
Controlled functionalization of carbon nanodots for targeted intracellular production of reactive oxygen species†
Ding-Kun
Ji
a,
Giacomo
Reina
 a,
Shi
Guo
a,
Matilde
Eredia
b,
Paolo
Samorì
b,
Cécilia
Ménard-Moyon
a and
Alberto
Bianco
*a
a,
Shi
Guo
a,
Matilde
Eredia
b,
Paolo
Samorì
b,
Cécilia
Ménard-Moyon
a and
Alberto
Bianco
*a
aCNRS, Immunology, Immunopathology and Therapeutic Chemistry, UPR 3572, University of Strasbourg, ISIS, 67000 Strasbourg, France. E-mail: a.bianco@ibmc-cnrs.unistra.fr
bUniversité de Strasbourg, CNRS, ISIS, 67000 Strasbourg, France
First published on 10th June 2020
Abstract
Controlled intracellular release of exogenous reactive oxygen species (ROS) is an innovative and efficient strategy for cancer treatment. Well-designed materials, which can produce ROS in targeted cells, minimizing side effects, still need to be explored as new generation nanomedicines. Here, red-emissive carbon nanodots (CNDs) with intrinsic theranostic properties are devised, and further modified with folic acid (FA) ligand through a controlled covalent functionalization for targeted cell imaging and intracellular production of ROS. We demonstrated that covalent functionalization is an effective strategy to prevent the aggregation of the dots, leading to superior colloidal stability, enhanced luminescence and ROS generation. Indeed, the functional nanodots possess a deep-red emission and good dispersibility under physiological conditions. Importantly, they show excellent targeting properties and generation of high levels of ROS under 660 nm laser irradiation, leading to efficient cell death. These unique properties enable FA-modified carbon nanodots to act as a multifunctional nanoplatform for simultaneous targeted imaging and efficient photodynamic therapy to induce cancer cell death.
New conceptsDuring the last two decades, various low-dimensional nanomaterials with tailored composition, structure and properties have been developed, and applied as photosensitizers. However, their exploitation to efficiently produce reactive oxygen species in targeted cells, minimizing undesired side effects, has not been demonstrated yet. Carbon nanodots are highly promising nanomaterials for future clinical translations as they combine numerous characteristics including high photostability, low cytotoxicity, and superior biocompatibility. Here, we have conceived and synthesized multifunctional carbon nanodots with deep-red emission properties through their controlled chemical modification using the folic acid ligand. These carbon nanodots, endowed with a high colloidal stability and an enhanced luminescence, are suitable for targeted intracellular production of reactive oxygen species by laser irradiation leading to efficient cancer cell death. Our approach is radically novel and of general applicability; thus it will open new pathways towards the development of a large variety of other multifunctional carbon nanomaterials for phototherapies. In particular, the ad-hoc synthesis of nanomaterials with intrinsic therapeutic properties offers multiple opportunities for combined therapies by taking full advantage of the combination of appropriate anticancer drugs, targeting and fluorescent probes, and photothermal agents. |
1. Introduction
Reactive oxygen species (ROS) are well-established key signaling species of cellular stress, damage and death.1 They include hydrogen peroxide, singlet oxygen, superoxide anion radicals, and hydroxyl, peroxyl, alkoxyl and hydroperoxyl radicals.2,3 Since ROS cause oxidative damage of the fatty acids in lipids, the nucleobases in DNA and the amino acids of proteins, leading to cell death by necrosis and/or apoptosis, the controlled intracellular release of exogenous ROS has been employed as an effective strategy for cancer treatment.4 Light-induced ROS generation can trigger necrosis or apoptosis of cancer cells by eliciting an adequate level of oxidative stress.5,6 Despite the success of several well-designed materials to achieve this goal,7,8 the production of ROS in targeted cells, minimizing their damaging effect, has been elusive.Folate receptor (FR), also known as the high-affinity membrane folate-binding protein, is a glycosylphosphatidylinositol-linked membrane glycoprotein. Two membrane-bound isoforms of FR have been identified as α and β with high affinity to folic acid (FA) (KD ∼ 0.1 nM).9,10 FRs are highly expressed in various types of human cancers, and generally absent in most normal cells.11,12 Therefore, FRs are ideal biomarkers for targeted cancer diagnosis and therapy. Taking into account the affinity between FA and FRs, it has been well confirmed that nanoparticles functionalized with FA can improve endocytosis of cancer cells.13 In this context, FA-modified nanoparticles have been thoroughly explored for intracellular payload delivery leading to a minimum drug dose administration, less side effects and an improved therapeutic efficacy.14,15 Consequently, FA-modified nanoparticles show great application potential for targeted disease theragnosis.
Carbon nanodots, as rising stars in the carbon nanomaterial family, have drawn tremendous attention in the last few years,16 by virtue of their numerous merits including a tunable emission, a high photostability, a low toxicity, a superior biocompatibility and a rather inexpensive cost in large scale production.17–19 Thanks to the presence of abundant reactive groups on their surface, the properties of CNDs can be well regulated through chemical functionalization.20,21 CNDs have been demonstrated to be a promising candidate in various biomedical applications.22–24 In particular, near-infrared (NIR)-emissive or red-emissive CNDs (RCNDs), which can be excited by long-wavelength lights, show great potential in the biomedical field because of their good tissue penetration.25,26 Recent literature witnesses an extension of RCNDs to the field of phototherapy because of their unique photophysical properties and biocompatibility.27–30 However, it is still highly desired to develop synthetic methods to prepare and functionalize RCNDs as their potential for targeted intracellular production of ROS and possible use in photodynamic therapy (PDT) has been rarely explored.
PDT is a medical modality that is currently widely used in clinics.31 However, the current FDA approved photosensitizers lack specificity (low targeting) and require high light dose irradiation to be effective. In this context, new photosensitizers able to target tumor cells and requiring low energy irradiation are highly desirable.
Here, we explored a method to prepare RCNDs and we covalently functionalized RCNDs with an FA ligand for the targeted generation of ROS in a cell line that highly expresses FA receptors (Fig. 1). A certain number of techniques were used to monitor the preparation process, validating our controllable strategy to prepare multifunctional RCND nanoparticles with improved properties. We demonstrate that the covalent modification of RCNDs with FA effectively suppressed the aggregation of the starting carbon nanodots, leading to an enhanced fluorescence emission and increased production of ROS. The as-prepared functional RCNDs display a broad UV absorption from 300 to 800 nm and a red emission from 500 to 800 nm. Finally, they exhibit a high capacity to target cells via ligand-receptor interaction and they produced ROS upon irradiation at 660 nm, thereby inducing effective cell death, ultimately confirming their biomedical potential as a versatile, highly efficient photosensitizer for photodynamic cancer therapy.
 | ||
| Fig. 1 Schematic illustration of the covalent functionalization of RCNDs and their targeted intracellular production of ROS through (I) FR-mediated endocytosis and (II) light irradiation. FL stands for fluorescence. | ||
2. Results and discussion
2.1. Design, synthesis and characterization of RCND–TEG–FA
RCNDs were prepared through the hydrothermal treatment of conjugated polythiophene phenylpropionic acid (PPA) as illustrated in Scheme 1. To obtain the precursor PPA molecule, a Suzuki coupling between 3-(4-bromophenyl) propanoic acid and 3-thiophene boronic acid was performed. The carboxyl group in 3-(4-bromophenyl) propanoic acid was first protected by benzyl bromide. The resulting compound 1 further reacted with 3-thiophene boronic acid via a successful Suzuki coupling yielding the protected monomer (2) of PPA. The benzyl group was next hydrolyzed under basic conditions to obtain the desired deprotected compound 3. RCND precursor PPA was synthesized via oxidative polymerization of 3 and then the polymer was hydrothermally carbonized to form water-dispersible RCNDs. The high resolution transmission electron microscopy (HRTEM) images show that RCNDs possess a size below 5 nm with a well-resolved crystal lattice (Fig. 2a). The selected area diffraction (SAED) pattern further confirmed the high crystalline nature of the RCNDs. In addition, the presence of oxygen-containing groups (OH, COO−) was demonstrated by FT-IR analysis (Fig. 2c). The percentages of C, H, S and O were measured via element analysis and corresponded to 56.1%, 3.9%, 12.4%, and 27.6%, respectively. All results indicated that S-doped RCNDs with COOH groups on their surface were well prepared. | ||
| Scheme 1 Synthetic pathway to prepare RCNDs. | ||
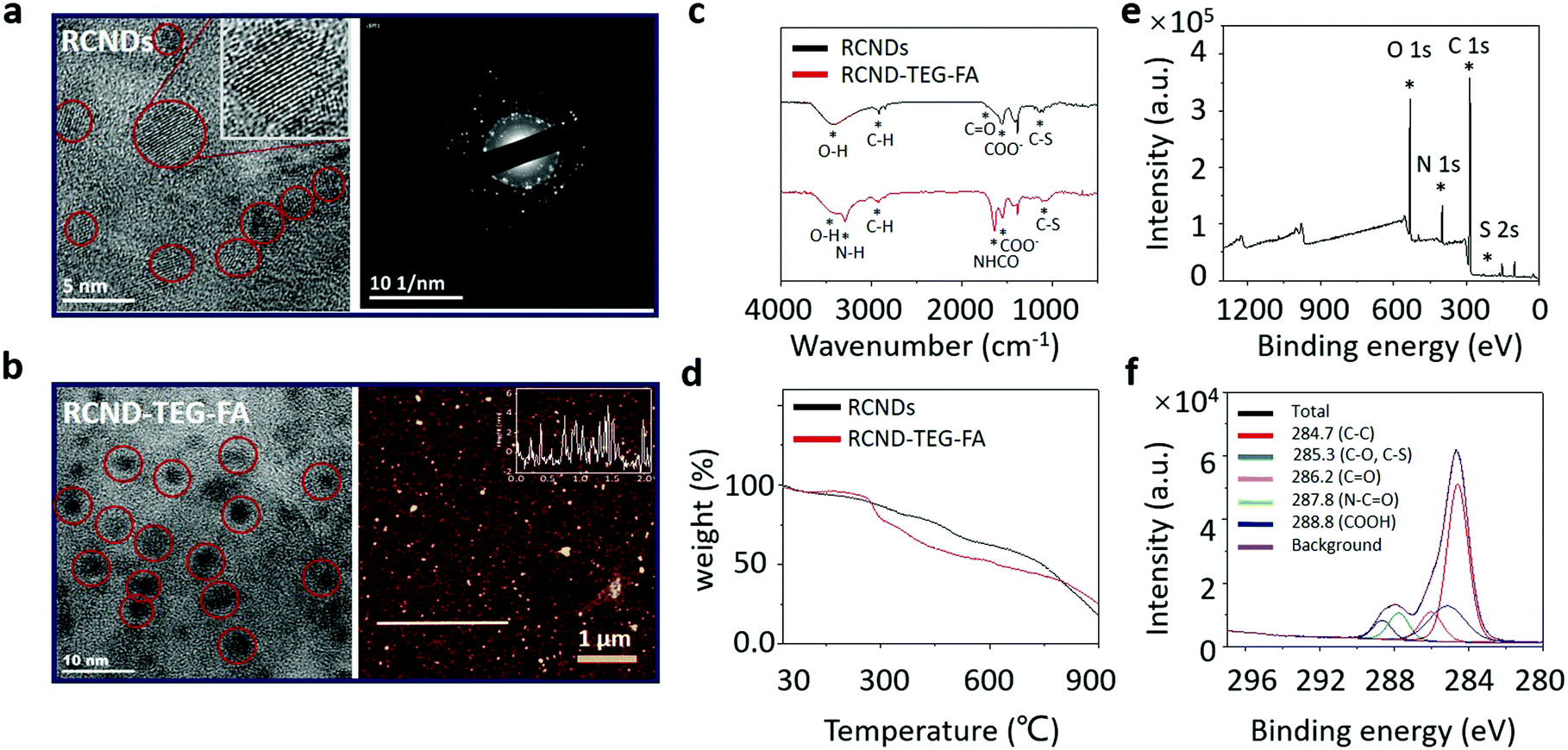 | ||
| Fig. 2 (a) HRTEM image (left) and SAED pattern (right) of RCNDs; (b) HRTEM (left) and AFM images (right) of RCND–TEG–FA; (c) FTIR spectra of RCNDs, FA ligand and RCND–TEG–FA; (d) TGA of RCNDs and RCND–TEG–FA; and (e) XPS spectra of survey and (f) high resolution C1s for RCND–TEG–FA. | ||
It is widely recognized that the hydrothermal technology is a powerful protocol to prepare nanomaterials.32,33 The hydrothermal treatment is an easy way to transform different conjugated polymers into small-size carbon nanodots endowed with different types of functional groups. The abundant oxygenated groups present on their surface allow a wide range of reactions for the post-modification of CNDs. However, the use of conjugated polymers as precursors to prepare RCNDs with specific functional groups is still not completely understood and fully explored. Here, we attempted to explore tailored RCNDs for biomedical applications.
After obtaining the starting RCNDs, we designed and synthesized the FA ligand according to the path outlined in Scheme 2. A short amine-terminated triethylene glycol (TEG) was chosen as a linker to connect FA and RCNDs. Its hydrophilic character can enhance the FA water-dispersibility upon functionalization. The use of short ethylene glycol chains offers also the advantage of minimizing adsorption phenomena and facilitating the removal of unreacted precursors, compared to longer ethylene glycol chains.34 Compound 4 was synthesized by Boc-monoprotection of commercially available 2,2′-(ethylenedioxy)bis(ethylamine).35 Folic acid was conjugated to compound 4 by amidation through N-hydroxysuccinimide (NHS)/1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) activation. After Boc cleavage using trifluoracetic acid, FA ligand 6 was obtained in 27% total yield. Though FA has two free COOH groups in the glutamic acid moiety, it has already been reported that the γ-COOH is more prone to amidation than the α-COOH due to its higher reactivity.36,37
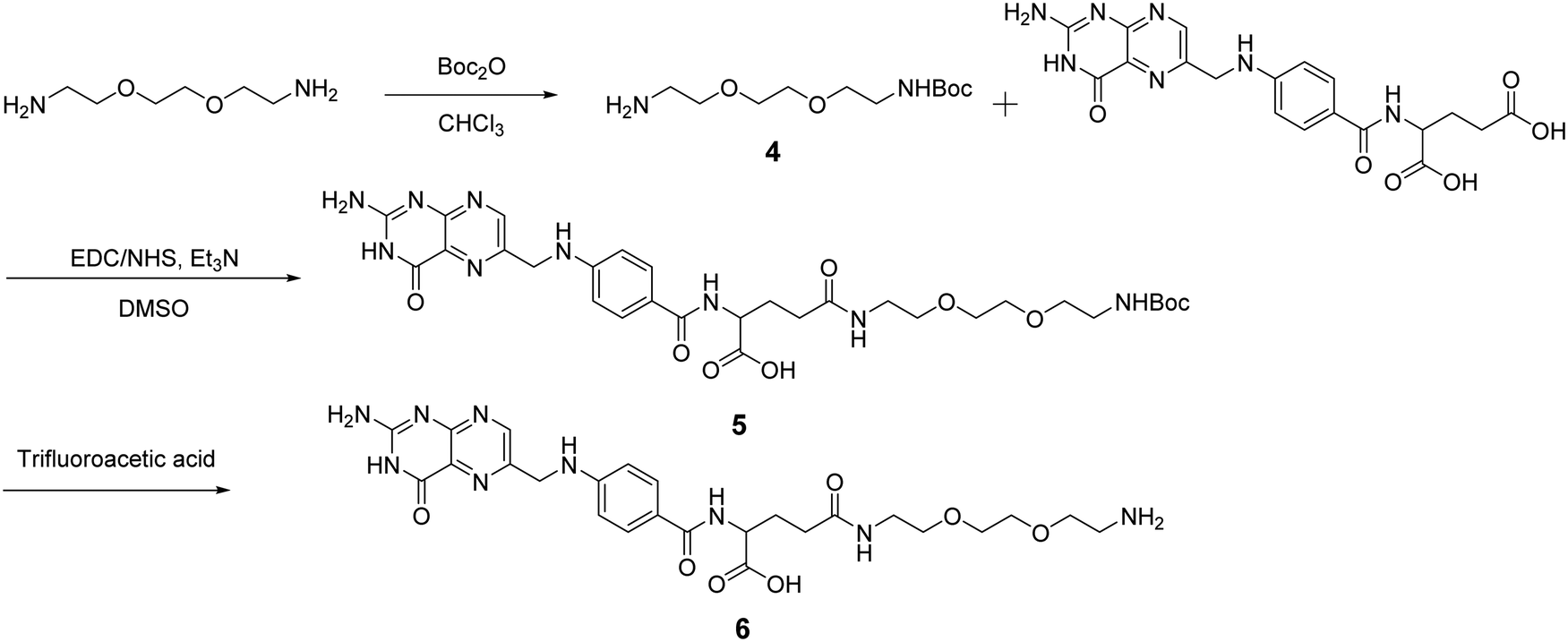 | ||
| Scheme 2 Synthetic pathway to prepare an FA ligand. For simplicity we have represented only the structure of the ligand with the aminated γ-COOH. | ||
RCND–TEG–FA was prepared via amidation of the carboxyl groups of RCNDs with the primary amine of FA derivative in the presence of EDC. The formation of RCND–TEG–FA was confirmed by FT-IR, UV-Vis, and X-ray photoelectron spectroscopy (XPS). The XPS survey spectra evidenced the presence of carbon, sulfur, oxygen, and nitrogen in the RCND–TEG–FA (Fig. 2e). Compared to the XPS of RCNDs (Fig. S1, ESI†), the additional peak of nitrogen confirmed the conjugation with FA ligand. The deconvolution of the C1s peak displays different peaks of carbon atoms, namely the graphitic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, the aliphatic C–C, the various oxygenated C–O, CO, and COO, the sulfur C–S and the amide (NH–CO) bonds (Fig. 2f). FT-IR confirmed the presence of N–H and the amide I stretching at 3296 cm−1 and 1640 cm−1, respectively (Fig. 2c). Thus, these results collectively confirm that FA ligand was successfully covalently linked onto the surface of the RCNDs. Thermogravimetric analysis (TGA) of RCND–TEG–FA performed in N2 showed an increase of 10% weight loss at 600 °C in comparison to the RCNDs, also confirming the efficiency of FA functionalization (Fig. 2d). Finally, the size of the as-prepared RCND–TEG–FA was measured by HRTEM and atomic force microscopy (AFM) (Fig. 2b). The obtained nanoparticles display a nearly round shape with a lateral size of ∼5 nm and a height of 1 to 5 nm, respectively. Such a narrow size distribution is consistent with graphene quantum dots,38 and PEG-modified carbon nanodots.39
C, the aliphatic C–C, the various oxygenated C–O, CO, and COO, the sulfur C–S and the amide (NH–CO) bonds (Fig. 2f). FT-IR confirmed the presence of N–H and the amide I stretching at 3296 cm−1 and 1640 cm−1, respectively (Fig. 2c). Thus, these results collectively confirm that FA ligand was successfully covalently linked onto the surface of the RCNDs. Thermogravimetric analysis (TGA) of RCND–TEG–FA performed in N2 showed an increase of 10% weight loss at 600 °C in comparison to the RCNDs, also confirming the efficiency of FA functionalization (Fig. 2d). Finally, the size of the as-prepared RCND–TEG–FA was measured by HRTEM and atomic force microscopy (AFM) (Fig. 2b). The obtained nanoparticles display a nearly round shape with a lateral size of ∼5 nm and a height of 1 to 5 nm, respectively. Such a narrow size distribution is consistent with graphene quantum dots,38 and PEG-modified carbon nanodots.39
2.2. Colloidal stability of RCNDs and RCND–TEG–FA
A high colloidal stability in an aqueous medium is essential for evaluating the biological applications of nanoparticles and nanomaterials.40 In fact, their aggregation can jeopardize many desirable therapeutic performances, including high drug loading capacity and efficient cellular uptake.41,42 Even worse, the aggregated nanoparticles can cause hemolysis and tissue damage.43–45 To verify the colloidal stability, 100 μg of RCNDs and RCND–TEG–FA were dispersed in 1 mL of deionized (DI) water, phosphate-buffered saline (PBS) without Ca2+ and Mg2+, saline solution (0.9%), and cell culture medium (Fig. S2, ESI†). A high colloidal stability of RCND–TEG–FA in both simple and complex physiological media was found (Fig. S2a, ESI†). No precipitates were observed after storing the solutions for 4 days. The concentration of RCND–TEG–FA can reach 1 mg mL−1 in cell culture medium without any aggregation (Fig. S2c, ESI†), while pristine RCNDs exhibited a bad colloidal stability in the different media (Fig. S2b, ESI†). We further confirmed the aggregation and the increase of the size of the RCNDs using HRTEM, UV-Vis spectra and dynamic light scattering (DLS). From the HRTEM images (Fig. S3, ESI†), we observed a strong aggregation of RCNDs in water, while no aggregation was observed in an RCND–TEG–FA sample. We also observed a red shift in UV-Vis absorbance of redispersed RCNDs (Fig. 3a), which is a sign of aggregation. DLS showed that monodisperse RCNDs possessed a good dispersibility with a narrow size distribution of 4–10 nm (Fig. 3b). However, after lyophilization the redispersed RCNDs revealed a strong aggregation with an abnormal size range from 400 to 700 nm by DLS (Fig. 3b). The RCND–TEG–FA displays an increased size compared to monodisperse RCNDs. All the results indicated that pristine RCNDs without modification are easy to aggregate in neutral solution. The large number of hydroxyl, carbonyl, and carboxyl groups on the surface of RCNDs strongly interact via hydrogen bonding, thus reducing the repelling forces between the dots, and causing the nanoparticles to cluster into large aggregates.46 In addition, the lyophilization process likely enhances the aggregation through π–π stacking between RCNDs. Since it is widely accepted that the size and morphology of nanomaterials play crucial roles in the biomedical field,47–49 there is a real risk that the aggregation may affect their performance similarly to other carbon materials. Fortunately, the aggregated architecture can be prevented by a covalent modification of RCNDs. After amidation with FA ligand, the nanoparticles became well-dispersed in water, even after lyophilization, with a slightly increased size to 15–30 nm and similar UV-Vis spectra as monodispersed RCNDs. Indeed, the covalent functionalization of RCNDs with FA ligand greatly inhibits their aggregation in a physiological environment, allowing stable solutions that have potential biomedical applications to be obtained.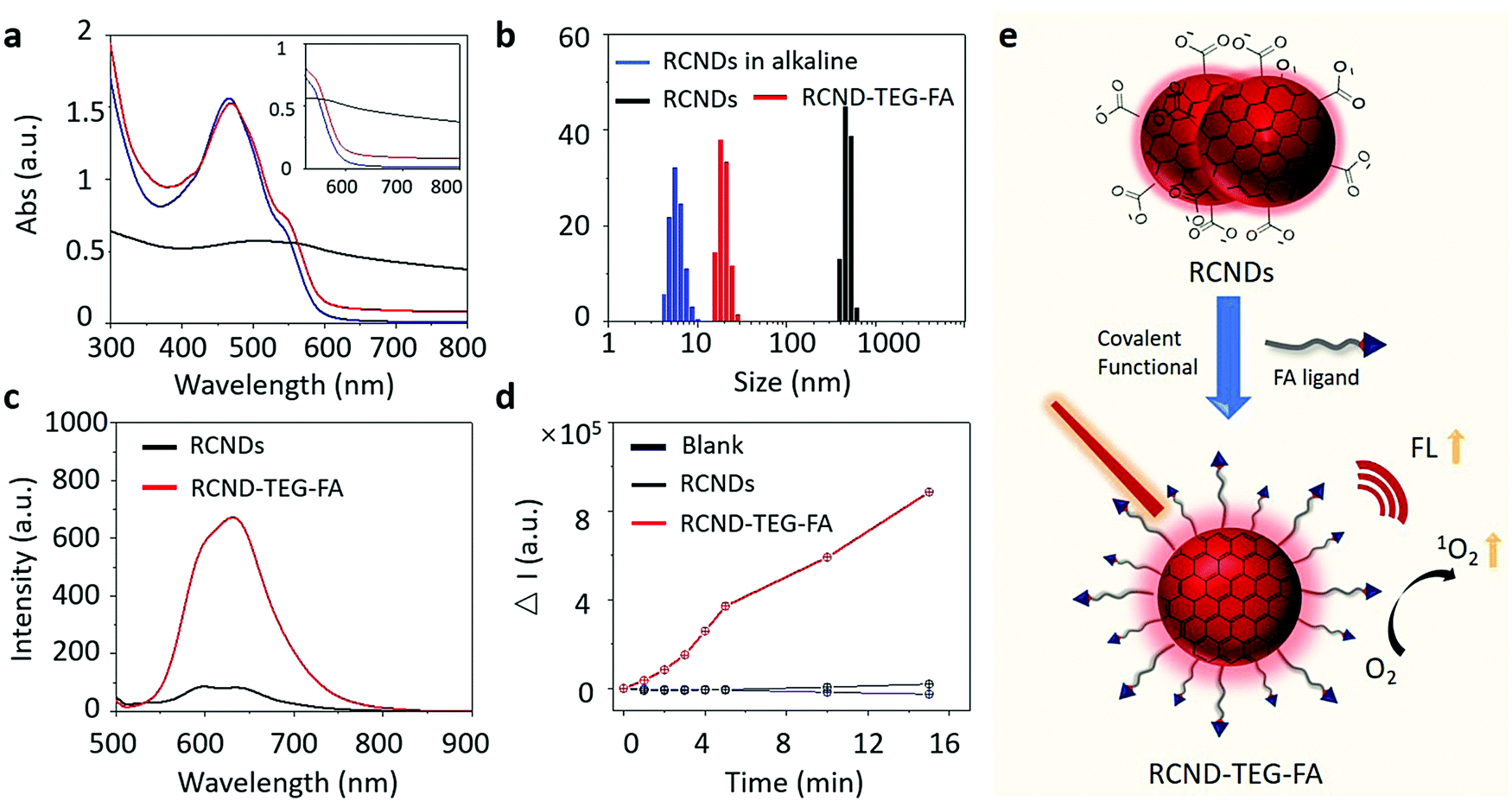 | ||
| Fig. 3 (a) Stacked UV-vis spectra and (b) DLS of RCNDs in alkaline solution at pH = 12 (blue line), RCNDs in water (black line) and RCND–TEG–FA in water (red line); (c) the FL spectra of RCND (25 μg mL−1) and RCND–TEG–FA (25 μg mL−1 of RCNDs) in water; (d) plotting the fluorescence intensity of DHR123 at 525 nm in the presence of RCNDs (25 μg mL−1) and RCND–TEG–FA (25 μg mL−1 of RCNDs) upon 660 nm laser irradiation (0.1 W cm−2) with time; (e) scheme of the covalent functionalization of carbon nanodot aggregates. | ||
2.3. Fluorescence detection and ROS generation
With RCND–TEG–FA in hand, we decided to investigate their fluorescence emission for cell imaging and their ROS production. Fig. 3c displays the fluorescence spectra of RCNDs and RCND–TEG–FA aqueous solutions. Both samples exhibited a wide range emission spectrum from 500 to 800 nm with a strong peak at 640 nm (excitation: 485 nm). However, RCND–TEG–FA showed a remarkably stronger fluorescence emission, being ∼6-fold higher than that of RCNDs in water (at 25 μg mL−1). Furthermore, the fluorescence quantum yield of RCNDs was measured using a spectrometer attached to an integrating sphere and corresponded to 0.4% in water, while the fluorescence quantum yield of RCND–TEG–FA increased to 1%. This quantum yield is higher than that of Se-doped CDs (0.2%) and CD nanoparticles (0.15%) prepared from polythiophene derivatives.50,51 Although recently reported F-doped carbon dots were endowed with a high fluorescence quantum yield,52 the low quantum yield of our RCNDs does not prevent their use for biomedical applications, including fluorescence imaging.51,52 This enhancement of RCND fluorescence is clearly due to the presence of the FA ligand preventing RCND aggregation and improving the solubility. These results clearly indicate that the chemical functionalization of the RCNDs improved their fluorescence properties. The enhanced fluorescence intensity could make RCND–TEG–FA useful for cell imaging.Then, we focused our attention on the generation of ROS by RCND–TEG–FA. ROS were measured by using dihydrorhodamine 123 (DHR123) as the indicator (Fig. S4, ESI†). This non-fluorescent dye becomes rhodamine 123 with a high red emission upon reaction with ROS. The aqueous solution containing RCND–TEG–FA (25 μg mL−1) showed an increase of the fluorescence intensity at 530 nm upon exposure to a 660 nm low power laser (0.1 W cm−2) at different irradiation times, which was not the case with RCNDs (Fig. 3d). The remarkable fluorescence intensity was ∼40-fold higher than that generated by the same concentration of pristine RCNDs. As a control, the exposure of an aqueous solution of DHR123 to a 660 nm laser did not induce obvious fluorescence changes, indicating fast and efficient ROS generation by RCND–TEG–FA. This notable increase of the ROS generation can be ascribed to the higher quantum yield of the functionalized material compared to pristine RCNDs. Altogether, these results reveal that controlled functionalization of FA ligands is an effective strategy to prevent the aggregation of nanodots, leading to a superior colloidal stability, enhanced luminescence and ROS generation (Fig. 3e).
To investigate the mechanism of ROS generation, we used thiourea and glutathione (GSH) as inhibitors of ˙OH and O2˙−, respectively.53–55 As shown in Fig. S5 (ESI†) neither thiourea nor GSH were able to inhibit DHR123 oxidation. These results suggest that the hydroxyl radical and the superoxide were not generated during the photo-irradiation of RCND–TEG–FA. Moreover, in de-aerated solution, RCND–TEG–FA are also not able to generate ROS indicating that DHR123 oxidation is mediated by O2 radical formation. Radical oxygen formation (1O2) was evaluated using the specific probe 9,10-anthracenediyl-bis dimalonic acid (ABDA) (Fig. 4 and Fig. S6, ESI†). RCND–TEG–FA were able to efficiently oxidize ABDA after laser irradiation. The singlet oxygen quantum yield was further calculated in water by using methylene blue as the standard photosensitizer (1O2 quantum yield ΦMB = 0.52 in water) and corresponded to 0.40 (Fig. S7, ESI†).56,57 The yield is similar to the recently reported CD nanopheres (0.45).58 An inhibition test irradiating RCND–TEG–FA in the presence of NaN3 as a singlet oxygen inhibitor was also performed (Fig. 4c and d). Negligible ABDA oxidation was recorded in the presence of the inhibitor. The ABDA oxidation mechanism is normally triggered by singlet oxygen (via an energy transfer mechanism) or superoxide (via a charge transfer mechanism).59 As NaN3 quenches only singlet oxygen, the results suggest that only 1O2 is formed during the irradiation, while superoxide is again absent, confirming the data described above using GSH inhibitor. Singlet oxygen is commonly produced via type 2 photosensitizers through an energy transfer mechanism. In our case, we used a radical trap (ABDA) well-known to be specific for singlet oxygen detection. The mechanism can be ascribed to the energy transfer as described by other type 2 photosensitizers. From these results we can conclude that 1O2 is generated via the typical energy transfer mechanism between the triplet state of RCND–TEG–FA and 3O2 that is excited to its singlet state.22
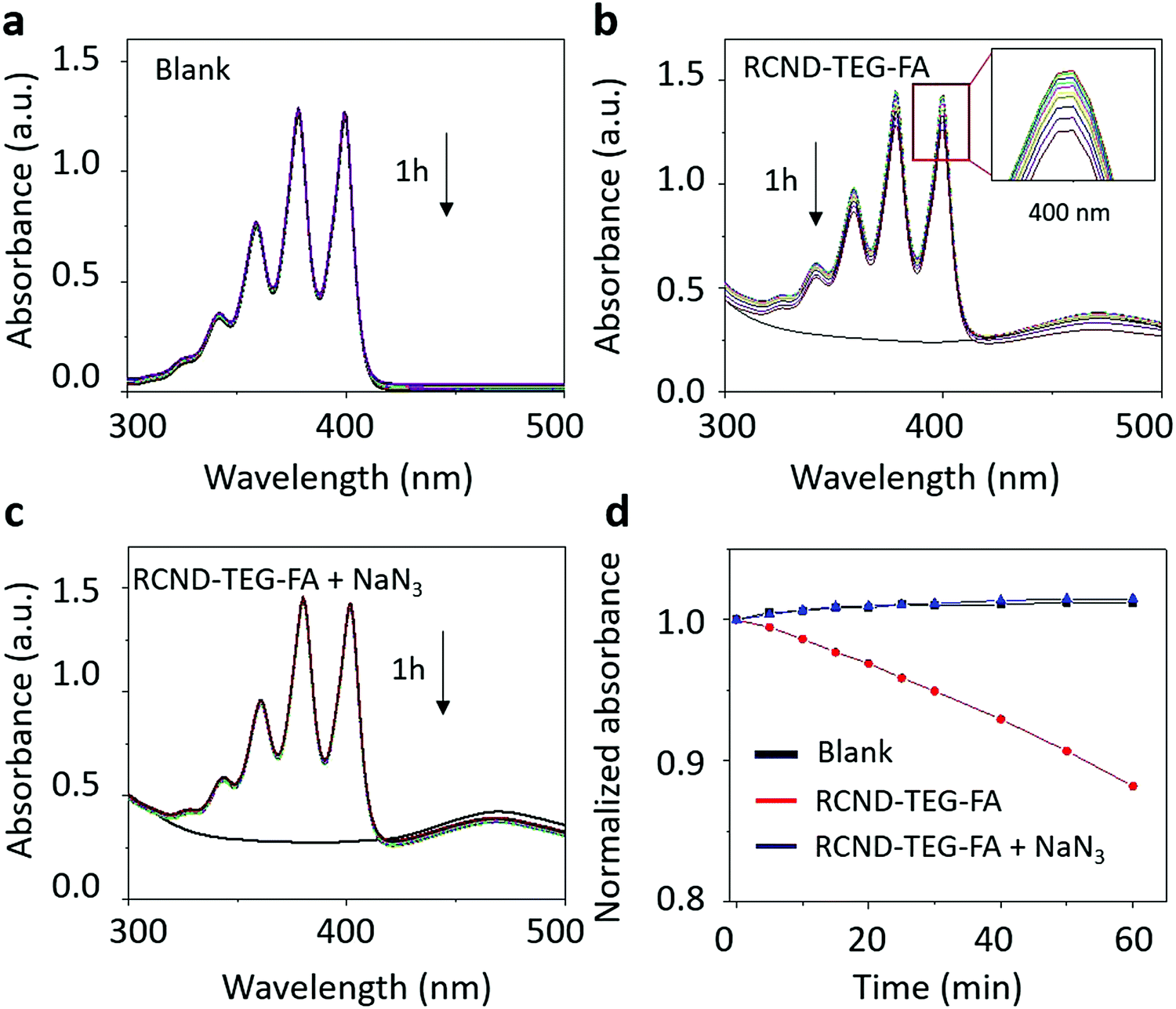 | ||
| Fig. 4 Singlet oxygen characterization. Time-dependent absorbance of the ABDA in different conditions. (a) Blank; (b) RCND–TEG–FA (25 μg mL−1), insert image to show the peak at 400 nm; (c) RCND–TEG–FA with adding 5 mg NaN3; (d) normalized absorbance of ABDA at 400 nm in the presence of RCND–TEG–FA under different conditions. | ||
2.4. Cellular uptake and fluorescence cell imaging
To test the receptor-targeted imaging capacity of RCND–TEG–FA, HeLa cells were chosen as a model because of their high expression level of FRs. HeLa cells were incubated with RCND–TEG–FA at the concentration of 20 μg mL−1 or 100 μg mL−1 at different time points (0, 4, 8, 24 h). Because of their red emission, RCND–TEG–FA were easily visualized by confocal microscopy. CellMask green was used to observe the morphology of the cell membranes. As shown in Fig. 5, RCND–TEG–FA were able to be internalized in HeLa cells already after 4 h, displaying a remarkable red emission. After 8 h incubation, the cytoplasm of the cells was full of red dots. RCND–TEG–FA displayed a stable fluorescent signal, which was clearly observed in the cells even after 24 h incubation. To prove the FR-mediated penetration, a competition assay showed that the pretreatment of HeLa cells with FA resulted in a minimized uptake of RCND–TEG–FA, confirming FR-mediated endocytosis (Fig. 6). These results are consistent with previous reports.60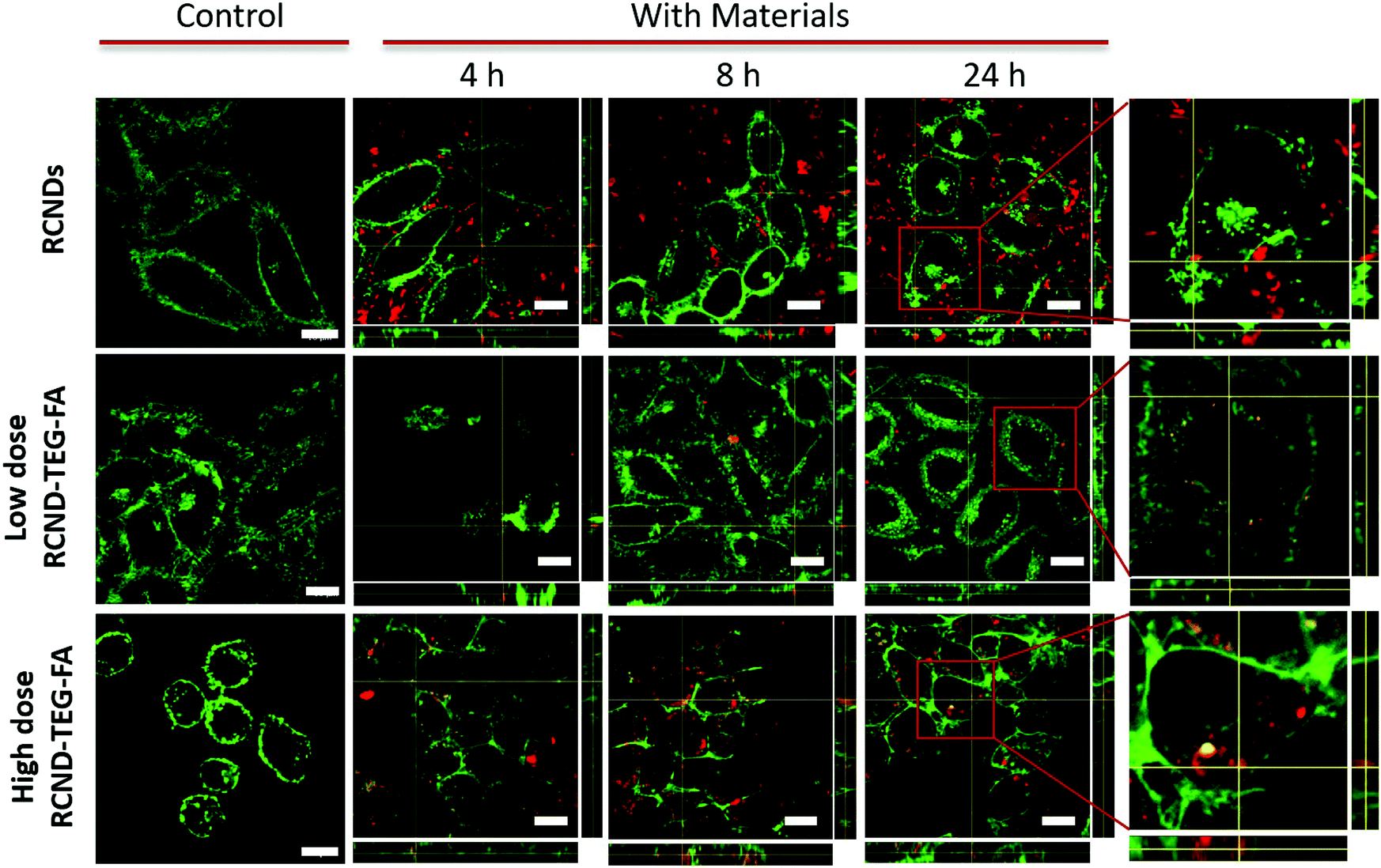 | ||
| Fig. 5 z-Stacking analysis with xz and yz orthogonal views on live HeLa cells incubated with 20 μg mL−1 of RCNDs, and 20 μg mL−1 or 100 μg mL−1RCND–TEG–FA (upper panel) and UT (lower panel) after 4, 8, and 24 h. In green, membranes stained with CellMask (excitation channel: 488 nm, emission: 500–550 nm); in red, RCND–TEG–FA (excitation channel: 405 nm, emission: 665–715 nm). The scale bar is 10 μm. | ||
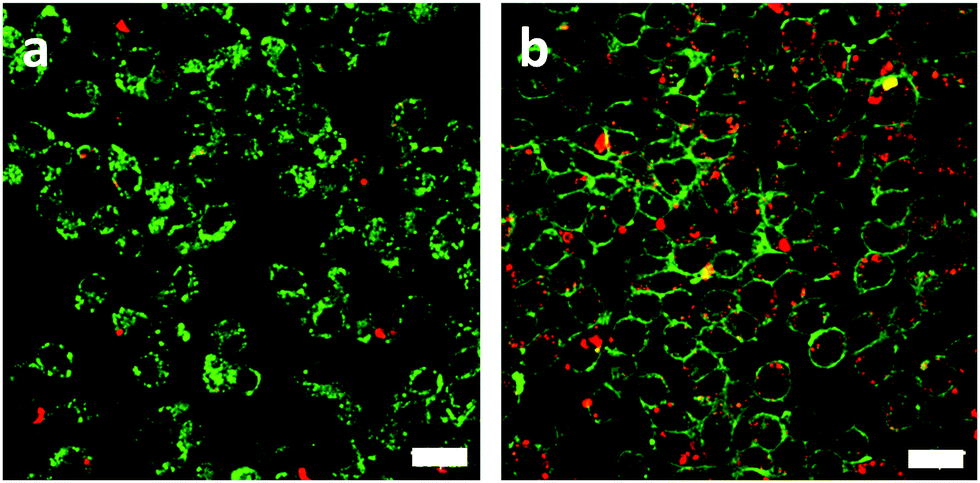 | ||
| Fig. 6 Fluorescence images of HeLa cells incubated with 100 μg mL−1 of RCND–TEG–FA in the presence (a) or the absence (b) of free FA pretreatment. In green, membranes stained with CellMask (excitation channel: 488 nm, emission: 500–550 nm); in red, RCND–TEG–FA. The scale bar is 20 μm. | ||
We also found that the pristine RCNDs strongly aggregated outside the cells even at a low concentration (20 μg mL−1), which was not the case for RCND–TEG–FA under similar conditions. This is in agreement with the limited colloidal stability of the RCNDs, demonstrating again the importance of the covalent modification of RCNDs.
2.5. Intracellular detection of ROS
Inspired by the above results, we attempted to assess the ROS producing ability of RCND–TEG–FA in the targeted cells. To capture the ROS in HeLa cells, CellROX reagent was chosen as an indicator. This dye is initially non-fluorescent in a reduced state and becomes fluorescent (yellow color) upon oxidation by ROS.61 The HeLa cells were incubated with RCND–TEG–FA (at 100 μg mL−1) for 24 h at 37 °C. After washing to remove RCND–TEG–FA outside the cells, the CellROX reagent was added to monitor the intracellular generation of ROS. We observed that the ROS level was negligible until the laser irradiation was switched on (Fig. 7a and b). HeLa cells showed strong fluorescence after 10 min irradiation using a low power laser (0.1 W cm−2) at 660 nm. As a control, HeLa cells without RCND–TEG–FA incubation were also exposed to the same laser irradiation, and no fluorescence changes were observed (Fig. S8, ESI†). These results provide evidence that RCND–TEG–FA are able to produce a high level of ROS under irradiation in the visible region, resulting in potentially promising tools for PDT.62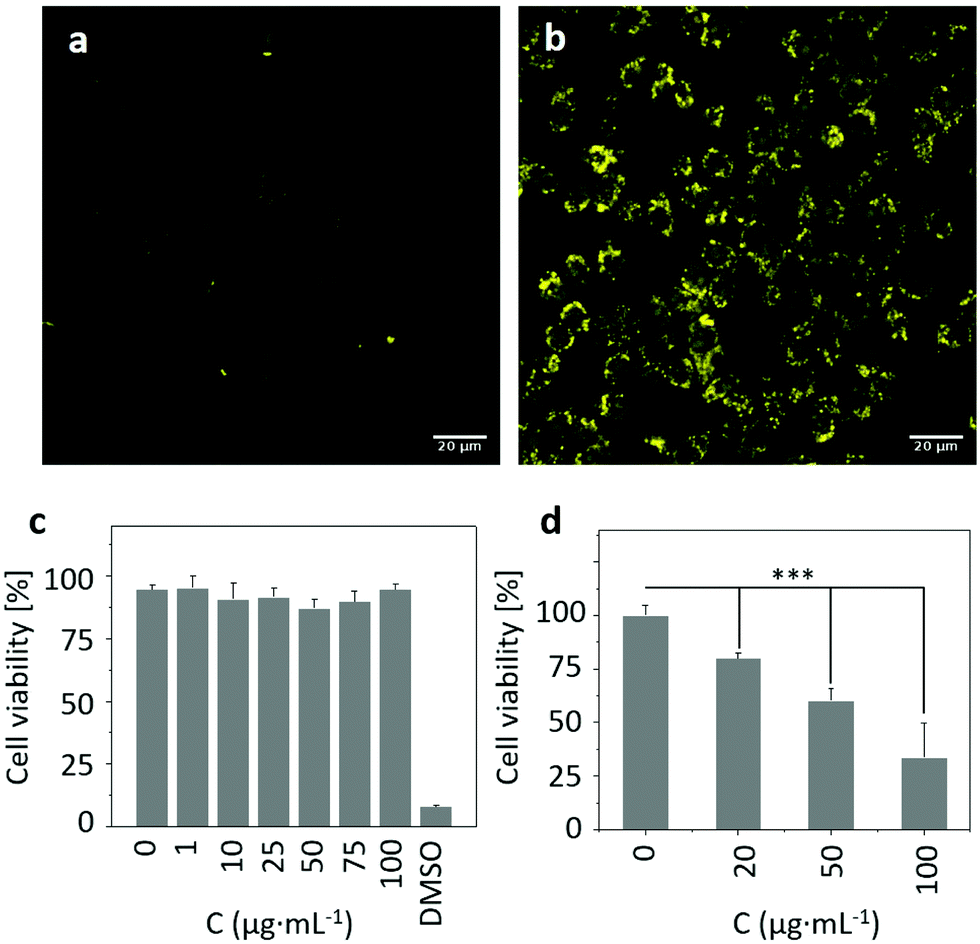 | ||
| Fig. 7 Intracellular ROS formation without (a) or with (b) low-dose 660 nm laser irradiation (0.1 W cm−2). HeLa cells were incubated with CellROX reagent (excitation channel: 488 nm, emission: 500–550 nm) for 30 min before irradiation. The scale bar is 20 μm. Cell viability of HeLa cells incubated with increased concentrations of RCND–TEG–FA (0–100 μg mL−1) (c) without laser irradiation or (d) under 660 nm laser irradiation (0.1 W cm−2). Data are the mean ± sd of three independent experiments with each treatment performed in three wells (one-way analysis of variance followed by Tukey's post-test: ***p < 0.001). | ||
2.6. In vitro red-light responsive PDT
To explore the potential of functionalized RCNDs for in vitro red-light responsive PDT, the cytotoxicity and PDT efficacy of RCND–TEG–FA were quantitatively analyzed by an alamarBlue™ cell viability assay. RCND–TEG–FA tested at different concentrations (0–100 μg mL−1) without laser irradiation exhibited negligible toxicity to HeLa cells (Fig. 7c). This result indicated that RCND–TEG–FA cannot induce cancer cell death, confirming good biocompatibility. However, the viability of the cells incubated with RCND–TEG–FA (at 0, 20, 50 and 100 μg mL−1) under laser irradiation at 660 nm (0.1 W cm−2) gradually decreased as RCND–TEG–FA concentration increased (Fig. 7d). A statistically significant mortality rate of ∼70% was observed at 100 μg mL−1 of RCND–TEG–FA, indicating that RCND–TEG–FA produce a sufficient amount of ROS to kill the cancer cells. The cell viability assay clearly demonstrated that RCND–TEG–FA can be applied as a versatile, highly efficient photosensitizer for simultaneous in vitro red-light responsive fluorescence imaging and PDT.A few studies have reported the synthesis of carbon nanodots with intrinsic properties for ROS generation and used as a PDT agent.22,63 However, the targeted release of toxic levels of ROS produced by red-emissive carbon nanodots without loading additional photosensitizers, leading to cell death, has not been reported so far. FA-modified RCNDs showed great capacity to target FR overexpressing cells. Our strategy is flexible as a wide variety of other multifunctional carbon nanodots could be developed for targeted disease imaging and therapy by appropriate covalent functionalization. Other targeting agents such as glycoligands, peptides, aptamers, and small molecules, might expand the use of carbon nanodots.
3. Conclusions
In summary, we have developed a new method to synthesize red-emissive carbon nanodots using conjugated polythiophenes as precursors, and further modified the nanomaterial with FA for targeted cell imaging and intracellular production of ROS. Many techniques were used to monitor the preparation process, validating our controlled strategy to prepare multifunctional carbon nanodots. We discovered that the covalent functionalization of RCNDs can effectively prevent the aggregation of the dots, leading to superior colloidal stability, enhanced luminescence and ROS generation. Conjugation with folic acid has resulted in a material with very good cancer cell targeting capabilities. The collective properties of FA-modified RCNDs make them a multifunctional nanoplatform for simultaneous targeted imaging and a highly efficient PDT agent for cancer treatment. Our approach might open new routes for the development of a large variety of highly dispersible multifunctional carbon nanodots for phototherapies. The synthesis of nanomaterials with intrinsic therapeutic properties offers opportunities for combined therapies through the conjugation of anticancer drugs, nucleic acids, or photothermal agents.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We gratefully acknowledge the Centre National de la Recherche Scientifique (CNRS), the International Center for Frontier Research in Chemistry (icFRC), and financial support from the Agence Nationale de la Recherche (ANR) through the LabEx project Chemistry of Complex Systems (ANR-10-LABX-0026_CSC) within the Investissement d’Avenir program (ANR-10-120 IDEX-0002-02). We wish to thank A. Aliprandi for the quantum yield measurements, and C. Royer and V. Demais for help with TEM analyses at the “Plateforme Imagerie in vitro” at the Center of Neurochemistry (INCI, Strasbourg, France).References
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579 CrossRef CAS PubMed.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597 RSC.
- X. Chen, F. Wang, J. Y. Hyun, T. Wei, J. Qiang, X. Ren, I. Shin and J. Yoon, Chem. Soc. Rev., 2016, 45, 2976 RSC.
- (a) D.-K. Ji, C. Ménard-Moyon and A. Bianco, Adv. Drug Delivery Rev., 2019, 138, 211 CrossRef CAS PubMed; (b) S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990 CrossRef CAS PubMed.
- D.-K. Ji, Y. Zhang, Y. Zang, J. Li, G.-R. Chen, X.-P. He and H. Tian, Adv. Mater., 2016, 28, 9356 CrossRef CAS PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380 CrossRef CAS PubMed.
- R. Kurapati, K. Kostarelos, M. Prato and A. Bianco, Adv. Mater., 2016, 28, 6052 CrossRef CAS PubMed.
- (a) L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869 CrossRef CAS PubMed; (b) S. Gai, G. Yang, P. Yang, F. He, J. Lin, D. Jin and B. Xing, Nano Today, 2018, 19, 146 CrossRef CAS.
- J. Sudimack and R. J. Lee, Adv. Drug Delivery Rev., 2000, 41, 147 CrossRef CAS PubMed.
- C. P. Leamon and J. A. Reddy, Adv. Drug Delivery Rev., 2004, 56, 1127 CrossRef CAS PubMed.
- Y. Lu, E. Sega, C. P. Leamon and P. S. Low, Adv. Drug Delivery Rev., 2004, 56, 1161 CrossRef CAS PubMed.
- Y. Lu and P. S. Low, Adv. Drug Delivery Rev., 2012, 64, 342 CrossRef.
- L. Huang, Z. Li, Y. Zhao, J. Yang, Y. Yang, A. I. Pendharkar, Y. Zhang, S. Kelmar, L. Chen, W. Wu, J. Zhao and G. Han, Adv. Mater., 2017, 29, 1604789 CrossRef PubMed.
- E. Nogueira, A. C. Gomes, A. Preto and A. Cavaco-Paulo, Nanomedicine, 2016, 12, 1113 CrossRef CAS PubMed.
- Y. Liu, Y. Xu, Z. Zhang, Y. Huo, D. Chen, W. Ma, K. Sun, G. Y. Tonga, G. Zhou, D. S. Kohane and K. Tao, Nano Lett., 2019, 19, 5515 CrossRef CAS PubMed.
- M. Shamsipur, A. Barati and S. Karami, Carbon, 2017, 124, 429 CrossRef CAS.
- S. Baker and G. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726 CrossRef CAS PubMed.
- X. T. Zheng, A. Ananthanarayanan, K. Q. Luo and P. Chen, Small, 2015, 11, 1620 CrossRef CAS PubMed.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362 RSC.
- S. A. Chechetka and E. Miyako, ChemistrySelect, 2016, 1, 608 CrossRef CAS.
- S. A. Chechetka and E. Miyako, Chem. Lett., 2016, 45, 854 CrossRef CAS.
- J. Ge, M. Lan, B. Zhou, W. Liu, L. Guo, H. Wang, Q. Jia, G. Niu, X. Huang, H. Zhou, X. Meng, P. Wang, C. Lee, W. Zhang and X. Han, Nat. Commun., 2014, 5, 4596 CrossRef CAS PubMed.
- D. Gao, H. Zhao, X. Chen and H. Fan, Mater. Today Chem., 2018, 9, 103 CrossRef CAS.
- K. Hola, Y. Zhang, Y. Wang, E. P. Giannelis, R. Zboril and A. L. Rogach, Nano Today, 2014, 9, 590 CrossRef CAS.
- Y.-H. Chien, K. K. Chan, T. Anderson, K. V. Kong, B. K. Ng and K.-T. Yong, Adv. Ther., 2019, 2, 1800090 CrossRef.
- L. Guo, J. Ge and P. Wang, Photochem. Photobiol., 2018, 94, 916 CrossRef CAS PubMed.
- L. Guo, G. Niu, X. Zheng, J. Ge, W. Liu, Q. Jia, P. Zhang, H. Zhang and P. Wang, Adv. Sci., 2017, 4, 1700085 CrossRef PubMed.
- Q. Jia, J. Ge, W. Liu, X. Zheng, S. Chen, Y. Wen, H. Zhang and P. Wang, Adv. Mater., 2018, 30, 1706090 CrossRef PubMed.
- D. Li, P. Jing, L. Sun, Y. An, X. Shan, X. Lu, D. Zhou, D. Han, D. Shen, Y. Zhai, S. Qu, R. Zbořil and A. L. Rogach, Adv. Mater., 2018, 30, 1705913 CrossRef PubMed.
- G. Xu, X. Bao, J. Chen, B. Zhang, D. Li, D. Zhou, X. Wang, C. Liu, Y. Wang and S. Qu, Adv. Healthcare Mater., 2019, 8, 1800995 CrossRef PubMed.
- M. Triesscheijn, P. Baas, J. H. M. Schellens and F. A. Stewart, Oncologist, 2006, 11, 1034 CrossRef CAS PubMed.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734 CrossRef CAS PubMed.
- K. Byrappa and T. Adschiri, Prog. Cryst. Growth Charact. Mater., 2007, 53, 117 CrossRef CAS.
- C. Spinato, A. Perez Ruiz de Garibay, M. Kierkowicz, E. Pach, M. Martincic, R. Klippstein, M. Bourgognon, J. T.-W. Wang, C. Ménard-Moyon, K. T. Al-Jamal, B. Ballesteros, G. Tobias and A. Bianco, Nanoscale, 2016, 8, 12626 RSC.
- G. Baier, D. Baumann, J. M. Siebert, A. Musyanovych, V. Mailänder and K. Landfester, Biomacromolecules, 2012, 13, 2704 CrossRef CAS PubMed.
- A. Gabizon, A. T. Horowitz, D. Goren, D. Tzemach, F. Mandelbaum-Shavit, M. M. Qazen and S. Zalipsky, Bioconjugate Chem., 1999, 10, 289 CrossRef CAS PubMed.
- S. D. Boss, T. Betzel, C. Müller, C. R. Fischer, S. Haller, J. Reber, V. Groehn, R. Schibli and S. M. Ametamey, Bioconjugate Chem., 2016, 27, 74 CrossRef CAS PubMed.
- Y. Li, W. Zhang, L. Zhang, J. Li, Z. Su and G. Wei, Adv. Mater. Interfaces, 2017, 4, 1600895 CrossRef.
- P. Huang, J. Lin, X. Wang, Z. Wang, C. Zhang, M. He, K. Wang, F. Chen, Z. Li, G. Shen, D. Cui and X. Chen, Adv. Mater., 2012, 24, 5104 CrossRef CAS PubMed.
- T. L. Moore, L. Rodriguez-lorenzo, V. Hirsch, S. Balog, D. Urban, C. Jud, B. Rothen-rutishauser and A. Petri-fink, Chem. Soc. Rev., 2015, 44, 6287 RSC.
- C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2002, 54, 631 CrossRef.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2016, 17, 20 CrossRef PubMed.
- M. Mutlu, G. R. S. Budinger, A. A. Green, D. Urich, S. Soberanes, S. E. Chiarella, G. F. Alheid, D. R. Mccrimmon, I. Szleifer and M. C. Hersam, Nano Lett., 2010, 10, 1664 CrossRef PubMed.
- L. Yildirimer, N. T. K. Thanh, M. Loizidou and A. M. Seifalian, Nano Today, 2011, 6, 585 CrossRef CAS PubMed.
- A. Albanese and W. C. W. Chan, ACS Nano, 2011, 5, 5478 CrossRef CAS PubMed.
- D. A. Kurdyukov, D. A. Eurov, M. K. Rabchinskii, A. V. Shvidchenko, M. V. Baidakova, D. A. Kirilenko, S. V. Koniakhin, V. V. Shnitov, V. V. Sokolov, P. N. Brunkov, A. T. Dideikin, Y. M. Sgibnev, L. Y. Mironov, D. A. Smirnov, A. Y. Vul’ and V. G. Golubev, Nanoscale, 2018, 10, 13223 RSC.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218 RSC.
- M. Zhu, G. Nie, H. Meng, T. Xia, A. Nel and Y. Zhao, Acc. Chem. Res., 2013, 46, 622 CrossRef CAS PubMed.
- H. Heinz, C. Pramanik, O. Heinz, Y. Ding, R. K. Mishra, D. Marchon, R. J. Flatt, I. Estrela-Lopis, J. Llop, S. Moya and R. F. Ziolo, Surf. Sci. Rep., 2017, 72, 1 CrossRef CAS.
- M. Lan, S. Zhao, Z. Zhang, L. Yan, L. Guo, G. Niu, J. Zhang, J. Zhao, H. Zhang, P. Wang, G. Zhu, C.-S. Lee and W. Zhang, Nano Res., 2017, 10, 3113–3123 CrossRef CAS.
- L. Guo, G. Niu, X. Zheng, J. Ge, W. Liu, Q. Jia, P. Zhang, H. Zhang and P. Wang, Adv. Sci., 2017, 4, 1700085 CrossRef PubMed.
- L. Jiang, H. Ding, M. Xu, X. Hu, S. Li, M. Zhang, Q. Zhang, Q. Wang, S. Lu, Y. Tian and H. Bi, Small, 2020, 16, 2000680 CrossRef CAS PubMed.
- Y. Guo and B. Li, Carbon, 2015, 82, 459 CrossRef CAS.
- J. R. Harbour and S. L. Issler, J. Am. Chem. Soc., 1982, 104, 903 CrossRef CAS.
- W. Wang, M. N. Schuchmann, H.-P. Schuchmann, W. Knolle, J. von Sonntag and C. von Sonntag, J. Am. Chem. Soc., 1999, 121, 238 CrossRef CAS.
- Z. Li, D. Wang, M. Xu, J. Wang, X. Hu, S. Anwar, A. C. Tedesco, P. C. Morais and H. Bi, J. Mater. Chem. B, 2020, 8, 2598–2606 RSC.
- L. V. Lutkus, S. S. Rickenbach and T. M. McCormick, J. Photochem. Photobiol., A, 2019, 378, 131–135 CrossRef CAS.
- Q. Jia, J. Ge, W. Liu, L. Guo, X. Zheng, S. Chen, M. Chen, S. Liu, L. Zhang, M. Wang, H. Zhang and P. Wang, Adv. Healthcare Mater., 2017, 6, 1601419 CrossRef PubMed.
- C. Felip-León, M. Puche, J. F. Miravet, F. Galindo and M. Feliz, Mater. Lett., 2019, 251, 45 CrossRef.
- (a) Y. Choi, S. Kim, M. Choi, S. Ryoo, J. Park, D. Min and B. Kim, Adv. Funct. Mater., 2014, 24, 5781 CrossRef CAS; (b) Y. Song, W. Shi, W. Chen, X. Li and H. Ma, J. Mater. Chem., 2012, 22, 12568 RSC.
- T. Kushibiki, Y. Tu, A. O. Abu-Yousif and T. Hasan, Sci. Rep., 2015, 5, 13114 CrossRef CAS PubMed.
- X. Shi, H. Meng, Y. Sun, L. Qu, Y. Lin, Z. Li and D. Du, Small, 2019, 15, 1901507 CrossRef CAS PubMed.
- J. Ge, Q. Jia, W. Liu, M. Lan, B. Zhou, L. Guo, H. Zhou, H. Zhang, Y. Wang, Y. Gu, X. Meng and P. Wang, Adv. Healthcare Mater., 2016, 5, 665 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00300j |
| This journal is © The Royal Society of Chemistry 2020 |