
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast chemical force microscopy demonstrates that glycopeptidolipids define nanodomains of varying hydrophobicity on mycobacteria†
Albertus
Viljoen
 a,
Felipe
Viela
a,
Laurent
Kremer
bc and
Yves F.
Dufrêne
*ad
a,
Felipe
Viela
a,
Laurent
Kremer
bc and
Yves F.
Dufrêne
*ad
aLouvain Institute of Biomolecular Science and Technology, UCLouvain, Croix du Sud, 4-5, bte L7.07.07, B-1348 Louvain-la-Neuve, Belgium. E-mail: yves.dufrene@uclouvain.be
bInstitut de Recherche en Infectiologie de Montpellier (IRIM), Université de Montpellier, CNRS UMR 9004, Montpellier, France
cINSERM, IRIM, 34293 Montpellier, France
dWalloon Excellence in Life sciences and Biotechnology (WELBIO), Belgium
First published on 21st April 2020
Abstract
Mycobacterium abscessus is an emerging multidrug-resistant bacterial pathogen causing severe lung infections in cystic fibrosis patients. A remarkable trait of this mycobacterial species is its ability to form morphologically smooth (S) and rough (R) colonies. The S-to-R transition is caused by the loss of glycopeptidolipids (GPLs) in the outer layer of the cell envelope and correlates with an increase in cording and virulence. Despite the physiological and medical importance of this morphological transition, whether it involves changes in cell surface properties remains unknown. Herein, we combine recently developed quantitative imaging (QI) atomic force microscopy (AFM) with hydrophobic tips to quantitatively map the surface structure and hydrophobicity of M. abscessus at high spatiotemporal resolution, and to assess how these properties are modulated by the S-to-R transition and by treatment with an inhibitor of the mycolic acid transporter MmpL3. We discover that loss of GPLs leads to major modifications in surface hydrophobicity, without any apparent change in cell surface ultrastructure. While R bacilli are homogeneously hydrophobic, S bacilli feature unusual variations of nanoscale hydrophobic properties. These previously undescribed cell surface nanodomains are likely to play critical roles in bacterial adhesion, aggregation, phenotypic heterogeneity and transmission, and in turn in virulence and pathogenicity. Our study also suggests that MmpL3 inhibitors show promise in nanomedicine as chemotherapeutic agents to interfere with the highly hydrophobic nature of the mycobacterial cell wall. The advantages of QI-AFM with hydrophobic tips are the ability to map chemical and structural properties simultaneously and at high resolution, applicable to a wide range of biosystems.
New conceptsThere is a need in current cell biology and microbiology for advanced nanoimaging techniques that can investigate molecular interactions and properties in living cells, at high spatial and temporal resolutions. Here we show that fast quantitative imaging (QI) atomic force microscopy (AFM) combined with hydrophobic tips is a powerful tool to quantitatively map hydrophobic properties of bacterial pathogens, at high spatiotemporal resolution (∼10 min for 128 × 128 pixel images). We focus on Mycobacterium abscessus, a multidrug-resistant bacterial pathogen causing severe lung infections in cystic fibrosis patients. We discover that the transition from a smooth to a rough colony morphology, caused by the loss of cell envelope associated glycopeptidolipids (GPLs), leads to a dramatic change in surface hydrophobicity, smooth bacteria displaying unusual nanodomains with varying degrees of hydrophobicity. These results show that GPLs modulate the nanoscale distribution of hydrophobicity of M. abscessus, which is critical for regulating bacterial adhesion and aggregation, as well as virulence and pathogenicity. This study demonstrates the power of QI-AFM as a nanoimaging tool for probing the hydrophobic properties of cell surfaces in relation to function, at high speed and spatial resolution. |
Introduction
Mycobacteria, including the clinically-relevant pathogens Mycobacterium tuberculosis and Myobacterium leprae are known to possess a highly hydrophobic cell surface. This is largely due to the presence of the mycomembrane, an atypical outer membrane containing extremely hydrophobic lipid components, such as mycolic acids.1 Cell surface hydrophobicity plays important functional roles, including acting as an effective permeation barrier to most common antibiotics,2 favouring cell–surface interactions such as adhesion to solid surfaces3 and host tissues – the first step leading to infection,4–6 favouring bacterium-to-bacterium adhesion allowing mycobacterial cord formation and immune evasion,7 and favouring aerosol transmission.8–10 Although the synthesis, structure and function of the cell envelope components have been widely studied, how they are spatially distributed and how exactly they contribute to surface hydrophobicity remains elusive and somewhat controversial.Mycobacterium abscessus is an emerging human-transmissible multidrug-resistant pathogen causing severe lung infections in cystic fibrosis patients.11,12 This mycobacterial species exhibits a peculiar feature, the ability to form morphologically rough (R) and smooth (S) colonies. The smooth (S) to rough (R) transition occurs due to irreversible genetic lesions that occur in a cluster of genes that control the biosynthesis and export of glycopeptidolipids (GPLs) which are extractible lipids covering the bacterial surface (Fig. 1a).13,14 GPLs are known to mask phosphatidyl inositol mannosides (PIM), another lipid species present in the mycomembrane and surface-exposed in R variants as well as a large array of lipoproteins, both being strong TLR2 agonists participating in the inflammatory response.15,16 Hence, it is possible that GPLs play a role in regulating surface hydrophobicity: with their hydrophilic di- or tri-glycosylated amino alcohol head groups, GPLs may mask the hydrophobic mycolic acids in the outer membrane (mycomembrane) of S variants (Fig. 1a), leading to a lower relative hydrophobicity compared to R variants. So far, a direct demonstration of the role of GPLs in controlling cell surface hydrophobicity has never been established.
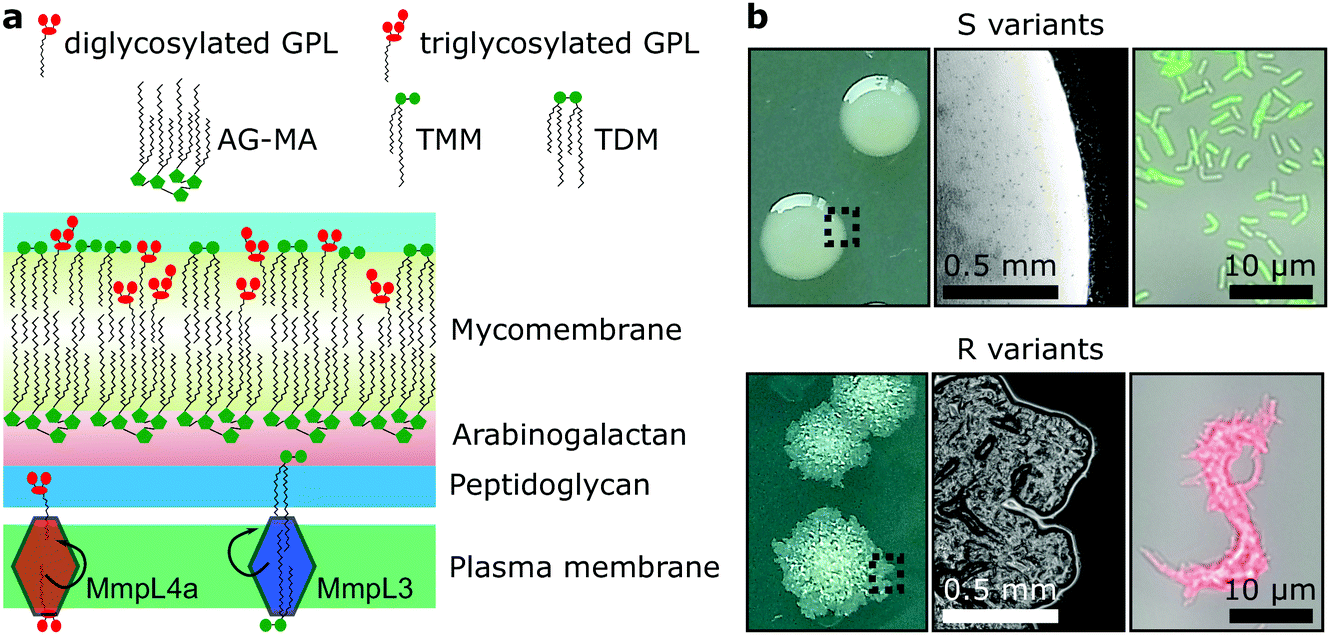 | ||
| Fig. 1 Dramatic morphological differences in Mycobacterium abscessus are determined by lipid composition of the mycomembrane. (a) Schematic view of the M. abscessus cell wall. Smooth (S) morphotypes produce large quantities of glycopeptidolipids (GPLs), which are absent in the outer membranes of rough (R) morphotypes, usually due to mutations in genes that encode enzyme effectors or transporters. The red diamond shape in the plasma membrane represents the GPL transporter MmpL4a and the blue diamond shape the mycolic acid transporter, MmpL3. (b) S variants produce smooth mucoid colonies on solid media (left panel – picture taken with a camera; and middle panel – a colony observed under a 10× objective) and disperse liquid cultures (right panel – cells from a liquid culture were deposited in a hydrophobic Petri dish and allowed to settle before observation with a 100× oil objective), while R variants grow as rough, wrinkled and dry colonies and as serpentine corded structures in broth culture. AG-MA – arabinogalactan mycolic acids, TMM – trehalose monomycolate, TDM – trehalose dimycolate. | ||
GPLs offer S variants the capacity to form mature biofilms as well as a form of communal motility through sliding, properties that allow mycobacteria to colonize surfaces in the environment or the airways of the infected host.17–19 During infection of macrophages S variants suppress phagosomal acidification as well as apoptosis.20,21 On the other hand, R variants, which are almost exclusively isolated from infected patients, do not block phagosomal acidification or apoptosis, causing the death and lysis of their host macrophages and consequentially their release into the extracellular environment.20,22 They hyper aggregate and hence organize as they grow into serpentine cord-like structures – seen both in vitro (Fig. 1b) and during infection.7 These cords are too large to be phagocytosed by macrophages providing R variants a measure to evade innate immunity.7,20 Indeed, evidence points to a greater capacity of the M. abscessus R variant than the S variant to colonise the airways chronically23 and the R variant is generally isolated from patients suffering from more severe disease than those infected by pure S variant populations.24 Cording of R cells can thus be considered a critical virulence factor in M. abscessus and probably more generally in all pathogenic mycobacteria.25,26
While GPLs and mycolic acids are key components of the M. abscessus cell envelope that have been widely investigated, we still know little about how their occurrence and spatial distribution control the physical and chemical properties of the cell surface. Specifically, it is unclear how changes in GPL and mycolic acid contents during the S-to-R transition play a role in modulating the hydrophobic character of the cells spatially and how this affects their ability to form cords. To address these questions, we have used advanced atomic force microscopy (AFM)-based quantitative imaging (QI) with tips functionalized with specific chemical groups4,5,27 to map the hydrophobic properties of the M. abscessus cell surface. While recently developed QI-AFM enables to image the structure and physical properties of biosystems simultaneously at high speed and high resolution, it has never been combined with hydrophobic tips to quantitatively map biophysical properties and interactions. Using QI-AFM, we compare S and R variants as well as a mutant originating from the S variant background, in which the GPL transporter gene mmpL4a was disrupted.19,28 In addition, to investigate the contribution of the mycolic acid layers to M. abscessus S and R variant surface hydrophobicity, we studied bacteria treated with a specific inhibitor of the trehalose monomycolate (TMM) transporter MmpL3.29,30 TMM is the final product of mycolic acid synthesis in the mycobacterial cytosol and MmpL3 is the flippase that transports TMM across the plasma membrane to the periplasm,30 where it is subsequently modified to produce another abundant extractable mycolic acid species, trehalose dimycolate (TDM).31 In addition, TMM is used to esterify the peptidoglycan–arabinogalactan complex that forms the skeleton of the cell wall of mycobacteria.31 Here, mycolic acids attached to arabinogalactan form the inner leaflet of the mycomembrane, while extractable mycolates, TMM and TDM, along with a variety of other extractable lipids, including GPLs, form the outer leaflet32 (Fig. 1a).
Results
M. abscessus S and R variants exhibit smooth surface ultrastructure
M. abscessus S and R variants show very different phenotypes in vitro.7,14 On solid agar, S variant colonies are smooth, shiny, dome shaped and have a mucoid texture, while R variant colonies are wrinkled and non-mucoid (Fig. 1b, left). In planktonic culture, S variant bacilli disperse, while the R variant hyper aggregates and appears to grow as cords of cells adhered to each other (Fig. 1b, right). We first asked whether these phenotypes would correlate with differences in cell surface ultrastructure. We imaged the surface topography of single living bacteria using contact mode, in which the AFM tip is scanned across the cell while applying a very small force (∼150 pN). To this end, cells were immobilized by depositing them into a hydrophobic culture dish and allowing them to adhere overnight in culture medium without detergents, thus with minimal treatment that could alter their cell wall state. For S cells, even though we frequently observed them in microcolonies of the size shown in Fig. 2a (left panel), they were mostly dispersed as single cells over the surface of the dish, indicating that some cells of this variant may detach from the hydrophobic surface and re-adhere elsewhere. For R and mmpL4a::pUX1 cells, on the contrary, the majority of bacteria were in cords (microcolonies) of various sizes, ranging from small ones that were easy to image (Fig. 2b and c, left panel) to very large ones that were impossible to image. That the R variant formed cords in the dishes (like they do under most in vitro growth conditions) indicates that their growth on these surfaces is appropriate for studying cord formation, thus supporting the relevance of our conclusions in in vivo environments where mycobacterial cords are found.7,24 High-resolution topography images recorded on top of M. abscessus S (Fig. 2a, right panel) and R (Fig. 2b, right panel) morphotypes as well as on the mmpL4a::pUX1 mutant (Fig. 2c, right panel) revealed even, featureless surfaces for all strains. Calculation of the average arithmetical mean deviation of measured heights provides a measure of the regularity or roughness (Ra) within an AFM topographic image. The surface roughnesses (from 300 nm × 300 nm scan areas) were very similar, with Ra = 0.30 ± 0.02 nm, 0.41 ± 0.09 nm and 0.37 ± 0.02 nm (mean ± s.e.m.) for M. abscessus S (n = 9 images from 6 cells), M. abscessus R (n = 4 images from 4 cells) and the mmpL4a::pUX1 mutant (n = 11 images from 6 cells), respectively, and did not test significantly different from each other using a one-tailed Student's t-test (p > 0.05). These topographic images, comparable to those reported for Mycobacterium bovis,5 demonstrate that S and R phenotypes have very even surface structures which are not influenced by the presence or absence of GPLs. They also suggest that the M. abscessus macroscopic adhesive behaviour is not determined by differences in cell surface structural or morphological properties.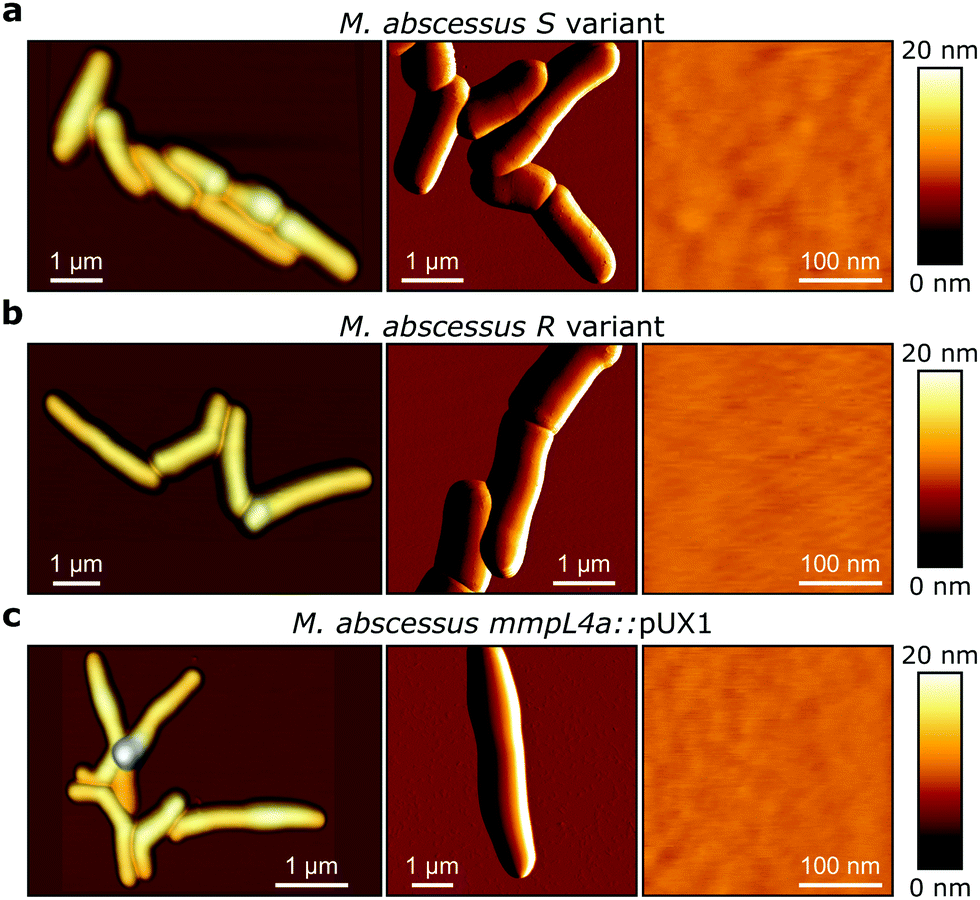 | ||
| Fig. 2 Topographic AFM images of Mycobacterium abscessus. (a, left) 3-D height image obtained in QI mode of M. abscessus S variants. (Middle) Deflection image of M. abscessus S variants. (right) High-magnification (300 nm × 300 nm) height images obtained in contact mode of the surface topology of the M. abscessus S variant. (b and c) Images obtained under the same conditions of the R variant (b) and of mmpL4a::pUX1 (c). | ||
Chemical force microscopy demonstrates major differences in the surface hydrophobicity of M. abscessus S and R variants
Loss of GPLs leads to cell aggregation and formation of serpentine cords, structures that protect the bacteria from the onslaught by the immune system during infection and potentially shield bacilli in the core from antibiotics.7 We postulated that this switch in adhesive phenotype is controlled by changes in the distribution of specific cell wall components, especially hydrophobic mycolic acids. To test this, we used multiparametric AFM imaging with hydrophobic probes33,34 to map the nanoscale structure and hydrophobicity of mycobacteria at high resolution. Traditionally, bacterial cell surface properties have been investigated using AFM force-volume (FV) imaging, in which arrays of force curves are recorded across the cell surface,35,36 usually at low speeds limiting the size of the arrays composing the images (∼32 × 32 pixels in ∼35 minutes). Recently developed multiparametric imaging, such as peak force tapping (PFT) and quantitative imaging (QI) with functionalized tips,34,37 enables to simultaneously image the structural, chemical and biophysical properties of living cells at higher lateral speeds allowing easy, less time-consuming generation of high resolution images (∼128 × 128 pixels in ∼10 minutes).We initially used QI imaging with hydrophobic methyl-terminated tips to study the surface of mycobacteria lacking GPLs. Adhesion maps recorded on R variant cells revealed large adhesion forces of 1.41 ± 0.21 nN magnitude (mean ± s.e.m., n > 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 adhesive curves from 12 cells) that were uniformly distributed on the surface (mean binding frequency = 94.1 ± 3.9%, Fig. 3a, d and Fig S1a–d, ESI†). Similarly, large adhesion forces of 1.33 ± 0.2 nN and high binding frequencies of 96.8 ± 2.4% (n > 10000 adhesive force curves from 9 images of 9 cells) were found for the mmpL4a::pUX1 mutant (Fig. 3b and d). We attribute these forces to strong hydrophobic interactions between mycolic acids and the hydrophobic tip. To further substantiate this, we performed additional experiments with hydrophobic (CH3 group-exposing) and hydrophilic (OH group-exposing) model surfaces. As can be seen in Fig. S2 (ESI†), hydrophobic adhesive forces were practically lacking on hydrophilic surfaces (Fig. S2a, ESI†), while Fig. S2b (ESI†) shows that they were clearly present on hydrophobic surfaces, both in FV mode (0.04 ± 0.02 nN magnitude and 11 ± 6% adhesive frequency vs. 0.97 ± 0.24 nN and 100 ± 0% for CH3 surfaces; n = 3 model surface–tip combinations) and in QI mode (0.03 ± 0.02 nN and 8 ± 4% vs. 1.01 ± 0.21 nN and 100 ± 0% for CH3 surfaces; n = 3). Our hydrophobic forces (∼1 nN) are smaller than those we reported earlier (∼7 nN),27 which is easily explained by differences in tip radii (∼60 nm vs. ∼30 nm here). Also, the adhesion forces probed on mycobacterial surfaces are smaller than those previously reported for Mycobacterium bovis BCG, which exhibits hydrophobic forces of approximately 3 nN.4,5,27 Again, this apparent discrepancy results from differences in probe geometry, leading to differences in contact area,38 and perhaps from differences in probing parameters as well. Another explanation for lower surface hydrophobicity in our study compared to previous studies on mycobacteria may rely on the mycomembranes of M. tuberculosis/M. bovis BCG and M. abscessus, that largely differ both with regards to the type of mycolic acids present and the types and composition of extractable lipids.40,41M. tuberculosis for instance harbors a large panoply of hydrophobic lipids such as phthiocerol dimycocerosate, acylated trehaloses and sulfoglycolipids, that are lacking in M. abscessus.10 Overall, these observations indicate that M. abscessus R variant cells exhibit a surface hydrophobicity similar to that of pure CH3 surfaces, and that hydrophobicity is conferred by surface exposed mycolic acids when GPLs are lacking.
000 adhesive curves from 12 cells) that were uniformly distributed on the surface (mean binding frequency = 94.1 ± 3.9%, Fig. 3a, d and Fig S1a–d, ESI†). Similarly, large adhesion forces of 1.33 ± 0.2 nN and high binding frequencies of 96.8 ± 2.4% (n > 10000 adhesive force curves from 9 images of 9 cells) were found for the mmpL4a::pUX1 mutant (Fig. 3b and d). We attribute these forces to strong hydrophobic interactions between mycolic acids and the hydrophobic tip. To further substantiate this, we performed additional experiments with hydrophobic (CH3 group-exposing) and hydrophilic (OH group-exposing) model surfaces. As can be seen in Fig. S2 (ESI†), hydrophobic adhesive forces were practically lacking on hydrophilic surfaces (Fig. S2a, ESI†), while Fig. S2b (ESI†) shows that they were clearly present on hydrophobic surfaces, both in FV mode (0.04 ± 0.02 nN magnitude and 11 ± 6% adhesive frequency vs. 0.97 ± 0.24 nN and 100 ± 0% for CH3 surfaces; n = 3 model surface–tip combinations) and in QI mode (0.03 ± 0.02 nN and 8 ± 4% vs. 1.01 ± 0.21 nN and 100 ± 0% for CH3 surfaces; n = 3). Our hydrophobic forces (∼1 nN) are smaller than those we reported earlier (∼7 nN),27 which is easily explained by differences in tip radii (∼60 nm vs. ∼30 nm here). Also, the adhesion forces probed on mycobacterial surfaces are smaller than those previously reported for Mycobacterium bovis BCG, which exhibits hydrophobic forces of approximately 3 nN.4,5,27 Again, this apparent discrepancy results from differences in probe geometry, leading to differences in contact area,38 and perhaps from differences in probing parameters as well. Another explanation for lower surface hydrophobicity in our study compared to previous studies on mycobacteria may rely on the mycomembranes of M. tuberculosis/M. bovis BCG and M. abscessus, that largely differ both with regards to the type of mycolic acids present and the types and composition of extractable lipids.40,41M. tuberculosis for instance harbors a large panoply of hydrophobic lipids such as phthiocerol dimycocerosate, acylated trehaloses and sulfoglycolipids, that are lacking in M. abscessus.10 Overall, these observations indicate that M. abscessus R variant cells exhibit a surface hydrophobicity similar to that of pure CH3 surfaces, and that hydrophobicity is conferred by surface exposed mycolic acids when GPLs are lacking.
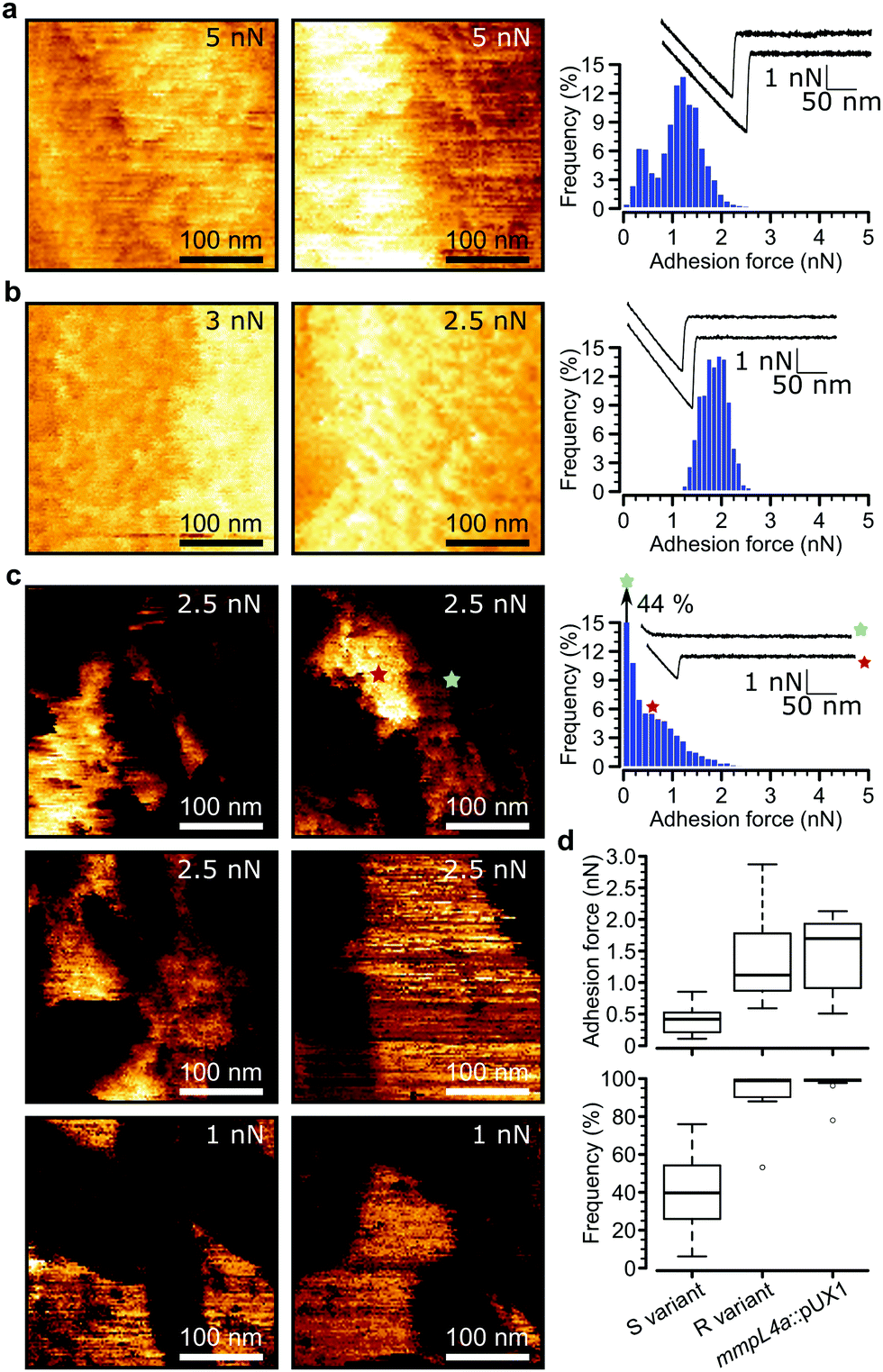 | ||
| Fig. 3 AFM with hydrophobic tips captures hydrophobic nanodomains on Mycobacterium abscessus. (a) Representative high-resolution adhesion map (300 nm × 300 nm, left) and accompanying histogram of adhesion forces with two representative force curves (right) obtained by the QI mode with hydrophobic tips for M. abscessus R. The z-range was from 0 nN to the value indicated in the top right corner of each map. (b and c) Similar conditions were used to produce adhesion maps and histograms of the mmpL4a::pUX1 mutant (b) and the M. abscessus S variant (c). Similar results were obtained for a total of 15 cells (S variant), 12 cells (R variant) and 9 cells (mmpL4a::pUX1 mutant) probed in three independent experiments. (d) Boxplots of adhesion forces (top) and frequencies (bottom). M. abscessus S (n = 15) and R (n = 12) variants as well as mmpL4a::pUX1 (n = 9). The bold bar within the boxes represents the median, while the bottom and top lines of the boxes represent the first and third quartiles, respectively, and the whiskers represent the range. | ||
Next, we tested the effect of increasing retraction speeds (loading rates) on adhesion forces measured by FV- and QI modes, both on model hydrophobic surfaces (Fig. S3a, ESI†) and on live M. abscessus R variant cells (Fig. S3b, ESI†). Higher loading rates in the range that we used for our QI experiments (5 × 102 nN s−1) did not lead to a significant change in the forces measured compared to when lower loading rates usually used in FV-mode (20 nN s−1) were used (Fig. S3, ESI†). In a recent dynamic force spectroscopy study on the nanoadhesion between bare silicon nitride tips and a model hydrophobic surface, significantly increased forces were reported over a very large loading rate range (101–107 nN s−1).39 However, forces were not much increased (<20%) in the loading range of 20–5 × 102 nN s−1, similar to our results (Fig. S3, ESI†).
Remarkably, multiparametric images of M. abscessus S cells revealed nanoscale variations of adhesive properties across the bacterial surface, while the topographic contrast was homogeneous (Fig. 3c and Fig. S1e–h, ESI†). Adhesive patches about 100–250 nm wide and covering 47 ± 5% (mean ± s.e.m. from n = 15 images on 15 different cells) of the cell surface were observed. These domains showed adhesion forces of 0.70 ± 0.07 nN and adhesion frequencies of 79 ± 3% (n > 10000 adhesive force curves from 15 images on 15 different cells), and were found to alternate with poorly adhesive areas (0.12 ± 0.01 pN; 5 ± 3%) (Fig. 3d, left). These hydrophobic nanodomains and poorly adhesive areas were also observed using the FV mode and standard parameters albeit at lower resolution (Fig. S4, ESI†), confirming the validity of the QI data. We attribute the strong forces to hydrophobic interactions between the hydrophobic tip and mycolic acids, and the weak forces to the presence of hydrophilic GPLs masking the mycolic acids. Presumably, the slightly smaller hydrophobicity observed here (0.7 vs. 1.4 nN for the R variant) suggests that some hydrophilic GPL molecules might be present in the nanodomains.
These results lead us to conclude that the presence and spatial distribution of GPLs play critical roles in controlling the surface hydrophobicity of M. abscessus. In S variants, mycolic acids are either sequestered into hydrophobic nanodomains surrounded by a hydrophilic matrix of GPLs or different classes of GPLs, i.e. less hydrophilic diglycosylated GPLs and more hydrophilic triglycosylated GPLs determine the presence of hydrophobic and hydrophilic nanodomains respectively; while loss of GPLs in R variants leads to fully homogeneous hydrophobic cell surfaces. We speculate that this major change in nanochemical properties promotes cell aggregation and formation of serpentine cords, a swap in phenotypes that has important implications for virulence and pathogenesis, i.e. protection from the immune system and drug tolerance.7
Inhibition of mycolic acid transport alters the surface ultrastructure and hydrophobicity of both morphotypes
To further investigate the contribution of mycolic acids to the surface hydrophobicity of the M. abscessus morphotypes, we analyzed bacterial cells treated with a specific inhibitor of the TMM transporter MmpL3, the antitubercular pyrrole derivative BM212 (Fig. 1a).29,30,42 Contact mode imaging of treated cells proved to be very difficult, probably due to mechanical fragility of the cells. By contrast, QI mode imaging yielded well-resolved topographic images of treated cells, showing that this dynamic mode is much less invasive than more traditional contact mode. As in contact mode, untreated cells showed smooth surface ultrastructure for both S (Fig. 4a, right) and R (Fig. 4e, right) variants, with Ra measurements for the S variant of 0.53 ± 0.11 nm (n = 10) and for the R variant of 0.58 ± 0.09 nm (n = 7) that were not significantly different (one-tailed Student's t-test, p > 0.05).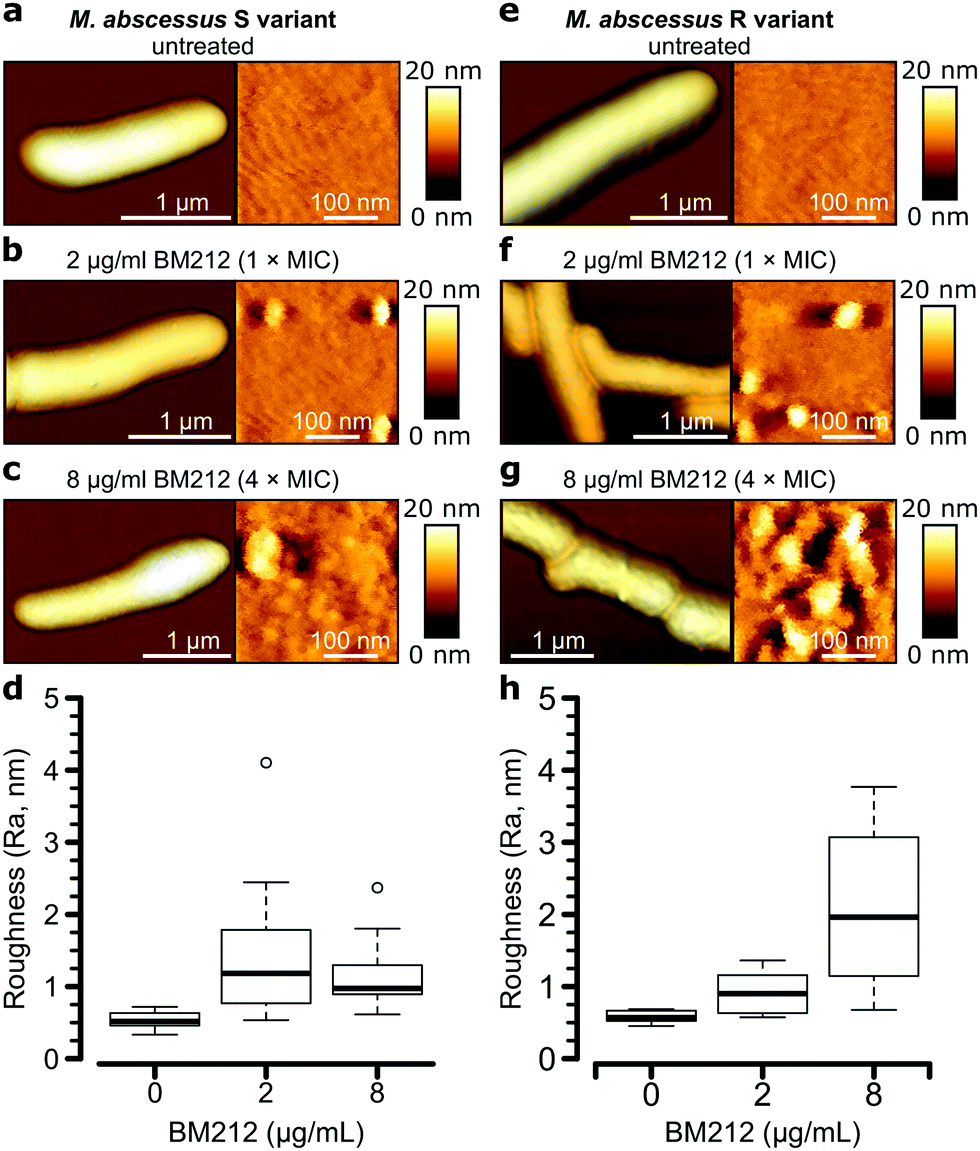 | ||
| Fig. 4 Effect of an inhibitor of mycolic acid transport on the M. abscessus surface topology. (a–c) 3-D topographical images at low (left) and high (right) magnification of the M. abscessus S variant untreated (a) or treated with BM212 at the MIC (b) or above the MIC (c). (d) Boxplots of roughness (Ra) for the S variant untreated or treated with BM212. The bold bar within the boxes represents the median, while the bottom and top lines of the boxes represent the first and third quartiles, respectively, the whiskers represent the range and open circles are outliers. (e–h) AFM images and boxplots generated under similar conditions for the M. abscessus R variant. Ra values from images of at least 7 bacilli per strain per treatment condition were used to draw the boxplots in (d and h). | ||
Notably, BM212 treatment of the cells at 1 × MIC (2 μg ml−1) and 4 × MIC (8 μg ml−1) resulted in major structural alterations. Images at 1 × MIC featured elongated and circular bumps of ∼20–40 nm in width, visible at both low and high resolutions in 83% of S variant cells and 36% of R variant cells (Fig. 4b and f, left) and produced Ra values of 1.45 ± 0.30 nm and 0.93 ± 0.09 nm (mean ± s.e.m.) for the S (Fig. 4d) and R (Fig. 4h) variants, respectively. High-resolution images revealed that the areas between the bumps remained even (Fig. 4b and f, right). At 4 × MIC, bumps were also observed (25% of S variant cells and 54% of R variant cells) but were clearly more pronounced in size on the R variant (∼40–80 nm). As a result, the overall surface roughness was higher for all treated cells (Fig. 4c and g, right), with the larger increase in Ra values being observed at 4 × MIC on the R variant (2.10 ± 0.32 nm compared to 1.16 ± 0.14 nm for S variant cells). As control experiments, we treated S and R variant cells at 4× the MIC with apramycin, an aminoglycoside that targets the bacterial ribosome and does not directly inhibit the synthesis of a cell wall or an outer membrane component. This led to moderate cell surface structure alterations that did not resemble those induced by BM212 (Fig. S5a and b, ESI†) and in comparatively smaller increases in the Ra values for the two variants (1.42 ± 0.29 nm for the S variant, n = 8, and 1.02 ± 0.31 nm for the R variant, n = 8 images from 8 cells).
Next, we asked whether BM212 alters the surface hydrophobicity of the M. abscessus morphotypes. Adhesion QI mapping with hydrophobic tips was used to probe the distribution and strength of hydrophobic forces on BM212-treated cells. For S cells, exposure to either 1× (Fig. 5a) or 4× (Fig. 5b) the MIC resulted in a complete loss of the hydrophobic nanodomains usually observed on native cells, as reflected in the very low mean adhesion forces and frequencies (0.12 ± 0.02 nN and 6 ± 3% at 1× BM212's MIC [n = 12 images from 12 cells]; 0.11 ± 0.01 nN and 6 ± 1.6% at 4× MIC [n = 11 images from 11 cells, each for which the map average adhesion was used]). Intriguingly, exposure of R cells at 1× BM212's MIC did not appreciably affect surface hydrophobicity (Fig. 5c), with mean adhesion forces of 2.45 ± 0.55 nN and frequencies of 71 ± 12% (n = 11 on 11 cells) although a small population of cells (36%) showed very low average adhesion forces of 0.1–0.2 nN. By contrast, treatment at 4× the MIC resulted in a substantial decrease in hydrophobic forces (0.52 ± 0.1 nN and 45 ± 8%; n = 13) (Fig. 5d). In sharp contrast to the BM212 results, apramycin had no appreciable effect on hydrophobicity (0.98 ± 0.17 nN and 85 ± 5.6% for the S variant and 1.12 ± 0.09 nN and 99.8 ± 0.07% for the R variant, n = 8 images from 8 cells of each variant) (Fig. S5c and d, ESI†). In summary, specific inhibition of mycolic acid transport could significantly decrease the hydrophobic forces detected on both S and R variants of M. abscessus, although a higher concentration of the inhibitor BM212 is necessary to achieve this in R variants.
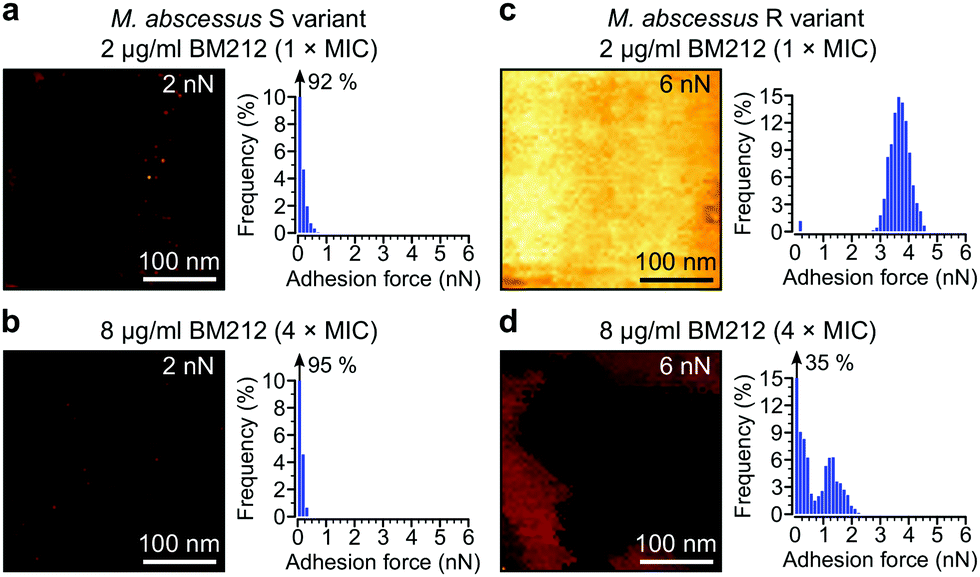 | ||
| Fig. 5 Inhibiting mycolic acid transport dramatically alters M. abscessus surface hydrophobicity. (a and b) Representative high-magnification adhesion map (300 nm × 300 nm, left) and corresponding histograms of adhesion forces (right) obtained with hydrophobic tips for the M. abscessus S variant treated with BM212 at 1× (a) or 4× its MIC (b). The z-range for each map is indicated in the top right corner. (c and d) Adhesion maps and histograms of the M. abscessus R variant obtained under similar conditions. At least eight cells from two independent experiments per strain and treatment condition showed similar results. | ||
Discussion
Bacterial pathogens use hydrophobic forces to promote their adhesion to surfaces and tissues, which usually represents the initial step leading to infection.43 Therefore, characterization of bacterial cell surface hydrophobicity is an important topic, which could also be helpful in designing efficient anti-adhesion molecules capable of blocking pathogen–host interactions. Traditionally, bacterial surface hydrophobicity has been assessed using ensemble assays such as water-contact angle measurements, adhesion to hydrocarbons, partitioning in aqueous two-phase systems and hydrophobic interaction chromatography. These methods are, however, limited because they do not probe surface hydrophobicity unambiguously. In addition, they can also be invasive and extract cell surface components, and they investigate whole populations of cells while we now know that bacteria, particularly mycobacteria, are heterogeneous, even at the subcellular level.44,45 In M. abscessus, a transition from a smooth (S) to rough (R) colony morphology, caused by the loss of GPLs in the outer cell wall layer, correlates with an increase in virulence.7,14,24 Despite the physiological and clinical importance of this transition, whether it involves changes in cell surface properties is not known. We have used an advanced AFM imaging method with hydrophobic probes to quantitatively map the surface hydrophobicity of M. abscessus on a scale of a few nanometres, and to assess how this parameter changes upon the S-to-R transition and under treatment with a drug targeting mycolic acids.Our main finding is that loss of GPLs during the S-to-R switch leads to major changes in nanoscale surface hydrophobicity, without any apparent change in cell surface ultrastructure. Despite the multiple phenotypic differences previously associated with this morphological transition,14,22 we found it remarkable that both variants exhibit the same uniform, featureless nanoscale surface architecture, indicating that GPLs are not major determinants of cell shape and surface topology in M. abscessus. In an earlier study on M. abscessus morphology, scanning electron micrographs showed similar featureless surfaces of M. abscessus S and R variants, while dramatic differences in communal bacterial organization were clear in lower magnification micrographs.46 While R cells are homogeneously hydrophobic, S cells feature variations of hydrophobic properties, i.e. hydrophobic nanodomains, alternating with hydrophilic regions. Hydrophobicity was also spread homogeneously on the mmpL4a::pUX1 mutant which carries a defined loss-off-function mutation in the GPL transporter and which does not exhibit GPLs on its surface.19,28 These observations, together with results gained using the mycolic acid transport inhibitor (see below) strongly support the notion that the hydrophobic surface of R variants is essentially made of mycolic acids while in S variants hydrophilic regions consist of GPLs with their large hydrophilic head groups. It can thus be inferred either (i) that GPLs are essentially localized within clusters on the M. abscessus S surface rather than being homogenously distributed all over the bacilli or (ii) that the different classes of GPLs, i.e. diglycosylated (less hydrophilic head group) and triglycosylated (more hydrophilic head group) GPLs,21 define nanodomains of varying hydrophobicity. To test either of these hypotheses would ultimately require a technique allowing to specifically probe GPLs, including different classes of GPLs that would rely on anti-GPL antibodies specific for each class of GPL (currently unavailable) or on specific mutants lacking either form of GPL. Such future studies are warranted and would further unravel nanoscale heterogeneity of mycobacterial surface properties. Interestingly, a polar-punctate localization of the protein complex responsible for GPL biosynthesis and transport in Mycobacterium smegmatis was observed by fluorescence microscopy,47 despite an absence of heterogeneities in the surface localization of GPLs detected by anti-GPL antibodies, presumably due to the vastly inferior resolution of immunofluorescence microscopy compared to AFM. We, however, failed to observe a punctate localization of hydrophilic nanodomains at the poles but rather heterogeneously spread across the entire length of the cells, suggesting that GPLs are not more concentrated at the site of their export. As mycolic acid is the major hydrophobic component of the mycomembrane, we believe that loss of GPLs in R cells mostly results in an increased exposure of the hydrophobic arabinogalactan-attached mycolic acid tails present in the inner leaflet. Alternatively, one may argue that the loss of GPLs leaves voids that are filled by other more hydrophobic extractable lipids, such as TMM and/or TDM. A thin layer liquid chromatography comparative analysis of the different mycolic acid species that are present in M. abscessus showed no major differences in the quantities of these lipids between the two variants (Fig. S6, ESI†). Therefore, the void created by the loss of abundant GPLs in the R variant does not appear to be compensated for by increased amounts of TMM and TDM. Whether this void is filled with other hydrophobic lipids remains however, to be investigated further.
We suggest that this major difference in hydrophobicity between the two variants plays an important role in modulating adhesion and aggregation, impacting upon virulence and pathogenicity. The S morphotype is adapted to an environmental lifestyle while the R morphotype hyper-aggregates, forming cords that protect them from phagocytosis by primary immune cells and likely also from the activity of antibiotics. Indeed, the transition of an S to R morphotype, linked to mutations in a cluster of genes that control synthesis and transport of GPLs, has been linked to a greater capacity of the bacteria to survive in their infected hosts and cause more severe disease.23,24 It is tempting to speculate that M. abscessus has evolved two different cell wall organizations to favour survival, growth and pathogenesis. Hydrophilic heterogeneities on S cells might be used to lower cell-to-cell adhesion forces, favouring dispersion and spreading of the bacteria in an aqueous environmental niche,8 while retaining the ability to colonize surfaces through sliding motility. Evidence that exposure to certain antibiotics could induce the phenotypic S–R transition without the occurrence of mutations in the GPL biosynthesis machinery48 supports the view that down- and up-regulation of GPL expression may be used by the bacteria to modulate their level of hydrophilicity during their life cycle. In addition, regulation of genes encoding enzymes that decorate GPLs, such as the various glycosyltransferases responsible for the synthesis of the glycosidic moiety of GPL, may by changing the structure of the GPLs, directly participate in controlling the surface hydrophobicity of the S variant. Strong and homogeneous hydrophobic adhesive forces of R cells would explain the high capacity of this variant to establish strong cell-to-cell adhesion, leading to the formation of mycobacterial cords.
The stronger hydrophobicity of R cells could also be important in promoting M. abscessus transmission through aerosols. An epidemiological report of recently emerged dominant circulating M. abscessus clones that have spread globally, highlighted that most M. abscessus infections are acquired through transmission, potentially via fomites and aerosols.12 Indeed, it has been observed that M. abscessus can associate with small diameter aerosol particles of 1–5 μm diameter, which could easily penetrate into the lower areas of the lung.49 It was recently proposed that M. tuberculosis uses this exact strategy to avoid the bacterial flora and antimicrobial immunity of the upper respiratory pathways, while recruiting permissive immune cells in the lower reaches of the lung.9,50 So strong hydrophobic properties could, through promoting preferential association of the bacteria with the water–air interface of a bubble,8,51 favor aerosolization and transmission of M. abscessus R morphotype cells, thus increasing their pathogenicity.
Treatment of both variants with BM212, an inhibitor of mycolic acid transport, resulted in substantial ultrastructural modifications, mostly in the form of nanoscale blebs, and concomitant increases in surface roughness. These results are partly in line with previous AFM studies on M. bovis BCG exposed to isoniazid (INH) and ethionamide (ETH), two antitubercular drugs that inhibit components of the type II fatty acid synthase responsible for mycolic acid elongation.5 Both drugs increased the surface roughness of M. bovis BCG, yet without any detectable blebs. Scanning electron micrographs of INH-treated M. smegmatis, a rapidly growing species more closely related to M. abscessus than M. bovis BCG, revealed a pattern of cell wall blebbing at randomly distributed locations all over the bacterial surface, showing similarity to our AFM observations on BM212-treated M. abscessus cells.52 Interestingly, a recent study on another class of MmpL3 inhibitors, the indoleamides, reported morphological changes in M. tuberculosis observed by electron microscopy.53 Together, these observations strongly implicate mycolic acids, but not GPLs, as factors that are required for the expression of a uniform mycobacterial cell shape and surface topology.
Interestingly, the influence of BM212 on surface hydrophobicity differs on S and R variants. At and above the MIC, the inhibitor completely abolishes the presence of hydrophobic nanodomains on S variant cells, confirming they are made of mycolic acids, while on R variant cells the antibiotic only appreciably decreased hydrophobic forces and their distribution at a concentration higher than the MIC. Is there a correlation between morphological changes and decreased hydrophobicity in BM212-treated cells? We do not attribute decreases in hydrophobicity to increased roughness of the BM212-treated cells, but rather to the specific action of this drug that leads to decreased mycolic acid levels in the mycomembrane. This conclusion is supported by (i) the intrinsic extreme hydrophobicity of mycolic acids and (ii) the fact that apramycin, an unrelated drug that affects protein synthesis in general, also increases surface roughness of both S and R variants, yet had no clear effects on surface hydrophobicity. That BM212-treatment significantly decreased the distribution and strength of hydrophobic adhesive forces on M. abscessus proves that the inhibitors of MmpL322,42 show promise as chemotherapeutic measures to interfere with the highly hydrophobic nature of the mycobacterial cell wall.
In conclusion, our experiments show that GPLs modulate the nanoscale surface hydrophobicity of M. abscessus, with S cells displaying hydrophobic nanodomains and R cells homogeneous surface hydrophobicity. These findings are likely to play critical roles in important physio(patho)logical processes. We demonstrate also that mycolic acids are absolutely required for expression of a hydrophobic cell surface in M. abscessus and that, by inhibiting the mycolic acid flippase MmpL3, mycobacterial hydrophobicity is severely diminished, thus opening avenues for future therapeutic interventions. As some other important mycobacterial species produce GPLs, including M. avium, M. chelonae and M. smegmatis, GPL-dependent nanodomains may be widespread among mycobacteria.
Methods
Mycobacterial strains, culture conditions and antibiotic treatments
M. abscessus CIP104536T S and R variants as well as the mmpL4a::pUX1 mutant (tdTomato+)28 was routinely cultured at 30 °C in Middlebrook 7H9 medium (Difco) supplemented with 0.2% v/v glycerol, 0.2% w/v glucose and 0.25% v/v tyloxapol (7H9-GGT). The three different strains were maintained separately as pure cultures and were not mixed prior to or during AFM experiments and could be differentiated from each other through fluorescence microscopy observation. Selective antibiotics used were kanamycin (200 μg ml−1) or hygromycin (500 μg ml−1). All antibiotics were purchased from Sigma, except for BM212 which was from MedChem Express. For AFM experiments, bacteria were first disaggregated by serial passage through a 26 GA syringe needle, collected by centrifugation and re-suspended in 7H9 medium without the detergent tyloxapol (7H9-GG). After another round of passages through a 26 GA syringe needle to break aggregates produced by centrifugation, the bacterial suspension was filtered through a 5 μm PVDF syringe filter that only allowed single bacteria or very small clumps of 2–3 bacteria to pass through. The OD600 of the filtered suspension was adjusted to 0.02 and 1 ml-aliquots of the suspension frozen at −80 °C. The day before an AFM experiment, an aliquot was thawed and transferred to a hydrophobic μ-fluorodish (iBidi), which was subsequently incubated overnight at 30 °C to allow adhesion of the bacteria to the surface under sterile conditions. For BM212 or apramycin (control antibiotic) treatment experiments, bacteria were seeded into the μ-fluorodishes and incubated at 30 °C for 8 h when they were washed and medium replaced with 7H9-GG supplemented with antibiotics at the concentrations indicated in the text. These dishes were incubated overnight (16 h) prior to AFM analysis.Preparation of AFM probes and model surfaces, and AFM parameters
AFM QI and FV modes were applied in 7H9-GG or deionized water using a NanoWizard® 4 NanoScience AFM (JPK). For topographic imaging, oxide-sharpened microfabricated Si3Ni4 cantilevers (MSCT, Bruker) were used and all imaging was done in 0.22 μm-membrane filtered Middlebrook 7H9 medium. Height images were processed by independently subtracting a third-degree polynomial fit from each line prior to Ra calculation using the JPK Data Processing software V.6.1.125. For adhesion mapping experiments, gold cantilevers (OMCL-TR4, Olympus) with a tip radius of 30 nm were first functionalized for 16 h in a 1 mM HSCH2(CH2)10CH3 (1-dodecanothiol) solution in ethanol prior to being rinsed with ethanol. Addition of the 1-dodecanothiol self-assembled monolayer to the gold surface of the AFM tip would theoretically increase the tip radius by ∼2 nm.54 Hydrophilic and hydrophobic model surfaces were prepared by first coating glass microscopy coverslips with a 5 nm thick Cr layer followed by a 30 nm thick Au layer using electron beam thermal evaporation and then immersing these in either an ethanolic solution of 1 mM HSCH2(CH2)9CH2OH (11-mercapto-1-undecanol, hydrophilic) or 1 mM 1-dodecanothiol (hydrophobic) for 16 hours. All experiments with hydrophobic AFM tips were performed in ultrapure water. The cantilevers’ spring constants were empirically determined by the thermal noise method prior to all measurements and ranged between 0.02 and 0.04 N m−1.55 Bacteria were first imaged in their entirety, at 256 × 256 pixels, with an applied force kept at 0.15 nN, and a constant approach/retract speed of 50 μm s−1 (z range of 300 nm). Zooms of 300 nm × 300 nm at 128 × 128 pixels were then recorded to obtain the high-resolution topography on top of the bacteria. For QI experiments with the functionalized probes, similar image size parameters were used, but the approach/retract speed was reduced to 25 μm s−1, the setpoint increased to 0.5 nN and the z-range increased to 500 nm. For force distance curves collected in FV mode (which only served as a control for QI experiments) on 300 nm × 300 nm areas at 16 × 16 pixels, a force of 0.5 nN, a constant approach/retract speed of 1 μm s−1 and a z-range of 1 μm was used. For dynamic force spectroscopy experiments, retractions speeds were varied (0.5, 1, 2.5, 5, 10, 25 and 50 μm s−1 for FV-mode and 1, 5, 10, 25 and 50 μm s−1 for QI mode), while approach speed was kept constant (1 μm s−1 for FV-mode and 25 μm s−1 for QI mode) on areas of 300 nm × 300 nm both on model surfaces and on bacterial cells. The effect of varying approach speed in either FV- or QI mode (either 1 or 25 μm s−1) on force measurements was also tested and no differences were observed.Author contributions
A. V. and Y. F. D. designed the experiments, analyzed the data and wrote the article. A. V. and F. V. collected the data. All authors critically revised the article and approved its content for publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work at UCLouvain was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. [693630]), the National Fund for Scientific Research (FNRS), the FNRS-WELBIO (Grant no. WELBIO-CR-2015A-05), and the Research Department of the Communauté française de Belgique (Concerted Research Action). Y. F. D. is a Research Director at the FNRS. L. K. acknowledges the support of the Fondation pour la Recherche Médicale (grant number DEQ20150331719).References
- M. Daffé and H. Marrakchi, Microbiol. Spectrum, 2019, 7 CrossRef PubMed , GPP3-0027-2018.
- V. Jarlier and H. Nikaido, FEMS Microbiol. Lett., 1994, 123, 11–18 CrossRef CAS PubMed.
- J. O. Falkinham, C. D. Norton and M. W. LeChevallier, Appl. Environ. Microbiol., 2001, 67, 1225–1231 CrossRef CAS PubMed.
- D. Alsteens, E. Dague, P. G. Rouxhet, A. R. Baulard and Y. F. Dufrêne, Langmuir, 2007, 23, 11977–11979 CrossRef CAS PubMed.
- D. Alsteens, C. Verbelen, E. Dague, D. Raze, A. R. Baulard and Y. F. Dufrêne, Pflugers Arch., 2008, 456, 117–125 CrossRef CAS PubMed.
- R. W. Stokes, R. Norris-Jones, D. E. Brooks, T. J. Beveridge, D. Doxsee and L. M. Thorson, Infect. Immun., 2004, 72, 5676–5686 CrossRef CAS PubMed.
- A. Bernut, J.-L. Herrmann, K. Kissa, J.-F. Dubremetz, J.-L. Gaillard, G. Lutfalla and L. Kremer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E943–E952 CrossRef CAS PubMed.
- J. O. Falkinham, Emerging Infect. Dis., 2003, 9, 763–767 CrossRef PubMed.
- C. J. Cambier, S. Falkow and L. Ramakrishnan, Cell, 2014, 159, 1497–1509 CrossRef CAS PubMed.
- M. Jankute, V. Nataraj, O. Y.-C. Lee, H. H. T. Wu, M. Ridell, N. J. Garton, M. R. Barer, D. E. Minnikin, A. Bhatt and G. S. Besra, Sci. Rep., 2017, 7, 1315 CrossRef PubMed.
- A. van Dorn, Lancet Respir. Med., 2017, 5, 15 CrossRef PubMed.
- J. M. Bryant, D. M. Grogono, D. Rodriguez-Rincon, I. Everall, K. P. Brown, P. Moreno, D. Verma, E. Hill, J. Drijkoningen, P. Gilligan, C. R. Esther, P. G. Noone, O. Giddings, S. C. Bell, R. Thomson, C. E. Wainwright, C. Coulter, S. Pandey, M. E. Wood, R. E. Stockwell, K. A. Ramsay, L. J. Sherrard, T. J. Kidd, N. Jabbour, G. R. Johnson, L. D. Knibbs, L. Morawska, P. D. Sly, A. Jones, D. Bilton, I. Laurenson, M. Ruddy, S. Bourke, I. C. Bowler, S. J. Chapman, A. Clayton, M. Cullen, T. Daniels, O. Dempsey, M. Denton, M. Desai, R. J. Drew, F. Edenborough, J. Evans, J. Folb, H. Humphrey, B. Isalska, S. Jensen-Fangel, B. Jönsson, A. M. Jones, T. L. Katzenstein, T. Lillebaek, G. MacGregor, S. Mayell, M. Millar, D. Modha, E. F. Nash, C. O’Brien, D. O’Brien, C. Ohri, C. S. Pao, D. Peckham, F. Perrin, A. Perry, T. Pressler, L. Prtak, T. Qvist, A. Robb, H. Rodgers, K. Schaffer, N. Shafi, J. van Ingen, M. Walshaw, D. Watson, N. West, J. Whitehouse, C. S. Haworth, S. R. Harris, D. Ordway, J. Parkhill and R. A. Floto, Science, 2016, 354, 751–757 CrossRef CAS PubMed.
- A. Pawlik, G. Garnier, M. Orgeur, P. Tong, A. Lohan, F. Le Chevalier, G. Sapriel, A.-L. Roux, K. Conlon, N. Honoré, M.-A. Dillies, L. Ma, C. Bouchier, J.-Y. Coppée, J.-L. Gaillard, S. V. Gordon, B. Loftus, R. Brosch and J. L. Herrmann, Mol. Microbiol., 2013, 90, 612–629 CrossRef CAS PubMed.
- A. V. Gutiérrez, A. Viljoen, E. Ghigo, J.-L. Herrmann and L. Kremer, Front. Microbiol., 2018, 9, 1145 CrossRef PubMed.
- E. R. Rhoades, A. S. Archambault, R. Greendyke, F.-F. Hsu, C. Streeter and T. F. Byrd, J. Immunol., 2009, 183, 1997–2007 CrossRef CAS PubMed.
- A.-L. Roux, A. Ray, A. Pawlik, H. Medjahed, G. Etienne, M. Rottman, E. Catherinot, J.-Y. Coppée, K. Chaoui, B. Monsarrat, A. Toubert, M. Daffé, G. Puzo, J.-L. Gaillard, R. Brosch, N. Dulphy, J. Nigou and J.-L. Herrmann, Cell. Microbiol., 2011, 13, 692–704 CrossRef CAS PubMed.
- S. T. Howard, E. Rhoades, J. Recht, X. Pang, A. Alsup, R. Kolter, C. R. Lyons and T. F. Byrd, Microbiology, 2006, 152, 1581–1590 CrossRef CAS PubMed.
- A. Martínez, S. Torello and R. Kolter, J. Bacteriol., 1999, 181, 7331–7338 CrossRef.
- A. Bernut, A. Viljoen, C. Dupont, G. Sapriel, M. Blaise, C. Bouchier, R. Brosch, C. de Chastellier, J.-L. Herrmann and L. Kremer, Mol. Microbiol., 2016, 99, 866–883 CrossRef CAS PubMed.
- A.-L. Roux, A. Viljoen, A. Bah, R. Simeone, A. Bernut, L. Laencina, T. Deramaudt, M. Rottman, J.-L. Gaillard, L. Majlessi, R. Brosch, F. Girard-Misguich, I. Vergne, C. de Chastellier, L. Kremer and J.-L. Herrmann, Open Biol., 2016, 6, 160185 CrossRef PubMed.
- J. Whang, Y. W. Back, K.-I. Lee, N. Fujiwara, S. Paik, C. H. Choi, J.-K. Park and H.-J. Kim, Cell Death Dis., 2017, 8, e3012 CrossRef CAS PubMed.
- A. Viljoen, J.-L. Herrmann, O. K. Onajole, J. Stec, A. P. Kozikowski and L. Kremer, Front. Cell. Infect. Microbiol., 2017, 7, 388 CrossRef PubMed.
- B. E. Jönsson, M. Gilljam, A. Lindblad, M. Ridell, A. E. Wold and C. Welinder-Olsson, J. Clin. Microbiol., 2007, 45, 1497–1504 CrossRef PubMed.
- E. Catherinot, A.-L. Roux, E. Macheras, D. Hubert, M. Matmar, L. Dannhoffer, T. Chinet, P. Morand, C. Poyart, B. Heym, M. Rottman, J.-L. Gaillard and J.-L. Herrmann, J. Clin. Microbiol., 2009, 47, 271–274 CrossRef PubMed.
- I. Halloum, S. Carrère-Kremer, M. Blaise, A. Viljoen, A. Bernut, V. Le Moigne, C. Vilchèze, Y. Guérardel, G. Lutfalla, J.-L. Herrmann, W. R. Jacobs and L. Kremer, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4228–E4237 CrossRef CAS PubMed.
- M. S. Glickman, J. S. Cox and W. R. Jacobs Jr., Mol. Cell, 2000, 5, 717–727 CrossRef CAS PubMed.
- E. Dague, D. Alsteens, J.-P. Latgé, C. Verbelen, D. Raze, A. R. Baulard and Y. F. Dufrêne, Nano Lett., 2007, 7, 3026–3030 CrossRef CAS PubMed.
- A. Viljoen, A. V. Gutiérrez, C. Dupont, E. Ghigo and L. Kremer, Front. Cell. Infect. Microbiol., 2018, 8, 69 CrossRef PubMed.
- V. L. Rosa, G. Poce, J. O. Canseco, S. Buroni, M. R. Pasca, M. Biava, R. M. Raju, G. C. Porretta, S. Alfonso, C. Battilocchio, B. Javid, F. Sorrentino, T. R. Ioerger, J. C. Sacchettini, F. Manetti, M. Botta, A. D. Logu, E. J. Rubin and E. D. Rossi, Antimicrob. Agents Chemother., 2012, 56, 324–331 CrossRef PubMed.
- Z. Xu, V. A. Meshcheryakov, G. Poce and S.-S. Chng, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7993–7998 CrossRef CAS PubMed.
- J. T. Belisle, V. D. Vissa, T. Sievert, K. Takayama, P. J. Brennan and G. S. Besra, Science, 1997, 276, 1420–1422 CrossRef CAS PubMed.
- L. Chiaradia, C. Lefebvre, J. Parra, J. Marcoux, O. Burlet-Schiltz, G. Etienne, M. Tropis and M. Daffé, Sci. Rep., 2017, 7, 12807 CrossRef PubMed.
- R. S. Dover, A. Bitler, E. Shimoni, P. Trieu-Cuot and Y. Shai, Nat. Commun., 2015, 6, 7193 CrossRef PubMed.
- Y. F. Dufrêne, D. Martínez-Martín, I. Medalsy, D. Alsteens and D. J. Müller, Nat. Methods, 2013, 10, 847–854 CrossRef PubMed.
- V. Dupres, F. D. Menozzi, C. Locht, B. H. Clare, N. L. Abbott, S. Cuenot, C. Bompard, D. Raze and Y. F. Dufrêne, Nat. Methods, 2005, 2, 515–520 CrossRef CAS PubMed.
- P. Hinterdorfer and Y. F. Dufrêne, Nat. Methods, 2006, 3, 347–355 CrossRef CAS PubMed.
- D. Alsteens, V. Dupres, S. Yunus, J.-P. Latgé, J. J. Heinisch and Y. F. Dufrêne, Langmuir, 2012, 28, 16738–16744 CrossRef CAS PubMed.
- E. W. van der Vegte and G. Hadziioannou, Langmuir, 1997, 13, 4357–4368 CrossRef CAS.
- H. Gojzewski, M. Kappl, H.-J. Butt and A. Ptak, Langmuir, 2016, 32, 4500–4508 CrossRef CAS PubMed.
- M. Daffé and P. Draper, Adv. Microb. Physiol., 1998, 39, 131–203 CrossRef.
- J. Pawelczyk, A. Viljoen, L. Kremer and J. Dziadek, Sci. Rep., 2017, 7, 42692 CrossRef CAS PubMed.
- W. Li, A. Yazidi, A. N. Pandya, P. Hegde, W. Tong, V. Calado Nogueira de Moura, E. J. North, J. Sygusch and M. Jackson, Front. Microbiol., 2018, 9, 1547 CrossRef PubMed.
- R. J. Doyle, Microbes Infect., 2000, 2, 391–400 CrossRef CAS PubMed.
- N. Dhar and J. D. McKinney, Curr. Opin. Microbiol., 2007, 10, 30–38 CrossRef CAS PubMed.
- N. Dhar, J. McKinney and G. Manina, Microbiol. Spectrum, 2016, 4 CAS , TBTB2-0021-2016.
- A. Sánchez-Chardi, F. Olivares, T. F. Byrd, E. Julián, C. Brambilla and M. Luquin, J. Clin. Microbiol., 2011, 49, 2293–2295 CrossRef PubMed.
- C. Deshayes, H. Bach, D. Euphrasie, R. Attarian, M. Coureuil, W. Sougakoff, F. Laval, Y. Av-Gay, M. Daffé, G. Etienne and J.-M. Reyrat, Mol. Microbiol., 2010, 78, 989–1003 CrossRef CAS PubMed.
- S.-H. Tsai, H.-C. Lai and S.-T. Hu, Antimicrob. Agents Chemother., 2015, 59, 6161–6169 CrossRef CAS PubMed.
- L. A. Hamilton and J. O. Falkinham, J. Med. Microbiol., 2018, 67, 1491–1495 CrossRef CAS PubMed.
- C. J. Cambier, K. K. Takaki, R. P. Larson, R. E. Hernandez, D. M. Tobin, K. B. Urdahl, C. L. Cosma and L. Ramakrishnan, Nature, 2014, 505, 218–222 CrossRef CAS PubMed.
- B. C. Parker, M. A. Ford, H. Gruft and J. O. Falkinham, Am. Rev. Respir. Dis., 1983, 128, 652–656 CAS.
- C. Vilchèze, H. R. Morbidoni, T. R. Weisbrod, H. Iwamoto, M. Kuo, J. C. Sacchettini and W. R. Jacobs, J. Bacteriol., 2000, 182, 4059–4067 CrossRef PubMed.
- S. Lun, R. Tasneen, T. Chaira, J. Stec, O. K. Onajole, T. J. Yang, C. B. Cooper, K. Mdluli, P. J. Converse, E. L. Nuermberger, V. S. Raj, A. Kozikowski and W. R. Bishai, Antimicrob. Agents Chemother., 2019, 63, e00343 CrossRef CAS PubMed.
- T. Kondo, M. Yanagida, K. Shimazu and K. Uosaki, Langmuir, 1998, 14, 5656–5658 CrossRef CAS.
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868–1873 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00736a |
| This journal is © The Royal Society of Chemistry 2020 |