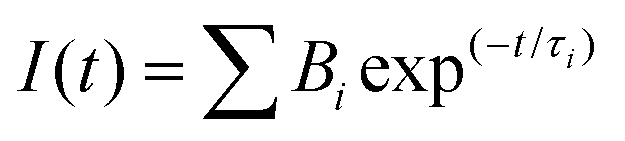DOI:
10.1039/C9NJ03693H
(Paper)
New J. Chem., 2020,
44, 218-230
Perturbing the AIEE activity of pyridine functionalized α-cyanostilbenes with donor substitutions: an experimental and DFT study†
Received
16th July 2019
, Accepted 21st November 2019
First published on 22nd November 2019
Abstract
A series of pyridine functionalized α-cyanostilbenes bearing different amino group donors, carbazole–phenyl (C-1), N-ethyl carbazole (C-3), dimethylamino (DM), julolidine (J-1) and triphenylamine (T-1), were synthesized, and their impact on the aggregation propensity of the α-cyanostilbenes was investigated. Distinct aggregation-induced emission (AIE) features have been obtained in water, where the emission intensity varied in the following order: C-1 > DM > C-3 > T-1 > J-1. The formation of aggregates was substantiated by fluorescence lifetime decay profiles and their average lifetime correlated with the rigidity of the molecular framework. The contributing factors behind the AIE phenomena are the bulkiness of the donor segments, their donating strength, planarity of the molecular framework, the distance between the α-cyano unit and the donors and its interaction with neighboring units. Based on the experimental observations from this study and previous research reports, we have used DFT and TDDFT methods to evaluate the donor–AIEE property relationship. Cyclic voltammetry techniques were used to examine the redox properties of the compounds. SEM and DLS studies provided us with a detailed picture of the morphology and hydrodynamic radii of the aggregates, respectively. The results obtained from this study would be helpful to understand the substituent dependent aggregation propensity of α-cyanostilbenes and provide design guidelines toward the development of AIE active materials for various optoelectronic and bioimaging applications.
Introduction
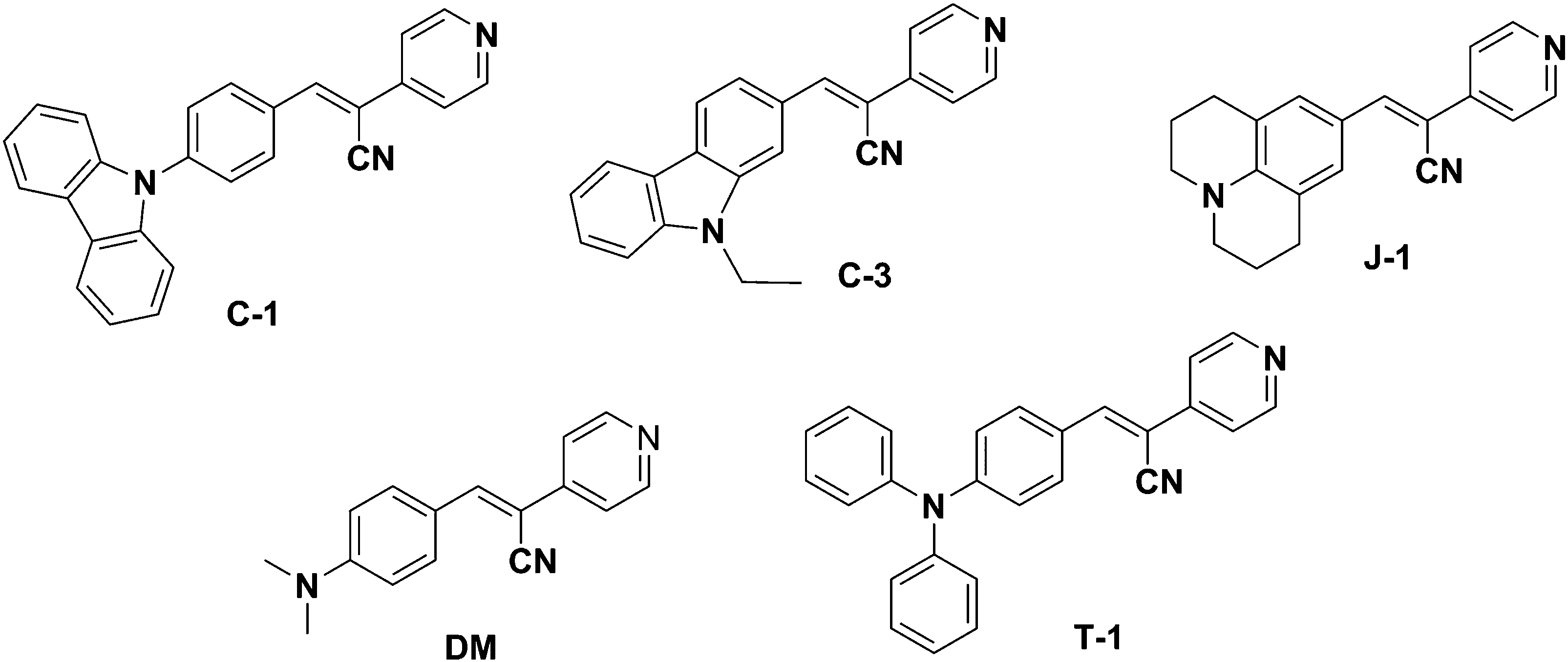
Aggregation-induced emission (AIE) is a unique phenomenon that induces emission intensity enhancement or wavelength shifts upon aggregation.1,2 Various mechanistic findings such as restriction of molecular motions (vibration and rotation), planarization, J-aggregate formation, E/Z isomerization, excited-state intramolecular proton transfer (ESIPT), etc. have been reported as contributing factors for the AIE phenomenon.3–5 A plethora of applications involving the AIE effect of several molecular scaffolds and their mechanism have been extensively investigated to date.4,6–9 Of these, the widely accepted mechanism is the restriction of intramolecular motions typically observed in tetraphenylethene (TPE) and hexaphenyl silole (HPS) derivatives where only emission intensity enhancement is noted. Hampering the rotation of free phenyl rings in TPE and HPS upon aggregation imparts rigidity to the system, dissipating the energy through radiative channels. The greater the number of free phenyl rings, the higher the emission intensity, i.e., the AIE effect in HPS is more pronounced than in TPE.10 Like TPE and HPS molecular scaffolds, α-cyanostilbene is a versatile motif that undergoes efficient intramolecular rotation (IMR) with large torsional distortions and is non-emissive in solution. Upon aggregation, the restricted IMR of the α-cyano moiety results in planarization, rendering excellent photophysical properties in its aggregated form through effective intermolecular interactions.8,11 Unlike HPS or TPE derivatives, α-cyanostilbenes influence both emission intensity and wavelength shifts upon aggregation. It should be noted that the aggregation type (H or J) is solely dependent on the positional substitution of α-cyanostilbenes.12 For instance, Y. Zhang et al. demonstrated the distinct photophysical features of two isomeric triphenylamine based acrylonitriles with respect to the positional substitution of the α-cyano unit to the donor and acceptor groups respectively.13 Isomer 1 (TPA-CNa) containing the α-cyano unit proximal to the donor exhibited a hypsochromic shift with H-type aggregation.14 Similar observations were noticed by Upamali and coworkers for carbazole substituted cyanostilbenes.15 On the other hand, substitution of the α-cyano unit proximal to the terminal acceptor (spacer) unit (TPA-CNb) results in a bathochromic shift. Recently, Zhang et al. also perturbed the aggregation behavior of α-cyanostilbenes by proximal substitution of bulkier tetraphenyl imidazole (TPI) units. The positional substitution of the bulkier TPI groups hampers the interaction of α-cyanostilbenes with neighboring units to a certain extent. Their aggregation abruptly increases the emission intensity but they show no wavelength shifts despite the presence of imidazole acceptor units. Thus, it can be stated that AIE mainly originates from the restriction of intramolecular rotation of tetraphenyl imidazole rings with minimal participation of the α-cyanostilbene motif.16 Typically, the incorporation of acceptor units into the cyanostilbene framework dramatically shifts the emission to longer wavelengths with a concomitant large Stokes shift. For example, our group developed a series of four coumarin-based AIE active α-cyanostilbenes by varying the spacers and terminal acceptors without perturbing the α-cyano moiety. The resultant aggregates shifted the emission wavelength from the visible to the far red/NIR region with a large Stokes shift of 158–213 nm.17 Similarly, insertion of an acrylonitrile group enables emission tunability, yielding fluorophores with a large Stokes shift.18,19 The unique emission characteristics of α-cyanostilbenes have opened a new platform towards development of various novel and smart emissive materials with applications as solid-state light emitters, non-linear optics, bioprobes, and chemosensors, among others.12,13,20–24 Recently, we also demonstrated the geometrical impact on the AIE characteristics by varying the structure from linear (mono-branched) to V-shaped (di-branched) and star-shaped (tri-branched) molecules.25 We have observed that the AIE effect gradually decreases upon increasing the number of branching units. This study reveals that each branch present in the star-shaped molecule hampers interactions of α-cyano units of other branches. Scrutiny of these reports shows the paramount importance of positional substitution of donor/acceptor units to the aggregation characteristics of α-cyanostilbenes. To study this fact, we have developed a series of pyridine functionalized acrylonitrile derivatives with various donor substitutions, carbazole (C-1), N-ethyl carbazole (C-3), N,N-dimethylamine (DM), julolidine (J-1) and triphenylamine (T-1) [Fig. 1]. Pyridine acts as an acceptor moiety and also aids in using these compounds as stimuli-responsive materials towards use in acid sensing, organogel formation, and non-linear optical (NLO) applications.26–28 The extent of the donor's influence on the AIEE activity of cyanostilbenes is studied using various experimental techniques and DFT/TDDFT methods. The distance between the donor units and α-cyano moiety distinctly differs for each compound. For instance, the carbazole donor and α-cyano moiety are separated by a phenyl spacer in C-1, whereas the donor units are directly connected to an α-cyano moiety for the remaining compounds. The steric repulsion and rigidity of the donor are expected to have a greater impact in determining the emission intensity and wavelength shift. Additionally, each donor unit has its distinguishing characteristics upon aggregation. Our observations are detailed in the following sections.
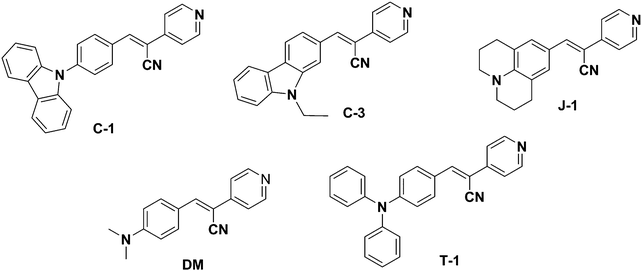 |
| | Fig. 1 Structures of the synthesized acrylonitrile derivatives. | |
Experimental details
All the reagents required for the synthesis of the pyridine functionalized acrylonitriles were purchased from Sigma-Aldrich, Alfa Aesar, Acros, and S. D. Fine Chem. The synthesized compounds were characterized using a Bruker Avance III-500 MHz NMR spectrometer and ITMS-probe electron spray ionization (pESI) mass spectrometer. UV-vis absorption spectra were recorded using an Analytik Jena Specord 210 Plus, and fluorescence emission studies were performed using a Horiba-Jobin-Yvon Fluorolog-3 spectrofluorimeter. The excitation wavelengths were set at the absorption maxima (λa) of the compounds. All the fluorescence spectra were recorded in 10 mm path length quartz cuvettes with a slit width of 2 nm. For the UV-vis absorption & fluorescence measurements, ∼10 μM concentrations of chromophores were used prepared from a stock solution of the compounds in THF or DMSO. The fluorescence lifetime measurements were performed using a picosecond time-correlated single photon counting (TCSPC) setup (Edinburgh Instruments Ltd, Lifespec II model) employing a picosecond light emitting diode laser at 405 nm for the excitation of dye solution in 99% water. A reconvolution procedure was used to analyze the decay by using a proper instrument response function (IRF) obtained by using LUDOX HS-40 colloidal silica particles in a 40 wt% suspension in water as the light scatterer. The fluorescence decays [I(t)] were analyzed in general as a sum of exponentials, eqn (1)| | 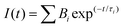 | (1) |
where Bi and τi are the pre-exponential factor and fluorescence lifetime of the ith component. Reduced chi-square (χ2) values and the random distribution of the weighted residuals among the data channels were used to judge the acceptance of the fits. The error associated with the lifetime studies is 0.30–13%. Scanning electron microscopy (SEM) analysis was carried out using a field emission SEM (JSM 7600F JEOL). 10 μL of the sample (∼10−5 M solution) was drop cast on a Si-wafer. The samples were dried at 35 °C for 16 h to ensure complete removal of any residual water and coated with platinum before being analyzed. Electrochemical measurements are performed using a Metrohm Autolab PGSTAT 101 potentiostat at room temperature. A three electrode system was used during the experiment, a 1 mm glassy carbon disc working electrode, a Pt-wire counter electrode, and Ag/AgCl (in saturated KCl) as a reference electrode. All experiments were performed in dry DMF using 0.1 M tetrabutylammonium tetrafluoroborate (TBAF) as a supporting electrolyte. All cyclic voltammetry data reported in this work are internally referenced against ferrocene.
Computational details
All theoretical calculations have been carried out in this study using the Gaussian 09 ab initio software package within the framework of density functional methods.29 The ground state geometry of the compounds was optimized in the gas and solution phase at the DFT/B3LYP/6-311G(d,p) level of theory with no symmetry constraints imposed. The optimized geometries are subjected to vibrational analysis to ensure global minima on the potential energy surface with no imaginary frequency. These structural coordinates were used as the input for further single point calculations to examine the electron density population, TDDFT, and PDOS analysis, etc. Singlet excited state calculations have been employed using time-dependent DFT (TDDFT) methods. To simulate the experimental absorption spectra of the compounds, three different density functionals, B3LYP, long-range corrected M062X30 and ωB97XD,31 have been used with a common basis set of 6-311G(d,p). As the absorption of the molecules was recorded in 1,4-dioxane, same solvent medium has been used under the conductor-like polarizable continuum model (C-PCM) integral equation formalism within self-consistent field theory was taken into consideration to mimic the real environment.32 Density of states (DOS) and projected density of states (PDOS) analysis has been employed to understand the percentage contribution of electron density from individual segments toward the charge transfer transition of the compounds against their respective molecular orbital density coefficients. The simulation of the major portion of the absorption spectrum, PDOS analysis, and interpretation of the nature of the transitions were executed using GaussSum 3.0.2.
General synthetic procedures
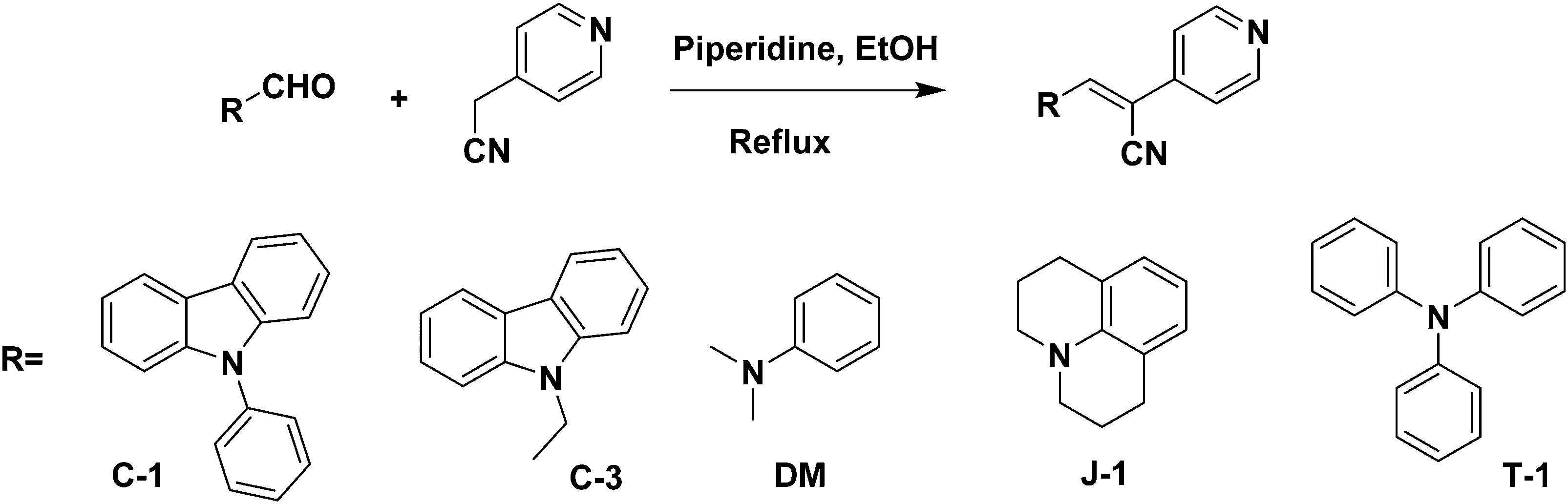
4-Pyridyl acetonitrile hydrochloride [1.6 mmol, 1 eq.] and 0.5 mmol of piperidine were dissolved in 10 mL anhydrous methanol. After 10–15 min of stirring, the desired aldehyde (1 eq., 1.6 mmol) was added to the reaction solution and it was heated to 60 °C for 5 h. The reaction was monitored by thin layer chromatography (TLC) to ensure the consumption of the aldehyde. Upon completion, the reaction solution was cooled to room temperature, and the solvent was evaporated to dryness. The obtained residue was washed with fresh methanol followed by diethyl ether. The precipitate obtained was filtered and dried under a vacuum for 1 h to afford the desired products as yellow to red solid α-acrylonitrile derivatives.
C-1
.
Yield 55% (150 mg): 1H-NMR (500 MHz, DMSO-d6), δ (ppm): 8.76–8.75 (d, 2H) 8.49 (s, 1H), 8.33–8.28 (dd, 4H), 7.92–7.90 (d, 2H), 7.83–7.82 (d, 2H), 7.56–7.54 (d, 2H), 7.50–7.47 (dd, 2H), 7.37–7.33 (dd, 2H) 13C NMR (125 MHz, DMSO-d6): δ (ppm) 151.06, 145.62, 141.59, 140.12, 139.98, 132.28, 132.03, 127.29, 126.96, 123.64, 121.10, 120.43, 117.56, 110.37, 108.68. ITMS (pESI): C26H17N3 (M), [M + H]+ cal. m/z 372.15, found m/z 372.17.
C-3
.
Yield 55% (150 mg): 1H-NMR (500 MHz, DMSO-d6), δ (ppm): 8.91 (s, 1H), 8.88–8.86 (d, 2H), 8.74 (s, 1H), 8.35–8.33 (d, 1H), 8.19–8.17 (d, 1H), 8.14–8.13 (d, 2H), 7.90–7.89 (d, 1H), 7.76–7.75 (d, 1H), 7.60–7.57 (dd, 1H), 7.37–7.34 (dd, 1H), 4.57–4.56 (q, 2H), 1.40–1.37 (t, 3H) 13C NMR (125 MHz, DMSO-d6) δ (ppm): 142.42, 140.82, 128.22, 127.46, 125.60, 124.14, 123.10, 122.52, 121.38, 120.91, 120.88, 118.19, 110.65, 79.71, 79.45, 79.19, 37.94, 14.26. ITMS (pESI): C22H17N3 (M), [M + H]+ cal. m/z 324.15, found m/z 324.17.
DM
.
Yield 80% (200 mg): 1H-NMR (500 MHz, DMSO-d6), δ (ppm): 8.63–8.61 (d, 2H), 8.11 (s, 1H), 7.96–7.95 (d, 2H), 7.66–7.65 (2H), 6.87–6.85 (d, 2H), 3.07 (s, 6H) 13C NMR (125 MHz, DMSO-d6): δ (ppm) 152.9, 150.7, 146.2, 142.8, 132.5, 120.5, 119.4, 119.01, 112.1, 99.64. ITMS (pESI): C16H15N3 (M), [M + H]+ cal. m/z 250.13, found m/z 250.17.
J-1
.
Yield 65% (150 mg): 1H-NMR (500 MHz, DMSO-d6), δ (ppm): 8.59–8.57 (d, 2H), 7.94 (s, 1H), 7.61–7.60 (d, 2H), 7.53 (2H, s), 3.30–3.32 (m, 2H), 2.72–2.70 (m, 4H) 1.91–1.89 (m, 4H) 13C NMR (125 MHz, DMSO-d6): δ (ppm) 150.64, 145.97, 130.12, 120.86, 119.19, 49.78, 27.65, 21.28. ITMS (pESI): C20H19N3 (M), [M + H]+ cal. m/z 302.16, found m/z 302.17.
T-1
.
Yield 75% (150 mg): 1H-NMR (500 MHz, DMSO-d6), δ (ppm): 8.67–8.66 (d, 2H), 8.20 (s, 1H), 7.93–7.92 (d, 2H), 7.71–7.70 (d, 2H), 7.44–7.41 (dd, 4H), 7.24–7.19 (m, 6H), 6.97–6.95 (d, 2H) 13C NMR (125 MHz, DMSO-d6) δ (ppm): 152.88, 150.72, 146.23, 142.82, 132.51, 120.59, 119.45, 119.01, 112.11, 99.62. ITMS (pESI): C26H19N3 (M), [M + H]+ cal. m/z 374.16, found m/z 374.33.
Results and discussion
Synthesis
A series of acrylonitrile derivatives were synthesized with varying donor substitutions via a condensation reaction of the corresponding aldehyde derivatives with 4-pyridine acetonitrile as depicted in Scheme 1. The corresponding aldehydes of C-1, J-1 and T-1 were synthesized using previously published literature procedures.19,33,34 The commercially obtained aldehydes of C-3 and DM were used without further purification. The structures of the cyanostilbenes investigated in this study are depicted in Fig. 1 and the molecules are coded as C-1 (N-phenyl carbazole), C-3 (N-ethyl carbazole), J-1 (julolidine), DM (dimethylamino) and T-1 (triphenylamine).
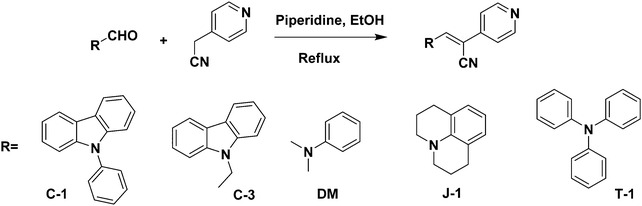 |
| | Scheme 1 General synthetic strategy adopted for the synthesis of acrylonitrile derivatives. | |
Absorption and emission studies
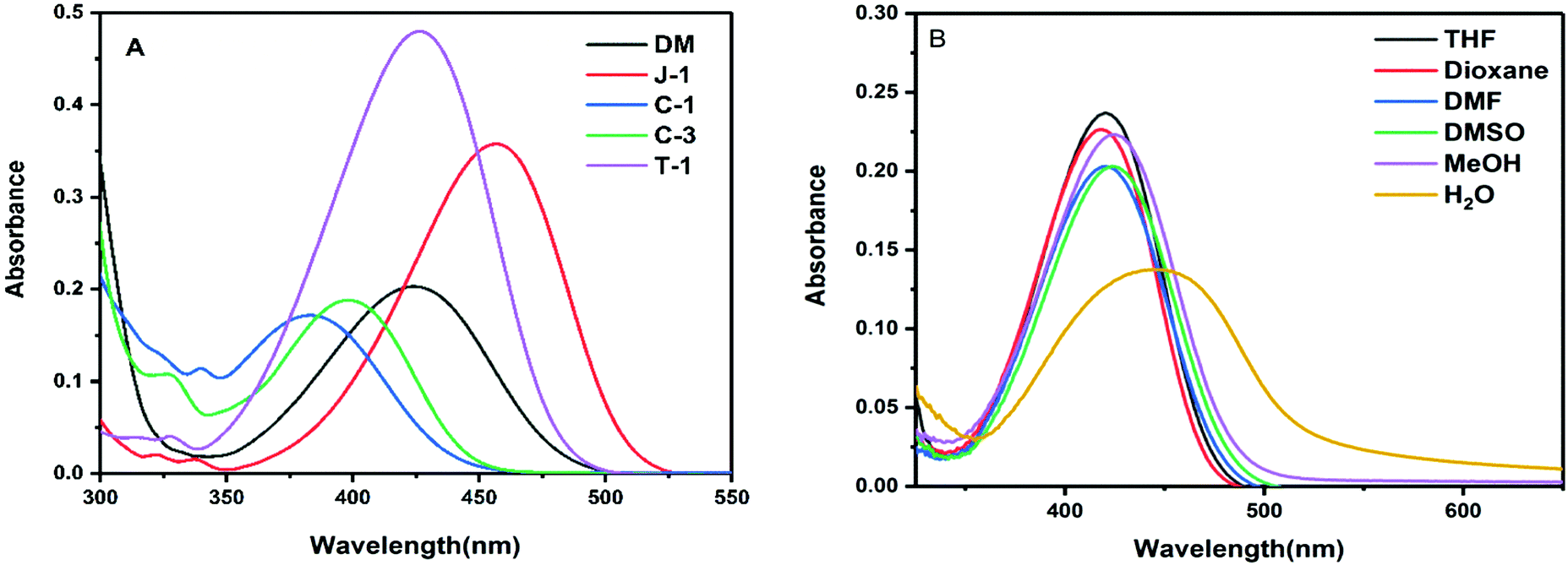
The absorption and emission spectra data of the compounds recorded in various solvents are shown in Table 1. Among the compounds studied, C-1 with N-carbazole substitution possesses the lowest absorption maximum, whereas J-1 with julolidine substitution yields the highest absorption maximum (Fig. 2A). The lower absorption of C-1 is attributed to the poor overlap of orbital interactions due to the large torsional distortion between the carbazole unit and phenyl group.35,36 The absorption of C-3 is slightly red-shifted due to the enhanced planarity achieved through direct conjugation of acrylonitrile with the third position of carbazole. Compounds DM and T-1 possessing dimethylamine (DM) and triphenylamine (TPA) donating segments respectively show similar absorption maxima. The larger absorption maximum (λa) for J-1 is ascribed to its increased planarity and the electron donating nature of the amino unit of julolidine. The overall order of absorption is: J-1 > T-1 ∼ DM > C-3 > C-1. Upon changing the polarity of the solvents, weaker solvatochromic absorption behavior was noted except for J-1, which exhibits greater solvatochromic shifts (Fig. 2B and Fig. S1, ESI†). The broad absorption profiles noted in water are an indication of aggregate formation. The absorption band of T-1 is weak with the band at 420 nm and a shoulder at 502 nm and J-1 shows two absorption bands at 462 nm and 533 nm (Fig. 2 and Fig. S1, ESI†).
Table 1 Absorption and emission data of the acrylonitrile derivatives investigated
| Solvents |
DM
|
J-1
|
C-1
|
C-3
|
T-1
|
|
λ
a
|
λ
f
|
ϕ
f
|
λ
a
|
λ
f
|
ϕ
f
|
λ
a
|
λ
f
|
ϕ
f
|
λ
a
|
λ
f
|
ϕ
f
|
λ
a
|
λ
f
|
ϕ
f
|
|
Relative quantum yields determined using quinine sulfate (quantum yield: 0.545 0.1 N H2SO4).
|
| THF |
420 |
546 |
0.28 |
442 |
502 |
0.15 |
384 |
514 |
0.65 |
392 |
450 |
0.13 |
413 |
487 |
0.03 |
| Dioxane |
418 |
520 |
0.26 |
436 |
499 |
0.17 |
386 |
488 |
0.62 |
389 |
442 |
0.11 |
408 |
484 |
0.03 |
| DMF |
420 |
580 |
0.20 |
451 |
525 |
0.10 |
381 |
552 |
0.67 |
395 |
458 |
0.19 |
422 |
502 |
0.14 |
| DMSO |
424 |
591 |
0.17 |
457 |
532 |
0.09 |
383 |
564 |
0.66 |
398 |
459 |
0.17 |
426 |
511 |
0.04 |
| MeOH |
425 |
586 |
0.11 |
455 |
526 |
0.06 |
383 |
571 |
0.25 |
398 |
461 |
0.13 |
422 |
500 |
0.01 |
| H2O |
445 |
564 |
0.35 |
462, 513 |
542 |
0.02 |
406 |
531 |
0.88 |
399 |
552 |
0.27 |
421 |
601 |
0.11 |
| Solid |
— |
609 |
|
— |
558 |
|
— |
548 |
|
— |
574 |
|
— |
627 |
|
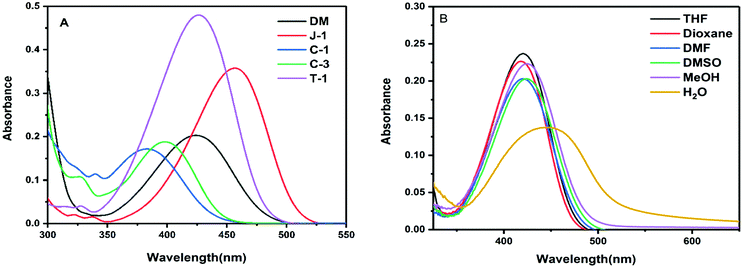 |
| | Fig. 2 (A) Absorption spectra of the investigated acrylonitrile derivatives in THF (10 μM); (B) absorption spectra of DM recorded in various solvents. | |
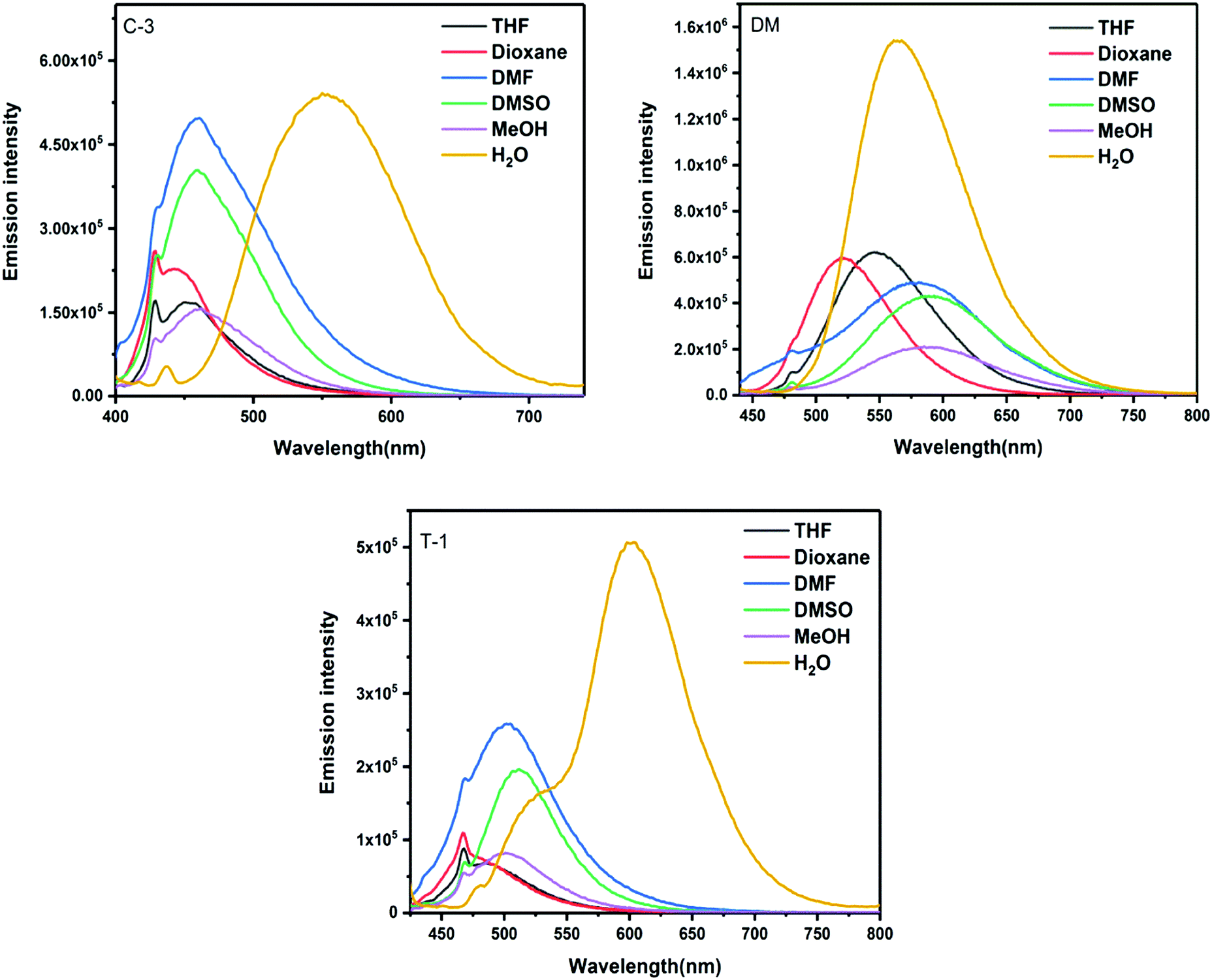
The variation in emission wavelength with changes in solvent polarity is stronger as compared to their spectral absorption changes. The emission profiles exhibit a positive solvatochromic behavior and substantiate the intramolecular charge transfer (ICT) from the amino donor group to the electron withdrawing cyano and pyridine units.37 Depending on the strength of the donating group, variable emission shifts were noted with DM and C-1 showing a >70 nm shift from dioxane to DMSO [Fig. 3 and Fig. S2, ESI†]. Moderate emission shifts have been noted for J-1 (36 nm), C-3 (17 nm) and T-1 (27 nm). The charge transfer nature of these compounds was also qualitatively shown using Lippert–Mataga plots [Fig. S2a, ESI†]. However, unique emission is noted in water where the emission intensity increased from 4.7 to 60 fold depending on the nature of the substituent. This unique emission in aqueous media is attributed to the formation of aggregates. The emission maximum observed in the solid state is greater than the solution phase due to the strong molecular packing in the solids (Fig. S3, ESI†).38,39 It is known that para-cyano substitution at the principal axis leads to intramolecular charge transfer, whereas in the current study, the cyano unit on the double bond is positioned vertical to the principal axis. Owing to the dynamic intramolecular rotations of the α-cyano unit with respect to the principal axis of the molecular framework in a vertical direction, its solvatochromic behavior distinctly varies for each compound. The reason behind the variations needs further investigation.
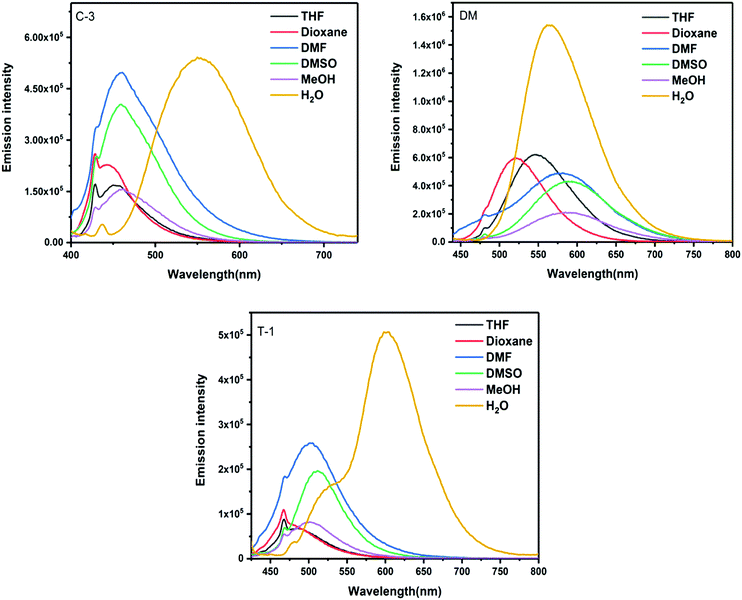 |
| | Fig. 3 Emission spectra of C-3, DM, and T-1 measured in various solvents. | |
Molecular geometry and frontier energy levels
To gain deeper insights into the structure and aggregation relationship of the acrylonitrile derivatives with respect to the donor substitution, we studied their geometrical, electronic and spectroscopic properties at the molecular level via DFT/TDDFT methods. In general, the charge transfer propensity is majorly dependent on the degree of π-conjugation that is measured via torsional distortion between the individual functional building blocks, the donor, π-conjugated bridge and acceptor units, present in the molecular framework. The higher the planarity, the more efficient the charge transfer due to the better overlap of π-orbital interactions. The molecular geometries of all the compounds have been optimized using the B3LYP/6-311G(d,p) level of theory.40
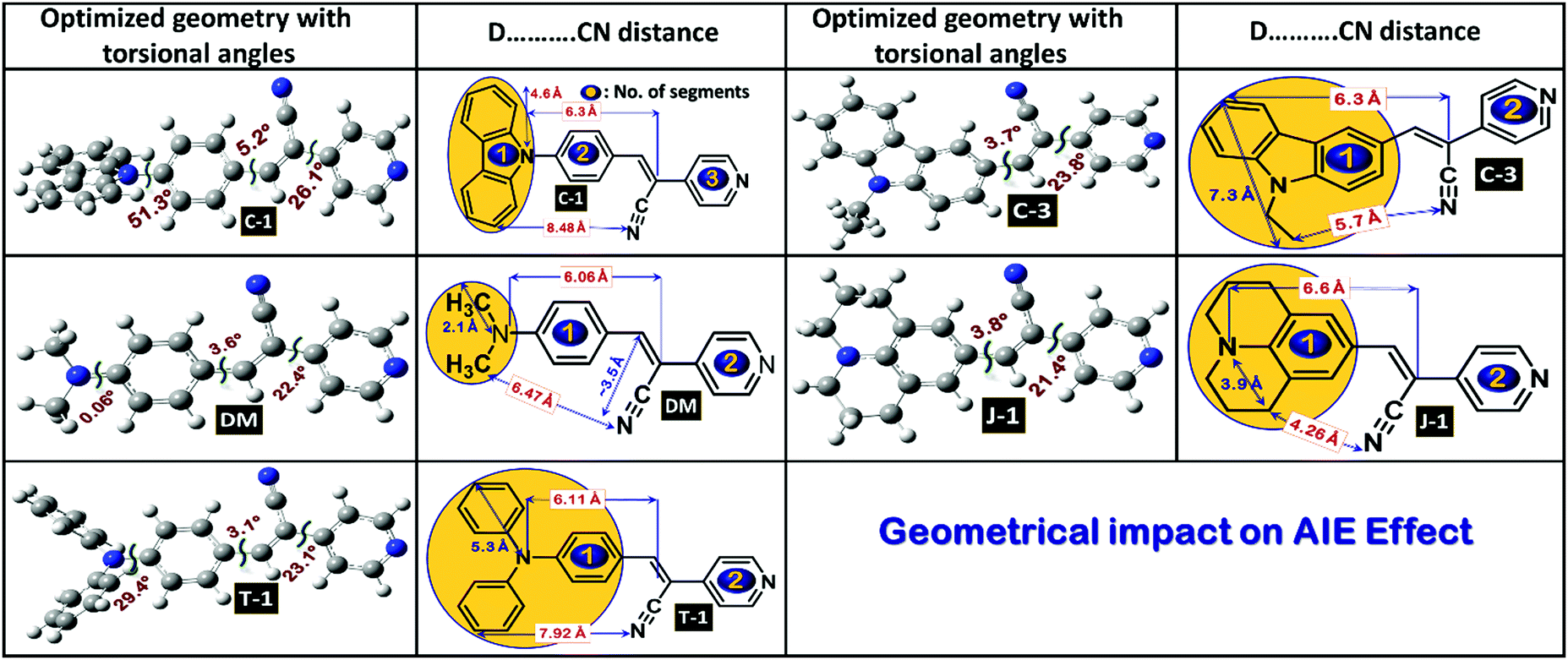
Compound C-1 contains three segments (carbazole, phenyl and the acceptor) as shown in Fig. 4 and the remaining compounds comprise two parts, i.e., donors linked to the pyridyl unit via an α-cyano moiety. In C-1, the phenyl unit connected to the carbazole is highly distorted in an out-of-plane fashion with a dihedral angle of 51.3° on one side and a slight twist of 5.3° with the vinylene part on the other side.35 The α-cyano moiety of the C-1 group deviates with a distortion of 26.1° from the terminal pyridyl acceptor unit. This mainly originates from the steric repulsion induced between the α-cyano unit and ortho-hydrogens of the terminal pyridyl unit. The other molecules C-3, DM, J-1, and T-1 are two-segmented; the donors maintain co-planarity with the vinylene chain with slight torsional distortions in the range of 3.1–3.8°. On the other hand, a significant deviation is seen between the α-cyano unit and pyridyl group for all these compounds in the range of 21.4–23.8°. The geometrical coordinates of the compounds are provided in Fig. S4 (ESI†). It can be seen from the molecular geometries that the N-atoms of the donor group and the α-cyano moiety are in the same line as the principal axis. The measured bond distances between the central styryl carbon and the cyano units are found to be around 3.5 Å. The bond distances measured between the N-atom and the terminal carbon of the donor group are in the following order (units in Å): C-3 (7.3) > T-1 (5.3) > C-1 (4.6) > J-1 (3.9) > DM (2.1). The steric environment caused by the donor units, the distance between the α-cyano unit and donors, and the flexibility and rigidity imparted by the fused aliphatic chains and aromatic groups may affect the aggregation propensity.
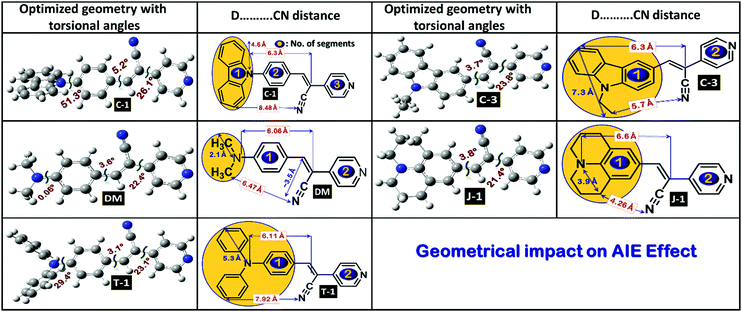 |
| | Fig. 4 Optimized molecular geometries of the acrylonitrile derivatives with selected torsional angles and bond distances. | |
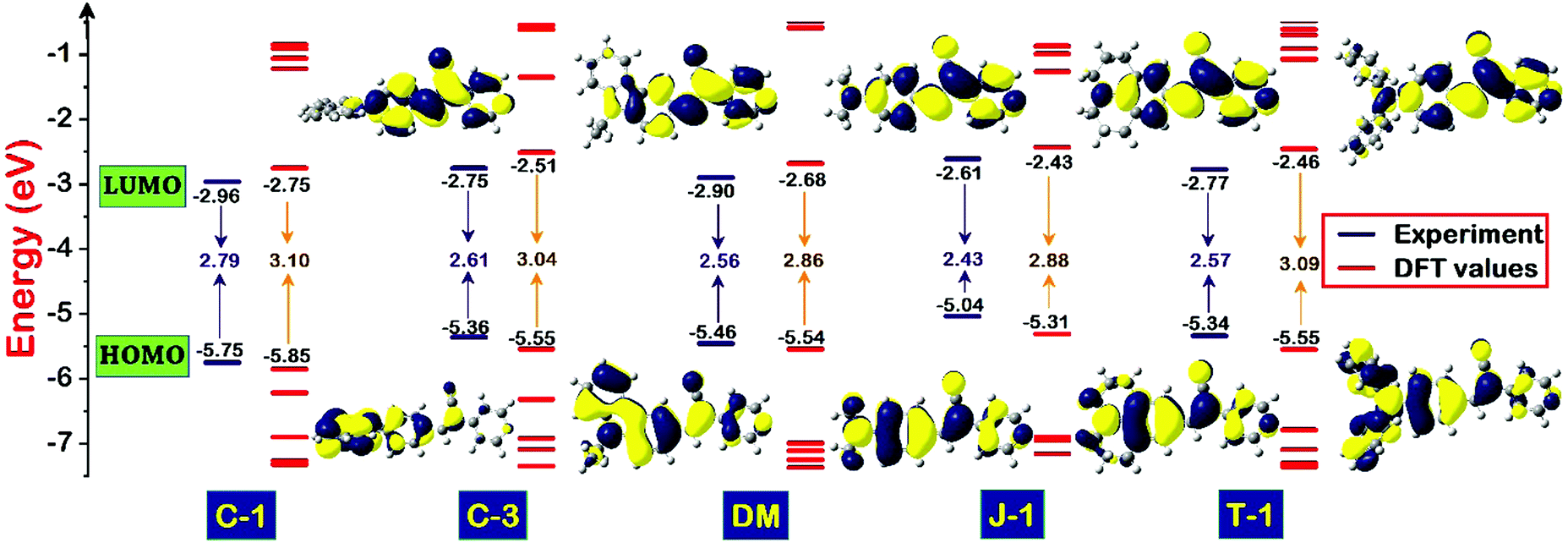
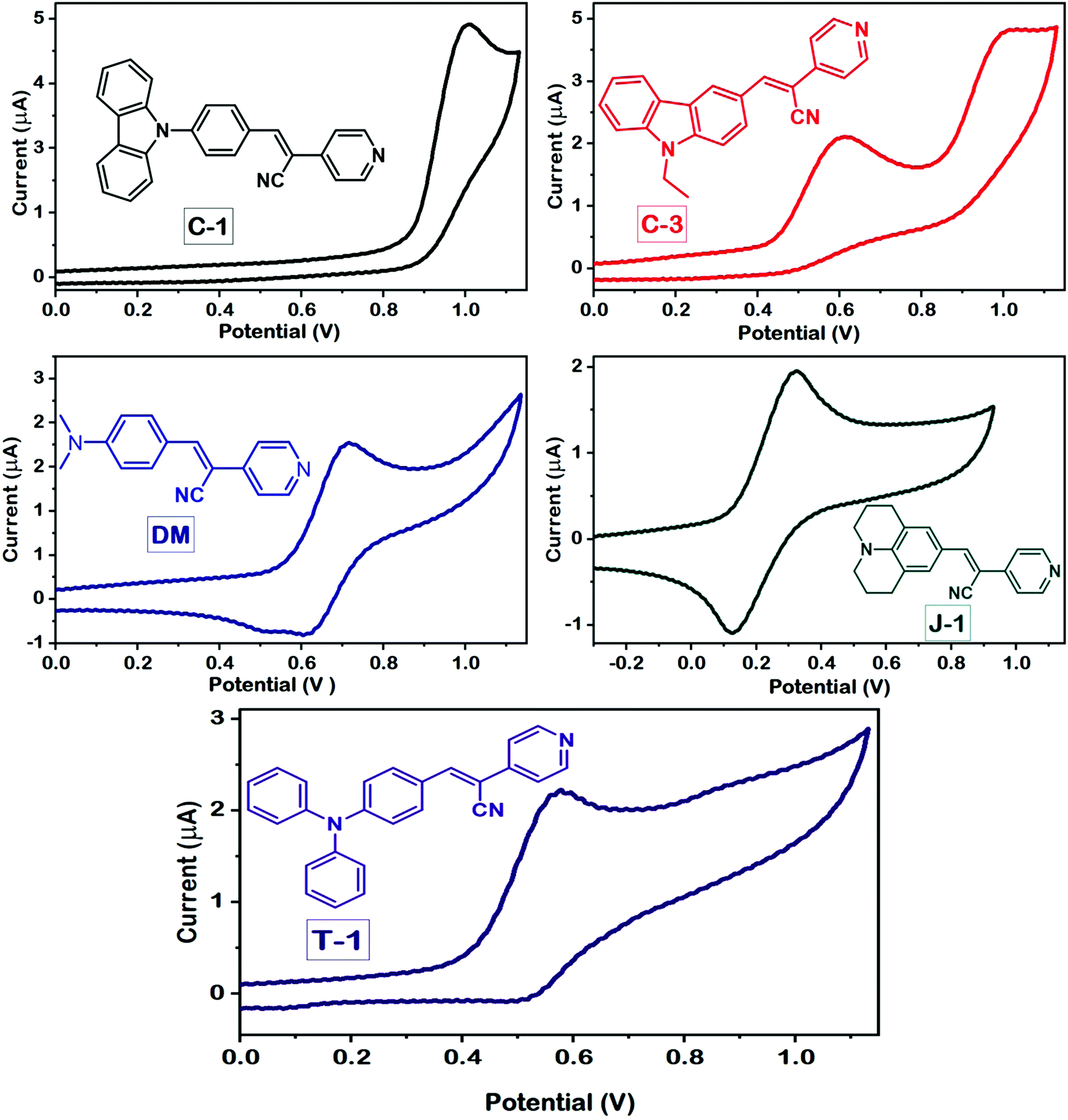
To evaluate the redox behavior and energy levels of the compounds, the cyclic voltammetry (CV) technique was performed in dry dimethylformamide (DMF) containing 0.1 M tetrabutylammonium tetrafluoroborate (TBAF) as a supporting electrolyte using a 1 mm diameter glassy carbon disc electrode as a working electrode, while a Pt wire and Ag/AgCl in DMF solution were used as a counter and reference electrode, respectively. Ferrocene was used as an internal standard during this experiment, and all the potential values were referenced against the ferrocene couple (FeCp2+/0). The frontier energy levels, the HOMO and LUMO, calculated from the oxidation potentials of the compounds measured using the CV technique and their comparison with DFT predicted results are displayed in Fig. 5 and 6, and the corresponding data are summarized in Table 2. All the compounds exhibited quasi-reversible oxidation peaks except J-1 where the oxidation process was found to be irreversible. The HOMO energy level of the molecules is calculated from the empirical formula EHOMO = −e[4.8 + Eox] eV. The optical band gap (E0-0) obtained from the absorption onset recorded in DMF and the LUMO is computed from the following equation: ELUMO = [E0-0 − EHOMO] eV.
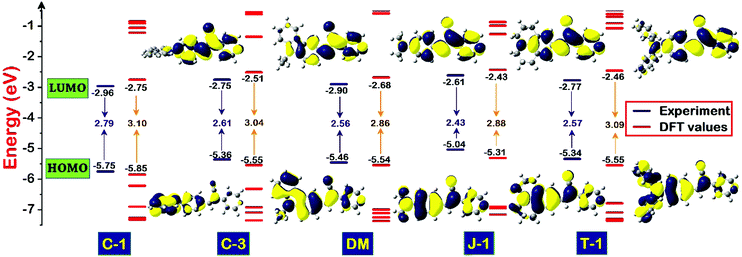 |
| | Fig. 5 Schematic diagram illustrating the energy level comparison of the acrylonitrile derivatives estimated from electrochemical-optical profiles and DFT predicted values along with their computed isodensity (0.02) surfaces of the frontier molecular orbitals involved in S0 → S1 transitions. | |
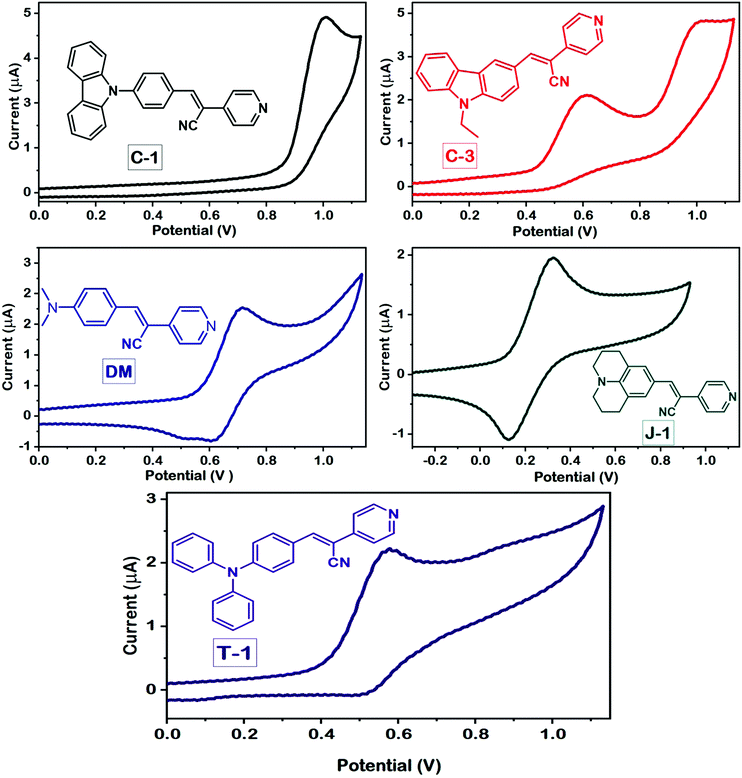 |
| | Fig. 6 Cyclic voltammograms of the compounds measured in DMF using a standard three-electrode configuration at a scan rate of 50 mV s−1. | |
Table 2 Data obtained from the optical-electrochemical profiles and energy levels obtained from DFT predictions
| Code |
λ
abs
(nm) |
λ
mono
(nm) |
λ
aggr
(nm) |
Emission intensityb (×106 a.u.) |
λ
em
shift (nm) |
Stokes shift (cm−1) |
E
ox
(V) |
HOMOe (eV) |
E
0-0
(eV) |
LUMOg (eV) |
|
Absorption and emission spectra of the monomers recorded in dioxane.
Emission spectra of the aggregates and their intensity.
Emission wavelength shift from monomers to aggregates.
Measured in DMF with 0.1 M tetrabutylammonium perchlorate (TBAPC) as a supporting electrolyte with a scan rate of 50 mV s−1.
Deduced from the formula HOMO = −e[4.8 + Eox].
Bandgap obtained from the absorption onset of the compounds.
LUMO = [E0-0 − EHOMO].
|
|
C-1
|
386 |
488 |
531 |
7.93 |
43 |
7074 |
0.95 |
−5.75 |
2.79 |
−2.96 |
|
C-3
|
389 |
554 |
558 |
0.79 |
4 |
7786 |
0.56 |
−5.36 |
2.75 |
−2.75 |
|
DM
|
420 |
522 |
560 |
1.97 |
38 |
4652 |
0.66 |
−5.46 |
2.90 |
−2.90 |
|
J-1
|
437 |
494 |
601 |
0.10 |
102 |
6244 |
0.24 |
−5.04 |
2.61 |
−2.61 |
|
T-1
|
408 |
474 |
603 |
0.68 |
136 |
7926 |
0.54 |
−5.34 |
2.77 |
−2.77 |
The pronounced torsional distortion of the carbazole–phenyl segment in C-1 abruptly lowers the HOMO. Connecting the third position of the carbazole in C-3 destabilized the HOMO to a certain extent due to planarity enhancement. It should be noted that the conjugation length is remarkably reduced from C-1 to C-3, which is reflected in their LUMO energy levels i.e., the LUMO of C-1 is lower than that of C-3. Compared to C-3, the HOMO of DM is further lowered because of slightly lower delocalization of the amino unit. The strong donating propensity of J-1 leads to pronounced HOMO destabilization and it has the smallest bandgap in the series. The HOMO of T-1 is nearly identical to that of C-3. The bandgaps of the compounds calculated from the optical profiles are in the following order (eV): J-1 (2.61) < C-3 (2.75) < T-1 (2.77) < C-1 (2.79) < DM (2.90) [Table 2]. Nevertheless, the HOMO and LUMO energy levels and the HOMO–LUMO gap predicted using the DFT/B3LYP/6-311G(d,p) level of theory undergo little variation, but follow a consistent trend with the experimental energy levels measured [Table 2].
Electron density distribution
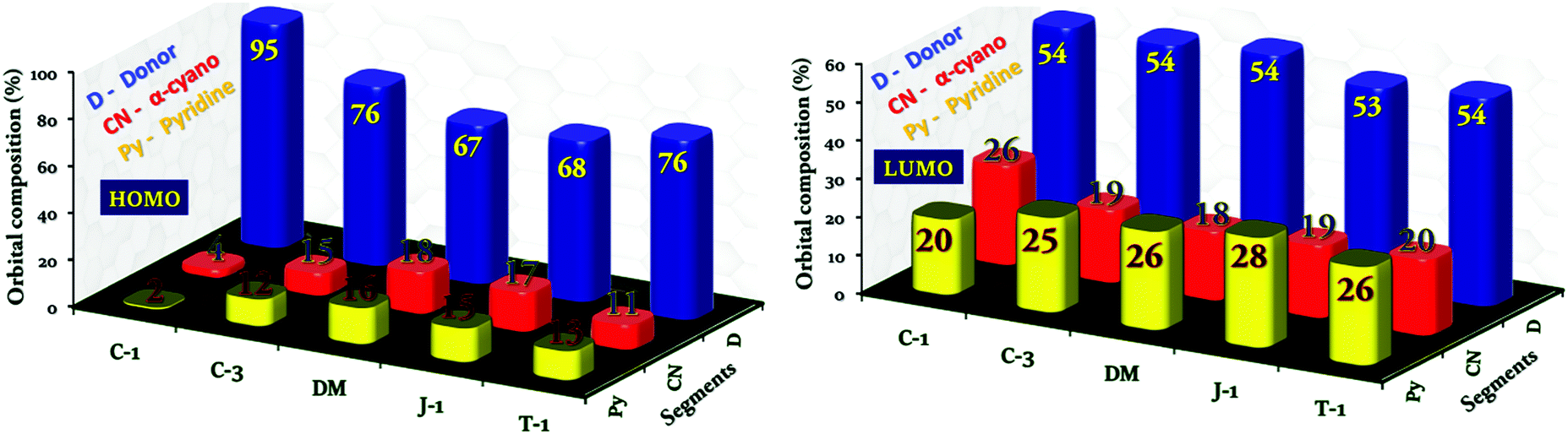
To delve into the type of charge transitions corresponding to the longer wavelength absorption, whether a π–π* or charge-transfer transition, we carried out electron density population analysis. The electron density of the HOMO in C-1 dominantly localizes on the carbazole unit, and the extent of delocalization was limited to the α-cyano unit without reaching the terminal pyridyl group. On the other hand, the electron density distribution is delocalized over the entire molecular framework for the remaining compounds in the series. In the LUMO, the electron density population is transferred from the donor unit to the α-cyano and terminal pyridyl groups, but the extent of electron density delocalization is majorly governed by the donor units attached. This indicates the presence of ICT transitions in the acrylonitrile derivatives. Unfortunately, it is hard to identify the donor's contribution toward the total charge population of the molecules. Hence, to compute the electron density contribution of the individual segments toward the charge density population of the units in terms of molecular orbital coefficients, we have carried out projected density of states (PDOS)41,42 analysis by grouping the atoms into three segments, donor (D), α-cyano (CN) and pyridyl (Py) units [Fig. 7]. In the HOMO, we found that the pronounced torsional distortion between the carbazole and phenyl groups is conducive for 94% electron density localization in C-1. A good electron delocalization characteristic feature has been found for the other compounds. The electron density distribution of the LUMO distinctly varies with respect to the donor units. For instance, a marginal increment of 26% electron density population is noticed on the α-cyano group in C-1, which in turn reduces the electron density of the terminal pyridyl segment. In the two-segment systems (C-3, DM, J-1, and T-1), the pyridyl unit prevails to retain the electron density over the α-cyano group. This analysis would be helpful to understand the donor's influence on the AIEE activity of α-cyano groups in the excited state.
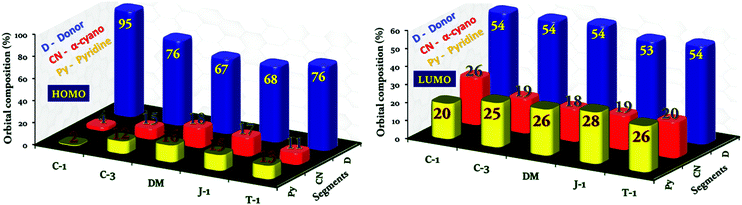 |
| | Fig. 7 Percentage molecular orbital density contribution from individual segments, the donor, α-cyano unit, and pyridine, to the charge density population of the acrylonitrile derivatives. | |
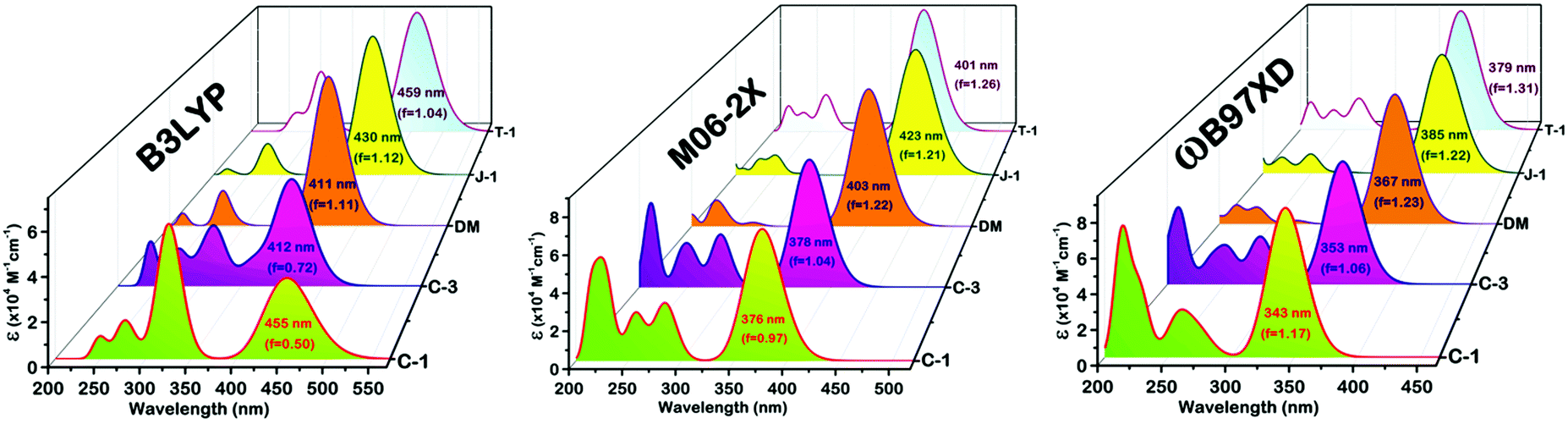
To simulate the photophysical features and major transitions involved corresponding to the maximum spectral region of the compounds, we used the TDDFT approach using different density functionals, TDDFT/B3LYP, M06-2X, and ωB97XD, with a common 6-311G(d,p) basis set.43 As the absorption spectra of the compounds were recorded in 1,4-dioxane, we have used the same solvent medium under the CPCM framework to mimic the real environment.44 The simulation of a significant portion of the absorption spectrum and the interpretation of the nature of the transitions were executed using GaussSum 3.0.2.45 Of these functionals, the B3LYP functional simulated values didn’t follow the trend of the experimental results and underwent significant deviation [Fig. 8]. Despite having the same pattern as the experimental values, the long-range corrected ωB97XD functional provides absorption values with a substantial underestimation of 30–60 nm. In comparison to the other functionals, the results obtained from M06-2X matched well with little underestimation, and the deviations are in the permissible range (Table 3). It is known that the oscillator strength is dependent on the wavefunction overlap between the frontier molecular orbitals HOMO and LUMO upon photoexcitation. If the overlap is weak, lower will be the oscillator strength (f).46 In our case, C-1 possesses a low f value due to the reduced overlap of orbital interactions between the highly twisted carbazole and phenyl groups. As a consequence, the major transition involved in the absorption maximum is also significantly reduced. C-3 and T-1 come second in the order [Fig. 8]. The transient dipole moment (μe) is found to be higher than the ground state. This observation corroborates the positive solvatochromism behavior of the α-cyanostilbene derivatives.
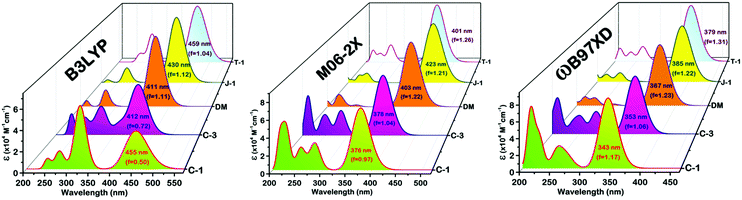 |
| | Fig. 8 TDDFT simulated optical absorption profiles along with their oscillator strength using various functionals, the B3LYP, M06-2X and ωB97XD/6-311G(d,p)/CPCM levels of theory. | |
Table 3 TDDFT simulated absorption values corresponding to major S0–S1 transitions and ground and transient dipole moment
| Code |
λ
B3LYP
(nm) |
λ
ωB97XD
(nm) |
λ
M06-2X
(nm) |
Major transitionsc |
HOMOb (eV) |
LUMOb (eV) |
E
H–L
(eV) |
μ
g
(D) |
μ
e
(D) |
Energy (Hartrees) |
|
Values obtained from various density functionals (B3LYP, M06-2X and ωB97XD)/6-311G (d, p)/C-PCM (1,4-dioxane) levels of theory, and oscillator strength values are given in parenthesis.
Values obtained from the DFT/B3LYP/-311G(d,p) level of theory.
Ground state dipole moment.
Transient dipole moment.
|
|
C-1
|
455 (0.50) |
343 (1.17) |
376 (0.97) |
HOMO → LUMO (72%), H−2 → LUMO (21%) |
−5.85 |
−2.75 |
3.10 |
3.99 |
4.93 |
−1165.53 |
|
C-3
|
412 (0.72) |
353 (1.06) |
378 (1.04) |
HOMO → LUMO (87%), H−2 → LUMO (5%), H−1 → LUMO (2%) |
−5.55 |
−2.51 |
3.07 |
7.73 |
9.65 |
−1013.08 |
|
DM
|
411 (1.11) |
367 (1.23) |
403 (1.22) |
HOMO → LUMO (94%), H−1 → LUMO (2%) |
−5.54 |
−2.68 |
2.86 |
8.63 |
10.69 |
−783.15 |
|
J-1
|
430 (1.12) |
385 (1.22) |
423 (1.21) |
HOMO → LUMO (94%), H−2 → LUMO (2%) |
−5.31 |
−2.43 |
2.88 |
9.28 |
11.79 |
−938.04 |
|
T-1
|
459 (1.04) |
379 (1.31) |
401 (1.18) |
HOMO → LUMO (87%), H−1 → LUMO (6%) |
−5.55 |
−2.46 |
3.09 |
7.09 |
8.20 |
−1166.71 |
AIEE studies
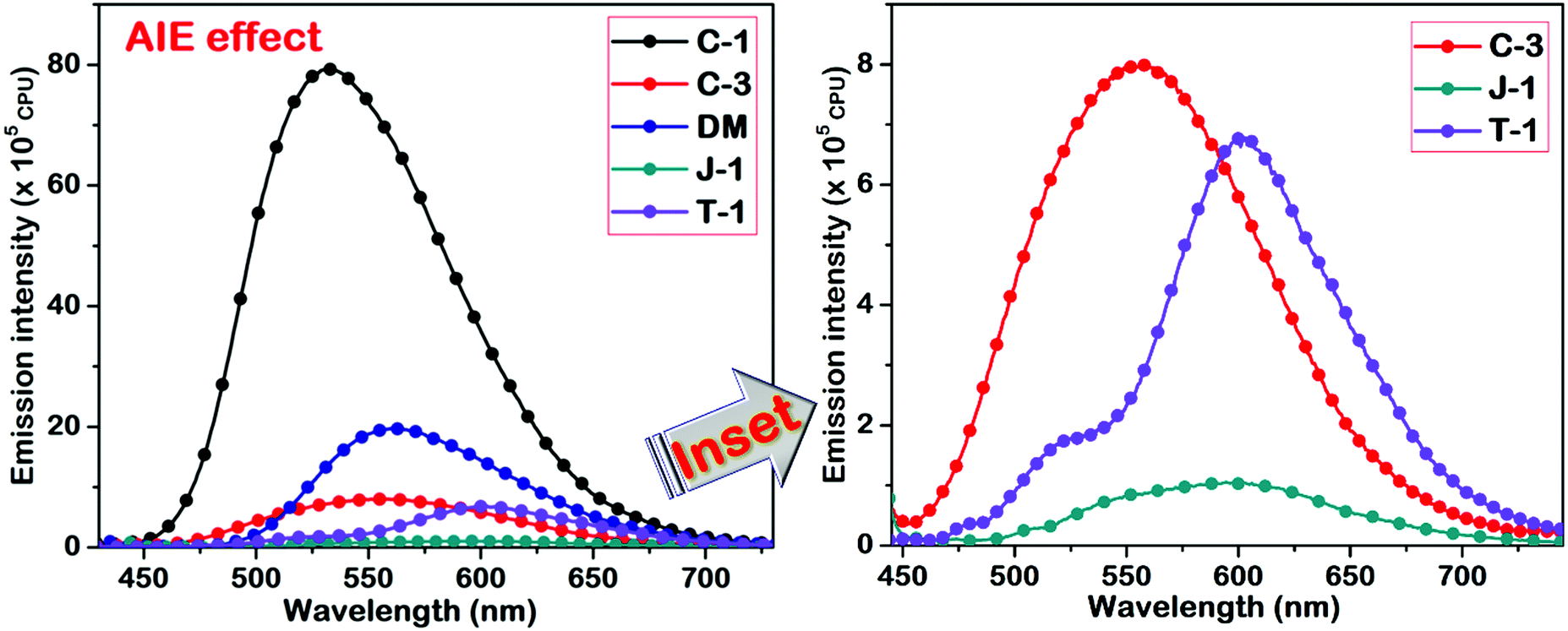
To investigate the influence of different amino donor groups on the AIE effect of acrylonitrile derivatives, we measured the emission profiles of the compounds by varying the composition of dioxane–water binary mixtures. All the synthesized derivatives exhibit the AIE effect in the dioxane–water binary combination, but the emission intensity and wavelength shift of the aggregates distinctly differ concerning the donor substitution. In the case of C-1, C-3, and DM, a gradual decrement was observed in the emission intensity along with a bathochromic shift upon the addition of a 70–80% water fraction into dioxane. At 100%, a new and broader band was noticed at a longer wavelength with higher intensity (C-1: 44 nm, C-3: 107 nm, DM: 38 nm, J-1: 101 nm; T-1: 122 nm) indicating the formation of J-aggregates [Fig. S5 (ESI†) and Fig. 9]. The nature of aggregation is not only dependent on the donor units but also on the positional substitution of the α-cyano moiety in the molecular framework. For instance, the α-cyano group placed at the central position mostly leads to H-type aggregation, and the placement at the terminal position renders J-type aggregation.2 It should be remembered that the AIE induced by tetraphenylethene (TPE) derivatives is mainly based on freezing the intramolecular motions of four dynamic free phenyl rings, resulting in emission intensity enhancement, but minimal variation in their emission wavelength shifts. Unlike the TPE or HPS derivatives, the AIE effect in acrylonitrile systems occurs via J or H-type aggregation through intermolecular interactions along with restriction of intramolecular rotation and intramolecular planarization in a synergistic way. As discussed earlier, the donor or acceptor substitutions introduced on the α-cyanostilbene scaffold are of paramount importance for the AIEE effect, i.e., the α-cyano moiety must interact freely with the neighboring molecules for efficient aggregation. Bulky donor/acceptor substitution closer to the α-unit might hamper the desired aggregation effect.47,48 Keeping these effects in mind, we examined the impact of the donor's substitution on the acrylonitrile scaffold.
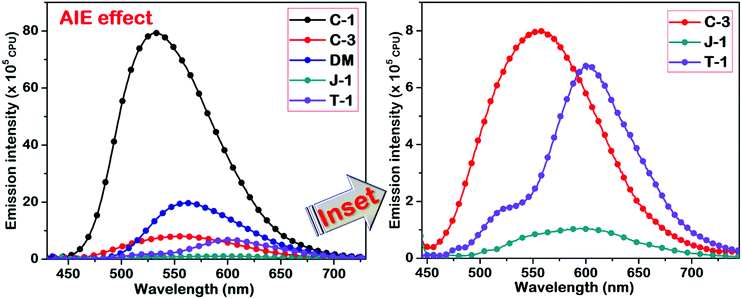 |
| | Fig. 9 AIE studies of the acrylonitriles performed in a dioxane–water binary solvent system. The inset represents the compounds with weak AIE characteristics. | |
In C-1, the phenyl π-linker keeps the α-cyano moiety away from the carbazole donor, thereby allowing efficient aggregation. The restriction of intramolecular rotation of the carbazole–phenyl segment upon aggregation increases the rigidity of the molecular system. As a result, the emission intensity of the C-1 aggregates is exceptionally high as compared to the other compounds. For C-3, the proximity of the α-cyano moiety to the carbazole group and the steric effect of N-alkyl substitution lead to minimal AIE effect/emission wavelength shifts. On the contrary, DM containing the smaller dimethylamino group didn’t perturb the α-cyano unit, and a comparable AIE intensity with a wavelength shift of 38 nm upon aggregation was observed. Among the compounds studied, julolidine possesses strong electron-donating propensity and undergoes efficient J-type aggregation but shows only minor emission intensity enhancement due to its cyclohexyl chain. The AIE peak of T-1 is higher in emission intensity as compared to J-1 and this can be assigned to the restriction of intramolecular rotation of the two phenyl rings that undergo restriction of molecular motions upon aggregation. J-1 and T-1 showed nearly the same AIE emission wavelength maxima (of ∼600 nm). A significant shift of 121 nm has been found for T-1 because of its absorption at a shorter wavelength over J-1. We also examined the stability of the compounds based on the energy obtained through DFT optimized results. The obtained values demonstrated that the energy of T-1 was found to be more stable due to its prominent delocalization over the triphenylamine segment, and for C-1 it can be assigned its extended conjugation of the molecular system.
The results obtained from this study reveal that the aggregation propensity distinctly varies with various donor substitutions such as carbazole, triphenylamine, julolidine, and the smaller dimethylamino group. Carbazole and triphenylamine have bulkier and rigid donor units, and julolidine is made of fused and flexible aliphatic chains. In C-1, although the carbazole donor is bulky, the cyano unit is distal to the donor group and can interact efficiently. However, the sizeable torsional deviation between the carbazole and phenyl segments leads to poor overlap of orbital interactions, enabling hypsochromic emission shifts as compared to the others in the series. It should be noted that the rigidity of the donor unit enhanced the AIE intensity but not the wavelength. Likewise, substitutional changes in C-3 result in dramatic reduction of the AIE emission intensity. In DM, bathochromic emission is observed where the size of the donating group has a weaker influence on the interactions of the cyano unit. Thus, free interactions of the cyano unit with the neighboring unit yield a bathochromic shift due to J-type aggregation. The crystal structure reported by Percino et al. also shows the propensity of the molecule to form J-aggregates.38 In T-1, the bulky triphenylamine unit hampers the aggregation of α-cyano groups and the same pattern is observed in the case of J-1. The more flexible side chains of J-1 reduced the AIE emission intensity and showed lower aggregation behavior. From these studies, we conclude that the emission in an organic/water mixture gradually shifts from aggregation-induced emission (AIE) characteristics to aggregation caused quenching (ACQ) in the following order C-1 > DM > C-3 > T-1 > J-1. It is solely based on the bulky nature and rigidity of the donor units and their interaction with the α-cyano motif.
In comparison to their emission profiles in organic solvents, the compounds exhibited a distinct bathochromic shift in water, which is a clear indication of molecules undergoing aggregation. The possible aggregation sites considered in these derivatives are the α-cyano and pyridyl units, which can interact with the positive sites of the donor parts from neighboring molecules. Hence, the aggregation is mainly determined by the negative charge density populated on the N-atom of the cyano and pyridyl units, respectively. For this purpose, we performed Mulliken population analysis to examine the charge density population on each atom of the title compounds (Fig. S6, ESI†).
This analysis is explicitly sensitive to the basis set choice but can provide the optimal results in a qualitative trend upon increasing the size of the basis set. For this purpose, we used the common but larger 6-311++G(d,p) basis set for all the resultant compounds, which produces reliable results. According to this study, the charge population on the N-atom of the cyano moiety in all the compounds is given in the following order: C-1 (−0.233) < T-1 (−0.242) ≈ C-3 (−0.243) < DM (−0.247) < J-1 (−0.252) and the order of the charge population on the N-atom of the pyridyl group is: C-1 (−0.285) < T-1 (−0.289) ≈ C-3 (−0.289) < DM (−0.291) ≈ J-1 (−0.252). In addition, the negative charge density on the N-atom from the amino donor part indicates its electron donation strength and the order obtained is: C-1 (−0.599) > J-1 (−0.558) > C-3 (−0.541) > T-1 (−0.494) > DM (−0.482). Despite the larger electron density on the N-atom in C-1, the large distortion between the carbazole and phenyl segments hampers its donating strength. We also performed electrostatic surface potential (ESP) analysis of all the compounds and the results are depicted in Fig. S7 (ESI†). Alkyl substituted amino group such as dimethylamino units, julolidine, N-ethyl unit of carbazole bears a positive density population whereas the carbazole of C-1 possesses neutral charge density.
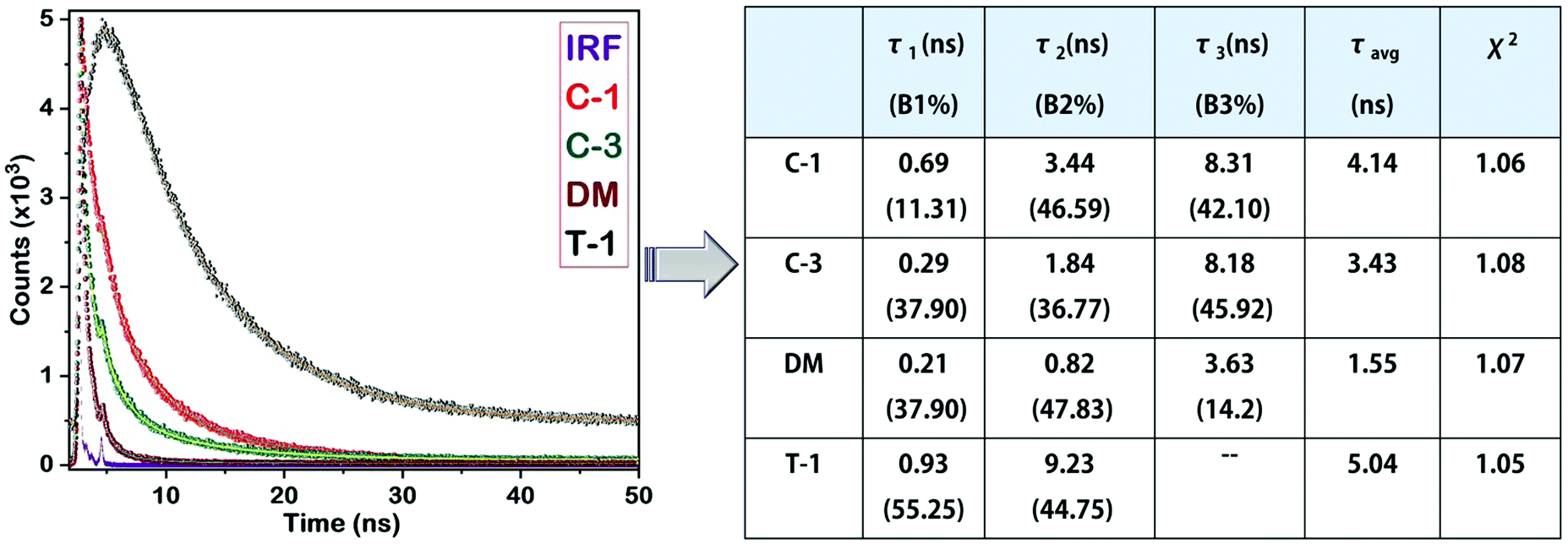
Lifetime study
As previously discussed, although various working mechanisms have been reported behind the AIE phenomena, we found that two factors mainly determine the AIE characteristics of the compounds in this study. One is freezing of molecular motions upon aggregation, helping to open up the radiative channels for energy dissipation. Second is restriction of intramolecular rotation of the α-cyano unit and its subsequent planarization, allowing the molecule to undergo efficient H or J-type aggregation. Previous research demonstrated that the fluorescence quantum yield produced by H-type aggregates is remarkably higher than that of the J-type counterparts, and it is reflected in their emission lifetimes.2 Before examining the aggregates, it is important to understand the emission lifetime of the monomers for better comparison. Measurement of the time-resolved fluorescence lifetime shows monomer emission lifetimes of a few tens of picoseconds, implying their preference for energy dissipation through non-radiative channels and due to the dynamic intramolecular rotation of α-cyanostilbenes. On the contrary, the emission lifetime of the aggregates is enhanced with a multi-exponential decay. The aggregates of C-1, C-3, and DM exhibited tri-exponential decays, whereas T-1 shows a two-exponential decay (Fig. 10). In the case of J-1, we were unable to perform the lifetime decay studies due to its minor emission enhancement. A minimal lifetime of the first exponential decay can be assigned to the possible existence of monomers in the solution of aggregates. The second exponential decays of the C-1, C-3 and DM aggregates are in the following order (ns): T-1 (9.23) > C-1 (3.44) > C-3 (1.84) > DM (0.82). These values mainly originated from the emission due to the restriction of intramolecular rotation of the donor segments. For instance, the greater emission lifetime of T-1 can be ascribed to freezing the dynamic rotations of the two phenyl rings of the triphenylamine donor segment. This helps to rigidify T-1 upon aggregation in an efficient way, yielding a longer emission lifetime. For C-1, the IMR of the carbazole–phenyl segment yields an average emission lifetime as compared to T-1. Compound C-3 bearing bulky and aromatic carbazole shows a considerably larger lifetime than DM. The third exponential decay is absent in the case of T-1, implying that the bulky phenyl rings of the TPA donor perturb the AIE activities of α-cyanostilbenes to a greater extent. On the contrary, the carbazole unit in C-1 keeps the cyano moiety away and allows the molecule to undergo efficient J-type aggregation. The emission lifetime of C-1 corresponding to the third exponential is found to be the largest in the series. In comparison to that of C-1, the emission lifetime of C-3 is marginally lowered. The smaller dimethylamino donor units' influence on the α-cyanostilbenes may be inferior in DM and subsequently it possesses the lowest emission lifetime (Fig. 10). The second and third exponential decays can be attributed to the RIR of the donor and α-cyanostilbene segments, respectively. The observation from this study demonstrated that the rigidification of the molecular framework is proportional to the emission lifetime.
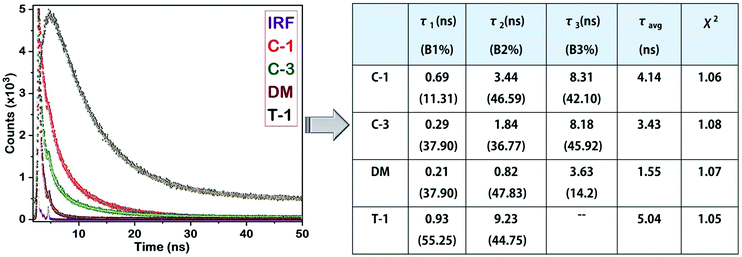 |
| | Fig. 10 Lifetime decay profiles of C-1, C-3, DM and T-1, and their corresponding data. | |
SEM and DLS study
The morphology of the compounds was assessed using scanning electron microscopy and dynamic light scattering measurements. The SEM images are shown in Fig. 11 and show disordered aggregates for J-1 and lump like structures for C-1, C-3, and DM. A network pattern is noted for T-1. Dynamic light scattering study is used to determine the size of the aggregated particles in solution [Table S1, ESI†]. All the α-cyanostilbenes have shown a polydispersity index (PdI) of more than 0.2, which is an indication of the polydisperse nature of the aggregated molecules (Fig. S9, ESI†). However, the mean diameter of the particles in C-1 is measured to be 172.2 nm, where the aggregation was defined by the α-cyano unit. In the case of T-1, this value is abruptly increased to 3940 nm where the aggregation originated from the restriction of intramolecular rotations of phenyl rings from the TPA donor. The PdI value of C-1 illustrated a monodisperse nature. The order of the Z-average value for all α-cyanostilbene molecules is in the following order (units in nm): T-1 > C-3 > J-1 > DM > C-1.
 |
| | Fig. 11 Drop-cast SEM of J-1 and T-1 [Fig. S8, ESI†]. | |
Conclusions
In summary, we synthesized and characterized a series of 4-pyridyl acrylonitriles bearing different donating groups [carbazole (C-1 and C-3), dimethylamino (DM), julolidine (J-1) and triphenylamine (T-1)]. The restrictions imposed due to the intramolecular rotation of the aromatic ring structure or donating groups can explain the changes in the AIE behavior. The energy levels obtained from the optical-electrochemical profiles distinctly vary with the nature of the donor substitution. The bulkiness of the donor substitution has a great impact on the aggregation propensity of the acrylonitrile derivatives. The AIE characteristics of the compounds gradually decreased in the order C-1 > DM > C-3 > T-1 > J-1. The rigid carbazole unit and distal substitution of the cyano unit (C-1) yield a greater AIE effect due to efficient interactions with neighboring units. The significant differences found between the AIE intensities in T-1 and J-1 reveals the impact of the rigidity of the donor unit and its influence on the α-cyano unit, i.e., moving from AIE to ACQ. The absence of distinct rigidity caused by the aromatic rings in DM reduced the AIE intensity but yielded a bathochromic shift. Electron density distribution and projected DOS analysis of the compounds obtained from DFT and TDDFT methods reinforced our experimental observations. The AIE effect and lifetime decay analysis of the compounds are scrutinized to probe the structure–property relationship of the acrylonitrile derivatives. Eventually, we found that the geometrical impact of substitutions, donors, and their substitutions such as alkyl/cyclic or aromatic units and their distance from the cyano motif determine the type of molecular interactions. We believe that this study gives a better understanding of the donor's influence on the AIE effect of acrylonitrile derivatives and may help in providing design guidelines to develop novel AIEgens for various optoelectronic applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the Bioinformatics Resources and Applications Facility (BRAF) C-DAC, Pune for the computational facilities, Board of Research in Nuclear Sciences (37(2)/14/05/2016) for the funding and IIT Gandhinagar for infrastructural facilities.
References
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544–554 CrossRef CAS PubMed.
- L. Zhu and Y. Zhao, J. Mater. Chem. C, 2013, 1, 1059–1065 RSC.
- Z. He, C. Ke and B. Z. Tang, ACS Omega, 2018, 3, 3267–3277 CrossRef CAS PubMed.
- N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 20, 15349–15353 CrossRef CAS PubMed.
- L. Mao, Y. Liu, S. Yang, Y. Li, X. Zhang and Y. Wei, Dyes Pigm., 2019, 162, 611–623 CrossRef CAS.
- X. Zhang, K. Wang, M. Liu, X. Zhang, L. Tao, Y. Chen and Y. Wei, Nanoscale, 2015, 7, 11486–11508 RSC.
- Y. Chen, J. W. Y. Lam, R. T. K. Kwok, B. Liu and B. Z. Tang, Mater. Horiz., 2019, 6, 428–433 RSC.
- F. Khan, A. Ekbote and R. Misra, New J. Chem., 2019, 43, 16156–16163 RSC.
- Y. Cai, L. Du, K. Samedov, X. Gu, F. Qi, H. H. Y. Sung, B. O. Patrick, Z. Yan, X. Jiang and H. Zhang, Chem. Sci., 2018, 9, 4662–4670 RSC.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- G. Fan and D. Yan, Sci. Rep., 2014, 4, 4933 CrossRef CAS PubMed.
- Y. Zhang, G. Zhuang, M. Ouyang, B. Hu, Q. Song, J. Sun, C. Zhang, C. Gu, Y. Xu and Y. Ma, Dyes Pigm., 2013, 98, 486–492 CrossRef CAS.
- Y. Zhang, J. Sun, X. Lv, M. Ouyang, F. Cao, G. Pan, L. Pan, G. Fan, W. Yu, C. He, S. Zheng, F. Zhang, W. Wang and C. Zhang, CrystEngComm, 2013, 15, 8998–9002 RSC.
- K. A. N. Upamali, L. A. Estrada, P. K. De, X. Cai, J. A. Krause and D. C. Neckers, Langmuir, 2011, 27, 1573–1580 CrossRef CAS PubMed.
- Y. Zhang, H. Li, G. Zhang, X. Xu, L. Kong, X. Tao, Y. Tian and J. Yang, J. Mater. Chem. C, 2016, 4, 2971–2978 RSC.
- M. Paramasivam and S. Kanvah, J. Phys. Chem. C, 2016, 120, 10757–10769 CrossRef CAS.
- P. O. Suntsova, A. K. Eltyshev, T. A. Pospelova, P. A. Slepukhin, E. Benassi and N. P. Belskaya, Dyes Pigm., 2019, 166, 60–71 CrossRef CAS.
- S. Redon, G. Eucat, M. Ipuy, E. Jeanneau, I. Gautier-Luneau, A. Ibanez, C. Andraud and Y. Bretonnière, Dyes Pigm., 2018, 156, 116–132 CrossRef CAS.
- M. Martínez-Abadía, R. Giménez and M. B. Ros, Adv. Mater., 2018, 30, 1704161 CrossRef PubMed.
- X. Wang, Z. Ding, Y. Ma, Y. Zhang, H. Shang and S. Jiang, Soft Matter, 2019, 15, 1658–1665 RSC.
- R. D. Fonseca, M. G. Vivas, D. L. Silva, G. Eucat, Y. Bretonnière, C. Andraud, L. De Boni and C. R. Mendonça, J. Phys. Chem. C, 2018, 122, 1770–1778 CrossRef CAS.
- H. Lai, Y. Xiao, S. Yan, F. Tian, C. Zhong, Y. Liu, X. Weng and X. Zhou, Analyst, 2014, 139, 1834–1838 RSC.
- N. Zhao, P. Li, J. Zhuang, Y. Liu, Y. Xiao, R. Qin and N. Li, ACS Appl. Mater. Interfaces, 2019, 11, 11227–11237 CrossRef CAS PubMed.
- B. Kumari, S. P. Singh, R. Santosh, A. Dutta, S. S. Mallajosyula, S. Ghosal and S. Kanvah, New J. Chem., 2019, 43, 4106–4115 RSC.
- J. Seo, J. W. Chung, I. Cho and S. Y. Park, Soft Matter, 2012, 8, 7617–7622 RSC.
- K. Mahesh, V. Priyanka, A. S. Vijai Anand and S. Karpagam, J. Mol. Struct., 2018, 1154, 445–454 CrossRef CAS.
- B. Wang and C. Wei, RSC Adv., 2018, 8, 22806–22812 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- E. G. Hohenstein, S. T. Chill and C. D. Sherrill, J. Chem. Theory Comput., 2008, 4, 1996–2000 CrossRef CAS PubMed.
- C. Walter, V. Krämer and B. Engels, Int. J. Quantum Chem., 2017, 117, e25337 CrossRef.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- Y. Wang, G. Lai, Z. Li, Y. Ma, Y. Shen and C. Wang, Tetrahedron, 2015, 71, 2761–2767 CrossRef CAS.
- K. Guzow, M. Czerwińska, A. Ceszlak, M. Kozarzewska, M. Szabelski, C. Czaplewski, A. Łukaszewicz, A. A. Kubicki and W. Wiczk, Photochem. Photobiol. Sci., 2013, 12, 284–297 RSC.
- M. Paramasivam, A. Gupta, N. J. Babu, K. Bhanuprakash, S. V. Bhosale and V. J. Rao, RSC Adv., 2016, 6, 66978–66989 RSC.
- O. Kwon, S. Barlow, S. A. Odom, L. Beverina, N. J. Thompson, E. Zojer, J.-L. Brédas and S. R. Marder, J. Phys. Chem. A, 2005, 109, 9346–9352 CrossRef CAS PubMed.
- J. T. Buck, R. W. Wilson and T. Mani, J. Phys. Chem. Lett., 2019, 10, 3080–3086 CrossRef CAS PubMed.
- M. J. Percino, M. Cerón, P. Ceballos, G. Soriano-Moro, O. Rodríguez, V. M. Chapela, M. E. Castro, J. Bonilla-Cruz and M. A. Siegler, CrystEngComm, 2016, 18, 7554–7572 RSC.
- M. J. Percino, V. M. Chapela, M. Cerón, G. Soriano-Moro, M. E. Castro and F. J. Melendez, J. Mol. Struct., 2013, 1034, 238–248 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. Paramasivam, R. K. Chitumalla, J. Jang and J. H. Youk, Phys. Chem. Chem. Phys., 2018, 20, 22660–22673 RSC.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, J. Phys. Chem. A, 2013, 117, 12590–12600 CrossRef CAS PubMed.
- Y. Takano and K. N. Houk, J. Chem. Theory Comput., 2005, 1, 70–77 CrossRef PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- J.-L. Brédas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691–1699 CrossRef PubMed.
- X. Feng, J. Zhang, Z. Hu, Q. Wang, M. M. Islam, J.-S. Ni, M. R. J. Elsegood, J. W. Y. Lam, E. Zhou and B. Z. Tang, J. Mater. Chem. C, 2019, 7, 2283–2290 RSC.
- Y. Dong, J. Qian, Y. Liu, N. Zhu, B. Xu, C.-L. Ho, W. Tian and W.-Y. Wong, New J. Chem., 2019, 43, 1844–1850 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and 1H and 13C NMR of the compounds are provided. See DOI: 10.1039/c9nj03693h |
| ‡ Equal contribution. |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Shikha
Khandelwal
,
Arnab
Dutta
*,
Shikha
Khandelwal
,
Arnab
Dutta