
Metal-oxide surface-enhanced Raman biosensor template towards point-of-care EGFR detection and cancer diagnostics†
Meysam
Keshavarz
 a,
Panagiotis
Kassanos
a,
Bo
Tan
bc and
Krishnan
Venkatakrishnan
*cde
a,
Panagiotis
Kassanos
a,
Bo
Tan
bc and
Krishnan
Venkatakrishnan
*cde
aThe Hamlyn Centre, Institute of Global Health Innovation, Imperial College London, Bessemer Building, South Kensington Campus, Exhibition Rd, Kensington, London SW7 2AZ, UK
bNanocharacterization Laboratory, Department of Aerospace Engineering, Ryerson University, 350 Victoria Street, Toronto, Ontario M5B 2K3, Canada
cNanoBioInterface facility, Department of Mechanical and Industrial Engineering, Ryerson University, 350 Victoria Street, Toronto, ON M5B 2K3, Canada. E-mail: venkat@ryerson.ca
dUltrashort Laser Nanomanufacturing Research Facility, Department of Mechanical and Industrial Engineering, Ryerson University, 9 350 Victoria Street, Toronto, ON M5B 2K3, Canada
eKeenan Research Centre for Biomedical Science, St. Michael's Hospital, Toronto, Ontario M5B 1W8, Canada
First published on 23rd September 2019
Abstract
Surface-enhanced Raman spectroscopy (SERS) is a non-invasive and powerful tool for identification of molecular species. Proposed SERS structures have thus far been restricted to noble metals such as Au and Ag. However, metal-oxide-based SERS represents a new frontier in the field of biosensing. While the implementation of semiconductor materials such as TiO2 as a SERS template would enormously widen the range of use for this technique due to its biocompatibility, the detection sensitivity is still seriously impeded by inferior SERS enhancement. Consequently, the application of TiO2 for molecular biosensing has been greatly hampered. Herein, we report on a novel concept of non-stoichiometric titanium dioxide, as well as the incorporation of self-assembled quantum scale structured (Q-structured) TiOx as an ultra-high sensitive template to intensify the Raman response of tumor biomarkers, such as epidermal growth factor receptors (EGFR), to diagnose breast cancer. We demonstrate the incorporation of oxygen vacancies as a strategy to significantly increase the SERS enhancement of Q-structured TiO2. We found that reducing the TiO2 particles to quantum scale can increase the EF by up to 3.5 × 105, while further enhancement can be achieved by inducing the oxygen vacancies to the Q-structured TiOx. The detection limit was as low as 1 nM and the maximum EF was 3.4 × 107, which, to our best knowledge, is within the highest sensitivities achieved among semiconducting materials, even comparable to noble metals. Measurements with breast and cervical cancer cells lines where used to demonstrate the clinical application of the proposed SERS template.
New conceptsWe report on the use of self-assembled quantum scale structured (Q-structured) TiOx as a non-plasmonic material for SERS diagnosis, and its novel attribution to achieve comparable sensitivity to that of plasmonic noble metals. We first demonstrate that quantum-scale-structured TiO2 can lead to Raman signal enhancement and subsequently, that the introduction of oxygen vacancies to obtain TiOx leads to significantly greater enhancement. The proposed SERS templates are first evaluated using a dye and the optimized template is then used for detection of the EGFR biomarker for breast cancer diagnosis and to reveal the SERS biomolecular fingerprint spectra in cervical cancer cell and mammalian fibroblast cells. Thus, we demonstrate that Q-structured TiOx can be used as an alternative to plasmonic noble materials, which are related to localized heating issues, hindering their use in biosensing applications. This work can establish Q-structured TiOx as a promising SERS template for in vivo diagnostics, with a significant impact to the field. |
Introduction
Raman spectroscopy is a powerful non-destructive method for chemical analysis.1,2 It relies on the inelastic scattering of monochromatic light.3 Nevertheless, signal enhancement is often needed because the scattering to excitation ratio is significantly low.4,5 Consequently, there is a need to enhance the weak Raman scattering. To achieve this, an active surface-enhanced Raman scattering (SERS) template is needed, to enable the use of Raman spectroscopy for precise identification of biological molecules such as proteins, DNA, RNA as well as cancer biomarkers.6–10 The employment and development of novel nanostructured materials plays a crucial role in these developments. SERS has been proven as an extremely sensitive technique for biomolecular detection. Of particular importance, is the diagnosis of breast adenocarcinoma (MDAMB 231) cancer by non-invasive tracing of the epidermal growth factor receptor (EGFR).11 Detection of the EGFR biomarker would be a breakthrough in early cancer diagnostics. This, however, is currently limited by two main factors: weak adsorptivity and hence inadequate capability of conventional SERS-templates to discern the biomarker as well as cellular aversions to the SERS-templates.1,11 To achieve this, research has mainly focused on enhancing Raman scattering by the use of plasmonic metals such as gold and silver.12,13 Such plasmonic nanomaterials (e.g. Ag and Au nanostructures or roughened substrates) have demonstrated their potential in their use as SERS templates for molecular detection.14–16Despite the supremacy of noble metals in this regard, noble metal-based SERS nanomaterials are related with several limitations, rendering them unattractive in many applications. Noble metals are expensive, and they often require specialized synthetic processes (chemical and electrochemical methods) and costly fabrication methods (e.g. nanosphere lithography, electron-beam lithography, focused ion beam milling) for the realization of well controlled geometrical patterns to engineer nanogaps between nanostructures for biomolecular detection.17–19 High sensitivity SERS with noble-metal nanostructures arises from plasmonic hotspots of localized high intensity electromagnetic field arising in the nanogaps between nanostructures.20 At these hotspots, optical excitation leads to localized heat generation due to electron transitions.21–23 This is detrimental in several applications. Local heating of the plasmonic structures can led to changes of their refractive indices via the thermo-optic effect.24–26 Depending on the illuminating intensity, the metallic based plasmonic nanostructures can even be distorted and/or even melted, while heating may induce molecular desorption or pyrolysis.27 They can thus vaporize the surrounding liquid/solvent media or degrade molecules and proteins in their vicinity, while also induce coagulation. Furthermore, such noble-metal nanomaterials are often described by poor biocompatibility and stability, and it is challenging to control their intrinsic material properties, while reactions of the metal due to its photocatalytic effect with adsorbed biomaterials further impede their use.1,24 Additionally, often to keep these hotspots discrete and to avoid agglomeration, specialized surfactants are employed, adding cost and processing steps. Consequently, they are often not suitable for non-destructive, cost-effective biomolecular detection.11,26,28,29
Non-plasmonic materials, such as various insulating or semiconducting oxides (e.g. SiO2, ZnO, CuO and TiO2) can address some of the aforementioned shortcomings of plasmonic noble metals. Their material properties (e.g. bandgap, stoichiometry, crystallinity and physical morphology) can be easily engineered via established methods of the semiconductor industry.29 Furthermore, they lead to significantly less localized heat generation upon optical excitation, due to the fewer charge carriers.24,30 However, their SERS capabilities are not comparable to those of noble metals. Therefore, there is a need for a non-plasmonic material that exhibits the plasmonic attribution of conventional noble metals. Amongst the prospective non-plasmonic materials to meet these requirements, titanium dioxide (TiO2) is a promising candidate.11,31
TiO2 nanostructures are a family of compositionally diverse biocompatible materials that can be of low cost, with high adsorptivity and chemical stability and with a wide range of applications in physics, chemistry, biology and medicine.32,33 Research activities have focused almost entirely on the extracellular matrix (ECM) attributions of TiO2 nanostructures in biological applications.34,35 Thus far, to the best of our knowledge, TiO2 nanostructures have been only used as a substrate decorated with plasmonic noble metals for SERS applications, as current TiO2 based nanomaterials do not exhibit SERS activity.31,32,36 However, tailoring its physicochemical properties, has allowed TiO2 to achieve plasmonic-material characteristics.37
In recent years, quantum-sized particles (1–10 nm in diameter) have attracted much attention due to their large surface area and, consequently, their high activity, that can potentially overcome the wavelength limitation of current TiO2 nanostructures towards broader photon absorption. This characteristic property is particle size dependent.12,30,38,39 Additionally, many physical and chemical properties of quantum-scale particles, have been found to be distinctive from those of bulk and nanoscale materials. These include the tunability of their fluorescence wavelength by the particle size, their high quantum yield, photostability, and high resistance to photobleaching, along with an ease to penetrate into the cellular membrane due to their physical properties, including size (<10 nm), surface charge, and chemical composition, which leads to their ability to covalently bioconjugate to proteins, antibodies, and to be electrostatically linked to DNA, eliminating the need for biomarkers and labeling.30,35,40
One approach to enhance Raman characteristics of the TiO2 based nanostructures is by elemental doping with either metallic or non-metallic materials.41,42 Doping has a major impact on the band structure and trap states of TiO2, affecting important properties such as the conduction band energy and charge transport, recombination and collection.31,41,43 Another approach to achieve similar effects, is by introducing defects in the TiO2 structure via self-doping to introduce oxygen vacancies (OVs).44,45 Engineering of oxygen defects has recently attracted attention as a novel approach to improve the electro-catalytic activity of metal-oxides. The presence of the OVs in metal-oxides has shown to improve their performance to be used in different fields such as regenerative fuel cells, electrolytic cells, and metal–air batteries, which is determined by electrocatalytic activity of the induced OVs.46 Moreover, OVs as an interstitial defect are found to have a critical impact on material properties, such as the electronic structure and the narrowing of the bandgap, ionic/electronic conductivity and magnetic properties, while it offers stronger exciton resonance.46,47 However, the influence of the OVs on SERS enhancement of TiO2 based nanostructures has not been fully studied.
The defect states of TiO2 are highly dependent on the homogeneity of the dopant within the matrix structure. The self-doping strategies in TiOx materials, not only help to retain the intrinsic crystal structure of TiO2 but also help to overcome many problems associated with elemental doping, such as toxicity. Nevertheless, the influence of self-doped OVs on the SERS-activation of TiO2 still remains unknown. Non-plasmonic attributions of TiO2 nanostructures are currently being investigated to improve the potential use in SERS applications. Size reduction of surface features to the quantum-scale, as well as inducing atomic defects such as OVs, have been theoretically proven to contribute to the plasmonic attribution of TiO2.48 To the best of our knowledge the incorporation of OVs in quantum-scale to induce TiO2 SERS-activity comparable to that of noble metals has never been demonstrated before.11,24
This paper presents a quantum-structured (Q-structured) TiOx template, that not only overcomes the limitation of nanoscale TiO2 on the range of wavelengths that can be effectively absorbed, but also whose self-doped OVs lead to an increase of charge carriers. This effectively reduces the bandgap energy of the material and intensifies the SERS effect. The concurrent incorporation of a quantum-structure and of OVs, mediates plasmonic activation of the inherently non-plasmonic TiO2 without any noble plasmonic metals. Moreover, the uniquely synthesized ‘self-doped’ TiO2 quantum particles with OVs are self-assembled and structurally formed as a three-dimensional structure that exhibits appreciable SERS in the order of 107 in magnitude at 785 nm Raman wavelength. Quantum scale TiO2 can only increase enhancement factors (EF) by up to 105 (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold), as opposed to OV-incorporated samples, which endow a limit of detection below 10−9 M. The use of this non-plasmonic Q-structured TiOx, is proposed as a means to address the restrictions imposed by plasmonic templates for molecular sensing, in an effort to address early diagnosis of breast cancer. The conceptual scheme of this paper interlaces the design, fabrication, and assessment of the proposed material for the detection of individual EGFR peptides in multicomponent mixtures, eliminating the limitation of SERS-biosensing where the signatures of numerous molecular components are interfering with each other causing ambiguity. This method allows detection of peptides composing the EGFR protein, with a sensitivity within the 10−9 M range. The significant SERS detection of EGFR biomarker demonstrated in this paper, is expected to advance the forefront of breast cancer diagnostics. Finally, in the presence of multiple peptides, a specificity down to the single molecule level and the ability to differentiate between healthy fibroblasts and cervical cancer HeLa cells is also demonstrated.
000-fold), as opposed to OV-incorporated samples, which endow a limit of detection below 10−9 M. The use of this non-plasmonic Q-structured TiOx, is proposed as a means to address the restrictions imposed by plasmonic templates for molecular sensing, in an effort to address early diagnosis of breast cancer. The conceptual scheme of this paper interlaces the design, fabrication, and assessment of the proposed material for the detection of individual EGFR peptides in multicomponent mixtures, eliminating the limitation of SERS-biosensing where the signatures of numerous molecular components are interfering with each other causing ambiguity. This method allows detection of peptides composing the EGFR protein, with a sensitivity within the 10−9 M range. The significant SERS detection of EGFR biomarker demonstrated in this paper, is expected to advance the forefront of breast cancer diagnostics. Finally, in the presence of multiple peptides, a specificity down to the single molecule level and the ability to differentiate between healthy fibroblasts and cervical cancer HeLa cells is also demonstrated.
Results and discussion
The TiOx quantum particles were self-assembled via a multiphoton ionization growth mechanism driven by the USLP on the Ti substrate. The plasma condensation mechanism was regulated, and the kinetics of the plume were modified through the alteration of the ionic content, which helped to optimize the ionization process and tune the plume temperature, enabling us to design the method of self-doping and self-assembly for the disordered multiphase TiOx at the atomic level, and thereafter engineer the three-dimensional Q-structured TiOx. The transformation from the ionized state of Ti to the self-assembled Q-structure was initiated by breaking the Ti atoms down to form ions within the plasma plume driven by the femtosecond laser. To deprave the plasma plume from oxygen ions and hence determine the influence of OVs in the atomic structure of the TiO2 Q-structures, followed by inducing non-stoichiometric ionization plume, nitrogen gas was introduced. This method of fabrication has an advantage of being highly programmed so that multiple variables can be manipulated concurrently. The strike of femtosecond laser pulses transfers the multiphoton energy to the Ti substrate above its ionization threshold, which results in the formation of a plasma plume composed of Ti ions that recombine to form a desired morphology and physicochemical properties, which can be tuned by the impinging laser pulse parameters.The morphological and compositional characterizations were carried out using FE-SEM, HR-TEM, XPS, and XRD analysis by which the influence of the laser parameters on the physiochemical properties of the fabricated Q-structured TiO2 and TiOx were revealed. Fig. 1(a-1) and (b-1) show FE-SEM micrographs, while Fig. 1(a-2), (a-3), (b-2) and (b-3) HR-TEM images of the fabricated Q-structured TiO2 and TiOx templates fabricated at a 214 fs laser pulse width. As it can be seen by the FE-SEM images, the fused quantum scale particles have formed self-assembled three-dimensional structures. Comparing the TiO2 and the TiOx FE-SEM images, it can be seen that inducing OVs does not affect the structural morphology of the generated templates. To determine the influence of deprived plasma plume from reactive oxygen ions on average particle size and morphology of the fabricated Q-structured TiO2, HR-TEM imaging was performed. As depicted in the Fig. 1(a) and (b) this unique self-assembled 3D Q-structures are comprised of spherical particles with an average diameter of 5–15 nm (Fig. 1(c)). As it can be seen from the data of Fig. 1(c), the average particle size remained approximately the same in the TiOx samples, supporting the FE-SEM results. However, this alters the stoichiometry of the TiO2. HR-TEM images (Fig. 1(b-2) and (b-3)) evidenced the presence and prevalence of OVs within the crystal lattice structures of the Q-structured synthesized in the oxygen-deprived environment, unlike their counterpart fabricated in ambient conditions (Fig. 1(a-2) and (a-3)). To further differentiate the observed phenomenon from other crystal point defects, such as vacancies and interstitial defects, XPS analysis was performed (Fig. 1(d-1)). The XPS elemental and compositional analysis (Fig. 1(d-1) and (d-2)) revealed the chemical and electronic state of the Q-structured TiO2 fabricated at atmospheric (red line) and the Q-structured TiOx fabricated at depleted oxygen atmosphere (black line). The effect of changes in oxygen stoichiometry due to the different atmospheric conditions during the fabrication process is evident when comparing the TiO2 and TiOx spectra of the synthesized Q-structures in Fig. 1(d-2). Deconvolution of the XPS spectrum within the oxygen band (Fig. 1(d-3)), verified the presence of OVs in the TiOx template.
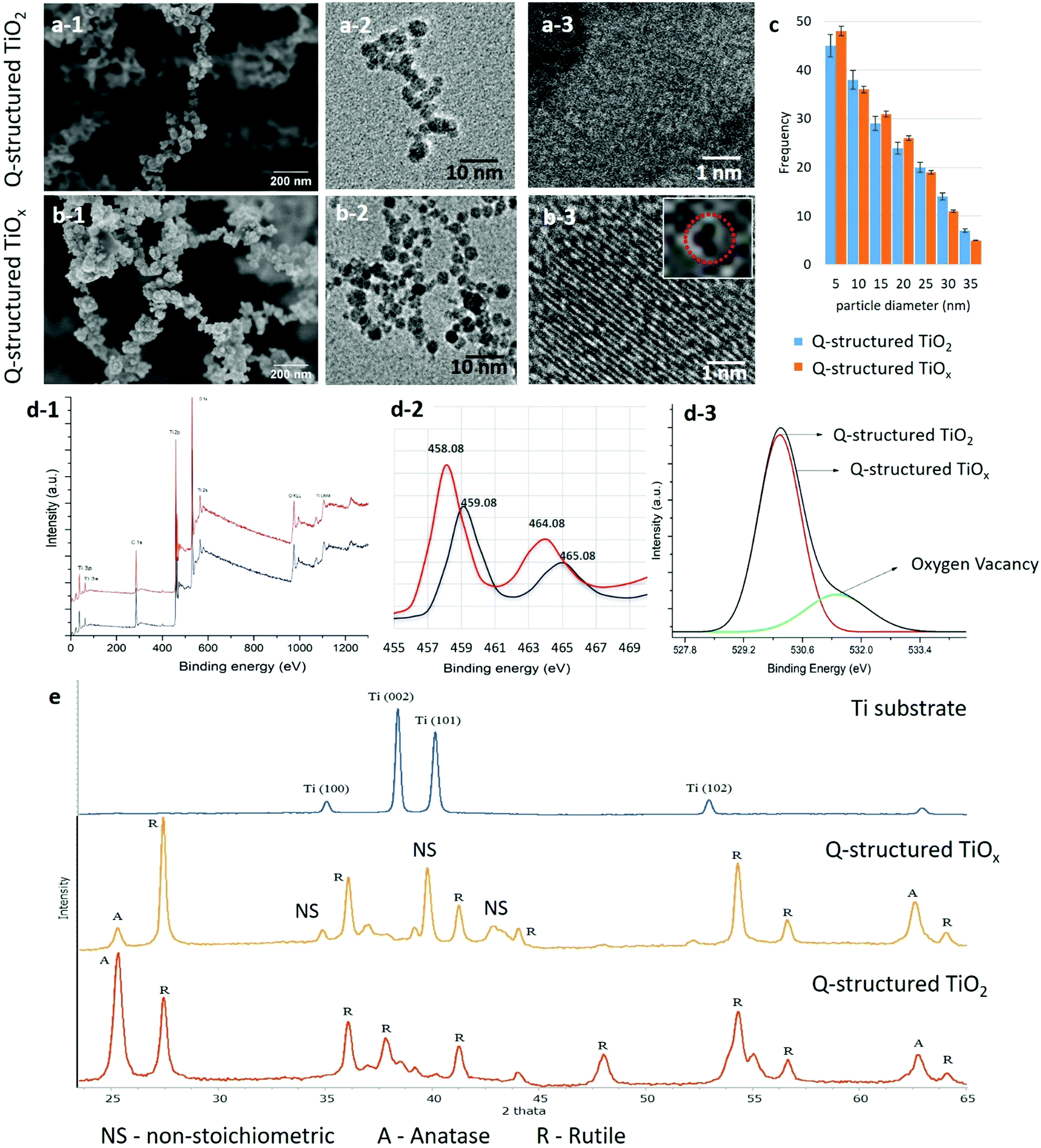 | ||
| Fig. 1 Material characterization of the synthesized self-doped Q-structured TiO2 and Q-structured TiOx. Depleted laser plasma plum from reactive oxygen ions resulted in dominant OVs in the crystal structure of the Q-structured TiOx. (a-1) FE-SEM micrographs of the synthesized Q-structured TiO2 and (b-1) self-assembled Q-structured TiOx. HR-TEM micrographs of the (a-2, a-3) Q-structured TiO2 and (b-2, b-3) the Q-structured TiOx quantum particles, with the latter showing the presence of OVs (inset corresponds to the TEM image of OVs (b-3)). (c) Comparison of the particle size distribution of Q-structured TiO2 and Q-structured TiOx obtained from the HR-TEM images with ImageJ. (d-1) XPS spectra of the Q-structures; (d-2) Ti 2P spectra and (d-3) deconvolution of O 1s spectra. (e) XRD patterns of the Ti substrate, Q-structured TiO2 and self-doped Q-structured TiOx depicted in section, where non-stoichiometric phases such as OVs and Ti3O are dominant in the matrix of the Rutile phase. | ||
The XRD spectra (Fig. 1(e)) obtained from the Q-structured TiO2 and Q-structured TiOx reveal the structural and phase changes at plasma plume saturated with oxygen ions and depleted plasma plume from reactive oxygen ions by introducing the nitrogen gas. The sharp and narrow XRD peaks obtained from Q-structured TiO2 and Q-structured TiOx imply the crystallinity and the small particle size of the fabricated Q-structures. However, the comparison of the XRD patterns of Fig. 1(e) obtained from the Ti substrate (blue), the Q-structured TiOx (yellow) and the Q-structured TiO2 (red), reveals that fabrication of the Q-structure at atmospheric condition plasma leads to the formation of a Rutile dominant phase (Fig. 1(e) red spectrum) whereas a non-stoichiometric oxide of titanium emerges in the Q-structured TiOx (Fig. 1(e) yellow spectrum). Therefore, the XRD analysis (Fig. 1(e)) indicates that by alteration of the atmospheric condition in which the Q-structures were fabricated, non-stoichiometric states of titanium oxide (Ti3O, Ti3O5, (TiO0.316)7.36) can be formed. The presence of the TiOx phase within the Q-structure evidenced with XRD, compliments the XPS results of Fig. 1(d-1).
Fig. 2(a-1) and (b-1) compare the UV-Vis spectra obtained from Q-structured TiO2 and TiOx samples. The photon absorption attribution of the samples was measured by using a spectrophotometer and a deuterium-halogen light source. The absorption spectrum of the base titanium substrate was also acquired as a reference and is shown for comparison in Fig. 2(a-1) and (b-1). Spectra obtained from samples fabricated with different laser pulse width parameters from 214 fs to 1428 fs, which lead to different achieved mean particle sizes from 5 nm to 12 nm are shown for comparison. A wide-range absorption spectrum ranging from 200 nm to 1000 nm was employed and higher absorption, particularly at lower wavelengths, was observed on the Q-structured TiOx. The average enhancement when compared with the base titanium substrate is five folds higher within the visible region and seven folds higher in the near infrared region (NIR) for both Q-structured TiOx and Q-structured TiO2. However, it was observed that the Q-structured TiOx exhibits a greater absorption enhancement in the 300–500 nm range. Higher absorption actively in Q-structured TiOx compared to Q-structured TiO2 is due to an increase in OVs, which results in a higher carrier density and thereby, enhances the electrical conductivity. Finally, as it can be seen in Fig. 2(b-1), as the particle size decreases the absorption of TiOx is enhanced.
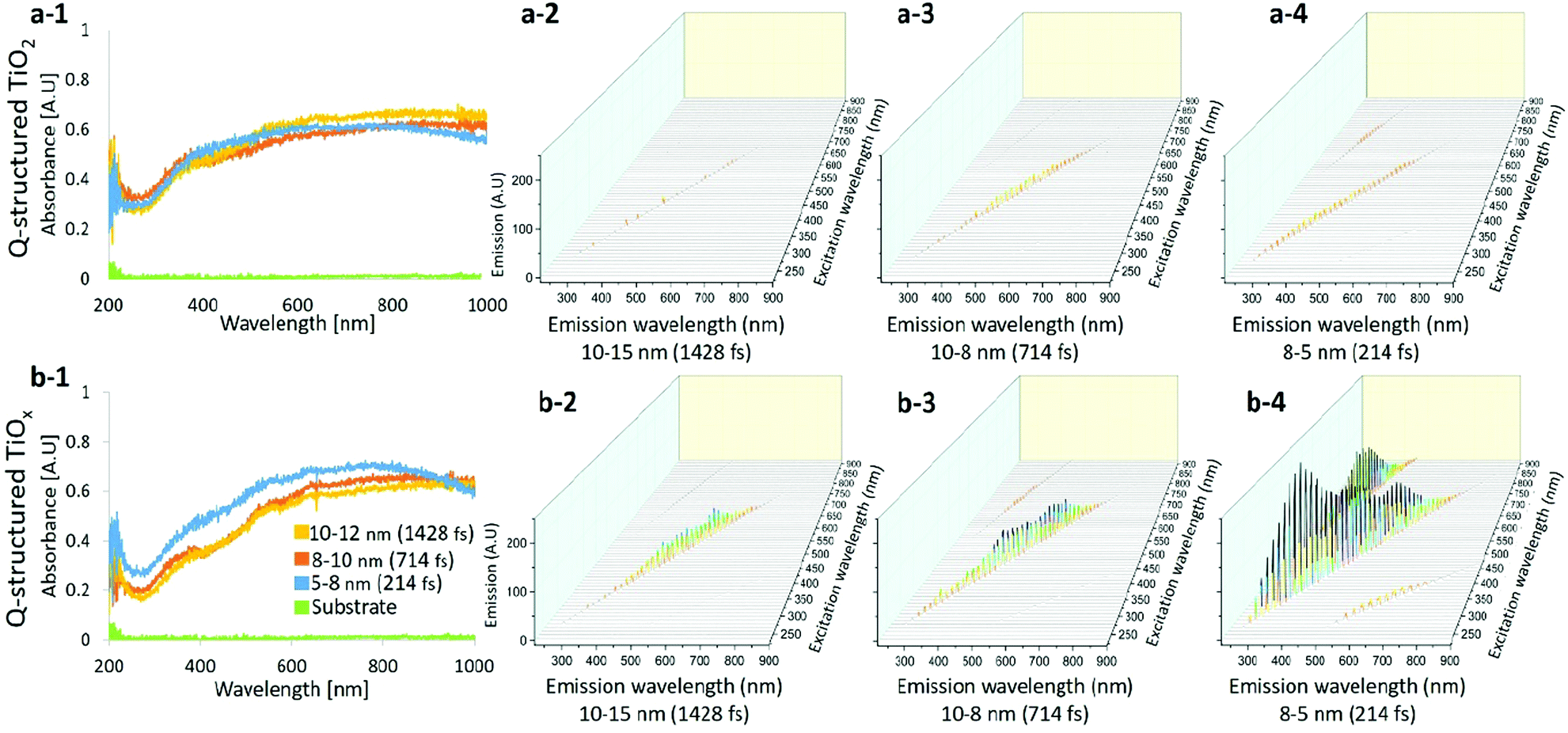 | ||
| Fig. 2 UV-Vis absorption and excitation/emission spectra obtained from the Q-structured TiOx and Q-structured TiO2 templates. UV-Vis (a-1) obtained from the Q-structured TiO2 and (b-1) from Q-structured TiOx show the absorption correlation with particle size; as the particle size decreases the absorption of TiOx is enhanced. Excitation/emission spectra exhibits higher excitation of (a-2–a-4) the Q-structured TiO2 compared to that of (b-2–b-4) the Q-structured TiOx. | ||
The excitation emission spectra of both the Q-structured TiOx compared to Q-structured TiO2 are plotted and shown in Fig. 2(a-2)–(a-4) and (b-2)–(b-4). As shown in Fig. 2(b-2)–(b-4), the Q-structured TiOx exhibits higher emission at smaller particle average size. Furthermore, it was observed that excitation at visible and NIR wavelengths ranging from 600 to 900 nm lead to emission spectra in the range of 350–400 nm wavelengths, which is due to an up-conversion attribution of the Q-structured TiOx that shall be further investigated as a future study.
Furthermore, the corresponding bandgap energy (eV) for Q-structured TiO2 and Q-structured TiOx were calculated to be 3.26 eV and 2.11 eV, respectively. The XPS survey also showed an average valence band (VB) of 2.43 eV for Q-structured TiO2 and 1.8 eV for Q-structured TiOx. To calculate the bandgap energy, the Kubelka–Munk (K–M or F(R)) method was used (more details can be found in the ESI†).
As SERS is highly dependent on amplification of an electromagnetic field on the surface of a material and plasmonic effects, increased absorption of incident light together with plasmonic coupling effects present in the nano gaps between Q-structured particles of the TiOx has propelled an attempt to exploit the Q-structured TiOx as a SERS template towards biosensing of biomarkers such as EGFR and potential cancer diagnosis.
To examine the SERS capability of the Q-structured TiO2 before and after incorporation of the OVs, the Raman excitation wavelengths of 532 and 785 nm were employed, and Raman spectra were obtained from the bare samples in the absence of any dyes. Fig. 3(a) and (b) show the SERS spectra acquired from the Ti substrate, self-assembled 3D structured quantum scale Ti particles before (Q-structured TiO2), and after introducing the OVs (Q-structured TiOx), fabricated with laser pulse width at 214, 714 and 1428 fs. The stability of the OVs induced into the Q-structured TiOx templates have been evaluated over long exposures of 360 s to the Raman excitation laser of 785 nm, at a high intensity of 250 mW μm−2 and for 120 cycles. These parameters are beyond the time and intensity that have been used in this study for SERS measurements (2.5 mW μm−2 for 3 s and 3 cycles). As evident from Fig. S2 of the ESI,† no shift and deterioration on the characteristic peaks of the Q-structured TiOx have been observed. As it is shown in Fig. 3(e), increasing the laser pulse width used in the fabrication of the proposed SERS-templates from 214 fs to 714 fs and 1428 fs, increases the average particle size. In turn and according to Fig. 3(c) and (d) smaller particles improve the SERS effect of the realized templates. This is true for both the TiO2 and the TiOx templates.
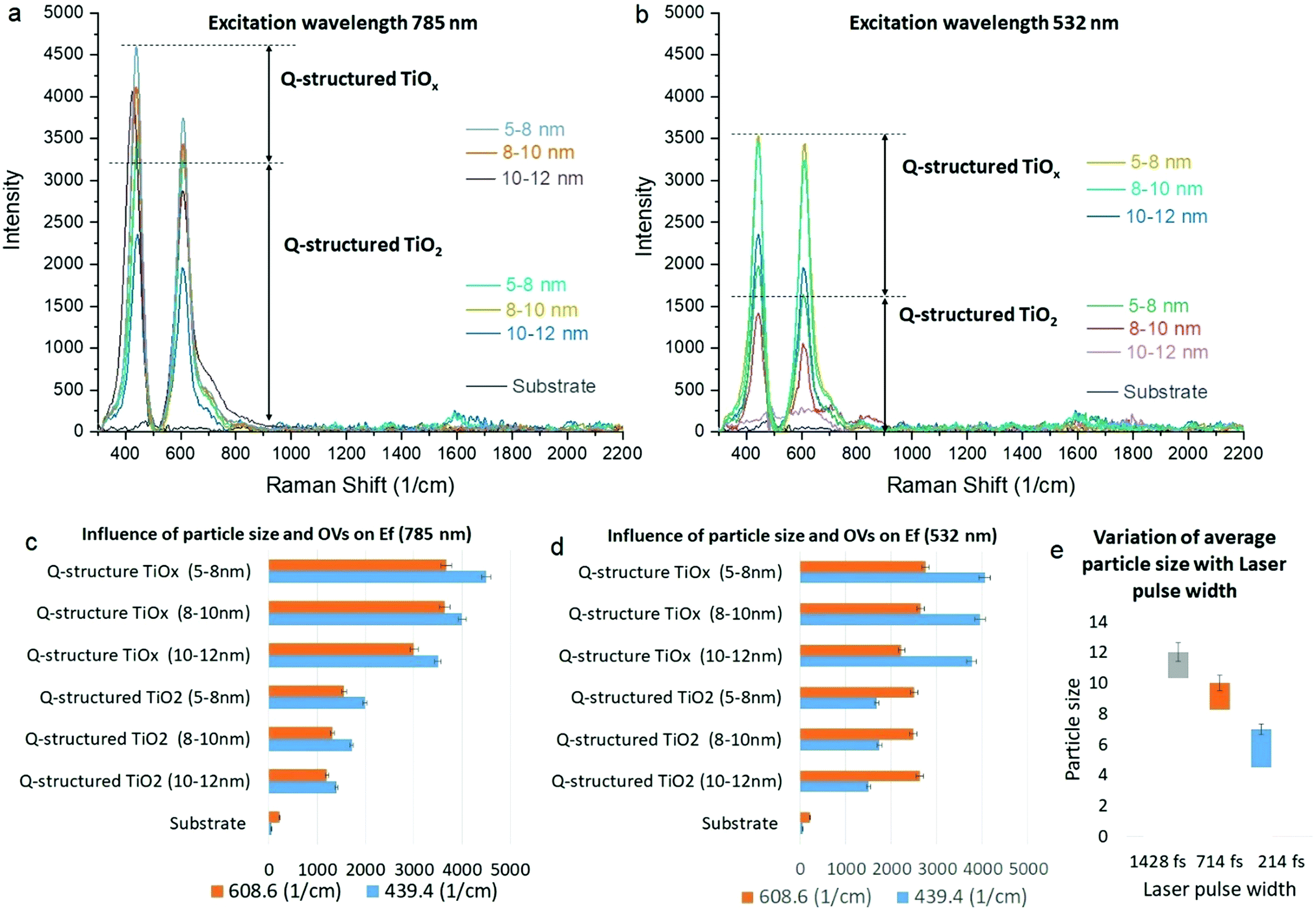 | ||
| Fig. 3 SERS spectra at (a) 785 nm and (b) 532 nm laser excitations acquired from the Ti substrate, the Q-structured TiO2 and after incorporation of OVs. The influence of particle size at (c) 785 nm and (d) 532 nm excitation wavelengths on the SERS behavior of the synthesized Q-structures at the two Raman peak locations of 439.4 and 608.6 cm−1. (e) The influence of laser pulse width on the average achieved particle size. | ||
The results of the enhancement fold (EF) obtained at 785 nm and 532 nm Raman excitation wavelengths presented in Fig. 3(c) and (d), respectively, were calculated by the ratio of the template's peak intensity vs. the bare substrate, to establish the SERS peak intensity amplification due to each template at each fabrication parameter. As it can be seen, slightly higher enhancement is achieved at 785 nm compared to 532 nm with TiOx, which is due to the higher absorption of the templates at longer wavelengths, as shown in the Fig. 2(a-1) and (b-1). At 785 nm, EF increases gradually with decreasing particle size, while at 532 nm particle size does not dramatically change the EF. Nevertheless, for both wavelengths higher EF is achieved with TiOx and with the smaller particle size. The highest EF is obtained with the 785 nm excitation. As evidenced by the Raman spectral studies (Fig. 3(a)), peaks of augmented intensity were observed with the Q-structured TiOx, further enhancing the induced SERS effect within the same range of laser intensities used for the realization of the templates, when compared with the fabricated TiO2 substrates. Although our results evidenced that by scaling the particle size to the range of quantum scale significant enhancement can be achieved, inducing OVs has demonstrated augmented SERS behaviour. It is well known that Raman enhancement is caused due to a highly amplified electromagnetic (EM) field developed on a nanostructured material surface, due to an increased absorption of incident light. Hence, a higher SERS effect evidenced with the quantum scaled structures can be inferred as an increased amplification of plasmonic effects, due to the gaps between quantum particles. Furthermore, our observation (Fig. 3) unveiled that the augmented SERS phenomenon has not only been attributed to the amplified EM field at the material surface, but that it is further improved by the induced atomic defects (OVs) within the crystal planes of the Ti-based Q-structure. Consequently, for all subsequent measurements, templates synthesized with a 214 fs laser pulse width were used, resulting in a Q-structured TiOx with a particle size of 5–8 nm.
As discussed, TiOx Q-structures synthesized at depleted oxygen environment exhibit a significant photon excitation and emission capability over a broad spectral range (Fig. 2(b-2)–(b-4)). This propelled an attempt to quantify the latent Raman sensing capability of TiOx with OVs. To achieve this, crystal violet (CV), a chemical dye was used to examine the Raman enhancement capability of the synthesized Q-structure. CV dye has a large Raman cross section and is thus commonly used to demonstrate SERS. To examine the SERS effect on the TiOx Q-structure, CV dye was dispensed on its surface and the Raman spectra were acquired. To aid in the EF calculation of the TiOx Q-structure, which is a value used to evaluate the Raman sensing capability of a material, the CV dye was also coated onto Q-structured TiO2 and bare titanium substrates and its Raman spectra were also measured. Fig. 4 shows the Raman spectra of the CV dye on the bare titanium substrate, Q-structured TiO2 and the Q-structured TiOx templates.
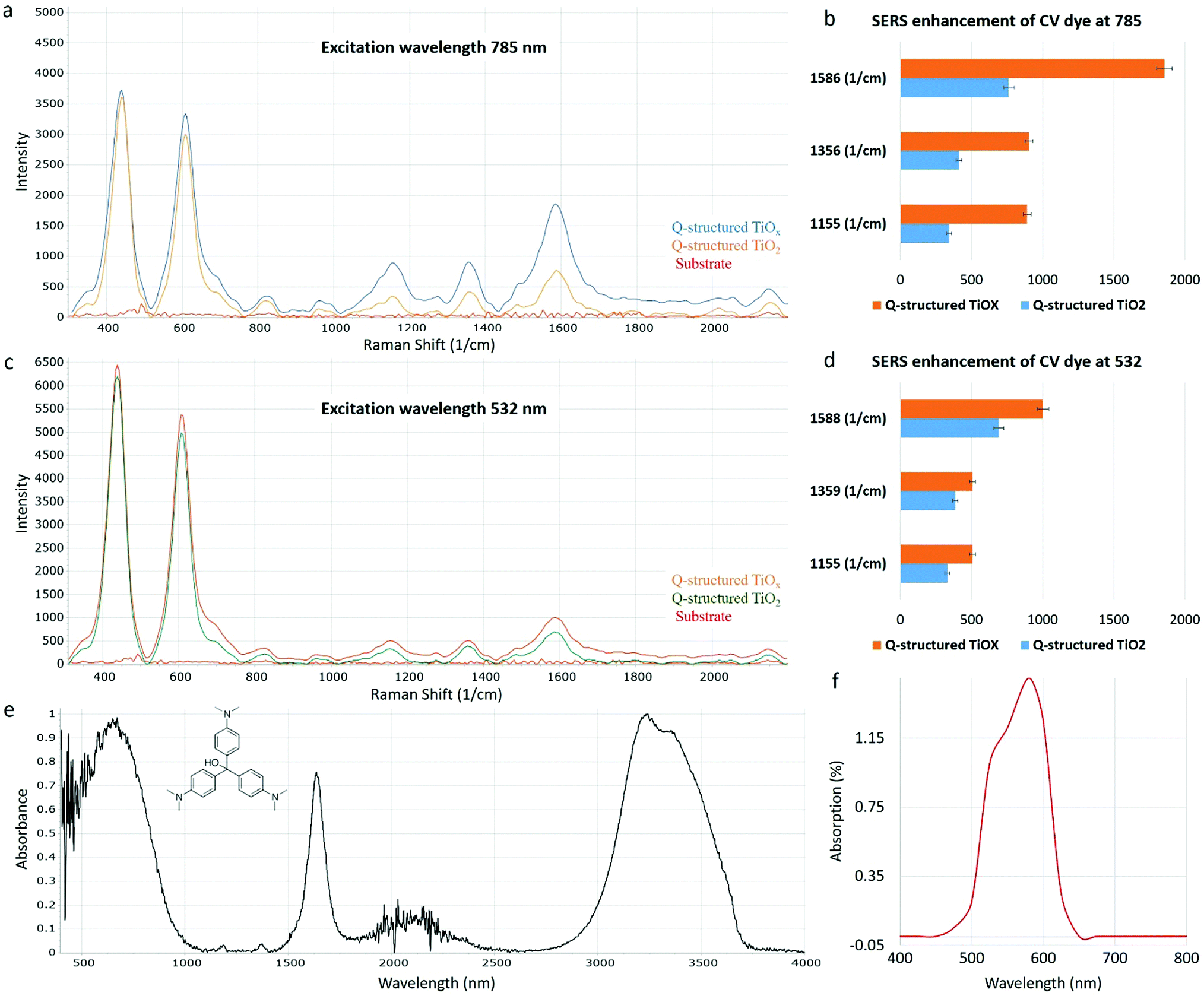 | ||
| Fig. 4 Raman spectra of 1 nM concentration CV dye solution acquired at (a) 785 and (c) 532 nm wavelength from the Q-structured TiO2 and Q-structured TiOx. The characteristic peaks of the CV dye re evident at (a) 1155, 1356 and 1586 cm−1 at 785 nm and (c) 1155, 1359 and 1588 cm−1 at 532 nm wavelengths. The EF of these characteristic peaks of CV dye on the Q-structured TiO2 and Q-structured TiOx are calculated and plotted for the (b) 785 and (d) 532 nm wavelengths. (e) Fourier transform infrared (FTIR) spectroscopy and (f) absorption spectrum of the 1 nM concentration CV dye. | ||
The SERS activities of the Ti-based Q-structures were further investigated by using CV dye. As depicted in Fig. 4, the SERS spectra of CV have been acquired at 785 (Fig. 4(a)) and 532 nm (Fig. 4(c)) Raman excitation wavelengths on the Q-structured TiO2 and on the Q-structured TiOx templates at a nanomolar (10−9 M) concentration, which represents the limit of detection. This was found experimentally by measuring dilutions of CV from mM to nM concentrations. The TiOx Q-structure exhibits higher photon absorption at a wavelength of 785 nm, as seen in Fig. 2, hence the stronger Raman response obtained in Fig. 4(a) when compared to those obtained in Fig. 4(c). Similarly, with the results of Fig. 3, the enhancement fold improvement on the peak intensity was measured for the Q-structured TiO2 and TiOx templates relative to the bare unmodified substrate. The SERS characteristic peaks of the CV dye at 1155, 1356 and 1586 cm−1 and 1155, 1359 and 1588 cm−1 obtained at 785 at 532 nm Raman excitation wavelengths are shown in Fig. 4(a) and (c) respectively. These peaks together with those from Fig. 3, were then used for the calculation of the EF of the CV dye at the two Raman excitation wavelengths of 785 at 532 nm, using eqn (2) and (3), with the results of Fig. 4 used as ISERS and those from Fig. 3(a) as Icontrol.
As opposed to a 3.5 × 105 enhancement achieved with the TiO2 template, an EF of 3.4 × 107 was determined for the Q-structured TiOx, which is comparable to those achieved with plasmonic noble metals. For instance, an EF of 1.72 × 105 has been achieved for Ag nano-stars, a maximum EF of 107 for gold nanoparticle arrays and an EF of 2 × 109 for Ag nanowire monolayers.51–53 Such a significant EF in the order of 107 calculated for Q-structured TiOx, is appreciably high, considering that with most plasmon-free TiO2-based semiconductor nanomaterials an EF of 104 has been reported.41,54 Presence of OVs and crystal defects is hypothesized to be the cause of high SERS sensitivity in the proposed Q-structured TiOx. This could explain the high orders of magnitude of the EF calculated for the Q-structured TiOx. To the best of our knowledge, this is the first time that ‘self-doping’ has been introduced to self-assembled quantum scale TiOx spherical particles in the form of a complex three-dimensional structure, exhibiting SERS capabilities similar to plasmonic materials.
When comparing the results of Fig. 3(a) and (b) with Fig. 4(a) and (c) it can be seen that the intrinsic Raman peaks of the substrate at ∼430 nm and ∼608 nm still remain when adding the CV dye (as expected), but more importantly that these are not overlapping and do not interfere with the Raman characteristic peaks of the CV dye. When comparing the results obtained with the two different wavelengths and the CV dye in Fig. 4, it is obvious that stronger dye peaks relevant to the intrinsic substrate peaks are obtained with the 785 nm laser. This is more evident from the EF calculations of the different peaks obtained from the two wavelengths used, shown in Fig. 4(b) and (d). As it can be seen, almost twice the EF can be achieved with the 785 nm laser excitation. In all cases the results obtained with the Q-structured TiOx template surpass those obtained from the Q-structured TiO2 substrate. In both plots, Raman spectra obtained from the unmodified substrate are shown for reference. The biological optical window is considered to be between 600 cm−1 to 1800 cm−1.55,56 Consequently, the intrinsic Raman spectral properties of the proposed templates do not interfere significantly. The above are further supported in the Raman spectra acquired from the cell lines in Fig. 5, where the peaks of the substrate are significantly suppressed and with most of the peaks being beyond 600 cm−1.
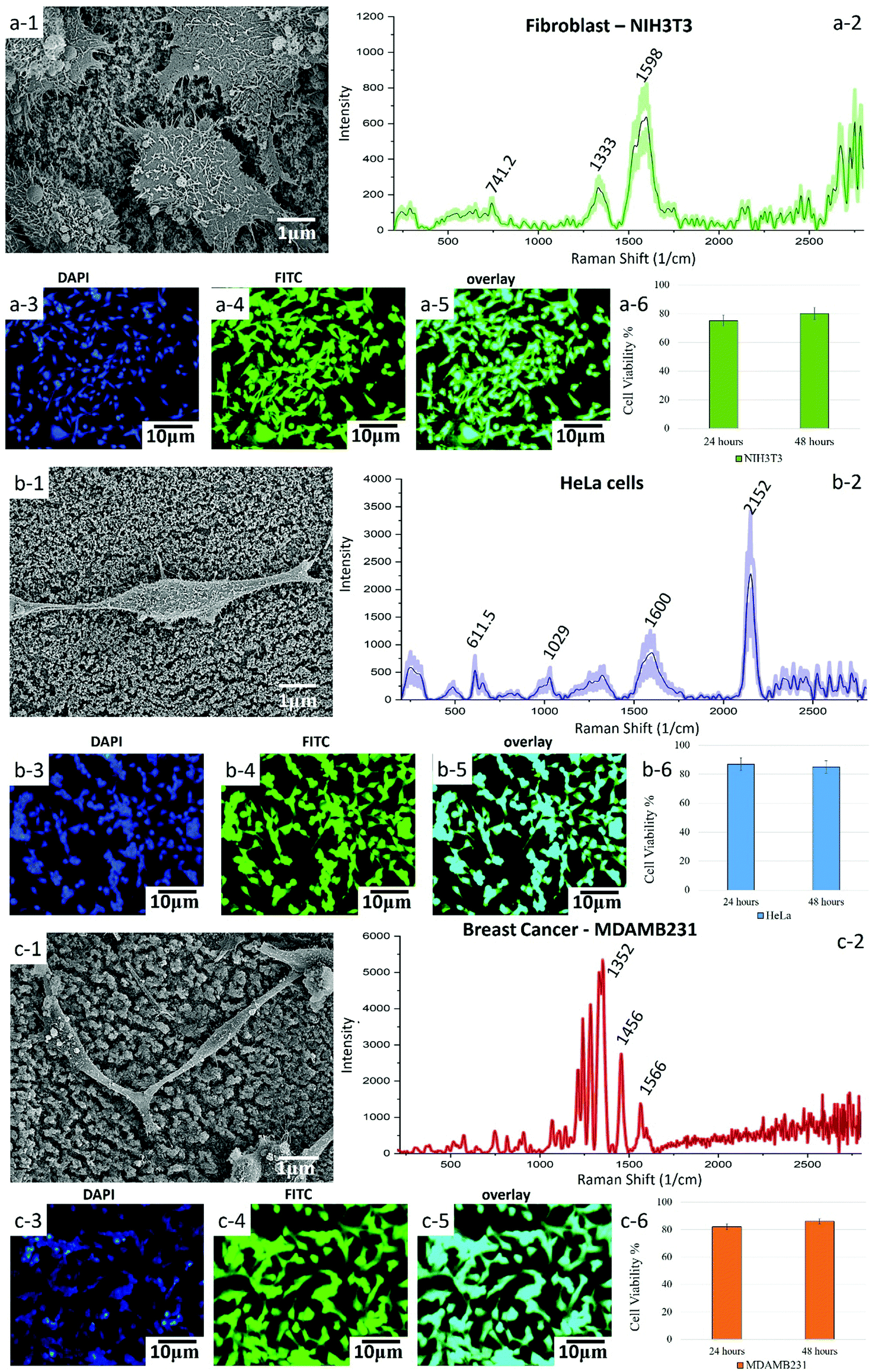 | ||
| Fig. 5 SEM, fluorescent microscopy, cell viability and SERS (at 785 nm) studies of fibroblast, HeLa and breast cancer cells with the Q-structured TiOx template fabricated with the 214 fs laser pulse width. (a-1, b-1 and c-1) SEM micrographs show the morphology and interaction of a cells with the Q-structured TiOx. SERS spectra depicts molecules within cells, such as proteins, nucleic acids, and lipids obtained from (a-2) fibroblast, (b-2) HeLa and (c-2) breast cancer cells seeded on the Q-structured TiOx after 12 hours. Confocal fluorescent microscopy images show in (a-3, b-3 and c-3) the nucleus stained in blue with DAPI, in (a-4, b-4 and c-4) the cytoskeleton stained in green with FITC and in (a-5, b-5 and c-5) the overlay images taken form fibroblast, HeLa and breast cancer cell lines, respectively. Cell viability and cytotoxicity assessment (a-6, b-6 and c-6) evidence the nontoxicity of the Q-structured TiOx on fibroblast, HeLa, and breast cancer cells after 24 and 48 h of culture. | ||
FTIR spectroscopy was also used, as a means of identifying the main characteristic peaks of the CV dye. Fig. 4(e) shows the FTIR spectrum of CV acquired in the range of 4000–400 cm−1. A broad peak at 3413 cm−1 is assigned to the overlap of N–H and O–H groups. The peaks at 1645 and 1524 cm−1 corresponds to the C O stretching band and the vibrating band of the N–H bond or the C–N. Fig. 4(f) shows the UV-Vis absorption spectrum of CV. The maximum absorption band of the CV dye solution is at λ = 586 nm. Using an excitation wavelength within about a 250 nm separation between the maximum resonant frequency of an analyte, leads to molecular resonance contributing to the overall Raman enhancement. This is known as surface-enhanced resonance Raman spectroscopy (SERRS). Although both wavelengths are within this range (with the 532 nm being closer to the resonant frequency of the dye), greater enhancements are achieved from the 785 nm excitation.57,58
Further investigation into the use of Q-structured TiOx for improved SERS can help in its prospective use in molecular detection and sensing. Therefore, to investigate the capability of the synthesized template for point-of-care diagnosis of Epidermal Growth Factor Receptors (EGFR) as a biomarker of breast cancer, breast cancer (MDAMB231) cells were cultured onto the Q-structured TiOx and compared with non-cancerous dermal fibroblast cells (NIH3T3). To further investigate the cancer diagnostic capabilities of the proposed SERS template, cervical cancer cells (HeLa) were also analyzed. The first and foremost requirements for a temple to be used for cellular analysis, is to determine its biocompatibility and to examine cell adhesion and proliferation. To assess these, SEM micrographs were obtained, as depicted in Fig. 5(a-1), (b-1) and (c-1). These images revealed that the cells were adhering and proliferating onto the Q-structured TiOx platform. This was evidenced by the expansion of the lamellipodia and filopodia extensions of the cells, which are known as the cellular anchors to the extracellular matrix.59 Furthermore, a cell viability assay has been carried out on each cell line for 24 and 48 h where no sign of cytotoxicity has been observed; an average of 70 percent viability in the first 24 and about 80 percent after 48 h have been achieved. The fact that after 48 h cell viability is higher is interpreted to be due to cell proliferation over the prolonged culturing time. This analysis together with fluorescent imaging and SEM observations indicated the biocompatibility of the template for both healthy and cancerous cells after 48 h of cell culture. The fluorescent images obtained by staining the nuclei and cytoskeleton in blue (DAPI) and green (FITC) are depicted in Fig. 5(a-3), (a-4), (b-3), (b-4), (c-3) and (c-4) for fibroblast, HeLa and breast cancer cells, respectively.
Having assessed biocompatibility, SERS spectra were collected. These are shown in Fig. 5(a-2), (b-2) and (c-2). As it can be seen in the SEM and fluorescent images of Fig. 5 and their scale bars, the average cell diameter is smaller than the spot size of the laser (10 μm) and it is large enough to cover a single cell. Consequently, we could not examine separately different regions of single cells. Raman spectra from 3 different single cells were collected and then averaged and the error bars represent the SD. The presence of the 574.5 and 611.5 cm−1 peaks in Fig. 5(a-2), (b-2) and (c-2) indicate phosphatidylinositol (phospholipid), which is a main constituent of the cell membrane. The peaks at 1029 cm−1 was assigned to RNA and peaks at 2400–2800 cm−1 region mainly represent the overexpression of C–H vibrations in lipids. The mammalian fibroblast cell (NIH3T3, Fig. 5(d)) demonstrated quinoid ring vibrations at around 1200 cm−1, as well as antisymmetric phosphate stretching vibrations at 1322 cm−1. The intensity of these peaks was, however, much smaller, when compared to the intensity of the corresponding peaks obtained from cancerous breast cells (MDAMB231, Fig. 5(c-2)).
The SERS spectrum acquired from the breast cancer cell line with the Q-structured TiOx depicted in Fig. 5(c-2) demonstrated the characteristic peaks of the EGFR peptide observed at 1134, 1242–1284 and 1566–1610 cm−1.60–64 Furthermore, spectral comparison between fibroblast (Fig. 5(a-2)) and breast cancer (Fig. 5(c-2)) shows that breast cancer cells (MDAMB231) are characterized by stronger vibrational modes of lipid peaks in the region of 2600–2800 cm−1. This excessive lipid presence can serve as an additional (together with the highly specific EGFR peaks) signature of malignancy in breast cancer cells.65 Moreover, the spectrum acquired from the breast cancer cell line showed a significantly intense peak at 1070 cm−1, which is assigned to phenylalanine. Additionally, breast cancer cells displayed CH2 deformation at 1456 cm−1, as well as a high concentration of nucleic acid at 1566 cm−1 and overexpression of cholesterol and cholesterol ester at 1200–1400 cm−1. High concentration of cholesterol is a signature of cancer cells, associated with damage to the mitochondrial membrane. Detection and differentiation of cancerous cells was done based on comparison of Raman intensities of breast cells and non-cancerous cells (fibroblast) at 1465 to 1322 cm−1 and 1566 to 1599 cm−1, respectively. Furthermore, the ratio of lipid-to-protein content was significantly higher for breast cells as compared to fibroblast cells, which serves as another indication of a high concentration of lipids among cancer cells. This is supported by immunological studies in the literature.64,66
Fig. 5(b-2) shows a SERS spectrum acquired from HeLa cells. The Raman peak at 611.5 cm−1 is assigned to a vibrational mode of the pyrrole ring in cytochrome c, which is crucial in ATP generation. The Raman signal observed at 611.5 cm−1 represents the mitochondria, as cytochrome is localized in mitochondria. The 1600 cm−1 band is assigned to the amide I vibrational mode of peptide bonds, while the 2717 cm−1 band ascribes to the symmetric stretching vibration of CH2, which is related to lipid droplets, as well as membranes of cell organelles, such as mitochondria and Golgi apparatus.14 However, further investigations and analysis are required, using e.g. principal component analysis, to establish the cervical cancer detection capabilities of the proposed template. This is since cervical cancer cells are not associated with a distinct specific biomarker as are breast cancer cells with EGFR.
A list of tentative spectral assignments, Raman shifts, and cellular functional groups and the relevant references for these, is provided in the ESI† (Table S1). Listed wavenumbers, however, may vary as the given peak position is directly related to the resolution of the used spectrometer. The Raman peaks assigned to vibrational modes are verified in the literature.
It has to be noted that the performance of the proposed SERS template has been evaluated in pH 7 environments only, as our work currently focuses on SERS biosensing in cell cultures, which are typically kept at a pH 7. Future work will examine the performance of the proposed template for strong alkali and acidic environments to explore other potential applications. The next steps in this work involve assessing the capabilities of the proposed template in cultures containing both healthy and cancer cells to differentiate between the two, as well as its use with biopsy samples in vitro, towards our ultimate goal of using such approaches for in vivo and in situ optical biopsies.
Conclusion
In summary, we have introduced a novel concept of concurrently inducing atomic-level defects (OVs), as well as self-assembled quantum scale structures for the realization of an ultra-high sensitivity noble metal-free SERS template for biomolecular detection. Multiphoton ionization used for the fabrication of the Q-structured TiOx provided the advantage of tuning the nano-structuring of the material and the particle dimensions in the quantum scale and at the same time inducing the OVs as atomic-level defects. The synthesis of the Q-structured TiOx demonstrated a limit of detection of 1 nM and an EF of 3.4 × 107 with the CV dye at the 785 nm excitation wavelength. In addition, the sensitivity of the Q-structured TiOx was examined using 3 different cell lines: fibroblast, HeLa and breast cancer cells. The biomolecular signatures of the intracellular compounds were unveiled after culturing the cells on the Q-structured TiOx for 12 hours which lead to identification of EGFR peptide on the breast cells and a higher lipid content when compared to fibroblasts. Similarly, spectra collected from HeLa cells demonstrated distinct features when compared to fibroblasts, but more work is necessary to evaluate the capabilities of the proposed SERS template in cervical cancer diagnostics. To the best of our knowledge, this is the first report on the incorporation of atomic defects into 3D structured Q-scaled TiOx as a SERS template for biomolecular detection. We believe that the proposed template is promising for early cancer detection.Materials and methods
Fabrication of Q-structured TiOx
A self-assembled Q-structured TiOx was achieved by multiphoton ionization through the unique interaction of an ultra-short pulsed laser (USPL) with commercially available grade 2 pure Ti samples in a partial pressure of oxygen. The laser pulse beam was generated using a direct diode pumped Yb-doped fiber amplified laser system (IM-PULSE series ultra-short pulse laser, Clark-MXR Inc., Dexter, Michigan USA). The central wavelength of the Gaussian shaped laser beam was 1040 nm and the pulse repetition rate varied from 200 kHz to 26 MHz. A dual-axis computer-controlled Galvano scanner (Clark-MXR Inc.) supported by EZCAD software was used to scan the laser beam (of 10 μm spot diameter) across the surface of the Ti samples at a scanning speed of 1 mm s−1. The average power of the laser beam was maintained at 16 W and the TiOx Q-structure was synthesized at laser intensity values ranging from 7.4 × 1013 W cm−2 to 7.2 × 1014 W cm−2, and laser pulse width durations of 714 fs and 1100 fs were selected.The Ti samples were cut with a precision diamond cutter to the dimensions of 10 mm × 10 mm × 2 mm and were ground finished and polished to remove minor surface defects, followed by their ultrasonic cleaning in acetone, ethanol and distilled water for 5 minutes each. The Ti samples were then mounted on a flat chuck placed in front of the Galvano scanner. As demonstrated in Fig. S1 in the ESI,† six tubes (Masterflex 6411.14) were arranged in a square bracket and were used to introduce nitrogen gas, which was used to deprive the ionization plasma from reactive oxygen ions. This bracket was placed in parallel and in contact with the chuck on which the Ti samples had been mounted on. This ensured that an uninterrupted and constant gas pressure was introduced at the site of the generated plasma plume. All experiments were conducted at a constant gas flow rate of 4 SCFH (standard cubic feet per hour).
The experiments were performed at a maximum average power of 16 W. A laser spot with a diameter of around 10 μm was scanned across the surface of the specimen using a two-axis galvo scanner with laser pulse width of 214, 714 and 1428 fs and a laser scanning speeds of 50 mm s−1.
It was found that varying the laser pulse duration has direct effects on the average particle size as particles ranging from 5–8, 8–10 and 10 to 12 nm were synthesized at 214, 714 and 1428 fs respectively (Fig. 1). The synthesis of the Q-scaled particles is based on nucleation and a subsequent growth mechanism during vapor condensation, governed by the multiphoton ionization within the laser plasma plume, that leads to self-assembly of 3D Q-structures. Furthermore, both the laser intensity and the density of Ti ions within the plume influence the rate of nucleation and growth, which eventually determines the physicochemical aspects of the synthesized Q-structures. It is known that nucleation initiates when the barrier energy due to surface tension is overcome by the surface energy, which are related to temperature and particle sizes, as described by:49
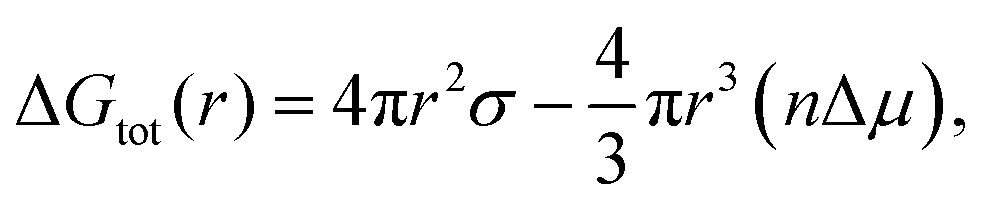 | (1) |
lnS is the difference in the chemical potential between condensed and uncondensed phases, σ is the surface tension, n is the atom number density, k is the Boltzmann constant, T is absolute temperature and S is entropy. Also, the nucleation rate is a function of condensation and the rate of volume growth, once the nuclei is formed, is proportional to its area cross section. The laser intensity and the density of ionic species can thus influence the nucleation and subsequent formation of the Q-structures.
Characterization
Elemental composition and crystallographic analysis of samples synthesized at ambient and partial pressures of oxygen was performed using a Bruker AXS D8 Advance microdiffraction system for X-ray diffraction (XRD) analysis, equipped with a Cu-K source and graphite monochromator to eliminate unwanted Cu-K-beta lines. A 2θ scanning range of 15–60° was used to acquire the relevant peaks associated with the Q-structures. To further characterize the samples X-ray photoelectron spectroscopy (XPS) was performed using a K-Alpha XPS instrument (Thermo Scientific, USA). The structural and surface morphology of the Q-structured TiOx was studied by a field emission scanning electron microscope (FE-SEM, Hitachi SU8230) and a high-resolution transmission electron microscope (HR-TEM, JOEL 2010). The particle size distribution of the quantum particles was measured by using ImageJ TM. The absorption spectrum was acquired for a broadened spectrum of 300 to 1000 nm by using a spectrometer (AvaSpec-2048 Fiber Optic Spectrometer). The excitation/emission spectra were measured using a UV-Vis-NIR spectrofluorophotometer (Shimadzu RF-5301PC). The Fourier transform infrared (FTIR) spectroscopy and absorption spectrum of the CV were acquired using ATR-FTIR (Thermo-Fisher Nicolet iS50) and a UV-vis-NIR spectrophotometer (Hitachi UV-3100), respectively.Cell culture and seeding
The templates were first exposed to UV light for 20 min prior to seeding the cells to sterilize them. Mammalian fibroblasts (NIH3T3), human cervical cancer (HeLa, ATCC, American Type Culture Collection (ATCC) No. CCL-2), and Breast cancer (MDAMB231) cell lines were employed in cell experiments to ascertain the comparative functionality of mammalian and cancer cell lines in response to the fabricated samples. Fibroblast cells were grown in DMEM containing 10% heat-activated Fetal Bovine Serum (FBS) with 1% penicillin–streptomycin antibiotics (Pen–strep). HeLa and breast cancer cells were grown in DMEM-F12 supplemented with 10% fetal bovine serum and 1% Pen–strep separately. Subsequently, the cells were separately cultured on the substrates placed in Petri dishes with a seeding density of 750000 cells per cm2 of substrate surface area. The Petri dishes were placed in an incubator for 12 hours at 37 °C. SERS acquisitions were subsequently performed after discarding the cell culture media and washing the templates three times with PBS and after drying.
Fluorescence microscopy
To stain the samples after cell seeding for fluorescence microscopy, they were fixed in methanol-free paraformaldehyde after which they were incubated in skim milk to prevent nonspecific binding. The actin, cytoskeleton, and nucleus of cells were stained with FITC and DAPI (both from Life Technologies, CA, USA) fluorophore dyes. The samples were then visualized using a Zeiss LSM 710 META upright confocal laser-scanning microscope.Cell imaging and morphology
The cellular morphology of cells seeded on the fabricated samples was observed using a Hitachi SU1510 scanning electron microscope. After the prescribed period, spent media were aspirated. This was followed by fixing of the samples in 2% glutaraldehyde in 0.1 M pH 7.3 sodium cacodylate buffer for 1 h. Next, the samples were immersed in 0.1 M sodium cacodylate buffer with 0.2 M pH 7.3 sucrose for 20 min, followed by dehydration at increasing concentrations of alcohol for 20 min. The samples were then critical point dried and coated by gold with Emitech Mini Sputter Coater Quorum SC7620; at this point, cells were ready to be directly observed using SEM. SEM and fluorescent microscopy were used for the qualitative and quantitative assessment of cell adhesion, morphology and viability analysis of cultured cells on the fabricated templates, as in our previous work.50Raman spectroscopy
Raman spectra of the as fabricated samples were obtained with a confocal Raman microscope at 532 nm and 785 nm at a laser power of 250 mW μm−2 and with an average spot size of 10 μm (SENTERRA II, Bruker, MA, USA) to characterize the fabricated Q-structures to determine the structural/compositional changes at different fabrication parameters. To test the Raman enhancement capability of the nanomaterial, a crystal violet (CV) (Sigma-Aldrich C0775) dye was used for the analysis, a popular dye for SERS analysis due to its large Raman cross-section. The CV dye was dissolved in deionized water to a concentration of 10−9 M and a single droplet of 1 μL volume was placed on the samples with a pipette. A handheld Raman system (Inc NanoRam®, B&W Tek, De, USA) with a wavelength 758 nm and with a maximum power of 2.5 mW μm−2 and an average spot size of 10 μm at the focal point was used to obtain the spectra from the self-assembled Q-structures before and after dispensing the dye onto the surface of the samples. The latter was also used for the cell culture measurements. All acquired Raman spectra were obtained at 3 s integration time and repeated in triplicate then averaged.Calculation of the EF
To investigate the SERS activities of each template, the EF was calculated by using the following equations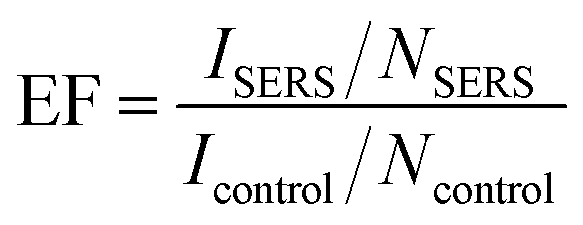 | (2) |
| NSERS = CVNAAARaman/Acontrol, | (3) |
| Ncontrol = MρhARamanNA, | (4) |
All experiments were repeated three times for each sample, as well as the assessment of three samples from each fabrication parameter. Consequently, all Raman measurements are averaged over 3 × 3 = 9 measurements. All error represent the SD.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Natural Science and Engineering Research Council of Canada (NSERC Discovery Grants 126042 and 119087).References
- M. Keshavarz, B. Tan and K. Venkatakrishnan, ACS Appl. Mater. Interfaces, 2018, 10, 34886–34904 CrossRef CAS.
- L. Yang, Y. Peng, Y. Yang, J. Liu, H. Huang, B. Yu, J. Zhao, Y. Lu, Z. Huang, Z. Li and J. R. Lombardi, Adv. Sci., 2019, 6, 1900310 CrossRef.
- D. Sebba, A. G. Lastovich, M. Kuroda, E. Fallows, J. Johnson, A. Ahouidi, A. N. Honko, H. Fu, R. Nielson, E. Carruthers, C. Diédhiou, D. Ahmadou, B. Soropogui, J. Ruedas, K. Peters, M. Bartkowiak, N. F. Magassouba, S. Mboup, Y. B. Amor, J. H. Connor and K. Weidemaier, Sci. Transl. Med., 2018, 10, eaat0944 CrossRef CAS.
- J. S. Teguh, F. Liu, B. Xing and E. K. L. Yeow, Chem. – Asian J., 2012, 7, 975–981 CrossRef CAS.
- K. Y. Goud, S. K. Kailasa, V. Kumar, Y. F. Tsang, S. E. Lee, K. V. Gobi and K.-H. Kim, Biosens. Bioelectron., 2018, 121, 205–222 CrossRef CAS.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS.
- V. S. P. K. Sankara, A. Jayanthi, A. B. Das and U. Saxena, Biosens. Bioelectron., 2017, 91, 15–23 CrossRef.
- A. Kowalczyk, J. Krajczewski, A. Kowalik, J. L. Weyher, I. Dzięcielewski, M. Chłopek, S. Góźdź, A. M. Nowicka and A. Kudelski, Biosens. Bioelectron., 2019, 132, 326–332 CrossRef CAS.
- M. Shi, J. Zheng, C. Liu, G. Tan, Z. Qing, S. Yang, J. Yang, Y. Tan and R. Yang, Biosens. Bioelectron., 2016, 77, 673–680 CrossRef CAS.
- N. N. Nam, T. L. Bui, S. J. Son and S.-W. Joo, Adv. Funct. Mater., 2019, 29, 1809146 CrossRef.
- P. Dharmalingam, K. Venkatakrishnan and B. Tan, Appl. Mater. Today, 2019, 16, 28–41 CrossRef.
- J. A. Powell, K. Venkatakrishnan and B. Tan, ACS Appl. Mater. Interfaces, 2017, 9, 40127–40142 CrossRef CAS.
- H.-Y. Wu, Y.-H. Lai, M.-S. Hsieh, S.-D. Lin, Y.-C. Li and T.-W. Lin, Adv. Mater. Interfaces, 2014, 1, 1400119 CrossRef.
- A. Rygula, K. Majzner, K. M. Marzec, A. Kaczor, M. Pilarczyk and M. Baranska, J. Raman Spectrosc., 2013, 44, 1061–1076 CrossRef CAS.
- J.-K. Yang, I.-J. Hwang, M. G. Cha, H.-I. Kim, D. Yim, D. H. Jeong, Y.-S. Lee and J.-H. Kim, Small, 2019, 15, 1900613 CrossRef.
- Y. Liu, Z. Gao, M. Chen, Y. Tan and F. Chen, Adv. Funct. Mater., 2018, 28, 1805710 CrossRef.
- X. Li, G. Chen, L. Yang, Z. Jin and J. Liu, Adv. Funct. Mater., 2010, 20, 2815–2824 CrossRef CAS.
- L. Guerrini and D. Graham, Chem. Soc. Rev., 2012, 41, 7085–7107 RSC.
- G. Demirel, H. Usta, M. Yilmaz, M. Celik, H. A. Alidagi and F. Buyukserin, J. Mater. Chem. C, 2018, 6, 5314–5335 RSC.
- H. K. Lee, Y. H. Lee, C. S. L. Koh, G. C. Phan-Quang, X. Han, C. L. Lay, H. Y. F. Sim, Y.-C. Kao, Q. An and X. Y. Ling, Chem. Soc. Rev., 2019, 48, 731–756 RSC.
- J. R. Lombardi and R. L. Birke, J. Phys. Chem. C, 2014, 118, 11120–11130 CrossRef CAS.
- M. Mochizuki, G. Lkhamsuren, K. Suthiwanich, E. A. Mondarte, T.-a. Yano, M. Hara and T. Hayashi, Nanoscale, 2017, 9, 10715–10720 RSC.
- E. L. Keller, N. C. Brandt, A. A. Cassabaum and R. R. Frontiera, Analyst, 2015, 140, 4922–4931 RSC.
- R. Haldavnekar, K. Venkatakrishnan and B. Tan, Nat. Commun., 2018, 9, 3065 CrossRef.
- E. L. Keller and R. R. Frontiera, ACS Nano, 2018, 12, 5848–5855 CrossRef CAS.
- M. Caldarola, P. Albella, E. Cortés, M. Rahmani, T. Roschuk, G. Grinblat, R. F. Oulton, A. V. Bragas and S. A. Maier, Nat. Commun., 2015, 6, 7915 CrossRef CAS.
- Z.-C. Zeng, H. Wang, P. Johns, G. V. Hartland and Z. D. Schultz, J. Phys. Chem. C, 2017, 121, 11623–11631 CrossRef CAS.
- J. A. Powell, K. Venkatakrishnan and B. Tan, J. Mater. Chem. B, 2016, 4, 5713–5728 RSC.
- J. A. Powell, K. Venkatakrishnan and B. Tan, Sci. Rep., 2016, 6, 19663 CrossRef CAS.
- K. Meysam, T. Bo and V. Krishnan, Adv. Sci., 2018, 5, 1700548 CrossRef.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798 CrossRef CAS.
- X. X. Han, C. Köhler, J. Kozuch, U. Kuhlmann, L. Paasche, A. Sivanesan, I. M. Weidinger and P. Hildebrandt, Small, 2013, 9, 4175–4181 CrossRef CAS.
- S. Lee, H. Chon, J. Lee, J. Ko, B. H. Chung, D. W. Lim and J. Choo, Biosens. Bioelectron., 2014, 51, 238–243 CrossRef CAS.
- I. Alessandri, J. Am. Chem. Soc., 2013, 135, 5541–5544 CrossRef CAS.
- K. K. Maiti, U. S. Dinish, C. Y. Fu, J.-J. Lee, K.-S. Soh, S.-W. Yun, R. Bhuvaneswari, M. Olivo and Y.-T. Chang, Biosens. Bioelectron., 2010, 26, 398–403 CrossRef CAS.
- M. Wang, H. Yin, Y. Zhou, C. Sui, Y. Wang, X. Meng, G. I. N. Waterhouse and S. Ai, Biosens. Bioelectron., 2019, 128, 137–143 CrossRef CAS.
- G.-C. Fan, X.-L. Ren, C. Zhu, J.-R. Zhang and J.-J. Zhu, Biosens. Bioelectron., 2014, 59, 45–53 CrossRef CAS.
- Y. Wang, J. Zhang, H. Jia, M. Li, J. Zeng, B. Yang, B. Zhao, W. Xu and J. R. Lombardi, J. Phys. Chem. C, 2008, 112, 996–1000 CrossRef CAS.
- J. Neng, M. H. Harpster, W. C. Wilson and P. A. Johnson, Biosens. Bioelectron., 2013, 41, 316–321 CrossRef CAS.
- H. Ilkhani, T. Hughes, J. Li, C. J. Zhong and M. Hepel, Biosens. Bioelectron., 2016, 80, 257–264 CrossRef CAS.
- S. Wen, Y. Su, R. Wu, S. Zhou, Q. Min, G.-C. Fan, L.-P. Jiang, R.-B. Song and J.-J. Zhu, Biosens. Bioelectron., 2018, 117, 260–266 CrossRef CAS.
- W. Zhou, B.-C. Yin and B.-C. Ye, Biosens. Bioelectron., 2017, 87, 187–194 CrossRef CAS.
- Y. Pang, C. Wang, J. Wang, Z. Sun, R. Xiao and S. Wang, Biosens. Bioelectron., 2016, 79, 574–580 CrossRef CAS PubMed.
- Z. Zhang, S. Zhang, L. He, D. Peng, F. Yan, M. Wang, J. Zhao, H. Zhang and S. Fang, Biosens. Bioelectron., 2015, 74, 384–390 CrossRef CAS.
- D. Qi, L. Lu, L. Wang and J. Zhang, J. Am. Chem. Soc., 2014, 136, 9886–9889 CrossRef CAS.
- X. Fan, M. Li, Q. Hao, M. Zhu, X. Hou, H. Huang, L. Ma, O. G. Schmidt and T. Qiu, Adv. Mater. Interfaces, 2019, 1901133 CrossRef CAS.
- Y. Zhu, L. Zhang, B. Zhao, H. Chen, X. Liu, R. Zhao, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Funct. Mater., 2019, 29, 1901783 CrossRef.
- S. Cong, Y. Yuan, Z. Chen, J. Hou, M. Yang, Y. Su, Y. Zhang, L. Li, Q. Li, F. Geng and Z. Zhao, Nat. Commun., 2015, 6, 7800 CrossRef CAS.
- A. Tavangar, B. Tan and K. Venkatakrishnan, J. Appl. Phys., 2013, 113, 023102 CrossRef.
- M. Keshavarz, B. Tan and K. Venkatakrishnan, ACS Appl. Mater. Interfaces, 2017, 9, 6292–6305 CrossRef CAS.
- A. Garcia-Leis, J. V. Garcia-Ramos and S. Sanchez-Cortes, J. Phys. Chem. C, 2013, 117, 7791–7795 CrossRef CAS.
- Y. Wang, H. Chen and E. Wang, Nanotechnology, 2008, 19, 105604 CrossRef.
- A. Tao, F. Kim, C. Hess, J. Goldberger, R. He, Y. Sun, Y. Xia and P. Yang, Nano Lett., 2003, 3, 1229–1233 CrossRef CAS.
- W. Song, Y. Wang and B. Zhao, J. Phys. Chem. C, 2007, 111, 12786–12791 CrossRef CAS.
- A.-I. Henry, B. Sharma, M. F. Cardinal, D. Kurouski and R. P. Van Duyne, Anal. Chem., 2016, 88, 6638–6647 CrossRef CAS PubMed.
- L. T. Kerr, H. J. Byrne and B. M. Hennelly, Anal. Methods, 2015, 7, 5041–5052 RSC.
- F. Nicolson, L. E. Jamieson, S. Mabbott, K. Plakas, N. C. Shand, M. R. Detty, D. Graham and K. Faulds, Analyst, 2018, 143, 5965–5973 RSC.
- G. McNay, D. Eustace, W. E. Smith, K. Faulds and D. Graham, Appl. Spectrosc., 2011, 65, 825–837 CrossRef CAS.
- M. Keshavarz, B. Tan and K. Venkatakrishnan, Sci. Rep., 2016, 6, 35425 CrossRef CAS.
- L. Li, M. Liao, Y. Chen, B. Shan and M. Li, J. Mater. Chem. B, 2019, 7, 815–822 RSC.
- G. Rusciano, E. Sasso, A. Capaccio, N. Zambrano and A. Sasso, Sci. Rep., 2019, 9, 1832 CrossRef.
- L. Litti, A. Ramundo, F. Biscaglia, G. Toffoli, M. Gobbo and M. Meneghetti, J. Colloid Interface Sci., 2019, 533, 621–626 CrossRef CAS.
- D. Lazaro-Pacheco, A. M. Shaaban, S. Rehman and I. Rehman, Appl. Spectrosc. Rev., 2019, 1–37, DOI:10.1080/05704928.2019.1601105.
- C. Nieva, M. Marro, N. Santana-Codina, S. Rao, D. Petrov and A. Sierra, PLoS One, 2012, 7, e46456 CrossRef CAS.
- A. Chakraborty, A. Ghosh and A. Barui, J. Raman Spectrosc., 2019 DOI:10.1002/jrs.5726.
- B. Brozek-Pluska, J. Musial, R. Kordek, E. Bailo, T. Dieing and H. Abramczyk, Analyst, 2012, 137, 3773–3780 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting Fig. S1, Table S1 and references. See DOI: 10.1039/c9nh00590k |
| This journal is © The Royal Society of Chemistry 2020 |