
Reversibly-regulated drug release using poly(tannic acid) fabricated nanocarriers for reduced secondary side effects in tumor therapy†
Chao
Chen‡
 *a,
Tonghao
Ma‡
a,
Wen
Tang
a,
Xiaoli
Wang
a,
Yibing
Wang
a,
Jiafeng
Zhuang
a,
Yucheng
Zhu
b and
Ping
Wang
*ac
*a,
Tonghao
Ma‡
a,
Wen
Tang
a,
Xiaoli
Wang
a,
Yibing
Wang
a,
Jiafeng
Zhuang
a,
Yucheng
Zhu
b and
Ping
Wang
*ac
aState Key Laboratory of Bioreactor Engineering, Shanghai Collaborative Innovation Center for Biomanufacturing, Biomedical Nanotechnology Center, School of Biotechnology, East China University of Science and Technology, Shanghai 200237, China. E-mail: chaochen@ecust.edu.cn
bShanghai Key Laboratory of Functional Materials Chemistry, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, P. R. China
cDepartment of Bioproducts and Biosystems Engineering, University of Minnesota, St Paul, MN 55108, USA. E-mail: ping@umn.edu
First published on 2nd April 2020
Abstract
Numerous nanocarriers with pH-responsive properties have been designed and fabricated to reduce the adverse side effects of traditional chemotherapeutics, but these traditional nanocarriers are rarely reversible; this may cause “secondary” side effects on normal tissues, because the nanocarriers cannot be sealed again to prevent the leakage of incompletely released drugs after re-entering blood circulation. To overcome these limitations, we report herein the synthesis of a reversibly pH-responsive drug delivery system, which can achieve regulated drug release in a “release-stop-release” manner corresponding to changes in pH. Specifically, poly(tannic acid) as the “gatekeeper” was firstly deposited and polymerized on the surface of mesoporous silica nanoparticles (MSNs) via a modified mussel-inspired method similar to dopamine, and the formed polymer shell can be easily decorated with a targeting ligand HER2 antibody for the selective delivery of drugs to specific cells. The resulting nanocomposites exhibited good colloidal stability, good biocompatibility, high drug loading capacity and accurate HER2 antibody mediated targeting ability. Interestingly, a series of experiments fully demonstrated that the fabricated nanocomposites possessed intelligent reversible pH-responsive controlled release behavior through adjusting the density of the “gatekeeper” under different pH conditions, thereby achieving reversible switching from “on” to “off”. Furthermore, in vitro and in vivo experiments verified that the fabricated targeting nanoparticles could efficiently inhibit tumor growth with minimal side effects. Meanwhile, these nanocarriers exhibited excellent reusability, in vitro cytotoxicity and minimal in vivo myocardial damage. Collectively, the reversible pH-operated nanovalve on the MSNs constructed here could serve as a nanoplatform to solve the problem of “secondary” side effects caused by residual drugs in irreversible “gatekeeper” systems.
New conceptsVarious drug delivery systems with pH-responsive properties have been designed and exploited to improve the treatment effect of chemotherapeutics. However, traditional “gatekeeper” systems are usually irreversible, which may cause “secondary” side effects for normal tissues because the “gatekeeper” systems usually cannot be closed again once they are open. Meanwhile, the drug loading of MSNs is usually very high and drug release takes quite a long time, so most of the residual drug will thus re-enter blood circulation. To overcome these limitations, we report herein the synthesis of a reversibly pH-responsive drug delivery system. Specifically, poly(tannic acid) as the “gatekeeper” was firstly polymerized on the surface of MSNs, and the formed polymer shell can be easily decorated with a targeting ligand HER2 antibody. Interestingly, subsequent experiments indicate that the synthesized nanoparticles could reversibly adjust the density of encapsulation in response to different pH conditions to reversibly regulate the release of loaded drugs. Thus, the nanoparticles possess reversible pH-responsive controlled release properties, thereby reducing the “secondary” side effects caused by residual drugs. Therefore, this strategy is expected to solve the problem of “secondary” side effects caused by residual drugs in irreversible “gatekeeper” systems. |
Introduction
Currently, cancer is one of the greatest threats to human health, and chemotherapy, as the most frequently used approach, has attracted great attention in cancer treatment.1–3 However, some unfavourable factors severely limit the application of chemotherapy drugs, such as non-specific cytotoxicity, leakage during transportation, and low concentrations in tumor tissues.4 Fortunately, with the development of nanomedicine, the emergence of a large number of original nano-based drug carriers with stimuli-responsive properties is expected to achieve the accurate release of drugs at tumor sites, thereby reducing the damage to normal cells.5–7 At present, frequently-used environmental stimuli-responsive factors are pH, redox, light, and enzymes.8–13 Among these, pH-triggered release has the most extensive applications due to the weak acidity of tumor sites, which is an attractive biological feature.14,15 Typically, it has been demonstrated that tumor tissue (pH 5.7–6.8), as well as lysosomal (pH 4.5–5.0) and endosomal (pH 5.5–6.0) compartments with acidic internal environments, possesses a lower pH microenvironment than normal tissue, leading to the autonomic responsiveness of pH-responsive controlled release.16–19Based on this fact, pH-responsive “gatekeepers” have attracted special attention for the construction and development of drug controlled release systems. To date, a variety of pH-sensitive nanocarriers with various pore blockers have been certified to be useful for delivering chemotherapeutic drugs. Regretfully, the pH-responsive release behavior of traditional nano drug carriers is barely reversible.20 In other words, these pH-sensitive “gatekeepers” usually cannot be closed again in response to physiological pH once they are open in the acidic tumor microenvironment. Meanwhile, nanocarriers tend to have considerable drug loading, leading to a certain amount of drug remaining in the nanocarrier after killing the tumor cells, and the drug release also takes a long time to complete. If the nanocarrier fails to respond to the physiological environment of the normal tissue and closes the “gatekeeper” again in time, a large amount of drug will re-enter blood circulation before being released completely at the target site, which will give rise to “secondary” killing effects on normal cells.20–22 In short, a reversibly responsive drug delivery system could effectively prevent the “secondary” killing of normal tissues by overloaded drugs. Therefore, it is desirable to design and fabricate an intelligent “gatekeeper” that can reversibly respond to the specific microenvironment of tumor tissue and the normal physiological environment, and achieve reversible switching from “on” to “off” under external stimulation, thereby reducing the “secondary” side effects on normal tissues.
To achieve this level of control, a system must be designed that is capable of resealing itself when a particular stimulus is removed. Dopamine can self-polymerize to form polydopamine (PDA) under alkaline conditions due to the catechol and amine groups contained in the molecule, and lots of studies have indicated that PDA layers are sensitive to external pH changes.19,23,24 Inspired by this, it can be safely deduced that molecules with catechol and amino groups may exhibit adhesive properties similar to PDA, which can be used to replace dopamine.25,26 Considering that tannic acid (TA), with abundant catechol groups, and tetraethylenepentamine (TEPA), with amine groups, possess similar chemical structures to dopamine, TA and TEPA binary systems can form a pH-sensitive polymer layer as a “gatekeeper” coat on the surface of supports through oxidative polymerization in weak alkaline conditions.15,27–29 Inspired by this thought, the polymer can be used as a “gatekeeper” to coat the surface of supports to obtain pH-responsive release drug carriers. In addition, the encapsulating layer of the nanocarrier formed only by tannic acid could spontaneously degrade under acidic conditions, which could be used to realize pH-triggered drug release in response to the acidic microenvironment of tumor cells.30 Simultaneously, the introduction of the crosslinker TEPA makes it possible for the self-assembled polymer “gatekeeper” to dissociate into a loose state under acidic conditions without degradation and for the dissociated polymer to reassemble into a closed state again when the solution is brought back to alkaline or neutral through adjusting the protonation and deprotonation of TEPA, which endows the synthesized nanocarrier with reversible pH-responsive controlled release capability.
The current work examines the possibility of replacing dopamine with a TA/TEPA binary system to design a reversible pH-responsive polymer “gatekeeper” system based on mesoporous silica nanoparticles (MSNs). Then human epidermal growth factor receptor-2 (HER-2) antibody is conjugated on the surface of reversible pH-responsive polymer modified MSNs to reduce the side effects of anticancer drugs. Afterwards, in vitro and in vivo experiments are conducted to investigate the biocompatibility and specific targeting of the nanocarriers, as well as the selective cytotoxicity and antitumor activity of the drug-loaded nanoparticles. In particular, the reversible pH-responsive controllable drug release behaviors of the nanoparticles are investigated under alternate pH conditions that mimic the changes in pH during operation in vivo. Besides, in vitro repeated cell killing efficiency and in vivo myocardial damage are studied to verify the reversible controlled release property of the nanocomposites, which can reduce the “secondary” side effects to unspecific normal tissues. Therefore, we hypothesize that the multifunctional nanoparticles can be applied as an effective reversibly pH-responsive platform for targeted tumor therapy.
Materials and experiments
Materials
Ethylene glycol (EG), cetyltrimethyl ammonium bromide (CTAB), tetraethyl orthosilicate (TEOS) and sodium hydroxide (NaOH) were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Fluorescein isothiocyanate (FITC) and 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR) were purchased from Sigma-Aldrich (Shanghai, China). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT), doxorubicin hydrochloride (DOX) and tris-(hydroxymethyl)-aminomethane (Tris) were obtained from Sangon Biotech (Shanghai, China). Tannic acid (TA) and tetraethylenepentamine (TEPA) were purchased from Macklin Reagent Company (Shanghai, China). All other chemicals and reagents were analytically pure and commercially available, and were used without further purification.The human epidermal growth factor receptor-2 (HER2) antibody was obtained by expression of the Pichia pastoris expression system that has been constructed in this laboratory.31
Synthesis of mesoporous silica nanoparticles (MSNs)
MSNs were prepared by a relatively mature and repeatedly reported sol–gel method.13,32 To put it succinctly, 500 mg of CTAB was first dispersed in a mixture of ultrapure water (200 mL), EG (40 mL) and NaOH (2 M, 1.75 mL). Subsequently, the mixture was continuously stirred at 80 °C for 1 h. TEOS (2.5 mL) was then quickly added to the above mixture, and the mixture was stirred at 80 °C for an additional 2 h. Thereafter, the mixture was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 rpm for 20 min, and the solid precipitate was collected. After washing several times with ultrapure water and ethanol, the precipitate was dried under vacuum. Finally, in order to remove the CTAB template, the dried sample was calcined at 550 °C for 6 h.
100 rpm for 20 min, and the solid precipitate was collected. After washing several times with ultrapure water and ethanol, the precipitate was dried under vacuum. Finally, in order to remove the CTAB template, the dried sample was calcined at 550 °C for 6 h.
DOX loading
In this work, doxorubicin hydrochloride (DOX) was selected as a model drug for follow-up studies. For anticancer drug loading, MSNs (100 mg) were firstly ultrasonically dispersed in 100 mL of PBS (0.1 M, pH 7.4). Afterwards, DOX (100 mg) was added to the above solution, and the mixture was stirred at room temperature for 24 h under dark conditions. The resulting product was then centrifuged to remove the unloaded drug.The supernatant was collected to calculate the drug entrapment efficiency and loading efficiency. The DOX standard curve was used to determine the DOX content in the supernatant. Afterwards, entrapment efficiency and loading efficiency were calculated via the following formula:
| Entrapment efficiency (%) = (A0 − A1)/A0 |
| Loading efficiency (%) = (A0 − A1)/A2 |
Synthesis of poly(tannic acid) coated MSNs (MSNs-PTA)
The poly(tannic acid)-wrapped MSNs were synthesized through a modified mussel-inspired method.26,29 First of all, MSNs (100 mg) were ultrasonically dispersed in Tris–HCl buffer (100 mL, pH 8.5). Thereafter, TA (50 mg) and TEPA (20 μL) were added to the above solution, and the mixture was stirred under dark conditions for 8 h. Ultimately, the dry solid product was obtained by the same centrifugation, washing and vacuum drying steps, and denoted MSNs-PTA.Synthesis of HER2 antibody functionalized MSNs-PTA (MSNs-PTA-HER2)
MSNs-PTA covalently modified with HER2 antibody were synthesized by the following procedure. First, the prepared MSNs-PTA (20 mg) were ultrasonically dispersed in 20 mL phosphate buffered saline (PBS) (0.1 M, pH 7.4) containing HER2 antibody, and then the mixture was stirred under dark conditions for 6 h. Next, the solid product was obtained by the same centrifugation and washing steps. Lastly, the dry solid product was obtained by freeze vacuum drying and denoted MSNs-PTA-HER2. If DOX was loaded, the product obtained was DOX/MSNs-PTA-HER2.Characterization
Transmission electron microscopy (TEM) images of MSNs and MSNs-PTA were observed on a JEM-1400 transmission electron microscope (JEOL, Japan) with an accelerating voltage of 120 kV. Zeta potentials, hydrodynamic diameters and polydispersity indices (PDI) of nanoparticles were determined with a ZetasizerNano ZS (Malvern, UK). Fourier transform infrared (FTIR) spectroscopy of nanoparticles was performed with an IFS 55 spectrometer (Bruker, Switzerland) using KBr pellets. X-ray diffraction (XRD) patterns of free DOX and nanoparticles were carried out on a RINT2000 X-ray diffractometer with Cu Kα radiation (Rigaku, Japan). Thermo-gravimetric analysis (TGA) was performed on a TGA-50 instrument (Shimadzu, Japan), and the heating rate was 10 °C min−1 under a nitrogen stream. Nitrogen adsorption–desorption isotherms were measured on an ASAP2010 sorptometer (Micromeritics, GA) by a Brunauer–Emmett–Teller (BET) approach and Barrett–Joyner–Halenda (BJH) method to obtain the specific surface areas and pore size distributions of the nanoparticles. X-ray photoelectron spectroscopy (XPS) measurements were collected on an ESCALAB 250Xi X-ray photoelectron spectrometer (Thermo Fisher, UK).In vitro drug release study
The in vitro stimuli-responsive controlled release property of the nanoparticles was investigated in different pH environments.33,34 Firstly, DOX-loaded MSNs-PTA-HER2 were suspended in 10 mL of PBS solution and the mixture was transferred to a dialysis bag (cut-off molecular weight: 7000 Da). Subsequently, the dialysis bag was placed in the release medium at different pH values (pH 5.0, 6.8 and 7.4) at 37 °C. At appropriate time intervals, 1 mL of the medium was removed to measure the amount of released drug, and the same volume of fresh buffer solution was added to keep the volume constant. The absorbance value of the removed solution was measured by a UV-vis spectrophotometer at a wavelength of 485 nm. Afterwards, the concentration of the released drug in the removed solution was calculated using the DOX standard curve.Selective endocytosis assay
In this experiment, confocal laser scanning microscopy (CLSM) and flow cytometry (FCM) were used to qualitatively and quantitatively investigate the endocytosis of nanoparticles by cells.35,36 For the qualitative CLSM study, L-02 and SK-BR-3 cells were seeded at a density of 1 × 105 cells per well in glass bottom culture dishes and incubated overnight. Afterwards, the original medium was discarded, fresh medium containing 1.0 μg mL−1 FITC-labeled MSNs-PTA-HER2 was added, and the cells were incubated for an additional 2, 6 and 12 h, respectively. Subsequently, the medium was discarded, and the cells were rinsed three times with fresh PBS. Thereafter, the cells were fixed with 4% paraformaldehyde for 10 min, and the nuclei were then stained with Hoechst 33258 for another 10 min. Finally, after removing the culture medium, the cells were rinsed three times with fresh PBS and observed by CLSM.For the quantitative FCM study, L-02 and SK-BR-3 cells were seeded in a 6-well plate at a density of 2 × 105 cells per well and then incubated with FITC-labeled MSNs-PTA-HER2 for different times. Subsequently, the cells were collected and washed three times with PBS. Finally, intracellular fluorescence intensity was measured by FCM.
In vitro biocompatibility and cytotoxicity assay
In this experiment, the biocompatibility of MSNs-PTA-HER2 with L-02 and SK-BR-3 cells was determined by a mature MTT assay.37,38 Briefly, L-02 and SK-BR-3 cells were seeded into 96-well plates at a density of 1 × 104 cells per well and incubated overnight. Subsequently, fresh DMEM medium containing different concentrations of MSNs and MSNs-PTA-HER2 was added to the 96-well plates, and the cells were incubated for an additional 24 h or 48 h, respectively. Thereafter, 20 μL of 5 mg mL−1 MTT was added to each well and the cells were incubated for another 4 h. Afterwards, the medium in each well was sucked out, and 150 μL of dimethyl sulfoxide was added to each well. Finally, the absorbance was measured by a microplate reader at 570 nm.Next, MTT assays were also performed to determine the cytotoxicity of free DOX and DOX-loaded nanocarriers (DOX/MSNs-PTA and DOX/MSNs-PTA-HER2). The cell pretreatment followed the above method, and the medium in each well was replaced with fresh medium (200 μL) containing the free DOX and DOX-loaded nanoparticles at different concentrations (equivalent to free DOX concentrations of 0.05, 0.1, 0.25, 0.5, 1 and 2 μg mL−1). Then the same procedure as above was performed to treat L-02 and SK-BR-3 cells and calculate the cell viability. The cytotoxicity was expressed as the percentage cell viability compared with the control group.
Acquisition of animals and establishment of tumor model
Female healthy BALB/c nude mice about 4–5 weeks old (weighing ∼20 g) were obtained from the Shanghai SLAC Laboratory Animal Co., Ltd. All the animal experiments were performed in compliance with the guidelines for the Care and Use of Research Animals established by the East China University of Science and Technology Animal Studies Committee. The tumor model of each mouse was established by subcutaneously injecting 100 μL PBS containing 2 × 106 SK-BR-3 cells in the right rear flank. Only after the tumor volume reached 100 mm3 could tumor-bearing mice be utilized for subsequent experiments, while the tumor volume was calculated according to the following formula: V = L·W2/2 (L: maximum length of the tumor; W: minimum width of the tumor).39,40In vivo tumor therapy assay
Mice with tumors up to a size of 100 mm3 with similar weights were randomly divided into five groups (n = 4 per group), and then the mice in the five groups were intravenously injected with 100 μL of PBS, MSNs-PTA-HER2, free DOX, DOX/MSNs-PTA and DOX/MSNs-PTA-HER2 (equivalent to free DOX concentrations of 3 mg kg−1, three times each week), respectively. Thereafter, the tumor size measured with a caliper and the body weight were recorded every two days for two weeks. Meanwhile, the relative tumor volume was normalized to its original volume before intravenous injection.Immunohistochemistry evaluations
After treatment for 14 days, all mice were sacrificed to perform histology assessment. Then, tumor and typical heart, kidney, liver, lung and spleen tissues of the mice from each group were obtained and dehydrated in 4% paraformaldehyde solution at 4 °C for 24 h. Subsequently, liquid paraffin was selected to embed the tumor sand tissues. Finally, the sliced tumors and tissues were stained with hematoxylin and eosin (H&E), followed by examination under a microscope.Statistical analysis
Statistical analysis was performed by one-way analysis of variance (ANOVA) using OriginPro 9.0 (OriginLab, MA, USA) software. The p-value of less than 0.05 (greater than 95% confidence interval) was considered to be statistically significant. All experiments were repeated at least twice under the same conditions. The results are expressed as mean ± standard deviation (SD).Results and discussion
Preparation and characterization of nanoparticles
A mesoporous silica nanoparticles based drug delivery system with targeting performance and reversibly pH-responsive property was prepared by step-by-step chemical modification. PTA layer and HER2 antibody were sequentially modified on the surface of mesoporous silica (Scheme 1). Specifically, MSNs were firstly synthesized according to the literature with TEOS as the silica source and CTAB as the surfactant template agent.41 Subsequently, the PTA layer was employed as a “gatekeeper” coating on the surface of MSNs to prevent premature drug release. It was formed by a TA/TEPA binary system through the Schiff base reaction of catechol in TA and amine groups in TEPA under alkaline conditions (Scheme S1, ESI†). Finally, in order to enhance the targeting effect of nanoparticles, HER2 antibody was selected as the targeting ligand and grafted on the surface of PTA modified MSNs through Michael addition and/or Schiff base reactions.42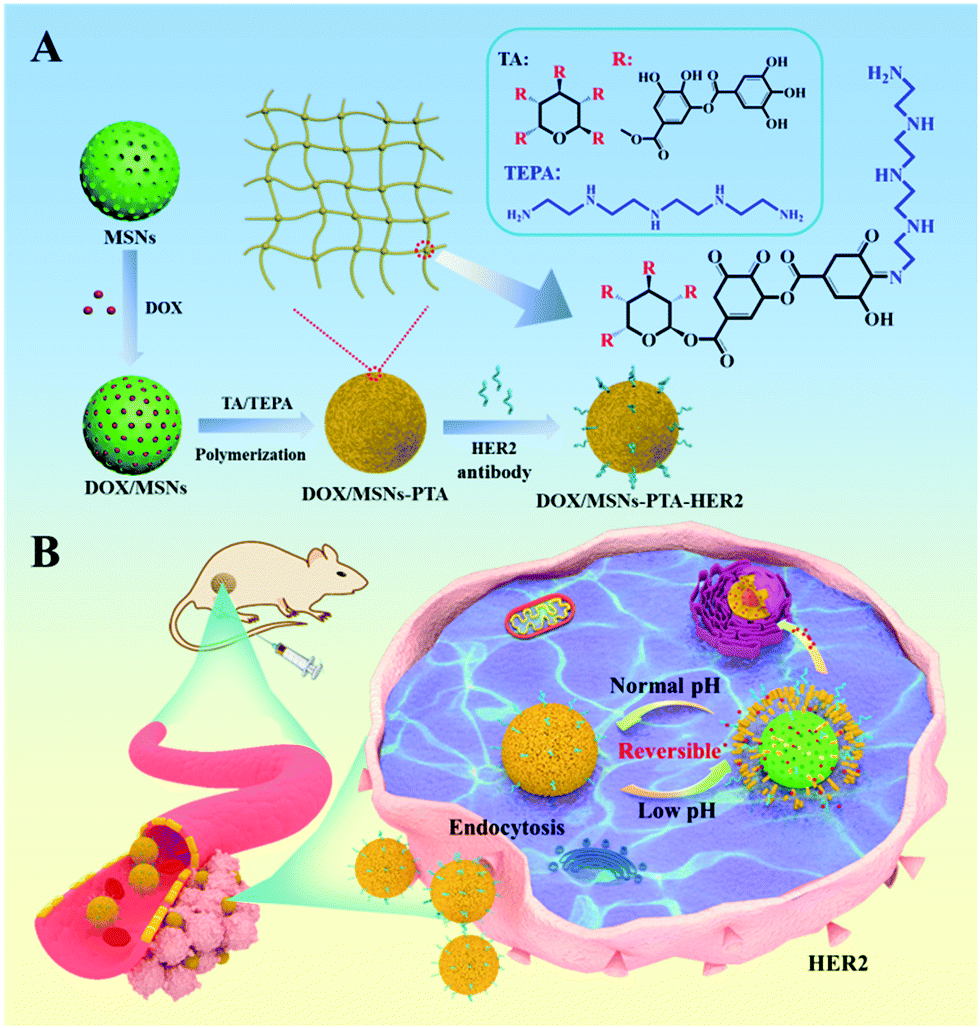 | ||
| Scheme 1 Schematic illustration of (A) synthesis of DOX/MSNs-PTA-HER2 and (B) drug delivery process of DOX/MSNs-PTA-HER2 in the acidic tumor microenvironment. | ||
According to TEM measurements (Fig. 1A and B), blank MSNs exhibited a relatively regular circular shape, and the specific mesoporous structure was clearly visible, which was consistent with previous studies. After oxidative polymerization of the TA/TEPA binary system on the surface of MSNs (MSNs-PTA), a layer of polymer was formed on the surface of the nanoparticles, and the edge was blurred and irregular. The hydrodynamic diameter of the synthesized nanoparticles was measured by a dynamic light scattering (DLS) method. As shown in Fig. 1C and Table S1 (ESI†), the average sizes of MSNs, MSNs-PTA and MSNs-PTA-HER2 gradually increased, which could be interpreted as the successful functionalization of PTA and HER2 antibody on the surface of MSNs. In addition, changes in the zeta potential also demonstrated the success of surface modification (Fig. 1D and Table S2, ESI†). Initially, due to the presence of a large number of Si–OH bonds on the surface of the MSNs, the lowest negative potential was exhibited (−26.2 mV). After the adhesion of the PTA layer, the zeta potential increased to −6.7 mV due to the introduction of the amine groups and the quaternary ammonium salts. Finally, after grafting with the targeting molecular HER2 antibody, TA was bound to the amine groups of the antibody and the excess carboxyl group was exposed, which caused the zeta potential to drop to −13.6 mV. Furthermore, Fig. S1 (ESI†) showed that MSNs-PTA-HER2 were stable in PBS and DMEM solution media over a long period without any precipitation, which agreed well with the variation of hydrodynamic diameter of MSNs-PTA-HER2 (Fig. S2, ESI†). The above observations directly indicated that the tumor selective theranostic agent MSNs-PTA-HER2 was successfully fabricated.
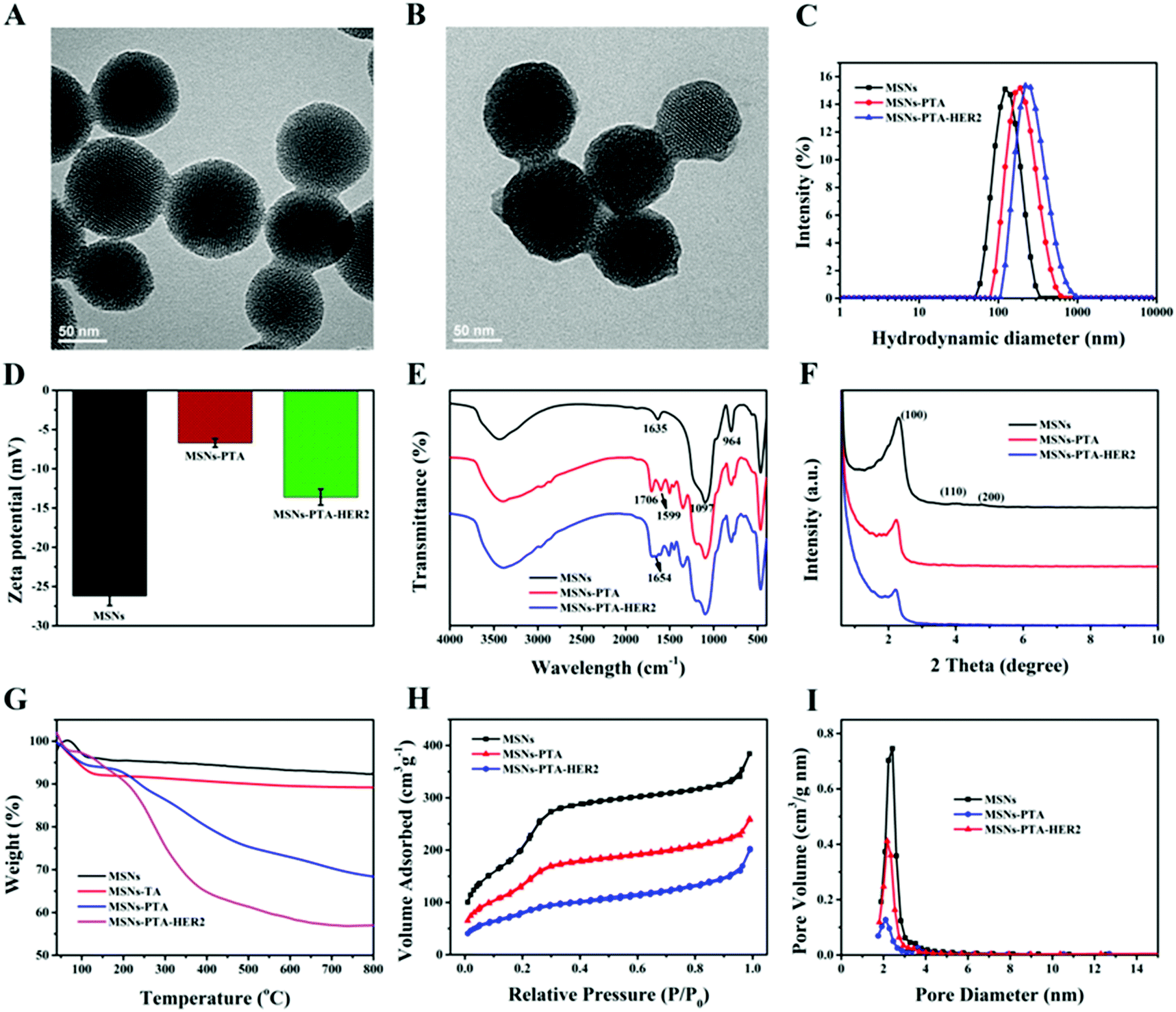 | ||
| Fig. 1 Characterizations of different MSNs nanoparticles. TEM images of (A) MSNs and (B) MSNs-PTA; (C) size distribution of MSNs, MSNs-PTA and MSNs-PTA-HER2; (D) zeta potential change of MSNs, MSNs-PTA and MSNs-PTA-HER2; (E) FTIR spectra of MSNs, MSNs-PTA and MSNs-PTA-HER2; (F) low-angle XRD patterns of MSNs, MSNs-PTA and MSNs-PTA-HER2; (G) TGA curves of MSNs, MSNs-TA, MSNs-PTA and MSNs-PTA-HER2; (H) BET nitrogen adsorption/desorption isotherms and (I) BJH pore size distributions of MSNs, MSNs-PTA and MSNs-PTA-HER2. | ||
Furthermore, the successful surface modification was also characterized by a different spectroscopy method. The Fourier transform infrared spectroscopy (FTIR) spectra of various nanoparticles are shown in Fig. S3 (ESI†) and Fig. 1E. Fig. S3 (ESI†) demonstrated the complete removal of template CTAB because of the disappearance of the three peaks at 2923, 2853 and 1484 cm−1, which were assigned to the C–H stretching vibrations and C–H deformation of CTAB.43Fig. 1E displayed that MSNs had a strong absorption peak at 1097 cm−1, which was mainly attributed to the vibration of Si–O–Si in silane. Compared with blank MSNs, MSNs-PTA exhibited two new absorption peaks at 1706 and 1599 cm−1, which were assigned to the stretching vibrations of the carbonyl and amine groups in TA and TEPA, respectively. This phenomenon illustrated the successful modification of PTA on the surface of MSNs. After modification with the targeting ligand HER2 antibody, a new absorption peak appeared at 1654 cm−1 in MSNs-PTA-HER2, which was the characteristic absorption peak of the amide I band in HER2 antibody. This result indicated the successful modification of HER2 antibody on the surface of the PTA layer. Moreover, the crystal form of MSNs was determined by small angle XRD analysis. As shown in Fig. 1F, all the nanoparticles exhibited three typical diffraction peaks, indexed as (100), (110) and (200) Bragg peaks, which represent the typical MCM-41 series of MSNs with hexagonal stacked channels.44 With the sequential addition of PTA and HER2 antibody, the intensity of the three diffraction peaks gradually decreased, which further illustrated the success of surface modification. Besides, TGA was performed to characterize the success of surface modification as well. Fig. 1G revealed that the curves of all the nanoparticles displayed a progressive tendency towards a decrease in weight with an increase in temperature. The final weight losses of different samples are shown in Table S3 (ESI†), indicating the successful step-by-step modification of PTA and HER2 antibody on the surface of MSNs; the content of HER2 antibody in the final nanoparticles (MSNs-PTA-HER2) was about 11%. Moreover, the weight loss of MSNs-TA (MSNs was encapsulated only by TA for 8 h, without TEPA) was visibly lower than that of MSNs-PTA, indicating the presence of TEPA could accelerate the polymerization ability of TA on MSNs. Furthermore, nitrogen adsorption–desorption isotherms, surface areas and pore size distributions of the various nanoparticles are shown in Fig. 1H, I and Table S4 (ESI†). The nitrogen adsorption–desorption isotherm was classified as a typical type IV isotherm, which represented the mesoporous structure of MSNs. With the sequential modification by PTA and HER2 antibody, the isotherm gradually became flat and the surface area, pore size and pore volume were gradually reduced, which demonstrated the successful modification of PTA and HER2 antibody. These results again demonstrated that the PTA layer and HER2 antibody were modified onto the surface of MSNs and sealed the mesopores of MSNs.
Next, to further determine the surface composition of the fabricated nanoparticles, XPS analysis was performed to characterize the change in the content of each element. The specific content of the four atoms in various nanoparticles is shown in Table S5 (ESI†), and the changes in elements between different nanoparticles strongly indicated the successful surface modification of MSNs with the PTA layer and HER2 antibody. Specifically, from Fig. 2A–C, compared with the spectra of the bare MSNs, the appearance of the N 1s peak at a binding energy of ∼399.3 eV corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C structure caused by Schiff base reaction in the spectra of MSNs-PTA and MSNs-PTA-HER2 illustrates the presence of the PTA layer. Furthermore, the nitrogen peak of MSNs-PTA-HER2 was more intense than that of MSNs-PTA, which verifies the grafting of HER2 antibody on the surface of the PTA layer. As shown in Fig. 2D–F, after oxidative polymerization of PTA on the surface of MSNs, a large proportion of the CO peak (∼288.3 eV) appeared. Meanwhile, the CO peak was further strengthened after grafting with targeting ligand HER2 antibody, which might be due to the large number of carboxyl and peptide bonds present in the antibody.45 In addition, Fig. S4 and S5 (ESI†) show that the amount of O and Si atoms obviously decreased with step-by-step modification, demonstrating the conjugation of the PTA layer and HER2 antibody onto the surface of MSNs. These results also support the successful modification of PTA and HER2 on MSNs.
N–C structure caused by Schiff base reaction in the spectra of MSNs-PTA and MSNs-PTA-HER2 illustrates the presence of the PTA layer. Furthermore, the nitrogen peak of MSNs-PTA-HER2 was more intense than that of MSNs-PTA, which verifies the grafting of HER2 antibody on the surface of the PTA layer. As shown in Fig. 2D–F, after oxidative polymerization of PTA on the surface of MSNs, a large proportion of the CO peak (∼288.3 eV) appeared. Meanwhile, the CO peak was further strengthened after grafting with targeting ligand HER2 antibody, which might be due to the large number of carboxyl and peptide bonds present in the antibody.45 In addition, Fig. S4 and S5 (ESI†) show that the amount of O and Si atoms obviously decreased with step-by-step modification, demonstrating the conjugation of the PTA layer and HER2 antibody onto the surface of MSNs. These results also support the successful modification of PTA and HER2 on MSNs.
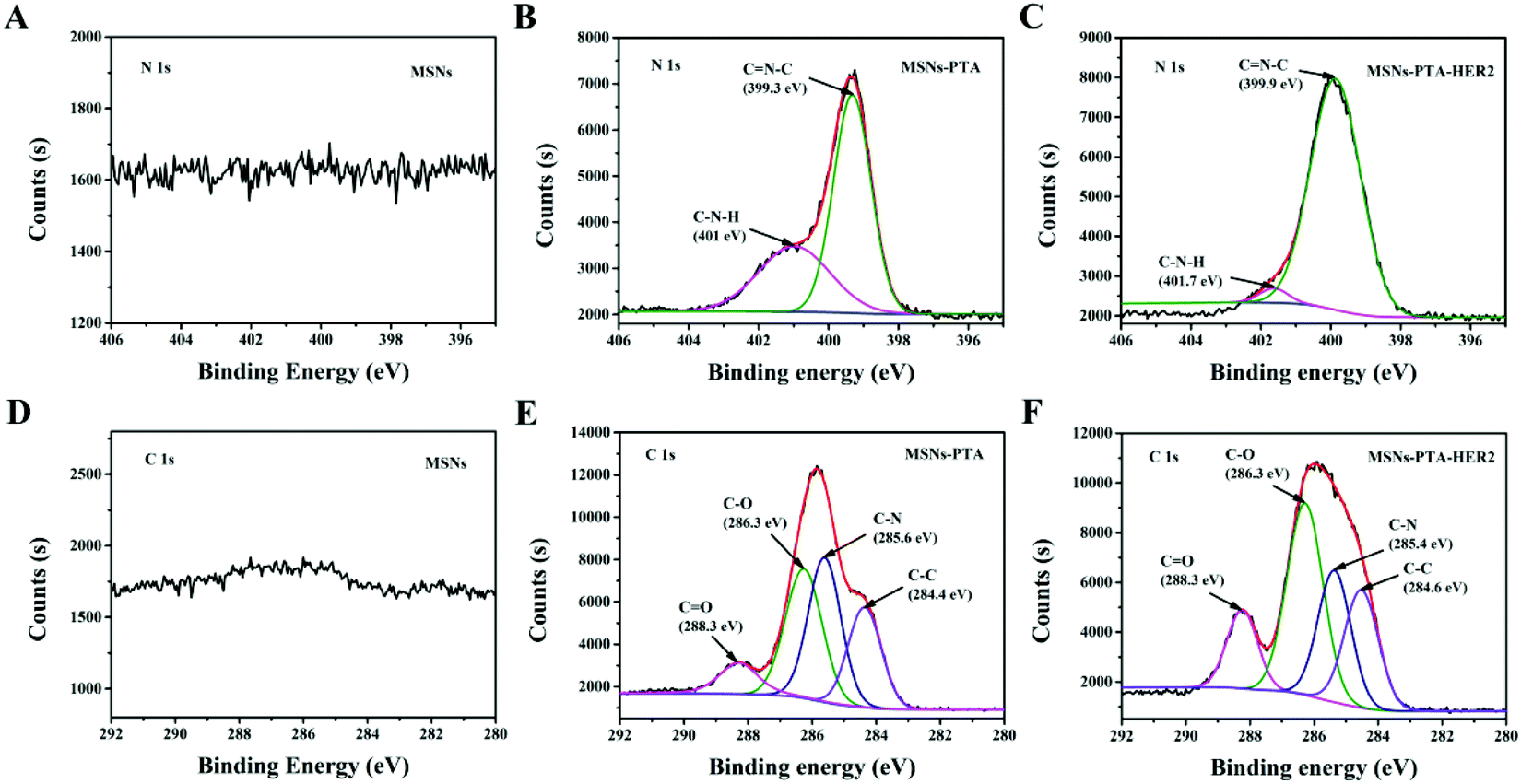 | ||
| Fig. 2 Narrow scan XPS N 1s spectra of (A) MSNs, (B) MSNs-PTA and (C) MSNs-PTA-HER2. Narrow scan XPS C 1s spectra of (D) MSNs, (E) MSNs-PTA and (F) MSNs-PTA-HER2. | ||
In vitro drug loading and reversible pH-responsive release study
Stimuli-responsive properties are essential for nanocarriers, so they can achieve accurate release of drugs at the tumor site without leakage at the site of normal tissues during transportation. Among the numerous stimuli-responsive conditions, pH is the most widely used due to its self-responsiveness because the pH of the tumor microenvironment is lower than that of the physiological environment. To investigate the reversible pH-triggered drug release behavior of the MSNs-PTA-HER2 system, DOX was selected as model drug and different pH values were employed as triggers in this study. Meanwhile, the DOX loading process depends mainly on the physical adsorption mechanisms of the pores in MSNs. UV-vis absorbance spectra were recorded to determine the successful loading of DOX (Fig S6, ESI†). Specifically, a characteristic absorbance peak at 485 nm appeared in free DOX and DOX/MSNs-PTA-HER2, but not in MSNs-PTA-HER2, verifying that DOX was successfully loaded into the mesopores of MSNs. The loading capacity of DOX determined through the standard curve drawn by measuring the DOX absorbance value at 485 nm (Fig. S7, ESI†) was 112 mg per 1000 mg MSNs-PTA-HER2 (10.1%), which was a relatively high loading efficiency. Furthermore, the physical state of the loaded DOX in the mesopores of MSNs was characterized by wide-angle XRD analysis. Fig. S8 (ESI†) shows that free DOX displayed characteristic and intense crystalline diffraction peaks, but no distinctive crystalline peaks were shown in either DOX/MSNs-PTA-HER2 or MSNs-PTA-HER2 samples, indicating that DOX in MSNs-PTA-HER2 existed in a non-crystalline state due to the confining effect of the mesopores of MSNs, which was conducive to the maintenance of drug activity.Subsequently, the in vitro pH-responsive controlled release property was investigated under different pH conditions. In this work, three different pH values were chosen: pH 7.4 represented the normal physiological environment, pH 6.8 represented the tumor microenvironment, and pH 5.0 represented some acidic organelles (e.g. lysosomes).16 From Fig. S9 (ESI†), little DOX was released from DOX/MSNs-PTA after 48 h at pH 7.4 (only 8.1%) without any stimulation, while a large amount of DOX leaked from DOX/MSNs (up to 88.2%), demonstrating the good stability of the PTA layer, which successfully trapped DOX in the pores of MSNs-PTA under physiological conditions. In addition, pure TA encapsulated DOX-loaded MSNs (DOX/MSNs-TA) also displayed a certain amount of drug release (34.1%) at pH 7.4, indicating that the layer formed by pure TA was unstable. However, DOX was readily released from MSNs-PTA-HER2 when exposed to different acidic environments. Meanwhile, Fig. 3A and Fig. S10 (ESI†) show that the cumulative release of DOX from DOX/MSNs-PTA-HER2 increased with the decrease in pH value. After 48 h, around 42.5% and 67.9% of DOX was released from the MSNs-PTA-HER2 system when the pH value decreased from 7.4 to 6.8 and 5.0, verifying the good pH-responsive controlled release ability of the fabricated nanocarriers. Based on this property, the fabricated DOX/MSNs-PTA-HER2 is likely to be further developed as a pH-sensitive release system that can target tumor cells and allow drug release within the tumor microenvironment and some acidic intracellular compartments such as lysosomes and endosomes, where the pH value is lower than that in normal tissue.
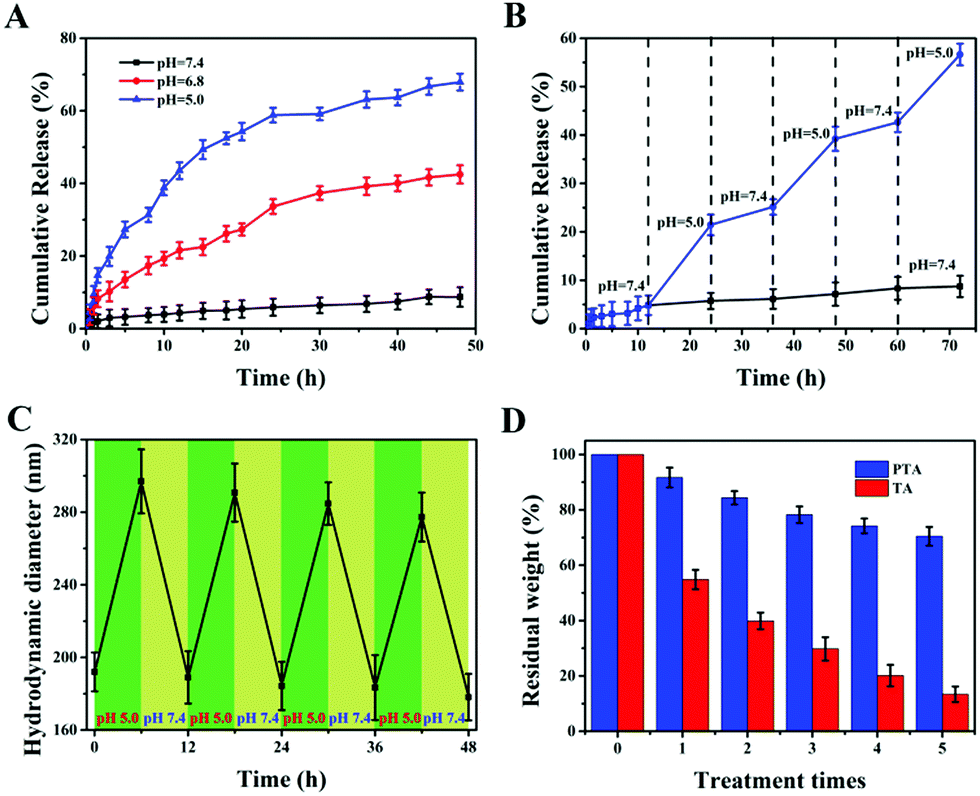 | ||
| Fig. 3 In vitro pH-responsive drug release behaviors. (A) The release profile of DOX from DOX/MSNs-PTA-HER2 under different pH conditions (pH 7.4, 6.8 and 5.0). (B) DOX release from DOX/MSNs-PTA-HER2 under alternate pH conditions (pH 7.4 and 5.0). (C) Changes in particle diameter under alternate pH conditions (pH 7.4 and 5.0). (D) Residual weight of PTA layer and TA layer coated on the surface of MSNs after repeated treatment with pH 5.0 and pH 7.4 buffer 5 times. | ||
In addition, reversible stimuli-responsive controlled release properties are also significant for nanocarriers in order to avoid possible “secondary” damage to normal tissues caused by the continuous release of overloaded drugs after the tumor cells are lysed. Drug-loaded nanoparticles tend to have a large drug loading amount, and therefore it is difficult to completely release the drug at the tumor site. After the tumor cells are lysed, if the nanocarrier is unable to close the “gatekeeper” again in response to the physiological environment, the residual drug will cause “secondary” damage to normal cells.20 Therefore, to mimic how the drug delivery system works in vivo and evaluate whether the synthesized drug-loaded nanoparticles could be used to reduce “secondary” side effects, we investigated reversible pH-responsive release behavior in an alternating pH environment (pH 7.4 and pH 5.0). As shown in Fig. 3B, repeated exposure of DOX-loaded MSNs-PTA-HER2 to pH 7.4 and pH 5.0 buffer solution caused the “gatekeeper” to alternately turn “off” and turn “on”, and the release rate of loaded DOX alternated between the “on” and “off” valve conditions. Furthermore, the “on” and “off” switching of DOX release could be repeated several times. This phenomenon confirmed that the controlled reversible nature of the “on” and “off” behavior of the PTA valve, which allowed the leakage of drugs, depended on the acidity of the surrounding environment and directly illustrated that our fabricated nanocarriers had reversible pH-responsive controlled release ability. The possible reason for the reversible pH-responsive property of the prepared nanoparticles is explained as follows. First of all, TA can spontaneously polymerize to form an encapsulation layer adhering to the surface of nanocarriers under physiological pH conditions to prevent the leakage of loaded drugs. Simultaneously, the degradation of the TA layer under acidic conditions makes it a suitable sealing agent for pH-responsive controlled release. Undesirably, the exfoliated TA layer can’t immediately polymerize after the pH changes from acidic to neutral or alkaline again. Therefore, the pH-responsive controlled release behavior of the TA layer is irreversible. Herein, we introduced TEPA as an intramolecular crosslinker to construct a TA/TEPA binary system. In the presence of TEPA, the oxidative polymerization of TA can be accelerated to form a PTA layer under alkaline conditions by the combination of catechol and amine groups, which is denser than the TA layer and allows for more efficient drug encapsulation. In addition, the TEPA molecule contains abundant amine groups that can be protonated. Hence, TEPA is easily protonated in an acidic environment, resulting in an increase in the hydrophilicity of the PTA layer, which in turn causes the swelling of the PTA layer and a large amount of small molecule drug leakage. However, since the tannic acid molecules are closely connected by TEPA, the PTA layer does not fall off under acidic conditions. After re-entering neutral or alkaline conditions, deprotonation of the amine groups in TEPA makes the PTA layer dense again to prevent residual drug from leaking. Therefore, the prepared nanocarriers possess a reversible pH-responsive controlled release property.
In order to further explore the reversible controlled release performance of the TA/TEPA binary system, a series of characterization methods were performed to investigate the change of the nanoparticles before and after acid treatment. First of all, the morphology of the nanoparticles was observed by TEM after acid treatment. It could be seen from the TEM image (Fig. S11, ESI†) that the surface of the nanoparticles returning to a neutral environment was still obviously covered with a layer of polymer encapsulant with a thinner thickness, indicating that the “gatekeeper” PTA hadn’t fallen off and became dense again to continue maintaining a good sealing effect after acid treatment. Besides, a DLS method was used to detect the changes in hydrodynamic diameter of the synthesized nanoparticles under alternate pH conditions (pH 5.0 and 7.4). As shown in Fig. 3C, the hydrodynamic diameter increased every time the nanocarriers entered an acidic environment and decreased after the nanocarriers returned to a neutral environment, and was only slightly decreased after each cycle. This indicated that the PTA layer could remain stable and reversibly adjust its density to switch between the “on” and “off” states of the “gatekeeper” under different pH conditions. Finally, the loss of the PTA layer after repeated treatment in acidic and neutral environments (MSNs-TA or MSNs-PTA were treated at pH 5.0 for 6 h and pH 7.4 for another 6 h in each round) was specifically investigated by TGA analysis (Fig. 3D). The total amount of weight of the PTA layer remained above 70% after 5 rounds of treatment. In sharp contrast, the weight of the TA layer decreased rapidly after each round of treatment, and only a small amount remained after five rounds of processing. This phenomenon verified the stability of the PTA layer under different pH conditions, which is beneficial for reversible controlled release behavior. In summary, all the characteristic results further indicated the reversibly pH-responsive performance and described the process of reversible switching between “on” and “off” in response to pH.
On the whole, the in vitro reversible pH-responsive release study shows that the prepared targeting nanocarriers (MSNs-PTA-HER2) have the capability to achieve reversible pH-responsive controlled release, which is essential for reduced secondary side effects in the practical application of nanocarriers.
Targeting uptake and intracellular release assays
Targeting nanocarriers require the ability to specifically recognize particular tumor cells, enabling the drug-loaded nanoparticles to be enriched around the surface of tumor cells and facilitating the endocytosis of nanoparticles by tumor cells. Considering that antigen–antibody binding possesses specificity and high efficiency and HER2 is an antigen that exists in multitudinous cancer cells, especially breast cancer cells, HER2 antibody was covalently grafted on the surface of PTA to achieve specific targeting of the nanocarriers to HER2 over-expressing breast cancer cells in this experiment.Herein, SK-BR-3 cells with over-expressed HER2 and normal L-02 cells with few HER2 were selected for research, and the endocytosis of the nanocarriers (MSNs-PTA-HER2) by both cells was qualitatively studied by CLSM. As displayed in Fig. S12 and S13 (ESI†), the amount of endocytosed targeting nanoparticles obviously increased with the increasing incubation time and concentration of FITC labeled MSNs-PTA-HER2 nanoparticles. Moreover, Fig. 4A–C also obviously show that the uptake amount of MSNs-PTA-HER2 by SK-BR-3 cells was higher than that of FITC labeled MSNs and MSNs-PTA nanoparticles. The possible reason could be explained by the fact that the HER2 receptor mediated cellular endocytosis, resulting in more MSNs-PTA-HER2 uptake by SK-BR-3 cells. Furthermore, the intracellular green fluorescence originating from MSNs-PTA-HER2 was greater in SK-BR-3 cells than in L-02 cells (Fig. 4D), demonstrating that MSNs-PTA-HER2 was readily taken up by SK-BR-3 cells. These results clearly revealed that the grafting of HER2 antibody could specifically increase the cellular uptake of MSNs-PTA-HER2 nanoparticles in HER2 receptor-positive tumor cells. Furthermore, when SK-BR-3 cells were pretreated with 100 μg mL−1 of free HER2 antibody for 2 h and then incubated with 1.0 μg mL−1 FITC labeled MSNs-PTA-HER2 for another 12 h (Fig. S14, ESI†), the intracellular green fluorescence was significantly reduced, corresponding to the greatly reduced cellular uptake of MSNs-PTA-HER2. This phenomenon provided direct evidence that the endocytosis of MSNs-PTA-HER2 was mediated by HER2 receptors, which were over-expressed on the membranes of SK-BR-3 cells. In addition, an energy-dependent endocytosis pathway was demonstrated through treating the SK-BR-3 cells with NaN3 at 4 °C, leading to the marked inhibition of cell uptake efficiency (Fig. S15, ESI†). The most reasonable mechanism suggests that NaN3 could block the synthesis process of adenosine 5-triphosphate (ATP), and the energy metabolism in the tumor cells could also be inhibited at 4 °C.5 These results indicated that the HER2 receptor-mediated uptake was ATP-dependent endocytosis and was relatively inactive at low temperatures. It is well known that an excellent drug delivery system can not only target tumor cells to reduce non-specific toxicity, but can also release drugs inside cancer cells to give good antitumor activity. Based on this, CLSM analysis was performed to study the intracellular drug release and whether the drug release from MSNs-PTA-HER2 was triggered by the acidic environment. As shown in Fig. 5A, the intracellular DOX red fluorescence was significantly increased with the decrease of pH values, indicating that DOX release from MSNs-PTA-HER2 was pH-sensitive. Furthermore, FCM quantitative analysis (Fig. 5B and C) also illustrated similar results. These pH-triggered release behaviors could be explained by the fact that the PTA layer was pH-sensitive, and the polymer layer would become loose in the acidic tumor environment, resulting in the quick release of DOX from the targeting nanoparticles. Taken together, these results demonstrated that DOX release from MSNs-PTA-HER2 was dependent on the intracellular acidity level.
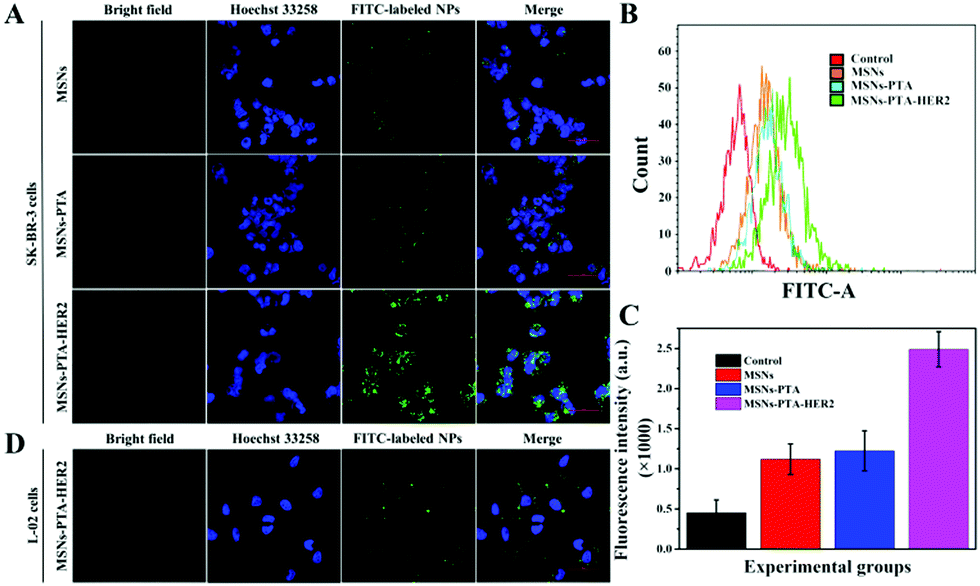 | ||
| Fig. 4 (A) CLSM images of SK-BR-3 cells incubated with MSNs, MSNs-PTA and MSNs-PTA-HER2 for 12 h. (B) FCM analysis of SK-BR-3 cells after treatment with MSNs, MSNs-PTA and MSNs-PTA-HER2, respectively. (C) Quantification analysis of FITC fluorescence intensity in SK-BR-3 cells after incubation with MSNs, MSNs-PTA and MSNs-PTA-HER2, respectively. (D) CLSM images of L-02 cells incubated with MSNs-PTA-HER2 for 12 h. FITC labeled nanoparticles are seen in the green channel and Hoechst 33258 stained nuclei are seen in the blue channel. Scale bar: 50 μm. | ||
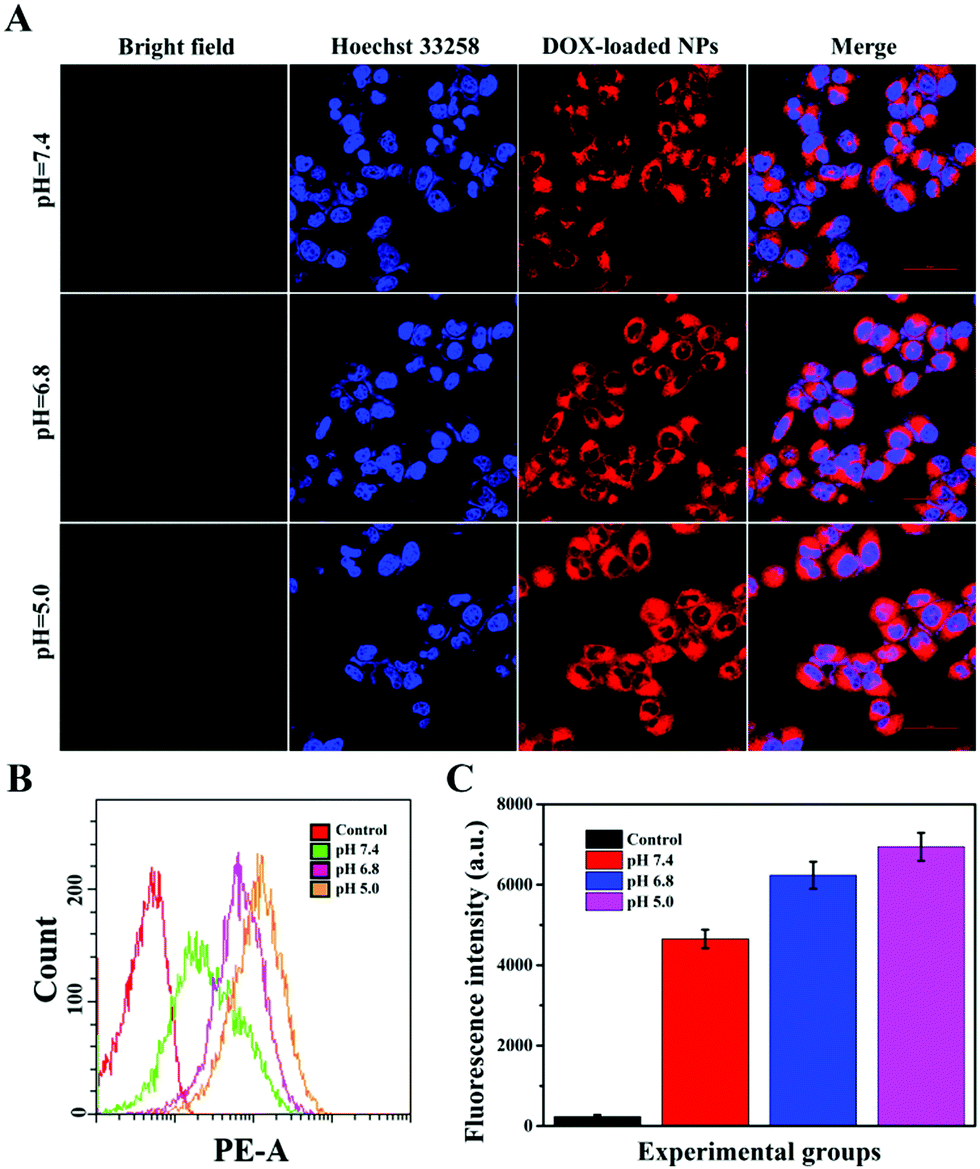 | ||
| Fig. 5 (A) CLSM images of SK-BR-3 cells exposed to DOX/MSNs-PTA-HER2 for 12 h at pH 7.4, pH 6.5 and pH 5.0, respectively. Scale bar: 50 μm. (B) FCM analysis of SK-BR-3 cells after treatment with DOX/MSNs-PTA-HER2 for 12 h at pH 7.4, pH 6.5 and pH 5.0, respectively. (C) Quantification analysis of DOX fluorescence intensity in SK-BR-3 cells after incubation for 12 h at pH 7.4, pH 6.5 and pH 5.0, respectively. Data are represented as mean ± SD (n = 3). | ||
Generally, the results of both CLSM and FCM demonstrated the superior targeting properties of the fabricated nanocarriers (MSNs-PTA-HER2), which enabled the nanocarriers to be specifically enriched around the surface of HER2 over-expressing breast cancer cells and enhanced the specific intracellular DOX release efficiency triggered by the acidic environment, thereby improving the selective killing ability of the drug-loaded nanoparticles.
In vitro biocompatibility and cytotoxicity assays
Excellent biocompatibility is key for a smart multifunctional nanocarrier to be used in various in vivo biomedical applications. To this end, a typical MTT method was performed to test the biocompatibility of the prepared drug-free nanocarriers. As displayed in Fig. S16 (ESI†), the survival rate of both SK-BR-3 and L-02 cells remained above 87%, even though the concentration of the incubated MSNs-PTA-HER2 reached 200 μg mL−1 and the co-culture time was up to 48 h. These results clearly demonstrated that the fabricated MSNs-PTA-HER2 was biocompatible and displayed no obvious toxicity towards cells, hence was suitable for biomedical applications. In addition, this result further indicated that the toxicity of drug-loaded nanoparticles was derived from DOX, but had nothing to do with the drug-free carriers.Next, the cytotoxic effects of free DOX and DOX-loaded nanoparticles to SK-BR-3 and L-02 cells were evaluated by a typical MTT method. Fig. 6A and B show that SK-BR-3 and L-02 cells treated with free DOX and DOX-loaded nanoparticles all displayed significant DOX dose-dependent cytotoxicity. In HER2 over-expressing SK-BR-3 cells, the combination of DOX/MSNs-PTA-HER2 displayed a higher cytotoxicity compared with that of free DOX and DOX/MSNs-PTA. Meanwhile, DOX/MSNs-PTA-HER2 exhibited higher cytotoxicity against SK-BR-3 cells than L-02 cells under the same conditions. This phenomenon can be explained by the specific recognition by MSNs-PTA-HER2 of HER2 over-expressed on the surface of SK-BR-3 cells, which enhanced the cellular uptake efficacy. Furthermore, analysis of the half inhibitory concentration (IC50) was performed to quantify the anticancer effects of free DOX and DOX-loaded nanoparticles. Table S6 (ESI†) shows that the anticancer effects of free DOX and DOX/MSNs-PTA exhibited similar IC50 to L-02 and SK-BR-3 cells. This result might be due to the non-specific cell recognition of free DOX and DOX/MSNs-PTA. Nevertheless, it was notable that DOX/MSNs-PTA-HER2 exhibited a higher cytotoxicity to SK-BR-3 cells; the IC50 against SK-BR-3 cells (IC50: 0.32 μg mL−1) was much lower than that against L-02 cells (IC50: 1.19 μg mL−1) after treatment for 24 h. Besides, the IC50 of DOX/MSNs-PTA-HER2 against SK-BR-3 cells was lower than that of free DOX (IC50: 0.42 μg mL−1). Therefore, this result demonstrated that the prepared drug-loaded nanoparticles (DOX/MSNs-PTA-HER2) had specificity and high efficiency for killing HER2 over-expressing cancer cells, and efficaciously reduced the cytotoxicity of DOX to normal tissue cells.
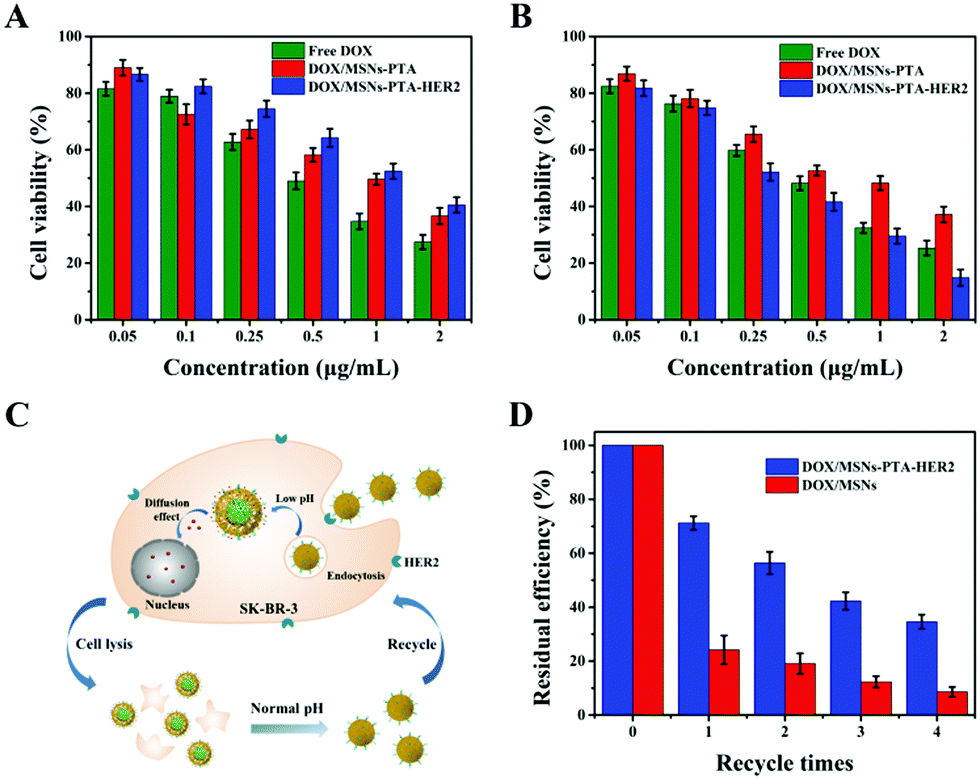 | ||
| Fig. 6 In vitro cytotoxicity of different drug-containing systems. Cell viability of (A) L-02 cells and (B) SK-BR-3 cells after 24 h incubation with different concentrations of free DOX, DOX/MSNs-PTA and DOX/MSNs-PTA-HER2. (C) Schematic illustration of recycling of the synthesized drug-loaded nanoparticles (DOX/MSNs-PTA-HER2) for cell killing. (D) Residual killing efficiency of DOX/MSNs-PTA-HER2 and DOX/MSNs to SK-BR-3 cells after 4 cycles, respectively. Data are represented as mean ± SD (n = 3). | ||
More importantly, the drug-loaded nanoparticles were supposed to possess reversible stimuli-responsive drug controlled release performance, which was assessed by repeated toxicity assays. Fig. 6C exhibits the process of recycling the drug-loaded nanoparticles (DOX/MSNs-PTA-HER2). Specifically, the nanoparticles were released and the “gatekeeper” was closed again in response to the neutral environment of the medium after the killing and lysis of cells. Subsequently, the nanoparticles were collected by centrifugation and reused for a new round of cytotoxicity experiments. Fig. 6D shows that the residual killing efficiency (compared to the first killing efficiency) of DOX/MSNs-PTA-HER2 decreased significantly more slowly than that of DOX/MSNs, which could be attributed to the reversible pH-responsive property of the PTA layer with the ability to realize a secondary sealing effect to prevent the continuous leakage of the remaining drug after the drug-loaded nanoparticles killed the tumor cells and were released, thereby maintaining the higher cell killing efficiency of the nanocarriers. This result indirectly demonstrated the superior reversible pH-responsive drug controlled release property of the nanocarriers, which could prevent the “secondary” killing effect on normal tissues caused by continuous drug leakage.
In vivo therapeutic effect of nanocomposite
In order to further investigate the therapeutic effect of the synthesized targeting MSNs-PTA-HER2 nanoparticles in vivo, a tumor-bearing nude mouse model was used in this study, and the tumor model was established by subcutaneous injection. First of all, the biodistribution of DiR-loaded MSNs-PTA-HER2 (DiR/MSNs-PTA-HER2) intravenously administrated in a tumor-bearing mouse was investigated by detecting the near infrared (NIR) images using an IVIS (Lumina XR Series III, PerkinElmer) to verify the HER2 antibody mediated targeting property (Fig. 7A). The fluorescence of the DiR-loaded nanoparticles originally spread all over the body of the mouse and accumulated at the tumor site 8 h post-injection, validating the specific targeting ability of the synthesized nanoparticles, which prevented the nanoparticles from being nonspecifically enriched at undesired normal tissue sites. Furthermore, high intensity persistent fluorescence could be maintained 24 h post-injection, illustrating the enhanced tumor retention effect of the nanocarriers which was essential for tumor therapy. Afterwards, the DiR/MSNs-PTA-HER2 treated mouse was sacrificed and the tumor and main organs (heart, kidney, spleen, liver, lung) were collected for ex vivo imaging after injection for 24 h (Fig. 7B). The fluorescence intensity of the DiR-loaded nanoparticles was visibly stronger in the tumor than in the other organs, confirming the specific targeting of the nanocarriers to SK-BR-3 tumor. Besides, the specific fluorescence intensities of tumors and main organs in free DiR and DiR/MSNs-PTA-HER2 treated mice were quantitatively analyzed and displayed in Fig. 7C. In the DiR/MSNs-PTA-HER2 treated mouse, the appearance of the strongest fluorescence intensity in the tumor reflected the HER2 antibody mediated targeting accumulation of the nanocarriers in the tumor. Meanwhile, the fluorescence intensity in the tumor of the DiR/MSNs-PTA-HER2 treated mouse was 2.2-fold higher than that in the tumor of the free DiR treated mouse, indicating the targeting ability of the synthesized nanocarriers. In summary, these results qualitatively and quantitatively prove the superior targeting ability of the fabricated nanoparticles. Finally, the DOX concentration in blood at different time intervals post-injection was detected by liquid chromatography to obtain the blood clearance curves, which were fitted to a typical two-compartment model. As shown in Fig. S17 (ESI†), the half-life of DOX/MSNs-PTA-HER2 was longer than that of free DOX, demonstrating the capability of the synthesized nanocarriers to prolong blood circulation.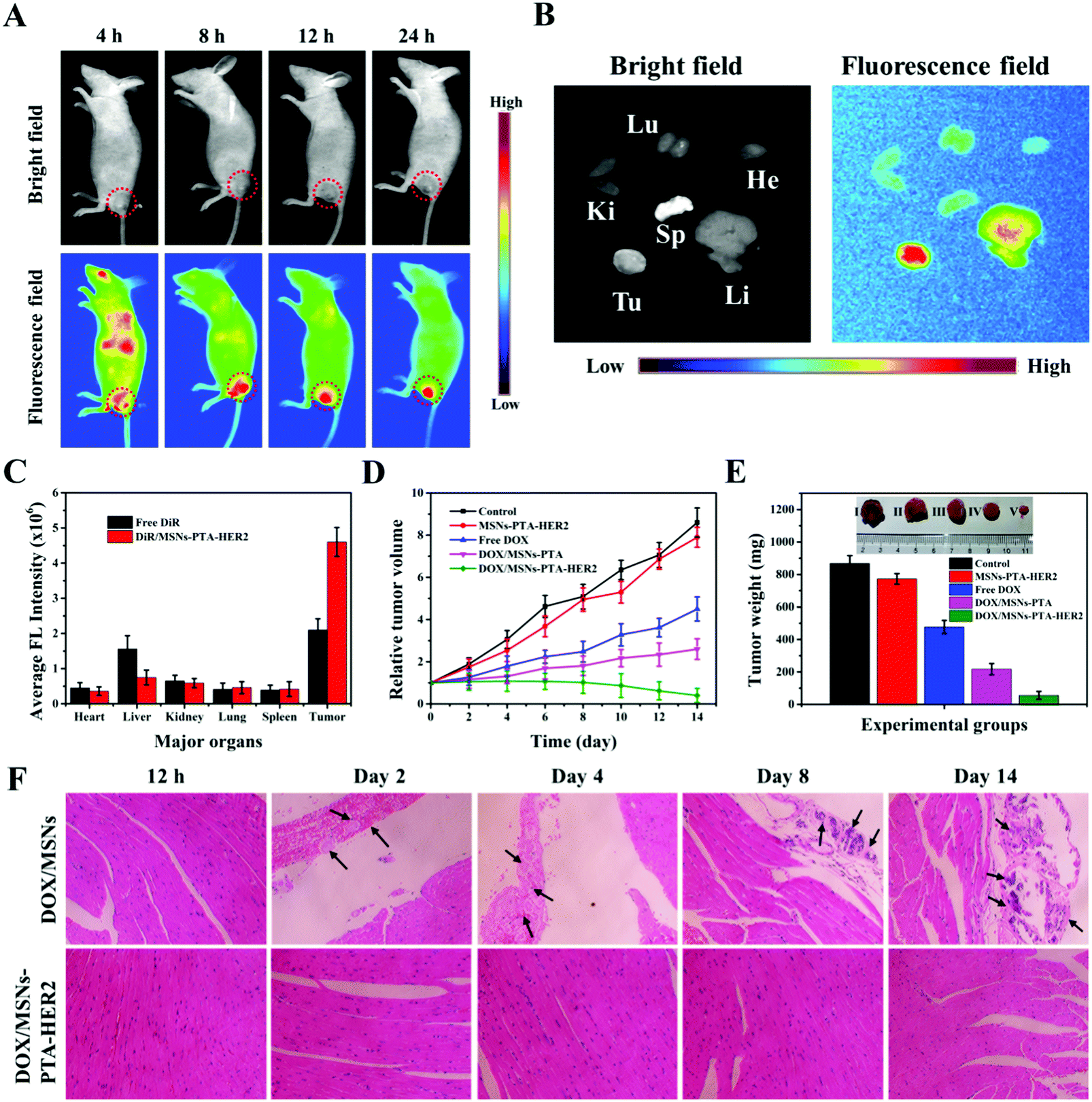 | ||
| Fig. 7 (A) Whole-body real-time fluorescence imaging after intravenous injection of DiR/MSNs-PTA-HER2 for 4, 8, 12, and 24 h, respectively. The dashed circles represent the tumor locations. (B) Representative ex vivo fluorescence images of dissected tumor and major organs (lung, kidney, spleen, heart, and liver) of DiR/MSNs-PTA-HER2 treated mice. (C) Fluorescence intensity of the tumor and major organs. (D) Average relative tumor volume of each group on different days. (E) Tumor weight of each group after treatment for two weeks. Inset: Photograph of excised tumor from each group. (F) Standard H&E staining of sliced typical heart tissue of DOX/MSNs and DOX/MSNs-PTA-HER2 groups at different time intervals. Data are represented as mean ± SD (n = 4); p-value is less than 0.05. | ||
Next, the in vivo therapeutic efficiency of DOX/MSNs-PTA-HER2 was investigated. The tumor volumes were firstly measured to reflect the curative effects of different treatments on tumor growth (Fig. 7D). After injection for 14 days, the tumors treated with PBS (control group) and the MSNs-PTA-HER2 group displayed rapid growth. However, compared to MSNs-PTA-HER2 (positive control) and PBS (negative control), the tumors treated by both free DOX and DOX/MSNs-PTA showed slow growth. Meanwhile, DOX/MSNs-PTA possessed stronger anticancer activity than free DOX due to the enhanced permeability and retention (EPR) effect, which made it easier for the nanoparticles to be passively enriched at the tumor site. As expected, the tumors treated with DOX/MSNs-PTA-HER2 had the smallest tumor volume among all the groups. This phenomenon could be explained by the fact that DOX/MSNs-PTA-HER2 could not only passively target tumor tissue through the EPR effect, but also actively target HER2 over-expressing tumor cells by the HER2 antibody, which made it easier for DOX/MSNs-PTA-HER2 to accumulate at the tumor site. Then, the release of sufficient DOX in response to the acidic tumor microenvironment can efficiently kill the tumor cells. This result clearly verified that the prepared DOX/MSNs-PTA-HER2 possessed the strongest antitumor effect, which was consistent with in vitro cytotoxicity assays. Moreover, the weight and size of tumors from each group were also measured after treatment for 14 days. As shown in Fig. 7E, the DOX/MSNs-PTA-HER2 group exhibited the lightest tumor weight and the smallest tumor size, which demonstrated the strongest antitumor activity of the prepared DOX/MSNs-PTA-HER2. Meanwhile, the body weight of mice in different groups on different days was also measured (Fig. S18, ESI†). Mice treated with different components showed similar healthy weight gain, illustrating that the prepared drug-loaded nanoparticles had no discernable short-term side effects on mouse bodies.
Subsequently, the in vivo biocompatibility of the nanocarriers and the specific toxicity of the drug-loaded nanoparticles were investigated more comprehensively by standard hematoxylin and eosin (H&E) staining of sliced tumor and typical normal tissues, in which the cytoplasm was stained red and the nuclei were stained blue. As for tumor tissue (Fig. S19, ESI†), compared to the control group, no obvious cell reduction was observed after treatment with MSNs-PTA-HER2. Simultaneously, a small number of cells were absent after treatment with both free DOX and DOX/MSNs-PTA. However, large areas of tumor cells were destroyed after treatment with DOX/MSNs-PTA-HER2, which was consistent with the previous in vivo experimental results, further demonstrating the extremely strong antitumor activity of the synthesized drug-loaded nanoparticles (DOX/MSNs-PTA-HER2) with active targeting ability. Furthermore, for typical heart, kidney, liver, lung and spleen tissues (Fig. S20, ESI†), except for the phenomenon of neutrophil accumulation in the hearts of mice treated with free DOX, which was possibly due to the acute cardiotoxicity of free DOX,46 no visible pathological abnormalities appeared in the other groups, demonstrating that the prepared drug-loaded nanoparticles (DOX/MSNs-PTA-HER2) possessed superior in vivo biocompatibility and controlled release ability; this could prevent the premature release of chemotherapeutic drugs during transportation in vivo to reduce the non-specific killing of normal tissues by chemotherapeutic drugs.
Furthermore, H&E staining of sliced heart tissue was selected to investigate the reversible drug controlled release property of the synthesized DOX-loaded nanocomposite, which could be used to reduce “primary” and “secondary” side effects (Fig. 7F). For mice treated with DOX/MSNs without encapsulation, the condition of myocardial damage gradually deteriorated over time, specifically by the exacerbation of thrombosis and the appearance and increase of calcium mass and hyperplastic connective tissue, accompanied by inflammatory cell infiltration. However, no visible myocardial damage appeared in DOX/MSNs-PTA-HER2 treated mice at any time. This phenomenon could be explained by the fact that the fabricated nanocarriers possess reversible pH-responsive controlled release performance, which could both keep the encapsulation stable in the process of reaching the tumor site through blood circulation to prevent the premature leakage of loaded drugs, and seal again in response to the neutral environment of normal tissue after release in the acidic tumor tissue to prevent the continuous leakage of residual drugs, thereby reducing the “primary” and “secondary” cytotoxicity of the synthesized therapeutic nanoplatforms. This result verified the reversible controlled release property of the fabricated nanocarriers.
Overall, in vivo animal experiments authenticated the outstanding biocompatibility, reversible drug controlled release property and active targeting ability of the fabricated drug-loaded nanoparticles (DOX/MSNs-PTA-HER2). The results were consistent with in vitro experimental results and mean that DOX/MSNs-PTA-HER2 is promising for further use in actual clinical treatment.
Conclusions
In summary, reversibly pH-responsive and HER2 targeted drug-loaded nanoparticles were successfully designed and fabricated by modifying PTA and HER2 antibody on the surface of MSNs with a high drug loading of 10.1 wt%. The in vitro release results demonstrated that such nanoparticles clearly displayed pH-sensitive drug release, which could effectively prevent drug leakage during blood circulation. Most importantly, the prepared nanocomposite achieved intelligent reversible pH-responsive release behavior by regulating the density of the “gatekeeper”, thereby realizing reversible rotation from “on” to “off” through the stimulation of external pH. Besides, the in vitro cellular uptake measurements of such nanoparticles exhibited that the cellular uptake efficacy of SK-BR-3 cells (HER2 positive) through HER2-mediated endocytosis was higher than that of L-02 cells (HER2 negative). Furthermore, the multifunctional drug-loaded nanoparticles grafted with HER2 antibodies could selectively target cancer cells over-expressing HER2 both in vitro and in vivo, and they significantly enhanced therapeutic effects and exhibited superior antitumor effects. Last but not least, both in vitro and in vivo assays proved that the pH-responsive controlled release property of fabricated nanocarriers was reversible, which was essential for applicable therapeutic nanoplatforms. This strategy is expected to construct a reversibly pH-responsive nanocomposite to solve the problem of “secondary” side effects caused by residual drugs in the irreversible “gatekeeping” system in the process of conventional nanoparticle-based pH-responsive drug delivery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (No. 21908059, 41907318 and 21636003), the China Postdoctoral Science Foundation (No. 2019M651419), Shanghai Sailing Program (No. 19YF1410900), the Fundamental Research Funds for the Central Universities (No. 22221818014), the Shanghai Post-doctoral Excellence Program (No. 2018011), and the Open Funding Project of the State Key Laboratory of Bioreactor Engineering. All animal experiments were conducted in conformity with Chinese legislation on the Use and Care of Research Animals (Document No. 55, 2001). This experiment was approved by the East China University of Science and Technology Animal Studies Committee, complying with established institutional guidelines for the Care and Use of Laboratory Animals.Notes and references
- Q. Zhang, F. Liu, K. T. Nguyen, X. Ma, X. Wang, B. Xing and Y. Zhao, Adv. Funct. Mater., 2012, 22, 5144–5156 CrossRef CAS.
- C. Chen, W. Sun, X. Wang, Y. Wang and P. Wang, Int. J. Biol. Macromol., 2018, 111, 1106–1115 CrossRef CAS PubMed.
- T. Li, X. Shen, Y. Geng, Z. Chen, L. Li, S. Li, H. Yang, C. Wu, H. Zeng and Y. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 13748–13758 CrossRef CAS PubMed.
- N. An, H. Lin, C. Yang, T. Zhang, R. Tong, Y. Chen and F. Qu, Mater. Sci. Eng., C, 2016, 69, 292–300 CrossRef CAS PubMed.
- L. Dai, Q. Zhang, J. Li, X. Shen, C. Mu and K. Cai, ACS Appl. Mater. Interfaces, 2015, 7, 7357–7372 CrossRef CAS PubMed.
- B. Sahoo, K. S. P. Devi, S. K. Sahu, S. Nayak, T. K. Maiti, D. Dhara and P. Pramanik, Biomater. Sci., 2013, 1, 647–657 RSC.
- D. Xiao, H.-Z. Jia, N. Ma, R.-X. Zhuo and X.-Z. Zhang, Nanoscale, 2015, 7, 10071–10077 RSC.
- D. Xiao, H. Z. Jia, J. Zhang, C. W. Liu, R. X. Zhuo and X. Z. Zhang, Small, 2014, 10, 591–598 CrossRef CAS PubMed.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S. Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 5038–5044 CrossRef CAS PubMed.
- C. H. Lee, S. H. Cheng, I. P. Huang, J. S. Souris, C. S. Yang, C. Y. Mou and L. W. Lo, Angew. Chem., Int. Ed., 2010, 49, 8214–8219 CrossRef CAS PubMed.
- B. Zhang, Z. Luo, J. Liu, X. Ding, J. Li and K. Cai, J. Controlled Release, 2014, 192, 192–201 CrossRef CAS PubMed.
- S. Angelos, Y. W. Yang, K. Patel, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2008, 47, 2222–2226 CrossRef CAS PubMed.
- J. Liu, B. Zhang, Z. Luo, X. Ding, J. Li, L. Dai, J. Zhou, X. Zhao, J. Ye and K. Cai, Nanoscale, 2015, 7, 3614–3626 RSC.
- W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharmaceutics, 2010, 7, 1913–1920 CrossRef CAS PubMed.
- H. Huang, P. Li, C. Liu, H. Ma, H. Huang, Y. Lin, C. Wang and Y. Yang, RSC Adv., 2017, 7, 2829–2835 RSC.
- E. R. Gillies and J. M. Fréchet, Bioconjugate Chem., 2005, 16, 361–368 CrossRef CAS PubMed.
- R. Mo, Q. Sun, J. Xue, N. Li, W. Li, C. Zhang and Q. Ping, Adv. Mater., 2012, 24, 3659–3665 CrossRef CAS PubMed.
- A. Popat, J. Liu, G. Q. M. Lu and S. Z. Qiao, J. Mater. Chem., 2012, 22, 11173–11178 RSC.
- Q. Zheng, T. Lin, H. Wu, L. Guo, P. Ye, Y. Hao, Q. Guo, J. Jiang, F. Fu and G. Chen, Int. J. Pharm., 2014, 463, 22–26 CrossRef CAS PubMed.
- X. Mei, S. Yang, D. Chen, N. Li, H. Li, Q. Xu, J. Ge and J. Lu, Chem. Commun., 2012, 48, 10010–10012 RSC.
- J. Lu, M. Liong, Z. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1794–1805 CrossRef CAS PubMed.
- N. Bertrand and J.-C. Leroux, J. Controlled Release, 2012, 161, 152–163 CrossRef CAS PubMed.
- S. H. Kim, I. In and S. Y. Park, Biomacromolecules, 2017, 18, 1825–1835 CrossRef CAS PubMed.
- F.-F. Cheng, J.-J. Zhang, F. Xu, L.-H. Hu, E. Abdel-Halim and J.-J. Zhu, J. Biomed. Nanotechnol., 2013, 9, 1155–1163 CrossRef CAS PubMed.
- H. Wang, J. Wu, C. Cai, J. Guo, H. Fan, C. Zhu, H. Dong, N. Zhao and J. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 5602–5608 CrossRef CAS PubMed.
- L. Wang, Y. Shi, R. Sa, N. Ning, W. Wang, M. Tian and L. Zhang, Ind. Eng. Chem. Res., 2016, 55, 12547–12556 CrossRef CAS.
- B.-S. Kim, H.-i. Lee, Y. Min, Z. Poon and P. T. Hammond, Chem. Commun., 2009, 4194–4196 RSC.
- N. Ninan, A. Forget, V. P. Shastri, N. H. Voelcker and A. Blencowe, ACS Appl. Mater. Interfaces, 2016, 8, 28511–28521 CrossRef CAS PubMed.
- X. Zhang, P.-F. Ren, H.-C. Yang, L.-S. Wan and Z.-K. Xu, Appl. Surf. Sci., 2016, 360, 291–297 CrossRef CAS.
- H. Shagholani and S. M. Ghoreishi, J. Drug Delivery Sci. Technol., 2017, 39, 88–94 CrossRef CAS.
- X. Cao, H. Yu, C. Chen, J. Wei and P. Wang, Biotechnol. Lett., 2015, 37, 1347–1354 CrossRef CAS PubMed.
- M. Xie, H. Shi, Z. Li, H. Shen, K. Ma, B. Li, S. Shen and Y. Jin, Colloids Surf., B, 2013, 110, 138–147 CrossRef CAS PubMed.
- Z. Zou, D. He, X. He, K. Wang, X. Yang, Z. Qing and Q. Zhou, Langmuir, 2013, 29, 12804–12810 CrossRef CAS PubMed.
- Z. Luo, K. Cai, Y. Hu, B. Zhang and D. Xu, Adv. Healthcare Mater., 2012, 1, 321–325 CrossRef CAS PubMed.
- L. Huang, K. Tao, J. Liu, C. Qi, L. Xu, P. Chang, J. Gao, X. Shuai, G. Wang and Z. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 6577–6585 CrossRef CAS PubMed.
- W. Feng, W. Nie, C. He, X. Zhou, L. Chen, K. Qiu, W. Wang and Z. Yin, ACS Appl. Mater. Interfaces, 2014, 6, 8447–8460 CrossRef CAS PubMed.
- Q. Zhao, J. Liu, W. Zhu, C. Sun, D. Di, Y. Zhang, P. Wang, Z. Wang and S. Wang, Acta Biomater., 2015, 23, 147–156 CrossRef CAS PubMed.
- J. Jiao, X. Li, S. Zhang, J. Liu, D. Di, Y. Zhang, Q. Zhao and S. Wang, Mater. Sci. Eng., C, 2016, 67, 26–33 CrossRef CAS PubMed.
- W.-H. Chen, G.-F. Luo, W.-X. Qiu, Q. Lei, L.-H. Liu, S.-B. Wang and X.-Z. Zhang, Biomaterials, 2017, 117, 54–65 CrossRef CAS PubMed.
- W. Zhao, J. Hu and W. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 23528–23535 CrossRef CAS PubMed.
- C. Chen, W. Yao, W. Sun, T. Guo, H. Lv, X. Wang, H. Ying, Y. Wang and P. Wang, Int. J. Biol. Macromol., 2019, 122, 1090–1099 CrossRef CAS PubMed.
- W. Cheng, J. Nie, L. Xu, C. Liang, Y. Peng, G. Liu, T. Wang, L. Mei, L. Huang and X. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 18462–18473 CrossRef CAS PubMed.
- J. Zhang, Y. Sun, B. Tian, K. Li, L. Wang, Y. Liang and J. Han, Colloids Surf., B, 2016, 144, 293–302 CrossRef CAS PubMed.
- M. Zhang, J. Liu, Y. Kuang, Q. Li, D.-W. Zheng, Q. Song, H. Chen, X. Chen, Y. Xu and C. Li, Int. J. Biol. Macromol., 2017, 98, 691–700 CrossRef CAS PubMed.
- W. Tao, X. Zeng, J. Wu, X. Zhu, X. Yu, X. Zhang, J. Zhang, G. Liu and L. Mei, Theranostics, 2016, 6, 470 CrossRef CAS PubMed.
- H. Yu, Z. Cui, P. Yu, C. Guo, B. Feng, T. Jiang, S. Wang, Q. Yin, D. Zhong and X. Yang, Adv. Funct. Mater., 2015, 25, 2489–2500 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00032a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |