
Single-molecule mechanical unfolding experiments reveal a critical length for the formation of α-helices in peptides†
Damien
Sluysmans‡
 *a,
Nicolas
Willet‡
ab,
Julie
Thevenot
b,
Sébastien
Lecommandoux
b and
Anne-Sophie
Duwez
*a
*a,
Nicolas
Willet‡
ab,
Julie
Thevenot
b,
Sébastien
Lecommandoux
b and
Anne-Sophie
Duwez
*a
aMolecular Systems Research Unit, University of Liège, Sart-Tilman B6a, 4000 Liège, Belgium. E-mail: damien.sluysmans@uliege.be; asduwez@uliege.be
bUniv. Bordeaux, CNRS, Bordeaux INP, LCPO, UMR 5629, F-33600, Pessac, France
First published on 31st January 2020
Abstract
α-Helix is the most predominant secondary structure in proteins and supports many functions in biological machineries. The conformation of the helix is dictated by many factors such as its primary sequence, intramolecular interactions, or the effect of the close environment. Several computational studies have proposed that there is a critical maximum length for the formation of intact compact helical structures, supporting the fact that most intact α-helices in proteins are constituted of a small number of amino acids. To obtain a detailed picture on the formation of α-helices in peptides and their mechanical stability, we have synthesized a long homopolypeptide of about 90 amino acids, poly(γ-benzyl-L-glutamate), and investigated its mechanical behaviour by AFM-based single-molecule force spectroscopy. The characteristic plateaus observed in the force–extension curves reveal the unfolding of a series of small helices (from 1 to 4) of about 20 amino acid residues connected to each other, rather than a long helix of 90 residues. Our results suggest the formation of a tertiary structure made of short helices with kinks, instead of an intact compact helical structure for sequences of more than 20 amino acid residues. To our knowledge, this is the first experimental evidence supporting the concept of a helical critical length previously proposed by several computational studies.
New conceptsThe conformation adopted by a protein is critical for its function in cells. α-Helices constitute the most abundant folded conformations and were intensively studied. However, so far, the influence of sequence length on the folding in an intact helical conformation remains elusive and was only investigated theoretically. Several computational studies have suggested a critical length value of 20 amino acid residues for all sorts of α-helices from Protein Data Bank. Here, we experimentally address this fundamental question of the sequence length influence on the intact helical winding by probing individual polypeptides by atomic force microscopy. Using single-molecule force spectroscopy, we probed the mechanical helix–coil transition of a very stable and robust polypeptide in a strong helicogenic solvent in real-time. The force profiles evidenced the unfolding of several α-helices in series, with a critical length of ∼20 AA residues. Longer sequences show a tendency to self-fold into tertiary structures with kinks, perfectly supporting previous simulations. This concept of critical length is a crucial parameter for the design of artificial molecular machinery and materials with high-performance mechanical properties. Our study opens the way for the synthesis of artificial helices with much more rigid backbones than peptides to circumvent this critical length limitation. |
Introduction
Helical structures are of highest importance in the structural organization and stability of biopolymers.1 Particularly, the conformation of proteins has a vital importance in biology, knowing that small defects in the folding can possibly lead to malfunctions of their biological roles, e.g. the biochemical transformation of enzymes.2–5 The secondary structure of a protein also holds its molecular shape, supports its tertiary structure and regulates its conformational changes essential to perform many biological functions.5,6 One of the most abundant secondary structure found in the Protein Data Bank is the α-helix. The arrangement of this right-handed helix is dictated by the primary sequence of amino acid residues, which are connected to each other in the backbone of the helix in an arrangement allowed by specific dihedral angles. α-Helices are stabilized by intramolecular hydrogen bonds connecting residue i (CO group) to residue i + 4 (NH group) and contain 3.6 amino acid residues per turn.5Understanding the basics of the formation of α-helices in proteins, as well as their stability under external stress, as constantly experienced by proteins in the turbulent environment of cells, is crucial.7,8 Many computational studies showed that the stability of α-helices depends on the primary sequence of amino acids, the number of amino acids, as well as the environment (solvent, pH, temperature, external force, etc.).9–14 Buehler and coworkers showed7 that the formation of an α-helix is also highly dependent on the sequence length: a short peptide has a tendency to adopt a coiled-coil conformation whereas a long α-helix tends to self-fold into shorter helices connected in series by loops or kinks. This tendency of a long α-helix to adopt a self-folded conformation made of multiple helices in series seems to be dominating for homopolypeptides and other semiflexible polymers.7,15–18 Additionally, the length of intact α-helices in proteins is known to be small, the majority of helical structures being constituted of less than 30 amino acid residues.19,20 Longer α-helices present a typical kink in their secondary structure.20
In order to gain insight into the underlying mechanisms sustaining conformational changes taking place in polypeptides, several simulations were performed focusing mainly on the helix–coil transition under external force or by thermal denaturation.13,16,21 Gao and coworkers employed steered molecular dynamics (SMD) to relate the extension of a peptide to its mechanical response during the helix–coil transition.16 They showed that homopolypeptides made of more than 20 amino acids (AA) residues display a well-defined conformational transition, occurring at constant unfolding force. Such theoretical works tend to simulate the behaviour of small peptides under an external force, as a parallel to the natural agitation experienced by proteins in cells or external tension in muscles.
Pulling experiments employing atomic force microscopy (AFM) or optical traps have been successfully used to probe the mechanical response of individual biopolymers when subjected to an external force.22 AFM-based single-molecule force spectroscopy (SMFS) consists in trapping a single molecule (e.g. a peptide) and measure its force response upon stretching.23–27 The precise force detection (picoNewton = pN) and high spatial resolution (sub-nanometer) of this technique have permitted the conformational study of proteins,27–34 DNA,35,36 sugars,35,37–39 artificial small molecules,40–43 synthetic polymers,23 and biological machines.24
Here, we probe the mechanically-induced helix–coil transition of a synthetic polypeptide in real-time using AFM-based SMFS. Poly(γ-benzyl-L-glutamate) (PBLG, 90 residues) was synthesized using a previously established strategy44,45 (see ESI†) and was trapped between a substrate and an AFM tip for single-molecule force spectroscopy experiments. PBLG has been chosen for its ability to robustly fold in a stable α-helix conformation in helicogenic solvents such as dioxane, as confirmed by circular dichroism (ESI†).44,46–48 If a peptide made of an amino acid with strong helical propensity (like glutamic acid here) in a strong helicogenic solvent (like dioxane) is shown to be limited by a critical length, we can reasonably expect that other amino acids with less propensity to form stable helices will be also limited by a critical length. As mentioned above, several computational studies have evidenced a critical length value of 20 AA for all sorts of α-helices (from Protein Data Bank).7 Here, PBLG is functionalized with a dithiopyridine on one end for its anchoring on a gold substrate using strong Au–S interactions and with a poly(ethylene glycol) (PEG) tether on the other end for its attachment to the AFM tip during the force experiments (Fig. 1a).
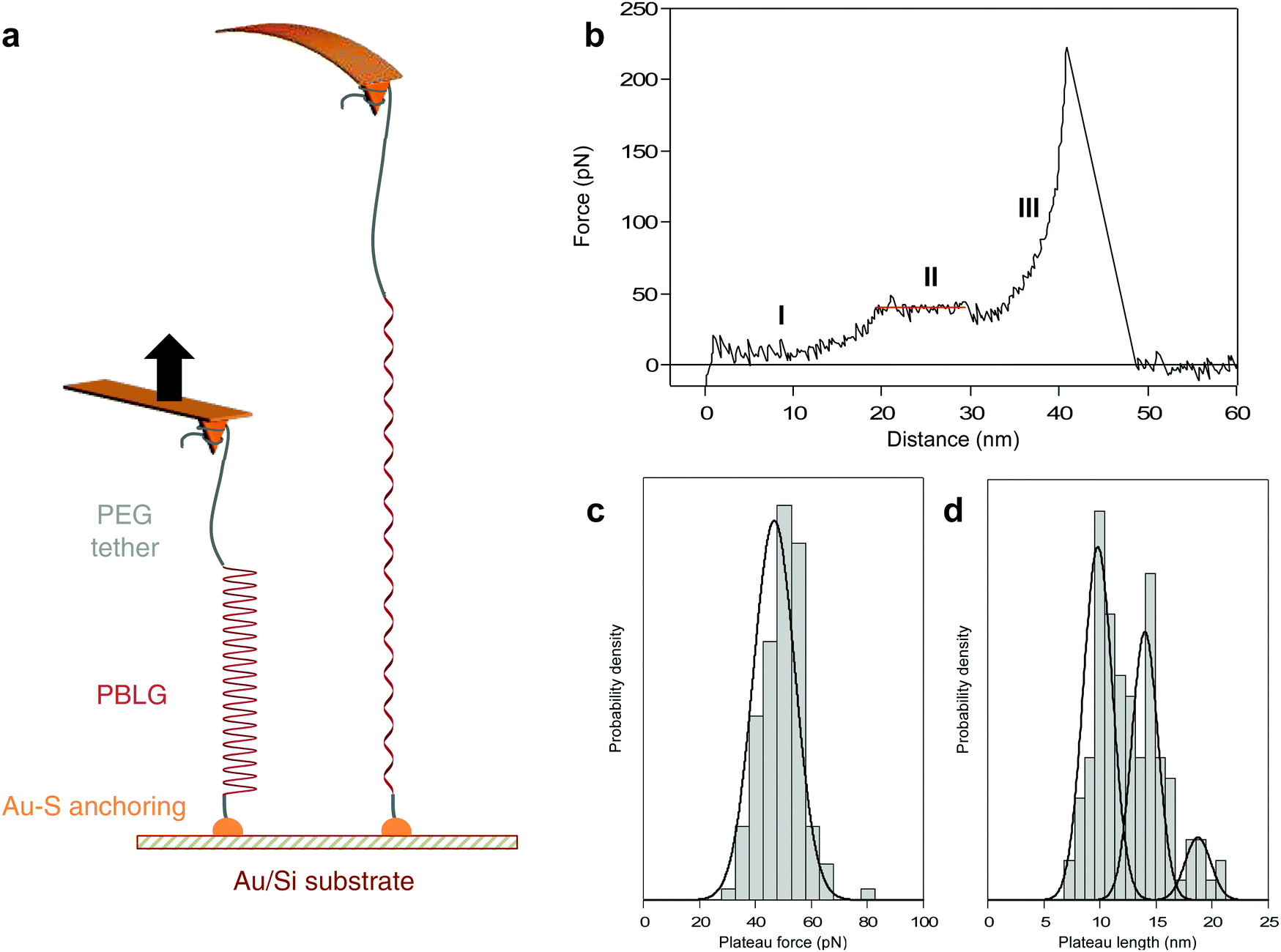 | ||
| Fig. 1 Mechanical pulling of a synthetic peptide displaying a characteristic unfolding plateau. (a) Scheme of the AFM-based force measurement of a PBLG attached to a gold-coated silicon substrate and stretched by an AFM tip via a PEG tether, leading to its unfolding and extension. (b) Characteristic force–distance curve associated with the stretching of a single PEG-PBLG at 200 nm s−1 in dioxane: (I) alignment and stretching of the PEG, (II) unfolding of the helical PBLG by sequential breaking of intramolecular H-bonds (linear fit in red), (III) stretching of PEG-PBLG until the last rupture point (i.e. detachment of molecule from the tip). (c and d) Distributions of plateau force (FP) and plateau length (ΔL): (c) one narrow population of FP (46.7 ± 1.4 pN) was observed, in agreement with the sequential breaking of intramolecular hydrogen bonds supporting the helical structure; (d) three ΔL populations centered at 9.8 ± 0.3 (54%), 14.0 ± 0.4 (37%) and 18.7 ± 0.7 nm (19%) correspond to the simultaneous unfolding of 2, 3 and 4 individual helices respectively (45 ± 1, 64 ± 2 and 85 ± 3 amino acid residues). | ||
Experimental
Experimental details are described in ESI.†Results and discussion
Signature of the mechanical helix–coil transition
PBLG was chemically attached onto gold-coated silicon substrate in highly-diluted grafting conditions40 in order to disperse the molecules and favour single-molecule interactions with the AFM probe. Silicon nitride tips were used to pick up the molecules individually and to probe the mechanical response upon stretching with high precision. The experiments were performed in dioxane to promote the formation of PBLG helical conformation in solution.44 A standard experiment consists in approaching the AFM tip towards the surface and apply a contact force to promote physisorption of PEG on the tip, then the tip is pulled away from the surface and the force exerted on the tip-bearing cantilever upon retraction is measured (Fig. 1a).When a molecule was trapped between the tip and the surface, we observed a characteristic force–extension curve showing a reproducible unfolding pattern, shown in Fig. 1b. First, the force increases with the extension following a worm-like chain49 (WLC) behaviour (I), characteristic of the stretching of a flexible polymer in a random coil conformation. Then, the force becomes constant over a definite extension, marked as a force plateau (II, red line), consistent with the sequential breaking of identical intramolecular interactions in series. Finally, after the plateau, the force increases again following a WLC behaviour (III) up to the last rupture peak that corresponds to the detachment of the molecule from the tip. We suggest that the WLC behaviours observed in the force–extension curves are due to the stretching of the PEG (I) or PEG-PBLG (III) in coiled-coil conformations while the plateau pattern (II) corresponds to the mechanical unwinding of the PBLG α-helix, signature of the helix–coil transition. It is noteworthy that the sequential rupture of intramolecular H-bonds maintaining the PBLG helical secondary structure is associated with a tiny revealed length (0.22 nm per amino acid, see ESI† for details), which is to small be resolved by AFM. However, the consecutive breaking of several weak noncovalent interactions can be evidenced by an unfolding plateau, i.e. rupture of similar interactions at identical force, that can be related to the total revealed length during the helix–coil transition. This observation is also in complete agreement with previous force spectroscopy studies describing unwinding plateaus50–52 and with simulations showing a theoretical plateau in force–extension curves during the helix–coil transition of a homopolypeptide.16
Evidencing an intact α-helix critical length
In order to correlate the force–extension characteristic profile with a conformational change of the probed molecule, we determined the length of the observed plateau (ΔL) as well as the associated plateau force (FP). The distribution of plateau force is shown in Fig. 1c and presents one narrow population at 46.7 ± 1.4 pN, in agreement with the rupture of weak noncovalent interactions in series such as H-bonds.26 The plateau length distribution (Fig. 1d) exhibits three distinct populations at 9.8 ± 0.3, 14.0 ± 0.4 and 18.7 ± 0.7 nm. Considering the revealed distance per amino acid (AA), we can relate the plateau length to the number of residues constituting the α-helix, respectively 45 ± 1, 64 ± 2 and 85 ± 3 AA. Remarkably, the most probable values of ΔL are associated with multiples of about 20 AA, which is the value mentioned by several simulations as the threshold for the folding as an intact α-helical conformation.7,16 Above this threshold value of 20 AA, the formation of a tertiary self-folded conformation made of multiple helices in series is favoured. Therefore, we attribute the three ΔL populations to the unfolding of 2, 3 and 4 helices in series. We did not observe the unfolding of a single helix of 20 AA. In a long polypeptide chain (90 AA) and in a helicogenic solvent, it is not surprising to have a very low probability of observing a single 20 AA helix in solution, mainly due to the stabilizing interaction between two helices as a double-helix bundle.17 Furthermore, a helix of 20 AA is subjected to an equilibrium between unfolded coiled-coil and folded helical conformations,7 which decreases the probability to probe it in real-time by AFM. However, when performing pulling–relaxing experiments (discussed below), we could evidence a few plateaus of about 5 nm (= 23 AA). Only a maximum value of 4 helices can be reached given the degree of polymerization of PBLG which is equal to 90 (see ESI†). The probability of observing 4 helices in series is dependent of the helix–coil dynamics of each α-helix in solution, and thus quite low. As shown in Fig. 1d, probing 4 α-helices in series occurs with the lowest probability (19% of the cases). Based on this distribution, we can conclude that most of the time we probe a double-helix bundle that is formed in solution17,50 (54% of the cases). It is important to note that the breaking of intermolecular interactions within the helix bundles is not detected during our pulling experiments given the very small inter-helix rupture force (and very small length increase) associated with this bundle opening, in agreement with previous force spectroscopy data on triple helix bundles of spectrin.50Mechanism of the force-induced peptide unfolding
The characteristic unfolding pattern is indicative of the polypeptide original conformation and behaviour during the stretching. We can exclude the possibility of an intact α-helix made of up to 90 residues, which would lead to a single population of plateau length. Here the plateau length distribution shows distinct populations. While we suggest that these populations correspond to the mechanical unfolding of 1 to 4 α-helices of 20 residues, only one plateau is observed in the force curves and not a series of consecutive plateaus. This observation arises from the alignment of the whole molecule backbone in the pulling direction, facilitating the increase of the helical content and the disappearance of the kink defects at the beginning of the stretching (Fig. 2), a behaviour that has been evidenced for homopolypeptides in pulling simulations.21 Indeed, the alignment of the polypeptide backbone in the pulling direction—e.g. two helices and one kink—favours the formation of an α-helix constituted of a higher number of AA connected by previously-absent hydrogen bonds. This fast refolding step (i.e. local rearrangement around the kink regions) is facilitated by the high flexibility of the main chain (polypeptide + PEG) and can possibly occur at low forces, as shown by simulations.21 Thus, when applying a small external force and aligning the 20-residues helices in the pulling direction, they undergo a fast refolding step to form a single long helix under the pulling force. This refolding step is indistinguishable here given the inherent noise of the AFM experiment and the small length variation. The α-helix is then subjected to a helix–coil transition under external force, appearing as a plateau in the force curves (Fig. 2). It is important to note that the number of intact α-helices segments aligned and probed during the pulling is dependent on the helix–coil transition dynamics in solution, decreasing the probability of observing the mechanical helix–coil transition of 4 α-helices during each pulling experiment.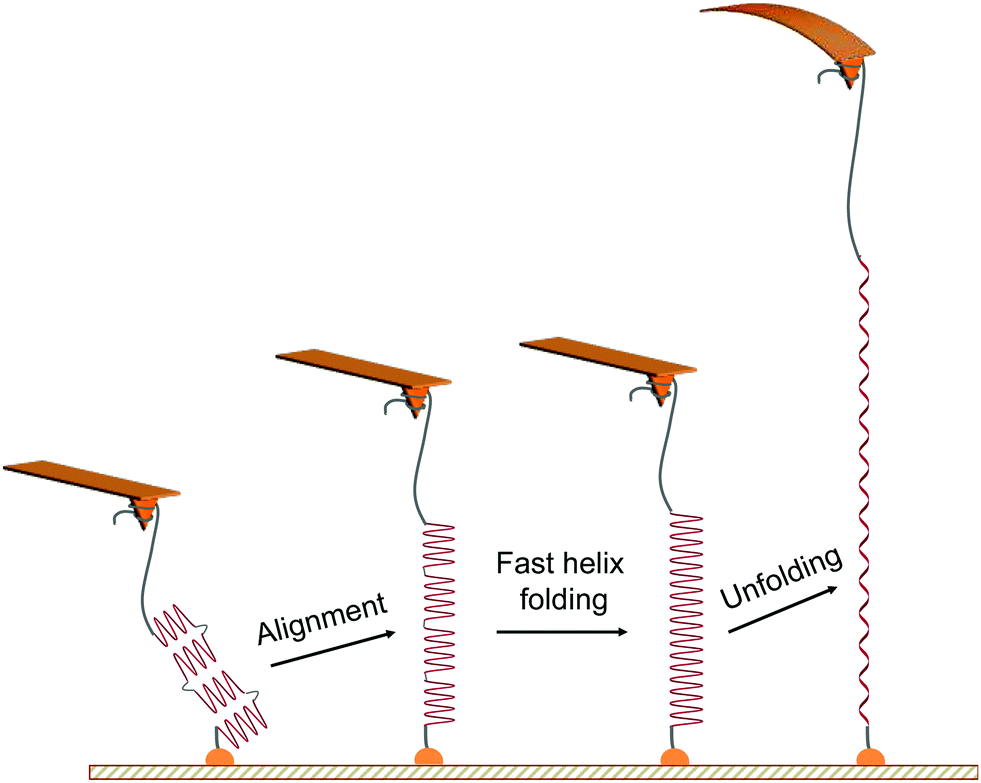 | ||
| Fig. 2 Scheme of the proposed mechanism for the mechanical unfolding of multiple α-helices in series. The mechanical pulling of the PEG-PBLG starts with the alignment of helical substructures (of about 20 amino acid residues each) along the stretching axis, favouring the fast local folding of a longer helix21via the formation of H-bonds between close residues formerly absent in the kink regions of the multi-helix structure. The mechanical stretching of this peptide is associated with a longer unfolding plateau instead of several sequential plateaus in force–distance curves. | ||
Modulation of the number of helical segments mechanically unfolded
As an additional evidence supporting the mechanical helix–coil transition of up to 4 α-helices, we considered the force curves corresponding to the detachment of the molecule from the tip before the final extension of the α-helix (rupture distance (DR) < 35 nm, corresponding to the maximum length of PBLG). It corresponds to a situation where the tip interacts with PBLG during the pulling. Thus, depending on the tip attachment point, the portion of polypeptide stretched during the force experiment varies. By analyzing the populations associated to DR < 35 nm and DR > 35 nm separately, we determined the proportion of 2, 3 and 4 helices stretched in both situations (tip attachment with PEG tether or with the PBLG chain). Pulling the molecule via the PBLG (Fig. S1, ESI†) leads to relative proportions of 2 and 3 helices stretched of 76 and 24% respectively (Fig. S3, ESI†). As expected, the stretching of the whole PBLG (4 helices = 90 AA) was not observed in this case due to the lower portion of PBLG trapped between the tip and the surface. For DR > 35 nm (i.e. when stretching the molecule by the PEG tether, Fig. S2, ESI†), the relative proportions of 2, 3 and 4 helices are respectively 17, 72 and 11% (Fig. S3, ESI†), showing a higher helical content available during the molecule stretching.Reversibility of the mechanical helix–coil transition for a single α-helix
The reversibility of the helix–coil transition in PBLG was also investigated by performing pulling–relaxing cycles on a single molecule (Fig. 3), i.e. a molecule trapped between the tip and the surface is subjected to small retraction and approach movements subsequently without breaking the tip-molecule interaction. During the pulling (blue), the force–extension curve displays a typical plateau pattern corresponding to the unfolding of one α-helix of PBLG (ΔL ≈ 5 nm). When decreasing the force applied on the molecule (relaxing curve in red), a similar refolding pattern was evidenced. Unfolding and refolding plateaus are characterized by identical forces (FP) pointing out the reversible conformational change behaviour of the PBLG α-helix under stress, i.e. the rupture and reformation of intramolecular interactions take place at identical external force. The presence of a small hysteresis at high extension is typical of a pulling experiment performed in out-of-equilibrium conditions.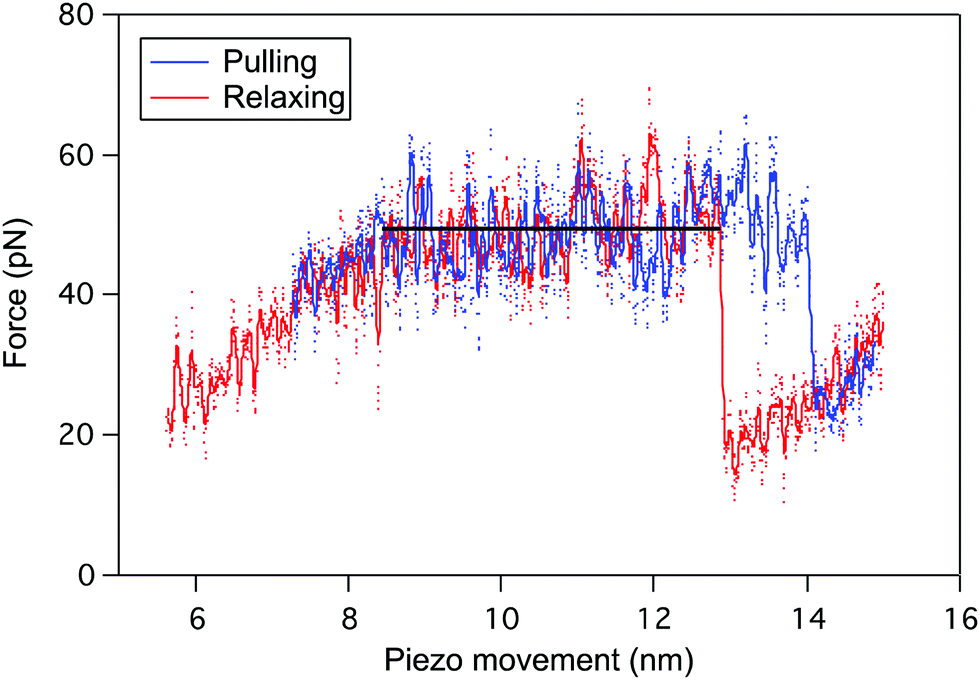 | ||
| Fig. 3 Reversible mechanical unfolding of PBLG and detection of a single 20-amino acid helix conformation as a 5 nm long unfolding plateau. Pulling-relaxing experiment: an individual PBLG is pulled (blue) in dioxane over a few nanometers and presents a characteristic unfolding plateau of about 5 nm (23 AA residues). Before reaching the detachment point, the external force is released and the relaxing curve (in red) exhibits a similar plateau reflecting the reformation of previously broken intramolecular interactions, indicating the fast recovery of the α-helix. A small hysteresis was observed between pulling and relaxing curves, typical of an experiment performed in out-of-equilibrium conditions. | ||
Loss of the characteristic unfolding pattern of α-helices in denaturating conditions
Finally, we performed AFM pulling experiments in dioxane in the presence of a denaturing agent, trifluoroacetic acid (TFA). During these experiments, the most reproducible pattern is a single-peak profile (64% of the cases, Fig. 4a), characteristic of the stretching of a polymer chain in a random coil conformation. It indicates the disruption of the helical structure of PBLG in TFA (chemical denaturation) before the mechanical pulling. Even if most of the probed molecules seem to have undergone the helix–coil transition in solution due to the denaturing agent, some molecules still present a characteristic unfolding plateau (only 36% of the cases, Fig. 4b). Interestingly, the plateau pattern associated with the unfolding of those molecules is characterized by a lower plateau force of 37.4 ± 3.8 pN (Fig. 4d), indicating a weakening of the intramolecular interactions in presence of TFA. Also, the relative proportion of 2 and 3 helices in TFA was 58 and 34% respectively (Fig. 4e), shifting towards an overall decrease of the helical content in these conditions (Fig. S5, ESI†). This observation highlights the destabilization of intramolecular helical interactions and the higher proportion of coiled-coil conformations amongst probed molecules. In this denaturing environment, we were also able to evidence the stretching of individual α-helices characterized by ΔL ≈ 4 nm (i.e. 18 AA), shown in Fig. 4c. In this case, inter-helical interactions were ruptured and a single helix was mechanically probed. The observation of such small plateaus in force–extension curves again supports the concept of a critical length in the formation of α-helical structure in peptides and proteins.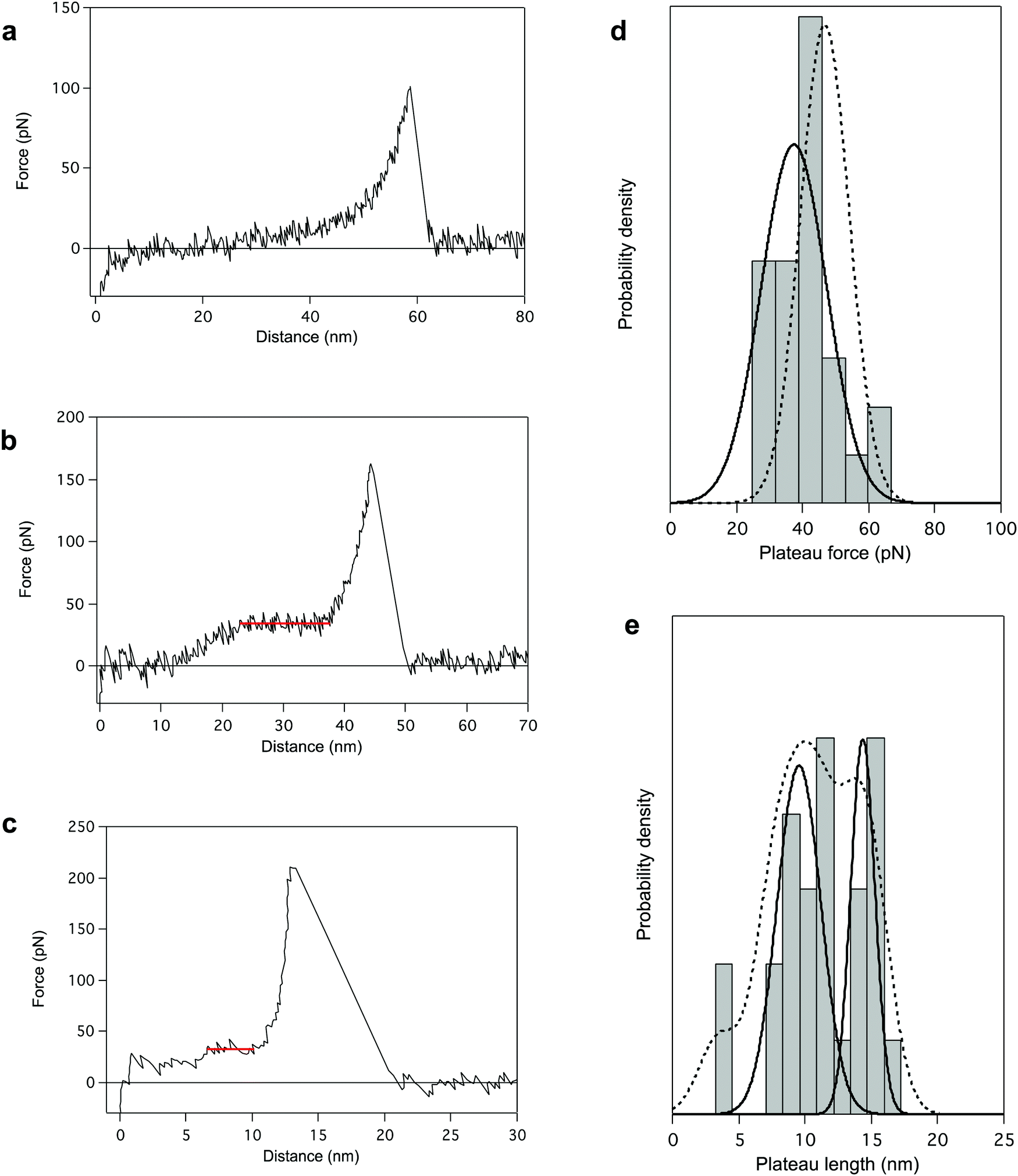 | ||
| Fig. 4 AFM pulling experiments of PBLG in denaturating environment (dioxane + trifluoroacetic acid (TFA)). The presence of TFA provokes either a chemical destabilization of the helical structure favouring a coiled-coil conformation (a), or a weakening of intramolecular interactions (b). The associated force–extension curves appear respectively as single-peak patterns (a, 64%) or plateau patterns with lower FP (b, 36%). (c) Force–distance curve for the standard pulling of PBLG in denaturating conditions presenting a 5 nm plateau corresponding to the unfolding of a single α-helix of about 20 AA residues. (d) The distribution of FP presents one population centered at 37.4 ± 3.8 pN (plain line), lower than the unfolding force previously observed in dioxane (population in Fig. 1c added here as a dotted line for comparison). (e) Distribution of the plateau length (ΔL) shows three populations centered at 3.5 ± 0.1 nm (8%), 9.6 ± 0.8 nm (58%) and 14.4 ± 0.6 nm (34%) corresponding to the unfolding of helices made of 16 ± 1 AA, 44 ± 4 AA and 65 ± 3 AA respectively. Probability density function (PDF) fit is added in dotted line. | ||
Conclusion
Even if α-helices are the most frequent secondary structures in proteins, the influence of a peptide length on its conformation had never been demonstrated experimentally. Here, using AFM-based single-molecule force spectroscopy, we showed the self-folding of a 90-amino acids helical synthetic polypeptide in solution and evidenced a critical length for intact α-helices. We designed a poly(γ-benzyl-L-glutamate) (DP = 90) functionalized with a chemical anchor on one end for its grafting on surface and with a polymer tether on the other end for AFM tip attachment during pulling experiments, and we probed its force-induced helix–coiled transition at the single-molecule level. The stretching of a single synthetic polypeptide showed a constant force plateau as characteristic unfolding pattern. The unfolding force in the plateau is consistent with a helix–coil transition, i.e. stepwise breaking of intramolecular hydrogen bonds maintaining the helical conformation of the peptide. We evidenced different plateau lengths corresponding to the unfolding of n α-helices of about 20 AA (n = 1, 2, 3 or 4) instead of an intact helix of 90 AA. This is the first time, to our knowledge, that the secondary structure of a polypeptide made of multiple α-helices in series is evidenced experimentally in real-time and in solution. We confirm here the presence of a critical α-helix length, as suggested by computational simulations. Our results are in complete agreement with the formation of a self-folded structure made of short helices instead of an intact compact helical structure for sequences of more than 20 amino acid residues. The reversibility of the helix–coil transition was evidenced by pulling-relaxing experiments which highlighted the high stability and fast refolding of PBLG α-helices in a helicogenic solvent.We have focused here on a homopolypeptide derived from poly(glutamic acid) in dioxane. We believe our strategy can now be used to study many other natural helices in physiological environment to study the influence of the nature of the amino acid sequence on the critical length. Indeed, such helices are known to present a helical critical length similar to the one presented here.7,15–18
We believe that the concept of critical length, previously envisioned by computational studies and hereby confirmed by our experiments, will also have an impact on the design of synthetic molecules made of artificial residues with much more rigid backbones than peptides (like backbones that possess aryl rings in their main chain),53 in a way to create stable helices of more than 20 units, thus leading to secondary structures with high conformational rigidity and possible unprecedented mechanical properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. S. thanks the Fonds de la Recherche Scientifique-Fonds National pour la Recherche Scientifique (FRS-FNRS) for his Chargé de Recherches position. S. L. is grateful to the Univ. Bordeaux and CNRS for their continuous support.References
- M. Gruber and A. N. Lupas, Historical review: Another 50th anniversary – new periodicities in coiled coils, Trends Biol. Sci., 2003, 28, 679–685 CrossRef CAS PubMed.
- C.-S. Goh, D. Milburn and M. Gerstein, Conformational changes associated with protein–protein interactions, Curr. Opin. Struct. Biol., 2004, 14, 104–109 CrossRef CAS PubMed.
- K. H. Nierhaus and D. N. Wilson, in Protein folding Handbook, ed. J. Buchner and T. Kiefhaber, 2005 Search PubMed.
- M. A. DePristo, D. M. Weinreich and D. L. Hartl, Missense meanderings in sequence space: a biophysical view of protein evolution, Nat. Rev. Genet., 2005, 6, 678–687 CrossRef CAS PubMed.
- Z. Weng and C. DeLisi, in Handbook of Proteins: Structure, Function and Methods, ed. M. M. Cox and G. N. J. Phillips, 2008, vol. 1 Search PubMed.
- T. Ackbarow, X. Chen, S. Keten and M. J. Buehler, Hierarchies, multiple energy barriers, and robustness govern the fracture mechanics of α-helical and β-sheet protein domains, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16410–16415 CrossRef CAS PubMed.
- Z. Qin, A. Fabre and M. J. Buehler, Structure and mechanism of maximum stability of isolated alpha-helical protein domains at a critical length scale, Eur. Phys. J. E: Soft Matter Biol. Phys., 2013, 36, 53 CrossRef PubMed.
- P. Palencar and T. Bleha, Buckling transition in long α-helices, J. Chem. Phys., 2014, 141, 174901 CrossRef PubMed.
- A. Buhot and A. Halperin, Extension of Rod-Coil Multiblock Co-polymers and the Effect of the Helix–Coil Transition, Phys. Rev. Lett., 2000, 84, 2160–2163 CrossRef CAS PubMed.
- A. Buhot and A. Halperin, Extension behavior of helicogenic polypeptides, Macromolecules, 2002, 35, 3238–3252 CrossRef CAS.
- H. Wada and R. R. Netz, Stretching helical nano-springs at finite temperature, Europhys. Lett., 2007, 77, 68001 CrossRef.
- A. R. Singh, D. Giri and S. Kumar, Force induced unfolding of biopolymers in a cellular environment: a model study, J. Chem. Phys., 2009, 131, 065103 CrossRef PubMed.
- S. Zhang, L.-J. Qu, T. Suo, Z. Liu and D. Yan, Multiple transitions between various ordered and disordered states of a helical polymer under stretching, J. Chem. Phys., 2017, 146, 174904 CrossRef PubMed.
- S. Sivaramakrishnan, B. J. Spink, A. Y. L. Sim, S. Doniach and J. A. Spudich, Dynamic charge interactions create surprising rigidity in the ER/K α-helical protein motif, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13356–13361 CrossRef CAS PubMed.
- M. J. Williams and M. Bachmann, The effect of surface adsorption on tertiary structure formation in helical polymers, J. Chem. Phys., 2017, 147, 024902 CrossRef PubMed.
- F. C. Zegarra, G. N. Peralta, A. M. Coronado and Y. Q. Gao, Free energies and forces in helix–coil transition of homopolypeptides under stretching, Phys. Chem. Chem. Phys., 2009, 11, 4019–4024 RSC.
- M. J. Williams and M. Bachmann, System-size dependence of helix-bundle formation for generic semiflexible polymers, Polymers, 2016, 8, 245 CrossRef PubMed.
- B. Chakrabarti and A. J. Levine, Nonlinear elasticity of an α-helical polypeptide, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 031905 CrossRef PubMed.
- S. Penel, R. G. Morrison, R. J. Mortishire-Smith and A. J. Doig, Periodicity in α-helix lengths and C-capping preferences, J. Mol. Biol., 1999, 293, 1211–1219 CrossRef CAS PubMed.
- S. Kumar and M. Bansal, Geometrical and sequence characteristics of α-helices in globular proteins, Biophys. J., 1998, 75, 1935–1944 CrossRef CAS PubMed.
- K. Torabi and G. C. Schatz, Tensile mechanics of α-helical polypeptides, Macromolecules, 2013, 46, 7947–7956 CrossRef CAS.
- K. C. Neuman and A. Nagy, Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy, Nat. Methods, 2008, 5, 491–505 CrossRef CAS PubMed.
- T. E. Fisher, P. E. Marszalek and J. M. Fernandez, Stretching single molecules into novel conformations using the atomic force microscope, Nat. Struct. Biol., 2000, 7, 719–724 CrossRef CAS PubMed.
- E. M. Puchner and H. E. Gaub, Force and function: probing proteins with AFM-based force spectroscopy, Curr. Opin. Struct. Biol., 2009, 19, 605–614 CrossRef CAS PubMed.
- A.-S. Duwez and N. Willet, Molecular Manipulation with Atomic Force Microscopy, CRC Press, 2012 Search PubMed.
- H. Clausen-Schaumann, M. Seitz, R. Krautbauer and H. E. Gaub, Force spectroscopy with single bio-molecules, Curr. Opin. Chem. Biol., 2000, 4, 524–530 CrossRef CAS PubMed.
- A. Borgia, P. M. Williams and J. Clarke, Single-molecule studies of protein folding, Annu. Rev. Biochem., 2008, 77, 101–125 CrossRef CAS PubMed.
- M. L. Hughes and L. Dougan, The physics of pulling polyproteins: a review of single molecule force spectroscopy using the AFM to study protein unfolding, Rep. Prog. Phys., 2016, 79, 076601 CrossRef PubMed.
- M. Rief, J. Pascual, M. Saraste and H. E. Gaub, Single molecule force spectroscopy of spectrin repeats: low unfolding forces in helix bundles, J. Mol. Biol., 1999, 286, 553–561 CrossRef CAS PubMed.
- I. Schwaiger, C. Sattler, D. R. Hostetter and M. Rief, The myosin coiled-coil is a truly elastic protein structure, Nat. Mater., 2002, 1, 232–235 CrossRef CAS PubMed.
- D. D. Root, V. K. Yadavalli, J. G. Forbes and K. Wang, Coiled-coil nanomechanics and uncoiling and unfolding of the superhelix and α-helices of myosin, Biophys. J., 2006, 90, 2852–2866 CrossRef CAS PubMed.
- R. Afrin, I. Takahashi, K. Shiga and A. Ikai, Tensile mechanics of alanine-based helical polypeptide: force spectroscopy versus computer simulations, Biophys. J., 2009, 96, 1105–1114 CrossRef CAS PubMed.
- T. Hoffmann and L. Dougan, Single molecule force spectroscopy using polyproteins, Chem. Soc. Rev., 2012, 41, 4781–4796 RSC.
- M. Kim, K. Abdi, G. Lee, M. Rabbi, W. Lee, M. Yang, C. J. Schofield, V. Bennett and P. E. Marszalek, Fast and forceful refolding of stretched α-helical solenoid proteins, Biophys. J., 2010, 98, 3086–3092 CrossRef CAS PubMed.
- Z. N. Scholl, Q. Li and P. E. Marszalek, Single molecule mechanical manipulation for studying biological properties of proteins, DNA, and sugers, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 211–229 CrossRef CAS.
- S. B. Smith, Y. Cui and C. Bustamante, Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules, Science, 1996, 271, 795–799 CrossRef CAS PubMed.
- Q. Zhang, Z. Lu, H. Lu, W. Yang and P. E. Marszalek, Direct detection of the formation of V-amylose helix by single molecule force spectroscopy, J. Am. Chem. Soc., 2006, 128, 9387–9393 CrossRef CAS PubMed.
- C. Liu, Z. Wang and X. Zhang, Force spectroscopy of single-chain polysaccharides: force-induced conformational transition of amylose disappears under environment of micelle solution, Macromolecules, 2006, 39, 3480–3483 CrossRef CAS.
- C. Liu, Z. Jiang, Z. Wang and X. Zhang, The unwinding of surfactant-induced helical structure of carboxymethyl amylose by single molecule force spectroscopy, Polymer, 2007, 48, 2030–2034 CrossRef CAS.
- P. Lussis, T. Svaldo-Lanero, A. Bertocco, C.-A. Fustin, D. A. Leigh and A.-S. Duwez, A single synthetic small molecule that generates force against a load, Nat. Nanotechnol., 2011, 6, 553–557 CrossRef CAS PubMed.
- A. Van Quaethem, P. Lussis, D. A. Leigh, A.-S. Duwez and C.-A. Fustin, Probing the mobility of catenane rings in single molecules, Chem. Sci., 2014, 5, 1449–1452 RSC.
- D. Sluymans, S. Hubert, C. J. Bruns, Z. Zhu, J. F. Stoddart and A.-S. Duwez, Synthetic oligorotaxanes exert high forces when folding under mechanical load, Nat. Nanotechnol., 2018, 13, 209–213 CrossRef PubMed.
- D. Sluysmans, F. Devaux, C. J. Bruns, J. F. Stoddart and A.-S. Duwez, Dynamic force spectroscopy of synthetic oligorotaxane foldamers, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9362–9366 CrossRef CAS PubMed.
- M. E. Martinez Barbosa, V. Montembault, S. Cammas-Marion, G. Ponchel and L. Fontaine, Synthesis and characterization of novel poly(γ-benzyl-L-glutamate) derivatives tailored for the preparation of nanoparticles of pharmaceutical interest, Polym. Int., 2007, 56, 317–324 CrossRef.
- W. Agut, A. Brûlet, D. Taton and S. Lecommandoux, Thermoresponsive micelles from Jeffamine-b-poly(L-glutamic acid) double hydrophilic block copolymers, Langmuir, 2007, 23, 11526–11533 CrossRef CAS PubMed.
- M. Losic, S. Kubowicz, B. Smarsly and H. Schlaad, Solid-state structure of polypeptide-based rod-coil block copolymers: folding of helices, Eur. Phys. J. E: Soft Matter Biol. Phys., 2004, 15, 407–411 CrossRef PubMed.
- H. Schlaad, B. Smarsly and I. Below, Solid-state structure of polystyrene-block-poly(γ-benzyl L-glutamate): helix folding vs. stretching, Macromolecules, 2006, 39, 4631–4632 CrossRef CAS.
- J. S. Crespo, S. Lecommandoux, R. Borsali, H.-A. Klok and V. Soldi, Small-angle neutron scattering from diblock copolymer poly(styrene-d8)-b-poly(γ-benzyl-L-glutamate) solutions: rod-coil to coil–coil transition, Macromolecules, 2003, 36, 1253–1256 CrossRef CAS.
- P. J. Flory, Statistical mechanics of chain molecules, Br. Polym. J., 1989, 2, 302–303 Search PubMed.
- H. Takahashi, F. Rico, C. Chipot and S. Scheuring, α-Helix unwinding as force buffer in spectrins, ACS Nano, 2018, 12, 2719–2727 CrossRef CAS PubMed.
- F. Berkemeier, M. Bertz, S. Xiao, N. Pinotsis, M. Wilmanns, F. Gräter and M. Rief, Fast-folding α-helices as reversible strain absorbers in the muscle protein myomesin, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14139–14144 CrossRef CAS PubMed.
- I. Schwaiger, C. Sattler, D. R. Hostetter and M. Rief, The myosin coiled-coil is a truly elastic protein structure, Nat. Mater., 2002, 1, 232–235 CrossRef CAS PubMed.
- G. Guichard and I. Huc, Synthetic foldamers, Chem. Commun., 2011, 47, 5933–5941 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00036a |
| ‡ These authors equally contributed to the work. |
| This journal is © The Royal Society of Chemistry 2020 |