
Electron accepting naphthalene bisimide ligand architectures for modulation of π–π stacking in nanocrystal hybrid materials†
Katherine C.
Elbert
 a,
Mohammad M.
Taheri
b,
Natalie
Gogotsi
c,
Jungmi
Park
a,
Jason B.
Baxter
*b and
Christopher B.
Murray
*ac
a,
Mohammad M.
Taheri
b,
Natalie
Gogotsi
c,
Jungmi
Park
a,
Jason B.
Baxter
*b and
Christopher B.
Murray
*ac
aDepartment of Chemistry, University of Pennsylvania, Philadelphia, PA 19104, USA. E-mail: cbmurray@sas.upenn.edu
bDepartment of Chemical and Biological Engineering, Drexel University, Philadelphia, PA 19104, USA. E-mail: jbaxter@drexel.edu
cDepartment of Materials Science and Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA
First published on 3rd September 2020
Abstract
Investigation of charge transfer in quantum dot (QD) systems is an area of great interest. Specifically, the relationship between capping ligand and rate of charge transfer has been studied as a means to optimize these materials. To investigate the role of ligand interaction on the QD surface for electron transfer, we designed and synthesized a series of ligands containing an electron accepting moiety, naphthalene bisimide (NBI). These ligands differ in their steric bulk: as one allows for π–π stacking between the NBI moieties at high surface coverages, while the other does not, allowing for a direct comparison of these effects. Once grafted onto QDs, these hybrid materials were studied using UV-Vis, fluorescence, and transient absorption spectroscopy. Interestingly, the sample with the fastest electron transfer was not the sample with the most NBI π–π stacking, it was instead where these ligands were mixed amongst oleic acid, breaking up H-aggregates between the NBI groups.
New conceptsCharge separation and transfer are critical areas of research for electronics and renewable energy applications. For quantum dot (QD) systems, previous research on surface ligand affects have been limited by what is commercially available, which includes many ligands that do not strongly couple to each other. This has led to results where the rate of electron transfer has a linear relationship with the number of electron acceptors on the surfaces on the QDs. However, many of the most interesting electron acceptors are made of large conjugated moities that would drive ligand–ligand interactions on the surfaces on the QDs, and probing how these interactions in turn affect electron transfer is key in understanding and developing these processes. It is well known in the field of organic electronics that configurations and differences in orientation of molecules change the HOMO/LUMO gap of the materials, but in this work, we specifically probe how π–π stacking of naphthalene bisimide effects electron tansfer from CdSe/CdS QDs. Due to the presence of H-aggregates between these groups, we find a nonmonotonic relationship between surface coverage and quenching dynamics. This work highlights the need for further study and consideration of ligand configurations on QD surfaces, specifically for electron dynamics studies. |
Combinations of quantum dots (QDs) and organic ligands have been studied for unique charge and energy transfer processes that occur between the inorganic and organic components, as each component is highly tunable. These QD-hybrid systems have potential in applications including biosensors,1,2 light emitting devices,3,4 photocatalytic systems,5,6 and solar cells.7,8 Typically, these investigations have been limited to commercially available ligands,9,10 which limits the potential optimization of these systems. Recently, studies on distances between QD and charge accepting groups have elucidated relationships between ligand length and charge transfer,11–13 while others have specifically tuned the HOMO/LUMO gaps of organic ligands to increase charge transfer rates.14
Currently, there are very few examples directly comparing how the arrangement of electronically active groups on the surface of QDs affect their charge transfer. It has been shown that clustering of ligands can change the band gaps of the QDs,15 as well as the band gaps of the ligands. It is well known that molecules such as naphthalene bisimide (NBI) can π–π stack, and this stacking can affect charge transfer;16,17 however, directly comparing molecules that contain the same electron accepting group organized on the surface of QDs in different arrangements has been a challenge. It has been shown that π–π stacking into J-aggregates can act as exciton bridges in between QDs, and have increased kinetics, however these are not directly bound to the QD surface.18
In this work, we have used logical building blocks to probe electron transfer in organic-QD systems. Cadmium selenide (CdSe) QDs are an ideal model system to study these effects due to their ease of synthesis, surface chemistry modification, bandgap tunability, and strong absorption and emission.19–21 The addition of a shell layer, such as cadmium sulfide (CdS), denoted CdSe/CdS, can help passivate surface traps and improve the fluorescence quantum yield.19,22 We designed a series of ligands to probe differences due to distance from the QD surface to the electron accepting moiety, as well as ligand interaction effects. By using a flexible, stepwise synthesis, we obtain a ligand backbone that can easily incorporate an electron accepting NBI moiety, while providing steric bulk to prevent interaction between the moieties. After coupling these ligands with CdSe/CdS QDs, we used uv-vis spectroscopy to calculate the amount of ligand on the surfaces of the QDs, and fluorescence and transient absorption spectroscopy to study electron transfer in these systems.
To study the effects on electron transfer of distance between an electron accepting moiety and a QD, and ligand assembly on the surface of the QD, two ligands were designed. These ligands vary in distance between the electron acceptor, NBI, and the sulfide anchoring unit, as well as steric bulk of the complete ligand, and are shown in Scheme 1 as molecules 14 and 15. Changing the bulk of the ligand backbone will vary the number of ligands, thus the number of NBI units, as well as how the NBI units can organize on the surface of the QDs. For the ligand with greater steric bulk, a gallic acid based backbone was designed to allow for incorporation of an electron accepting moiety during one of the final synthetic steps, providing synthetic flexibility.
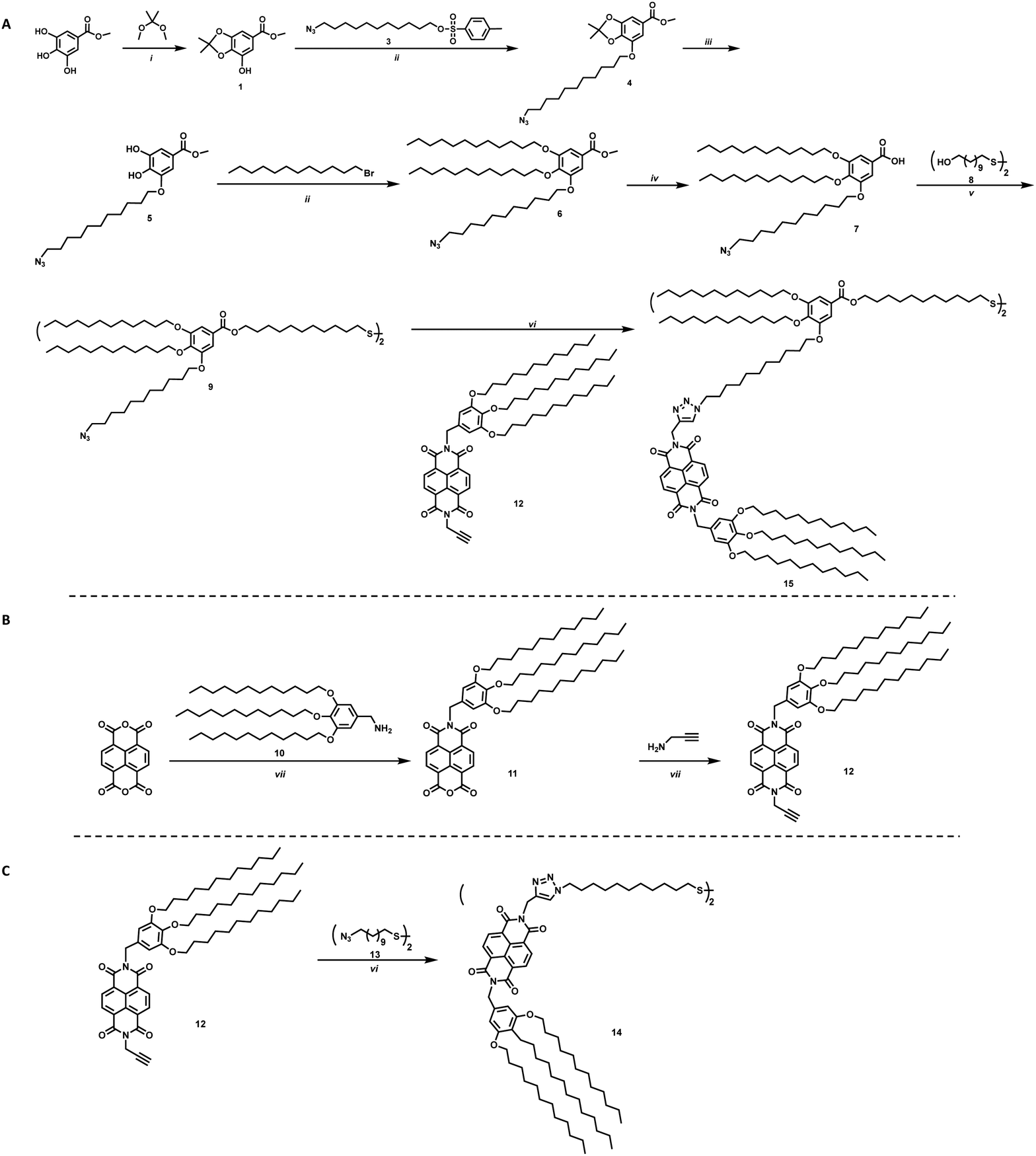 | ||
| Scheme 1 Synthetic steps for NBI-2 synthesis. Reagents and conditions: (i) 2,2-dimethoxypropane, p-toluene sulfonic acid, toluene, reflux, 24 h; (ii) K2CO3, KI, DMF, 80 °C, 12 h; (iii) TFA, CH2Cl2, rt, 4 h; (iv) KOH, THF/MeOH/H2O, 80 °C; (v) SOCl2, CH2Cl2, rt, N2 atm, 3 h, then Et3N, DMAP, CH2Cl2, −10 °C → rt, 18 h; (vi) CuSO4·5H2O, sodium ascorbate, THF/H2O, 75 °C, 20 h, under microwave irradiation; (vii) DMF, 75 °C (5 min) → 140 °C, 15 min, under microwave irradiation. | ||
The synthesis of the targeted ligands is shown in Scheme 1. For the ligand with greater steric bulk, the synthesis was started by reacting methyl gallate (methyl-3,4,5-trihydroxybenzoate) with 2,2-dimethoxypropane to install a protecting group on two of the alcohols to achieve 1, shown in Scheme 1a. An azide containing alkyl chain, 3, was added using Williamson etherification, where the product can be subsequently deprotected under acidic conditions without purification. Crystalline intermediate 5 reacts via an additional etherification with 1-bromododecane to provide ether 6, which can be readily hydrolyzed to 7. A one-pot two-step approach was employed to achieve the targeted disulfide molecule 9. This disulfide, or backbone, provides the ligand scaffold for an electron accepting group to be grafted onto NC surfaces. The incorporation of the azide group on one of the branches of the ligand allows for click chemistry to be utilized for straight-forward addition of NBI to the ligands, such as for synthesis of 15.
For the NBI moiety, 12, a gallic acid based group was also added to NBI to provide solubility in common organic solvents as well as improved stability to the QD-hybrid materials, as this group has shown to be an effective stabilizer in previous literature.23 Propargylamine was added to the other side of the NBI, as shown in Scheme 1b, to make it an intermediate where click chemistry can be used to make the final target molecules. This intermediate was used to synthesize molecules 14 and 15, where 14 is a simple disulfide bearing alkyl chain, highlighted in Scheme 1c, that should not limit interaction between the NBI moieties.
The final series of ligands is shown in Fig. 1. To achieve NBI1 and NBI2, molecules 14 and 15 were reduced with NaBH4 and a catalytic amount of ZrCl4 in THF. Crude NBI1 and NBI2 were used immediately after being reduced to sulfides, to limit oxidation. Two controls were selected for this study: oleic acid (OA), the ligand used during the QD synthesis, and polycatenar ligand (CD) that is structurally similar to the moiety added to the NBI to provide stability. As shown in Fig. 1a, the absorption spectra for NBI1 and NBI2 show that the peaks from NBI are unchanged when incorporated into the larger structure for NBI2. This highlights that the electronic properties of NBI are unchanged. The cyclic voltammetry data shown in Fig. 1b confirms this finding, as the estimated LUMO energy levels for both NBI1 and NBI2 are −4.23 and −4.22 eV, respectively. Both OA and CD have no optical activity beyond 300 nm, and it is expected that there will be no substantial changes in the photoexcited carrier dynamics of the QDs when these two ligands are compared.
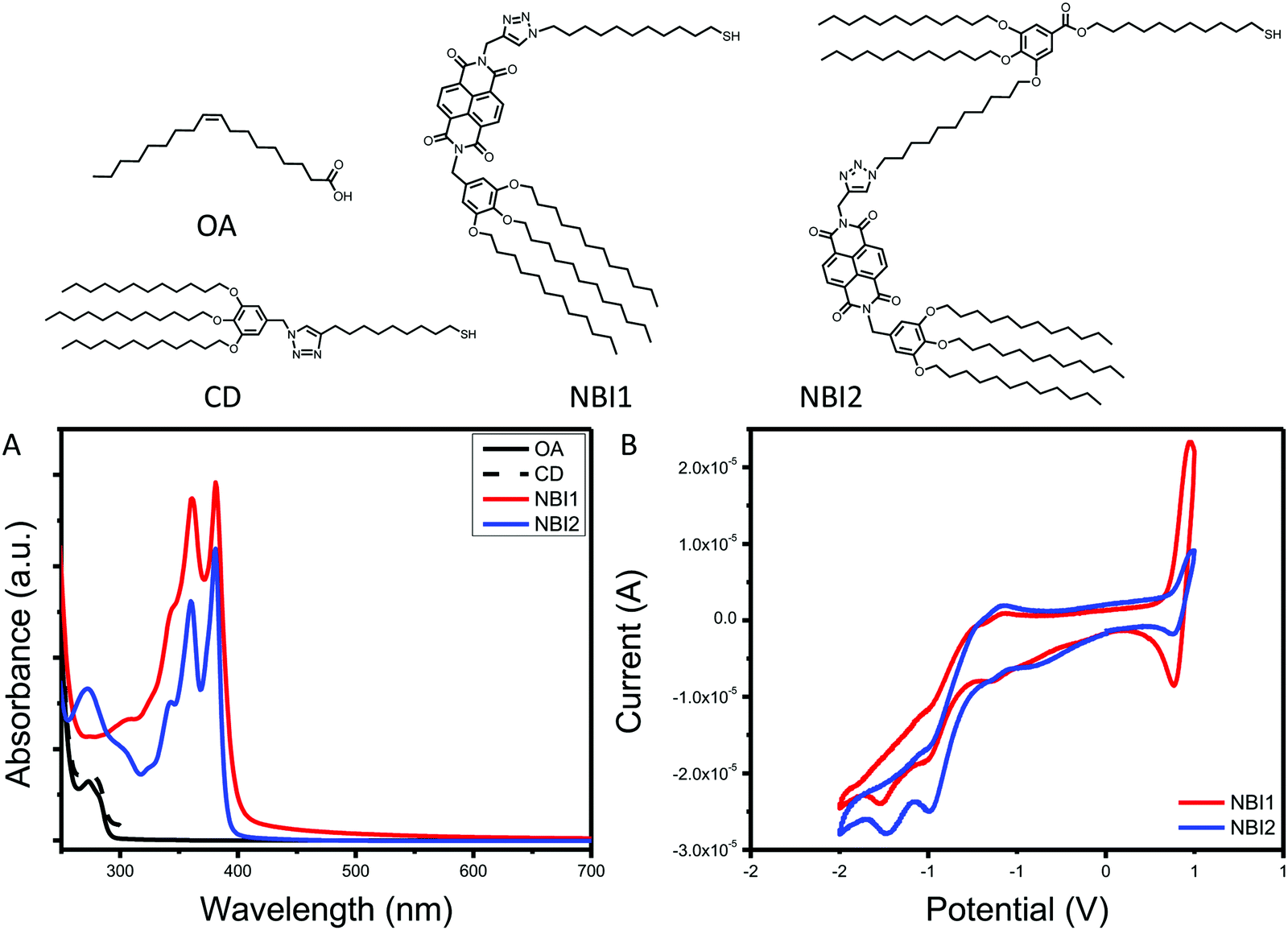 | ||
| Fig. 1 Complete series of ligands investigated with associated (A) absorption spectra and (B) cyclic voltammetry. | ||
The ligands were grafted to the QD surfaces (denoted as QD@ligand) through a simple ligand exchange procedure,24,25 allowing for the QDs to be synthesized by a known procedure19,26 while the amount of ligand used in the exchange can be tuned. The QDs used in this study were CdSe/CdS core/shell QDs, shown in Fig. S1 (ESI†), where the initial CdSe cores were 2.6 ± 0.4 nm, and the CdSe/CdS core/shell were 5.9 ± 0.6 nm based on TEM measurements. Multiple batches of CdSe/CdS from the same CdSe cores were synthesized to have access to enough QDs for the study, which accounts for slight shifts in absorption spectra across the samples. Initially, full ligand exchange for QD@NBI1 and QD@NBI2 was compared, as well as QD@CD for a control. There were no changes in QD composition after the ligand exchange procedure, as shown in Fig. S2 (ESI†). The QD@CD was used to determine whether the backbone used in the NBI2 structure, and the gallic acid based polycatenar moiety that was used to provide solubility and stability for the overall NBI group, had any effects on charge transfer. As can be seen from Fig. S3 (ESI†), the CD ligand is a good choice for a control, as no changes in the absorbance or fluorescence spectra are observed comparing QD@CD with QD@OA.
Prior to collecting optical spectra, samples were passed through a 0.2 μm PTFE syringe filter, to ensure no aggregates that were not observed in TEM imaging were present. Fig. 2a shows the relative absorbance spectra of the hybrid materials QD@NBI1, QD@NBI2, and QD@CD. All absorbance spectra were normalized for effective comparison. For the QD@CD case, there is no change in the spectra when compared to the as-synthesized QD@OA. In contrast, the spectral features from the NBI moieties dominate the UV spectrum for both QD@NBI1 and QD@NBI2, due to the significantly high number of ligands per QD. For clarity, the same functional form was applied to all samples to remove background scattering. Comparing the spectra of the two NBI containing ligands, the intensities of the two peaks at 360 nm and 380 nm can be compared to assess the assembly of the ligands on the surfaces of the NCs.27 For QD@NBI2, the relative intensities of the two peaks is similar to that for NBI2. However, QD@NBI1 shows a clear change in the relative intensities of the two peaks compared to NBI1. This type of altered absorption spectrum points towards electronic coupling between adjacent NBI units27,28 and indicates that there is π–π stacking between the NBI moieties of the ligands.29 Changes in absorbance peak ratios indicate that the NBI moieties are forming H-aggregates, as opposed to J-aggregates,30,31 which are less commonly observed on NC surfaces.32 The presence of a larger backbone breaks up this stacking, as there is no alteration of the NBI peaks for the QD@NBI2 case.
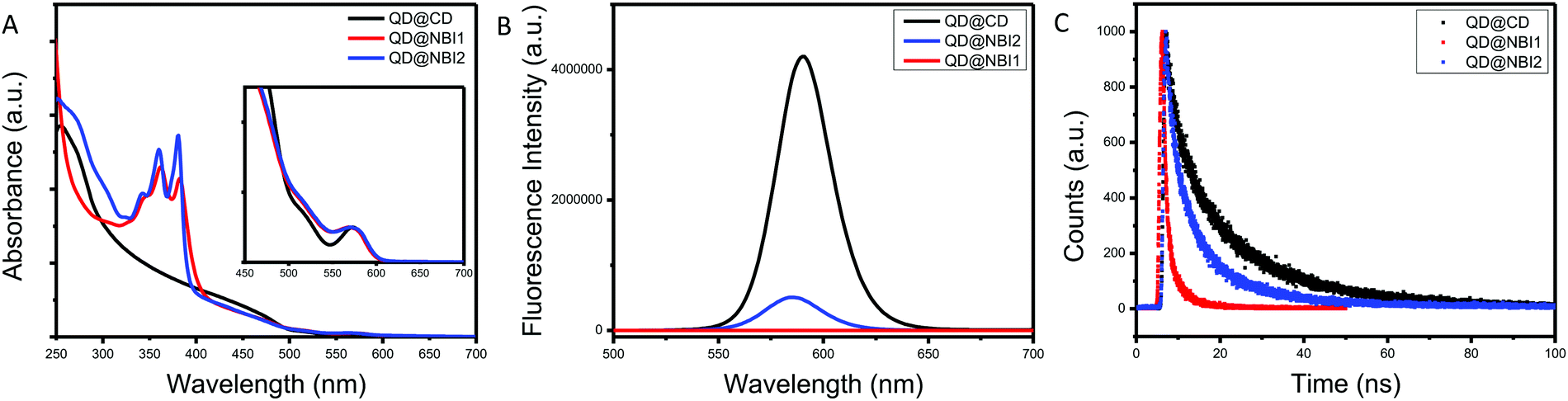 | ||
| Fig. 2 Optical spectra for NBI@QD hybrid materials: (A) absorption spectra, (B) fluorescence spectra, (C) lifetime fluorescence spectra. | ||
The fluorescence spectra of the hybrid materials are shown in Fig. 2b, where there is clear quenching of the fluorescence from the QD in the presence of an NBI-containing ligand. Comparing the different NBI ligands, greater quenching is observed for QD@NBI1 than QD@NBI2. Consistent with these measurements, the corresponding time-resolved fluorescence measurements in Fig. 2c show a shorter lifetime for QD@NBI1 compared to the larger ligand QD@NBI2, 0.87 ns and 3.79 ns, respectively. The QD@CD sample has a fluorescence lifetime of 4.55 ns. Comparing QD@NBI2 and QD@NBI1, the improved electron transfer process could be due to the difference in length between the two ligands, as the NBI moiety is much closer to the QD surface in NBI1 than in NBI2. Additionally, the size of each ligand could also play a role, as the larger NBI2 ligand limits the number of ligands on the surface of the QD compared to NBI1.
To explore the effects of number of NBI moieties on the surface of the QDs, the amount of ligand grafted on the surface of the QDs was varied by introducing specific amounts of NBI1 into each ligand exchange solution. The ligand used during QD synthesis, OA, does not exhibit any evidence of accepting electrons from the excited QD, and was used as the additional ligand on the surface. To confirm a mixture of OA and the selected NBI moiety, 1H NMR was used, shown in Fig. S4 (ESI†). The optical signatures of both the QD and NBI1 were used to determine the percent NBI1 present on the QDs.
Interestingly, when the amount of NBI1 ligands decreases, we can see changes in the absorption spectra, as shown in Fig. 3a. For the case of 32% NBI1@QD, a clear difference in the ratios of the peaks at 360 and 380 nm is observed compared to that of 100% NBI1@QD and 53% NBI1@QD. This change in ratios indicates differences in the arrangement of the NBI1 ligands on the surface of the QDs, possibly towards a degree of separation of the π–π stacking of the NBI moieties. As the number of NBI1 ligands decreases, to 19% and 15%, this trend continues, as shown in Fig. 3c, where the ratios of the peaks at 360 and 380 nm are plotted vs. the percent of ligand exchanged to highlight the relationship between number of ligands and presence of H-aggregates.
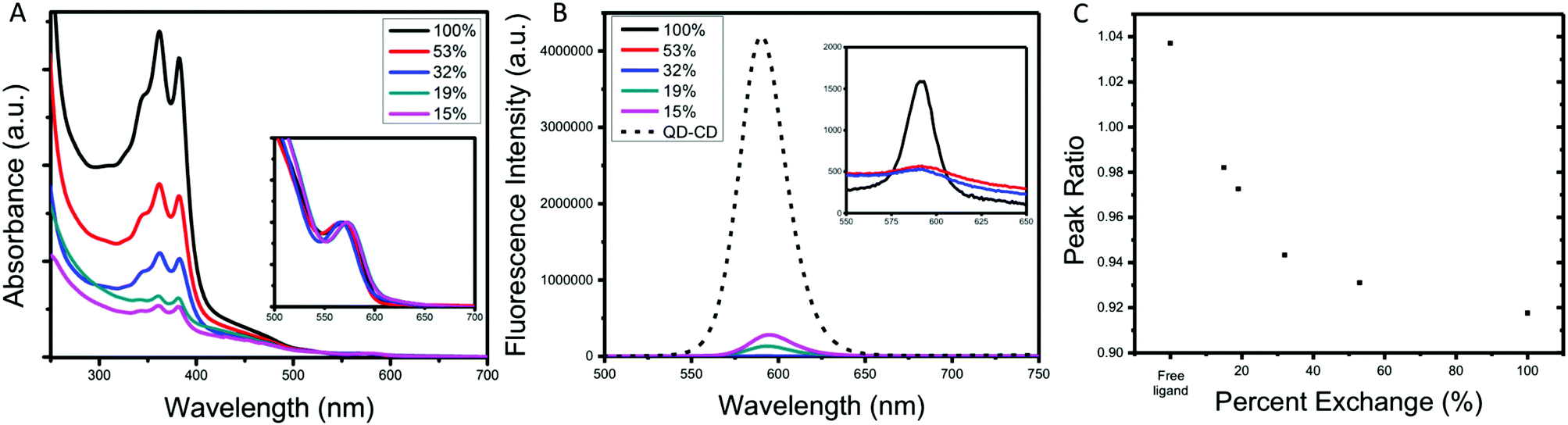 | ||
| Fig. 3 (A) Absorption spectra for different amounts of NBI1 on the QDs, (B) fluorescence spectra for the same series of samples, (C) percent NBI1 ligand exchange vs. peak ratio of NBI maxima peaks at 360 and 380 nm. | ||
The fluorescence data for this series, shown in Fig. 3b, exhibits significant quenching in the presence of the NBI1 ligand compared to the control QD@CD for all five samples of differing amounts of NBI1 ligand exchanged. Comparing these samples, there is a greater amount of quenching for the cases of QD@NBI1-53% and QD@NBI1-32% than for QD@NBI1-100%, which is highlighted by the inset in Fig. 3b. That there is increased fluorescence quenching when the H-aggregates between the NBI moieties are broken up is consistent with recent findings where the presence of H-aggregates can lead to less efficient charge transfer, particularly compared to J-aggregates.33 This effect is most likely due to the changes in the HOMO/LUMO gap between the aggregated and non-aggregated forms of the ligands, where H-aggregates cause an increase in the LUMO energy level, thus leading to a decrease in the efficiency of charge transfer.33,34
To further investigate these findings, charge transfer dynamics from QDs to NBI were quantified on picosecond timescales using transient absorption (TA) spectroscopy. Faster decay of the 1S bleach [1S3/2(h)–1S(e)]35 (Fig. 4a, corresponding to ∼575 nm in Fig. S5, ESI†) likely indicates electron transfer from QD to NBI. The QDs were photoexcited with a 535 nm pump pulse at a relatively low fluence of 154 μj cm−2 to generate 0.6 exciton/QD with negligible Auger recombination. Calculations are described in ESI,† Fig. S6 and S7. Comparison of the TA dynamics for aged and freshly prepared solutions showed no significant difference over one month, indicating that the samples are stable (Fig. S8, ESI†).
 | ||
| Fig. 4 TA dynamics at 1S state for (A) hybrid materials with 100% coverage, and (B) QD@NBI1 with varying amounts of NBI1 coverage. | ||
The trend illustrated in Fig. 4a is consistent with the time resolved fluorescence spectra shown in Fig. 2c, where the dynamics for QD@NBI structures are faster than the control sample. Faster decay for QD@NBI1 compared to QD@NBI2 could be due to shorter distance between NBI moiety and QD surface for QD@NBI1 case. There is no difference in kinetics between QD@OA and QD@CD, which indicates that the ligand exchange process does not induce surface states that cause faster dynamics. The kinetic traces were fit with a multiexponential function, 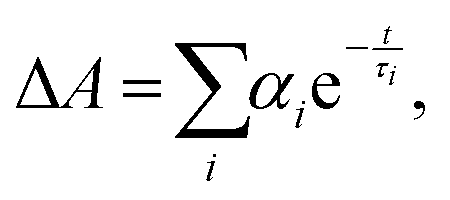 where αi and τi are the exponential coefficient and the decay time for each component, respectively. The average decay constant for each structure was found by:
where αi and τi are the exponential coefficient and the decay time for each component, respectively. The average decay constant for each structure was found by: 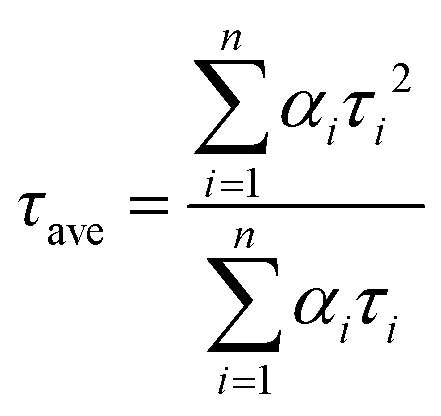 . The average time constants for QD@CD, QD@NBI2, and QD@NBI1 are 11 ns, 4.2 ns, and 122 ps, respectively (see Table S1, ESI† for αi and τi). The quantum efficiency of electron transfer can be calculated using QE = 1 − (τNBI/τCD), to give 0.99 and 0.62 for NBI1 and NBI2, respectively.
. The average time constants for QD@CD, QD@NBI2, and QD@NBI1 are 11 ns, 4.2 ns, and 122 ps, respectively (see Table S1, ESI† for αi and τi). The quantum efficiency of electron transfer can be calculated using QE = 1 − (τNBI/τCD), to give 0.99 and 0.62 for NBI1 and NBI2, respectively.
We also examined the effects of a different number of NBI moieties for NBI1, shown in Fig. 4b. The decay for QD@NBI1-100% is faster than QD@NBI1-19% and QD@NBI1-15%, where faster decay occurs because of increased number of NBI1 on the surface of the QD. Interestingly, kinetics are fastest for intermediate NBI loadings, with an average decay time of 12 ps for QD@NBI1-32%. This ordering is consistent with the fluorescence quenching in Fig. 3b and could be due to the differences in the NBI1 arrangement on the QD surface as explained previously, which could affect the charge transfer rate.
Conclusions
While it is known that differences between ligand architectures play a major role in many QD applications, specific probing of ligand conformation on the QD surface affecting properties such as electron transfer is less well understood. Our results show an expected trend where the fluorescence quenching is dependent on the distance between the QD and electron accepting moiety. However, interactions between the moieties, such as π–π stacking forming H-aggregates for the case of NBI1, is most likely responsible for the nonlinear trend between number of electron acceptors and charge transfer dynamics, observed in this work. These interactions will also cause ordered phases of the surface ligands, which has been shown to be a factor for decreasing photoluminescence, and could be an additional contribution for this system.36 Future studies to specifically probe the organization of these ligands on QD surfaces are required to probe these mechanisms further.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors acknowledge support from collaborative NSF grant CHE-1709827 (Penn)/CHE-1708991 (Drexel). K. C. E. acknowledges support from the NSF Graduate Research Fellowship Program under Grant No. DGE-1321851. C. B. M. acknowledges the Richard Perry University Professorship at the University of Pennsylvania. We acknowledge Professor Jessica Anna and Davit Jishkariani for helpful discussions.References
- W. R. Algar, M. H. Stewart, A. M. Scott, W. J. Moon and I. L. Medintz, J. Mater. Chem. B, 2014, 2, 7816–7827 RSC.
- N. Hildebrandt, C. M. Spillmann, W. R. Algar, T. Pons, M. H. Stewart, E. Oh, K. Susumu, S. A. Díaz, J. B. Delehanty and I. L. Medintz, Chem. Rev., 2017, 117, 536–711 CrossRef CAS.
- K. Wu, G. Liang, Q. Shang, Y. Ren, D. Kong and T. Lian, J. Am. Chem. Soc., 2015, 137, 12792–12795 CrossRef CAS.
- J. Zhao, J. A. Bardecker, A. M. Munro, M. S. Liu, Y. Niu, I.-K. Ding, J. Luo, B. Chen, A. K.-Y. Jen and D. S. Ginger, Nano Lett., 2006, 6, 463–467 CrossRef CAS.
- E. A. Weiss, ACS Energy Lett., 2017, 2, 1005–1013 CrossRef CAS.
- M. B. Wilker, K. E. Shinopoulos, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, J. Am. Chem. Soc., 2014, 136, 4316–4324 CrossRef CAS.
- P. R. Brown, D. Kim, R. R. Lunt, N. Zhao, M. G. Bawendi, J. C. Grossman and V. Bulović, ACS Nano, 2014, 8, 5863–5872 CrossRef CAS.
- H. Zhu, N. Song and T. Lian, J. Am. Chem. Soc., 2010, 132, 15038–15045 CrossRef CAS.
- D. A. Hines and P. V. Kamat, J. Phys. Chem. C, 2013, 117, 14418–14426 CrossRef CAS.
- J. T. DuBose and P. V. Kamat, J. Phys. Chem. C, 2020, 124, 12990–12998 CrossRef CAS.
- D. Dorokhin, N. Tomczak, A. H. Velders, D. N. Reinhoudt and G. J. Vancso, J. Phys. Chem. C, 2009, 113, 18676–18680 CrossRef CAS.
- M. Tagliazucchi, D. B. Tice, C. M. Sweeney, A. J. Morris-Cohen and E. A. Weiss, ACS Nano, 2011, 5, 9907–9917 CrossRef CAS.
- T. X. Ding, J. H. Olshansky, S. R. Leone and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 2021–2029 CrossRef CAS.
- J. H. Olshansky, T. X. Ding, Y. V. Lee, S. R. Leone and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 15567–15575 CrossRef CAS.
- R. D. Harris, V. A. Amin, B. Lau and E. A. Weiss, ACS Nano, 2016, 10, 1395–1403 CrossRef CAS.
- Y. Wu, M. Frasconi, D. M. Gardner, P. R. McGonigal, S. T. Schneebeli, M. R. Wasielewski and J. Fraser Stoddart, Angew. Chem., Int. Ed., 2014, 53, 9476–9481 CrossRef CAS.
- N. S. S. Kumar, M. D. Gujrati and J. N. Wilson, Chem. Commun., 2010, 46, 5464–5466 RSC.
- C. Wang and E. A. Weiss, Nano Lett., 2017, 17, 5666–5671 CrossRef CAS.
- O. Chen, J. Zhao, V. P. Chauhan, J. Cui, C. Wong, D. K. Harris, H. Wei, H.-S. Han, D. Fukumura, R. K. Jain and M. G. Bawendi, Nat. Mater., 2013, 12, 445–451 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 CrossRef CAS.
- H. Zhu, Y. Yang, K. Wu and T. Lian, Annu. Rev. Phys. Chem., 2016, 67, 259–281 CrossRef CAS.
- D. V. Talapin, I. Mekis, S. Götzinger, A. Kornowski, O. Benson and H. Weller, J. Phys. Chem. B, 2004, 108, 18826–18831 CrossRef CAS.
- K. C. Elbert, J. D. Lee, Y. Wu and C. B. Murray, Langmuir, 2018, 34, 13333–13338 CrossRef CAS.
- K. C. Elbert, D. Jishkariani, Y. Wu, J. D. Lee, B. Donnio and C. B. Murray, Chem. Mater., 2017, 29, 8737–8746 CrossRef CAS.
- A. Dong, X. Ye, J. Chen, Y. Kang, T. Gordon, J. M. Kikkawa and C. B. Murray, J. Am. Chem. Soc., 2011, 133, 998–1006 CrossRef CAS.
- L. Carbone, C. Nobile, M. De Giorgi, F. Della Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- C. C. Jumper, J. M. Anna, A. Stradomska, J. Schins, M. Myahkostupov, V. Prusakova, D. G. Oblinsky, F. N. Castellano, J. Knoester and G. D. Scholes, Chem. Phys. Lett., 2014, 599, 23–33 CrossRef CAS.
- M. Yamauchi and S. Masuo, Chem. – Eur. J., 2019, 25, 167–172 CrossRef CAS.
- S. Yagai, T. Seki, T. Karatsu, A. Kitamura and F. Würthner, Angew. Chem., Int. Ed., 2008, 47, 3367–3371 CrossRef CAS.
- S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343–11357 CrossRef CAS.
- M. Son, K. H. Park, C. Shao, F. Würthner and D. Kim, J. Phys. Chem. Lett., 2014, 5, 3601–3607 CrossRef CAS.
- M. Más-Montoya and R. A. J. Janssen, Adv. Funct. Mater., 2017, 27, 1605779 CrossRef.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS.
- V. I. Klimov, Annu. Rev. Phys. Chem., 2007, 58, 635–673 CrossRef CAS.
- A. D. Balan, J. H. Olshansky, Y. Horowitz, H. L. Han, E. A. O’Brien, L. Tang, G. A. Somorjai and A. P. Alivisatos, ACS Nano, 2019, 13, 13784–13796 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, full characterization of organic compounds, additional microscopy, and supporting spectroscopy. See DOI: 10.1039/d0nh00359j |
| This journal is © The Royal Society of Chemistry 2020 |