
Post-synthetic oriented attachment of CsPbBr3 perovskite nanocrystal building blocks: from first principle calculation to experimental demonstration of size and dimensionality (0D/1D/2D)†
Sanghyun
Jeon
 a,
Myung-Chul
Jung
b,
Junhyuk
Ahn
a,
Ho Kun
Woo
a,
Junsung
Bang
a,
Donggyu
Kim
c,
Sang Yeop
Lee
a,
Ho Young
Woo
d,
Jongchul
Jeon
e,
Myung Joon
Han
b,
Taejong
Paik
*d and
Soong Ju
Oh
*a
a,
Myung-Chul
Jung
b,
Junhyuk
Ahn
a,
Ho Kun
Woo
a,
Junsung
Bang
a,
Donggyu
Kim
c,
Sang Yeop
Lee
a,
Ho Young
Woo
d,
Jongchul
Jeon
e,
Myung Joon
Han
b,
Taejong
Paik
*d and
Soong Ju
Oh
*a
aDepartment of Materials Science and Engineering, Korea University, 145, Anam-ro Seongbuk-gu, Seoul, 02841, Republic of Korea. E-mail: sjoh1982@korea.ac.kr
bDepartment of Physics, Korea Advanced Institute of Science and Technology, 291, Daehak-ro, Yuseong-gu, 34141, Korea
cDepartment of Semiconductor Systems Engineering, Korea University, 145, Anam-ro Seongbuk-gu, Seoul, 02841, Republic of Korea
dDepartment of Integrative Engineering, Chung-Ang University, Seoul 06974, Republic of Korea. E-mail: paiktae@cau.ac.kr
eKorea Institute of Rare Metals, Korea Institute of Industrial Technology, Incheon, Republic of Korea
First published on 8th April 2020
Abstract
Post-synthesis engineering methods that employ oriented attachment to precisely control the size and dimensionality (0D/1D/2D) of as-synthesized CsPbBr3 nanocrystals (NCs) are demonstrated. We investigated the chemical effects of the properties of polar solvents, including their immiscibility, polarity, and boiling point, on the surfaces of NCs, as well as their effect on the structural and optical properties of NCs. Appropriate exploitation of the solvent properties made it possible to use a polar solvent to mildly affect the NCs indirectly such that they discarded their ligands and became attached to proximal NCs without being destroyed. Based on our observations, we developed a method whereby a solution of the NCs in a non-polar solvent is mixed with a polar solvent to form an immiscible phase to induce epitaxial growth of CsPbBr3 NCs. The method enables the size of NCs to be easily regulated from 5 to 50 nm by controlling the engineering time. Taking advantage of the minimal effect of a mild solvent, we also developed a self-assembly method that operates at the liquid–air interface to systematically control the dimensionality. At this interface, the NCs self-assemble in the horizontal direction and grow into micron-sized, single-crystalline, defect-free nanowires (1D) and nanoplates (2D) via oriented attachment. Finally, we discuss the origin of the non-destructive oriented attachment phenomenon and the surface chemistry of a perovskite NC using density functional theory (DFT) simulations and a proposed model system.
New conceptsRecently, all-inorganic lead halide perovskite has been attracting huge attention due to their outstanding optical properties. However, controlling size, dimension, and surface states of nano-sized halide perovskite materials remain as a great challenge, due to their unstable structures and the lack of the understanding in their surface states. This greatly inhibits the their practical applications. Here, we for the first time demonstrate that lead halide perovskite nanoparticles can be epitaxially grown and transformed into size-controlled 0-D nanocrystals, 1-D nanowire, and 2-D nanosheets through oriented attachment. This was done by investigating the effect of solvents on the ligands and surfaces of perovskite nanocrystals and developing the epitaxial and selective growth of nanocrystals. We conducted a first-principle study of surface chemistry and origin of oriented attachment by using density functional theory (DFT) calculation. We for the first time established a model system to explain the origin of the post-synthetic growth of NC and epitaxial growth. We believe that our study would make a significant contribution in nanomaterials and perovskite community as it provides a new method to effectively modulate nanocrystal surfaces and fundamental insight into the surface chemistry of nanomaterials. |
Introduction
Lead halide perovskites have attracted significant attention owing to their remarkable optical and electronic properties, such as the high quantum yield, narrow emission bandwidth, low trap density, and optical band gaps that are tunable by adjusting the compositions of the halide ions.1–7 These desirable properties offer unique opportunities for the application of lead halide perovskites to optoelectronic devices, including light emitting diodes (LEDs), lasing, photovoltaics, and photodetectors.2,7–12 Thus far, the performance of optoelectronic devices has been significantly improved by using organic–inorganic hybrid perovskites (CH3NH3PbX3, X = Cl, Br, and I). The power conversion efficiencies of perovskite-based photovoltaics have reached 24.2%,13 and the external quantum efficiencies (EQE) of PeLEDs have reached 22%,14,15 demonstrating the promising future of PeLEDs in optoelectronic technologies.However, the limited stability induced by the volatile organic components of hybrid perovskites has restricted the commercialization of these hybrid perovskites in many ways. This led to the emergence of the comparatively more stable all-inorganic perovskites, such as cesium lead halide perovskites (CsPbX3, X = Cl, Br, and I), as new promising candidates for optoelectronic devices.16–18 Despite the greater stability of all-inorganic perovskites, their optical properties are comparable to those of hybrid perovskites. Owing to the great potential of all-inorganic perovskites, numerous researchers have worked with these materials and attempted to further enhance their stability by modulating their surfaces and also to fabricate high-performance devices for various applications.12,18–23 Nanosized CsPbX3 crystals with different sizes and dimensionalities (0D/1D/2D) have been widely studied in many applications for their unique optical and electronic properties that depend on size and dimensionality. CsPbX3 nanocrystals (NCs), the sizes of which are smaller than the exciton Bohr radius, exhibit the quantum confinement effect. In this region, the carriers are squeezed into the physical size of the NCs, which causes the band gap to increase as the size of the NCs decreases.24–26 Further, the dimensionality of the perovskite NCs influences the band structures of CsPbX3, which significantly affects the optical and electronic properties.27–29 For example, Ying et al. reported a high-performance photodetector with high stability by exploiting the advantageous properties of the vapor-processed micrometer-scale CsPbBr3 platelet structure.30 Because the size and dimensionality are critical factors for tailoring the optical and electronic properties of NCs, it is important to develop a facile and controllable method to modulate the size and dimensionality of the perovskite NCs.
Post-synthetic chemical modifications of NCs have emerged as promising methods to control the optical and electronic properties of NCs. For example, anion, cation, and ligand exchange methods, phase transformation methods, and oriented attachment methods have been extensively developed to control the composition, shape, size, and dimensionality.31–43 Among these methods, those involving oriented attachment based on self-assembly, such as assembly at the liquid–air interface, is one of the most promising approaches to fabricate large-area, ordered NC-based superlattice structures or to control the size and dimensions of NC building blocks. Dipole–dipole interaction or the energies of different facets can induce the epitaxial fusion of assembled nanomaterials, thereby transforming 0D nanoparticles into 1D nanorods and nanowires (NWs) or 2D nanosheets.30,35–37,42,43 These modification methods provide a fundamental understanding of the kinetics, thermodynamics, and surface science of nanomaterials, and simultaneously offer technological advantages for various device applications.
Despite the benefits of these methods, post-synthetic approaches involving self-assembly-based oriented attachment to control the size, dimensionality, and physical properties of inorganic perovskite NCs have rarely been reported. A major disadvantage of these chemical methods is that they generally require the use of a polar solvent.28,31,39 However, perovskite NCs are vulnerable to polar solvents, as they are readily ionized or degraded and are decomposed and their structures destroyed.4,5 Even a small amount of polar solvent is able to remove ligands from the surface of NCs, leading to random and uncontrollable agglomeration of the NCs. Consequently, they lose their colloidal stability and all their desired properties. This limitation deprives of the opportunity to explore the post-synthetic chemical processing of perovskite NCs and control their size and dimensionality. Only few examples of oriented attachments based on reaction at the water interface, exposure to an electron beam, or light-induced reaction have been reported.44–46 These attempts require further development because most of them either failed to control the size and dimensionality precisely or the structures readily changed from cubic to orthorhombic phases during transformation, thus altering their optical and structural properties.
In this paper, we first introduce post-synthesis modification to tailor both the size and dimensionality of CsPbBr3 nanomaterials via the oriented attachment of NCs based on self-assembly induced by a rationally designed chemical process (Scheme 1). We systematically investigated the relationship between the properties of various polar solvents including their immiscibility, polarity, boiling point, and the chemical effects on chemically vulnerable CsPbBr3 NCs. Based on these results, we elucidated the origin of the chemical effects of each polar solvent on the destruction, degradation, and attachment of CsPbBr3 NCs. Two different strategies were developed to systematically control the size and dimensionality of CsPbBr3 NCs without destruction. First, we developed an immiscible solution phase mixing method (ISPM). Vigorous agitation of a combination of two immiscible solvents allows the polar solvent to mildly influence the NCs in the non-polar solvent. This approach enabled us to carefully modulate the ligands and atoms on the surfaces of the CsPbBr3 NCs without severe destruction of the crystals. In addition, the NCs could be attached to proximal NCs and systematically grown into larger NCs while preserving the highly crystalline structure. Second, a strategy involving self-assembly at the liquid–air interface was adopted to induce the post-synthetic growth of NCs in the confined dimension at the interface. This method was subsequently used to successfully fabricate highly crystalline nanowires (1D) and nanoplates (2D) via self-assembly followed by attachment of the NCs to each other at the interface. We conducted a first-principles study of the surface chemistry and origin of the oriented attachment of perovskite using Vienna ab initio simulation software. We employed density functional theory (DFT) to compute the surface energy of each surface and the overall change in energy during oriented attachment. Based on the DFT result, we proposed a model system to explain the origin of the post-synthetic epitaxial growth of the NCs. Our strategies enable the size and dimensionality of CsPbBr3 NCs to be finely controlled by taking advantage of the chemical effects of an immiscible polar solvent on the surface of as-synthesized NCs. Furthermore, we expect our discovery to not only boost the development of various applications of CsPbB3 but also to facilitate an understanding of the post-growth mechanism and surface chemistry of other perovskite materials.
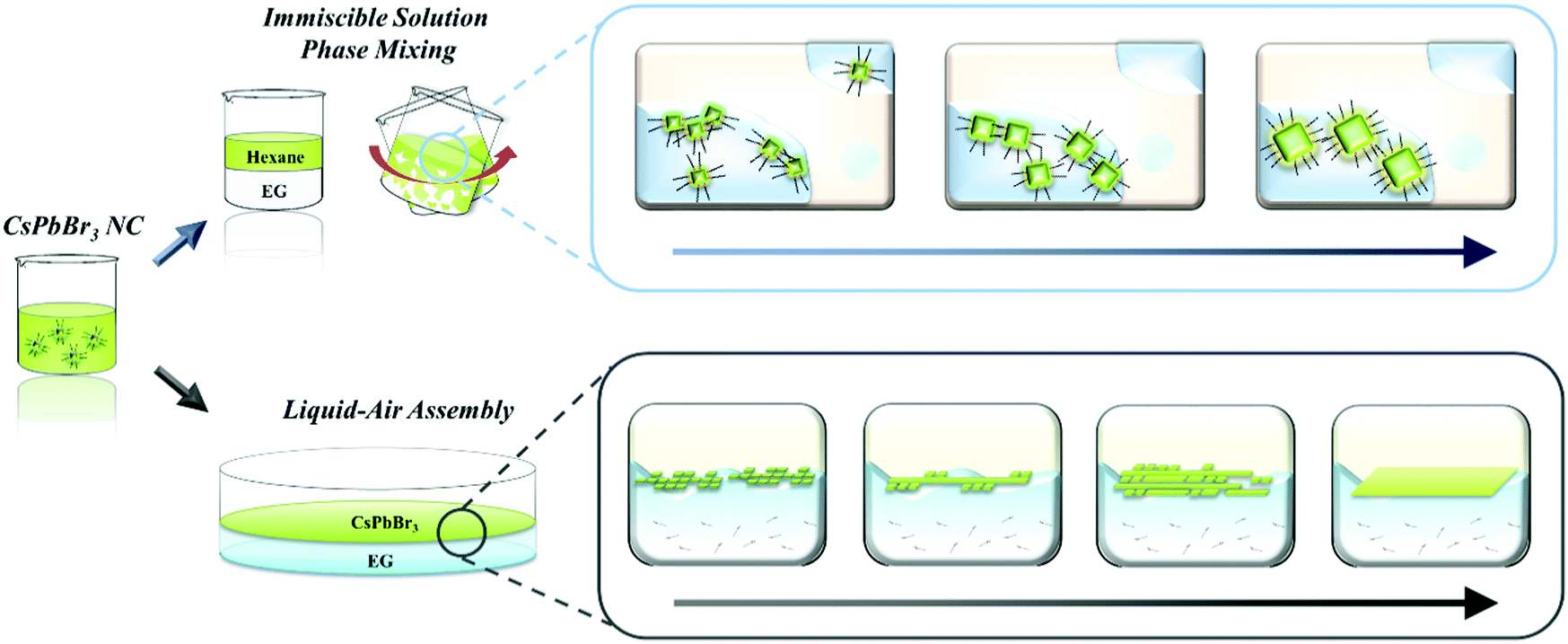 | ||
| Scheme 1 Schematic illustration of the immiscible solution phase mixing (ISPM) and self-assembly at the liquid–air interface methods to control the size and dimensionality of CsPbBr3 NCs, respectively. | ||
Results and discussion
CsPbBr3 NCs were synthesized via ligand-assisted re-precipitation at room temperature and ambient pressure. The as-synthesized NCs were dispersed in hexane.47 The transmission electron microscopy (TEM) image shows that CsPbBr3 NCs are 7 ± 1 nm in size (Fig. 1a). Unlike typical quantum dots that have spherical or pseudo-spherical shapes (e.g., truncated octahedral) to minimize the surface energy, as-synthesized CsPbBr3 NCs have a hexahedral structure that exposes the (100) plane in all directions. This is because the (100) plane is thermodynamically the most stable plane with the lowest surface energy.10,41 Owing to this unique characteristic, appropriate control of the surface ligands and atoms was expected to allow the NCs to bond together to form a highly crystalline structure.44 However, as discussed, CsPbBr3 is readily decomposed and destroyed when exposed to a polar solvent. This led us to systematically investigate the chemical effects of polar solvents on CsPbBr3 NCs. Specifically, we conducted a systematic study to examine the extent to which different solvents with different polarities, boiling points, and immiscibility affect the destruction and surface reconstruction of CsPbBr3 NCs. Five different polar solvents that contained the same hydroxyl functional group, i.e., isopropanol (IPA), ethanol (EtOH), methanol (MeOH), ethylene glycol (EG), and glycerol (Gly), were used in this study in the order of increasing polarity and boiling point (polarity/boiling point of IPA, EtOH, MeOH, EG, Gly: 0.617/97 °C, 0.654/78.5 °C, 0.762/64.6 °C, 0.790/197 °C, 0.812/290 °C). In this group, IPA and EtOH are miscible with a solution of CsPbBr3 NCs in hexane, whereas MeOH, EG, and Gly are immiscible with the NC solution.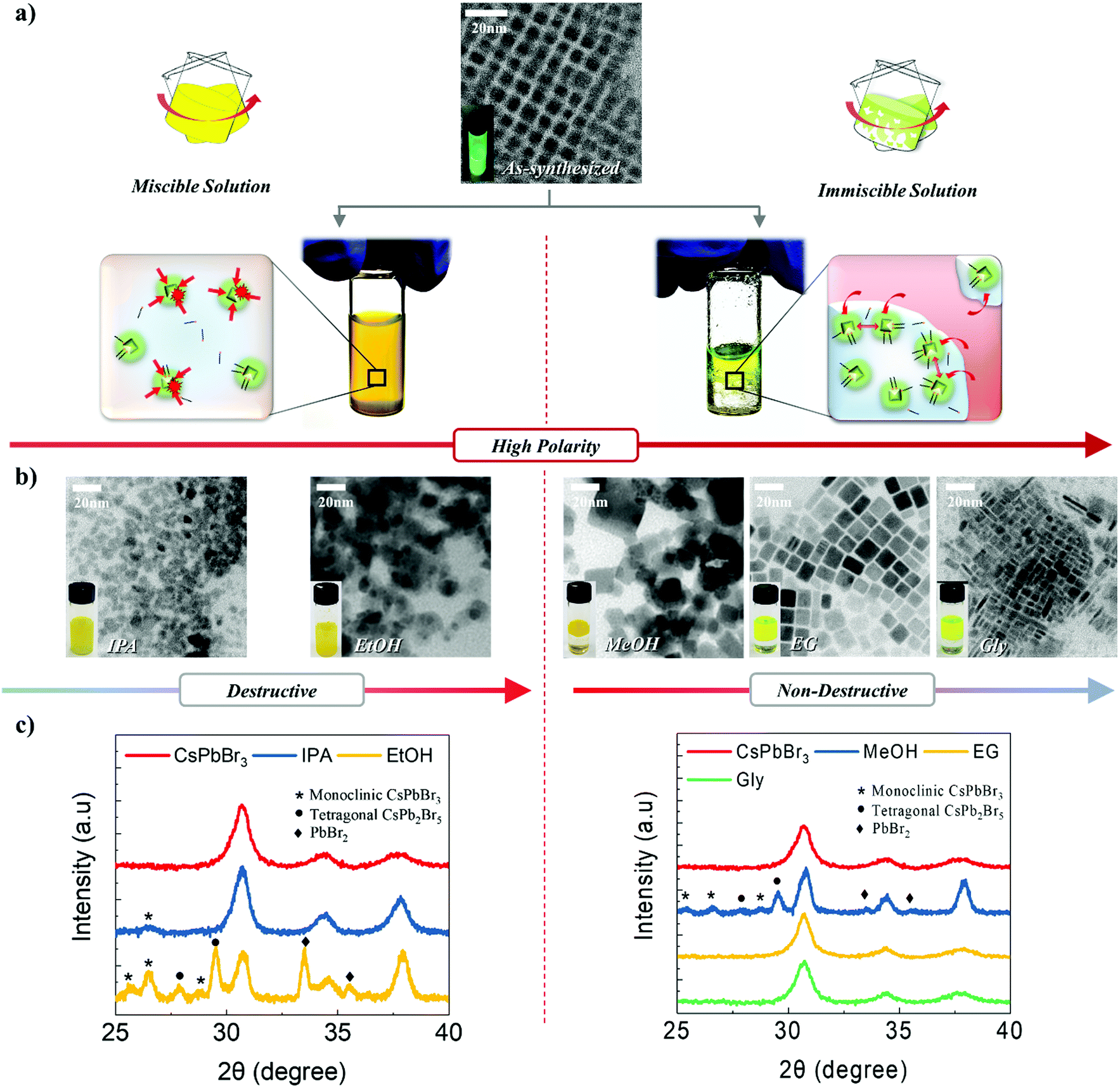 | ||
| Fig. 1 (a) TEM image of as-synthesized CsPbBr3 and schematic illustration of the direct and indirect effects of polar solvents on CsPbBr3 NCs during both miscible (left) and immiscible (right) solution phase mixing methods. (b) TEM images of CsPbBr3 solution exposed to different miscible (left) and immiscible (right) polar solvents. (c) XRD pattern of CsPbBr3 NCs exposed to different miscible (left) and immiscible (right) polar solvents. | ||
To investigate the effects of the polar solvents on the CsPbBr3 NCs, we conducted TEM measurements after exposing the NC solution to various polar solvents (Fig. 1b). When IPA and EtOH were mixed with the NC solution, the shape of the CsPbBr3 NCs became irregular and agglomeration of the structures appeared to occur. The addition of MeOH to the NC solution maintained the cubic shape of the NCs, although a broad distribution of sizes and shapes was subsequently observed. The addition of EG to the NC solution led to the clear observation of an increase in the size of the NCs; at the same time, the monodispersity and shape uniformity of the crystalline CsPbBr3 NCs was retained. A similar effect was observed when Gly was used as the polar solvent, in which case the acquired TEM images revealed the growth of the CsPbBr3 NCs into nanorods.
To further investigate the effect of solvents, we conducted X-ray diffraction (XRD) analysis (Fig. 1c). Untreated CsPbBr3 NCs show an intense single peak at 30.5°, attributed to the cubic CsPbBr3 (200) plane. The fact that this peak has the highest intensity represents that the (100) plane is dominant, as every surface in the (100) direction is exposed. Using the full width at half maximum (FWHM) of this peak, the crystallite size was calculated to be 7 nm, which was in good agreement with the TEM data (Fig. 1a). However, when the solution of NCs was exposed to IPA, a weak diffraction peak corresponding to a monoclinic CsPbBr3 structure appeared, and the FWHM value decreased. This result shows that the CsPbBr3 structure was partially destroyed and agglomerated upon exposure to IPA. With EtOH, intense peaks associated with monoclinic CsPbBr3, tetragonal CsPb2Br5, and PbBr2 appear, and the FWHM decreases significantly. This suggests that the CsPbBr3 NCs were significantly destroyed and ionized, and that significant agglomeration between NCs occurred. Upon exposure to MeOH, monoclinic CsPbBr3, tetragonal CsPb2Br5, and PbBr2 peaks appeared and the FWHM decreased; however, the amount of destruction was less than that caused by EtOH. When the NC solution was mixed with EG, the XRD data showed that the NCs almost have the same shape as that observed for as-synthesized NCs, except for a slight decrease in the FWHM value. This indicated that EG could be potentially utilized for modulating the size of CsPbBr3 NCs without destroying the crystals. The NCs that were exposed to Gly underwent negligible changes in their XRD patterns and FWHM. All of these results correspond exactly to the TEM data.
With regard to the miscible solvents, as their polarity increased, the degree of destruction and ionization of the NCs increased. Likewise, the amount of agglomeration due to the loss of surface ligands on the NCs increased. This demonstrates that an increase in the polarity enhances the effect of the solvent. This is in good agreement with the above-mentioned characteristics of CsPbBr3.5 In contrast, in the case of the immiscible solvents, an increase in the polarity or boiling point either destroyed the NCs or restricted their growth. This is because a high polarity and high boiling point increase the immiscibility of two solvents, in which case the effects of the solvents on the NCs would be reduced. As the solvents become increasingly immiscible, it becomes much more difficult for the molecules from the polar solvent to come into direct contact with and affect the NCs dissolved in the non-polar solvent. This is because the NCs spend most of their time in the non-polar solvent, with limited exposure time to the polar solvent. Only at the interface between the two solvents is a small portion of the NCs exposed to the polar solvent; however, only a facet or plane is exposed to a limited extent. Resultantly, this approach enables the effects of polar solvents to be minimized (more details are provided in Fig. S1–S3, ESI,† and the supporting discussions).
As the properties of nanomaterials are size-dependent,48–52 precise control of the size of the NCs is highly important. As described above, vigorously mixing the CsPbBr3 solution with EG, which has an appropriate polarity and boiling point, enabled the NCs to be epitaxially grown without destroying their structures. We named this method, which involves post-synthetic treatment and which induces NC growth, the “immiscible solution phase mixing method” (ISPM) (Fig. 2a). We approached the design of the method to control the NC size systematically by regulating the ISPM time. The entire process of controlling the size of NC assemblages is illustrated in Fig. 2b. TEM was used to investigate the effect of the duration of ISPM on the size, size distribution, and structural properties of the CsPbBr3 NCs (Fig. 2c). After 10 min of ISPM using EG, the size of the CsPbBr3 NCs increased to 14 ± 4 nm, which is approximately twice the size of the original NCs, and the morphology was hexahedral. After 30 min of mixing, the size further increased to 30 ± 10 nm. Neither the crystal structures nor the shapes of the NCs were destroyed in these two experiments and the narrow size distribution was maintained. The enlarged NCs had a highly crystalline structure with a lattice parameter of 0.58 nm, which corresponds to that of the cubic phase of CsPbBr3.53
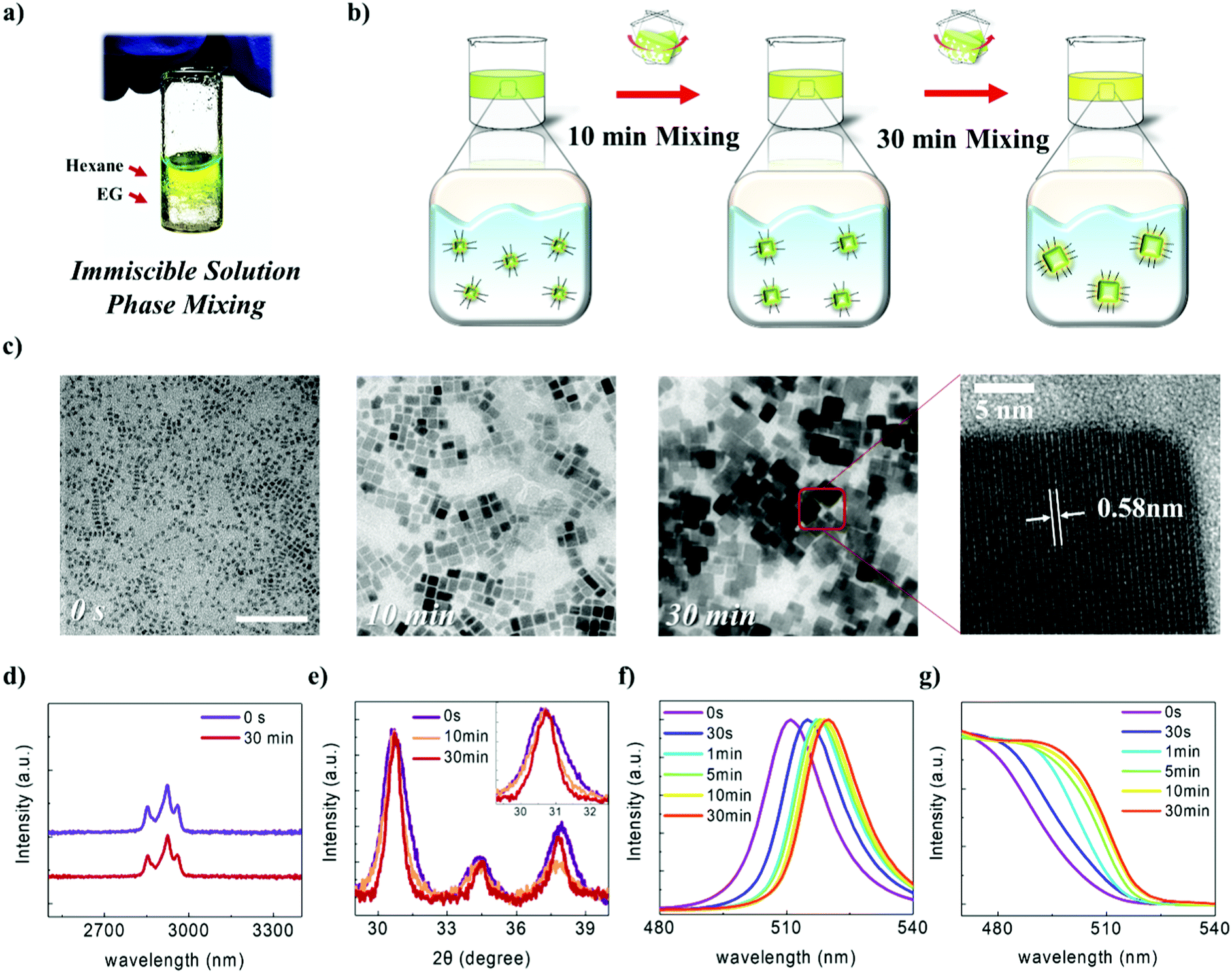 | ||
| Fig. 2 (a) Schematic of ISPM. (b) Schematic illustration of the size control of CsPbBr3 QD by using ISPM. (c) HR-TEM images of as-synthesized CsPbBr3 NCs after 10 min and after 30 min of mixing (scale bar = 100 nm). (d) FTIR spectra of CsPbBr3 NCs before (red) and after (purple) 30 min of exposure. (e) Multiple XRD spectra of CsPbBr3 and (f) PL intensity and (g) multiple UV-Vis spectra of CsPbBr3 after exposure to EG for differing periods of time. | ||
The surface chemistry of the NCs was examined using Fourier-transform infrared spectroscopy (FTIR) analysis (Fig. 2d). The as-synthesized CsPbBr3 NCs had a strong C–H stretching vibration band in the region 2800–3000 cm−1, which is consistent with the fact that the NCs are surrounded by hydrocarbon ligands. After exposure to EG, the intense peak in the 2800–3000 cm−1 region was retained, indicating that the organic ligands were still attached to the NC surface and that the NCs maintained their colloidal stability after their ISPM-induced growth.
XRD was used to quantitatively investigate the effects of the polar solvents on the structural properties (Fig. 2e). The as-synthesized NCs exhibited a single intense peak at 30.5°, which was attributed to the cubic CsPbBr3 (200) plane. The position of this peak or its relative intensity did not change after exposure to the polar solvent. Furthermore, peaks associated with decomposed or degraded materials, such as monoclinic CsPbBr3, tetragonal CsPb2Br5, or PbBr2, were not observed. The FWHM value of this peak, which gradually decreased, was used to calculate the crystallite size using the Scherrer equation. The calculated values were 7.93, 13.34, and 18.28 nm for the as-synthesized, 10 min, and 30 min processed CsPbBr3 NCs, respectively. Although the NC sizes obtained by XRD and TEM analysis differed slightly, the growth of NCs after ISPM is clearly observed in both measurements. Further, the unchanged intensity of the peak representing the (200) plane might suggest that the larger NCs that were fabricated also expose their (100) planes in all directions, much as the as-synthesized NCs.
The dependence of the optical properties on the processing time was additionally investigated using photoluminescence (PL) and ultraviolet-visible spectroscopy (UV-Vis) (Fig. 2f and g). The as-synthesized CsPbBr3 showed an intense PL peak at 511 nm with a FWHM of 18 nm, with the small FWHM value reflecting the small size distribution of the synthesized NCs. As the processing time increased, the PL peak gradually underwent red shift from 511 nm to 520 nm, and the FWHM decreased to 15 nm. These results can be interpreted as a reduction in the quantum confinement effect as the NC size increased. The Bohr exciton radius of CsPbBr3 is as high as 7–8 nm;26 as the NC size increased, the confinement effect began to weaken. Finally, the band gap changes to the fixed values of bulk materials when the effect vanishes entirely. Furthermore, the FWHM of the NCs is strongly affected by the quantum confinement effect. Therefore, as the size of the NCs exceeded that of the strong confinement region, the FWHM was narrower than that of small-sized NCs. This trend was also observed in the UV-Vis measurements. The CsPbBr3 NCs also showed strong UV-Vis absorbance with the onset wavelength near 511 nm, which corresponds to the band gap of CsPbBr3 NCs. The UV-Vis spectra exhibit red shift and saturation tendency as the processing time increases, which is in good agreement with the PL analysis. All these experimental results indicate that the size of the NCs is successfully controlled via ISPM without any structural degradation of the NCs.
Not only the size, but also the dimensionality (0D/1D/2D) is a highly important factor that determines the properties of nanomaterials.27–29 To control the NC dimensionality, we adopted self-assembly at the liquid–air interface in combination with our finding that EG can mildly detach the surface atoms and ligands of CsPbBr3. Nanowires (1D) and nanoplates (2D) were successfully fabricated via self-assembly at the liquid–air interface. The entire process whereby 1D and 2D assemblages are constructed is shown in Fig. 3a. A solution of CsPbBr3 NCs in hexane was added dropwise to the EG and formed a thin layer on the surface of the EG because of the difference in polarity between the two solvents. After the hexane evaporated, the NCs started to align because of their cubic-like shape and narrow size distribution (Fig. 3b). With time, the surface ligands gradually became detached because of the effects of EG. This caused the CsPbBr3 NCs to start combining. After 1 h, nanowires were formed. HR-TEM showed that the nanowires contained no defects, and the lattice parameter of the fabricated nanowires was approximately 0.58 nm, which is consistent with the CsPbBr3 cubic phase. The SAED pattern clearly shows that the structure was not destroyed (Fig. 3c).53
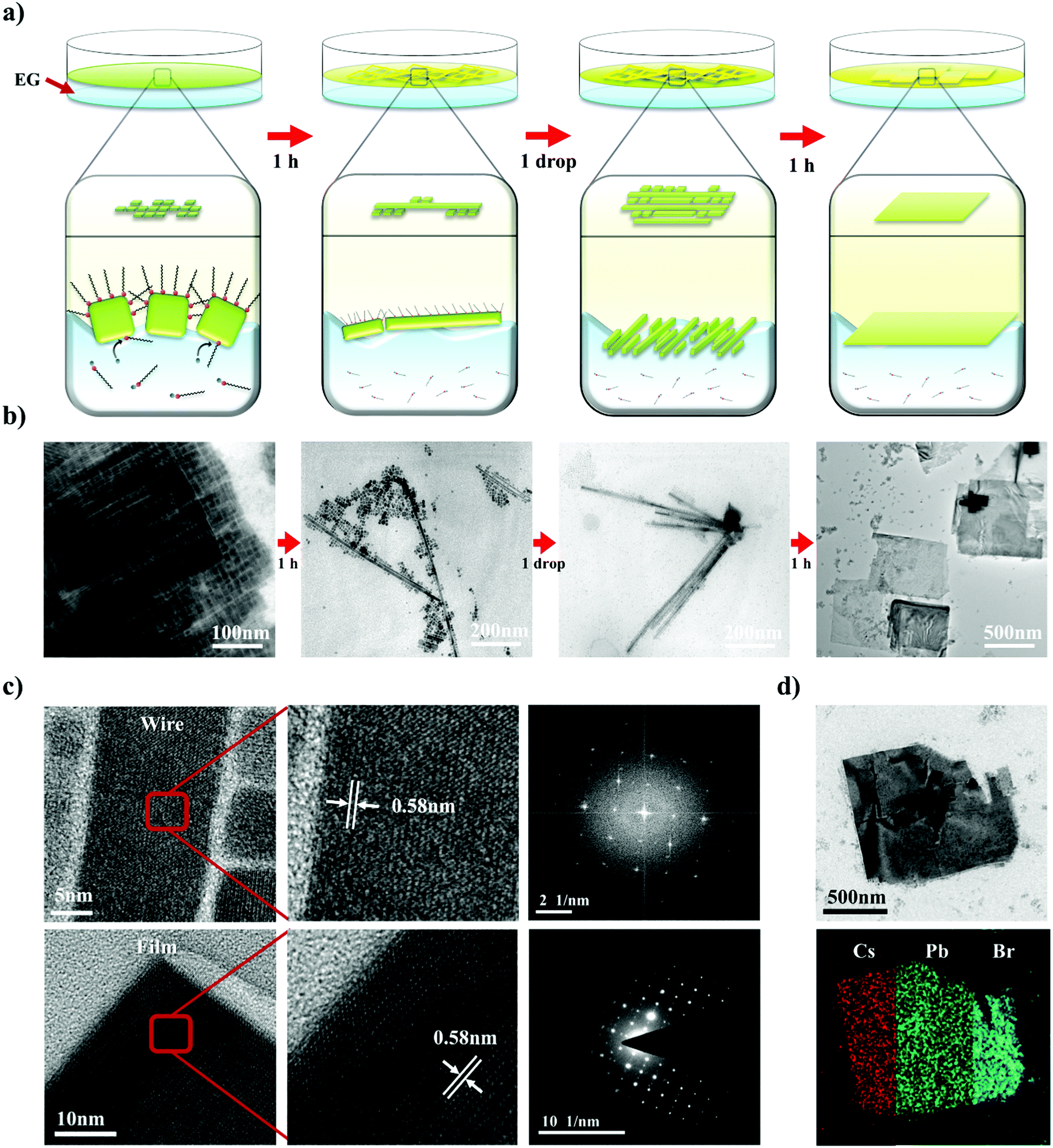 | ||
| Fig. 3 (a) Schematic illustration showing the formation of 1D and 2D structures via self-assembly at the liquid–air interface. (b) TEM images of each step shown in (a). (c) HR-TEM images and SAED pattern of a fabricated nanowire (upper row) and film (lower row). (d) EDX mapping of fabricated CsPbBr3 film. | ||
2D nanoplates were fabricated by slightly modified methods. A drop of a solution of NCs was added to the liquid interface, and a second drop was added after 1 h. One hour after addition of the first drop, numerous nanowires formed on the surface. However, the distance between the different nanowires was too large to enable the nanowires to combine with each other. Therefore, the second drop of CsPbBr3 solution was added to the EG. This time, the newly added NCs spontaneously aligned beside the formed nanowires and formed other nanowires next to them. One hour later, the adjacent NWs and NCs had combined into thin films. The properties of the fabricated thin films were investigated by HR-TEM and energy dispersive X-ray spectroscopy (EDX) mapping analysis (Fig. 3c). The thin films also had a highly crystalline structure with a lattice parameter of 0.58 nm, and the well-defined SAED patterns show that the structure was well preserved. Furthermore, the EDX analysis showed that the thin films were composed of Cs (red), Pb (green), and Br (blue) components, demonstrating that the thin films are CsPbBr3, rather than decomposed PbBr2 or CsBr clusters (Fig. 3d). Atomic force microscopy analysis shows that the thickness of 2D structure is 7 ± 1 nm, similar to the size of as-synthesized NCs (Fig. S4, ESI†). It indicates that fabricated nanoplatelets were composed of a single layer of NCs. These results confirmed that exploitation of the properties of solvents and the use of an alternative approach enabled 1D nanowires and 2D nanoplates of CsPbBr3 with highly crystalline structures to be successfully fabricated.
The effects of epitaxial growth on the photoluminescence quantum yield (PLQY), which is an important evaluation index for perovskite materials, were evaluated (Fig. S5, ESI†). An as-synthesized NC, a 1D nanowire, and a 2D nanoplate had PLQYs of 98.57%, 27.52%, and 12.33%, respectively, which shows a decreasing tendency. This can be understood by considering that, as the dimension increases, the probability of electron and hole recombination decreases because the confining effects weaken. This suggests that these materials would be suitable for optoelectronic devices that require efficient carrier transport, rather than those that rely on carrier recombination.
TEM analysis was also used to determine the origin of the epitaxial growth mechanism induced by ISPM and the dimensional transformation that occurred during self-assembly at the liquid–air interface. The effect of other immiscible solvents and the structure of the transition states were investigated to elucidate the growth mechanism induced by ISPM. Fig. 2c and 4a show that not only EG but also other immiscible solvents such as MeOH, Diethylene glycol (DEG), and Gly induced the NCs to grow into highly crystalline cubic or cuboid structures with a 0.58 nm lattice parameter (Fig. 4a). The growth mechanism was also studied by fabricating the transition states, which were monitored by intentionally modulating the ISPM time (Fig. 4b). These experiments showed that the transition states had a 90° inner edge. Furthermore, the size and shape of the empty spaces exactly fit those of pristine NCs, indicating that the fabricated nanostructures are formed by the combination of three or five pristine nanocrystals. The transition states in the liquid–air interface self-assembly process were also fabricated by modulating the processing time (Fig. 4c). These structures exhibited shapes that appeared to have chopsticks attached to them with 90° edges, which resembled the combination of fabricated nanowires. Additional TEM images of these transition states are shown in Fig. S6 (ESI†).
 | ||
| Fig. 4 (a) HR-TEM images of nanostructures fabricated via ISPM using MeOH, DEG, and Gly. (b and c) Transition states during the formation of fabricated nanocrystals and nanoplates. (d) Schematic of CsPbBr3 NCs surface conjugation. | ||
Taking all the data into account, the epitaxial growth phenomenon can be explained by the oriented attachment of pristine NCs rather than Ostwald ripening (Fig. 4d). Even though these growth mechanisms have similarities, in that they explain the growth of small particles into a larger crystal, the two principles differ fundamentally. Growth because of Ostwald ripening is mainly based on the migration of atoms between particles, whereas oriented attachment entails the self-organization of particles into a single crystal by sharing a common crystallographic orientation.54–58 Therefore, the formation of a 90° inner edge is typically associated with the oriented attachment process rather than Ostwald ripening where the NC grows to take on a shape with minimum surface energy such as a spherical or rectangular shape.59 The driving force behind oriented attachment is the strong coulombic interaction between two adjacent NCs. This interaction is generated when ligands are detached in the form of Cs-oleate, Pb-oleate, and oleylammonium bromide to expose specific surfaces of the NCs.4,5,60,62 In the case of ISPM, this exposure occurs in all directions because we vigorously agitated the solution and the NCs were forced to undergo Brownian motion. Therefore, oriented attachment could proceed in all directions, resulting in larger NCs. In the case of self-assembly at the liquid–air interface, exposure is confined to the bottom of the NCs, such that they are able to transform into 1D and 2D nanomaterials. Although this work focused on the type of solvent, it should be noted the type of ligand would also affect the growth behavior of the NCs, as the binding energies and preferential binding sites of the ligands would differ.
To further elucidate the proposed model system and to deepen our understanding of the surface chemistry of the perovskite NCs, we carried out surface energy calculations based on first-principles DFT. Evaluation of the preferential surface states under certain conditions and presumption of the ligand-binding dynamic was accomplished by specifying the unit cell of CsPbBr3 to enable us to compare the energy of each surface plane. For the (100) surface, we considered two different terminations, namely, CsBr rich (Fig. 5a), and PbBr2 rich (Fig. 5b). Similarly, the CsPbBr rich (Fig. 5c) and Br2 rich (Fig. 5d) terminations were calculated to simulate the (110) surface of CsPbBr3. The surface energy is expressed as follows.59,61
| Esurf = (Eslab − nEbulk)/2S |
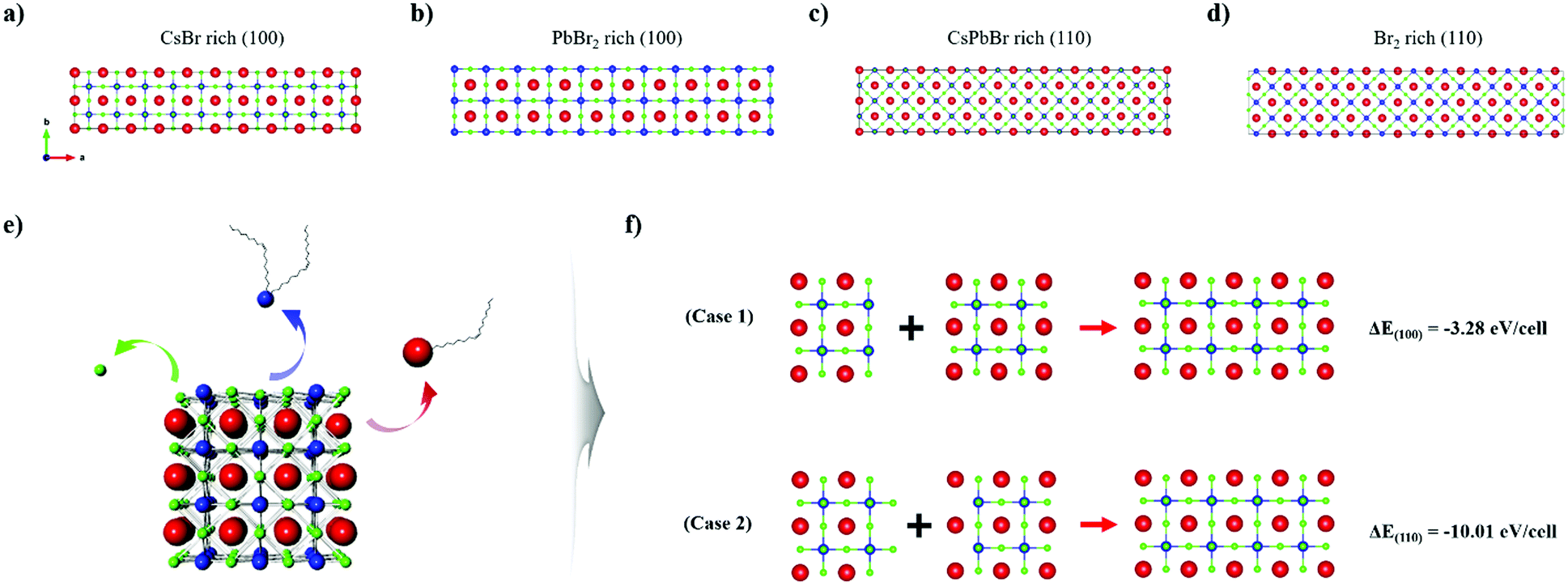 | ||
| Fig. 5 Unit cell structures for surface energy calculations of (a) CsBr rich (100), (b) PbBr2 rich (100), and (c) CsPbBr rich (110), and (d) Br2 rich surfaces. Red, blue, and green spheres represent Cs, Pb, and Br atoms, respectively. (e) Schematic of ligand detaching model system. (f) Calculated structures to simulate the attachment of two NCs via the (100) plane (case 1) and (110) plane (case 2). | ||
On the basis of these calculations we concluded that both the as-synthesized NCs and the nanomaterials fabricated via ISPM or self-assembly are all constructed to expose the (100) surface, and both CsBr rich and PbBr2 rich planes coexist, which is advantageous for and favors oriented attachment. On the surface of an NC, the ligand binding strength would be much lower than that of other planes with higher surface energy, such as the (110) plane. Therefore, a ligand attached to the (100) plane would be more easily detached if harsh extraneous conditions, such as solvent processing, were to be applied. Therefore, the (100) plane would be preferentially exposed. Moreover, removal of an atom from the surface by way of etching would cause the surface to reconstruct itself such that the (100) plane is exposed owing to its low energy. Consequently, we concluded that the oriented attachment process in perovskite NCs would dominantly occur via the exposed (100) plane. In fact, attachment via the (100) plane was directly observed with the high-energy electron beam of HR-TEM, which could likewise eliminate a ligand. The beam was applied to two adjacent NCs, thereby proving our assumption (Fig. S5, ESI†).
The total energy was also calculated using DFT to identify the origin of the oriented attachment mechanism and the formation of nanowires. Before constructing the DFT model system, we established a few situations in which surface atoms could be removed via etching (Fig. 5e). Cations in the NC can be readily removed in this way by detaching surface ligands. In particular, Cs atoms on the surface can be removed in the form of Cs-oleate, and Pb can be removed in the form of Pb-oleate. In the case of halide ions, they are highly dynamic,60,61 and could either readily depart or could be eliminated in the form of oleylammonium halide.36,62 As the number of surface states is infinite, we selected the two most plausible situations in which the surface is terminated along the (100) or along the (110) plane. This is because the contrary situation would require excessively high formation energy, which would not be able to prolong the state for sufficient time. We computed ΔEfacet = E4×2×2 − (E2×2×2 + E2×2×2′), which represents the total energy difference per atom between the 4 × 2 × 2 cluster and the sum of two separated clusters of 2 × 2 × 2, where each cluster exposed a different plane of a specific facet (Fig. 5f). For example, ΔE(100) represents the total energy differences between the 4 × 2 × 2 cluster and the sum of the energy of the PbBr2 rich (100) surface exposed 2 × 2 × 2 cluster and the CsBr rich (100) surface exposed 2 × 2 × 2 cluster (each cluster is surrounded by a vacuum layer on all six facets). The computed value of ΔE(100) was –50.81 meV per atom and that of ΔE(100) was –155.29 meV per atom. The values of both energy differences were negative, which implies that the attachment phenomenon is thermodynamically stable and would occur spontaneously, which is in good agreement with our experimental results. The higher value of ΔE(100) in comparison to that of ΔE(100) is derived from the significantly higher energy state of the (110) plane.
The formation of the nanowires could be understood in terms of the total energy of the system. The attachment of two NCs to each other and continued growth along the direction of initial attachment would reduce the total energy, as this would circumvent the additional edge state that generally requires high energy to form. The calculation shows that the fabrication of the nanowire itself could reduce the total energy of the system and could occur spontaneously. Details are provided in Fig. S8 (ESI†) and the discussion.
Conclusions
This study led to the proposal of new methods to precisely control the size and dimensionality of CsPbBr3via post-synthesis exposure to a polar solvent. Cube-like shaped CsPbBr3 NCs with narrow size distributions were used as building blocks. To trigger their assembly, a new method involving processing at the surface was developed by exploiting the interface between immiscible solvents. Furthermore, the chemical effect of the solvent properties on the NCs was elucidated by establishing a model system based on DFT calculation. It was possible to appropriately modify the CsPbBr3 NCs without any destruction, as our method reduces the aggressive action of polar solvents on the NCs. Consequently, as-synthesized NCs could be grown into larger sized NCs or 1D or 2D structures with highly crystalline architectures depending on the direction of interaction. We consider this study to be of significance, as it suggests a new way to modulate NC surfaces and provides fundamental insight into the surface chemistry of nanomaterials. Furthermore, our approach offers a new way to fabricate nanomaterials of different sizes and dimensionality post-synthetically and could be utilized in various applications, such as electronics, optoelectronics, and photovoltaics.Materials and methods
Materials
Cesium bromide (99.999%), lead(II) bromide (99.999%), N,N-dimethyl formamide (DMF) (anhydrous, 99.8%), oleic acid (90%), oleylamine (70%), toluene (anhydrous, 99.8%), methyl acetate (anhydrous, 99.5%), MeOH (anhydrous, 99.8%), ethanol (anhydrous, 99.5%), and IPA (anhydrous, 99.5%), di-ethylene glycol (99%), EG (anhydrous, 99.8%), and Gly (99.5%) were purchased from Sigma-Aldrich. All reagents were used without further purification.Synthesis of CsPbBr3 NCs
CsPbBr3 NCs were synthesized using a ligand-assisted re-precipitation method (LSPR) at room temperature. First, a precursor solution was prepared by the reaction of 147.6 mg of PbBr2 and 68.1 mg CsBr with 10 mL of DMF. After complete dissolution, 1 mL of oleic acid and 0.5 mL of oleylamine were added. A 2 mL volume of the precursor solution was injected into 50 mL of toluene under vigorous stirring. After centrifugation at 8000 rpm for one min and after discarding the precipitation, a bright green solution remained. The product was purified using toluene and methyl acetate as the solvent and anti-solvent by centrifugation at 8000 rpm for 3 min. The precipitated CsPbBr3 NCs were dispersed in hexane. All processes were conducted under ambient conditions.Control of size (ISPM)
The same volume of CsPbBr3 hexane solution and EG were mixed and shaken using a Voltax mixer (MIX-25P). After mixing for an appropriate time (depending on the target size), the separated hexane layer was collected.Control of dimensions (self-assembly at the liquid–air interface)
To control the dimensionality, we adopted a method that entails self-assembly at the liquid–air interface. One drop of a 20 mg mL−1 solution of CsPbBr3 in hexane was gently dropped into EG in a Teflon crucible. The crucible was covered with a glass slide, and the hexane was allowed to evaporate in a controlled manner. After 1 h, nanowires were formed via oriented attachment. One additional drop of the CsPbBr3 solution was added to this interface. After another hour, nanoplates formed.Characterization
The structural properties and morphologies of the nanomaterials were examined by XRD (S-4300, Hitachi High Technologies America, Inc.) and TEM (Tecnai G2 F30, FEI, Korea Basic Science Institute). The optical properties were investigated by UV-Vis (Cary 5000, Agilent Technologies) and photoluminescence spectroscopy (FA-356, Thermo). The surface chemistry of the processed CsPbBr3 NCs was analyzed using FTIR (LabRam ARAMIS IR2, Horiba Jobin Yvon) in attenuated total reflection mode.Computational details
Our total energy calculations were conducted with the Vienna ab initio simulation software package (VASP-5.4).63,64 We used the Perdew–Burke–Ernzerhof (PBE) form of the generalized gradient approximation (GGA) for our exchange–correlation energy functional.65 From the volume relaxation of cubic CsPbBr3 with 8 × 8 × 8 k-points and 500 eV energy cutoff, we obtained the lattice parameter of a = 5.989 Å. For the surface energy calculations, we adopted the slab-vacuum model with 10 × 2 × 2 supercells and a vacuum thickness of 15 Å. The surface atomic positions were then optimized, while the inner layers remained fixed. The total energy comparison to simulate nanowire growth was carried out by varying the depth of the vacuum layers while maintaining the same volume for all supercells; a = 26.748 Å and c = 61.992 Å. We adopted an energy cutoff of 500 eV and a Monkhorst–Pack grid with 2 × 6 × 6 k-points in the Brillouin zone. All supercells of CsPbBr3 were generated from the optimized cubic phase.Author contributions
S. Jeon conceived the idea and contributed to most aspects of the work. T. Paik and S. J. Oh supervised the project. M.-C. Jung and M. J. Han carried out DFT calculations. J. Ahn, H. K. Woo, and J. Bang helped to synthesize the material. D. Kim, S. Y. Lee, H. Y. Woo, and J. Jeon assisted with the material characterization. S. Jeon, M.-C. Jung, T. Paik, and S. J. Oh drafted and revised the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the Korea Electric Power Corporation (R17XA05-12), and the Creative Materials Discovery Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2018M3D1A1059001).References
- X. He, Y. Qiu and S. Yang, Adv. Mater., 2017, 29, 1–27 Search PubMed.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- J. Liang, J. Liu and Z. Jin, Sol. RRL, 2017, 1, 1700086–1700109 CrossRef.
- J. Pan, L. N. Quan, Y. Zhao, W. Peng, B. Murali, S. P. Sarmah, M. Yuan, L. Sinatra, N. M. Alyami, J. Liu, E. Yassitepe, Z. Yang, O. Voznyy, R. Comin, M. N. Hedhili, O. F. Mohammed, Z. H. Lu, D. H. Kim, E. H. Sargent and O. M. Bakr, Adv. Mater., 2016, 28, 8718–8725 CrossRef CAS PubMed.
- Y. Kim, E. Yassitepe, O. Voznyy, R. Comin, G. Walters, X. Gong, P. Kanjanaboos, A. F. Nogueira and E. H. Sargent, ACS Appl. Mater. Interfaces, 2015, 7, 25007–25013 CrossRef CAS PubMed.
- F. Zhang, H. Zhong, C. Chen, X. G. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- P. Ramasamy, D. H. Lim, B. Kim, S. H. Lee, M. S. Lee and J. S. Lee, Chem. Commun., 2016, 52, 2067–2070 RSC.
- H. Li, Y. Qian, X. Xing, J. Zhu, X. Huang, Q. Jing, W. Zhang, C. Zhang and Z. Lu, J. Phys. Chem. C, 2018, 122, 12994–13000 CrossRef CAS.
- F. Palazon, F. Di Stasio, S. Lauciello, R. Krahne, M. Prato and L. Manna, J. Mater. Chem. C, 2016, 4, 9179–9182 RSC.
- J. Haruyama, K. Sodeyama, L. Han and Y. Tateyama, J. Phys. Chem. Lett., 2014, 5, 2903–2909 CrossRef CAS PubMed.
- Y. Dong, Y. Gu, Y. Zou, J. Song, L. Xu, J. Li, J. Xue, X. Li and H. Zeng, Small, 2016, 12, 5622–5632 CrossRef CAS PubMed.
- Y. Li, Z. F. Shi, S. Li, L. Z. Lei, H. F. Ji, D. Wu, T. T. Xu, Y. T. Tian and X. J. Li, J. Mater. Chem. C, 2017, 5, 8355–8360 RSC.
- S. Yang, S. Chen, E. Mosconi, Y. Fang, X. Xiao, C. Wang, Y. Zhou, Z. Yu, J. Zhao, Y. Gao, F. De Angelis and J. Huang, Science, 2019, 365, 473–478 CrossRef CAS PubMed.
- T. Chiba, Y. Hayashi, H. Ebe, K. Hoshi, J. Sato, S. Sato, Y. J. Pu, S. Ohisa and J. Kido, Nat. Photonics, 2018, 12, 681–687 CrossRef CAS.
- W. Xu, Q. Hu, S. Bai, C. Bao, Y. Miao, Z. Yuan, T. Borzda, A. J. Barker, E. Tyukalova, Z. Hu, M. Kawecki, H. Wang, Z. Yan, X. Liu, X. Shi, K. Uvdal, M. Fahlman, W. Zhang, M. Duchamp, J. M. Liu, A. Petrozza, J. Wang, L. M. Liu, W. Huang and F. Gao, Nat. Photonics, 2019, 13, 418–424 CrossRef CAS.
- W. Chen, H. Chen, G. Xu, R. Xue, S. Wang, Y. Li and Y. Li, Joule, 2019, 3, 191–204 CrossRef CAS.
- B. Han, Q. Shan, H. Zeng, J. Li, L. Xu, J. Li, F. Zhang and J. Song, Adv. Mater., 2018, 30, 1800764 CrossRef PubMed.
- H. Yuan, Y. Zhao, X. Yang, Y. Wang, J. Duan, Q. Tang and B. He, Sol. RRL, 2019, 3, 1800284 CrossRef.
- F. Zhang, Z. Shi, S. Li, Z. Ma, Y. Li, L. Wang, D. Wu, Y. Tian, G. Du, X. Li and C. Shan, ACS Appl. Mater. Interfaces, 2019, 11, 28013–28022 CrossRef CAS PubMed.
- N. K. Kumawat, A. Swarnkar, A. Nag and D. Kabra, J. Phys. Chem. C, 2018, 122, 13767–13773 CrossRef CAS.
- J. B. Hoffman, G. Zaiats, I. Wappes and P. V. Kamat, Chem. Mater., 2017, 29, 9767–9774 CrossRef CAS.
- S. Ye, M. Yu, W. Yan, J. Song and J. Qu, J. Mater. Chem. C, 2017, 5, 8187–8193 RSC.
- M. Kulbak, S. Gupta, N. Kedem, I. Levine, T. Bendikov, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2016, 7, 167–172 CrossRef CAS PubMed.
- T. Takagahara and K. Takeda, 1992, 46, 578–581.
- A. P. Alivisatos, Science, 1996, 271(5251), 933–937 CrossRef CAS.
- S. K. K. Prasad, J. E. Halpert, G. Laufersky, D. Z. Metin, J. K. Gallaher, J. M. Hodgkiss, K. Chen, J. Butkus, P. Vashishtha and N. Gaston, Chem. Mater., 2017, 29, 3644–3652 CrossRef.
- , Nat. Nanotechnol., 2009, 4, 135, DOI:10.1038/nnano.2009.24.
- T. Sadhasivam, H. T. Kim, S. Jung, S. H. Roh, J. H. Park and H. Y. Jung, Renewable Sustainable Energy Rev., 2017, 72, 523–534 CrossRef CAS.
- J. Yao, A. Peng, H. Fu, Y. S. Zhao, Y. Ma and D. Xiao, Adv. Mater., 2008, 20, 2859–2876 CrossRef.
- Y. Li, Z. Shi, L. Lei, F. Zhang, Z. Ma, D. Wu, T. Xu, Y. Tian, Y. Zhang, G. Du, C. Shan and X. Li, Chem. Mater., 2018, 30, 6744–6755 CrossRef CAS.
- S. Jeon, J. Ahn, H. Kim, H. K. Woo, J. Bang, W. S. Lee, D. Kim, M. A. Hossain and S. J. Oh, J. Phys. Chem. C, 2019, 123, 11001–11010 CrossRef CAS.
- H. K. Woo, H. Kim, S. Jeon, W. S. Lee, J. Ahn, J. Bang, M. S. Kang and S. J. Oh, J. Mater. Chem. C, 2019, 7, 5059–5066 RSC.
- C. Van Overbeek, J. L. Peters, S. A. P. Van Rossum, M. Smits, M. A. Van Huis and D. Vanmaekelbergh, J. Phys. Chem. C, 2018, 122, 12464–12473 CrossRef CAS PubMed.
- C. S. S. Sandeep, J. M. Azpiroz, W. H. Evers, S. C. Boehme, I. Moreels, S. Kinge, L. D. A. Siebbeles, I. Infante and A. J. Houtepen, ACS Nano, 2014, 8, 11499–11511 CrossRef CAS PubMed.
- J. Z. Jiang, J. Mater. Sci., 2004, 39, 5103–5110 CrossRef CAS.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- D. H. Son, Y. Yin, S. M. Hughes and A. P. Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed.
- J. Ahn, S. Jeon, W. S. Lee, H. K. Woo, D. Kim, J. Bang and S. J. Oh, J. Phys. Chem. C, 2019, 123, 18087–18094 CrossRef CAS.
- J. Bang, W. S. Lee, B. Park, H. Joh, H. K. Woo, S. Jeon, J. Ahn, C. Jeong, T. Kim and S. J. Oh, Adv. Funct. Mater., 2019, 29, 1–8 CrossRef.
- A. Taleb, C. Petit and M. P. Pileni, J. Phys. Chem. B, 2002, 102, 2214–2220 CrossRef.
- J. H. Warner and R. D. Tilley, Adv. Mater., 2005, 17, 2997–3001 CrossRef CAS.
- C. Schliehe, B. H. Juarez, M. Pelletier, S. Jander, D. Greshnykh, M. Nagel, A. Meyer, S. Foerster, A. Kornowski, C. Klinke and H. Weller, Science, 2010, 74, 550–554 CrossRef PubMed.
- K. M. Gattás-Asfura, C. A. Constantine, M. J. Lynn, D. A. Thimann, X. Ji and R. M. Leblanc, J. Am. Chem. Soc., 2005, 127, 14640–14646 CrossRef PubMed.
- L. Gomez, J. Lin, C. De Weerd, L. Poirier, S. C. Boehme, E. Von Hauff, Y. Fujiwara, K. Suenaga and T. Gregorkiewicz, ACS Appl. Mater. Interfaces, 2018, 10, 5984–5991 CrossRef CAS PubMed.
- J. Liu, K. Song, Y. Shin, X. Liu, J. Chen, K. X. Yao, J. Pan, C. Yang, J. Yin, L. J. Xu, H. Yang, A. M. El-Zohry, B. Xin, S. Mitra, M. N. Hedhili, I. S. Roqan, O. F. Mohammed, Y. Han and O. M. Bakr, Chem. Mater., 2019, 31, 6642–6649 CrossRef CAS.
- Q. Jing, Y. Su, X. Xing and Z. Lu, J. Mater. Chem. C, 2019, 7, 1854–1858 RSC.
- X. Du, G. Wu, J. Cheng, H. Dang, K. Ma, Y. W. Zhang, P. F. Tan and S. Chen, RSC Adv., 2017, 7, 10391–10396 RSC.
- Y. Zhao, L. Samad, Y. Fu, J. Chen, S. Shen, L. Guo, L. Dang and S. Jin, Nano Lett., 2016, 17, 460–466 Search PubMed.
- M. S. Kang, A. Sahu, D. J. Norris and C. D. Frisbie, Nano Lett., 2010, 10, 3727–3732 CrossRef CAS PubMed.
- M. A. El-Sayed, Acc. Chem. Res., 2004, 37, 326–333 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- Y. P. He, Y. M. Miao, C. R. Li, S. Q. Wang, L. Cao, S. S. Xie, G. Z. Yang, B. S. Zou and C. Burda, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 1–9 Search PubMed.
- M. Zhang, Z. Zheng, Q. Fu, Z. Chen, J. He, S. Zhang, L. Yan, Y. Hu and W. Luo, CrystEngComm, 2017, 19, 6797–6803 RSC.
- W. Lv, W. He, X. Wang, Y. Niu, H. Cao, J. H. Dickerson and Z. Wang, Nanoscale, 2014, 6, 2531–2547 RSC.
- H. J. Lee, U. J. Yang, K. N. Kim, S. Park, K. H. Kil, J. S. Kim, A. M. Wodtke, W. J. Choi, M. H. Kim and J. M. Baik, Nano Lett., 2019, 19, 4306–4313 CrossRef CAS PubMed.
- Y. Yang, J. Wu, X. Wang, Q. Guo, X. Liu, W. Sun, Y. Wei, Y. Huang, Z. Lan, M. Huang, J. Lin, H. Chen and Z. Wei, Adv. Mater., 2019, 32(7), 1904347 CrossRef PubMed.
- J. Zhang, Y. Wang, J. Zheng, F. Huang, D. Chen, Y. Lan, G. Ren, Z. Lin and C. Wang, J. Phys. Chem. B, 2007, 111, 1449–1454 CrossRef CAS PubMed.
- M. Lin, Z. Y. Fu, H. R. Tan, J. P. Y. Tan, S. C. Ng and E. Teo, Cryst. Growth Des., 2012, 12, 3296–3303 CrossRef CAS.
- Z. Łodziana, N. Y. Topsøe and J. K. Nørskov, Nat. Mater., 2004, 3, 289–293 CrossRef PubMed.
- G. Nedelcu, P. Geiregat, I. Van Driessche, Z. Hens, M. Ibáñez, W. Walravens, J. C. Martins, J. De Roo, J. Maes and M. V. Kovalenko, ACS Nano, 2016, 10, 2071–2081 CrossRef PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- B. A. Koscher, J. K. Swabeck, N. D. Bronstein and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 6566–6569 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TEM, XRD, and calculation data, and further discussion of the effect of the polarity and boiling point on the CsPbBr3 NCs with respect to nanowire formation. This material is available free of charge. See DOI: 10.1039/d0nh00029a |
| This journal is © The Royal Society of Chemistry 2020 |