
Rational integration of spatial confinement and polysulfide conversion catalysts for high sulfur loading lithium–sulfur batteries†
Qingfei
Zhang
a,
Zhensong
Qiao
a,
Xinrui
Cao
b,
Baihua
Qu
 *a,
Jin
Yuan
a,
Tian-E
Fan
c,
Hongfei
Zheng
a,
Jinqing
Cui
a,
Shunqing
Wu
b,
Qingshui
Xie
*a and
Dong-Liang
Peng
*a
*a,
Jin
Yuan
a,
Tian-E
Fan
c,
Hongfei
Zheng
a,
Jinqing
Cui
a,
Shunqing
Wu
b,
Qingshui
Xie
*a and
Dong-Liang
Peng
*a
aDepartment of Materials Science and Engineering, College of Materials and Pen-Tung Sah Institute of Micro-Nano Science and Technology, State Key Lab of Physical Chemistry of Solid Surface, Collaborative Innovation Center of Chemistry for Energy Materials, Fujian Provincial Key Laboratory of Materials Genome, Xiamen University, Xiamen 361005, P. R. China. E-mail: bhqu@xmu.edu.cn; xieqsh@xmu.edu.cn; dlpeng@xmu.edu.cn
bCollaborative Innovation Center for Optoelectronic Semiconductors and Efficient Devices, Department of Physics, Xiamen University, Xiamen 361005, P. R. China
cCollege of Automation, Chongqing University of Posts and Telecommunications, Chongqing, 400065, P. R. China
First published on 8th February 2020
Abstract
Spatial confinement is a desirable successful strategy to trap sulfur within its porous host and has been widely applied in lithium–sulfur (Li–S) batteries. However, physical confinement alone is currently not enough to reduce the lithium polysulfide (Li2Sn, 4 ≤ n ≤ 8, LIPSs) shuttle effect with sluggish LIPS-dissolving kinetics. In this work, we have integrated spatial confinement with a polar catalyst, and designed a three-dimensional (3D) interconnected, Co decorated and N doped porous carbon nanofiber (Co/N-PCNF) network. This Co/N-PCNF film serves as a freestanding host for sulfur trapping, which could effectively facilitate the infiltration of electrolyte and electron transport. In addition, the polar Co species possess strong chemisorption with LIPSs, catalyzing their reaction kinetics as well. As a result of this rational design and integration, the Co/N-PCNF@S cathode with a sulfur loading of 2 mg cm−2 exhibits a high initial discharge capacity of 878 mA h g−1 at 1C, and maintains a discharge capacity of 728 mA h g−1 after 200 cycles. Even with high sulfur loading of 9.33 mg cm−2, the cathode still keeps a stable areal capacity of 7.16 mA h cm−2 at 0.2C after 100 cycles, which is much higher than the current areal capacity (4 mA h cm−2) of commercialized lithium-ion batteries (LIBs). This rational design may provide a new approach for future development of high-density Li–S batteries with high sulfur loading.
New conceptsIt is noteworthy that extensive research efforts have focused on designing conductive porous carbon as a sulfur host material to both improve the conductivity of the electrodes, and entrap sulfur/lithium polysulfides (LIPSs) within confined spaces in lithium–sulfur (Li–S) batteries. However, the physical constraint of ionic LIPSs is limited because the interaction between polar LIPSs and nonpolar carbon is weak. Herein, we propose a new concept to overcome the above issues that involves rational integration of spatial confinement with a polar catalyst, where the Co/N-PCNF film serves as a freestanding host for the sulfur trapping, and which could effectively facilitate the infiltration of electrolyte and electron transport. In addition, the polar Co species possess strong chemisorption with LIPSs, catalyzing their reaction kinetics as well. This rational design may provide a new approach for future development of high-energy density Li–S batteries with high sulfur loading. |
Introduction
With the rapidly increasing demand for electric vehicles and consumer electronics, rechargeable battery systems with improved energy density are expected to expand the traditional family of lithium-ion batteries (LIBs). Among the proposed candidates, lithium–sulfur (Li–S) batteries have become a promising next-generation energy storage option due to their high theoretical specific capacity (1675 mA h g−1), high theoretical energy density (2567 Wh kg−1) and low material cost.1–8 Nevertheless, the insulating nature of both sulfur and its discharging products (Li2S2, Li2S) causes a slow dynamic process, and hence poor rate capabilities.9,10 More challenging, the excellent solubility of ionic lithium polysulfides (Li2Sn, 4 ≤ n ≤ 8, LIPSs) in the electrolyte results in the loss of electrochemically active sulfur and generates the “shuttle effect”.11–14 To address these issues, extensive research efforts have focused on designing conductive porous carbon as a sulfur host material to both improve the conductivity of the electrodes, and entrap sulfur/LIPSs within confined spaces.15–18 Porous carbon structures possess large specific surface area, providing sufficient contact between sulfur and the electronic percolation, and numerous pores further imprison LIPSs through physical adsorption. Both functions could improve the utilization of the active materials.19,20 However, the physical constraint of ionic LIPSs is limited because the interaction between polar LIPSs and nonpolar carbon is weak. While the slow conversion of dissolved LIPSs into insoluble Li2S2/Li2S only further worsens the “shuttle effect” in long-term cycling.21–23Some recent research has proved that polar materials can greatly improve the cycling stability of sulfur cathodes by anchoring LIPSs via strong chemical bonding.24–29 For example, Zhang et al. reported a composite of carbon nanotubes and sulfur decorated with polar TiN nanoparticles which delivered a high capacity of 1269 mA h g−1 at 0.05C and showed long cycling stability with 1C for 400 cycles.30 Polar materials not only realize the uniform adsorption of LIPSs, but can also be used as a catalyst to accelerate the conversion of LIPSs into discharge products (Li2S/Li2S2).31–34 Nazar et al. reported that polar MnO2 served as a highly efficient polysulfide mediator binding LIPSs and accelerating their conversion.31 More recently, Lee et al. elucidated the catalytic activity of polar WO3−x and MoS2−x/rGO, which significantly enhanced the LIPS conversion kinetics in Li–S batteries.33,34 These results suggest that the introduction of a catalyst to accelerate the conversion of dissolved LIPSs into Li2S/Li2S2 presents a highly efficient method for improving electrochemical performance in Li–S batteries. Ideally, rational integration of a LIPS conversion catalyst with the spatial confinement effect of host materials appears interesting and promising; however, this idea has not yet been explored or reported. In addition, it is still difficult to realize the theoretical energy-density in practical applications of Li–S batteries as high areal sulfur loading remains an enormous challenge.35,36 Freestanding cathodes, eliminating non-energy carrier materials such as the metal collector, conductive agents or polymer binders, have natural advantages in increasing sulfur loading compared with cathodes prepared from the conventional slurry-coating method. Therefore, freestanding 3D current collectors have been used to fabricate high-energy sulfur cathodes.
In this work, we developed a freestanding three-dimensional (3D) network consisting of polar Co species and nitrogen dopant porous carbon nanofibers (Co/N-PCNF) as a sulfur host via electrospinning. The fabricated Co/N-PCNF@S cathode possesses several advantages. Firstly, due to the enormous specific surface area from the 3D conductive network, Co/N-PCNF@S is easily infiltrated by electrolyte and provides abundant active sites. Secondly, the engineered structural pores could not only provide space for the volume changes of sulfur, but also provide physical confinement for LIPSs. Meanwhile the polar Co species and nitrogen dopant will generate strong chemical interactions with LIPSs. Most importantly, the polar Co species could effectively promote the redox reaction of LIPSs and reduce the “shuttle effect”. Finally, a high sulfur-loading cathode was obtained by a facile stacking layer-by-layer of freestanding Co/N-PCNF film strategy for developing high-energy batteries. With these advantages, the freestanding Co/N-PCNF@S realized high capacity and excellent rate capability in Li–S batteries, delivering a remarkably high capacity of 926 mA h g−1 at 0.2C with a high sulfur loading of 9.33 mg cm−2, corresponding to an areal capacity of 7.16 mA h cm−2, which is almost two times that of the commercial LIBs.
Results and discussion
The Co/N-PCNF film was prepared with a simple electrospinning procedure as schematically drawn in Fig. 1. Firstly, monodispersed ZnCo-ZIF nanoparticles (Fig. S1a, ESI†) were mixed with an N,N-dimethylformamide (DMF) solution of polyacrylonitrile (PAN) to form a homogeneous turbid liquid. Then, this bluish liquid mixture was electrospun into ZnCo-ZIFs@PAN nanofibers (Fig. S1c, ESI†) that assembled into a purple film (Fig. S1d, the size of 9 × 15 cm, ESI†). Next, ZnCo-ZIFs@PAN nanofibers would be converted into a black Co/N-PCNF film (Fig. S2a, ESI†) after a carbonizing treatment. The N doped porous carbon nanofiber (N-PCNF) network was prepared as a control sample by using Zn-ZIF as a self-sacrificing template. Upon 180° bending (Fig. S2b, ESI†), the Co/N-PCNF film shows no breakage, revealing good mechanical strength and flexibility. Finally, sulfur was infiltrated into the carbonized Co/N-PCNF. The whole structure was termed as Co/N-PCNF@S and directly used as a cathode.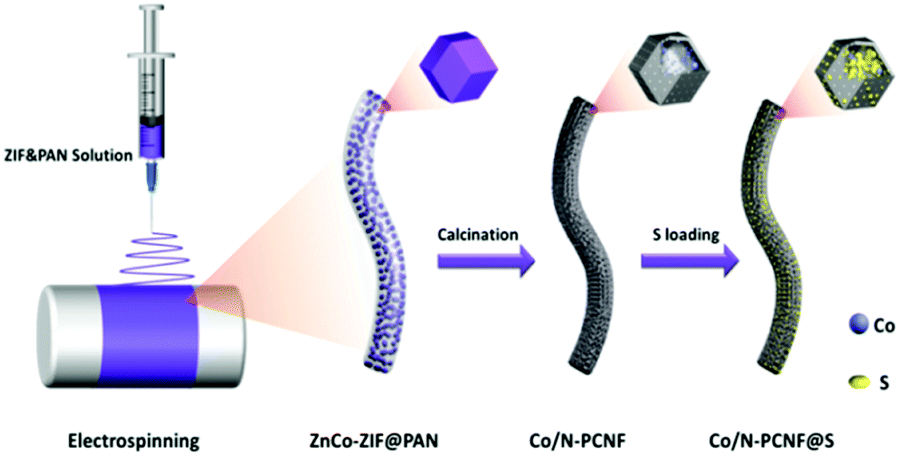 | ||
| Fig. 1 Schematic fabrication process of the Co/N-PCNF@S hybrid composite. | ||
The micro-structure and elemental mappings of the Co/N-PCNF film were characterized with scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Carbon fibers in random directions interweave with each other forming a 3D interconnected network structure (Fig. 2a). The spaces allow good contacts with the electrolyte and enhance the rapid ionic transfer. The diameter of Co/N-PCNF fibers is about 500–800 nm, and the length reaches several hundreds of micrometers. This high length-to-diameter ratio endows the interweaving film with robust texture and excellent conductivity. In the SEM and TEM images (Fig. 2b–d), hollow nanocages are clearly visible over the fibers. It is clear that the ZnCo-ZIF nanoparticles dispersed in the fibers and acted as sacrificial templates. During the carbonizing treatment, organics of ZIF and PAN decomposed to a N doped carbon substrate. The ionic Zn in ZIFs was converted by carbon to metallic Zn and subsequently evaporated from the fibers at high temperature,37 forming void space, and pores. Around the cages, carbon shells converted from PAN prevented the collapse of the ZIF polyhedron and some solid particles exist in hollow nanocages or voids (Fig. 2d). The HRTEM image (Fig. 2e) confirms the existence of graphitic carbon, as the annular inter-stripe distance of 0.34 nm is the typical distance between graphitic carbon (002) crystal planes.38 Indeed in Fig. 2f, solid particles were identified as metallic Co given the lattice fringes, as the space between the particles was measured as 0.177 nm, agreeing with the (200) crystal plane of metallic Co.39 High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) further confirms the porous structure of the Co/N-PCNF fibers (Fig. 2g), and the black dots on that image correspond to Co nanoparticles, which is further well consistent with the high light dots in Co elemental mapping (Fig. 2 Co). The signals of the C map and N map are coincident, revealing that N homogeneously distributed in the C fiber. In addition, the dispersive signal of the Co map indicated that there is considerable Co dispersed in the N doped carbon substrate in a smaller form. The morphology of N-PCNF is similar to Co/N-PCNF (Fig. S3–S5 ESI†) except that no Co nanoparticles exist in the hollow nanocages, which illustrates that introducing Co will not change the porous structure of the nanofibers.
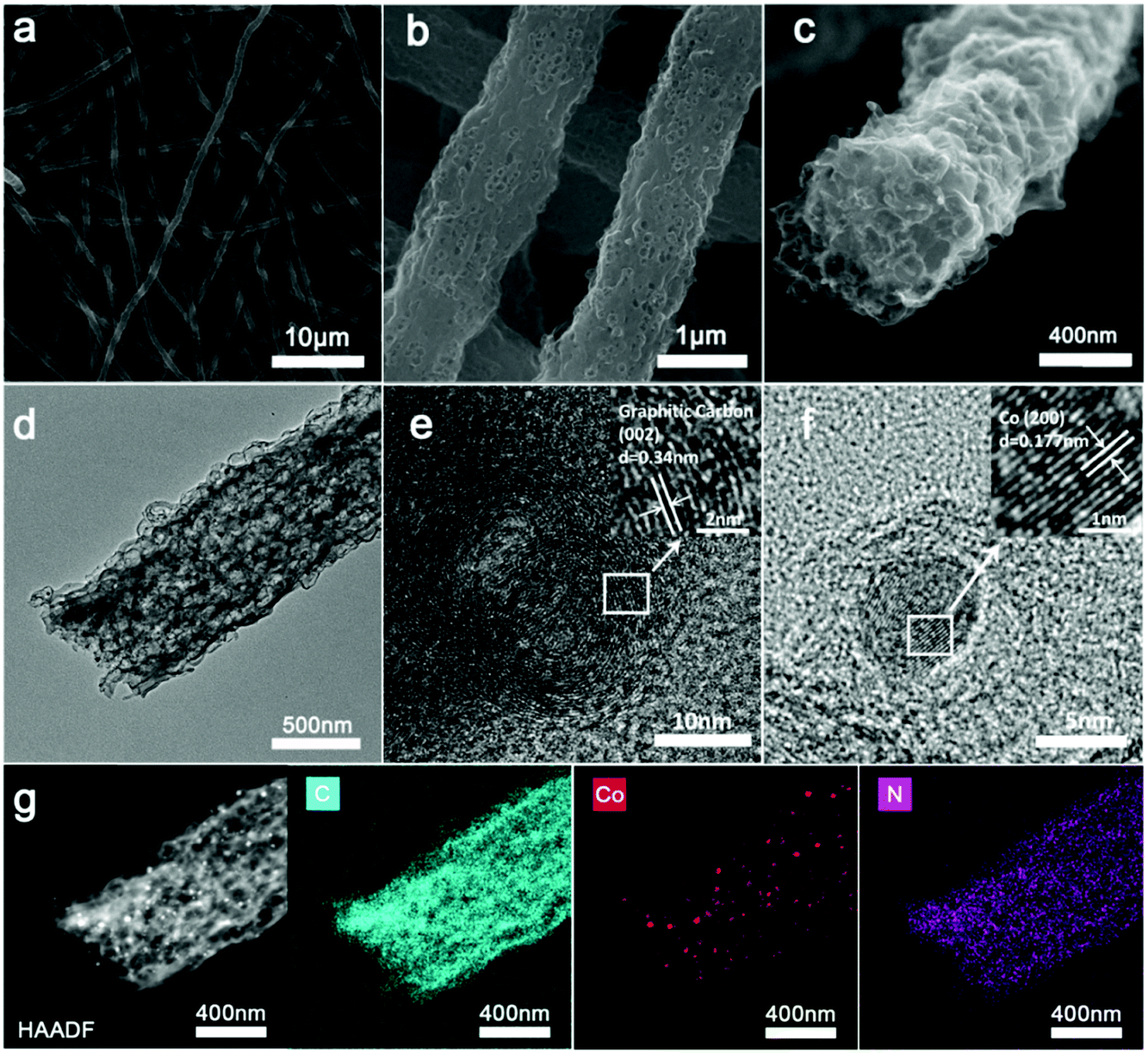 | ||
| Fig. 2 (a–c) SEM image of Co/N-PCNF; (d) TEM image of Co/N-PCNF; (e and f) HRTEM image of Co/N-PCNF; (g) HAADF-STEM image of a single Co/N-PCNF fiber and the elemental mappings of C, Co, and N; the corresponding colours are cyan, red and purple, respectively. | ||
X-ray photoelectron spectroscopy (XPS) (Fig. S6 and S7 ESI†) was used to explore the element state of Co/N-PCNF and N-PCNF. From the Zn2p spectrum in Fig. S6d and S7b (ESI†) we found that no characteristic peaks of Zn appeared in the positions of 1022 eV (Zn2p3/2) and 1045 eV (Zn2p1/2).40 The residual Zn content in the Co/N-PCNF and N-PCNF samples is infinitesimal and difficult to be detected, therefore, we believe that the influence of Zn on this system is very small and didn't take Zn into consideration. Signals for N1s have been detected around 400 eV. N is doped in the form of pyridinic N (398.5 eV), graphitic N (401 eV), pyrrolic N (400 eV) and in both Co/N-PCNF and N-PCNF. Meanwhile, the Co–N (399.2 eV) bond only appeared in Co/N-PCNF, and the corresponding peak can be found in the Co2p spectra. The Co–N bond indicates the existence of Co–Nx moieties, which were formed in the thermal decomposition of ZnCo-ZIF.
The N2 adsorption/desorption measurement was carried out to investigate the porosity of the Co/N-PCNF and N-PCNF. The N2 adsorption/desorption isotherms (Fig. S8a, ESI†) are typical H4-type hysteresis loops, indicating the existence of micropores and mesopores in both Co/N-PCNF and N-PCNF.41 Co/N-PCNF and N-PCNF carry high Brunner–Emmett–Teller (BET) specific surface areas of 684 m2 g−1 and 662 m2 g−1, respectively. There is no big different between the two samples because of the same porous structure. The density functional theory (DFT) method was employed to analyze the pore size distribution. Results from Fig. S8b (ESI†) also proved the presence of mesopores and micropores. The high specific surface areas grant the materials with good physical adsorption for sulfur species and large interfaces for sulfur conversion, which could enhance the sulfur utilization. Fig. S9 (ESI†) is the Raman spectrum of Co/N-PCNF. The peak located around 1348 cm−1 (D band) is associated with the disorder-induced asymmetric vibration mode and the one at 1598 cm−1 (G band) is related to the in-plane stretching motion of sp2 carbons. The value of IG/ID both samples is less than 1, meaning that rich defects exist in the carbon matrix.42 A higher IG/ID is observed for Co/N-PCNF(0.963) than N-PCNF(0.864) resulting from the graphitization catalytic effect of Co nanoparticles.43
To get Co/N-PCNF@S cathodes, the host material Co/N-PCNF films were cast-dropped with S/CS2 solution and subjected to a melt-diffusion process at 155 °C. Upon diffusion, Xray diffraction (XRD) was used to determine the structures of both Co/N-PCNF and Co/N-PCNF@S. Before S-loading, Co/N-PCNF (Fig. 3a) showed a broad peak at around 23.5°, which was characteristic for amorphous carbon.42 Two weak peaks at 26° and 43° confirm that some of the carbon has been graphitized.41 While in the Co/N-PCNF@S spectrum, peaks at 23.3°, 26.0°, 26.8°, 27.9°, 28.9°, and 31.6° corresponding to elemental sulfur (PDF 42-1278) were all detected, indicating that sulfur had been successfully loaded into the porous carbon fibers.44 According to the TGA analysis curves in Fig. 3b, the content of sulfur in the Co/N-PCNF@S is 62.2% and in N-PCNF@S it is 59.6%, which is consistent with the quantity we introduced in the Experiment section. When the temperature increases above 165 °C, sulfur begins to sublime and causes weight loss of Co/N-PCNF@S. Between 200 °C and 265 °C, the Co/N-PCNF@S suffers a quick weight loss of 58.6%. Then there is a gentle slope of 3.6% weight loss between 265 °C and 320 °C, because S embedded in the micropores of Co/N-PCNF is more stable and needs a higher temperature to evaporate away.
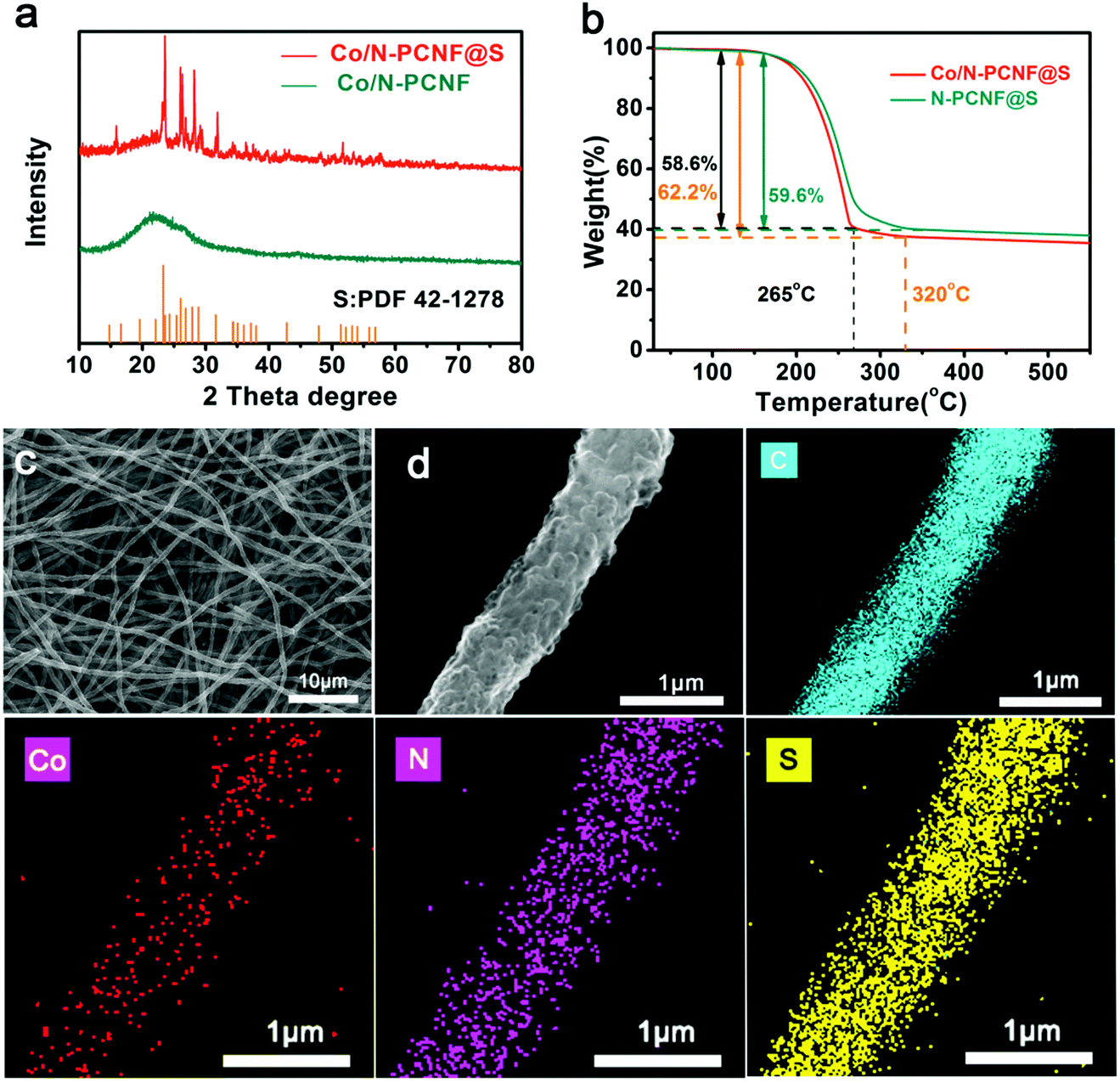 | ||
| Fig. 3 (a) XRD patterns of Co/N-PCNF and Co/N-PCNFS; (b) TGA curves of Co/N-PCNF@S and N-PCNF@S; (c) and (d) SEM images of the Co/N-PCNF@S film; element mappings of the Co/N-PCNF@S; the corresponding colors of C, Co, N, and S element are cyan, red, purple and yellow, respectively. | ||
After the sulfur loading, the Co/N-PCNF@S electrode still shows good flexibility and the freestanding nature (Fig. S10a and b, ESI†). The cross section of Co/N-PCNF@S shown in Fig. S10c (ESI†), has a thickness around 200 μm, with uniform and long fibers intertwined together suggesting that the fiber structures are well conserved. The corresponding elemental mapping analysis of Co/N-PCNF@S reveals the uniform distribution of S as shown in Fig. 3. More importantly, there is no obvious sulfur aggregation within the 3D network or on the surface of the fibers (Fig. 3c and d). The abundant pores in Co/N-PCNF provided space and large interface area, and sulfur can diffuse into the inside hollow carbon nanocages at 155 °C. The morphology of the N-PCNF@S electrode is also shown in Fig. S11 and S12 (ESI†). The distribution of sulfur in N-PCNF@S exhibits no difference to Co/N-PCNF@S, as N-PCNF has the same porous structure.
The Co/N-PCNF@S film was directly used as a freestanding cathode with lithium foil acting as an anode. The N-PCNF@S freestanding film cathode was also prepared as the contrast sample and tested under the same conditions. The cyclic voltammetry (CV) curves of the two cathodes, obtained within the voltage window 1.7–2.8 V at a scan rate of 0.1 mV s−1, are shown in Fig. 4a. The curve shows two pairs of redox peaks. The cathodic peaks appear at about 2.00 and 2.19 V corresponding to the conversion of element S to the soluble long-chain LIPSs and the following transition of LIPSs to the solid-state Li2S2/Li2S, respectively. Meanwhile, the two oxidative peaks at 2.32 and 2.40 V associated with the reverse reactions.21 The nearly overlapped CV curves in the initial 5 cycles (Fig. S13, ESI†) reveal that the electrochemical process of Co/N-PCNF@S is well reversible. Compared with the contrast sample (N-PCNF@S), the Co/N-PCNF@S cathode shows a smaller gap between the cathodic and anodic peaks in its curve, demonstrating a less severe potential polarization which suggests a lower energy barrier on the electrochemical conversion. Furthermore, as shown in Fig. 4b and Table S1, (ESI†), the Co/N-PCNF@S experiences a smaller charge-transfer resistance measured from electrochemical impedance spectroscopy31 than the N-PCNF@S, confirming the faster transformation of LIPSs. Improved sulfur transition kinetics suggests better rate capacities.
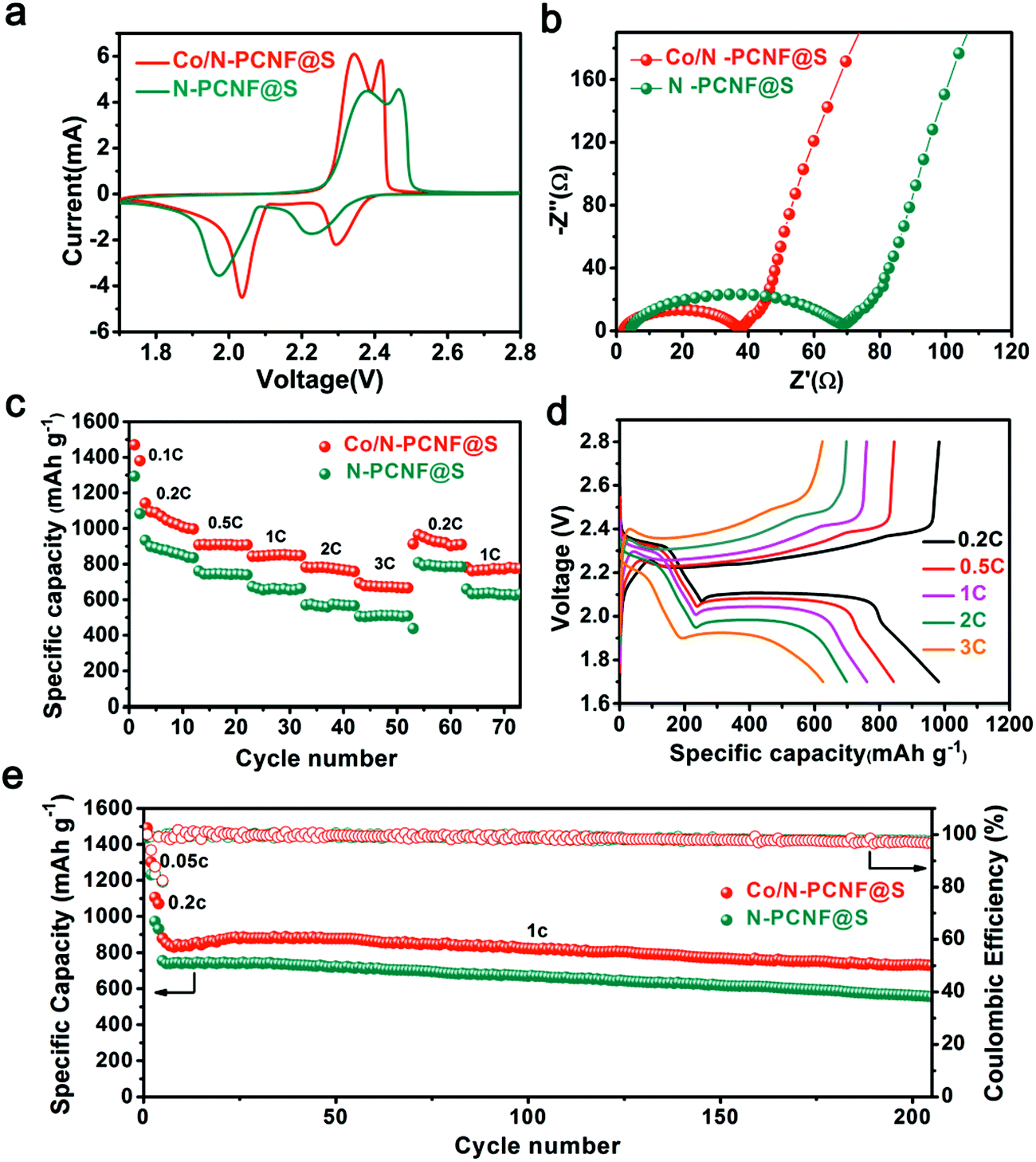 | ||
| Fig. 4 (a) CV curves of the Co/N-PCNF@S and N-PCNF@S cathodes at 0.1 mV s−1 between 1.7 and 2.8 V; (b) EIS spectra of the Co/N-PCNF@S and N-PCNF@S cathodes; (c) rate performance of the Co/N-PCNF@S and N-PCNF@S cathodes; (d) charge and discharge curves of the Co/N-PCNF@S cathode; (e) cycling performance of Co/N-PCNF@S and N-PCNF@S cathodes at 1C rate after two cycles activation at 0.05C and 0.2C, respectively. | ||
The rate capabilities of Co/N-PCNF@S and N-PCNF@S cathodes, each with a sulfur loading of 2 mg cm−2, are compared at various discharge/charge current densities (Fig. 4c). The electrodes were used for 10 cycles at each current density between 1.8 and 2.8 V. The Co/N-PCNF@S electrode gives the discharge capacities of 1048, 909, 850, 780, and 672 mA h g−1 at different rates of 0.2C, 0.5C, 1C, 2C and 3C, respectively. When the current density is reduced back to 0.2C, it exhibits a great reversibility, and the discharge capacity still reaches 952 mA h g−1. In comparison, the N-PCNF@S electrode shows much lower discharge capacities of 877, 746, 663, 558, and 512 mA h g−1, respectively. The Co/N-PCNF@S cathode owes its better rate performance to the Co species that greatly improved the redox kinetics of LIPSs. Fig. 4d shows typical galvanostatic discharge–charge curves of the Co/N-PCNF@S cathode at the tested current densities. At all current densities, both the discharge curves and charge curves have two plateaus that are associated with the electrochemical conversion of sulfur, and agree with the positions of the redox peaks in the cathode's CV profiles. The cycling performances of Co/N-PCNF@S and N-PCNF@S are also compared (Fig. 4e). Before cycling at a high rate of 1C, the electrodes were activated for 2 cycles at 0.05C and 0.2C, respectively. The Co/N-PCNF@S cathode shows a high discharge capacity 878 mA h g−1 at the fifth cycle, and gradually decreases to 728 mA h g−1 after 200 cycles. The capacity retention is 83%; this means that the decay ratio per cycle is 0.07% on average. While the N-PCNF@S cathode delivers an inferior performance achieving capacity retention of 74% after 200 cycles.
Higher areal loading of sulfur is a straightforward way to achieve high energy density, yet still a critical challenge due to the irreversible aggregation of sulfur. Herein, by facilely stacking layer-by-layer the freestanding Co/N-PCNF film (each film about 2 mg cm−2 of sulfur), we also increased sulfur loading and tested the cycling performances of the Co/N-PCNF@S cathodes with different sulfur loadings of (two-layer film) 4.67, (three-layer film) 6.41, and (four-layer film) 9.33 mg cm−2, respectively. As shown in Fig. 5a, these Co/N-PCNF@S cathodes can deliver high areal capacities of 4.38, 6.84 and 8.35 mA h cm−2, respectively, at a current density of 0.2C. The high areal capacities are considerably better than that of commercial LIBs (4 mA h cm−2). We also compared the performance of the Co,N-PCNF/S cathode with other freestanding sulfur cathodes. It can be seen from the Table S3 (ESI†) that the sulfur loading of Co,N-PCNF/S is not the highest, but it delivers the biggest areal capacities among the several carbon nanofiber-based cathodes which were prepared by the electrospinning route. This means that the areal energy density of Co,N-PCNF/S is superior.
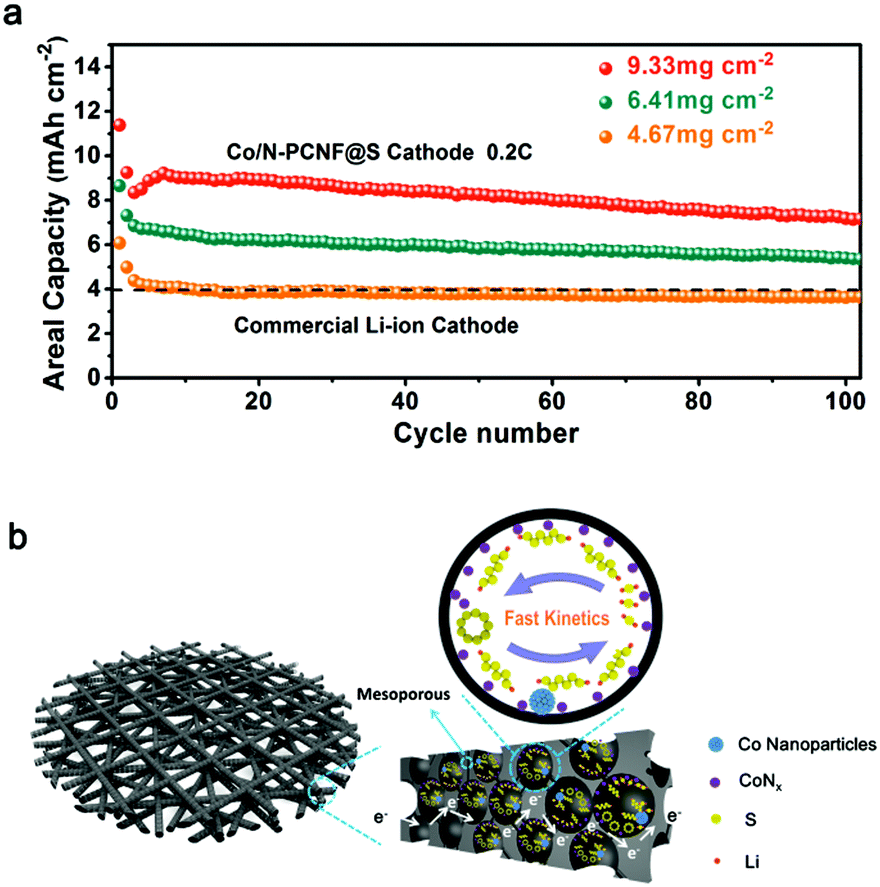 | ||
| Fig. 5 (a) Areal capacities of the N-PCNF@S cathode with 4.67, 6.41, and 9.33 mg cm−2 of sulfur at 0.2C. (b) Schematic of the 3D conductive network and the catalysis function of Co species on LIPS conversion. | ||
The superior performance is due to the integration of LIPS conversion catalysts with the spatial confinement effect due to the rational design of the 3D Co/N-PCNF network host materials as depicted in Fig. 5b. Firstly, the interlinked carbonaceous nanofibers act not only as an efficient current collector, but also as a wonderful sulfur host. After carbonization, the polar carbon substrate derived from Zn/Co-ZIF exists in the internal surface of the in situ formed hollow carbon nanocages. Sulfur could diffuse into the inside hollow carbon nanocages at 155 °C through the abundant pores and have a good contact with the polar N dopants and Co species. The 3D Co/N-PCNF network with a tremendous number of micro- and mesopores promotes the infiltration of electrolyte, and speeds up the transportation of electrons and Li+, which eventually guarantees improved performances even at high sulfur loadings and large charge/discharge rates. Then, the efficient confinement and the good catalytic activity of Co species promote the redox kinetics of soluble intermediate LIPSs. These benefits would not only guarantee a reduced loss of active material, but also alleviate the “shuttle effect” as the life of soluble LIPSs is shortened.
Further experiments and theoretical calculation have confirmed the above points. The ability to trap soluble LIPSs was examined with simple liquid phase adsorption experiments. Li2S6 (2 mmol L−1) in 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL) solvents (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume) was used as the representative LIPS. The same amount of Co/N-PCNF and N-PCNF was immersed in the Li2S6 solution for two days at room temperature, individually. The color changes of the Li2S6 solution after exposure to blank, Co/N-PCNF and N-PCNF composites are shown in the inset of Fig. 6a. The blank Li2S6 was barely faded after exposure for two days, and the one with N-PCNF still appears slightly yellowish. Meanwhile, a colorless Li2S6 solution was observed for that exposed to Co/N-PCNF, suggesting a significantly enhanced LIPS affinity with Co/N-PCNF. Fig. 6a shows the strong capability of Co/N-PCNF to adsorb Li2S6, which was further demonstrated by UV-vis spectroscopy, with a reduced LIPS concentration for the strongest absorbance in the 400–500 nm region for the polysulfide solution.
:1 volume) was used as the representative LIPS. The same amount of Co/N-PCNF and N-PCNF was immersed in the Li2S6 solution for two days at room temperature, individually. The color changes of the Li2S6 solution after exposure to blank, Co/N-PCNF and N-PCNF composites are shown in the inset of Fig. 6a. The blank Li2S6 was barely faded after exposure for two days, and the one with N-PCNF still appears slightly yellowish. Meanwhile, a colorless Li2S6 solution was observed for that exposed to Co/N-PCNF, suggesting a significantly enhanced LIPS affinity with Co/N-PCNF. Fig. 6a shows the strong capability of Co/N-PCNF to adsorb Li2S6, which was further demonstrated by UV-vis spectroscopy, with a reduced LIPS concentration for the strongest absorbance in the 400–500 nm region for the polysulfide solution.
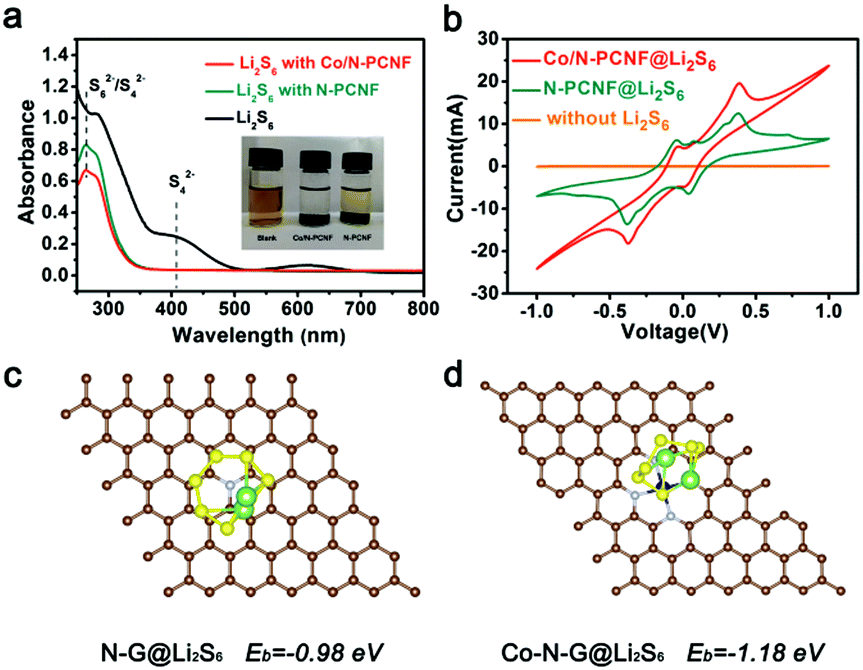 | ||
| Fig. 6 (a) UV-Vis spectra of the Li2S6 solution after exposure to blank, Co/N-PCNF and N-PCNF; (inset is the optical photograph of Li2S6 solution after addition of samples); (b) CV of symmetric cells of N-PCNF and Co/N-PCNF electrodes with Li; the DFT calculation results of interaction between Li2S6 and different substrates: (c) N-graphene, (d) Co@N-graphene. | ||
We have also studied the adsorption energy (Ead) of Li2S6 on different substrates, which was calculated based on density functional theory (DFT). Since adsorption is an important step of catalysis, the adsorption energy can also reflect the catalyst affinity between different catalytic sites and the polysulfide intermediates. It can be found from Fig. 2g that the amount of Co particles is limited. Generally, one hollow carbon nanocage only has one Co particle. Meanwhile, Co–Nx species are numerous and uniformly disperse on the internal surface of the hollow carbon nanocages, which have adequate contact with the sulfur species. Therefore, we take the Co–N4 mode in our calculation. The Co–N-graphene delivers an Ead of −1.18 eV (Fig. 6d) to Li2S6 which is more negative than that of N-graphene (−0.98 eV, Fig. 6c). The above results confirm that the introduction of Co species can enhance the interaction between N-doping graphene and LIPSs, also indicating that the Co–N-graphene is more active for the catalysis conversion of Li2S6. As the “shuttle effect” is mainly caused by the soluble long chain polysulfides (Li2Sx, 4 ≤ x ≤ 8), the DFT calculations of the interactions between Co–N-graphene and Li2S8 and Li2S4, respectively, were also performed. The results are shown in Fig. S14 (ESI†). The Co–N-G substrate displays a similar adsorption energy (−1.17 eV and −1.15 eV, respectively) of Li2S6 for both Li2S8 and Li2S4, which indicates that Co–N-graphene could fix soluble polysulfides effectively through strong bonding.
The cycled coin cells were disassembled to characterize the structure of the electrode after cycling and evaluate the migration of LIPSs from the sulfur cathodes to the Li anodes. In Fig. S15 (ESI†), the continuity of the fibers and the porous structure Co/N-PCNF and N-PCNF are well maintained after 300 cycles, which reveals the good structural stability of the films when used as a freestanding cathode on Li–S batteries. SEM images, elemental mapping of S (Fig. S16, ESI†) and energy dispersive spectroscopy (EDs) (Table S2, ESI†) of the Li anodes after 200 cycles offer more evidence. Polysulfides dissolved in the organic electrolytes would be transported to the anode side and corrode the surface of lithium, which will destroy the SEI protective layer, resulting in the terrible situation of a lithium anode. The morphology of the lithium anode surface paired with the Co/N-PCNF@S cathode is flat and lithium dendrites can’t be observed. The S content is only 5.65% (wt%) and it mainly exists in the crannies corroded by polysulfides. Meanwhile, lithium dendrites and more S content, 16.65% (wt%), nearly distribute in the whole surface of the lithium anode which was paired with the N-PCNF@S cathode. With the Co/N-PCNF@S cathode, even during long cycles much less LIPSs were dissolved in the organic electrolytes transported to the anode (Li foil) side and causing loss of active materials.42 These results reveal that the high specific surface area of the porous carbon nanofiber can effectively trap Li2S6 through physical absorption. The introduction of Co species can greatly strengthen the adsorption effect through chemical bonding.
To confirm the electrocatalysis of the LIPSs, we studied the kinetics reactions of LIPSs in the liquid phase on N-PCNF and Co/N-PCNF. The symmetric cells were assembled with Co/N-PCNF (or N-PCNF) disks as both working and counter electrodes in Li2S6 (0.2 M) electrolytes (Fig. 6b). In the blank electrolyte without Li2S6 there is only very low current occurring in the symmetric cell and no apparent peak is detected.45,46 The Co/N-PCNF system displays larger current peaks than the N-PCNF electrode, suggesting the boosted redox reactions and accelerated conversion of LIPSs for the former.
The CV curves under different scan rates are described in Fig. S17 (ESI†), and one can find that as the scan rates increase, both the Co/N-PCNF@S curves and N-PCNF@S curves show stronger redox peaks, but the redox peaks in the Co/N-PCNF@S curves are higher and more distinct, which further proves the superiority of Co/N-PCNF@S. Furthermore, previous reports have suggested that there is a linear relationship between the current of the redox peaks and the square root of the scan rate, which could reflect the diffusion process of lithium ions. The lithium ion diffusion process follows the Randles–Sevcik equation:45,46Ip = (2.69 × 105)n1.5AD0.5Cv0.5 (25 °C).
In this equation Ip is the peak current (A), n is the number of charge transfers, A is the active electrode area (cm2), D is the lithium ion diffusion coefficient (cm2 s−1), C is the concentration of Li ions (mol cm−3), and v is the scan rate (V s−1). As n, A, and C in the cell are kept constant, the slope of the plot (Ip/v0.5) is directly proportion to the diffusion rate of Li+D. Therefore, we investigated the behavior of the Co/N-PCNF@S and N-PCNF@S electrodes at different scanning rates to determine the Li+ diffusion rates in the two structures. For all scanning rates tested, the redox peaks for the Co/N-PCNF@S cathode are higher and more distinct from each other. The peak currents were plotted versus the square roots of the scan rates and obviously, the slopes for Co/N-PCNF@S are larger than those for N-PCNF@S in all three major redox peaks (Fig. S18, ESI†). The comparison indicates a faster diffusion of Li+ occurring in the Co/N-PCNF@S electrode. During the electrochemical cycling, the release of LIPSs into the electrolyte will increase its viscosity, resulting in restricted Li+ diffusion. Therefore, the faster diffusion, important for the rapid conversion of LIPSs, also implies the excellent LIPSs catalyzing capability and strong adsorption for LIPSs near the Co/N-PCNF@S surface.
To understand the reaction mechanisms of Co or N and LIPSs, the chemistry structure of Co/N-PCNF after the adsorption of Li2S6 was clarified with XPS. In the survey spectra (Fig. S19, ESI†), the characteristic peaks at around 164, 285, 400, 532, and 779 eV confirm the existence of S, C, N, O and Co elements. The detailed chemical states of C, Co, N, and S are described in the high-resolution XPS spectra. Fig. 7a shows that the C1s spectrum can be divided into three peaks, with the ones at 284.7 eV and 285.6 eV corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and CN bond, respectively. The peaks confirm the N heteroatoms in the carbon matrix. The high-resolution XPS spectrum of N1s (Fig. 7b) could be divided into four peaks corresponding to pyridinic N (398.4 eV), graphitic N (401 eV), pyrrolic N (400 eV) and Co–N (399.2 eV), and further prove the doping of N.47,48 According to previous reports, pyridinic N and pyrrolic N enhance the polarity of the carbon matrix through forming strong chemical affinity with LIPSs. The four distinct peaks in the high-resolution Co2p spectrum can be assigned to metallic Co, Co–N, Co2+ and satellites as demonstrated in Fig. 7c. It has been verified that Co–Nx moieties can effectively catalyze reduction and oxidation in Li–S batteries.41,48–50Fig. 7d possesses two dominating peaks in the S2p spectrum at 163.9 and 165.1 eV, corresponding to the terminal S and bridging S. Moreover, the peak located at 162.6 eV is assigned to the Co–S bonding, further confirming the chemical bonding between polysulfides and Co/N-PCNF.41,42
C bond and CN bond, respectively. The peaks confirm the N heteroatoms in the carbon matrix. The high-resolution XPS spectrum of N1s (Fig. 7b) could be divided into four peaks corresponding to pyridinic N (398.4 eV), graphitic N (401 eV), pyrrolic N (400 eV) and Co–N (399.2 eV), and further prove the doping of N.47,48 According to previous reports, pyridinic N and pyrrolic N enhance the polarity of the carbon matrix through forming strong chemical affinity with LIPSs. The four distinct peaks in the high-resolution Co2p spectrum can be assigned to metallic Co, Co–N, Co2+ and satellites as demonstrated in Fig. 7c. It has been verified that Co–Nx moieties can effectively catalyze reduction and oxidation in Li–S batteries.41,48–50Fig. 7d possesses two dominating peaks in the S2p spectrum at 163.9 and 165.1 eV, corresponding to the terminal S and bridging S. Moreover, the peak located at 162.6 eV is assigned to the Co–S bonding, further confirming the chemical bonding between polysulfides and Co/N-PCNF.41,42
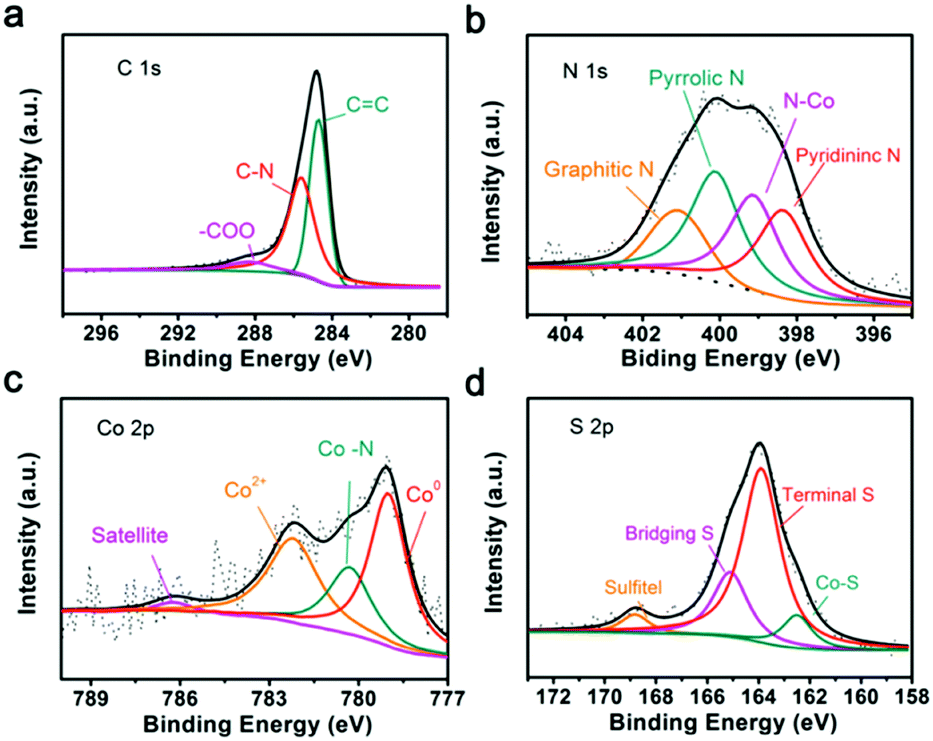 | ||
| Fig. 7 High-resolution XPS spectra of Co/N-PCNF after the adsorption of Li2S6: (a) C1s spectrum, (b) N1s spectrum, (c) Co2p3/2 spectrum and (d) S2p spectrum. | ||
Conclusions
In this work, we developed a freestanding network of Co decorated and N doped porous carbon nanofibers as advanced host materials for sulfur. It is confirmed by a series of experiments that the 3D structure network and high porosity of Co/N-PCNF can effectively trap Li2S6 through physical confinement. In particular, the introduction of Co nanoparticles can greatly strengthen the adsorption effect with LIPSs through chemical bonding, but also catalyze and promote the redox kinetics of LIPSs. Furthermore, the Co not only shows a strong interaction to LIPSs but also promoted redox kinetics of LIPSs. The results suggest that Co/N-PCNF can confine and catalyze the soluble LIPSs, leading to enhanced cycling stability of Li–S batteries. The Co/N-PCNF@S cathode can deliver a high areal capacity of 7.16 mA h cm−2 at 0.2C for 100 cycles even when the sulfur loading is increased to 9.33 mg cm−2. The rational integration paves the way for high energy density Li–S batteries with high sulfur loading.Experimental section
Synthesis of materials
000, Sigma-Aldrich) were added into 6 mL of N,N-dimethylformamide (DMF), followed by magnetically stirring overnight at room temperature then sonication for 1 h. Then, the homogeneous liquid mixture was loaded into a syringe with a steel needle and electrospun onto a steel roller receiver wrapped with a flow rate of 0.3 mL h−1. The distance between the needle and the receiver is 15 cm and a working voltage of 12 kV was provided with a high voltage DC power supply. The as-woven ZnCo-ZIFs@PAN nanofibers were pre-oxidized in air in a quartz tube heated to 220 °C at the rate of 1 °C min−1 and kept at that temperature for 2 h. Then, these pre-oxidation products were heated to 800 °C at a heating rate of 2 °C min−1 and calcined for 4 h under an Argon atmosphere. For comparison, plain N-PCNF was also synthesized from a similar process employing ZIF8 as the precursor.
Electrochemical measurements
CR2032-type coin cells were assembled in an M Braun glove box (MB-10-G-V2A, H2O and O2 < 0.5 ppm) filled with argon. The Co/N-PCNF@S or N-PCNF@S disks were used as cathodes directly. The anodes were lithium chips with a thickness and diameter of 0.45 mm and 16 mm, respectively. The electrolyte consisted of LiTFSI (1 mol L−1) dissolved in DME/DOL (1:1 vol) with LiNO3 (2 wt%) additive, and the ratio of electrolyte to sulfur was about 20 μL mg−1. The separator was a Celgard 2400 polypropylene membrane. A NEWARE BTS battery charger system (Shenzhen, China) was used to test the cells at a voltage range of 2.8–1.7 V. For the coin cells of symmetric electrochemical measurements, 50 μL of LiTFSI [1 mol L−1 in DME/DOL (1:1 vol)] and Li2S6 [0.2 mol L−1 in DME/DOL (1:1 vol)] in a 1:1 (v/v) ratio was used as the electrolyte and the Co/N-PCNF or N-PCNF disks were used as both the working and counter electrodes. Cyclic voltammetry (CV) were performed with a CHI660D electrochemical workstation (CH Instruments Co., Ltd, Shanghai, China) at the scanning rate of 0.1 mV s−1. The electrochemical impedance spectroscopy31 was measured with the same electrochemical workstation for a frequency range of 100 kHz to 0.1 Hz.
Characterization of materials
The structural information was measured by X-ray diffraction (XRD, Rikagu Ultima IV, Cu Kα radiation, 40 kV, 40 mA). The morphology and microstructure of the samples were collected by field-emission scanning electron microscopy (FESEM, SUPRA-55, ZEISS, Germany) and field emission transmission electron microscopy (FETEM, TECNAI-F30, Philips-FEI, Netherlands). The high-resolution transmission electron microscopy (HRTEM) and elemental mapping were carried out with a F30 at a working voltage of 200 kV. Raman spectra were recorded using a micro-Raman 2000 system (Renishaw, Britain). The specific surface area measurements were performed with 3H-2000PM2 (BeiShiDe, China) equipment with N2 as the adsorption–desorption gas. And thermogravimetric analysis (TGA, SDTQ600) was applied to analyse the sulfur contents in the composites within a temperature range of 25–700 °C with a heating rate of 10 °C min−1 in nitrogen. X-ray photoelectron spectroscopy (XPS) analysis was carried out on a PHI QUANTUM 2000 (monochromatic Al K X-ray source).First-principles calculations
The calculations were performed via the density functional theory (DFT) method by using the Vienna ab initio simulation package (VASP).52,53 To describe electron–ion interactions, we employed the projector augmented wave (PAW) pseudopotentials54 and used the gradient-corrected Perdew–Burke–Ernzerhof (GGA-PBE) functional to calculate electron exchange – correlation effects.55 A supercell containing 6 × 6 unit graphene cells with a vacuum spacing of 16 Å was used to mimic the N-graphene and Co@N-graphene. A cutoff energy of 400 eV for the plane-wave basis set was used; moreover, the total energy convergence and the force convergence for geometric optimization were set to be 1 × 10−5 eV and 0.02 eV Å−1, respectively. The Brillouin zone was sampled by k-point grids of 5 × 5 × 1 for geometric optimization. The adsorption energy (Ead) of Li2S6 on the substrate is defined as:| Ead = ELi2S6+Sub − ELi2S6 − ESub |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 51901031 and 51931006), NSF of Fujian Province, China (No. 2017J01103) and the National Key R&D Program of China (No. 2016YFA0202602). This work was also supported by Fundamental Research Funds for the Central Universities of China (Xiamen University: No. 20720190013), the Guangdong Basic and Applied Basic Research Foundation (No. 2019A1515011070) and the “Double-First Class” Foundation of Materials and Intelligent Manufacturing Discipline of Xiamen University. The State Key Laboratory for Physical Chemistry of Solid Surfaces at the Xiamen University is acknowledged for the supercomputer resources.Notes and references
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- J. Zhang, H. Huang, J. Bae, S.-H. Chung, W. Zhang, A. Manthiram and G. Yu, Small Methods, 2018, 2, 1700279 CrossRef.
- L. Qie and A. Manthiram, Adv. Mater., 2015, 27, 1694–1700 CrossRef CAS PubMed.
- Z.-W. Zhang, H.-J. Peng, M. Zhao and J.-Q. Huang, Adv. Funct. Mater., 2018, 28, 1707536 CrossRef.
- J. He, Y. Chen, W. Lv, K. Wen, C. Xu, W. Zhang, Y. Li, W. Qin and W. He, ACS Nano, 2016, 10, 10981–10987 CrossRef CAS PubMed.
- J. Zhou, X. Liu, L. Zhu, J. Zhou, Y. Guan, L. Chen, S. Niu, J. Cai, D. Sun, Y. Zhu, J. Du, G. Wang and Y. Qian, Joule, 2018, 2, 2681–2693 CrossRef CAS.
- M. Wild, L. O'Neill, T. Zhang, R. Purkayastha, G. Minton, M. Marinescu and G. J. Offer, Energy Environ. Sci., 2015, 8, 3477–3494 RSC.
- F. Wu, H. Lv, S. Chen, S. Lorger, V. Srot, M. Oschatz, P. A. van Aken, X. Wu, J. Maier and Y. Yu, Adv. Funct. Mater., 2019, 29, 1902820 CrossRef.
- S. Evers, T. Yim and L. F. Nazar, J. Phys. Chem. C, 2012, 116, 19653–19658 CrossRef CAS.
- L. Zhang, X. Chen, F. Wan, Z. Niu, Y. Wang, Q. Zhang and J. Chen, ACS Nano, 2018, 12, 9578–9586 CrossRef CAS PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- F. Pei, T. An, J. Zang, X. Zhao, X. Fang, M. Zheng, Q. Dong and N. Zheng, Adv. Energy Mater., 2016, 6, 1502539 CrossRef.
- F. Wu, E. Zhao, D. Gordon, Y. Xiao, C. Hu and G. Yushin, Adv. Mater., 2016, 28, 6365–6371 CrossRef CAS PubMed.
- Z. Qiao, F. Zhou, Q. Zhang, F. Pei, H. Zheng, W. Xu, P. Liu, Y. Ma, Q. Xie, L. Wang, X. Fang and D.-L. Peng, Energy Storage Mater., 2019, 23, 62–71 CrossRef.
- G. Li, X. Wang, M. H. Seo, M. Li, L. Ma, Y. Yuan, T. Wu, A. Yu, S. Wang, J. Lu and Z. Chen, Nat. Commun., 2018, 9, 705 CrossRef PubMed.
- Y. Tao, Y. Wei, Y. Liu, J. Wang, W. Qiao, L. Ling and D. Long, Energy Environ. Sci., 2016, 9, 3230–3239 RSC.
- H. Al Salem, G. Babu, C. V. Rao and L. M. Arava, J. Am. Chem. Soc., 2015, 137, 11542–11545 CrossRef CAS PubMed.
- N. Xu, T. Qian, X. Liu, J. Liu, Y. Chen and C. Yan, Nano Lett., 2017, 17, 538–543 CrossRef CAS PubMed.
- C. Zhang, J. J. Biendicho, T. Zhang, R. Du, J. Li, X. Yang, J. Arbiol, Y. Zhou, J. R. Morante and A. Cabot, Adv. Funct. Mater., 2019, 29, 1903842 CrossRef.
- Y. Li, J. Fan, J. Zhang, J. Yang, R. Yuan, J. Chang, M. Zheng and Q. Dong, ACS Nano, 2017, 11, 11417–11424 CrossRef CAS PubMed.
- X. Lu, Q. Zhang, J. Wang, S. Chen, J. Ge, Z. Liu, L. Wang, H. Ding, D. Gong, H. Yang, X. Yu, J. Zhu and B. Lu, Chem. Eng. J., 2019, 358, 955–961 CrossRef CAS.
- Y. Wu, X. Zhu, P. Li, T. Zhang, M. Li, J. Deng, Y. Huang, P. Ding, S. Wang, R. Zhang, J. Lu, G. Lu, Y. Li and Y. Li, Nano Energy, 2019, 59, 636–643 CrossRef CAS.
- T. Chen, L. Ma, B. Cheng, R. Chen, Y. Hu, G. Zhu, Y. Wang, J. Liang, Z. Tie, J. Liu and Z. Jin, Nano Energy, 2017, 38, 239–248 CrossRef CAS.
- W. Chen, T. Qian, J. Xiong, N. Xu, X. Liu, J. Liu, J. Zhou, X. Shen, T. Yang, Y. Chen and C. Yan, Adv. Mater., 2017, 29, 1605160 CrossRef PubMed.
- H. Li, S. Ma, H. Cai, H. Zhou, Z. Huang, Z. Hou, J. Wu, W. Yang, H. Yi, C. Fu and Y. Kuang, Energy Storage Mater., 2019, 18, 338–348 CrossRef.
- J. Zhang, C. You, W. Zhang, J. Wang, S. Guo, R. Yang and Y. Xu, Electrochim. Acta, 2017, 250, 159–166 CrossRef CAS.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- W. G. Lim, S. Kim, C. Jo and J. Lee, Angew. Chem., Int. Ed., 2019, 58, 18746–18757 CrossRef CAS PubMed.
- H. Lin, S. Zhang, T. Zhang, H. Ye, Q. Yao, G. W. Zheng and J. Y. Lee, Adv. Energy Mater., 2018, 8, 1801868 CrossRef.
- H. Lin, L. Yang, X. Jiang, G. Li, T. Zhang, Q. Yao, G. W. Zheng and J. Y. Lee, Energy Environ. Sci., 2017, 10, 1476–1486 RSC.
- L. Qie, C. Zu and A. Manthiram, Adv. Energy Mater., 2016, 6, 1502459 CrossRef.
- W. Xue, Z. Shi, L. Suo, C. Wang, Z. Wang, H. Wang, K. P. So, A. Maurano, D. Yu, Y. Chen, L. Qie, Z. Zhu, G. Xu, J. Kong and J. Li, Nat. Energy, 2019, 4, 374–382 CrossRef CAS.
- S.-K. Park, J.-S. Park and Y. C. Kang, J. Mater. Chem. A, 2018, 6, 1028–1036 RSC.
- S. Liu, J. Li, X. Yan, Q. Su, Y. Lu, J. Qiu, Z. Wang, X. Lin, J. Huang, R. Liu, B. Zheng, L. Chen, R. Fu and D. Wu, Adv. Mater., 2018, 30, e1706895 CrossRef PubMed.
- Q. Bai, F. C. Shen, S. L. Li, J. Liu, L. Z. Dong, Z. M. Wang and Y. Q. Lan, Small Methods, 2018, 2, 1800049 CrossRef.
- S. Wang, F. Gao, R. Ma, A. Du, T. Tan, M. Du, X. Zhao, Y. Fan and M. Wen, Metals, 2018, 8, 755 CrossRef CAS.
- D. Fang, Y. Wang, C. Qian, X. Liu, X. Wang, S. Chen and S. Zhang, Adv. Funct. Mater., 2019, 29, 1900875 CrossRef.
- W. Cai, G. Li, D. Luo, G. Xiao, S. Zhu, Y. Zhao, Z. Chen, Y. Zhu and Y. Qian, Adv. Energy Mater., 2018, 8, 1802561 CrossRef.
- J. Meng, C. Niu, L. Xu, J. Li, X. Liu, X. Wang, Y. Wu, X. Xu, W. Chen, Q. Li, Z. Zhu, D. Zhao and L. Mai, J. Am. Chem. Soc., 2017, 139, 8212–8221 CrossRef CAS PubMed.
- R. Liu, Z. Liu, W. Liu, Y. Liu, X. Lin, Y. Li, P. Li, Z. Huang, X. Feng, L. Yu, D. Wang, Y. Ma and W. Huang, Small, 2019, 15, 1804533 CrossRef PubMed.
- Y.-T. Liu, D.-D. Han, L. Wang, G.-R. Li, S. Liu and X.-P. Gao, Adv. Energy Mater., 2019, 9, 1803477 CrossRef.
- G. Zhou, H. Tian, Y. Jin, X. Tao, B. Liu, R. Zhang, Z. W. Seh, D. Zhuo, Y. Liu and J. Sun, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 840 CrossRef CAS PubMed.
- J. Xu, W. Zhang, Y. Chen, H. Fan, D. Su and G. Wang, J. Mater. Chem. A, 2018, 6, 2797–2807 RSC.
- S.-K. Park, J.-K. Lee and Y. C. Kang, Adv. Funct. Mater., 2018, 28, 1705264 CrossRef.
- Y.-J. Li, J.-M. Fan, M.-S. Zheng and Q.-F. Dong, Energy Environ. Sci., 2016, 9, 1998–2004 RSC.
- P. Wang, Z. Zhang, X. Yan, M. Xu, Y. Chen, J. Li, J. Li, K. Zhang and Y. Lai, J. Mater. Chem. A, 2018, 6, 14178–14187 RSC.
- W. Zhang, X. Jiang, X. Wang, Y. V. Kaneti, Y. Chen, J. Liu, J. S. Jiang, Y. Yamauchi and M. Hu, Angew. Chem., Int. Ed., 2017, 56, 8435–8440 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00663j |
| This journal is © The Royal Society of Chemistry 2020 |