-tetra-thiophene-substituted aza-BODIPY showed absorption in the deep near infrared region.28 With this knowledge, our research has focused on the structural constraint of 3,5-thiophene rings through intramolecular B–O chelation. Past reports involving cyclic fixation of 3,5-aryl moieties through the formation of bonds with β-pyrrole carbons29–33 or chelation with boron centers34–36 have been proposed to produce redshifts in the absorption bands (Fig. 1). However, to the best of our knowledge, aza-BODIPYs substituted with thieno[1,3,2]oxazaborinine are yet to be reported.
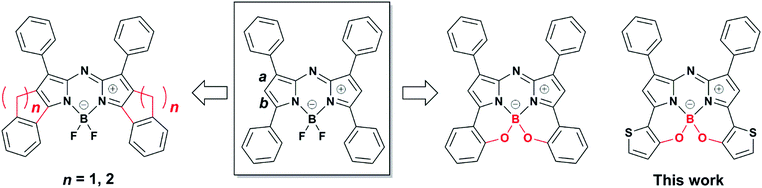 |
| | Fig. 1 Synthesis strategy for structural constraint of aza-BODIPYs. | |
Our continuous efforts to prepare NIR dyes for application in optoelectronics37 have motivated us to investigate intramolecular B–O chelation of 3,5-dithiophene-substituted aza-BODIPYs. Currently, organic NIR dyes applicable to solar cells, photodetectors, and various electronic devices have been extensively studied.38 Notably, photodetectors that can translate incident NIR photons to an electrical signal have attracted considerable attention due to their tremendous applications involving image sensing, optical communication, night surveillance, etc.39,40 To this end, development of sensitive photodetectors capable of capturing very weak NIR signals are necessary. Narrow-band gap π-conjugated polymers that serve as organic semiconductors make it possible to achieve high-performance photodetectors with spectral responses.41–46 Given the advantages of small molecules, including their high purities and well-defined molecular structures, it is worthwhile to investigate NIR dyes for such applications. In this work, the synthesis and characterization of thieno[1,3,2]oxazaborinine-containing aza-BODIPYs 1–3 are reported (Fig. 2). These new dyes have λmax values in the NIR range of 768–780 nm in CH2Cl2, with significant redshifts relative to their 3,5-benzo[1,3,2]oxazaborinine type counterpart, BO1.34 Among these, 3-thiophene-containing N2O–boron chelate analogue 3 was isolated as a stable NIR dye and investigated for possible applicability as an organic photodetector.38 The 3-containing film absorbed NIR light at 827 nm, had a long spectral width, and a λonset value of 897 nm. As a preliminary result, the related indium tin oxide (ITO)/3/Al device allowed photocurrent detection under light irradiation at 850 nm (130 μW cm−2), which demonstrated for the first time that an aza-BODIPY-based NIR dye could serve as an organic NIR photodetector for application in optoelectronics. In this way, a new family of B–O constrained aza-BODIPYs were introduced for use as NIR materials.
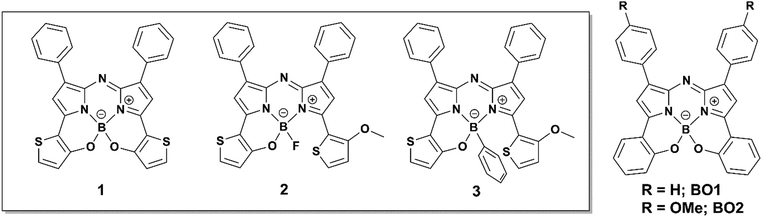 |
| | Fig. 2 Chemical structures of thieno[1,3,2]oxazaborinine-containing aza-BODIPYs and related dyes, BO1 and BO2. | |
Experimental section
General
NMR spectra were measured on a Bruker Avance 500 spectrometer (1H: 500 MHz, 13C: 126 MHz, 11B: 160 MHz, and 19F: 471 MHz). In 1H and 13C NMR measurements, chemical shifts (δ) are reported downfield from the internal standard Me4Si. BF3·OEt2 and C6F6 were used as the external standard for 11B NMR and internal standard for 19F NMR, respectively. Mass spectrometry data were obtained by using a JEOL JMS-700 fast atom bombardment (FAB) spectrometer where m-nitrobenzylalcohol was used as a matrix or a Bruker micrOTOF II-SDT1 spectrometer with atmospheric pressure chemical ionization (APCI) method. The absorption and fluorescence were measured using a Shimadzu UV-3600 UV/vis/NIR spectroscope and a JASCO FP-6500 spectrofluorometer, respectively. Elemental analyses were performed on an Exeter Analytical, Inc. CE-440F elemental analyzer. Ionization potential data were measured using a Riken photoelectron spectroscopy instrument, AC-3. An AFM-5400 (Hitachi High-Technologies Corporations) atomic force microscope (AFM) was used in dynamic force mode using silicon probes with a resonant frequency of 70 kHz and a force constant of 2 N m−1.
Materials
Reagents used for the synthesis were commercially available and used as supplied. Dry CH2Cl2 and dry toluene were prepared according to standard procedures. 3,5-Bis(o-methylthiophene)-substituted aza-BODIPY 4 was synthesized in line with the reaction path in Scheme S1 (ESI†). 3,5-Benzo[1,3,2]oxazaborinine type counterpart BO1 was synthesized by reference to the method reported previously.34
Synthesis
Boron chelated [5-(3-hydroxythiophen-2-yl)-3-phenyl-1H-pyrrol-2-yl]-[5-(3-hydroxythiophen-2-yl)-3-phenyl-2H-pyrrol-2-ylidene]amine 1.
To a solution of 4 (296 mg, 0.52 mmol) in dry CH2Cl2 (75 mL) was added 1 M BBr3 (2.5 mL, 2.5 mmol) under ice cooling conditions. The mixture was stirred for 1 h under ice cooling conditions and was then stirred at room temperature. After that the solution was poured into a saturated aqueous NaHCO3 solution and the organic layer was washed with water and evaporated. The residue was washed with water, CH2Cl2 and hexane in a step-by-step manner. 235 mg of the resultant solid was obtained as a dark green solid, being assignable to crude aza-dipyrrin 5 (FAB-MS: m/z = 493 [M]+). To a solution of the solid (235 mg) in dry toluene (50 mL) was added B(OMe)3 (0.53 mL, 4.74 mmol). The mixture was stirred at 100 °C. After that the solution was poured into hexane, and the precipitated solid was dissolved in CH2Cl2. The solution was filtrated with silica gel, evaporated, and dispersed in hexane. In this way, 21.6 mg of 1 was obtained with a 8% yield from 4. 1H NMR (500 MHz, CDCl3): δ (ppm) 8.14 (4H, d, J = 7.25 Hz), 7.49 (2H, d, J = 5.12 Hz), 7.47 (4H, t, J = 7.57 Hz), 7.40 (2H, tt, J = 7.33 and 1.49 Hz), 7.01 (2H, s), 6.85 (2H, d, J = 5.28 Hz). 13C NMR (126 MHz, CDCl3): δ (ppm) 159.8, 146.0, 144.4, 141.0, 132.5, 130.7, 129.1, 128.9, 128.7, 120.8, 112.1, 77.9. 11B NMR (CDCl3, 160 MHz): δ (ppm) −0.104 (s). APCI-MS: m/z = 502 [M + H]+; elemental analysis: calcd for C28H16BN3O2S2·0.25H2O: C, 66.48; H, 3.29; N, 8.31, found: C, 66.37; H, 3.22; N, 8.33.
Fluoroboron chelated [5-(3-hydroxythiophen-2-yl)-3-phenyl-1H-pyrrol-2-yl]-[5-(3-methoxythiophen-2-yl)-3-phenyl-2H-pyrrol-2-ylidene]amine 2.
To a solution of 4 (1.22 g, 2.15 mmol) in dry CH2Cl2 (300 mL) under a N2 atmosphere was added 1 M BBr3 solution (10.0 mL, 10.0 mmol). The resulting mixture was stirred for 10 min under ice cooling conditions. After quenching by adding saturated aqueous NaHCO3 solution, the resulting organic layer was washed with water and was almost evaporated. The residue was then dispersed in hexane. The collected solid was dissolved in dry toluene (200 mL). To the solution was added DIPEA (3.8 mL, 22.4 mmol). The solution was stirred for 30 min at room temperature under a N2 atmosphere. After adding BF3·OEt2 (3.8 mL, 30.79 mmol) dropwise to the solution, the resulting mixture was refluxed overnight. The reaction was quenched by saturated aqueous NaHCO3 solution, extracted with toluene and chloroform, washed with water, and evaporated. After washing with MeOH, the residue was chromatographed on silica gel (Wakogel C-300) using CHCl3 as an eluent and reprecipitated with CH2Cl2 and hexane to give 2 (0.672 g, 62%) as a brown solid. Further purification was thus achieved by reprecipitation with CH2Cl2 and hexane because a little contamination was present. 1H NMR (500 MHz, CDCl3): δ (ppm) 8.17–8.14 (2H, m), 8.07–8.05 (2H, m), 7.69 (1H, s), 7.58 (1H, d, J = 5.28 Hz), 7.49 (1H, d, J = 5.52 Hz'), 7.48–7.43 (4H, m), 7.41–7.37 (2H, m), 6.98 (1H, d, J = 5.60 Hz), 6.97 (1H, d, J = 5.28 Hz'), 6.90 (1H, s), 4.06 (3H, s). 13C NMR (126 MHz, CDCl3): δ (ppm) 161.5, 159.9, 146.0, 145.8, 145.0, 143.0, 142.7, 139.5, 133.3, 132.4, 132.2, 129.4, 129.3, 128.7, 128.7, 128.4, 128.4, 128.2, 120.8, 119.1, 115.2, 111.7, 111.6, 110.5, 59.0. 11B NMR (CDCl3, 160 MHz): δ (ppm) 0.674 (d, J = 46.5 Hz). 19F NMR (CDCl3, 471 MHz): δ (ppm) 26.4 (q, J = 46.5 Hz). APCI-MS: m/z = 536 [M + 1]+. Elemental analysis: calcd for C29H19BFN3O2S2·0.3CH2Cl2: C, 62.74; H, 3.52; N, 7.49. Found: C, 62.79; H, 3.56; N, 7.52.
[5-(3-Hydroxythiophen-2-yl)-3-phenyl-1H-pyrrol-2-yl]-[5-(3-methoxythiophen-2-yl)-3-phenyl-2H-pyrrol-2-ylidene]amine 6.
To a solution of 4 (0.102 g, 0.19 mmol) in dry CH2Cl2 (25 mL) under a N2 atmosphere was added 1 M BBr3 solution (0.88 mL, 0.88 mmol). The resulting mixture was stirred for 10 min under ice cooling conditions. After quenching by adding saturated aqueous NaHCO3 solution, the resulting organic layer was washed with water. After evaporation, the residue was chromatographed on silica gel (Wakogel C-300) using CH2Cl2 as an eluent to give 6 as a red-purple solid as well as 2 as a brown solid with yields of 74% and 9%, respectively. For 6, 1H NMR (500 MHz, CDCl3): δ (ppm) 10.30 (1H, br. s), 8.03–8.01 (2H, m), 7.93 (2H, d, J = 7.57 Hz), 7.68 (1H, d, J = 5.44 Hz), 7.42–7.41 (3H, m), 7.37 (2H, t, J = 7.68 Hz), 7.29 (1H, t, J = 6.93 Hz), 7.20 (1H, d, J = 5.36 Hz), 7.08 (1H, s), 6.96 (1H, d, J = 5.44 Hz), 6.77 (1H, s), 6.72 (1H, d, J = 5.52 Hz,), 4.15 (3H, s). 13C NMR (126 MHz, CDCl3): δ (ppm) 200.0, 156.9, 149.2, 147.7, 146.3, 137.0, 134.4, 134.3, 132.4, 132.0, 131.6, 129.6, 129.0, 128.8, 128.3, 128.2, 127.1, 123.6, 122.2, 121.6, 116.4, 111.5, 107.8, 107.7, 59.6. APCI-MS: m/z = 508 [M + 1]+.
Phenylboron chelated [5-(3-hydroxythiophen-2-yl)-3-phenyl-1H-pyrrol-2-yl]-[5-(3-methoxythiophen-2-yl)-3-phenyl-2H-pyrrol-2-ylidene]amine 3.
Aza-dipyrrin 6 (101 mg, 0.20 mmol) and phenylboronic acid (51.3 mg, 0.42 mmol) were dissolved in CH2Cl2 (30 mL). The resulting mixture was refluxed for 2 days. After silica gel filtration, the solvent was removed in vacuo. The resultant residue was treated with hexane to give 3 (90.4 mg) with a yield of 76%. 1H NMR (500 MHz, CDCl3): δ (ppm) 8.17–8.15 (2H, m), 8.13–8.11 (2H, m), 7.59 (1H, d, J = 5.20 Hz), 7.56 (1H, s), 7.49–7.43 (4H, m), 7.41 (1H, d, J = 5.60 Hz), 7.40–7.37 (2H, m), 7.14 (2H, dd, J = 7.56 and J = 1.82 Hz), 7.04 (1H, d, J = 5.20 Hz), 7.03–6.99 (3H, m), 6.86 (1H, d, J = 5.44 Hz), 6.82 (1H, s), 3.87(3H, s); 13C NMR (126 MHz, CDCl3): δ (ppm) 162.6, 160.4, 146.7, 145.9, 145.3, 143.3, 142.2, 139.0, 133.6, 133.1, 132.4, 131.4, 129.3, 129.1, 128.7, 128.6, 128.3, 128.2, 128.1, 127.0, 126.8, 120.5, 119.6, 115.3, 112.6, 111.9, 110.4, 59.0. 11B NMR (CDCl3, 160 MHz): δ (ppm) 2.77 (s). APCI-MS: m/z = 594[M + H]+. Elemental analysis: calcd for C35H24BN3O2S2·1H2O: C, 68.74; H, 4.29; N, 6.87. Found: C, 68.96; H, 4.12; N, 6.89.
Determination of fluorescence quantum yield
The fluorescence quantum yield (Φexp) was calculated using eqn (1).3| | 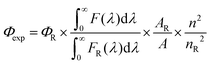 | (1) |
where F(λ) and FR(λ) describe the measured fluorescence intensities of the dye and the reference, respectively, and A and AR describe the corresponding absorbances at the excitation wavelength. The reference used was indocyanine green (ΦR = 0.23 in DMSO)47 for 1–3 when excited at 730 nm and zinc phthalocyanine (ΦR = 0.25 in THF)48 for 4 and BO1 when excited at 650 nm. The refractive indexes are n = 1.407 for THF, n = 1.424 for CH2Cl2 and n = 1.479 for DMSO, respectively.
Theoretical calculation
All geometries of the dyes at the ground state were fully optimized by means of the B3LYP/6-31G(d,p) level method, and density functional theory (DFT) calculations at the B3LYP/6-31G(d,p) level were performed in the Gaussian 09 package49 (Gaussian 09. Rev C.01). The molecular orbitals in Fig. 4 were visualized using the Gauss view 6.0.16 program.
Device fabrication
The dye films with a thickness of 85 nm were formed on the surface of the indium tin oxide (ITO) glass substrate using a resistance heating type vacuum deposition method. Subsequently, the devices were completed by evaporating a 100 nm thick aluminum film as an electrode. Current–voltage (J–V) measurements of the dye-loaded devices were performed using a Keithley 4200-SCS source meter under irradiation at 850 nm (130 μW cm−2) by means of a PVL-3300 (Asahi Spectra Co., Ltd) for the device with 3.
Results and discussion
Synthesis
The key synthon, 3,5-bis(o-methoxythiophene)-substituted aza-BODIPY 4, was prepared by a straightforward reaction of 3-methoxythiophene involving a chalcone intermediate (Scheme S1, ESI†).
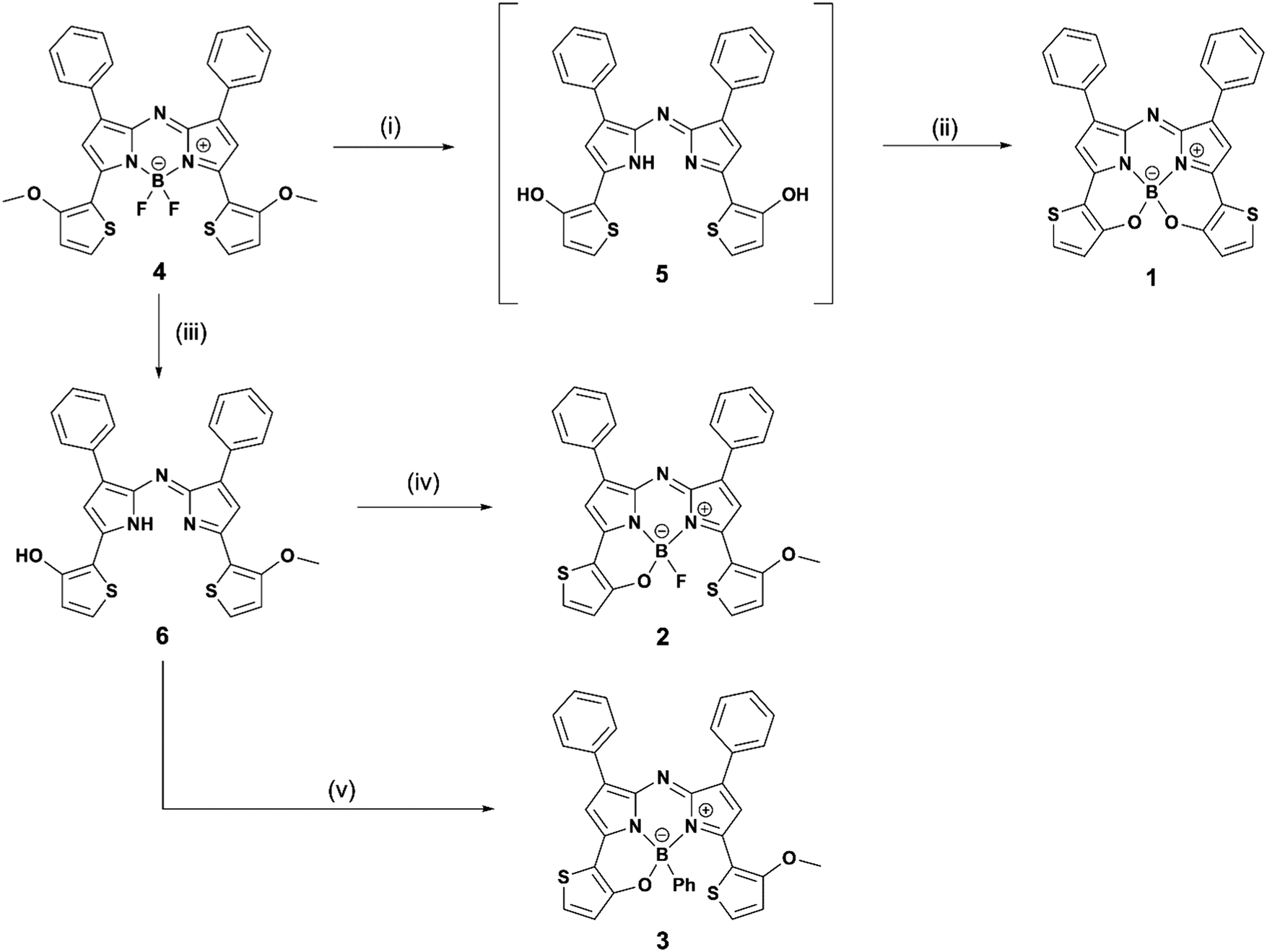
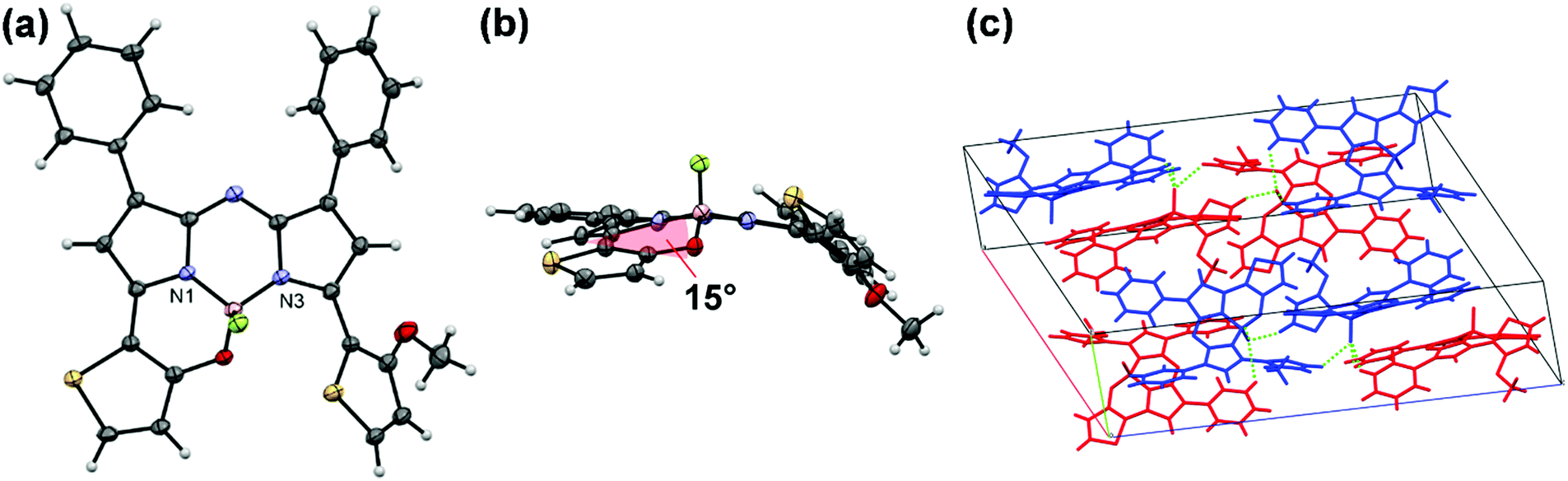
An initial reaction, involving intramolecular B–O chelation of 4 with BBr3 in dry CH2Cl2 at room temperature, was attempted to obtain 1 (Scheme 1). After quenching with saturated aqueous NaHCO3 solution, work-up of the reaction never produced the target dye. Instead, various products involving in situ generated aza-dipyrrin 5 (m/z = 493 [M]+) were isolated. This may have been due to the instability of 1 as the result of hydrolysis under the acidic conditions generated by HBr produced during the reaction. To avoid this, a two-step one-pot reaction involving demethylation with BBr3, followed by boron coordination using B(OMe)3, was conducted. Assuming that 1 would be easily hydrolyzed, a work-up of the reaction was carried out without washing or column purification. Instead, reprecipitation of the resultant sample with CH2Cl2 and hexane enabled isolation of pure 1 from 4 in a yield of 8%. To gain insight into the water-sensitivity of 1, time-course measurements of absorption spectra in water-saturated CH2Cl2 were carried out (Fig. S1, ESI†). The absorption band with a λmax of 780 nm decreased as time passed, whereas new bands at 664 nm and 730 nm appeared with isosbestic points at 747 nm, suggesting that the transformation of 1 to the corresponding aza-dipyrrin 5 occurred via hydrolysis.50 Once the water-sensitivity of 1 was determined to hamper the efficiency of the work-up of the synthetic reaction, an alteration of target dyes was made. As a result, demethylation of 4 with BBr3 was conducted for only 10 min in an ice bath, followed by boron coordination by BF3·OEt2 in the presence of diisopropylethylamine (DIPEA) to give monothiophene-involved B–O constrained 2 following chromatography, with 2 identified analytically. An assessment of BBr3-induced demethylation for 10 min in an ice bath enabled the detection of 2 and 1-(o-hydroxythiophene)-9-(o-methoxythiophene)-substituted aza-dipyrrin 6, in 9% and 74% yields, respectively. The facile isolation of 6 allowed the preparation of a corresponding 3 analogue bearing a phenyl on boron by reacting 6 with phenylboronic acid (76% yield). Unlike 1, thiophene-containing N2O-type aza-BODIPYs 2 and 3 were stable in solution. Characterization of these dyes was conducted by 1H and 13C NMR spectroscopies, mass spectrometry, and elemental analysis. A single crystal of 2 was obtained by the solvent evaporation technique of CHCl3/hexane, which had a monoclinic crystal system in the I2/a space group. The structure was analyzed by X-ray crystallography (CCDC No. 1948374†). The Oak Ridge thermal ellipsoid plot (ORTEP) diagram is shown in Fig. 3a. Boron binds to the two nitrogen atoms of the pyrrole rings, the thiophene-linked oxygen, and a fluorine atom, to form a tetrahedral geometry. The N1–B–N3 and O–B–N1 bite angles are 104.06 and 108.19°, respectively, and the B–O distance is 1.470 Å. These data are similar to those of the 3,5-bis(benzo[1,3,2]oxazaborinine) aza-BODIPY derivative BO2, previously reported by Burgess, et al.34 The thiophene ring was found to be tilted with respect to the aza-BODIPY plane by 15° (Fig. 3b). This value is half as long as that of BO2. This flattening of the structure may be advantageous not only to its stability against hydrolysis but also its increase in ε relative to 1, as described in the discussion of photophysical properties in Fig. 4 (vide infra). Fig. 3c shows the packing structure of 2, where two dyes are present as an enantiomer pair in the I2/a space group to form a dimer in which a fluorine atom at the boron center formed intermolecular hydrogen bonds with phenyl-H at the 1,7 positions (C–H⋯F distances of 2.622 Å and 2.624 Å) and methoxythiophene-H (C–H⋯F distance = 2.437 Å) of an adjacent dye with the same enantiomeric structure.
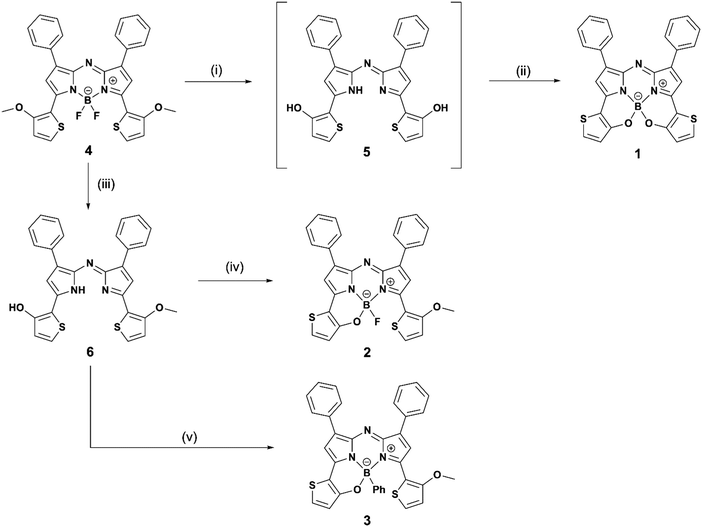 |
| | Scheme 1 Synthesis of target dyes (1–3). Reaction conditions: (i) 1 M BBr3 in dry CH2Cl2, and 1 h ice bath followed by room temperature for 14 h; (ii) B(OMe)3 in dry toluene, 100 °C, and overnight; (iii) 1 M BBr3, and 10 min ice bath; (iv) BF3·OEt2 and DIPEA in dry toluene; and (v) phenylboronic acid in CH2Cl2. | |
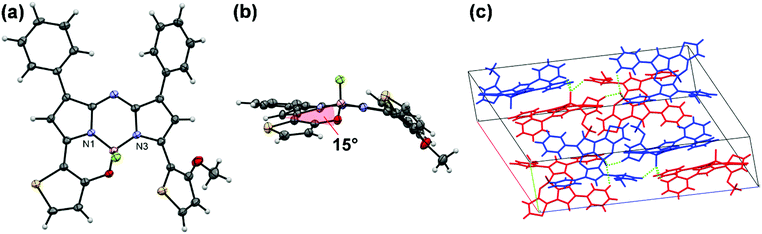 |
| | Fig. 3 ORTEP structures of 2, where thermal ellipsoids are drawn at the 50% probably levels. Front view (a), side view (b), and packing structure (c). | |
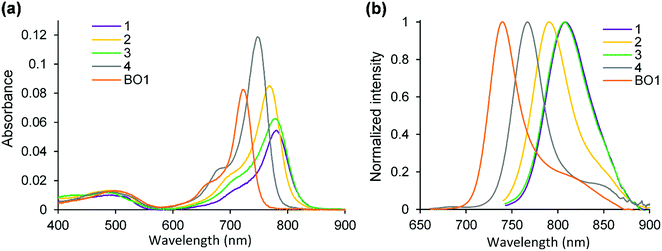 |
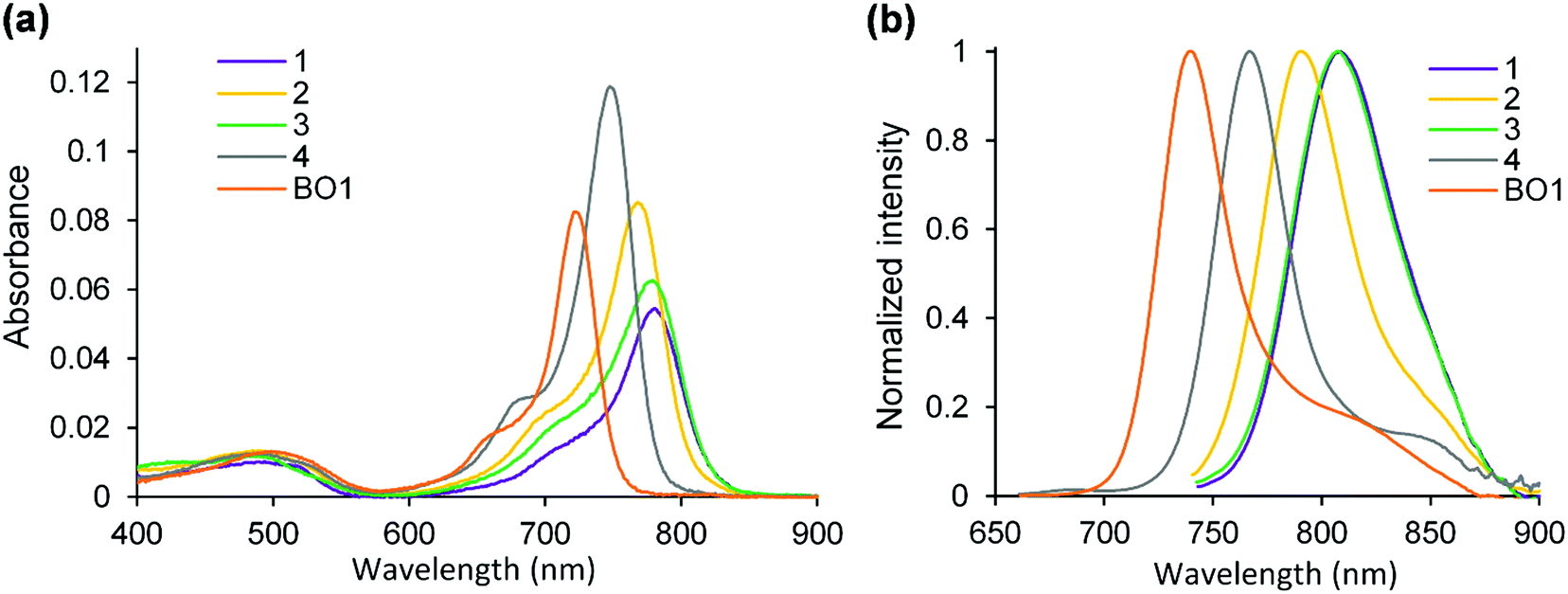
| | Fig. 4 Absorption (a) and fluorescence (b) spectra of dyes in CH2Cl2. λex = 650 nm for 4 and BO1. λex = 730 nm for 1–3. | |
Photophysical properties
Fig. 4 and Table 1 summarize the series of dyes prepared in this work, with 3,5-benzo[1,3,2]oxazaborinine type dye, BO1, as a reference. Target 1 absorbed NIR light at 780 nm, with a molar extinction coefficient (εmax) of 5.51 × 104 M−1 cm−1, and a bathochromic shift of 32 nm relative to 4 as a result of intramolecular B–O chelation. Comparatively, the absorption band of 1 was redshifted by 59 nm relative to that of BO1, due to replacement of the benzo[1,3,2]oxazaborinine unit with its corresponding thiophene counterpart. This structural perturbation led to a change in the shape of the absorption band, with 1 having a longer spectral width ranging from 670 to 820 nm. The shoulder peak observed in the range of 700–750 nm is related to a vibrational mode S0–S1 transition. The mono(thiophene)-containing N2O–boron chelated dye, 2, was found to absorb NIR light at 768 nm (εmax = 8.62 × 104 M−1 cm−1), suggesting that a significant bathochromic shift in the absorption band was attained even by introducing one thiophene B–O chelate in the aza-BODIPY core. It should be noted that 3, containing a phenyl unit at the boron, showed a relatively longer absorption band, centered at 779 nm with a λonset value of 820 nm, being close to the λmax value of 1. The εmax value of the absorbance (6.32 × 104 M−1 cm−1) was somewhat larger than that of 1. Unexpectedly, replacement of the fluorine with a phenyl unit led to a redshift of 11 nm in the absorption band, which contradicted the trend for diallyl aza-BODIPYs, reported previously.51 This interpretation was made based on the electrochemical measurements discussed later (electrochemistry section). We found that the absorption band of 3 was insensitive to solvent polarity; almost no shifts in the absorption or fluorescence spectra were observed for various solvents (Fig. S2a, ESI†). Use of a Lippert–Mataga plot52 allowed determination of a transition moment (D) of 2.07 Debye (Fig. S2b, ESI†), suggesting that 3 has a typical π–π* transition.
Table 1 UV-vis absorption properties and emission properties of the dyes (1 μM) in CH2Cl2
| Dye |
λ
max/nm |
ε/104 M−1 cm−1 |
λ
abs onset/nm |
λ
em
/nm |
Φ
f
|
|
λ
ex = 650 nm for 4 and BO1. λex = 730 nm for 1–3.
|
|
1
|
780 |
5.51 |
822 |
808 |
0.04 |
|
2
|
768 |
8.62 |
805 |
791 |
0.07 |
|
3
|
779 |
6.32 |
820 |
808 |
0.02 |
|
4
|
748 |
11.8 |
780 |
767 |
0.17 |
|
BO1
|
721 |
8.29 |
751 |
740 |
0.25 |
Fluorescence spectra (Fig. 4b) showed a typical mirror-image relationship53 for 2 and 4. The fluorescence maxima of the thiophene B–O chelated dyes were redshifted with reference to that of BO1 by 51–68 nm. Subsequently, 1 and 3 emitted NIR light at 808 nm. However, their fluorescence quantum yields were lower (Φf < 0.05) than that of BO1 (Φf = 0.25), possibly due to increased nonradiative deactivation as the result of a decreased energy gap.2 Interestingly, the spectral shape of 3 was consistent with that of 1, supporting the idea that the photophysical properties observed are mainly due to the presence of at least one thieno[1,3,2]oxazaborinine in the dye structure.
Electrochemistry
The electrochemical properties of the dyes were investigated using cyclic voltammetry (CV). The formal potential of Fc/Fc+ (E1/2Fc/Fc+) was 0.191 V versus Ag/Ag+. Two reversible reduction waves were observed with half-wave potentials (Ered1/2) of ≈−1.7 and −1.0 V, with an irreversible oxidation wave also detected (Fig. S3, ESI†). A summary of the data is presented in Table 2. As a result of the electron-donor properties of thiophene, incorporation of thiophene units in the dye cores led to reduced oxidation and first reduction potentials compared to those of BO1. Additionally, by varying the substituent groups at the 3,5-positions, oxidation potentials for the thiophene-containing dyes (1–4) were more sensitive than those of reduction. Further analysis of the CV and absorption data enabled determination of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels (Table 2).54 It was found that the HOMO levels were energetically higher in the following order: 3 > 1 > 2 > 4. Higher-lying HOMO energy levels led to smaller HOMO–LUMO band gaps (ΔE), resulting in bathochromic shifts in the absorption bands (Table 1). Unexpectedly, the LUMO energy level of 3 had the highest value; this can be interpreted as the replacement of the fluorine with an aryl substitution group on the boron creating instability in the LUMO level.51 However, the significant high-lying of the corresponding HOMO energy level resulted in a smaller ΔE value relative to 2. This insight rationalized the replacement of the fluorine with a phenyl substituent on the boron which led to a redshift in the absorption band (Table 1). It is interesting to note that the substitution effect on the boron was opposite to that of conventional 1,3,5,7-tetraphenyl aza-BODIPYs.51 It is thought that steric hindrance between the thieno[1,3,2]oxazaborinine unit and the phenyl substituent on the boron leads to significant perturbation of the chromophore system.
Table 2 Electrochemical properties of the dyes in CH2Cl2
|
|
E
IIred
(1/2)/V |
E
red(1/2)/V |
λ
onset
/eV |
E
LUMO
/eV |
E
HOMO
/eV |
|
Optical band gap (Eg) was estimated from the onset wavelength in the absorption spectra measured in CH2Cl2.
E
LUMO [eV] = −(4.8 + Ered(1/2) [V vs. Fc/Fc+]).
HOMO = LUMO − Eg(eV).
The peak was too broad to determine the value.
|
|
1
|
—d |
−1.01 |
1.51 |
−3.79 |
−5.30 |
|
2
|
−1.69 |
−0.99 |
1.54 |
−3.81 |
−5.35 |
|
3
|
−1.78 |
−1.08 |
1.51 |
−3.72 |
−5.24 |
|
4
|
−1.71 |
−0.97 |
1.59 |
−3.83 |
−5.42 |
|
BO1
|
−1.65 |
−0.91 |
1.65 |
−3.89 |
−5.54 |
Theoretical analysis
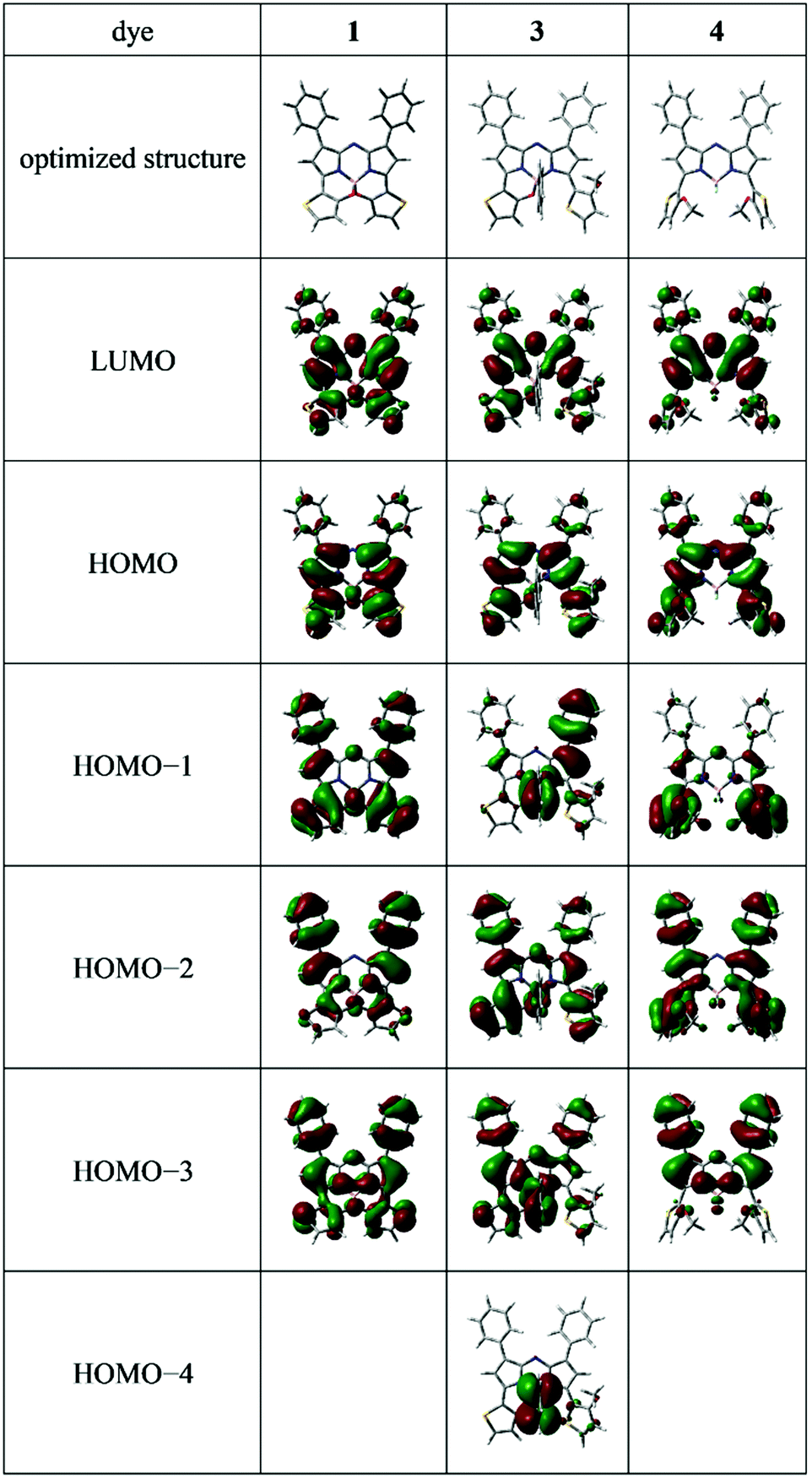
Time-dependent density functional theory (TD-DFT) and DFT analyses at the B3LYP/6-31G(d,p) level49 (Gaussian 09 Rev. C.01) were conducted to better understand the optical properties observed. Table 3 summarizes the absorption and theoretical data of 3,5-dithiophene-containing N2O2-type aza-BODIPY 1, the related N2O-type dye having a phenyl unit on the boron 3 and 3,5-bis(o-methoxythiophene) aza-BODIPY 4 along with transition energies (λcalcd) and oscillator strengths (f). Based on the f values listed, the major absorption bands were representative of S0–S1 transitions. Corresponding electron-density distributions are illustrated in Fig. 5.
Table 3 Theoretical data for the dyes
| Dye |
State |
λ (calcd.)/nm |
f/a.u. |
MO transition assignmenta |
|
Data given in parentheses are (CI coefficient)2 × 100%.55
|
|
1
|
S0 → S1 |
643 |
0.4007 |
HOMO → LUMO (51%) |
| S0 → S2 |
469 |
0.1868 |
HOMO–1 → LUMO (47%) |
| S0 → S3 |
452 |
0.0357 |
HOMO–2 → LUMO (49%) |
| S0 → S4 |
424 |
0.0734 |
HOMO–3 → LUMO (48%) |
|
|
|
3
|
S0 → S1 |
654 |
0.4053 |
HOMO → LUMO (50%) |
| S0 → S2 |
515 |
0.0259 |
HOMO–1 → LUMO (49%) |
| S0 → S3 |
477 |
0.0560 |
HOMO–3 → LUMO (48%) |
| S0 → S4 |
458 |
0.2091 |
HOMO–4 → LUMO (5%) |
| HOMO–2 → LUMO (42%) |
|
|
|
4
|
S0 → S1 |
627 |
0.4078 |
HOMO → LUMO (46%) |
| S0 → S2 |
529 |
0.1348 |
HOMO–1 → LUMO (49%) |
| S0 → S3 |
505 |
0.0768 |
HOMO–2 → LUMO (46%) |
| S0 → S4 |
465 |
0.1556 |
HOMO–3 → LUMO (49%) |
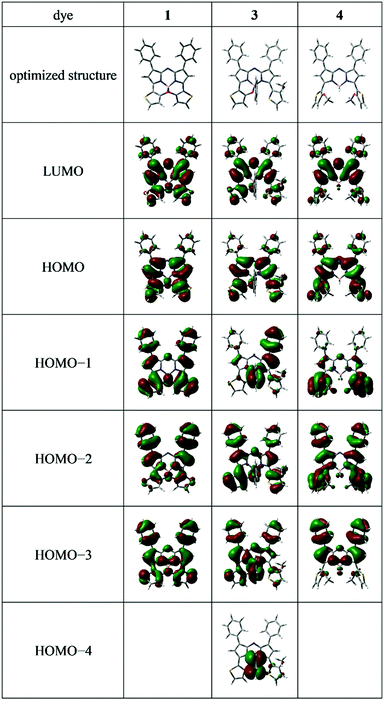 |
| | Fig. 5 The frontier molecular orbitals of the optimized structures of the dyes. | |
The electron-density distributions of both the HOMO and LUMO levels spread over the aza-BODIPY core, with substituents at the 3,5 positions being mainly responsible for π–π* transitions, as supported by the small Stokes shifts of the dyes (Table 1). For aza-BODIPY 4, a decrease in the electron-density distribution of the methoxythiophene units at the 3,5 positions through the HOMO → LUMO transition was observed (Fig. 5). However, based on the surface plots of the HOMO and LUMO energy levels, intramolecular charge transfer within the chromophore upon photoexcitation was not detected. This was supported by the transition moment (D) of 4 being 2.04 Debye, calculated based on the solvent-dependency of its absorption and fluorescence properties (Fig. S4, ESI†) being similar to those of 3 (Fig. S2, ESI†). This suggests that 4 still has π–π* transition characteristics in its photophysical properties. Conversely, formation of thieno[1,3,2]oxazaborinine through intramolecular B–O chelation, mainly contributed to the expansion of the π–π* transition of the dye chromophore. In this context, the broad absorption band at ≈500 nm may include plural transitions of the HOMO–1 → LUMO, HOMO–2 → LUMO, and HOMO–3 → LUMO. Based on these findings, the optical properties of 3 are similar to those of 1, strongly suggesting that 3 would be the most advantageous for device application from a structural stability point of view.
Photodetector fabrication
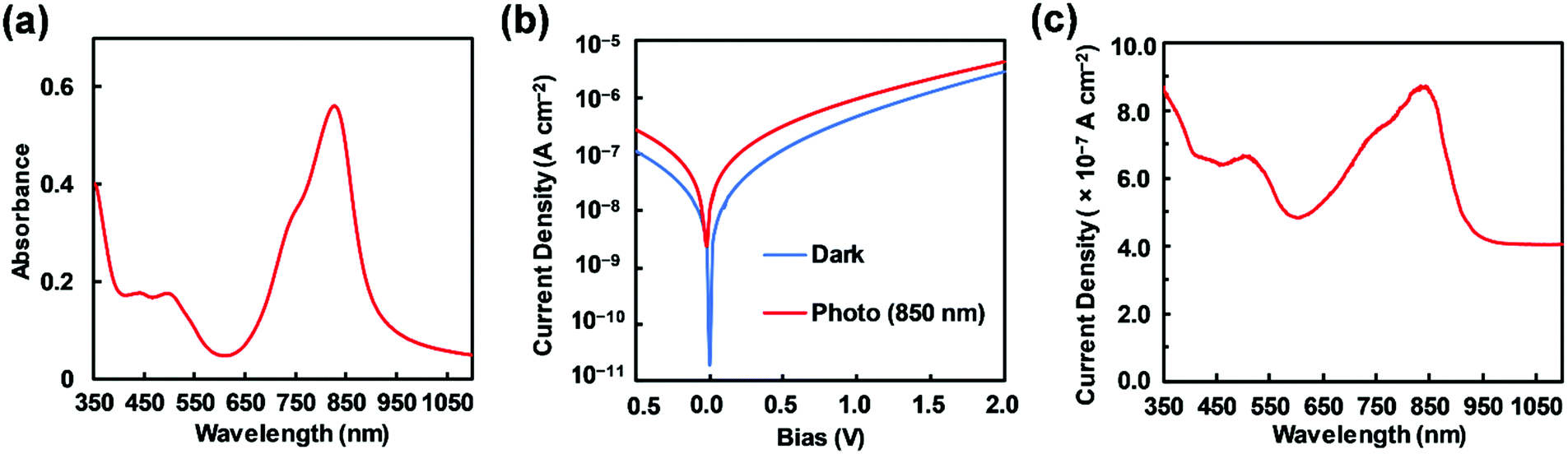
An investigation of 3 was performed to determine its applicability as a NIR photodetector. Prior to device fabrication, the thermal properties of 3 were investigated by differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) (Fig. S5, ESI†); a melting point of 273 °C was clearly observed. With consideration to this thermal property, resistance-heating type vacuum deposition was employed to prepare the dye film on an ITO glass substrate (Experimental section). Subsequently, an amorphous film was successfully formed on the glass with a sublimation yield of 40%. An atomic force microscopy (AFM) image of a 1 × 1 μm area of the dye-coated ITO substrate suggested a flat amorphous film with a root mean square (RMS) thickness of 0.6 nm (Fig. S6, ESI†). A film on quartz substrate was used to measure the absorption spectrum of the material, having a near infrared absorption band at 827 nm and a λonset value of 897 nm (Fig. 6a). Compared to its absorption spectrum in CH2Cl2, a bathochromic shift of 48 nm was observed. The HOMO energy level of the thin film was determined to be −5.42 eV, based upon photoelectron spectroscopy measurements under ambient conditions,56 which was lower than that of the ITO electrode (−5.0 eV). Taking into account the energy level of the work function of Al (−4.3 eV), it was expected that the electron and hole injection would be energetically favorable enough to provide photodetector functionality. Fig. 6b shows the current–voltage (J–V) measurements of the dye-loaded device under irradiation at 850 nm (130 μW cm−2). The device with 3 produced photocurrents of 3.19 × 10−7 A cm−2 and 9.57 × 10−7 A cm−2 at bias potentials of 0.5 and 1 V, respectively. On-to-off current ratios of 2.65 and 2.08 were calculated at bias potentials of 0.5 and 1.0 V, respectively, relative to dark current values at the same bias potentials. The increase in the forward injection of current under NIR light irradiation was ascribed to direct dissociation of photoexcited electron–hole pairs, although the on-to-off current ratio was not very large. To support this phenomenon, the wavelength-dependent photocurrent densities of the ITO/3 (85 nm)/Al (100 nm) device were measured, exhibiting a λmax of 838 nm, with a broad spectral response under a bias potential of 1 V (Fig. 6c). This clearly shows that infrared photons absorbed by 3 were converted to a photocurrent in the single-component device.
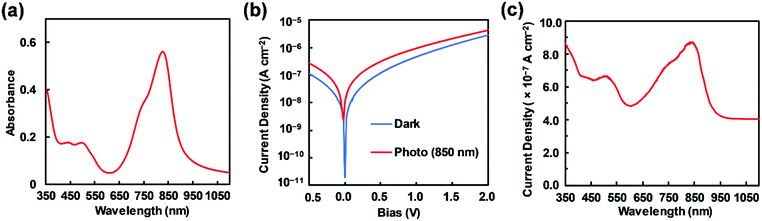 |
| | Fig. 6 (a) Absorption spectra of film 3 on a quartz substrate. (b) J–V curves of a 3-loaded device (ITO/3 (85 nm)/Al (100 nm)) measured in the dark (blue) and under illumination (red) at 850 nm (130 μW cm−2). (c) Photocurrent densities of the 3-loaded device at a 1 V applied potential as a function of time. | |
Conclusion
In this work, for the first time, structurally constrained aza-BODIPYs with thieno[1,3,2]oxazaborinine have been prepared. Although N2O2-type 1 was easily hydrolyzed in solution, 3-thiophene-containing N2O-type analogues 2 and 3 were isolated as stable dyes. Compared to benzo[1,3,2]oxazaborinine congener BO1, introduction of thiophene units in place of phenyl rings led to significant bathochromic shifts in the absorption bands of the dyes (λmax = 768–780 nm) and longer wavelength absorption edges beyond 800 nm. These characteristics were attributed to higher shifts in the HOMO energy levels compared to those of the LUMO levels. As a result, these dyes had longer absorption bands than those of all of the structurally constraining aza-BODIPYs reported thus far. Notably, the dye-loaded film (3) on quartz showed a NIR absorption band at 827 nm, with a wide spectral range, and a λonset value of 897 nm, thus enabling its evaluation as a NIR photodetector through fabrication of a single-component device. Preliminary results showed that the ITO/3/Al device produced a photocurrent under NIR light illumination at 850 nm (130 μW cm−2). This suggests that a sophisticated combination of a 3-thiophene-containing N2O-type aza-BODIPY with a n-type acceptor would allow for NIR photodiodes with a p–n interface,39,40 and deserves further investigation. Alternatively, from a synthetic point of view, reactions of 1-(o-hydroxythiophene)-9-(o-methoxythiophene)-substituted aza-dipyrrin 6 with several types of boronic acids could provide related aza-BODIPYs with NIR absorption bands, suggesting that 6 could be a valuable intermediate for the synthesis of organic NIR materials.
Conflicts of interest
There are no conflicts to declare.
References
- Y. Ge and D. F. O'Shea, Chem. Soc. Rev., 2016, 45, 3846–3864 RSC.
- G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006–1029 CrossRef CAS PubMed.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197–1226 CrossRef CAS.
- L. Beverina and P. Salice, Eur. J. Org. Chem., 2010, 1207–1225 CrossRef CAS.
- H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC.
- D. Ho, R. Ozdemir, H. Kim, T. Earmme, H. Usta and C. Kim, ChemPlusChem, 2019, 84, 18–37 CAS.
- K. Flavin, K. Lawrence, J. Bartelmess, M. Tasior, C. Navio, C. Bittencourt, D. F. O’Shea, D. M. Guldi and S. Giordani, ACS Nano, 2011, 5, 1198–1206 CrossRef CAS PubMed.
- J. Meiss, F. Holzmueller, R. Gresser, K. Leo and M. Riede, Appl. Phys. Lett., 2011, 99, 193307 CrossRef.
- S. Shimizu, T. Iino, A. Saeki, S. Seki and N. Kobayashi, Chem. – Eur. J., 2015, 21, 2893–2904 CrossRef CAS.
- G. Fan, L. Yang and Z. Chen, Front. Chem. Sci. Eng., 2014, 8, 405–417 CrossRef CAS.
- L. Liu, Y. Yuan, Y. Yang, M. T. McMahon, S. Chen and X. Zhou, Chem. Commun., 2019, 55, 5851–5854 RSC.
- N. Adarsh, P. S. S. Babu, R. R. Avirah, M. Viji, S. A. Nair and D. Ramaiah, J. Mater. Chem. B, 2019, 7, 2372–2377 RSC.
- S. Liu, Z. Shi, W. Xu, H. Yang, N. Xi, X. Liu, Q. Zhao and W. Huang, Dyes Pigm., 2014, 103, 145–153 CrossRef CAS.
- C. Staudinger, J. Breininger, I. Klimant and S. M. Borisov, Analyst, 2019, 144, 2393–2402 RSC.
- V. F. Donyagina, S. Shimizu, N. Kobayashi and E. A. Lukyanets, Tetrahedron Lett., 2008, 49, 6152–6154 CrossRef CAS.
- H. Lu, S. Shimizu, J. Mack, Z. Shen and N. Kobayashi, Chem. – Asian J., 2011, 6, 1026–1037 CrossRef CAS.
- Y. Wu, C. Cheng, L. Jiao, C. Yu, S. Wang, Y. Wei, X. Mu and E. Hao, Org. Lett., 2014, 16, 748–751 CrossRef CAS.
- J. Cui, W. Sheng, Q. Wu, C. Yu, E. Hao, P. Bobadova-Parvanova, M. Storer, A. M. Asiri, H. M. Marwani and L. Jiao, Chem. – Asian J., 2017, 12, 2486–2493 CrossRef CAS PubMed.
- W. Sheng, J. Cui, Z. Ruan, L. Yan, Q. Wu, C. Yu, Y. Wei, E. Hao and L. Jiao, J. Org. Chem., 2017, 82, 10341–10349 CrossRef CAS.
- E. M. A. Al-Imarah, P. J. Derrick and A. Partridge, J. Photochem. Photobiol., A, 2017, 337, 82–90 CrossRef CAS.
- N. Balsukuri, M. Y. Lone, P. C. Jha, S. Mori and I. Gupta, Chem. – Asian J., 2016, 11, 1572–1587 CrossRef CAS PubMed.
- L. Jiao, Y. Wu, S. Wang, X. Hu, P. Zhang, C. Yu, K. Cong, Q. Meng, E. Hao and M. G. H. Vicente, J. Org. Chem., 2014, 79, 1830–1835 CrossRef CAS.
- W.-J. Shi, P.-C. Lo and D. K. P. Ng, Dyes Pigm., 2018, 154, 314–319 CrossRef CAS.
- R. Gresser, H. Hartmann, M. Wrackmeyer, K. Leo and M. Riede, Tetrahedron, 2011, 67, 7148–7155 CrossRef CAS.
- Q. Bellier, F. Dalier, E. Jeanneau, O. Maury and C. Andraud, New J. Chem., 2012, 36, 768–773 RSC.
- X. Zhang, H. Yu and Y. Xiao, J. Org. Chem., 2012, 77, 669–673 CrossRef CAS PubMed.
- H. Yamane, K. Tanaka and Y. Chujo, Polym. J., 2018, 50, 271–275 CrossRef CAS.
- W. Zhao and E. M. Carreira, Angew. Chem., Int. Ed., 2005, 44, 1677–1679 CrossRef CAS.
- W. Zhao and E. M. Carreira, Chem. – Eur. J., 2006, 12, 7254–7263 CrossRef CAS.
- X.-D. Jiang, D. Xi, J. Zhao, H. Yu, G.-T. Sun and L.-J. Xiao, RSC Adv., 2014, 4, 60970–60973 RSC.
- X.-D. Jiang, D. Xi, C.-l. Sun, J. Guan, M. He and L.-J. Xiao, Tetrahedron Lett., 2015, 56, 4868–4870 CrossRef CAS.
- X.-D. Jiang, X. Liu, T. Fang, C. Sun and L. Xiao, Tetrahedron Lett., 2018, 59, 546–549 CrossRef CAS.
- A. Loudet, R. Bandichhor, K. Burgess, A. Palma, S. O. McDonnell, M. J. Hall and D. F. O’Shea, Org. Lett., 2008, 10, 4771–4774 CrossRef CAS PubMed.
- S. Y. Leblebici, L. Catane, D. E. Barclay, T. Olson, T. L. Chen and B. Ma, ACS Appl. Mater. Interfaces, 2011, 3, 4469–4474 CrossRef CAS PubMed.
- E. Bodio and C. Goze, Dyes Pigm., 2019, 160, 700–710 CrossRef CAS.
- Y. Kubo, S. Tobinaga, Y. Ueno, T. Aotake, H. Yakushiji and T. Yamamoto, Chem. Lett., 2018, 47, 300–303 CrossRef CAS.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- Q. Li, Y. Guo and Y. Liu, Chem. Mater., 2019 DOI:10.1021/acs.chemmater.9b00966.
- R. D. Jansen-vanVuuren, A. Armin, A. K. Pandey, P. L. Burn and P. Meredith, Adv. Mater., 2016, 28, 4766–4802 CrossRef CAS PubMed.
- Y. Yao, Y. Liang, V. Shrotriya, S. Xiao, L. Yu and Y. Yang, Adv. Mater., 2007, 19, 3979–3983 CrossRef CAS.
- X. Gong, M. Tong, Y. Xia, W. Cai, J. S. Moon, Y. Cao, G. Yu, C.-L. Shieh, B. Nilsson and A. J. Heeger, Science, 2009, 325, 1665 CrossRef CAS.
- K. H. Hendriks, W. Li, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 12130–12136 CrossRef CAS PubMed.
- X. Wang, L. Lv, L. Li, Y. Chen, K. Zhang, H. Chen, H. Dong, J. Huang, G. Shen, Z. Yang and H. Huang, Adv. Funct. Mater., 2016, 26, 6306–6315 CrossRef CAS.
- X. Zhou, D. Yang, D. Ma, A. Vadim, T. Ahamad and S. M. Alshehri, Adv. Funct. Mater., 2016, 26, 6619–6626 CrossRef CAS.
- S. Li, X. Deng, L. Feng, X. Miao, K. Tang, Q. Li and Z. Li, Polym. Chem., 2017, 8, 1039–1048 RSC.
- C. Würth, J. Pauli, C. Lochmann, M. Spieles and U. Resch-Genger, Anal. Chem., 2012, 84, 1345–1352 CrossRef PubMed.
- E. T. Saka, M. Durmuş and H. Kantekin, J. Organomet. Chem., 2011, 696, 913–924 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. L. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.01, Gaussiain, Inc., Wallingforld, CT, 2010 Search PubMed.
- Isolated 2 (1H NMR (500 MHz, DMSO-d6): δ (ppm) 8.05–8.03 (m, 4H), 7.65 (d, J = 5.36 Hz, 2H), 7.43 (t, J = 7.53 Hz, 4H), 7.35 (tt, J = 7.31 and 1.38 Hz), 6.88 (d, J = 5.36 Hz, 2H) showed an absortion band with two λ values of 665 and 729 nm, and a lower absorption band at 341 nm in water-saturated CH2Cl2.
- X.-D. Jiang, Y. Fu, T. Zhang and W. Zhao, Tetrahedron Lett., 2012, 53, 5703–5706 CrossRef CAS.
- S. Schwedler, D. Eickhoff, R. Brockhinke, D. Cherian, L. Weber and A. Brockhinke, Phys. Chem. Chem. Phys., 2011, 13, 9301–9310 RSC.
- J. B. Prieto, F. L. Arbeloa, V. Martinez, T. A. López, F. Amat-Guerri, M. Liras and I. L. Arbeloa, Chem. Phys. Lett., 2004, 385, 29–35 CrossRef.
- The electrical gap of those dyes based on CV measurements did not correlate with absorption bands observed in Fig. 4a, a transition other than HOMO–LUMO one may be involved to some extent in the absorption band.21.
- Y. Kubo, K. Yoshida, M. Adachi, S. Nakamura and S. Maeda, J. Am. Chem. Soc., 1991, 113, 2868–2873 CrossRef CAS.
- J. Sworakowski and K. Janus, Org. Electron., 2017, 48, 46–52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data of compounds, additional information, detailed single crystal X-ray diffraction study and calculation results. CCDC 1948374. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj04612g |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Takuma
Shimada
a,
Kentaro
Maeda
b and
Yuta
Hashimoto
b
*a,
Takuma
Shimada
a,
Kentaro
Maeda
b and
Yuta
Hashimoto
b