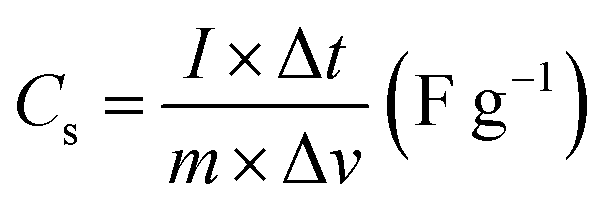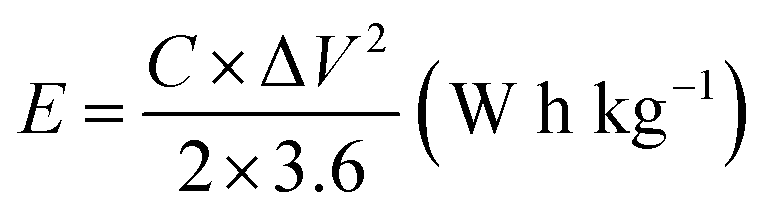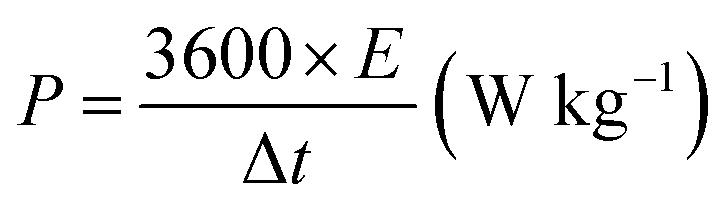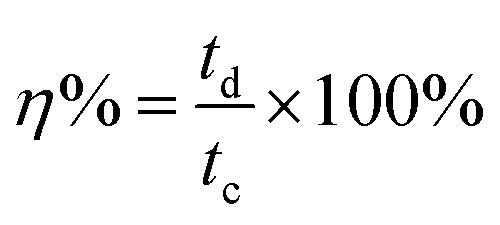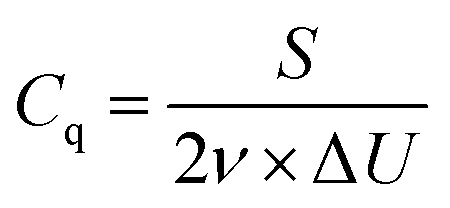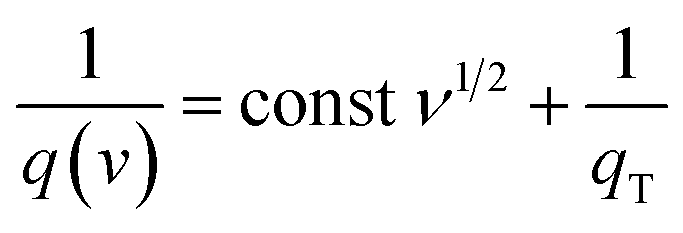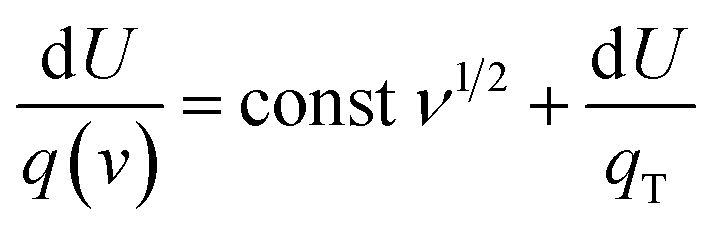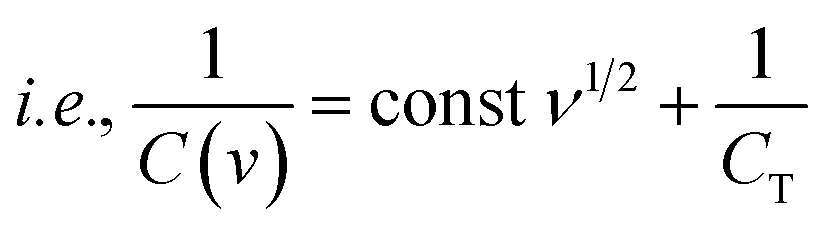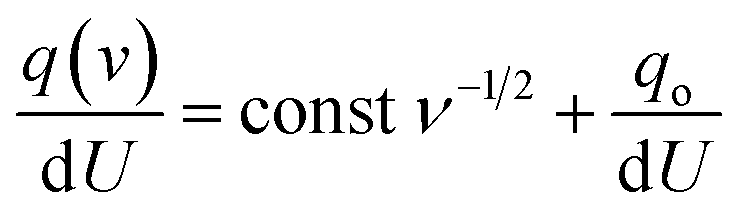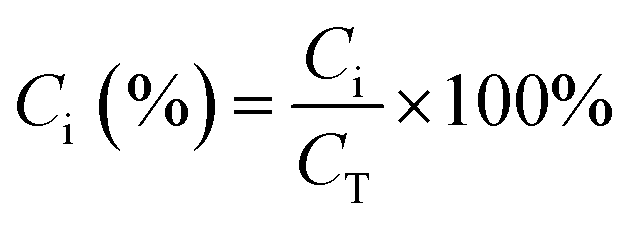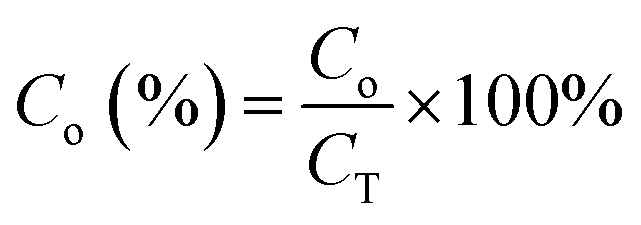DOI:
10.1039/D0NA00775G
(Paper)
Nanoscale Adv., 2021,
3, 486-498
Superior supercapacitive performance of Cu2MnSnS4 asymmetric devices
Received
16th September 2020
, Accepted 9th November 2020
First published on 19th November 2020
Abstract
Renewable energy sources are considered the cornerstone of achieving a sustainable future for today's modern world. In the current research on energy storage devices, transition metal sulfides are being explored as an excellent electrode material. In this study, the electrochemical performance of Cu2MnSnS4 and its micro/nano-structural changes due to variations in the structure directing agents used in its fabrication were investigated. Synthesis of the sulfide sediments was confirmed via standard characterization techniques. The charge-storage electrochemical activity was explored via electrochemical characterization techniques. Furthermore, the electrochemical charge-storage behavior was enhanced by Cu2MnSnS4. The best cyclic stabilities obtained were 83%, 87%, and 89% of capacitive retention over 5000 cycles for AA, CA, and OA electrodes in three-electrode arrangements, respectively, while an 80.35% capacitance retention and 97.54% coulombic efficiency were achieved after 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles in a fabricated device. The as-fabricated device exhibited an energy density of 27 W h kg−1 and power density of 759 W kg−1 at a current density of 1 A g−1 in the two-electrode setup.
000 cycles in a fabricated device. The as-fabricated device exhibited an energy density of 27 W h kg−1 and power density of 759 W kg−1 at a current density of 1 A g−1 in the two-electrode setup.
1. Introduction
Eco-friendly and efficient energy storage systems are emerging as an urgent need to meet the ever-increasing energy demands. Supercapacitors (SCs) in particular are attracting unparalleled attention because of their low weight, high power density, rapid charge discharge, extended cyclic ability, and robustness.1,2 However, the current carbon-based SCs are affected by a low energy density when compared to batteries. It is therefore attractive to have a wide range of applications for supercapacitors that can deliver high energy density, which has evolved into an essential requirement. It is still believed today for these supercapacitors that, it would be possible to improve the energy density by increasing the cell potential, which can be achieved by electrode combinations.3–5 Metal sulfides have many redox reactions to form good quality SCs with battery-type electrode materials and they are considered as positive electrode materials. Depending on the device, they may exhibit an extended potential window and higher energy density than conventional supercapacitors.6–10 Based on their electrochemical charge storage, generally pseudocapacitors (PCs) exhibit higher specific capacitance values than electric double layer capacitors (EDLCs).11,12 The reason is that in EDLCs, the charges between the electrode and electrolyte interfaces are stored by processes such as the diffusion/charge accumulation process, whereas in pseudocapacitors, the charge is stored by redox reactions in the active substance (mass), such as the faradaic process.13,14 Carbon-aerogel, carbon nanotubes, carbon cloth, graphene, and activated carbon are used in EDLCs, whereas metal oxides/hydroxides/sulfides are extensively used as electrode materials in pseudocapacitors.15,16 Metal oxides/sulfides, such as MnCo2O4, NiCo2O4, FeCo2O4, CoS, and Ni3S2, have attracted more research attention because they provide significant beneficial characteristics, such as a high energy and power density, and more specific capacitance thanks to their rapid and reversible rich redox reactions on the electrode/electrolyte interface. Various transition metal sulfide electroactive materials have been explored for electrochemical capacitors (ECs) due to their rich valence and desirable chemical stability.17,18 Sulfides are considered to be excellent pseudocapacitive materials, especially thanks to their multiple redox reactions, improved electrical conductivity, and low band gap, which lead to their superior performance, better oxidation state, and longer cycling capability when compared to metal oxides/hydroxides.19,20 In general, the pseudocapacitance capabilities of these sulfides in alkaline solutions are exhibited in the faradaic reactions that are associated with the mutual transformation from ion diffusion into electrode materials with rich electroactive sites.
Cu2XSnS4 (X = Mn, Fe, Co, Ni, Zn, and Cd) (CXTS) materials have excellent structural and optical properties, absorption coefficients, and catalytic efficacy in redox reactions due to their multiple oxidation states.21,22 Among their most interesting benefits in practice are the production of materials with a high surface area, good porosity, and production of comparatively labor-intensive planar and conformal nanostructured layers using a simple, low cost, quick, and measurable process. The metal chalcogenides have attracted great research interest due to their enhanced electrochemical properties. Molecular metal chalcogenide species can be broken down from the bulk chalcogenide complex and formed as a result of the basic nucleophilic reactions involving the chalcogenide anions.23–25 The hydrothermal method is the best technique for designing a good quality micro/nano structure compared to all other methods. During the initial nucleation in the hydrothermal reaction, the acceleration of the endothermic reaction results in a disruption of the nuclei, which dissolves the urea ions, resulting in an insufficient activation surface energy and subsequent defects.26,27 Acidic acid, citric acid, and oxalic acid are all well-known structure directing agents that can be classified from dicarboxylic acid. They are also considered to be powerful reducing and chelating agent for metal cations. The acidic strength of oxalic acid is much greater than the other two, and therefore, it can lead to a better structure; thereby creating more ionic transport at the electrode/electrolyte interface, resulting in an improved electrochemical behavior.28,29 In this study, a solvothermal process was pursued utilizing a ethanol-mixed de-ionized water solution as the solvent. The structure directing agent's role in creating the Cu2MnSnS4 nanostructure could be visualized from the FE-SEM analysis. Characterization of the materials was performed by several physicochemical, chemical, and electrochemical techniques. Here, superior supercapacitive performances of Cu2MnSnS4 asymmetric devices could be reported. To the best of our knowledge, this was also the first study to investigate and report the use of Cu2MnSnS4 nanostructures fabricated based on the use of different directing agents for fabricating a positive electrode for supercapacitor applications.
2. Experimental
Copper(II) chloride dihydrate (CuCl2·2H2O, 99%), manganese(II) chloride tetrahydrate (MnCl2·4H2O, 99%), citric acid (C6H8O7, 99%), thiourea (NH2CSNH2, 99%), and tin(IV) chloride pentahydrate (SnCl4·5H2O, 98%) were purchased. The solvothermal reaction was adapted to synthesize the Cu2MnSnS4 nanostructure. In a typical synthesis route, copper(II) chloride dehydrate (1 mole), manganese(II) chloride tetrahydrate (0.5 mole), tin(IV) chloride pentahydrate (0.5 mole), and structure directing acidic (AA), citric (CA), or oxalic (OA) acid (1 mole) were dissolved in 80 mL in a 1:1 ratio with ethanol and de-ionized water solvents under magnetic stirring until an homogeneous solution appeared. Thereafter, thiourea (0.4 mole) solution was added and the mixture was put in an autoclave at 180 °C for 12 h. The samples were collected after a thorough wash with de-ionized water, ethanol, and methanol with centrifugation at 4000 rpm. Finally, the collected samples were separately placed in a hot air oven for drying. The resultant products were used in the different characterizations.
The working electrode was formulated by mixing Cu2MnSnS4 (prepared with AA, CA, or OA) as an active cathode material, activated carbon as the anode material, and polyvinylidene fluoride (PVDF) as a binder in a 75:20:5, weight ratio. Then, all three materials were ground for at least 30 min. Thereafter, the mixed electrode material was dispersed in the solvent N-methyl-2-pyrrolidone (NMP) and ground for 1 h to prepare the electrode slurry. Well-cleaned nickel foam was employed as a substrate (1 × 2 cm2). A 1 × 1 cm2 section of nickel foam was coated with the prepared slurry using a mercury premium art small size brush. The coated nickel foam was dried in a hot air oven at 80 °C for 12 h and the loading material was adjusted to 2 mg cm−2 on the electrode surface. These Cu2MnSnS4 (AA, CA, OA) electrodes, platinum (Pt) wire, and Ag/AgCl were utilized as the working, counter, and reference electrodes. Electrochemical measurements in 2 M KOH electrolyte were performed using a BioLogic potentiostat. CVs at different scan rates ranging from 10 to 100 mV s−1 were recorded at a 0.6 V constant potential, and 0.5 to 5A g−1 current densities at 0.5 V potential were utilized for the galvanostatic charge discharge (GCD) readings. The electrochemical impedance spectroscopy (EIS) study results were analyzed by Nyquist plots and fitted by Z fit analysis. Eqn (1) was employed to estimate the specific capacitance from the GCD profile, and similarly the two-electrode (full-cell) system was set up with the two equal loadings mass balanced using the formula 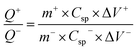 and then coated on to the positive and negative electrodes. The full-cell device parameters such as energy (E) and power (P) density, were measured using eqn (2) and (3). Eqn (4) was used to calculate the coulombic efficiency of the materials from the GCD curves.
and then coated on to the positive and negative electrodes. The full-cell device parameters such as energy (E) and power (P) density, were measured using eqn (2) and (3). Eqn (4) was used to calculate the coulombic efficiency of the materials from the GCD curves.
| | 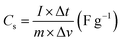 | (1) |
| | 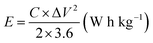 | (2) |
| | 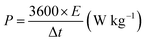 | (3) |
| | 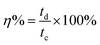 | (4) |
where,
td and
tc are the discharging and charging time, respectively.
30,31
2.1 Trasatti method analysis
The Trasatti method was first used to identify the EDLC behavior and PC behavior by the eminent scientist S. Trasatti in 1990 using the formula qT = qi + qo; CT = Ci + Co, where qT is the total voltammetric charge, qi is the charge stored at the inner surface, qo is the charge stored at the outer surface, and CT is the total capacity, which is the sum of the pseudocapacity and the electric double layer capacity. In general, in the outer surface V → ∞; therefore it just allows the surface process to happen. Hence, the more accessible area is in the electrode electrolyte interface. In the same way, at the inner surface, V → 0; therefore it gives sufficient time for the diffusion and for more ions to react. Hence, it has less accessible area.
Step 1.
Collect cyclic voltammogram at various scan rates (10 to 100 mV s−1).
Step 2.
Calculate the specific capacity (C g−1) at each scan rate using the following formula,| | 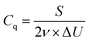 | (5) |
where Cq denotes the specific capacity (C g−1), S is the area enclosed by the CV curve, ν is the scan rate (mV s−1), and m is the active mass in the electrode surface.
Step 3.
Estimation of the total capacity (CT).
Let us assume semi-infinite linear ion diffusion (i.e., ions randomly diffuse from the bulk electrolyte to the electrode/electrolyte interface, when V → 0, q → qT). Based on the Cottrell equation, a linear correlation between the reciprocal of the calculated specific capacity (1/Cq) and the square root of scan rates (ν1/2) gives the maximum total capacity by extrapolation to the Y intercept.
| | 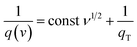 | (6) |
Multiply both sides by d
U:
| | 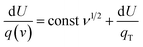 | (7) |
| | 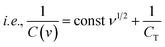 | (8) |
Step 4.
Estimation of the charge stored at the outer surface (Co).
In the same way, assuming semi-infinite linear ion diffusion (i.e., ions randomly diffuse from the bulk electrolyte to the electrode/electrolyte interface, when V → ∞, q → q0), based on the Cottrell equation, a linear correlation between specific capacity (Cq) and the reciprocal of square root of scan rates (ν−1/2) gives the charge stored at outer surface (Co) by extrapolation of the intercept to the Y axis.
| | | q(v) = const ν−1/2 + qo | (9) |
Divide both sides by d
U| | 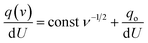 | (10) |
| | | i.e., C(v) = const ν−1/2 + Co | (11) |
Step 5.
Estimation of the inner surface (Ci).
Based on the Cottrell equation, the maximum capacity (CT) and charge stored at the outer surface (Co) could be calculated. The maximum capacity (CT) is the sum of the inner (Ci) and outer surface (Co), i.e., CT = Ci + Co, where CT is the total capacitance, Ci is responsible for the maximum pseudocapacitance (Ci), and C0 corresponds to the maximum electric double layer capacitance. Subtracting Co from CT gives the maximum pseudocapacitance (Ci).
Step 6.
Estimation of the percentage capacitive contribution.| | 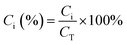 | (13) |
| | 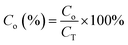 | (14) |
3. Results and discussion
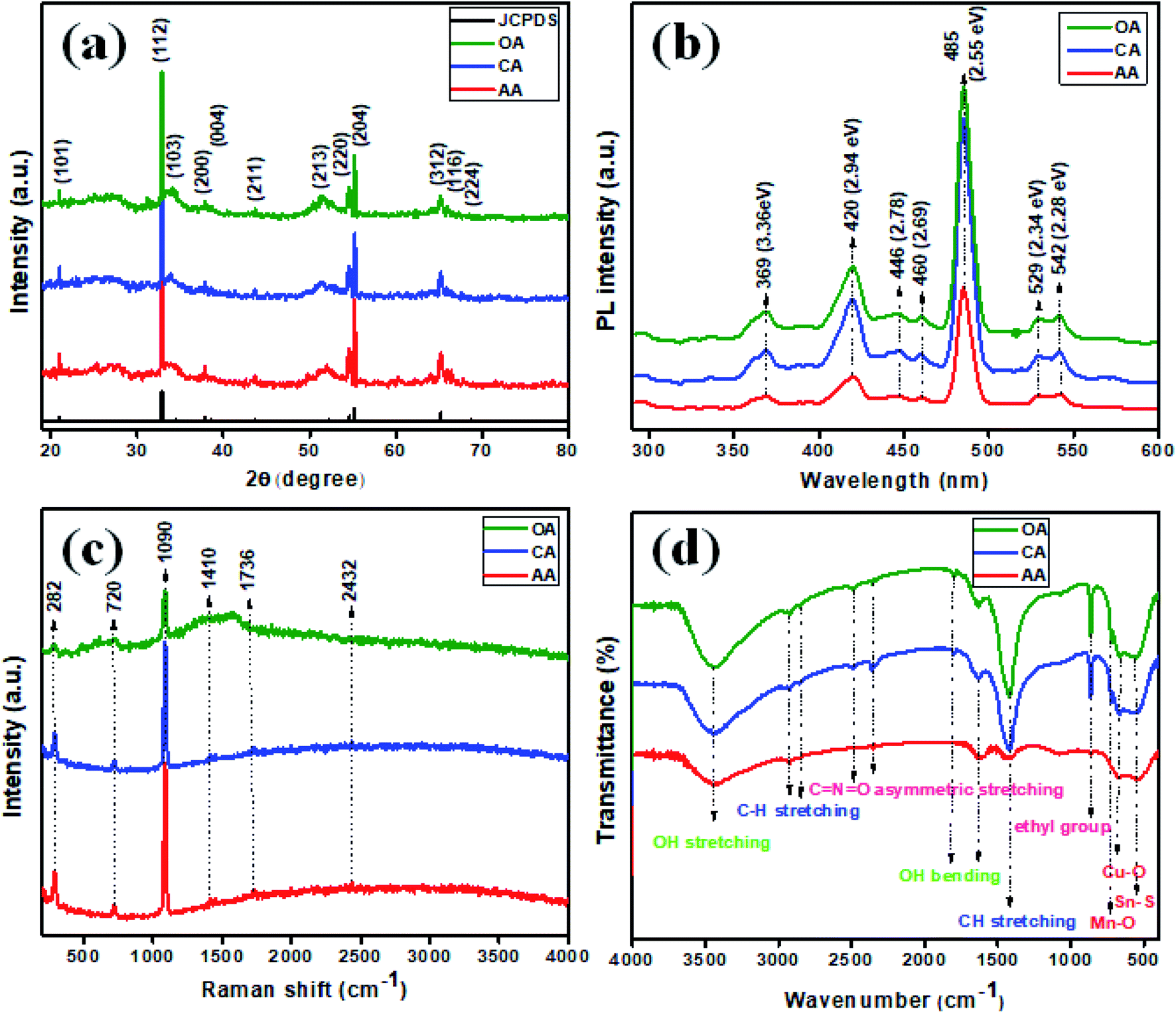
The obtained material crystal structure was characterized using X-ray diffraction analysis (Fig. 1(a)), wherein the 2θ values of 20.99°, 32.89°, 34.17°, 37.83°, 38.62°, 43.66°, 51.57°, 54.54°, 55.23°, 65.21°, 66.21°, and 68.77° corresponded to the (hkl) planes of (101), (112)*, (103), (200), (004), (211), (213), (220), (204), (312), (116), and (224) respectively (JCPDS card: 51-0757). The good crystalline nature was evidenced in all the samples, as suggested by the peaks observed. The high intensity peak present at 32.89° with the plane orientation of (112)* inferred the as-fabricated Cu2MnSnS4 had a polycrystalline nature. The structural parameters of the Cu2MnSnS4 microstructure are tabulated in Table 1.
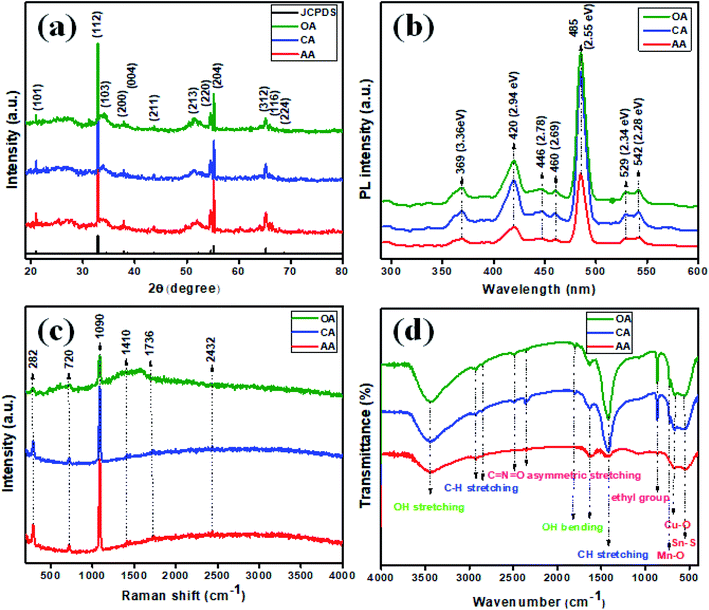 |
| | Fig. 1 (a) XRD, (b) photoluminescence, (c) Raman, (d) FTIR spectra of Cu2MnSnS4. | |
Table 1 Structural parameters of the Cu2MnSnS4 microstructure
| Physical Quantity (Symbol) (Units) |
Value |
| Lattice parameter (a) (Å) |
a = 5.513, c = 10.826 |
| Crystalline size (nm) |
56 |
| Microstrain (ε) × 10−3 |
0.618 |
| Dislocation density (δ) × 1014 |
3.18 |
| Cell volume (V) (nm3) |
329.11 |
The growth mechanism of the Cu2MnSnS4 product in the presence of the chloride precursor in the solvothermal reaction mixture in the well-closed Teflon tube produced Cu2+, Mn2+, Sn4+, and S2−. Most importantly, when the compounds were combined with thiourea, sulfur ions were released in the autoclave during the reaction mixture. Here, the S2− ions reacted with the metal ions Cu, Mn, and Sn during the nucleation formation of Cu2MnSnS4, as suggested by the following equations.32,33
| | | 2Cu2+ + Sn2+ → 2Cu+ + Sn4+ | (15) |
| | | Cu+ + en → [Cu(en)]+; Mn2+ + en → [Mn(en)]2+; Sn4+ + en → [Sn(en)]4 + | (16) |
| | | (NH2)2CS + 2H2O → S2− + 2NH4+ + CO2↑ | (17) |
| | | 2[Cu(en)]+ + [Mn(en)]2+ + [Sn(en)]4+ + 4S2− → Cu2MnSnS4 | (18) |
The small crystalline size and poor crystallinity of the material were explored for improving the electrochemical performance. This is because of the fact that poor crystalline materials create more ion transport channels than crystalline materials, which is an important factor in electrochemical supercapacitors. Photoluminescence was utilized as an eminent tool to investigate the emission behavior of the synthesized Cu2MnSnS4 (AA, CA, OA). In Fig. 1(b), PL emission bands could be observed at the wavelengths: 369 (3.36), 420 (2.94), 446 (2.78), 460 (2.69), 485 (2.55), 529 (2.34), and 542 nm (2.28 eV). Here, a higher intensity suggests a faster recombination rate. Seven photoluminescence peaks could be detected and with almost the same PL emission bands, expect for the intensity. The strong and broad blue emission band located at 485 nm was in good agreement with the Cu–S hierarchical structures, which absorb more photons to produce electron–hole pairs, which may start from the excitonic band edge or free-charge carrier transition at 2.55 eV. Weak green emission bands were observed at 529 and 542 nm. The blue emission band at 420 nm had a slightly broad band, which represented defects present in the prepared samples. The emission peaks assigned at 446 and 460 nm indicated different defects, which largely depended on the high density of sulfur and tin vacancies.34,35 The lattice vibrations of a material can be measured using Raman spectroscopy and so was used here, as shown in Fig. 1(c). The characteristic Raman band occurred at 282 cm−1, which was attributed to the Sn–S phases in Cu2MnSnS4. The Cu–S stretching mode vibration was detected at 449 cm−1. The small peak located at 720 cm−1 revealed a second-order Raman scattering mode of the prepared samples. In the synthesized product, the main broad peak existed in the Vis-NIR region at 1090 cm−1. C–C stretching vibrations were observed for the remaining bands present at 1736 and 2432 cm−1.36 FTIR spectrometry is a well-known technique used to characterize the molecular fingerprint region of synthesized products, and so was used here (Fig. 1(d)). Initially, a small quantity of the sample was mixed with potassium bromide (KBr) and finely grounded and then pressed well into a hydraulic press to be converted into a pellet shape. The acquired spectrum was within the range of 4000 to 400 cm−1. The characteristic spectrum 3448 cm−1 corresponded to the OH stretching vibration of water molecules observed on the sample. The vibrations at 1639 and 1820 cm−1 were because of the bending vibrations of OH water molecules. The absorption bands located at 2853 and 2929 cm−1 were due to the CH stretching vibration mode. The asymmetric stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NO was indicated by the vibration band at 2491 cm−1. The absorption vibration bands that existed in the fingerprint region indicated the metal oxide and metal sulfide vibrations. The bands at 729, 664, and 554 cm−1 signified the Mn–O, Cu–O, and Sn–S vibrations.37,38
NO was indicated by the vibration band at 2491 cm−1. The absorption vibration bands that existed in the fingerprint region indicated the metal oxide and metal sulfide vibrations. The bands at 729, 664, and 554 cm−1 signified the Mn–O, Cu–O, and Sn–S vibrations.37,38
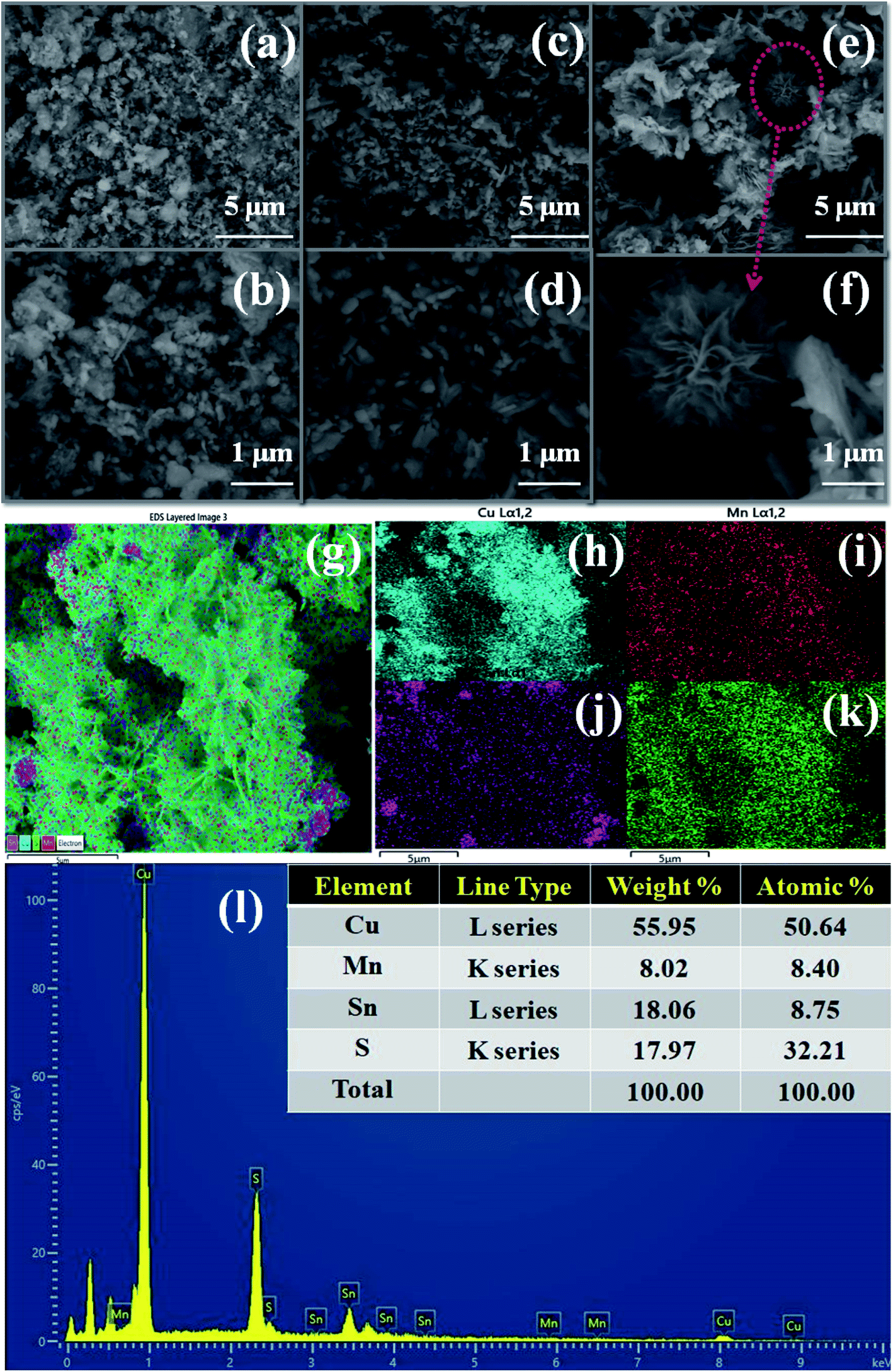
Fig. 2 shows the morphological images of the prepared Cu2MnSnS4 with using different structure directing agents, i.e., acidic acid (AA), citric acid (OA), and oxalic acid (OA), as studied using FE-SEM analysis. Irregular shapes with non-uniform size morphologies were found in the AA and CA samples, whereas well-formed flower shapes were found with the addition of oxalic acid, which exhibited a strong role in the flower-shape formation. The thickness of a few micrometer of the 2D thick plate-like petals was loosely covered with rose-like thrones. The structure of the flower shape facilitates high ionic transport and increases the accessible specific surface area. However, the petals in the flower were tightly stacked and appeared as thin pieces.39 It was clearly evidenced that when using the structure directing agents AA and CA in constant hydrothermal reactions, the particles agglomerated to form irregular flakes (Fig. 2(a–d)), while OA exhibited a fine flower-shaped morphology, as shown in Fig. 2(e and f). When compared to the AA and CA samples, the OA samples in the ethanol and distilled water mixture in the solvothermal route co-ordinate metal ions to urea and so are stronger combined. However, the petals in the flowers are stacked tightly, resembling thin slices. Initially, nanocrystals of Cu2MnSnS4 are formed as the seeds for microcrystal formation. The aggregated nanocrystals become larger and they coalesce to grow into flakes due to the CA addition in the hydrothermal reactions. The hydrogen bond interacted with urea to form self-assembled flower-like microstructures by the addition of oxalic acid at a constant temperature. By the proper selection of structure directing agents, the sheet-like structures self-assembled and coalesce into a flower-shaped morphology. During this formation, the surface energy decreased.40
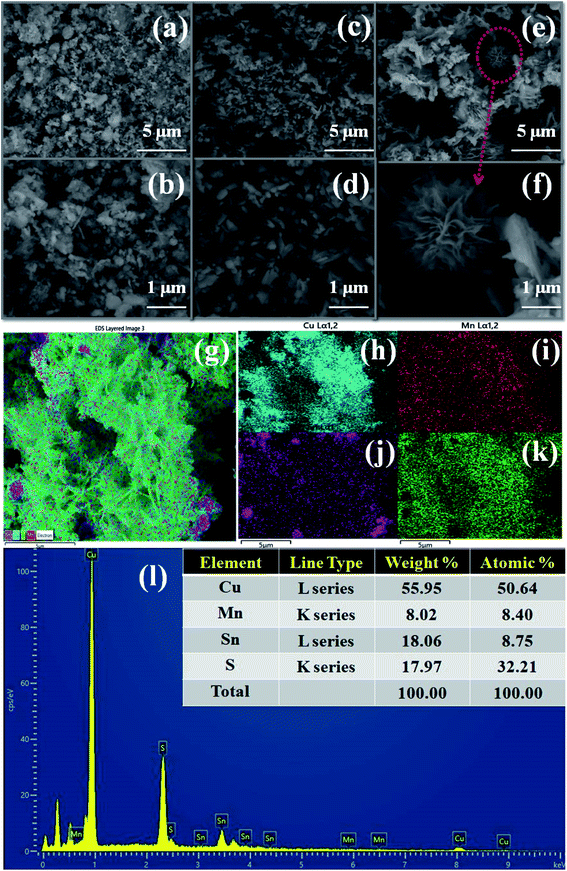 |
| | Fig. 2 Cu2MnSnS4 morphology (a and b) AA, (c and d) CA, (e and f) OA, (g–k) EDS mapping, (l) EDAX spectrum of the Cu2MnSnS4–OA sample. | |
Elemental analysis or chemical characterization of an area can be studied using EDS analysis. Fig. 2(g–k) shows the EDS SEM mapping images of the Cu2MnSnS4–OA sample. The higher peak in a spectrum revealed a higher concentration in the elemental composition. Fig. 2(l) shows the EDAX spectrum of the Cu2MnSnS4–OA electrode, which was composed of Cu, Mn, Sn, and S elements with good atomic and weight percentages as shown in the inset of Fig. 2(l). The elemental purity of the samples was confirmed by the absence of impurity peaks.41Fig. 3 presents the TEM images of the synthesized Cu2MnSnS4 nanostructure with the addition of oxalic acid. Non-uniform flower-like structures were located in many places with sizes in the 1–3 μm range. The well-resolved lattice fringe distances were approximately 0.31 nm, which well matched with the (112) crystal plane for the Cu2MnSnS4–OA electrode, as shown in Fig. 3(c). The selected area electron diffraction (SAED) pattern of the Cu2MnSnS4–OA electrodes showed the good crystallinity of the sample (Fig. 3(c)), which correlated with the X-ray results.
 |
| | Fig. 3 (a and b) TEM images, (c) lattice fringes, (d) SAED pattern of Cu2MnSnS4–OA. | |
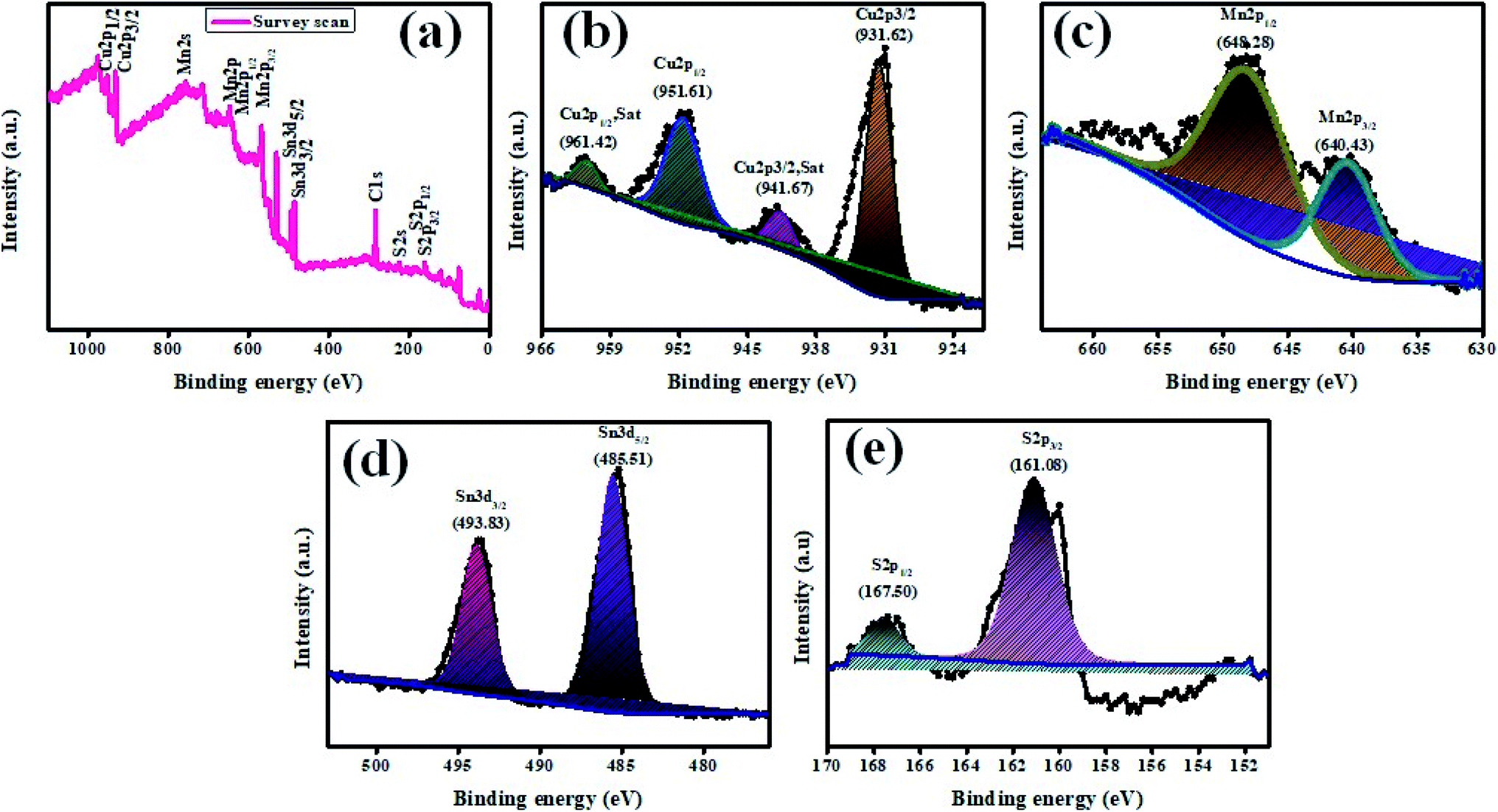
The elemental constituents present in the product Cu2MnSnS4 were studied using XPS (Fig. 4). Fig. 4(a) presents the survey spectrum of the as-obtained Cu2MnSnS4 compound. The Cu binding energy species are shown by major peaks at 931.62 eV (Cu2p3/2) and 951.61 eV (Cu2p1/2) and the corresponding satellite peaks at 941.67 eV (Cu2p3/2, Sat) and 961.42 eV (Cu2p1/2, Sat) (Fig. 4(b)). Here, the peak splitting of 19.99 eV represents the Cu(I) configurations. Two 2p peaks appeared at 648.28 eV for Mn2p1/2 and at 640.43 eV for Mn2p3/2 (Fig. 4(c)). The peak splitting of 7.85 eV indicated the Mn(II) configuration. The binding energies located at 485.51 and 493.83 eV were due to Sn3d5/2 and Sn3d1/2, respectively (Fig. 4(d)). The peak splitting of 8.52 eV indicates the Sn(IV) configuration. The binding energies from 160 to 164 eV were related to the sulfide phases (Fig. 4(e)). S2p3/2 and S2p1/2 showed peaks at 161.08 and 160.01 eV, respectively, with the peak splitting of 1.07 eV.42
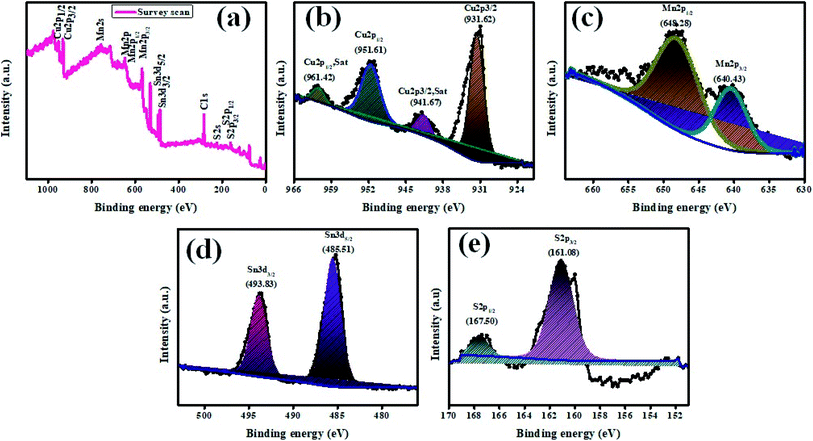 |
| | Fig. 4 XPS images of Cu2MnSnS4. | |
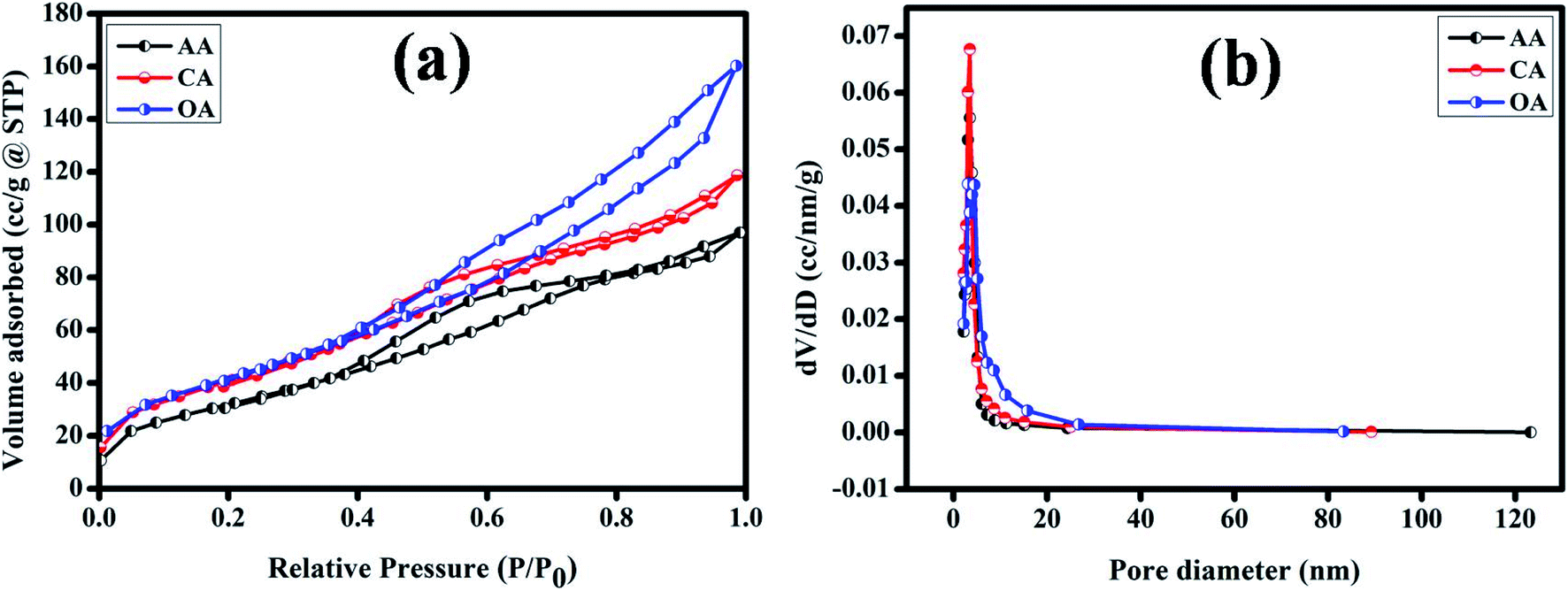
The surface area (m2 g−1) and pore-size (cm3 g−1) distribution of the synthesized Cu2MnSnS4 electrodes were measured using Brunauer–Emmett–Teller (BET) analysis, as displayed in Fig. 5(a). All the fabricated electrodes exhibited type IV isotherms, which are a general distinctive isotherm of mesoporous specimens (Table 2). The Cu2MnSnS4–OA electrodes provide high electrolyte contact at the electrode and electrolyte interface because of its large surface area. This leads to better ionic transport for faradaic reactions than the other two electrodes. The pore-size distribution was observed employing the Barrett–Joyner–Halenda (BJH) approach, which suggested that the entire prepared electrode had pore sizes in the 3 to 50 nm range, which clearly signified that the three samples were in the mesoporous range (Fig. 5(b)). Furthermore, the high specific surface area and mesoporous nature of the Cu2MnSnS4–OA electrode allows more specific parts of the electrolyte to diffuse effectively at the electrode with minimum internal resistance.43
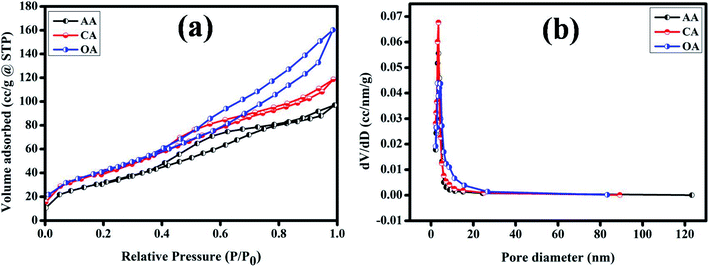 |
| | Fig. 5 (a) N2 adsorption–desorption (b) pore-size distribution of the Cu2MnSnS4–(AA, CA, OA) electrodes. | |
Table 2 BET analysis parameters
| Product |
BET surface area (m2 g−1) |
Cumulative pore volume (cm3 g−1) |
Pore diameter (nm) |
| Cu2MnSnS4–AA |
143.369 |
0.157 |
3.479 |
| Cu2MnSnS4–CA |
166.025 |
0.186 |
3.492 |
| Cu2MnSnS4–OA |
179.276 |
0.254 |
3.091 |
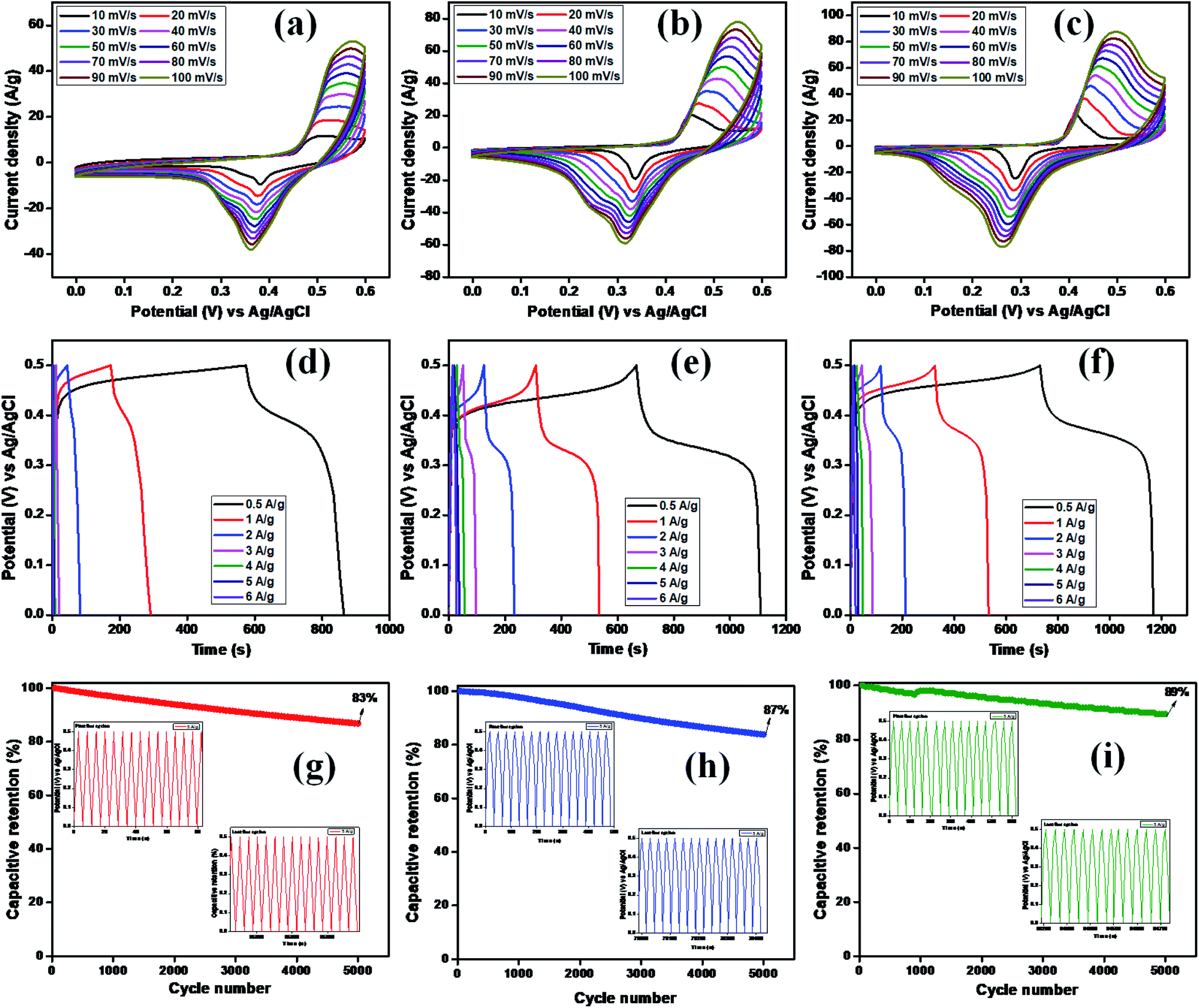
Electrochemical studies, such as CV, GCD, and EIS, were used to investigate the charge-storage performances of Cu2MnSnS4 (AA, CA, OA) in three-electrode setups. Fig. 6(a–c) show typical CV plots from 10 to 100 mV s−1. The pair of redox reactions that are invisible in electrical double layer capacitors are evident in the CV curves and confirmed its battery-type behavior, which is the key characteristic of the pseudocapacitive mechanism.44 The good redox peaks indicated charge storage derived from the Faraday process in battery-type electrodes. The apparent changes at high scan rates showed the improved rate performances of the electrode.45 A pair of oxidation and reduction reactions of the anodic and cathodic voltage peaks at 10 mV s−1 scan rate was observed at Ea = 0.50, 0.45, 0.41 V and Ec = 0.38, 0.33, 0.28 V for AA, CA, and OA, respectively. When the scan rate was increased from 10 to 100 mV s−1, the redox peaks moved toward higher and lower energies and the corresponding peak current (ip) also increased.
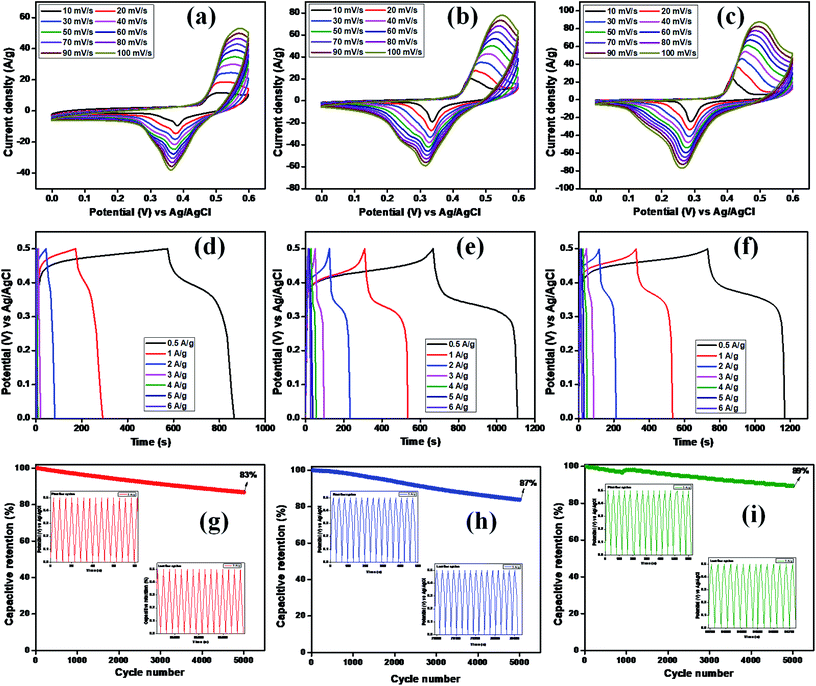 |
| | Fig. 6 (a–c) CV, (d–f) GCD, (g–i) capacitive retention of Cu2MnSnS4 (AA, CA, OA). | |
At low scan rates, the electrolyte ions and active species have sufficient time to interact with each other. This increases the probability of redox reactions occurring; nevertheless, at high scan rates, the interaction of ions is not perfect due to their short residence time, which limits the different reactions between them. Cyclic voltammetric curves are affected by diffusion-controlled and kinetic-controlled processes. If a redox process is only affected by diffusion, the peak potential should generally be independent of the scan rate. Nevertheless, in contrast, if the electrode kinetics is dominant, the peak potential is affected by the voltammetric changes due to the increasing scan rates. Furthermore, the nature of the redox process can be easily identified by the cathodic and anodic shifts. In view of the fact that this electrode material exhibited a battery-like behavior, there should be an apparent change in the peak shift.44
Furthermore, this reduces the reaction kinetics because of the material polarization and ohmic resistance during the redox process, resulting in the formation of reversible peaks. The anodic peak current increased when the scan rate increased, which also led to an increase in the CV curves area directly proportional to the specific capacity. In all three samples, the lower scan rate (10 mV s−1) exhibited a higher capacity, which gives sufficient time for the ions to diffuse and react in the electrode/electrolyte interface. As a result, there is less accessible area occupied at the lower surface of the electrode for the electrochemical reaction process to occur.46 The calculated specific capacity values are summarized in Table 3. The OA electrode displayed the highest specific capacity, which may be due to the well-arranged structure of the samples as proven in the FE-SEM analysis. This is because of the obtained nanostructures, which enhance the electrode specific surface area and rational pore-size distribution and greatly enhances the specific capacity, as clearly evidenced from the BET surface analysis.
Table 3 Specific capacity of the different scan rates
| Scan rate (mV s−1) |
10 |
20 |
30 |
40 |
50 |
60 |
70 |
80 |
90 |
100 |
| Specific capacitance (C g−1)–AA |
268 |
198 |
165 |
143 |
127 |
114 |
103 |
95 |
88 |
83 |
| Specific capacitance (C g−1)–CA |
294 |
237 |
225 |
217 |
207 |
194 |
182 |
170 |
159 |
149 |
| Specific capacitance (C g−1)–OA |
302 |
285 |
279 |
272 |
262 |
249 |
237 |
224 |
213 |
204 |
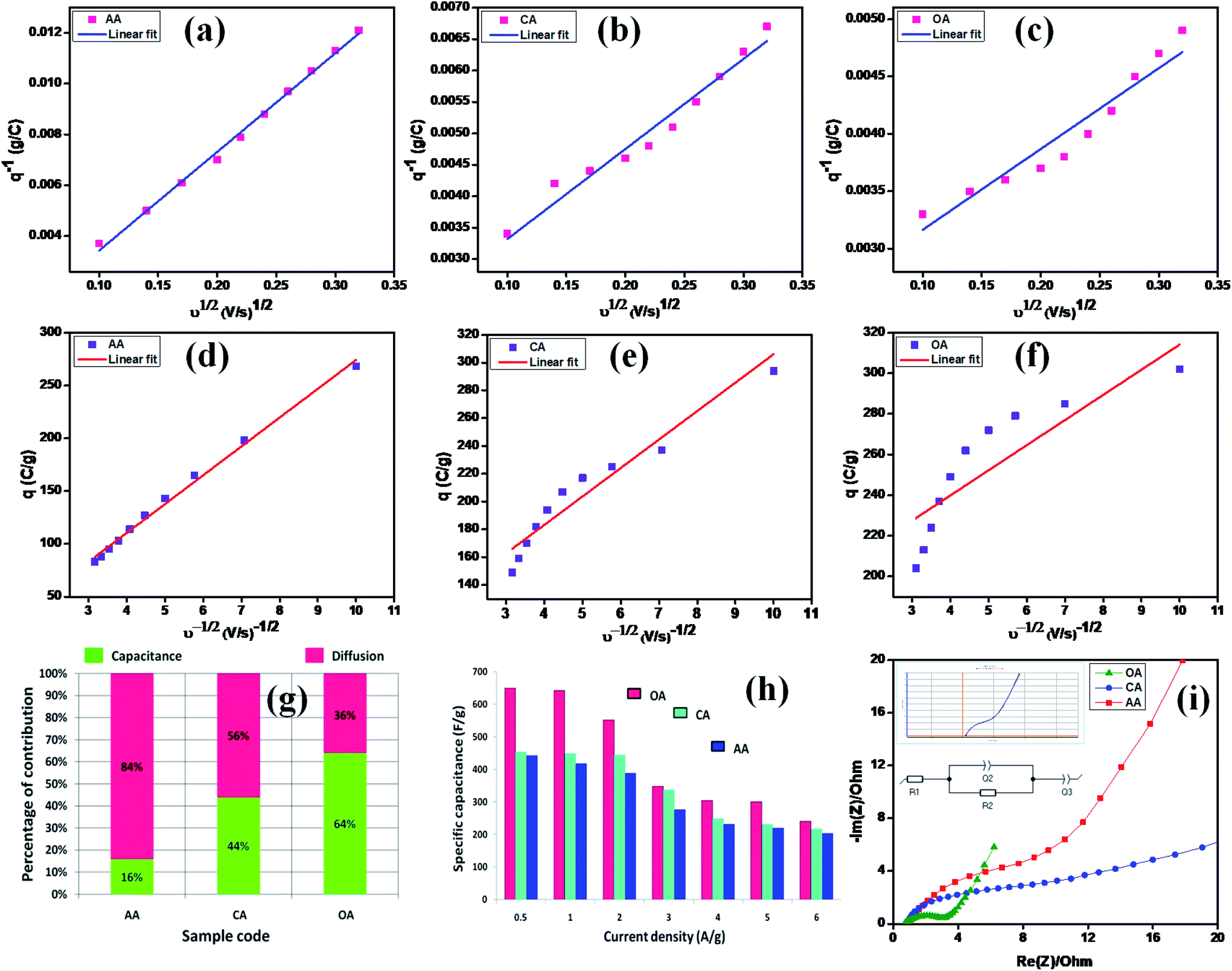
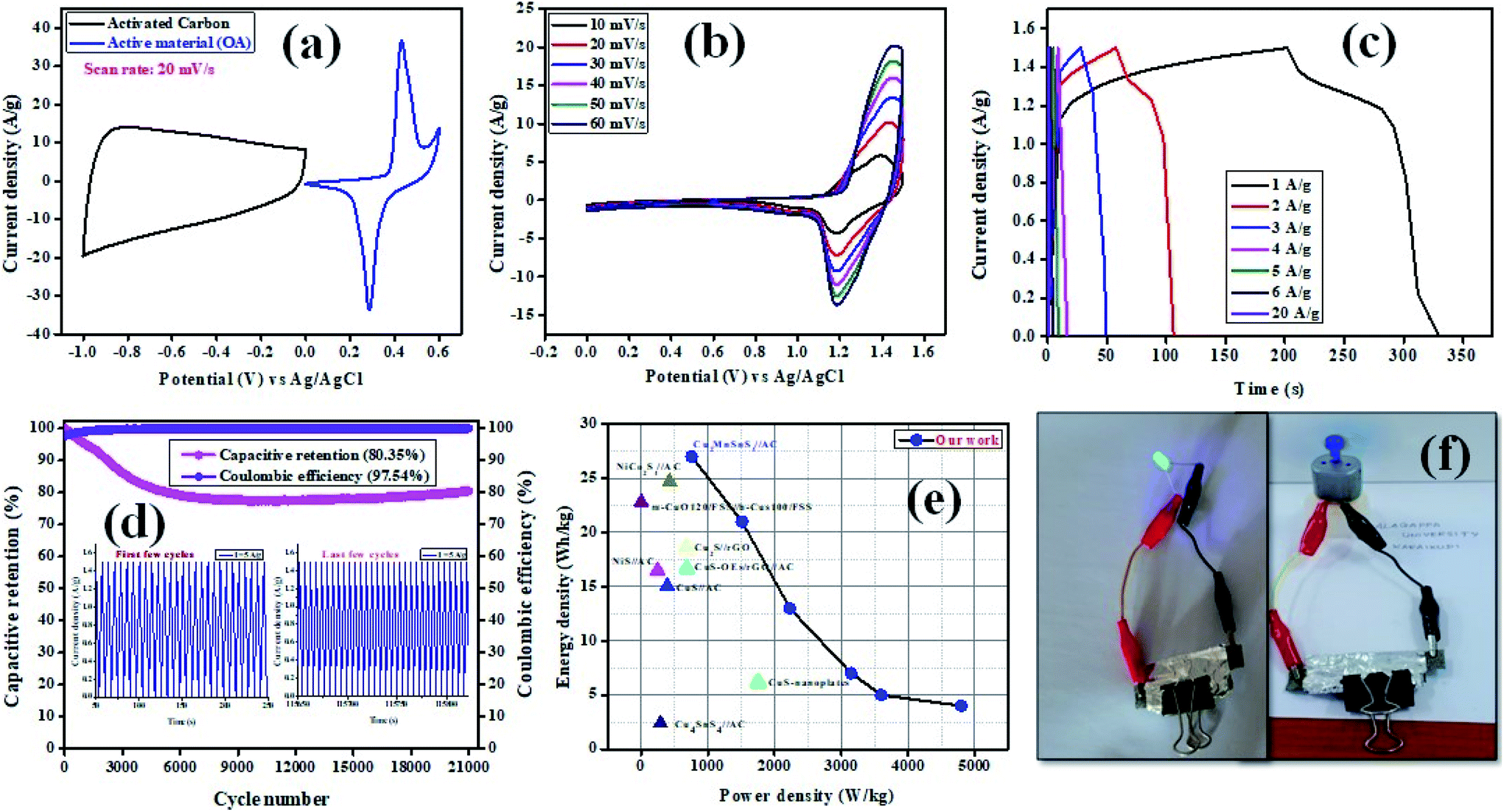
The specific capacity, stability, and time reversibility of the Cu2MnSnS4 electrode material were studied by GCD characterization study. Fig. 6(d–f) display the GCD curves comparison of the prepared as-fabricated Cu2MnSnS4 electrode materials. The highest reversible time was observed for the OA sample than AA and CA. The calculated specific capacitance values for OA, CA, and AA were 649, 453, and 442 F g−1 at 0.5 A g−1. The cone diagram displays the specific capacitance values at different current density ranges. The long-term cyclic stability up to 5000 cycles was assessed and capacitive retentions of 83%, 87%, and 89% were achieved for the AA, CA, and OA electrodes, respectively (Fig. 6g–i). Due to the electrode properties, such as internal resistance, polarization, and electrolytic ions insertion, and removal due to mechanical stress, its specific capacitance value decreases due to a high current density.47 According to the CV and GCD studies, the Cu2MnSnS4–OA electrode may be preferred as a suitable electrode material.
The CV studies further indicated that the redox reaction is a diffusion-controlled process, which may be the main cause of the pesudocapactive behavior with the increasing scan rates. The two differentiation methods, namely (i) Trasatti method and (ii) Dunn method, were then used to analyze the diffusion-controlled faradaic and non-diffusion-controlled battery-type processes of the synthesized electrodes.48 In the Dunn method, the peak current (ip) vs., the scan rate gives the power law ip = avb. The linear fit slope values give the ‘b’ value. A value of b = 0.5 means diffusion-controlled faradaic process, while b = 1 means a non-diffusion-controlled process due to surface redox reactions. Among the two studied methods, the Trasatti method is the simplest one to describe the mechanism of the electrode. Fig. 7(a, d, b, e, c and f) show the linear fit of C vs. ν−1/2 and C−1vs. ν1/2 for the AA, CA, and OA samples, respectively. The non-linear behavior of the graphs explored was due to the ohmic drop of the intrinsic resistance.49 The specific capacities of the AA, CA, and OA samples with their EDLC (capacitive) and pseudocapacitive (diffusive) behavior are shown in Fig. 7(g). The AA sample exhibited 16% and 84% capacity derived from the EDLC and PC contributions, while CA offered 44% and 56% capacity derived from the EDLC and PC contributions, and OA delivered 64% and 36% capacity for the EDLC and PC.
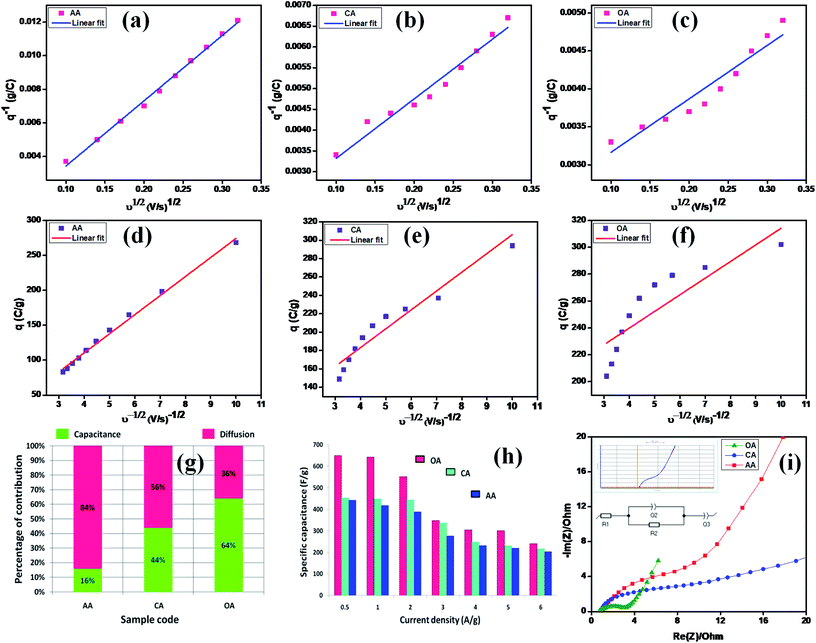 |
| | Fig. 7 (a and d), (b and e), and (c and f) linear fits of C vs. ν−1/2 and C−1vs. ν1/2 for AA, CA, and OA, (g) capacitive (EDLC) and diffusive (PC) charging derived from Trasatti's method, (h) specific capacitance from GCD, and (i) Nyquist plot. | |
The interfacial structure and its kinetic response due to electrical variation were studied by electrochemical impedance spectroscopy. The opposition force to an electrical current is known as impedance and it is denoted by the unit of resistance (Ω). There are three factors that determine the concepts of resistance used in an AC circuit: (i) the alternating current (AC) and voltage in phase, (ii) frequency independent, and (iii) all current and voltages. Nevertheless, the majority of these failed because the phase angle was not zero and almost in all cases capacitive and/or inductive effects were present at almost all the frequencies. Hence, it becomes an issue of frequency dependence. In these cases, the concept of impedance was used to denote the electrical current opposition in AC circuits.50 Here, EIS spectra were plotted by the real vs. the imaginary part of the impedance, which determines the inter phase resistance between the working electrode and the electrolyte. Fig. 7(i) shows Nyquist plots of the OA, CA, and AA samples with the consequent equal circuit model. The overall impedance contribution was simplified to transform the EIS spectrum into its corresponding equivalent circuit and the physical process involved in the samples was determined using equivalent circuit parameters. The following fitted equivalent circuit (R1 + Q2/R2 + Q3) was used to assess the different components of the cell resistance, where R1 corresponds to the ohmic drop in the electrolyte, R2 refers to the charge ion displacement through the electrode mass interface, and Q2 and Q3 represent the constant phase components.51 All the electrical components values are summarized in Table 4 and clearly show that the value of resistance decreases while the capacitance increases gradually, which significantly improve the bias potential. This behavior enhances the electron-transfer dynamics and causes significant changes, leading to improvements in the charge-transfer process. Also, from Table 4, the OA sample exhibited lower Rct values than the other two, which further supported the CV and GCD results.
Table 4 EIS plot Z fit analysis
| Parameter |
Equivalent circuit: R1 + Q2/R2 + Q3 |
| OA |
CA |
AA |
Unit |
|
R
1 = Rs |
0.623 |
0.740 |
0.940 |
Ohm |
|
Q
2
|
0.009 |
0.002 |
0.006 |
F s (a−1) |
|
a
2
|
0.467 |
0.706 |
0.739 |
— |
|
R
2 = Rct |
3.106 |
6.850 |
8.231 |
Ohm |
|
Q
3
|
0.209 |
0.079 |
0.079 |
F s (a−1) |
|
a
3
|
0.749 |
0.401 |
0.718 |
— |
3.1 Two-electrode fabrication of an ASC device
The electrochemical performance of Cu2MnSnS4//AC was evaluated under a two-electrode configuration via a specifically assembled asymmetric supercapacitor (ASC), with Cu2MnSnS4 as the working positive electrode and activated carbon (AC) as the negative electrode in 2 M KOH as the electrolyte. The operation voltage of the full cell was set to 0–1.5 V for all the studies. The loading mass of an electrode was estimated employing the formula: m+/m− = c− × 1V−/c+ × 1V+. Fig. 8(a) displays a comparative graph of the positive and negative electrodes. In order to analyze a proper voltage window for the ASC device, the potential windows were varied in the CV study, as shown in Fig. 8(b), which revealed both the battery-type and electric double layer effects.52 Very quick charge-transfer performance was exhibited by Cu2MnSnS4//AC ASC when the current density was increased from 10 to 60 mV s−1 and there was no distortion of the CV shape found. Fig. 8(c) presents GCD curves of Cu2MnSnS4//AC ASC, from which the electrochemical performance, such as energy and power density, was calculated (Table 5). For the highest current density of 20 A g−1, 2.5 W h kg−1 and 15000 W kg−1 energy and power density were obtained, respectively. Here in order to remove the stability barriers, we used a switch lock-type device to test the stability behavior. Initially the stability decreased due to the fast reaction kinetics of the electrode/electrolyte interactions. Then after a few cycles more (1000 cycles), constant stability was reached, which may be due to the stabilized reaction kinetics. The best cyclic stability test was carried out for 21000 cycles and 80.35% capacity as well as 97.54% coulombic efficiency were retained, thus indicating the quality of the chosen electrode, as shown in Fig. 8(d). Fig. 8(e) displays a Ragone plot of the energy vs. the power density with other comparative literature values, and these are summarized in Table 6. Further, for the practical demonstration of real-time application, two Cu2MnSnS4//AC ASCs were cascaded and were able to light up a blue LED and run a small-sized motor (Fig. 8(f)). Hence, Cu2MnSnS4//AC ASC devices may be suitable for electrochemical storage applications.
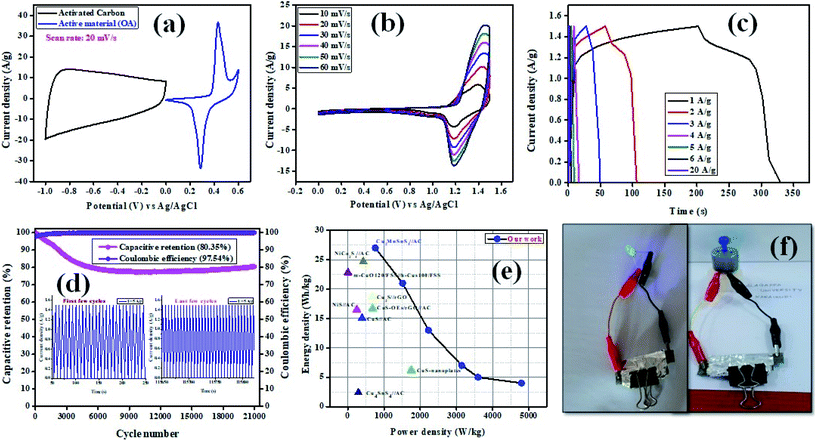 |
| | Fig. 8 (a) CV curves of Cu2MnSnS4–OA and AC electrodes at 20 mV s−1 in a three-electrode system, (b) CV curves, (c) GCD curves, (d) capacitive retention and coulombic efficiency of the ASC device, (e) Ragone plot with other literature values, (f) practical demonstration of an ASC device. | |
Table 5 Capacitive parameters from the GCD plot in a two-electrode configuration
| Potential window (V) |
Current (A) |
Discharge time (s) |
Specific capacitance (F g−1) |
Energy density (W h kg−1) |
Power density (W kg−1) |
| 1.5 |
1 |
128 |
85 |
27 |
759 |
| 2 |
50 |
67 |
21 |
1512 |
| 3 |
21 |
42 |
13 |
2229 |
| 4 |
8 |
21 |
7 |
3150 |
| 5 |
5 |
17 |
5 |
3600 |
| 6 |
3 |
12 |
4 |
4800 |
| 20 |
0.6 |
8 |
2.5 |
15000 |
Table 6 Comparison of the electrochemical performances between the assembled Cu2MnSnS4//AC ASC device with other literature values
| Positive material//negative material |
Cell configuration |
Electrolyte |
Specific capacitance (F g−1) @ GCD |
Energy density (W h kg−1) |
Power density (W kg−1) |
References |
| CuS-nanoplatelets |
Thin film (three-electrode) |
1 M LiClO4 |
55.55 @ 3 A g−1 |
6.23 |
1750 |
53
|
| CuS//AC |
Two-electrode (asymmetric) |
2 M KOH |
48.2 @ 0.5 A g−1 |
15.06 |
392.9 |
54
|
| CuS-OEs/rGO//AC |
Two-electrode (asymmetric) |
3 M KOH |
37.2 @ 10 A g−1 |
16.7 |
681 |
55
|
| NiS//AC |
Two-electrode (asymmetric) |
2 M KOH |
119@ 1 A g−1 |
16.5 |
250 |
56
|
| Cu2S//rGO |
Two-electrode (asymmetric) |
6 M KOH |
93.8 @ 1 A g−1 |
18.6 |
681.2 |
57
|
| NiCo2S4//AC |
Two-electrode (asymmetric) |
2 M KOH |
— |
24.7 |
428 |
58
|
| Cu4SnS4//AC |
Two-electrode (solid state) |
1 M PVA + NAOH |
34.9 |
2.4 |
291 |
59
|
| m-CuO120/FSS//h-CuS100/FSS |
Two-electrode (solid state) |
2 M PVA + KOH |
123 |
22.8 |
2.5 |
60
|
| Cu2MnSnS4//AC |
Two-electrode (asymmetric) |
2 M KOH |
85 |
27 |
759 |
This work |
In this study, a Cu2MnSnS4 electrode with a high working potential for aqueous asymmetric supercapacitors was proposed. The structure directing agent's role in fabrication of the Cu2MnSnS4 nanostructure was investigated. Here, OA electrodes showed the highest capacity and best cyclic stability. Moreover, the fabricated device exhibited a high energy density and power density.
4. Conclusions
In summary, Cu2MnSnS4 electrode materials fabricated with the use of varying structure directing agents (AA, CA, OA) in a three-electrode configuration were successfully developed. Two electrode devices and their electrochemical measurements were tested to determine the best electrode. The calculated specific capacitance values for electrodes fabricates with the use of OA, CA, and AA were 649, 453, and 442 F g−1 at 0.5 A g−1. The electrodes demonstrated 83%, 87%, and 89% capacity retention and cycling stability after 5000 cycles for the AA, CA, and OA electrodes. An asymmetric device was designed and exhibited 27 W h kg−1 energy and 759 W kg−1 power density at 1 A g−1. Moreover, the device provided 80.35% capacity retention and 97.54% Coulombic efficiency after 21000 cycles. Thus excellent electrochemical effects were obtained for the Cu2MnSnS4–OA//AC electrode.
Conflicts of interest
We hereby declare that there are no conflicts of interest.
Acknowledgements
This work was supported by RUSA, UGC-SAP, DST-FIST, DST-PURSE grants. The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through General Research Project under grant number GRP-180-41.
References
- Z. Liu, S. Zhang, L. Wang, T. Wei, Z. Qiu and Z. Fan, Nano Select, 2020, 1, 244–262, DOI:10.1002/nano.202000011.
- E. Hossain, H. M. R. Faruque, M. Sunny, S. Haque, N. Mohammad and N. Nawar, Energies, 2020, 13, 3651, DOI:10.3390/en13143651.
- S. Kumar, G. Saeed, L. Zhu, K. N. Hui, N. H. Kim and J. H. Lee, Chem. Eng. Trans., 2020, 403, 126352, DOI:10.1016/j.cej.2020.126352.
- Z. Yang, J. Tian, Z. Ye, Y. Jin, C. Cui, Q. Xie, J. Wang, G. Zhang, Z. Dong, Y. Miao and X. Yu, Energy Storage Materials, 2020, 33, 18–25, DOI:10.1016/j.ensm.2020.07.020.
- W. Liu, M. Li, G. Jiang, G. Li, J. Zhu, M. Xiao, Y. Zhu, R. Gao, A. Yu, M. Feng and Z. Chen, Adv. Energy Mater., 2020, 10, 2001275, DOI:10.1002/aenm.202001275.
- S. Biswas, V. Sharma, D. Mandal, A. Chowdhury, M. Chakravarty, S. Priya, C. C. Gowda, P. De, I. Singh and A. Chandra, CrystEngComm, 2020, 22, 1633–1644, 10.1039/C9CE01547G.
- T. Wang, F. Yu, X. Wang, S. Xi, K. J. Chen and H. Wang, Electrochim. Acta, 2020, 334, 135586, DOI:10.1016/j.electacta.2019.135586.
- A. M. Zardkhoshoui and S. S. H. Davarani, Chem. Eng. J., 2020, 402, 126241, DOI:10.1016/j.cej.2020.126241.
- C. Xiong, Q. Yang, W. Dang, M. Li, B. Li, J. Su, Y. Liu, W. Zhao, C. Duan, L. Dai and Y. Xu, J. Power Sources, 2020, 447, 227387, DOI:10.1016/j.jpowsour.2019.227387.
- Y. Wang, X. Wang, X. Li, Y. Bai, H. Xiao, Y. Liu and G. Yuan, Chem. Eng. J., 2021, 405, 126664, DOI:10.1016/j.cej.2020.126664.
- K. Robert, D. Stiévenard, D. Deresmes, C. Douard, A. Iadecola, D. Troadec, P. Simon, N. Nuns, M. Marinova, M. Huvé and P. Roussel, Energy Environ. Sci., 2020, 13, 949–957, 10.1039/C9EE03787J.
- M. Isacfranklin, R. Yuvakkumar, G. Ravi, S. I. Hong, D. Velauthapillai, M. Thambidurai, C. Dang, T. S. Algarni and A. M. Al-Mohaimeed, Carbon, 2020, 172, 613–623, DOI:10.1016/j.carbon.2020.10.081.
- K. Thiagarajan, T. Bavani, P. Arunachalam, S. J. Lee, J. Theerthagiri, J. Madhavan, B. G. Pollet and M. Y. Choi, J. Nanomater., 2020, 10, 392, DOI:10.3390/nano10020392.
- R. Wang and Y. Li, Energy Storage Materials, 2020, 31, 230–251, DOI:10.1016/j.ensm.2020.06.012.
-
T. Okamura and K. Nakagawa, ECSarXiv, 2020, DOI:10.1149/osf.io/9gjym.
- Y. Liu, X. Xu and Z. Shao, Energy Storage
Materials, 2020, 26, 1–22, DOI:10.1016/j.ensm.2019.12.019.
- K. N. Hui, K. San Hui, Z. Tang, V. V. Jadhav and Q. X. Xia, J. Power Sources, 2016, 330, 195–203, DOI:10.1016/j.jpowsour.2016.08.116.
- S. Liu, D. Ni, H. F. Li, K. N. Hui, C. Y. Ouyang and S. C. Jun, J. Mater. Chem. A, 2018, 6, 10674–10685, 10.1039/C8TA00540K.
- Q. Zhang, W. B. Zhang, P. Hei, Z. Hou, T. Yang and J. Long, Appl. Surf. Sci., 2020, 527, 146682, DOI:10.1016/j.apsusc.2020.146682.
- R. Pothu, R. Bolagam, Q. H. Wang, W. Ni, J. F. Cai, X. X. Peng, Y. Z. Feng and J. M. Ma, Rare Met., 2020, 258, 1–21, DOI:10.1007/s12598-020-01470-w.
- H. Hua, S. Liu, Z. Chen, R. Bao, Y. Shi, L. Hou, G. Pang, K. N. Hui, X. Zhang and C. Yuan, Sci. Rep., 2016, 6, 20973, DOI:10.1038/srep20973.
- X. Wang, R. Cao, S. Zhang, P. Hou, R. Han, M. Shao and X. Xu, J. Mater. Chem. A., 2017, 5, 23999–24010, 10.1039/c7ta06809c.
- J. S. Wei, T. B. Song, P. Zhang, Z. Y. Zhu, X. Y. Dong, X. Q. Niu and H. M. Xiong, ACS Appl. Energy Mater., 2020, 3, 6907–6914, DOI:10.1021/acsaem.0c00990.
- R. S. Gohar, I. Ahmad, A. Shah, S. Majeed, M. Najam-Ul-Haq and M. N. Ashiq, J. Electrochem. Energy Convers. Storage, 2020, 31, 101621, DOI:10.1016/j.est.2020.101621.
- P. Wonner, T. Steinke, L. Vogel and S. M. Huber, Chem.–Eur. J., 2020, 26, 1258–1262, DOI:10.1002/chem.201905057.
- R. Henríquez, C. Vásquez, E. Muñoz, P. Grez, F. Martín, J. R. Ramos-Barrado and E. A. Dalchiele, Phys. E Low-dimens. Syst. Nanostruct., 2020, 118, 113881, DOI:10.1016/j.physe.2019.113881.
- J. H. M. van Gils, E. van Dijk, A. Peduzzo, A. Hofmann, N. Vettore and M. P. Schützmann, PLoS Comput. Biol., 2020, 16(5), e1007767, DOI:10.1371/journal.pcbi.1007767.
- T. T. Trinh, K. Q. Tran, X. Q. Zhang, R. A. van Santen and E. J. Meijer, Phys. Chem. Chem. Phys., 2015, 17, 21810–21818, 10.1039/c5cp02068a.
- J. Qin, Q. Li, Y. Liu, A. Niu and C. Lin, Ecotoxicol. Environ. Saf., 2020, 201, 110873, DOI:10.1016/j.ecoenv.2020.110873.
- S. Asaithambi, P. Sakthivel, M. Karuppaiah, G. U. Sankar, K. Balamurugan, R. Yuvakkumar, M. Thambidurai and G. Ravi, J. Electrochem. Energy Convers. Storage, 2020, 31, 101530, DOI:10.1016/j.est.2020.101530.
- M. Isacfranklin, G. Ravi, R. Yuvakkumar, P. Kumar, D. Velauthapillai, B. Saravanakumar, M. Thambidurai and C. Dang, Ceram. Int., 2020, 46, 16291–16297, DOI:10.1016/j.ceramint.2020.03.186.
- L. Chen, H. Deng, J. Tao, H. Cao, L. Huang, L. Sun, P. Yang and J. Chu, RSC Adv, 2015, 5, 84295–84302, 10.1039/c5ra14595c.
- C. Sripan, D. Alagarasan, S. Varadharajaperumal, R. Ganesan and R. Naik, Curr. Appl. Phys., 2020, 20, 925–930, DOI:10.1016/j.cap.2020.05.003.
- M. Devika, N. Koteeswara Reddy, M. Prashantha, K. Ramesh, S. Venkatramana Reddy, Y. B. Hahn and K. R. Gunasekhar, Phys. Status Solidi, 2010, 207, 1864–1869, DOI:10.1002/pssa.200925379.
- S. Varadharajaperumal, C. Sripan, R. Ganesan, G. Hegde and M. N. Satyanarayana, Cryst. Growth Des., 2017, 17, 5154–5162, DOI:10.1021/acs.cgd.7b00632.
- L. Chen, H. Deng, J. Tao, W. Zhou, L. Sun, F. Yue, P. Yang and J. Chu, J. Alloys Compd., 2015, 640, 23–28, DOI:10.1016/j.jallcom.2015.03.225.
- S. Riyaz, A. Parveen and A. Azam, Perspectives in Science, 2016, 8, 632–635, DOI:10.1016/j.pisc.2016.06.041.
- Y. Liu, X. Mi, J. Wang, M. Li, D. Fan, H. Lu and X. Chen, Inorg. Chem. Front., 2019, 6, 948–954, 10.1039/C9qi00020h.
- Y. Zuo, J. Li, X. Yu, R. Du, T. Zhang, X. Wang, J. Arbiol, J. Llorca and A. Cabot, Chem. Mater., 2020, 32, 2097–2106, DOI:10.1021/acs.chemmater.9b05241.
- D. Sharma, A. V. Menon and S. Bose, Nanoscale Adv, 2020, 2, 3292–3303, DOI:
.
- X. Liang, P. Guo, G. Wang, R. Deng, D. Pan and X. Wei, RSC Adv, 2012, 2, 5044–5046, 10.1039/c2ra20198d.
- Y. Xie, C. Zhang, G. Yang, J. Yang, X. Zhou and J. Ma, J. Alloys Compd., 2017, 696, 938–946, DOI:10.1016/j.jallcom.2016.12.043.
- Y. A. Kumar, K. D. Kumar and H. J. Kim, Electrochim. Acta, 2020, 330, 135261, DOI:10.1016/j.electacta.2019.135261.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950, 10.1039/c5cs00580a.
- T. S. Mathis, N. Kurra, X. Wang, D. Pinto, P. Simon and Y. Gogotsi, Adv. Energy Mater., 2019, 9, 1902007, DOI:10.1002/aenm.201902007.
- P. Himasree, I. K. Durga, T. N. V. Krishna, S. S. Rao, C. V. M. Gopi, S. Revathi, K. Prabakar and H. J. Kim, Electrochim. Acta, 2019, 305, 467–473, DOI:10.1016/j.electacta.2019.03.041.
- J. Theerthagiri, K. Karuppasamy, G. Durai, A. U. H. S. Rana, P. Arunachalam, K. Sangeetha, P. Kuppusami and H. S. Kim, J. Nanomater., 2018, 8, 256, DOI:10.3390/nano8040256.
- D. Guragain, C. Zequine, R. K. Gupta and S. R. Mishra, Processes, 2020, 8, 343, DOI:10.3390/pr8030343.
- S. Kaipannan, K. Govindarajan, S. Sundaramoorthy and S. Marappan, ACS Omega, 2019, 4, 15798–15805 CrossRef CAS.
- W. Chen, X. Yu, Z. Zhao, S. Ji and L. Feng, Electrochim. Acta, 2019, 298, 313–320, DOI:10.1016/j.electacta.2018.12.096.
- G. M. Kumar, F. Xiao, P. Ilanchezhiyan, S. Yuldashev and T. W. Kang, RSC Adv, 2016, 6, 99631–99637 RSC.
- X. He, X. Mao, C. Zhang, W. Yang, Y. Zhou, Y. Yang and J. Xu, J. Mater. Sci.: Mater. Electron., 2020, 31, 2145–2152, DOI:10.1007/s10854-019-02737-4.
- C. J. Raj, B. C. Kim, W. J. Cho, W. G. Lee, Y. Seo and K. H. Yu, J. Alloys Compd., 2014, 586, 191–196, DOI:10.1016/j.jallcom.2013.10.056.
- J. Zhang, H. Feng, J. Yang, Q. Qin, H. Fan, C. Wei and W. Zheng, ACS Appl. Mater. Interfaces, 2015, 7, 21735–21744, DOI:10.1021/acsami.5b04452.
- Y. Cui, J. Zhang, G. Li, Y. Sun, G. Zhang and W. Zheng, Chem. Eng. J., 2017, 325, 424–432, DOI:10.1016/j.cej.2017.05.069.
- S. Nandhini and G. Muralidharan, Appl. Surf. Sci., 2018, 449, 485–491, DOI:10.1016/j.apsusc.2018.01.024.
- T. Zhao, X. Peng, X. Zhao, J. Hu, W. Yang, T. Li and I. Ahmad, Compos. B Eng., 2019, 163, 26–35, DOI:10.1016/j.compositesb.2018.11.023.
- Y. Zhu, X. Ji, Z. Wu and Y. Liu, Electrochim. Acta, 2015, 186, 562–571, DOI:10.1016/j.electacta.2015.10.176.
- A. C. Lokhande, A. Patil, A. Shelke, P. T. Babar, M. G. Gang, V. C. Lokhande, D. S. Dhawale, C. D. Lokhande and J. H. Kim, Electrochim. Acta, 2018, 284, 80–88, DOI:10.1016/j.electacta.2018.07.170.
- A. M. Patil, V. C. Lokhande, T. Ji and C. D. Lokhande, Electrochim. Acta, 2019, 307, 30–42, DOI:10.1016/j.electacta.2019.03.108.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
G.
Ravi
*a,
Dhayalan
Velauthapillai
b,
Mehboobali
Pannipara
c and
Abdullah G.
Al-Sehemi
c
*a,
G.
Ravi
*a,
Dhayalan
Velauthapillai
b,
Mehboobali
Pannipara
c and
Abdullah G.
Al-Sehemi
c
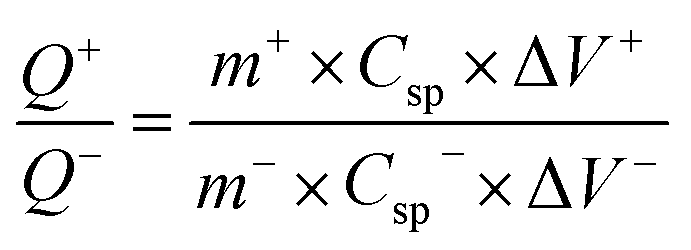 and then coated on to the positive and negative electrodes. The full-cell device parameters such as energy (E) and power (P) density, were measured using eqn (2) and (3). Eqn (4) was used to calculate the coulombic efficiency of the materials from the GCD curves.
and then coated on to the positive and negative electrodes. The full-cell device parameters such as energy (E) and power (P) density, were measured using eqn (2) and (3). Eqn (4) was used to calculate the coulombic efficiency of the materials from the GCD curves.