
Interface and grain boundary passivation for efficient and stable perovskite solar cells: the effect of terminal groups in hydrophobic fused benzothiadiazole-based organic semiconductors†
Yanbo
Gao‡
a,
Yanjie
Wu‡
a,
Yue
Liu
a,
Min
Lu
a,
Lili
Yang
b,
Yinghui
Wang
c,
William W.
Yu
 ad,
Xue
Bai
*a,
Yu
Zhang
*a and
Qilin
Dai
*e
ad,
Xue
Bai
*a,
Yu
Zhang
*a and
Qilin
Dai
*e
aState Key Laboratory of Integrated Optoelectronics, College of Electronic Science and Engineering, Jilin University, 2699 Qianjin Street, Changchun, 130012, China. E-mail: baix@jlu.edu.cn; yuzhang@jlu.edu.cn
bKey Laboratory of Functional Materials Physics and Chemistry of the Ministry of Education, Jilin Normal University, Siping 136000, Jilin, China
cFemtosecond Laser Laboratory, State Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun 130012, P. R. China
dDepartment of Chemistry and Physics, Louisiana State University, Shreveport, LA 71115, USA
eDepartment of Chemistry, Physics, and Atmospheric Sciences, Jackson State University, Jackson, Mississippi 39217, USA. E-mail: qilin.dai@jsums.edu
First published on 21st September 2020
Abstract
The defects at the interface and grain boundaries (GBs) of perovskite films limit the performance of perovskite solar cells (PSCs) seriously. Herein, organic semiconductors with different terminal groups including a ladder-type electron-deficient-core-based fused structure (DAD) fused core with 2-(3-oxo-2,3-dihydro-1H-inden-1 ylidene)malononitrile (BTP-4H), DAD with 2-(5,6-dichloro-3-oxo-2,3-dihydro-1H-inden-1 ylidene)malononitrile (BTP-4Cl), and DAD with 2-(5,6-difluoro-3-oxo-2,3-dihydro-1H-inden-1 ylidene)malononitrile (BTP-4F) are introduced into perovskite films to study the effects of the terminal groups on the PSC performance. A physical model is proposed to understand the effects of the terminal groups on the perovskite growth and energy level alignment of devices. Compared with BTP-4H and BTP-4Cl, BTP-4F can more effectively delay the crystallization rate and increase the crystal sizes due to hydrogen bonding of F and FA. BTP-4F can also provide more efficient charge transport channels due to the optimal energy level alignment. Most importantly, BTP-4F can promote charge transport from the perovskite film to spiro-OMeTAD and to SnO2, thus realizing simultaneous up-bottom passivation of perovskite films. Finally, the BTP-4F passivated PSCs exhibit a remarkable PCE of 22.16%, and the device can maintain ∼86% of the initial PCE after 5000 h. Therefore, this work presents significant potential of organic semiconductors in PSCs toward high efficiency and high stability due to the terminal groups.
New conceptsIn this work, organic semiconductors with different terminal groups of BTP-4H, BTP-4Cl, and BTP-4F are introduced into perovskite films to study the effects of the terminal groups on the PSC performance. A physical model is proposed to understand the effects of the terminal groups on the perovskite growth and energy level alignment of devices. Compared with BTP-4H and BTP-4Cl, BTP-4F can more effectively delay the crystallization rate and increase the crystal sizes due to hydrogen bonding of F and FA. BTP-4F can also provide more efficient charge transport channels due to the optimal energy level alignment. Most importantly, BTP-4F can promote charge transport from the perovskite film to spiro-OMeTAD and to SnO2, thus realizing simultaneous up-bottom passivation of perovskite films. Finally, the BTP-4F passivated PSCs exhibit a remarkable PCE of 22.16%, and the device can maintain ∼86% of the initial PCE after 5000 h. Therefore, this work presents significant potential of organic semiconductors in PSCs toward high efficiency and high stability due to the terminal groups. |
1. Introduction
The PCE of PSCs has been boosted rapidly from 3.8% to 25.2% in recent years by chemical composition engineering and interface manipulation of charge generation and transport layers.1–4 Although significant progress has been made in PSCs, there is still some space between the experimental PCE and the theoretical PCE (over 30%).5 Moreover, the stability of PSCs lags far behind the requirements of practical commercialization and it is still challenging to further improve the PCE and stability of devices by solution-processing techniques.6–8 Previous results showed that the PCE and stability of PSCs are significantly influenced by the recombination loss at interfaces and GBs of perovskite films.9–11 In addition, it has been reported that degradation of perovskite films occurs preferentially on GBs due to trap-assisted non-radiative recombination.12–18 These indicate that the stability and PCE of PSCs can be improved by increasing the grain size and reducing the GB density via regulating the perovskite crystallization process. In addition to the perovskite film quality, the energy level offsets between the perovskite layer and charge transport layers also play an important role in the stability and efficiency of PSCs. Research efforts have confirmed that the energy level alignment in devices has a significant impact on the PCE, which is also related to the device stability.19–21 Therefore, investigation of optimizing the energy level alignment of charge transport films and improving the crystallinity quality of perovskite films by passivating the GBs and interfaces is very necessary for the development of PSCs toward high efficiency and high stability.Some strategies have been demonstrated to optimize the energy level alignment and improve the crystallinity of perovskite films. For instance, Wu et al. reported an effective method to improve the crystallization quality of perovskite films by introducing [6,6]-phenyl-C61-butyric acid methyl ester into the anti-solvent.22 D. Bi et al. added an insulator into the anti-solvent solution to facilitate the nucleation process of perovskite films to obtain perovskite films with fewer defects.23 Polymer poly(vinylpyrrolidone) was also employed to improve the PCE and stability of PSCs.24–28 Some novel nonfullerene organic semiconductors with high electron mobility have been demonstrated to improve the performance of organic solar cells due to carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and cyano (C
O) and cyano (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) electron-withdrawing groups.29,30 It is reported that CO and CN groups in other small molecule materials have been proved to improve the crystallinity quality of the perovskite films through Lewis acid–base reactions.11 In spite of numerous studies, moreover, there is a lack of important guidelines for selecting and designing organic semiconductors to enhance the device performance due to poor understanding of the mechanisms of perovskite crystallization and molecule passivation. The fundamental understanding of the influence of organic molecules on perovskite films is still not clear. The interactions between the organic molecules and the perovskite lattice are still not well explained. The locations of the introduced organic molecules remain vague. The mechanisms of the modified perovskite crystal growth pathways, defect passivation, energy level offset, and charge transport by the organic molecules are still unknown.
N) electron-withdrawing groups.29,30 It is reported that CO and CN groups in other small molecule materials have been proved to improve the crystallinity quality of the perovskite films through Lewis acid–base reactions.11 In spite of numerous studies, moreover, there is a lack of important guidelines for selecting and designing organic semiconductors to enhance the device performance due to poor understanding of the mechanisms of perovskite crystallization and molecule passivation. The fundamental understanding of the influence of organic molecules on perovskite films is still not clear. The interactions between the organic molecules and the perovskite lattice are still not well explained. The locations of the introduced organic molecules remain vague. The mechanisms of the modified perovskite crystal growth pathways, defect passivation, energy level offset, and charge transport by the organic molecules are still unknown.
In this work, three nonfullerene organic semiconductors with different terminal groups are successfully introduced into perovskite films to analyze the effect of the terminal groups on the perovskite crystal growth and device performance. This new type of non-fullerene n-type organic semiconductors has attracted much attention in organic solar cells because of their great potential to obtain high PCEs. The organic molecules contain a large number of S, O and N atoms with lone pair electrons, which can passivate under-coordinated Pb2+ ions to improve charge transfer and collection, resulting in improved device efficiency. In addition, the hydrophobic properties of BTP-4X molecules can protect perovskite films from moisture, which can greatly enhance the moisture stability of PSCs. Meanwhile, BTP-4X has a series of optimal electrical properties, which are beneficial for electron extraction. They are novel organic semiconductors that have not been previously studied for passivation in perovskite optoelectronic devices. Finally, these three organic molecules indeed can passivate the Lewis acid traps of perovskite films effectively due to the same DAD fused core for the three organic molecules, which has been proved to passivate the Lewis acid traps.29,31 Furthermore, BTP-4F molecules have a strong hydrogen-bonding interaction with perovskite films, which can delay the crystallization process of the perovskite films by immobilizing FA to further improve the film quality. In addition, these molecules can diffuse to the interface of SnO2/perovskite to passivate interface defects to decrease charge recombination in the devices. Note that BTP-4F molecules can also provide more efficient charge transport channels compared to BTP-4H and BTP-4Cl due to the optimal energy level offset between BTP-4F and the perovskite layer. Most importantly, BTP-4F molecules not only promote the charge transport from the perovskite film to the hole transport layer (HTL), but also facilitate the charge transport from the perovskite to the SnO2 layer, thus realizing the simultaneous up-bottom interfacial passivation of perovskite films. Finally, the BTP-4F passivated devices reach an optimized PCE of 22.16%, and the device without any encapsulation could maintain nearly 86% of its initial efficiency after 5000 h storage under ambient conditions. It can keep ∼72% of its initial PCE after 30 h heating at 100 °C in air.
2. Results and discussion
Herein, three organic semiconductors (BTP-4H, BTP-4F and BTP-4Cl) are introduced into perovskite films to enhance the efficiency of PSCs. The effects of different terminal groups on the performance of the PSCs are also systematically studied. The structures of BTP-4F, BTP-4Cl and BTP-4H are very similar to each other except the terminal groups.32–34 The molecular structures of BTP-4H, BTP-4F and BTP-4Cl are shown in Fig. 1a. The organic molecules contain a large number of S, O and N atoms with lone pair electrons, which can passivate under-coordinated Pb2+ ions to improve charge transfer and collection, resulting in improved device efficiency.35 In addition, the hydrophobic properties of BTP-4X molecules can protect perovskite films from moisture, which can greatly enhance the moisture stability of PSCs.36 Therefore, the introduction of these molecules not only improves the device efficiency but also enhances the device stability. To fabricate a successful BTP-4X–perovskite device, BTP-4X should not damage the perovskite crystal structure. Therefore, BTP-4X can be introduced into perovskite films during the anti-solvent step. Fig. 1b displays the detailed preparation process of perovskite films modified by BTP-4X. Firstly, SnO2 layers are fabricated on FTO substrates by spin coating, and then the SnO2 layers are annealed at 150 °C for 30 min. Then, BTP-4X in chlorobenzene is dropped on the top of the perovskite films during the anti-solvent process, and then the films are crystallized by annealing at 150 °C for 15 minutes. Finally, perovskite films modified with BTP-4X are obtained. It is very possible that the BTP-4X molecules are located at the perovskite GBs to facilitate charge transport and improve the film crystallinity.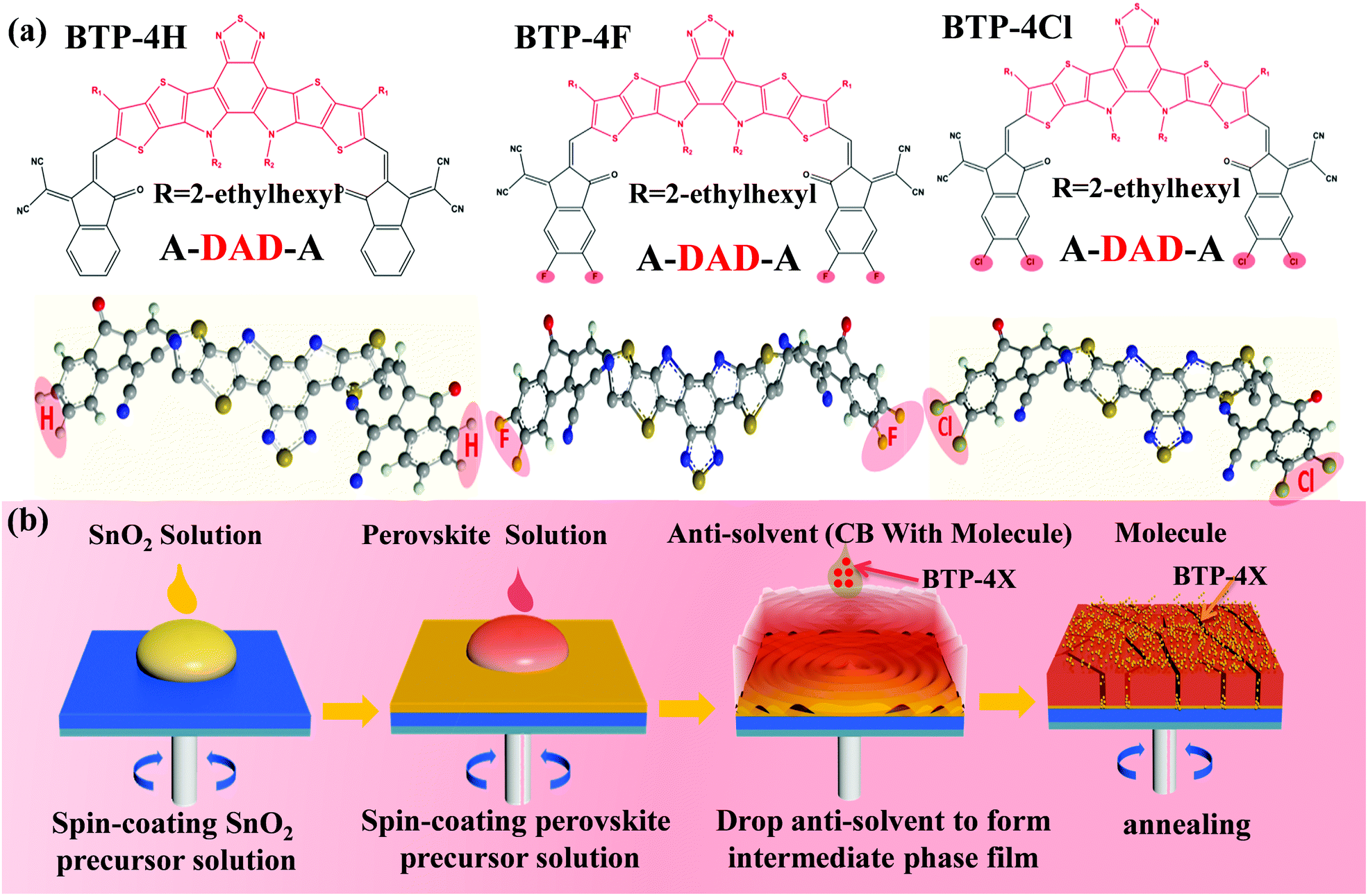 | ||
| Fig. 1 (a) Molecular structures of BTP-4X. (b) Schematic illustration of the growth process of perovskite films modified with BTP-4X. | ||
The effects of BTP-4X on the morphology and optical properties of perovskite films are systematically studied. Firstly, the morphology of the perovskite films modified with BTP-4X is investigated by scanning electron microscopy (SEM). Fig. 2a–d display the SEM images of perovskite films modified with BTP-4X. The control perovskite film (without BTP-4X) exhibits some voids and pinholes with a grain size of 100–600 nm. The voids and pinholes are detrimental to charge transport and collection. Surprisingly, the perovskite films with BTP-4X show increased grain sizes and appear to be more compact. These results show that the addition of BTP-4X, a Lewis base, could modify the crystallization process of the perovskite films toward large grain sizes.37–39 The average grain size of the films modified with BTP-4H, BTP-4Cl and BTP-4F is ∼500 nm, ∼500 nm and ∼800 nm, respectively (Fig. S1, ESI†). The control film shows a mean size of ∼400 nm. Therefore, the perovskite film with BTP-4F exhibits a larger grain size than that of BTP-4Cl and BTP-4H. It is possible that the fluoride terminal groups in BTP-4F influence the crystallization kinetics of the perovskite films toward a slow process, leading to large grain sizes.40
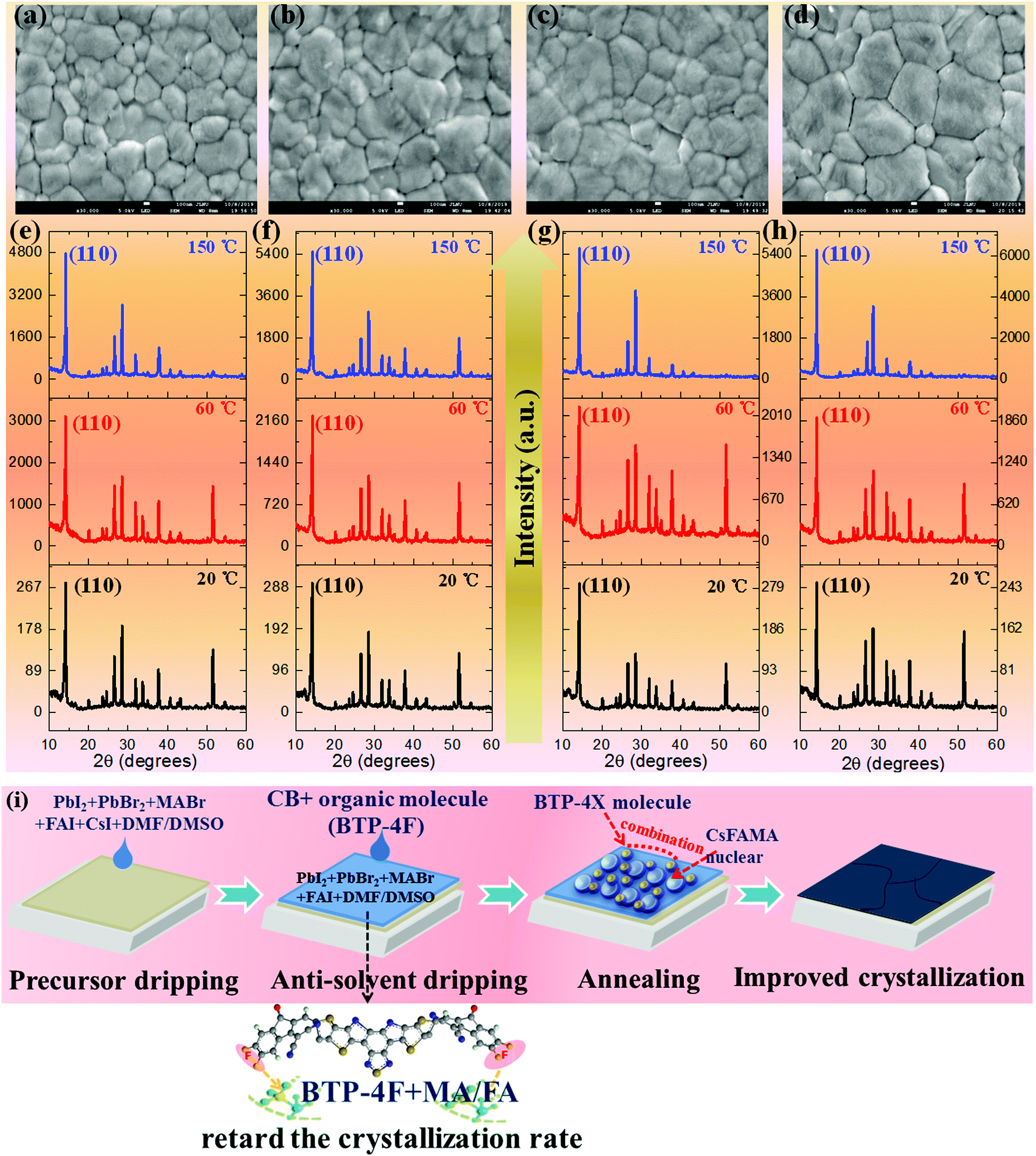 | ||
| Fig. 2 (a–d) Top-view SEM images of the perovskite films modified with different organic molecules: (a) the control perovskite film, (b) the perovskite film modified with BTP-4H, (c) the perovskite film modified with BTP-4Cl, and (d) the perovskite film modified with BTP-4F. Temperature-dependent XRD data of the perovskite films modified with different organic molecules: (e) the control perovskite film, (f) the perovskite film modified with BTP-4H, (g) the perovskite film modified with BTP-4Cl, and (h) the perovskite film modified with BTP-4F. (i) Schematic diagram of the improved perovskite film crystallinity by BTP-4F. | ||
To further study the effects of different organic molecules on the crystallization kinetics of perovskite films, the in situ X-ray diffraction (XRD) patterns of four kinds of perovskite films measured during annealing are shown in Fig. 2e–h and Fig. S2 (ESI†). It can be seen that all the unannealed perovskite films (labeled 20 °C in Fig. 2) show the same crystal structure. The diffraction peaks at 14.2°, 28.3° and 32.5° can be indexed to the crystal planes of (110), (220), and (310), respectively,38 indicating that all the perovskite films display the α black phase. This also indicates that the introduction of organic molecules does not affect the perovskite film phase and crystal structure. As the annealing temperature increases to 60 °C, all the XRD peak intensities increase, and the films display preferential growth along the (110) direction, which is evidenced by the increased relative intensity of the (110) crystal planes to other crystal planes. More importantly, the diffraction peak intensities of the (110) planes of the films modified with BTP-4H, BTP-4Cl and BTP-4F are estimated to be 2248, 2166 and 1915, respectively. The diffraction peak intensity of the (110) plane of the control film is 3122. This indicates that the small organic molecules delay the crystallization process of the perovskite films. The perovskite films modified with small molecules present higher diffraction peak intensities compared with the control perovskite film as the annealing temperature increases to 150 °C. The diffraction peak intensities of the (110) planes of the films without and modified with BTP-4H, BTP-4Cl and BTP-4F are estimated to be 4782, 5523, 5686 and 6300, respectively. This indicates that the perovskite film with BTP-4F exhibits the best crystallinity. Furthermore, as shown in Fig. S3 (ESI†), the full width at half maximum (FWHM) values of the (110) planes of BTP-4X are smaller than that of the control film. The perovskite film with BTP-4F shows the smallest FWHM value. This also implies that the three organic molecules can improve the crystallinity of the perovskite films, and the perovskite film with BTP-4F shows the best crystallinity, which is agreement with the SEM results. Therefore, the introduction of BTP-4X in the anti-solvent step could slow down the crystallization process of the perovskite films. It is reported that the hydrogen bonding of F and FA can lower the crystallization rate, leading to enhanced crystalline quality of perovskite films.40 In order to further study the interactions between BTP-4X and the perovskite films, the Fourier transform infrared (FTIR) spectra of BTP-4F, the perovskite, the perovskite film with BTP-4H, the perovskite film with BTP-4Cl and the perovskite film with BTP-4F were measured. As shown in Fig. S4 (ESI†), the peak at ∼3250 cm−1 is attributed to stretching vibration N–H in the perovskite film.41 As BTP-4F is introduced into the perovskite film, the stretching vibration peaks of N–H shift to low frequency due to the formation of hydrogen bonding between F and H–N.41 There is a strong hydrogen bonding interaction between BTP-4F and perovskite materials. The F in BTP-4F can immobilize FA ions, which slows down the crystallization process of the perovskite films and improves the crystalline quality. However, as BTP-4H and BTP-4Cl are introduced into the perovskite film, the stretching vibration peaks of N–H at ∼3250 cm−1 do not shift obviously, implying that the interactions between H/Cl and H–N are very weak. Therefore, the types of terminal groups of the molecules are very critical to control the perovskite film growth toward high crystallinity. The mechanism of the improved perovskite film crystallinity by BTP-4F is shown in Fig. 2i.
Cross-sectional SEM images and corresponding Energy Dispersive X-ray (EDX) mapping images are used to examine the locations of BTP-4F in the perovskite films. Fig. 3a and b display cross-sectional SEM images of the control and BTP-4F devices (PSCs modified with BTP-4F). The perovskite film with BTP-4F shows large crystalline grains compared to that of the control film. The large crystalline grains lead to a suppressed defect state density and enhanced carrier transport. Almost no GBs are observed, and the GBs are parallel to the substrate for the perovskite film with BTP-4F. These perovskite monolayer grains could be a perfect structure for a fast charge transfer process in the vertical direction. EDX mapping images further confirm the locations of BTP-4F in devices. The Cs element shows a uniform distribution, which indicates the uniform structure of the perovskite films. Moreover, the F element is distributed uniformly in the whole film. It is very possible that BTP-4F may be located at the GBs of the perovskite films. To further confirm the position of BTP-4F existing in the perovskite film, transmission electron microscopy (TEM) images were collected and are shown in Fig. 3c and d. We analyze the two highlighted regions of Fig. 3c and d; the two highlighted regions are marked by a red dotted frame and named regions 1 and 2. The regions of 1 and 2 are the interior and grain boundaries of the perovskite film, respectively. Fast Fourier transform (FFT) analysis in region 1 shows a lattice spacing of 0.32 nm, which is larger than that (0.31 nm) of a MAPbI3 film.42 The increased lattice spacing is attributed to the introduction of FA+ to replace part of MA+ to increase the spacing of the crystal planes.40 In region 2, compared with the control film, the inverse fast Fourier transform (IFFT) image of the perovskite film modified with BTP-4F clearly shows the formation of a small molecule–perovskite coating structure, which clearly shows that small molecules are introduced into the perovskite film and mainly distributed on the grain boundaries of the perovskite materials.
 | ||
| Fig. 3 Cross-sectional SEM images and the EDX element mapping images of the PSCs: (a) control device, and (b) BTP-4F device. (c) and (d) HRTEM images of the CsFAMA films without and with the BTP-4F additive. | ||
The influences of BTP-4X on the perovskite film surface and interfacial properties are investigated by Kelvin probe force microscopy (KPFM). The contact potential difference (CPD) between the tip and sample can be obtained by KPFM (see eqn (1))41,43,44
| vCPD = (ϕtip − ϕsample)/e | (1) |
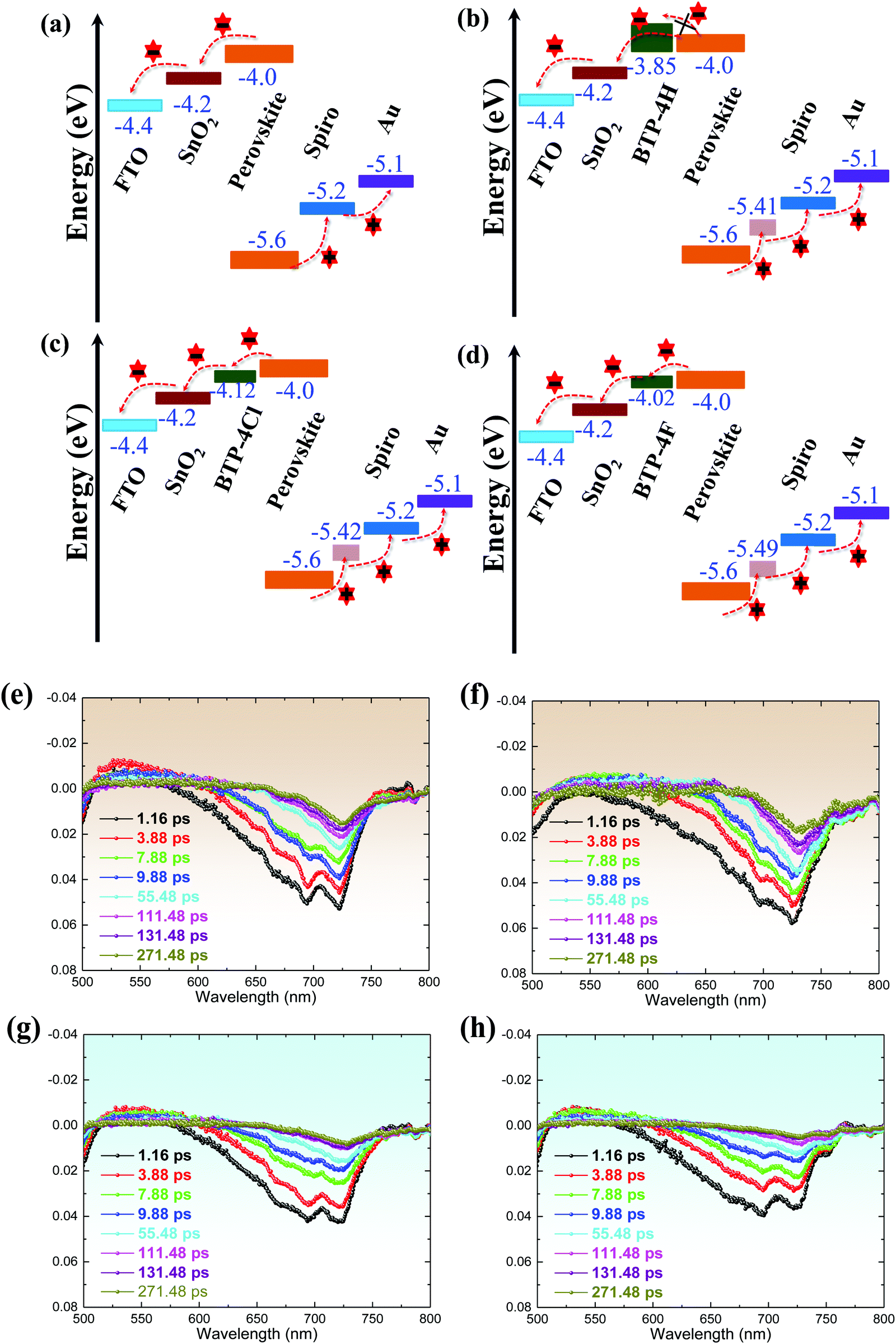 | ||
| Fig. 4 (a–d) Energy-level diagrams of the PSCs with different organic molecules. TAS spectral evolution at the FTO/SnO2/perovskite interface: (e) the control, (f) the perovskite film with BTP-4H, (g) the perovskite film with BTP-4Cl, and (h) the perovskite film with BTP-4F, measured under excitation light of 405 nm from the glass side. | ||
The charge separation and recombination processes of FTO/SnO2/perovskite and FTO/SnO2/perovskite with BTP-4X films are studied by Femto-second transient absorption spectroscopy (Fig. 4e–h). The photo-bleaching (PB) negative peak at ∼720 nm is attributed to the absorption of the perovskite films. Compared to the control perovskite film, it can be found that the perovskite films modified with BTP-4F and BTP-4Cl exhibit a faster decrease of the peak intensity at 720![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm than the other films, which indicates that the introduction of BTP-4F and BTP-4Cl can increase the charge transfer efficiency from the perovskite film to the SnO2 layer.45 However, the perovskite film modified with BTP-4H exhibits a slower decrease compared with the control perovskite film. Therefore, the introduction of BTP-4H hinders the charge transfer from the perovskite film to SnO2. The transient absorption data can be explained by the energy level positions of BTP-4X shown in Fig. 4a–d. The perovskite film modified with BTP-4F shows a better energy level alignment compared with BTP-4Cl and BTP-4H. Therefore, the introduction of BTP-4F should be beneficial to carrier transport in the devices compared to BTP-4Cl and BTP-4H, which is consistent with the results in Fig. 4a–d.
nm than the other films, which indicates that the introduction of BTP-4F and BTP-4Cl can increase the charge transfer efficiency from the perovskite film to the SnO2 layer.45 However, the perovskite film modified with BTP-4H exhibits a slower decrease compared with the control perovskite film. Therefore, the introduction of BTP-4H hinders the charge transfer from the perovskite film to SnO2. The transient absorption data can be explained by the energy level positions of BTP-4X shown in Fig. 4a–d. The perovskite film modified with BTP-4F shows a better energy level alignment compared with BTP-4Cl and BTP-4H. Therefore, the introduction of BTP-4F should be beneficial to carrier transport in the devices compared to BTP-4Cl and BTP-4H, which is consistent with the results in Fig. 4a–d.
In order to further study the influence of the types and the concentrations of organic semiconductors on the performance of PSCs, the influence of the doping concentrations of different organic semiconductors in chlorobenzene on the device performance is investigated and device parameters are listed in Table S1 (ESI†). As the content of BTP-4X in chlorobenzene increased from 0 mg mL−1 to 5 mg mL−1 the device efficiency increases and then it decreases with further increasing the concentration to 7 mg mL−1. Therefore, the optimal concentration of BTP-4H, BTP-4Cl and BTP-4F is 5 mg mL−1 for all three cases. Fig. 5a shows the J–V curves of cells modified with BTP-4X. The devices modified with BTP-4X are named BTP-4X devices. The control device shows relatively inferior performance (PCE = 19.25%, Jsc = 23.39 mA cm−2, Voc = 1.116 V and FF = 73.74%). The BTP-4H device shows a slightly increased PCE of 19.88% (Jsc = 23.02 mA cm−2, Voc = 1.148 V and FF = 75.26%). The slightly decreased Jsc for the BTP-4H device may be ascribed to the decreased carrier extraction caused by the mismatched energy level alignment. The increased FF and Voc of the BTP-4H device are attributed to the improved crystal quality and reduced recombination loss and surface defects of the perovskite thin films. The BTP-4Cl device exhibits an improved PCE of 20.99%, with a Jsc of 23.79 mA cm−2, a Voc of 1.16 V and an FF of 76.06%. The BTP-4F device exhibits the champion PCE in this work, reaching up to 22.16%, with a Jsc of 24.39 mA cm−2, Voc of 1.16 V and FF of 78.32%. The increased Voc values of the BTP-4Cl and BTP-4F device are attributed to the improved energy level alignment, which is beneficial to charge separation and transport in the devices. The increased FF and Jsc of the BTP-4Cl and BTP-4F devices are attributed to the improved crystal quality, and the reduced surface defects of the perovskite films. Compared with the other devices, the obvious increased PCE of the BTP-4F device may be attributed to (1) the defect passivation due to hydrogen bonding between F and FA and (2) the optimal energy level alignment between the perovskite layer and BTP-4F, which can improve the carrier extraction. Table S2 (ESI†) lists a comparison of reported studies for the application of various organic materials in PSCs. The present work obtains the highest PCE for PSCs based on various organic materials.4,47–51Fig. 5b presents the corresponding incident photon-to-electron conversion efficiency (IPCE) spectra and the integrated Jsc for the control, BTP-4H, BTP-4Cl and BTP-4F devices. The integrated Jsc values are 22.47, 21.98, 22.93 and 23.53 mA cm−2 for the control, BTP-4H, BTP-4Cl and BTP-4F devices, respectively.52
 | ||
| Fig. 5 (a) J–V curves of PSCs without and with molecules. (b) IPCE spectra of PSCs without and with molecules and corresponding integrated Jsc values of the devices. (c) and (d) Statistical histograms of Voc, Jsc, FF and PCE values obtained from over 50 devices modified with different molecules. (e) Steady-state current density and corresponding power conversion efficiency (PCE) with different devices. (f) J–V curves of devices obtained with different scan directions. | ||
The reason for the decreased Jsc for the BTP-4H device may be ascribed to the decreased carrier extraction caused by the mismatched energy level alignment.50 Devices were fabricated by the same process to confirm the reproducibility of the device performance. In order to study whether the introduction of organic semiconductors will affect the IPCE of PSCs, the IPCE spectra of PSCs without and modified with BTP-4X ranging from 750–850 nm are enlarged in Fig. S7 (ESI†). We do not observe IPCE enhancement corresponding to the absorption of BTP-4X (Fig. S6, ESI†), which is explained by the limited amount of organic semiconductor in the films.
The statistical distributions of the J–V parameters are shown in Fig. 5c and d. The distributions of all parameters are narrow, indicating that the device fabrication process is highly repeatable.52 The steady-state current density and PCE for the control, BTP-4H, BTP-4Cl and BTP-4F are shown in Fig. 5e. Jsc and the PCE of the control, BTP-4H, BTP-4Cl and BTP-4F devices are 22.35 mA cm−2, 21.5 mA cm−2, 22.95 mA cm−2, and 24.01 mA cm−2 and 18.45%, 18.93%, 20.48%, and 21.50%, respectively, which are very close to the results of the J–V curve. J–V hysteresis characterization is performed to study the stability of the PSCs. As shown in Fig. 5f, the J–V hysteresis effect is significantly eliminated by BTP-4X modification compared to the control device. The BTP-4F device shows two nearly overlapped J–V curves for the reverse and forward scan directions. More specifically, the PCE obtained from the reverse scan is 22.16%, while the PCE measured using the forward scan direction is 21.51%. The hysteresis index of the cells is described by
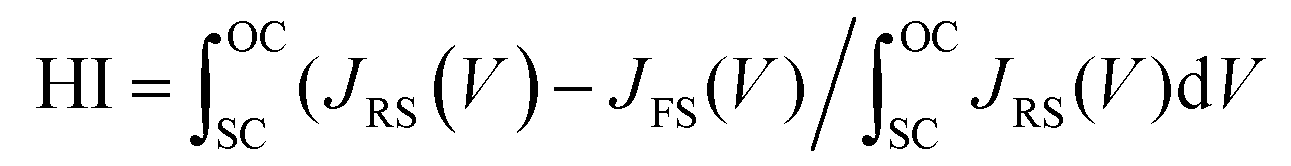 | (2) |
The hysteresis index for the BTP-4F device is 0.0293, lower than that of the control device (0.1896). The hysteresis index values for the four devices are summarized in Table S3 (ESI†). The BTP-4X devices exhibit decreased hysteresis, which is ascribed to the diminished trap density of perovskite films modified by organic molecules. These results confirm the excellent carrier transport properties of the modified films.
A series of optoelectronic measurements on the perovskite films is carried out to understand the passivation mechanisms of the molecules. Fig. 6a shows the steady-state photoluminescence (PL) spectra of the perovskite films on glass. The PL intensities of the perovskite films modified with organic molecules increase significantly compared with the control film, which indicates that the introduction of small organic molecules inhibits non-radiative recombination.53 In addition, a PL test was also carried out to study the carrier transfer process between the perovskite layer and the HTL. As shown in Fig. S8 (ESI†), the perovskite film shows strong fluorescence with an emission peak located at 775 nm. Obvious PL quenching is observed for the perovskite film modified with small molecules, which is attributed to the inhibited electron–hole pair recombination of the perovskite film by improving extraction of holes. This is consistent with the results obtained in Fig. 4. Electrical impedance spectroscopy (EIS) provides more information about the recombination process. The recombination resistance (Rrec) values of devices are evaluated. The equivalent circuit used to fit the EIS data is listed in the inset of Fig. 6b. The Rrec values tested in the dark and at 0 V bias are 73.85, 339.75, 626.92, and 898.14 Ω for the control, BTP-4H, BTP-4Cl and BTP-4F devices, respectively. These results indicate that the organic molecules can reduce the carrier recombination in the perovskite films, which is beneficial to the device performance. The dark-current curves show the leakage current formed by carrier recombination in the PSCs (Fig. 6c). The BTP-4F device exhibits the lowest dark current density compared to the other devices, indicating that the BTP-4F device inhibits charge carrier recombination and leakage current. The ideality factor (k) values of the devices are studied to reveal more information about charge recombination on GBs and interfaces in the PSCs modified by BTP-4X. The k values are calculated by the following equation:
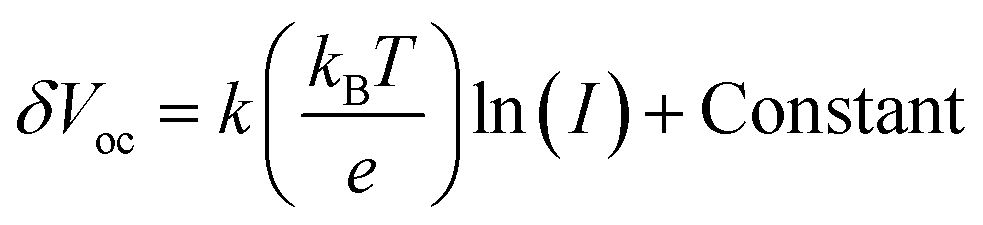 | (3) |
 | (4) |
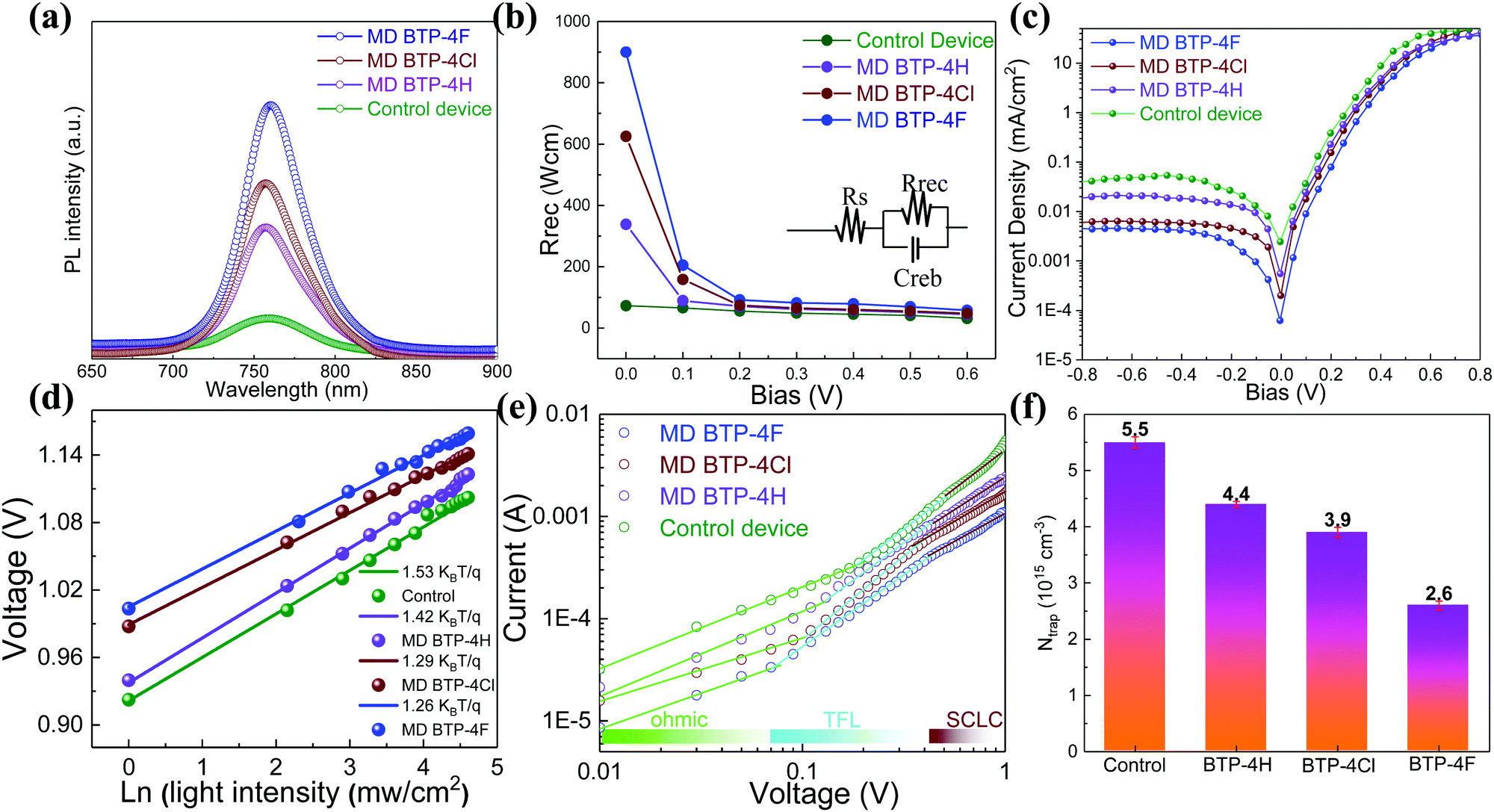 | ||
| Fig. 6 (a) Steady state PL spectra of CsFAMA films without and with organic molecules on the glass substrates. (b) Rrec obtained from fitting Nyquist plots through the equivalent circuit in the inset for PSCs without and with organic molecules. (c) Dark J–V curves of PSCs without and with organic molecules. (d) Voc dependence on the light intensity for the PSCs without and with organic molecules. (e) Current–voltage characteristics of the devices with an ITO/SnO2/CsFAMA/PCBM/Au configuration. (f) Corresponding defect density values obtained from (e). | ||
It is well known that BTP-4X molecules have hydrophobic properties, which can prevent moisture from permeating into perovskite films, leading to improved device stability. Fig. 7a shows the long-time stability of the four PSCs. The PSCs were stored in the dark in ambient air (humidity: 20–30%, temperature: ∼25 °C). The stability of the modified device is improved obviously compared to the control device. The PCE value of the control device decreases to ∼25% of the initial value over 2000 h. However, the PCE values of the modified devices with BTP-4H and BTP-4Cl can keep ∼60% of their initial values over 2000 h. The BTP-4F device can still maintain 86% of its initial PCE after 5000 h aging, indicating that the BTP-4F device has the best stability. To further study the effects of different organic molecules on the long-term stability of PSCs, we also compare the hydrophobicity of the perovskite films with different organic molecules. The contact angle values of the perovskite film with BTP-4F, perovskite film with BTP-4Cl, perovskite film with BTP-4H and pristine perovskite film are 71.9°, 48.2°, 46.9° and 37.7°, respectively (Fig. S9, ESI†). This indicates that the perovskite films modified by BTP-4F are more hydrophobic than the others. The improved stability is mainly attributed to the hydrophobicity of BTP-4F and hydrogen bonding of F and FA, which can immobilize FA and inhibit the decomposition of the perovskite film. These results further prove that BTP-4X can improve the moisture stability of the devices and the BTP-4F device exhibits the best stability. In order to prove the hydrophobicity of the organic small molecules, we conducted water droplet infiltration tests on FTO and BTP-4F/FTO substrates, and the corresponding pictures are shown in Fig. S10 (ESI†). The same amount of water (60 μL) is dropped onto the two substrates, and it is obvious that the water drop diffuses well and evenly on the FTO surface, while the water drop diffuses over only a small area on the substrate of BTP-4F/FTO. Therefore, this indicates that BTP-4F has strong hydrophobicity. Fig. 7b shows the normalized PCE value of PSCs aged at different temperatures and times in air. The humidity is 20%–30%. The PCE of the BTP-4F device retains 80% of the initial value after annealing at 85 °C for 30 h, and the PCE retains 72% of its initial value after annealing at 100 °C for 30 h. In contrast, the control device exhibits fast degradation. It can only maintain 15% of its initial PCE after annealing at 100 °C for 30 h. The above results confirm the strong interaction between hydrophobic BTP-4F and FA, which could inhibit the decomposition of perovskite films under thermal conditions. The XRD patterns of the control and BTP-4F-modified perovskite films under thermal treatment (100, 120, 140, or 150 °C) for 2 h are displayed in Fig. 7c. The BTP-4F perovskite film displays negligible diffraction peaks of PbI2, while the degradation of the control film is obvious. The influence of the annealing time on the XRD peaks is shown in Fig. 7d. The BTP-4F perovskite film shows excellent thermal stability. Fig. 7e and f also show the photographs of two perovskite films annealed at different temperatures and times. The BTP-4F perovskite film can retain a black color after high temperature annealing, while the control film exhibits a yellow color. This indicates that the BTP-4F perovskite film exhibits better heat tolerance. PL spectra of the control and BTP-4F perovskite films influenced by the light illumination time are depicted in Fig. S11a (ESI†). The PL intensity of the control perovskite film gradually decreases with the increase of the light illumination time. In contrast, the PL of the BTP-4F perovskite film shows very stable PL intensity for 160 min light illumination (Fig. S11b, ESI†), indicating that ion migration is effectively hindered. The light stability of the control and BTP-4F devices is measured under continuous light irradiation and the results are shown in Fig. S12 (ESI†). The PCE of the control device degrades completely within 300 h, while the BTP-4F device still retains 80% of its initial PCE even after 1000 h. Therefore, the introduction of BTP-4F into perovskite films can not only enhance the PCE of PSCs, but also improve the stability, including aging, moisture, thermal, and light stability. It shows significant potential toward commercialization of PSCs.
 | ||
| Fig. 7 (a) Normalized PCE values versus aging time of unsealed devices for testing the air stability of PSCs. (b) The normalized PCE of PSCs aged at different temperatures and times. (c) The XRD patterns of the perovskite films without and with BTP-4F treated at different annealing temperatures for 2 h. (d) The XRD data of the perovskite films without and with BTP-4F annealed at 150 °C with different treatment times. (e) and (f) The photographs of two perovskite films annealed at different temperatures and time. | ||
3. Conclusion
In conclusion, we present a unique strategy to improve the efficiency and stability of PSCs by introducing three novel organic semiconductors with different terminal groups into the perovskite films. Several interesting findings in this work should be highlighted. Firstly, a physical model is proposed to understand the effects of the three different terminal groups on the perovskite film growth and energy level alignment of devices. Secondly, compared with BTP-4H and BTP-4Cl, BTP-4F can more effectively increase the crystal size of the perovskite films due to the hydrogen bonding of Fand FA and also provide more efficient charge transport channels due to the optimal energy level offset. Most importantly, BTP-4F molecules not only promote the charge transport from the perovskite film to the HTL, but also facilitate the charge transport from the perovskite to the ETL, thus realizing the simultaneous up-bottom interfacial passivation of perovskite films. Finally, the optimal BTP-4F passivated PSC exhibits a remarkable PCE of 22.16%, and the device without any encapsulation can maintain ∼86% of the initial PCE after 5000 h under ambient conditions. Thus, this work represents GB and interface passivation by organic molecules, which is a promising strategy to overcome the efficiency and stability problems of PSCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (61822506, 11974142, 11674127, 61722505, 61935009, 61675086, 51772123), the National Key Research and Development Program of China (2017YFB0403601), Science and Technology Development Program of Jilin Province (20190101016JH, 20200401059GX), and the Special Project of the Province-University Constructing Program of Jilin University (SXGJXX2017-3).References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- P. Docampo, J. M. Ball, M. Darwich, G. E. Eperon and H. J. Snaith, Nat. Commun., 2013, 4, 761 Search PubMed.
- Y. Li, L. Meng, Y. Yang, G. Xu, Z. Hong, Q. Chen, J. You, G. Li, Y. Yang and Y. Li, Nat. Commun., 2016, 7, 10214 CrossRef CAS.
- L. Liu, S. Huang, Y. Lu, P. Liu, Y. Zhao, C. Shi, S. Zhang, J. Wu, H. Zhong, M. Sui, H. Zhou, H. Jin, Y. Li and Q. Chen, Adv. Mater., 2018, 30, 1800544 CrossRef.
- National Renewable Energy Laboratory (NREL) Efficiency Chart, https://www.nrel.gov/pv/cell-effciency.html, accessed: June 2019.
- J. Chen and N. G. Park, Adv. Mater., 2018, 7, 1803019 Search PubMed.
- P. Lu, M. Lu, H. Wang, N. Sui, Z. F. Shi, W. W. Yu and Y. Zhang, InfoMat, 2019, 1, 430–459 CrossRef CAS.
- C. Chen, H. Li, J. Jin, X. Chen, Y. Cheng, Y. Zheng, D. Liu, L. Xu, H. Song and Q. Dai, Adv. Energy Mater., 2017, 7, 1700758 CrossRef.
- C. Chen, D. Liu, Y. Wu, W. Bi, X. Sun, X. Chen, W. Liu, L. Xu, H. Song and Q. Dai, Nano Energy, 2018, 53, 849–862 CrossRef CAS.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef.
- Q. Wang, B. Chen, Y. Liu, Y. Deng, Y. Bai, Q. Dong and J. Huang, Energy Environ. Sci., 2017, 10, 516–522 RSC.
- H. Tan, A. Jain, O. Voznyy, X. Lan, F. P. García de Arquer, J. Z. Fan, R. Quintero-Bermudez, M. Yuan, B. Zhang, Y. Zhao, F. Fan, P. Li, L. N. Quan, Y. Zhao, Z.-H. Lu, Z. Yang, S. Hoogland and E. H. Sargent, Science, 2017, 355, 722–726 CrossRef CAS.
- A. A. Mamun, Y. Mohammed, T. T. Ava, G. Namkoong and A. A. Elmustafa, Mater. Lett., 2018, 229, 163–169 CrossRef.
- K. Domanski, E. A. Alharbi, A. Hagfeldt, M. Grätzel and W. Tress, Nat. Energy, 2018, 3, 61–67 CrossRef CAS.
- F. U. Kosasih and C. Ducati, Nano Energy, 2018, 47, 243–256 CrossRef CAS.
- S. Seo, S. Jeong, C. Bae, N. G. Park and H. Shin, Adv. Mater., 2018, 30, 1801010 CrossRef.
- X. Zhao and N.-G. Park, Photonics, 2015, 2, 1139–1151 CrossRef CAS.
- F. Zhang, W. Shi, J. Luo, N. Pellet, C. Yi, X. Li, X. Zhao, T. J. S. Dennis, X. Li, S. Wang, Y. Xiao, S. M. Zakeeruddin, D. Bi and M. Graetzel, Adv. Mater., 2017, 29, 606806 Search PubMed.
- L. Xie, H. H. Wang, M. Kim and K. Kim, Phys. Chem. Chem. Phys., 2017, 19, 1143–1150 RSC.
- X. Zhao, L. Tao, H. Li, W. Huang, P. Sun, J. Liu, S. Liu, Q. Sun, Z. Cui, L. Sun, Y. Shen, Y. Yang and M. Wang, Nano Lett., 2018, 18, 2442–2449 CrossRef CAS.
- D. Yang, R. Yang, X. Ren, X. Zhu, Z. Yang, C. Li and S. Liu, Adv. Mater., 2016, 28, 5206–5213 CrossRef CAS.
- D. Yang, P. Fu, F. Zhang, N. Wang, J. Zhang and C. Li, J. Mater. Chem. A, 2014, 2, 17281 RSC.
- Y. Wu, X. Yang, W. Chen, Y. Yue, M. Cai, F. Xie, E. Bi, A. Islam and L. Han, Nat. Energy, 2016, 1, 16148 CrossRef CAS.
- D. Bi, C. Yi, J. Luo, J. D. Decoppet, F. Zhang, S. M. Zakeeruddin, X. Li, A. Hagfeldt and M. Gratzel, Nat. Energy, 2016, 1, 5 Search PubMed.
- L. Xie, J. Chen, P. Vashishtha, X. Zhao, G. S. Shin, S. Mhaisalkar and N.-G. Park, ACS Energy Lett., 2019, 4, 2192–2200 CrossRef CAS.
- W. J. Nimens, S. J. Lefave, L. Flannery, J. Ogle, D. M. Smilgies, M. T. Kieber-Emmons and L. Whittaker-Brooks, Angew. Chem., Int. Ed., 2019, 58, 13912–13921 CrossRef CAS.
- J. C. Yu, S. Badgujar, E. D. Jung, V. K. Singh, D. W. Kim, J. Gierschner, E. Lee, Y. S. Kim, S. Cho, M. S. Kwon and M. H. Song, Adv. Mater., 2019, 31, 1805554 Search PubMed.
- P. Guo, Q. Ye, X. Yang, J. Zhang, F. Xu, D. Shchukin, B. Wei and H. Wang, J. Mater. Chem. A, 2019, 7, 2497–2506 RSC.
- P. Guo, X. Yang, Q. Ye, J. Zhang, H. Wang, H. Yu, W. Zhao, C. Liu, H. Yang and H. Wang, Adv. Energy Mater., 2019, 9, 1901341 CrossRef.
- L. Zuo, H. Guo, D. W. de Quilettes, S. Jariwala, N. De Marco, S. Dong, R. DeBlock, D. S. Ginger, B. Dunn, M. Wang and Y. Yang, Sci. Adv., 2017, 3, e1700106 CrossRef.
- J. Zhou, X. Yin, Z. Dong, A. Ali, Z. Song, N. Shrestha, S. Bista, Q. Bao, R. J. Ellingson, Y. Yan and W. Tang, Angew. Chem., Int. Ed., 2019, 58, 13717–13721 CrossRef CAS.
- T. H. Han, J. W. Lee, C. Choi, S. Tan, C. Lee, Y. Zhao, Z. Dai, N. De Marco, S. J. Lee, S. H. Bae, Y. Yuan, H. M. Lee, Y. Huang and Y. Yang, Nat. Commun., 2019, 4, 520–529 CrossRef.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H. Yip, T. Lau, X. Lu, C. Zhu, H. Peng, P. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1–12 CrossRef.
- J. S. Wu, S. W. Cheng, Y. J. Cheng and C. S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 RSC.
- H. Hu, K. Jiang, G. Yang, J. Liu, Z. Li, H. Lin, Y. Liu, J. Zhao, J. Zhang and F. Huang, J. Am. Chem. Soc., 2015, 137, 14149–14157 CrossRef CAS.
- Y. Cui, H. F. Yao, J. Q. Zhang, T. Zhang, Y. M. Wang, L. Hong, K. H. Xian, B. W. Xu, S. Q. Zhang, J. Peng, Z. X. Wei, F. Gao and J. H. Hou, Nat. Commun., 2019, 10, 1–8 CrossRef CAS.
- S. Fu, X. Li, L. Wan, Y. Wu, W. Zhang, Y. Wang, Q. Bao and J. Fang, Adv. Energy Mater., 2019, 9, 1901852 CrossRef.
- Y. Shao, Y. Fang, T. Li, Q. Wang, Q. Dong, Y. Deng, Y. Yuan, H. Wei, M. Wang, A. Gruverman, J. Shield and J. S. Huang, Energy Environ. Sci., 2016, 9, 1752–1759 RSC.
- D. B. Khadka, Y. Shirai, M. Yanagida, T. Sustain and K. Miyano, Energy Fuels, 2017, 1, 755–766 CAS.
- S. Wang, Z. Ma, B. Liu, W. Wu, Y. Zhu, R. Ma and C. Wang, Sol. RRL, 2018, 2, 1800034 CrossRef.
- J. Yang, C. Liu, C. Cai, X. Hu, Z. Huang, X. Duan, X. Meng, Z. Yuan, L. Tan and Y. Chen, Adv. Energy Mater., 2019, 9, 18 Search PubMed.
- M. Abdi-Jalebi, M. I. Dar, A. Sadhanala, S. P. Senanayak, M. Franckevičius, N. Arora, Y. Hu, M. K. Nazeeruddin, S. M. Zakeeruddin, M. Grätzel and R. H. Friend, Adv. Energy Mater., 2016, 6, 1502472 CrossRef.
- Z. Chen, H. Li, Y. Tang, X. Huang, D. Ho and C.-S. Lee, Mater. Res. Express, 2014, 1, 039501 CrossRef.
- Q. Chen, H. Zhou, Y. Fang, A. Z. Stieg, T. B. Song, H. H. Wang, X. Xu, Y. Liu, S. Lu, J. You, P. Sun, J. McKay, M. S. Goorsky and Y. Yang, Nat. Commun., 2015, 6, 7269 CrossRef CAS.
- R. Ranjan, A. Prakash, A. Singh, A. Singh, A. Garg and R. K. Gupta, J. Mater. Chem. A, 2018, 6, 1037–1047 RSC.
- W. Q. Wu, Z. Yang, P. N. Rudd, Y. Shao, X. Dai, H. Wei, J. Zhao, Y. Fang, Q. Wang, Y. Liu, Y. Deng, X. Xiao, Y. Feng and J. Huang, Sci. Adv., 2019, 5, 18 Search PubMed.
- D. Xin, S. Tie, R. Yuan, X. Zheng, J. Zhu and W. H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 44233–44240 CrossRef CAS.
- K. Chen, J. Wu, Y. Wang, Q. Guo, Q. Chen, T. Cao, X. Guo, Y. Zhou, N. Chen, M. Zhang and Y. Li, J. Mater. Chem. A, 2019, 7, 21140–21148 RSC.
- T. Wu, Y. Wang, Z. Dai, D. Cui, T. Wang, X. Meng, E. Bi, X. Yang and L. Han, Adv. Mater., 2019, 31, e1900605 CrossRef.
- Y. Guo, J. Ma, H. Lei, F. Yao, B. Li, L. Xiong and G. Fang, J. Mater. Chem. A, 2018, 6, 5919–5925 RSC.
- Y. Wu, Y. He, S. Li, X. Li, Y. Liu, Q. Sun, Y. Cui, Y. Hao and Y. Wu, J. Alloys Compd., 2020, 823, 153717 CrossRef CAS.
- S. Chen, L. Lei, S. Yang, Y. Liu and Z. S. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 25770–25776 CrossRef CAS.
- Y. Gao, Y. Wu, Y. Liu, C. Chen, X. Bai, L. Yang, Z. Shi, W. W. Yu, Q. Dai and Y. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 3631–3641 CrossRef CAS.
- F. Wang, W. Geng, Y. Zhou, H.-H. Fang, C.-J. Tong, M. A. Loi, L.-M. Liu and N. Zhao, Adv. Mater., 2016, 28, 9986–9992 CrossRef CAS.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- N. Tripathi, Y. Shirai, M. Yanagida, A. Karen and K. Miyano, ACS Appl. Mater. Interfaces, 2016, 8, 4644–4650 CrossRef CAS.
- Y. B. Gao, Y. J. Wu, H. B. Lu, C. Chen, Y. Lu, X. Bai, L. L. Yang, W. W. Yu, Q. L. Dai and Y. Zhang, Nano Energy, 2019, 59, 517–526 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00374c |
| ‡ Yanbo Gao and Yanjie Wu contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |