
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable dialdehyde/dicarboxylate nanocelluloses by stoichiometrically optimized sequential periodate–chlorite oxidation for tough and wet shape recoverable aerogels†
Gabriel
Patterson
 and
You-Lo
Hsieh
*
and
You-Lo
Hsieh
*
Biological and Agricultural Engineering, University of California, Davis, California 95616, USA. E-mail: ylhsieh@ucdavis.edu; Tel: +1 530 752 0843
First published on 4th November 2020
Abstract
Sequential periodate–chlorite (PC) oxidation has been optimized stoichiometrically according to the non-crystalline content in cellulose to generate a variety of versatile C2,C3 dialdehyde/dicarboxylate nanocelluloses (NCs) while economizing chemical and shear force inputs. The robust primary sodium periodate (NaIO4) oxidation not only regioselectively cleaved the C2–C3 carbon bond to oxidize the vicinal hydroxyls to aldehydes, but also governed the lengths of NCs, i.e., cellulose nanofibrils (PC-CNFs) at near-equal NaIO4 to non-crystalline anhydroglucose unit (AGU) stoichiometry and cellulose nanocrystals (PC-CNCs) at a doubled ratio. Secondary sodium chlorite (NaClO2) oxidation facilely converted C2,C3 dialdehydes to dicarboxylates and, upon deprotonation, facilitated defibrillation to NCs, irrespective of extents of carboxylation or charges. The optimal 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 NaIO4/AGU and 1:1 NaClO2/AGU oxidation produced highly uniform 1.26 nm thick, 3.28 nm wide, and ca. 1 μm long PC-CNFs with tunable surface aldehyde (0.71–0.0 mmol g−1) and carboxylate (0.64–1.35 mmol g−1) content at 94–98% yields. The C2–C3 glucosidic ring opening and oxidation along the 110 or 1
:1 NaIO4/AGU and 1:1 NaClO2/AGU oxidation produced highly uniform 1.26 nm thick, 3.28 nm wide, and ca. 1 μm long PC-CNFs with tunable surface aldehyde (0.71–0.0 mmol g−1) and carboxylate (0.64–1.35 mmol g−1) content at 94–98% yields. The C2–C3 glucosidic ring opening and oxidation along the 110 or 1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0 crystalline surfaces increased the heterogeneity of the hydrophilic surfaces and flexibility of PC-CNFs to influence their self-assembling into fibrils and amphiphilic superabsorbent aerogels. The ultra-light (ρ = 10.3 mg cm−3) aerogels showed an ultra-high dry specific compression modulus (50.2 kPa mg−1 cm−3) and specific stress (8.2 kPa mg−1 cm−3 at 0.8 strain), cyclic wet compressive behavior, and excellent water-activated shape recovery following 0.8 strain dry compression.
0 crystalline surfaces increased the heterogeneity of the hydrophilic surfaces and flexibility of PC-CNFs to influence their self-assembling into fibrils and amphiphilic superabsorbent aerogels. The ultra-light (ρ = 10.3 mg cm−3) aerogels showed an ultra-high dry specific compression modulus (50.2 kPa mg−1 cm−3) and specific stress (8.2 kPa mg−1 cm−3 at 0.8 strain), cyclic wet compressive behavior, and excellent water-activated shape recovery following 0.8 strain dry compression.
Introduction
Nanocelluloses are among the most unique one-dimensional biologically derived nanomaterials1 and have garnered considerable attention due to their exceptional strength2 and low thermal expansion coefficient.3 Nanocelluloses are typically obtained by top-down separation of crystalline domains in cellulose, the most abundant polymer in nature, from wood pulp. Among the most studied are cellulose nanocrystals (CNCs) and cellulose nanofibrils (CNFs). While CNC yields are typically 30% or less, CNFs can be produced by 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)-mediated oxidation of cellulose aided by magnetic stirring,4 blending,4,5 sonication,6 or twin-screw extrusion7 at much higher yields of 80 (ref. 7) to 97–98%.4–6 Other than woody sources, the abundant under-utilized agricultural biomass represents an attractive feedstock of cellulose. Rice straw, the world's largest crop residue,8 contains a significant amount of cellulose similar to woody biomass and has been isolated to generate CNCs9 and CNFs10 while the other major components have been converted into silica11,12 and carbon12 nanomaterials. Optimal TEMPO-oxidation followed by high-speed blending has been shown to convert 96.8% rice straw cellulose into super-fine CNFs (2.09 nm wide, 1.52 nm thick, and up to 1 μm long) with 1.29 mmol g−1 surface charge.10While TEMPO regioselectively oxidizes the C6 cellulose hydroxyl, another long-recognized regioselective reaction involves sodium periodate oxidation that cleaves the C2–C3 carbon bond and oxidizes the vicinal hydroxyls. Oxidation of starch13 and cellulose14,15 with sodium periodate (NaIO4) followed by sodium chlorite (NaClO2) produced water-soluble derivatives13–15 to enable conjugation with a platinum anticancer drug15 at near full conversion or produce CNCs.16 This periodate–chlorite oxidation has also been studied for producing nanocelluloses (Table S1†).16–20 At 0.5:1 NaIO4/anhydroglucose unit (AGU) and 1:1 NaClO2/AGU ratios, these reactions produced 10–20 nm thick nanofibrils, but gave yields only up to 50%, leaving the rest soluble.16 The yields of nanocelluloses could be improved to 84–100% with excess oxidants at 0.8:1 NaIO4/AGU17–19 and 2:1 NaClO2/AGU17–20 ratios and aided by homogenization17,19,20 or microfluidization,18 to produce thinner and varied levels of carboxylation by extending the periodate reaction time.16–19
In periodate–chlorite oxidation of cellulose, the primary periodate oxidation breaks the C2–C3 bond in the glucosidic ring to oxidize the vicinal hydroxyls into dialdehydes, and the secondary chlorite oxidation converts the dialdehydes to dicarboxylic acids. The sequential nature of periodate–chlorite oxidation thus offers two levels of control over this oxidative approach to nanocelluloses. In principle, not only does each of these oxidations control the respective hydroxyl-to-aldehyde and aldehyde-to-carboxylic acid conversion, but the primary periodate oxidant can be streamlined based on the stoichiometric quantity of non-crystalline cellulose of the feedstock. However, the potential of each of these oxidations to regulate the properties of nanocelluloses produced has yet to be fully elucidated and exploited. Furthermore, economized use of these chemicals adds benefits to the already proven sodium periodate recyclability21–23 and chlorine-containing effluent treatments.24
This study is rationally designed to optimize the sequential NaIO4–NaClO2 oxidation of cellulose to facilitate the generation as well as to diversify the quality attributes of dialdehyde/dicarboxyl nanocelluloses by tuning the respective reactions. As oxidation occurs in the non-crystalline domains of cellulose, primary oxidation was first optimized by targeting the periodate quantity to be near the stoichiometric range of non-crystalline AGU in rice straw cellulose with a crystallinity index of 61.8%.9 Upon determining the optimal stoichiometric NaIO4/AGU ratio, secondary chlorite oxidation was streamlined to a 1:1 NaClO2/AGU ratio and 0.5 M CH3COOH, both half of what has been reported.17–20 The effects of charges that facilitate the disintegration and dispersion of individualized nanocelluloses were discerned. Periodate–chlorite (PC) oxidized cellulose was disintegrated by the same optimized high-speed blending and centrifugation process for TEMPO-CNFs10 to collect the CNF-containing supernatant fraction. Reaction optimization was determined by yield as well as dimensional and surface chemistry attributes of PC-CNFs. Freezing induced self-assembled PC-CNFs were characterized in terms of their morphology, chemical and crystalline structures, and thermal stability to relate to nanocellulose surface attributes of open-ring and tunable C2,C3 dialdehyde/dicarboxyl chemistry. The porous structure, physical properties, liquid absorption properties, and dry and wet compressive behaviors of ice-templated aerogels were elucidated and compared to those of TEMPO-CNFs from the same cellulose source.
Experimental section
Materials
Cellulose was purified from rice straw (RS, Calrose variety) by 2:1 v/v toluene/ethanol extraction of epicuticular lipids, acidified sodium chlorite (NaClO2) dissolution of lignin (1.4%, 70 °C, 5 h), and alkaline dissolution of hemicellulose and silica (5% KOH, 24 h; 90 °C, 2 h) to give 36% yield and 61.8% crystallinity.9 Sodium periodate (NaIO4, 99.9%, Sigma-Aldrich), sodium chlorite (NaClO2, 80%, Sigma-Aldrich), glacial acetic acid (CH3COOH, 99.9%, Fisher Scientific), and sodium hydroxide (NaOH, 1 N, Certified, Fisher Scientific) were used as received. All water used was from a Milli-Q water purification system (Millipore Corporation, Billerica, MA).
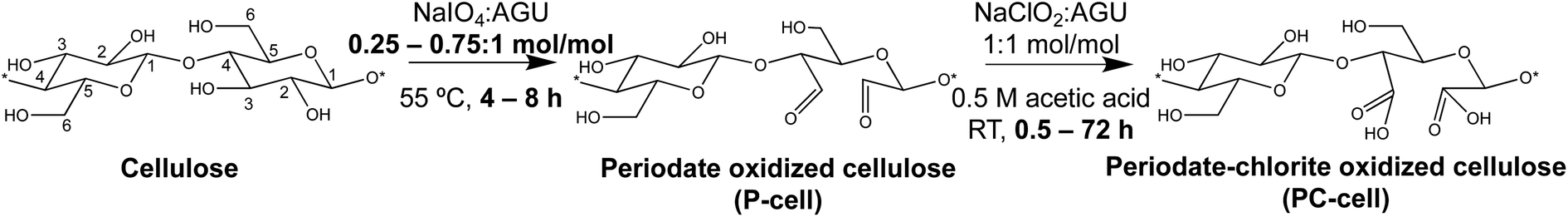
Sequential periodate–chlorite oxidation toward C2,C3-nanocellulose production
RS cellulose (0.5 g) was added to 100 mL water with vigorous mechanical stirring in an oil bath. Primary oxidation was conducted at 0.25, 0.5, and 0.75:1 NaIO4/anhydroglucose unit (AGU) molar ratios near the stoichiometric (ca. 38%) non-crystalline cellulose content at 55 °C for 4 or 8 h (in the dark). The never-dried periodate oxidized cellulose (P-cell) was dialyzed against water until conductivity lowered to <10 μS cm−1, or 24–48 h. Secondary oxidation was conducted at 1:1 NaClO2/AGU ratio (0.5 M CH3COOH, RT) for 0.5 to 72 h to yield periodate–chlorite oxidized cellulose (PC-cell). The never-dried PC-cell was dialyzed and blended (Vitamix 5200) at 37.5k rpm for 15, 30, 60, or 90 min intervals and centrifuged (Thermo Fisher Scientific, Megafuge 1.6 L) at 5k rpm for 15 min. PC-cell was deprotonated with 0.5 M NaOH (pH 7.5) to convert surface carboxylic acid groups to charged sodium carboxyl(ate), and the carboxylated PC-cell was then blended and centrifuged in a similar way to collect charged nanofibrils (PC-CNFs) in the aqueous supernatant. PC-CNFs were denoted by their chlorite reaction time, i.e., PC-CNF-0.5 h, PC-CNF-1 h, etc.
Characterization of aqueous nanocelluloses
An appropriate amount of 1 N HCl was added to 50 mL of 0.05% PC-CNF suspension to protonate all the carboxyl groups, and then titrated with 0.01 M NaOH solution. The charge of PC-CNFs (σ, in mmol g−1 cellulose) was recorded using an OAKTON pH/Con 510 series meter and calculated as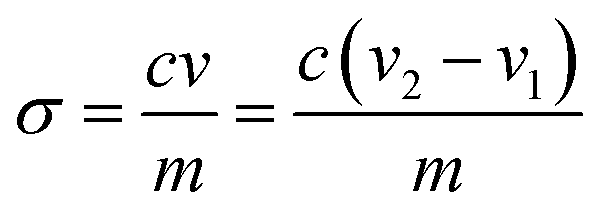 | (1) |
Atomic force microscopy (AFM) was used to characterize PC-CNF thickness, while transmission electron microscopy (TEM) was used for width and length dimensions. PC-CNF dispersions (10 μL, 0.0005 wt%) were deposited onto a fresh mica surface, air-dried, and scanned (Asylum-Research MFP-3D) under ambient conditions using tapping mode with OMCL-AC160TS standard silicon probes (26 N m−1 nominal spring constant, 7 nm nominal tip radius). The scan rate was 1 Hz and image resolution 512 × 512 pixels. The height images and profiles were processed using Igor Pro 6.21 loaded with MFP3D 090909 + 1409. Similarly, dispersions were deposited onto glow-discharged carbon-coated TEM grids (300-mesh copper, formvar–carbon, 100 kV, Ted Pella Inc., Redding, CA). Specimens were stained with 2% uranyl acetate solution, blotted, and ambient dried.
The least charged PC-CNF-1 h was used to observe the effects of concentrations from 0.1 to 1% on UV-Vis transmittance from 300 to 800 nm wavelengths using an Evolution 600 UV-vis spectrophotometer, and its gelation was observed.
Solid-state characterization
Purified cellulose, P-cell, PC-cell, and PC-CNF aqueous dispersions (0.1%) were frozen in liquid N2 (−196 °C, 15 min) and lyophilized (−50 °C, 0.05 mbar, 2 days) in a freeze-drier (FreeZone 1.0L Benchtop Freeze-Dry System, Labconco, Kansas City, MO) for all solid-state characterization experiments. Fourier Transform Infrared (FTIR) spectra were collected for samples in transparent KBr pellets (1:100, w/w) on a Thermo Nicolet 6700 spectrometer, under ambient conditions, in transmittance mode, from an accumulation of 128 scans at a 4 cm−1 resolution over the regions of 4000 to 400 cm−1. Thermal Gravimetric Analysis (TGA) was performed on 5–10 mg samples at 10 °C min−1 up to 600 °C under purging N2 (50 mL min−1) using a TGA-50 thermogravimetric analyzer (Shimadzu, Japan). Cellulose samples were compressed by hand between two glass slides and scanned from 5 to 40° 2θ diffraction angles using an X-ray diffractometer (Panalytical XRD) with Ni-filtered Cu Kα radiation (λ = 0.1548 nm) at 45 kV and 40 mA. The crystallinity index (CrI) was calculated using the Segal equation,25 incorporating the maximum intensity of the 200 crystallographic lattice peak (I200 = 2θ = 22.7°) and the intensity minimum between the 200 and 110 peaks (Iam = 2θ = 18.7°).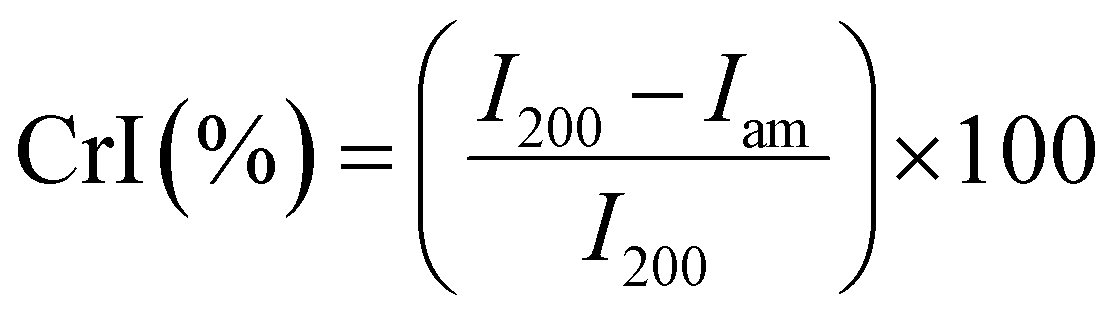 | (2) |
Fully carboxylated PC-CNF-72 h at varying concentrations (0.001–1.0%, 6 mL) was frozen at −196 °C for 15 min and the self-assembled fibrillar structures were sputter-coated with gold (15–20 Å, 2.5 kV, 5 mA, 30 s) and examined by scanning electron microscopy (SEM-FEG, QUATTRO Thermo Fisher Environmental) at 5 kV accelerating voltage with a 10 mm working distance. The widths of the assembled fibers (n = 30) were measured using the QUATTRO image analyzer. To probe the interfaces in the freezing induced self-assembled PC-CNF-72 h, solids from 1.0% aqueous suspension were redispersed in 30 mL water at 0.07, 0.1, and 0.13% by 5 min vortexing (Fisherbrand Analog Vortex Mixer) at 100% amplitude.
Aerogels were fabricated by freezing aqueous PC-CNF dispersions, 6 mL each in 14 mm inner diameter glass tubes, at −20 °C for 9 h (ThermoFisher, IsoTemp, Model HF-5017), and then lyophilized. The effect of concentration (0.1–1.0%, 6 mL) on the aerogel structure and properties was investigated using the least carboxylated PC-CNF-1 h, while the effect of charges was assayed using PC-CNF-4 h, -8 h, -12 h and -18 h. The density (ρa) of aerogels was calculated from the weighed mass and volume of 1 cm sections of cylindrical samples, and the porosity (ϕ) of the aerogel calculated as
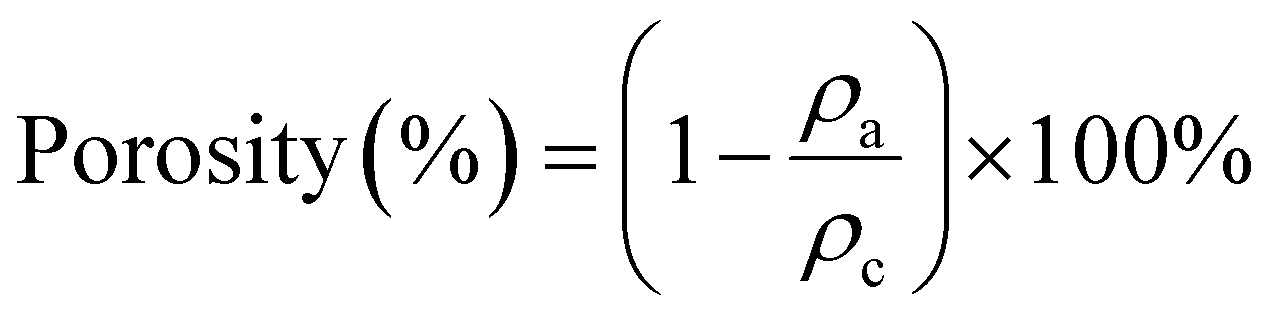 | (3) |
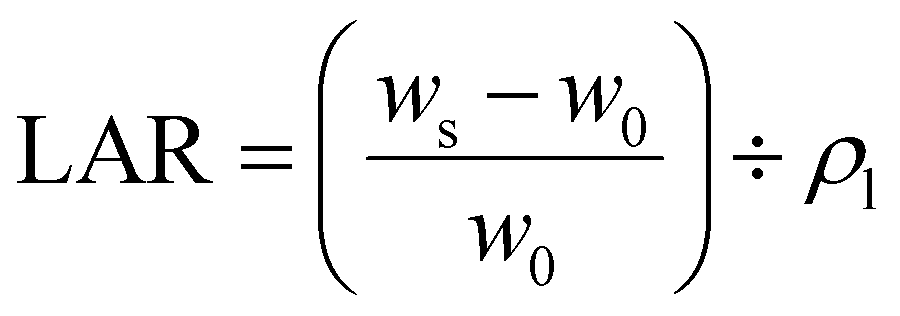 | (4) |
Results and discussion
Optimization of primary oxidation
Rice straw (RS) is the largest agricultural residue by mass globally,8 and therefore represents not only the highest quantity agricultural biomass but also the most significant non-wood lignocellulosic source. To optimize primary oxidation and facilitate the disintegration of RS cellulose into nanocelluloses, oxidation was conducted with periodate (NaIO4) near the stoichiometric range of the non-crystalline AGU in RS cellulose. Sodium periodate oxidation with 0.25, 0.5, and 0.75:1 NaIO4/anhydroglucose unit (AGU) molar ratios at 55 °C (4 or 8 h) was followed by secondary sodium chlorite oxidation (1:1 NaClO2/AGU, 0.5 M CH3COOH, RT, 48 h; Scheme 1) to produce the respective periodate oxidized cellulose (P-cell) and periodate–chlorite oxidized cellulose (PC-cell), which were then defibrillated by high speed blending (37.5k rpm) for intervals of either 15 or 30 min.
 | ||
| Scheme 1 Sequential periodate–chlorite oxidation of rice straw (RS) cellulose. | ||
The PC-nanocelluloses or PC-NCs in the aqueous supernatant fractions (5k rpm, 15 min) were characterized in terms of their gravimetric yields, surface carboxyl contents, and fibrillar attributes by AFM (Fig. 1). As expected, increasing quantities of PC-NCs were produced at higher NaIO4 oxidant levels and with longer blending (Fig. 1a). Below the stoichiometric level of 0.25:1 NaIO4/AGU (4 h), PC-NC yields increased linearly with longer blending and reached only 53.0% with the longest 90 min blending (Fig. 1b–d). Conversely, higher periodate levels of 0.5:1 and 0.75:1 NaIO4/AGU (4 h) produced similar (52.2%) and higher (58.6%) amounts of PC-NCs with much shorter 30 min or 15 min blending, respectively (Fig. 1e and h). Clearly, the primary NaIO4 oxidant at near-equal and higher stoichiometries of AGU in non-crystalline cellulose was key in producing PC-NCs. While longer blending produced additional PC-NCs (Fig. 1f, g and i), those produced in the first blending intervals had higher yield, and were finer, individualized, and 2.1 to 3.6 times more carboxylated (1.09–1.48 mmol g−1 charge), consistent with the highest UV-Vis transmittance of their dispersions (Fig. S1†). In fact, oxidation with excess periodate of 0.75:1 NaIO4/AGU for 8 h longer yielded 80.1% PC-NCs with only 15 min blending, which is significantly higher (Fig. 1j). However, these PC-NCs were also much shorter, more rod-like or resembled CNCs. These PC-CNCs were similar to those reported for wood pulp with a similar level of primary oxidant (0.8:1 NaIO4/AGU, 4.5 h) but with doubled secondary oxidant (2:1 NaClO2/AGU and 1 M CH3COOH) as well as longer time (40 h)19 (Table S1†).
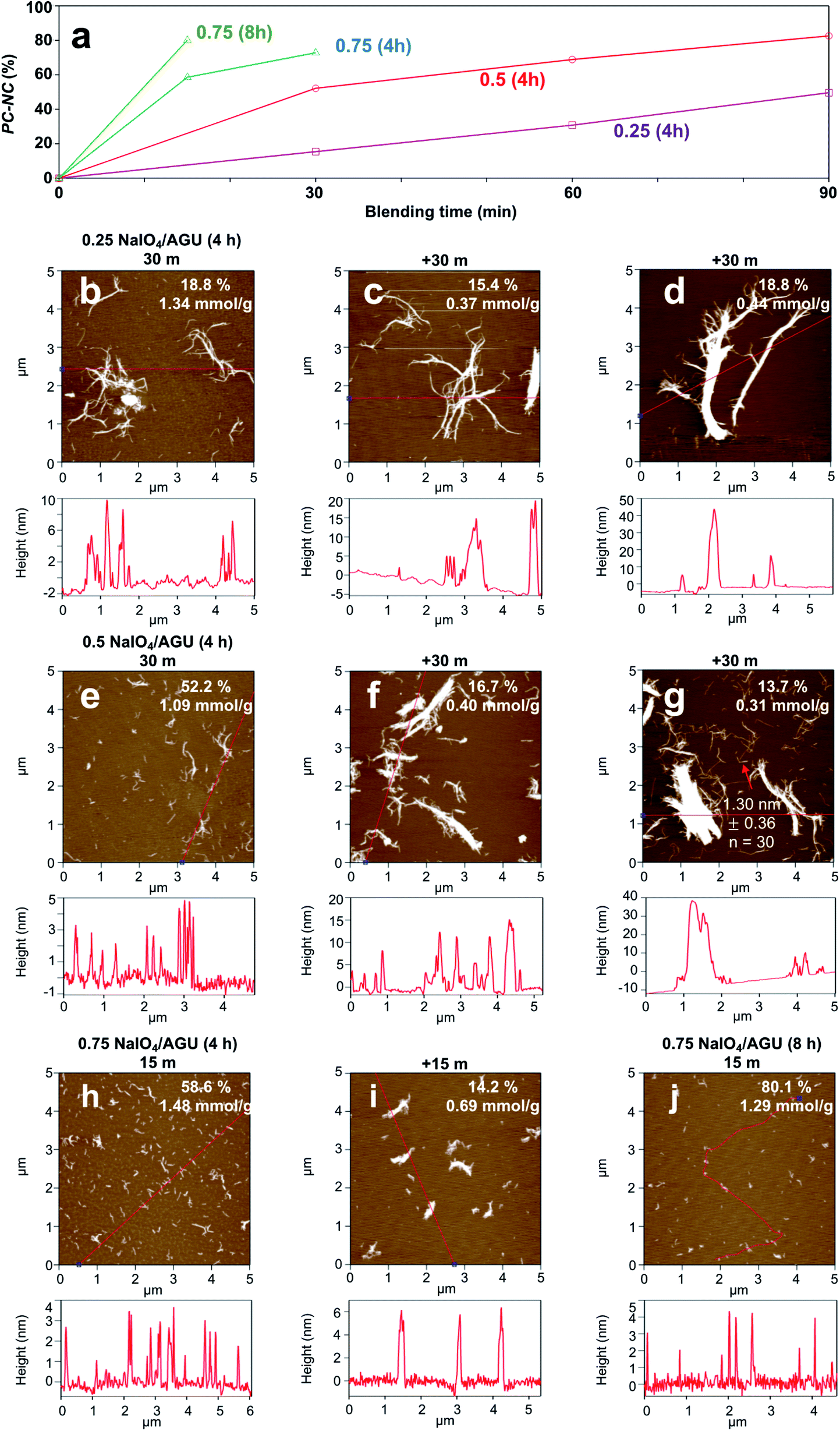 | ||
| Fig. 1 Primary periodate oxidation at varied NaIO4/AGU ratios (55 °C) and initial and additional blending time (37.5k rpm) on aqueous dispersible periodate–chlorite oxidized nanocellulose (PC-NC): (a) yield; AFM phase images and the corresponding height profiles of PC-NCs under the following conditions: (b–d) 0.25:1 (4 h); (e–g) 0.5:1 (4 h); (h and i) 0.75:1 (4 h); (j) 0.75:1 (8 h). The yield and carboxyl values of each population were noted. | ||
Notably, more primary oxidant and longer reaction time not only produce PC-NCs with less blending, but also to reduce their lengths (Fig. 1hvs.1e; 1jvs.1h). Evidently, more intensive primary periodate oxidation causes chain scissions, in addition to cleaving the C2–C3 bond of the glucosidic ring to form vicinal hydroxyls. With a 0.5:1 NaIO4/AGU ratio and 90 min blending, 1.3 nm (±0.36, n = 30) thick and about 1 μm long PC-CNFs were produced at 72.6% yield (Fig. 1g). Considering both PC-NC qualities and yields, the 0.5:1 NaIO4/AGU ratio was the optimal primary oxidation condition for further optimization of chlorite oxidation. This slightly higher primary oxidant than non-crystalline AGU stoichiometrically is anticipated when taking into account the AGU at the crystalline–amorphous interfaces of crystallite surfaces.
Secondary oxidation at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 NaClO2/AGU
:1 NaClO2/AGU
Secondary sodium chlorite (1:1 NaClO2/AGU) oxidation was conducted on optimally sodium periodate oxidized (0.5:1 NaIO4/AGU, 4 h) cellulose for varied lengths of time (ca. pH 3.5), and then blended for 30 min (Fig. 2). Chlorite oxidation for 4 to 48 h produced only 32 to 48% PC-CNFs showing little time dependence (Fig. 2a). The role of surface charge was found critical to disintegration to nanocelluloses. Specifically, converting the surface carboxylic acid on PC-cell to carboxylate with a base (pH 7.5, 0.5 N NaOH) significantly increased the yield by blending to as high as 98.6% PC-CNFs irrespective of the duration of secondary oxidation or charge levels (Table S2†). Deprotonation of optimal PC-CNFs increased charges exponentially from 0.64 mmol g−1 at 0.5 h to 1.29 mmol g−1 at 18 h, reaching a steady state at around 48 h and an ultimate charge of 1.35 mmol g−1 at 72 h (Fig. 2a). As blending of P-cell did not yield any P-NCs (Fig. S2a and b†), both secondary chlorite oxidation and deprotonation to add electrostatic repulsion are clearly critical to fully disintegrate and disperse the charged PC-CNFs. In fact, brief chlorite oxidation for only 0.5 h conferred near half of the maximum surface carboxylate content to produce PC-CNF-0.5 h at 94% yield (Table S2†). While extending the chlorite oxidation time from 0.5 to 18 h doubled charges, it only slightly improved the yield by ca. 5%. All PC-CNFs produced had essentially the same thickness of 1.26 ± 0.44 nm (n = 376) and length of 1.01 ± 0.15 μm (n = 69), irrespective of the duration of secondary chlorite reaction or charges (Fig. 2b and c, and S2c†). Thus, brief secondary oxidation is sufficient to generate PC-CNFs at 95.6 ± 1.6% so long as they are charged. Even a minor PC-cell precipitate fraction (6.5%, Table S2†), containing supposedly larger fibrils, also displayed carboxyl sodium salts (Fig. S2d†), confirming the effectiveness of 1 h secondary reaction.
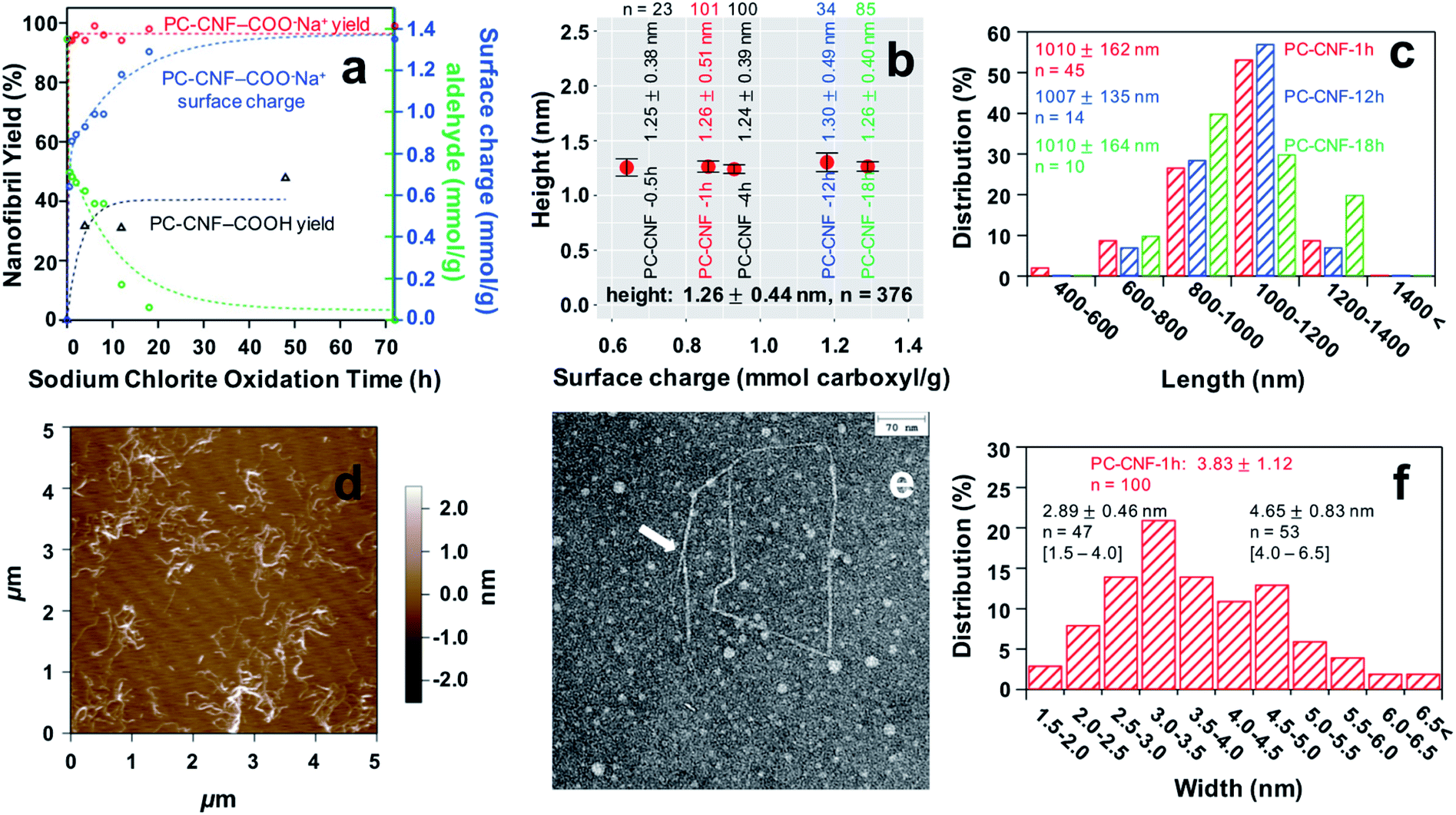 | ||
| Fig. 2 PC-CNFs from varied chlorite times of 0.5–72 h: (a) yield and surface chemistry; (b) PC-CNF thickness; (c) length distributions; (d) AFM phase image, (e) TEM image, and (f) width distribution of PC-CNF-1 h. | ||
Clearly, varying the duration of secondary chlorite oxidation of optimal P-cell with a fixed dialdehyde content easily tailors the extent of dicarboxylate on PC-CNF surfaces within the first 20 h, offering a simple way to tune the relative proportions of dual dialdehyde and dicarboxylate functionality. Furthermore, this optimized reaction generates much longer PC-CNFs with an impressive (ca. 800) length–thickness aspect ratio, significantly higher than all previously reported values (Table S1†). Among the least chlorite oxidized, PC-CNF-1 h showed a uniform and narrowly distributed thickness of 1.26 ± 0.51 nm (n = 101), width of 3.83 ± 1.12 nm (n = 100), and length of 1.01 ± 0.16 μm (n = 45) (Fig. 2d–f). Close observations by AFM and TEM showed numerous kinks along the nanofibrils (Fig. 2e and S3†), reasonably attributed to C2–C3 glucosidic ring opening along the main chain on fibril surfaces. Also observed were splitting/fraying at ends (Fig. S3†), and seemingly bimodally distributed widths that peaked at 2.89 ± 0.46 nm (n = 47) and 4.65 ± 0.83 nm (n = 53) (Fig. 2f), both of which may be linked to chain scissions.
Furthermore, blending could be shortened to 15 min to produce 88% PC-CNF-18 h with 1.15 mmol g−1 charge. The optimal 1:1 NaClO2/AGU chlorite oxidation was also confirmed as yields were diminished to only a few % at a halved chlorite level of 0.5:1 NaClO2/AGU (Table S3†). Therefore, by further economizing chemical and shear force inputs, this optimized periodate–chlorite oxidation becomes even more environmentally and energetically advantageous with the proven periodate recoverability21–23 and chlorine byproduct treatments.24
While regioselective, i.e., PC oxidation creates C2,C3 dicarboxylates and TEMPO oxidation generates C6 carboxylates, both are capable of producing various CNF attributes of charges and dimensions. Between the lowest 0.64 mmol g−1 charged PC-CNF-0.5 h (0.5 NaIO4/AGU, 94% yield) (Table S2†) and 0.45 mmol g−1 charged TEMPO-CNFs (0.24 mol mol−1 NaClO/AGU, 41% yield) (Table 1), the yield of PC-CNFs is more than double and consistent with the notion that glucosidic ring opening at C2 and C3 confers less rigid and fragmented cellulose segments to facilitate fibrillar disintegration aided by the same 30 min blending shear force. Between the highest 1.35 mmol g−1 charged PC-CNF-72 h (98% yield) and 1.29 mmol g−1 charged TEMPO-CNFs (5 mmol NaClO per g, 0.81 mol mol−1 NaClO/AGU, 97% yield), PC-CNFs (3.83 nm wide, 1.26 nm thick, 1 μm long) were nearly 2× wider than TEMPO-CNFs (2.09 nm wide, 1.52 nm thick, 1 μm long)10 with comparable thickness, length, and yield. As CNFs were deposited on hydrophilic surfaces for AFM and TEM imaging, the CNF surfaces along the width are assumed to be the hydrophilic 110 or 10 lattice planes and those in the height direction to be the hydrophobic 200 lattice planes (Fig. S4a†). Since PC-CNFs have two charges per oxidized AGU compared to one per oxidized AGU of TEMPO-CNFs, the highest charged counterparts have similar respective charges of 1.35 and 1.29 mmol g−1, equivalent to the respective oxidation of 11 and 21% of the 6.17 mmol AGU per gram cellulose. Given the same blending shear force applied for disintegration, the difference between the 3:1 lateral anisotropic cross-sectional PC-CNFs and in essence laterally isotropic TEMPO-CNFs may be associated with the distinct spacings of the adjacent C2–C3 dialdehyde/dicarboxylate vs. C6 carboxylate on the oxidized 110 or 10 lattice planes for nanofibril disintegration by the same shear force. While sequential PC oxidations take longer time and require dialysis in between, the advantages over TEMPO-oxidation are tunability of the surface aldehyde and carboxylate and more versatility in varying charges (Table 1).
| Oxidation (wt%) | Reagent (mol mol−1) | Time (h) | Water use (L) | Blending (min) | Yield (%) | Carboxyl (mmol g−1) | Ref. |
|---|---|---|---|---|---|---|---|
| a Also 0.74 to 0 aldehyde. | |||||||
| Periodate–chlorite (0.5) | 0.5:1 NaIO4/AGU |
4 | 8–10 | 15 | 72.0–88.3 | 0.92–1.16 | This study |
| 1:1 NaClO2/AGU |
0.5–72 | 30 | 93.7–98.6 | 0.64–1.35a | |||
| TEMPO (1.0) | 0.81:1 NaClO/AGU |
1–2 | 6–8 | 0 | 5.3 | 1.29 | 10 |
| 0.16:1 NaBr/AGU |
10 | 90.1 | |||||
| 0.02:1 TEMPO/AGU |
30 | 96.8 | |||||
During surface charge measurement by conductometric titration with NaOH, the aqueous conductivity initially declines due to the consumption of free HCl by the NaOH titrant, and then reaches a plateau corresponding to the removal of protons from mono- and di-dissociated C2,C3 surface carboxylic acids (pKa 3.66 and 4.76, respectively),27 followed by a final increase due to accumulated NaOH (Fig. 3a). The total surface charge was determined from the quantity of the titrate in the plateau region (Δv = v2 − v1). As C2,C3 dicarboxyl groups are weak acids, PC-CNFs at pH 7.5 would show completely dissociated carboxylate sodium salts, also confirmed by titration without HCl charge screening (Fig. S4b†). Conversely, the pKa of C6-carboxyl groups has been reported at 2.8–3.7 (more acidic). In comparison, C6-regioselective TEMPO oxidation showed CNFs with 14% surface carboxylic acid at pH 7.5, or 86% charged carboxylate sodium salts.10
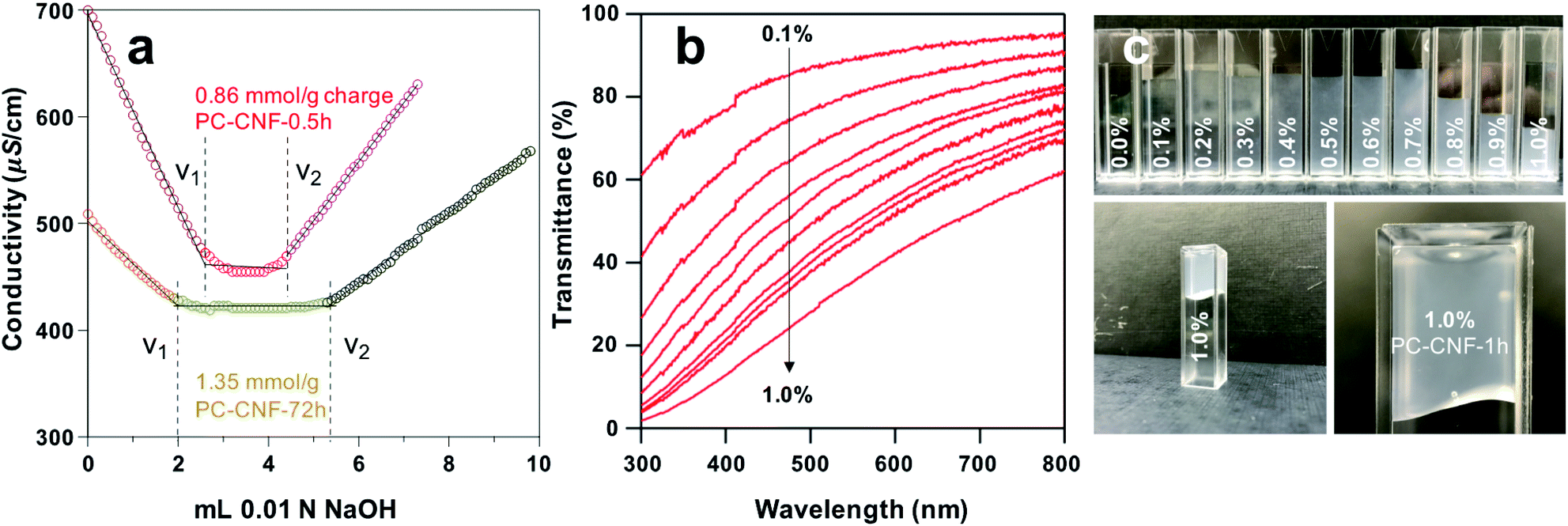 | ||
| Fig. 3 Characteristics of aqueous PC-CNFs: (a) conductometric titration with NaOH (0.01 N); (b) UV/Vis transmittance of PC-CNF-1 h dispersions; (c) presence of aq. PC-CNF-1 h at increasing concentrations. | ||
UV-Vis transmittance of aqueous PC-CNF-1 h reduced with increasing concentrations as expected (Fig. 3b). The more significant reduction in the lower wavelength region may be attributed to reduced polarity from the weaker dipole of surface aldehydes (0.71 mmol g−1) and UV absorption of typical carbonyl π–π* (bonding electrons) and n–π* (lone pair nonbonding electrons) transitions at around 343 nm. Despite their high (ca. 800) aspect ratios, PC-CNFs remained fluidic and showed shear thinning at up to 0.9% and gelled at 1.0% (Fig. 3c), which are significantly higher concentrations in comparison to those of TEMPO-CNFs that gelled at 0.4–0.5%. This is attributed to the greater flexibility of PC-CNF surface chains from glucosidic ring-opening, consistent with previously reported observations.19
Self-assembling
The most highly charged PC-CNF-72 h was self-assembled by freezing at −196 °C and freeze-drying into a white fluffy mass (Fig. 4), as hundreds of nm wide and tens of μm long fibrils from 0.001 to 0.1% (Fig. 4a–c and S5a–c†), tens of μm wide film-like structures at 0.5% (Fig. 4d and S5d†), and a lamellar structure aligned along the freezing direction at 1.0% (Fig. 4e and f). The 307 nm width of 0.1% PC-CNF-72 h is significantly higher than the width of those assembled from other RS CNFs under the same conditions, i.e., coupled TEMPO-blending at 5 mmol g−1 NaClO (137 nm)28 and 1.5 mmol g−1 (125 nm),10 blending (153 nm),29 and aqueous counter-collision (135 nm)30 (Table 2). The more than double ϕ of the assembled PC-CNF-72 h herein is indicative of greater inter-fibril association despite the high charge of 1.35 mmol g−1 carboxylate on wider 110 hydrophilic planes (Fig. S4a†). This may imply dominant self-association of CNFs via 200 hydrophobic planes. Furthermore, the most assembled lamellar structure from 1.0% CNF could be redispersed in water by vortexing to similar UV-vis transmission as the original 0.1% and even the CNFs in the less translucent 0.13% redispersion had a similar 1.25 ± 0.33 nm (n = 23) width as the original (Fig. 1b and S6†). The reduced UV-vis transmission of redispersions at >0.1% concentrations indicated higher non-dispersible fractions. This suggests dominant hydrophobic inter-nanofibril associations along the 200 hydrophobic planes at higher concentrations to be further elucidated later.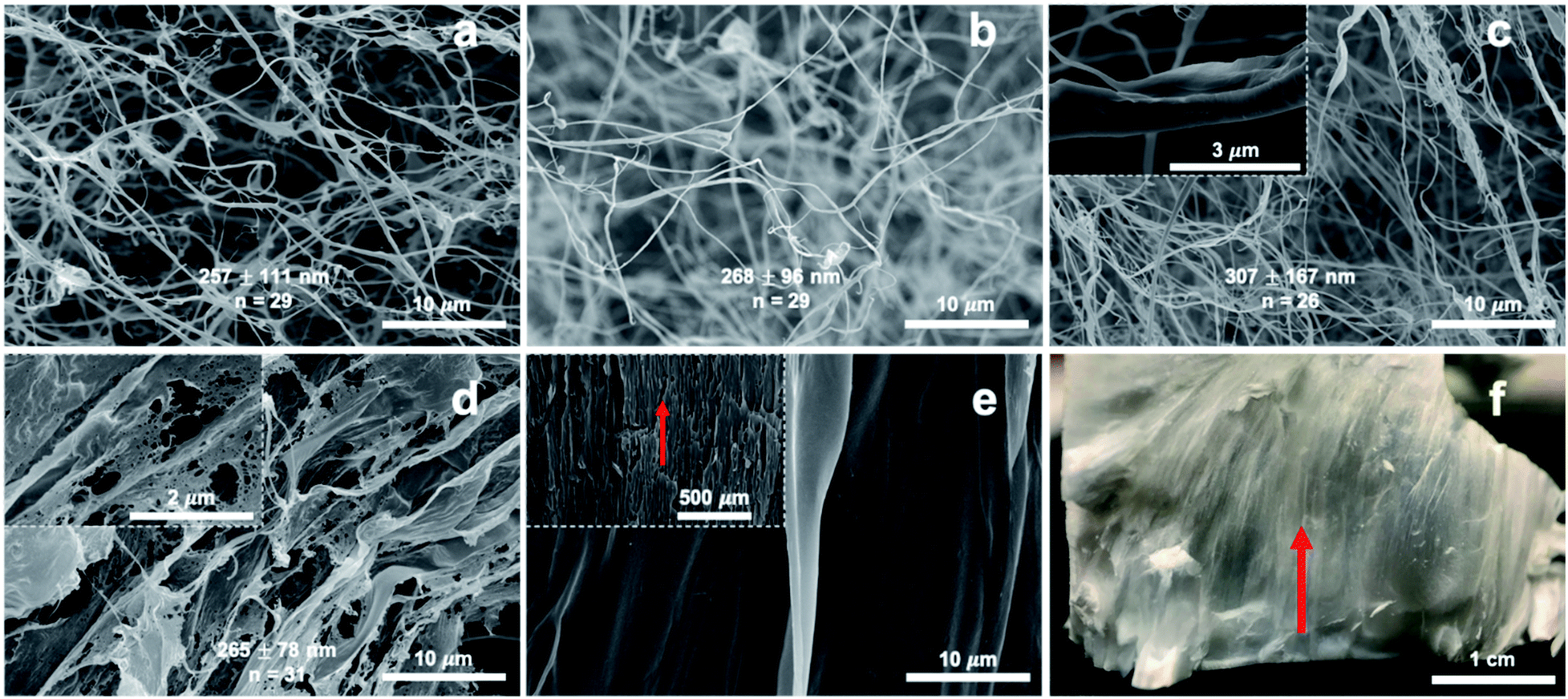 | ||
| Fig. 4 SEM images of the assembled PC-CNF-72 h: (a) 0.001%; (b) 0.01%; (c) 0.1%; (d) 0.5%; (e) 1.0%; (f) photographic image of the lamellar structure with the red arrow indicating the freezing direction. Fiber widths are included in (a–d). | ||
| Method | Yielda (%) | CNFs | Fibrilsb | Ref. | ||
|---|---|---|---|---|---|---|
| Charge (mmol g−1) | Thickness by AFM (nm) | Width by TEM (nm) | Width by SEM (nm) | |||
| a In the supernatant from centrifugation (5k rpm, 15 min). b From freezing (−196 °C, 15 min) and freeze-drying (−50 °C, 2 days) of 0.1% aqueous dispersions. | ||||||
| TEMPO (0.81:1 mol mol−1 NaClO/AGU), 30 min blending |
96.8 | 1.04 | 1.5 | 2.1 | 137 | 28 |
| TEMPO (0.24:1 mol mol−1 NaClO/AGU), 60 min blending |
41.0 | 0.45 | 2.8 | 3.4 | 125 | 10 |
| Blending, 120 min | 12.0 | 0.0 | 2.7 & 8.5 (bimodal) | NA | 153 | 29 |
| Aqueous counter-collision (180 MPa, 30 passes) | 78.7 | 0.0 | 3.7 | 5.5 | 135 | 30 |
| Periodate–chlorite, 30 min blending | 98.6 | 1.35 | 1.3 | 3.8 | 307 | This study |
FTIR spectra illustrated the chemical changes to the cellulose structure following primary periodate and secondary chlorite oxidations (Fig. 5a). The 897 cm−1 absorption band characteristic of the hemiacetal C1–O–C4 beta-glycosidic bond of native cellulose was retained throughout. The slight aldehydic shoulder at 1740 cm−1 of P-cell along with the more prominent 1640 cm−1 and 875 cm−1 absorption bands of hemiacetal C–O–C bridges suggests alternative hydrated and cross-linked aldehyde structures (Fig. S7†). The distinct carboxylic carbonyl band at 1740 cm−1 is indicative of chlorite oxidation to PC-cell. More intense bands at ca. 1620 cm−1 of assembled PC-CNFs are clearly evident for sodium carboxylates and overlapped O–H deformation absorption.
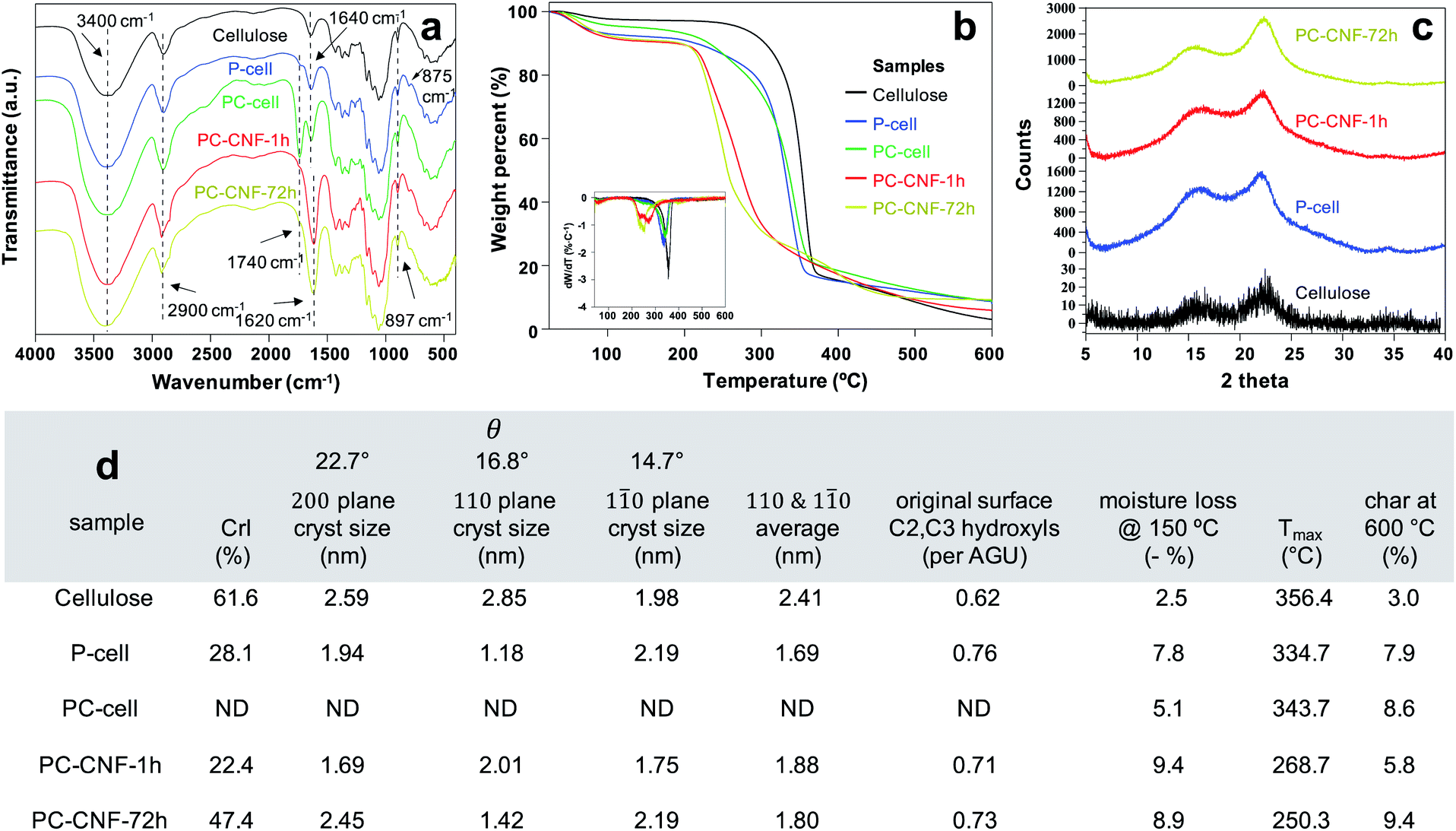 | ||
| Fig. 5 Characterization of freeze-dried (−196 °C, 0.1%) cellulose, P-cell, PC-cell, and PC-CNFs: (a) FTIR spectra, (b) TGA and dTGA curves, (c) XRD patterns, and (d) crystalline and thermal properties. | ||
While both oxidations lowered the thermal stability of P-cell and PC-cell, mechanical defibrillation reduced that of PC-CNFs further (Fig. 5b). The significant (ca. 9%) moisture losses of PC-CNFs reflect their carboxylated and higher specific surfaces. Intriguingly, P-cell with weaker dipoles of surface aldehydes absorbed more moisture but was consistent with the presence of aldehydic hydrates (Fig. S7†). Both assembled PC-CNFs and P-cell showed the characteristic cellulose Iβ XRD patterns with peaks at 2θ = 14.7°, 16.8° and 22.7° assigned to the respective 10, 110, and 200 crystallographic planes of the monoclinic lattice (Fig. 5c). Periodate oxidation alone significantly reduced cellulose crystallinity and crystal sizes (Fig. 5d), supporting glycol cleavage of crystal surfaces. The assembled PC-CNF-1 h was only slightly less crystalline than P-cell, indicative of the dominant effect of periodate oxidation on crystallinity but less from chlorite oxidation and blending. The assembled PC-CNF-72 h with the highest charge and no aldehyde showed increased CrI to 47.4% and a 45% increase in 200 crystal size relative to PC-CNF-1 h, supporting CNF association or interfacial recrystallization along the 200 crystallographic planes, i.e., the most homogeneous PC-CNF surfaces. This increased crystallization supports the dominant hydrophobic associations discussed earlier.
Aerogels
Aerogels were produced by freezing at −20 °C and freeze-drying of aq. PC-CNF-1 h at 0.1–1.0% concentrations (Fig. 6a) and variously charged (0.86–1.29 mmol g−1) PC-CNF-1 h, -4 h, -8 h, -12 h, and -18 h (Fig. 6b–d and Table S4†), all showing no appreciable lateral dimension change. Aerogels from PC-CNF-1 h had ultra-low densities (ρ = 1.7–10.4 mg cm−3) and high porosities (ϕ = 99.9–99.4%), showing strong respective positive and negative dependence of increasing concentrations (Fig. 6a), but a similar cellular structure with 100–300 μm diameter cellular pores (Fig. 6d) and super thin sub-micrometer cellular walls (Fig. S8†). At a fixed 1.0% concentration, aerogel densities slightly lowered from 10.9 to 9.1 mg cm−3 and the pore volume increased from 91 to 109 cm3 g−1 with increasing charges from 0.86 to 1.29 mmol g−1 (Fig. 6b and Table S4†), consistent with decreasing association among the more highly charged PC-CNFs. All aerogels were amphiphilic, absorbing water (up to 87 mL g−1) as well as hydrophobic chloroform (up to 72 mL g−1) and decane (up to 90 mL g−1) to fill 80, 79, and 90% of pore capacity, respectively. Similar absorption of water and chloroform (PC-CNF-1 h, -4 h, -8 h) as well as that of water and decane (CNF-12 h, -18 h) (Table S4†) confirm the dual polar and nonpolar nature of aerogel cellular wall surfaces, indicative of inter-CNF assembling via both polar–polar and nonpolar–nonpolar PC-CNF interfaces. As expected from a lower concentration, the less dense (6.2 mg cm−3) and more porous (99.6%) aerogel from 0.6% PC-CNF-12 h had a significantly higher pore volume of 160.3 cm3 g−1 to absorb more water (103 mg mL−1) and decane (129 mg mL−1), filling 64% and 80% of the pores, respectively (Table S4†). This higher decane absorption over water is indicative of more hydrophobic cellular wall surfaces, suggesting higher hydrophilic associations. Lastly, the less dimensional reduction of this PC-CNF-12 h aerogel led to a less dense, more porous structure to absorb 40% more decane but a similar amount of water to the denser (8.0 mg cm−3) but lower pore volume (125.1 cm3 g−1) aerogel from TEMPO-CNFs.31,32 Note that the absorption of the two comparable amounts of water is linked to their similar surface carboxylate contents (ca. 1.2 mmol g−1) (Table S4†). The ability of PC-CNF aerogels to absorb higher and varied quantities of non-polar liquids clearly reflects the exposure of hydrophobic CNF surfaces on cellular wall surfaces, or in other words, the inter-fibril association is influenced by the more heterogeneous oxidized nanofibril surfaces.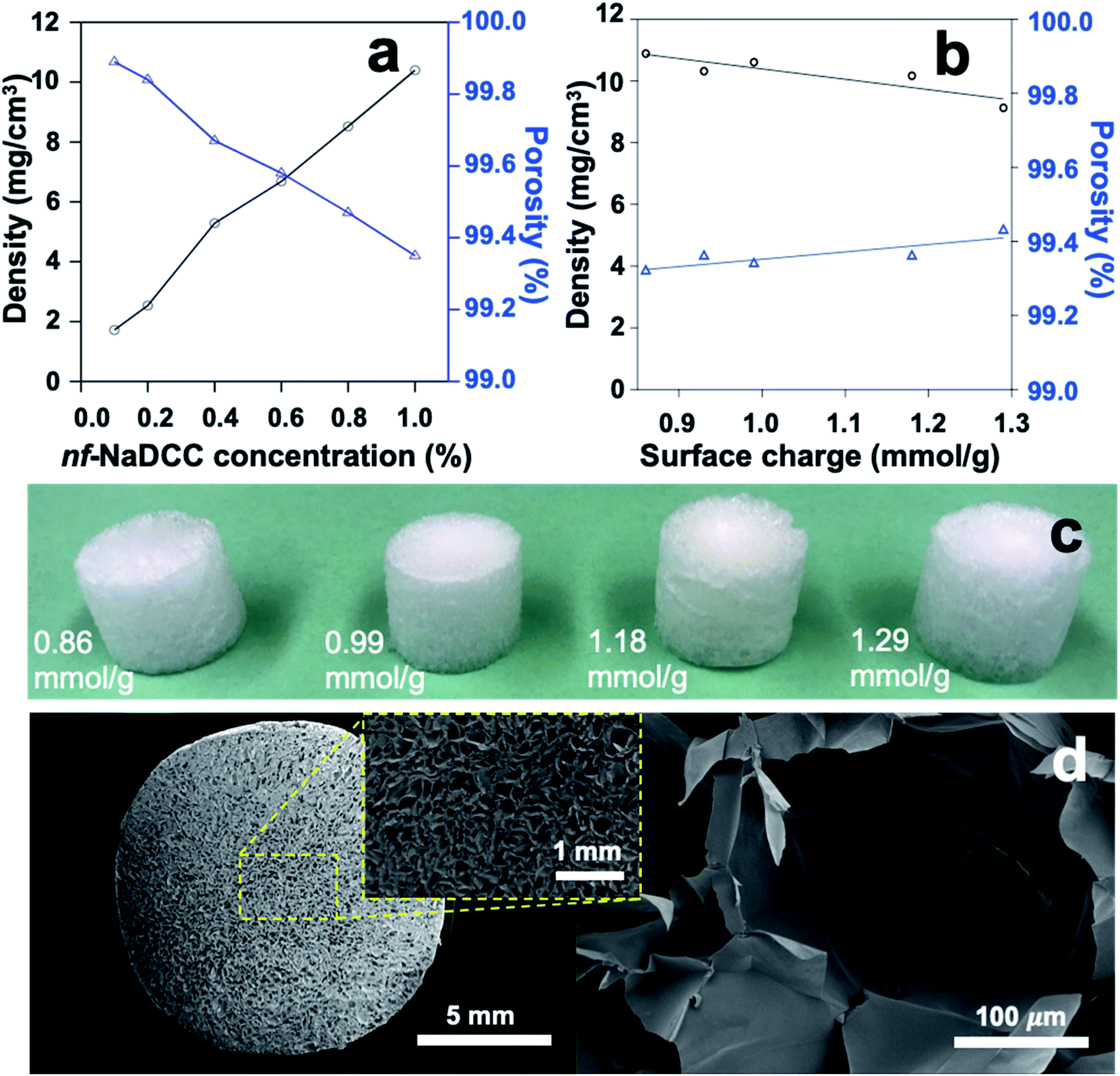 | ||
| Fig. 6 PC-CNF aerogel characteristics: (a) PC-CNF-1 h concentration effect; (b) PC-CNF charge effect; (c) photographs from variously charged PC-CNFs; (d) SEM images of PC-CNF-1 h (1.0%) radial cross-sections. | ||
Aerogels showed remarkable toughness and high dry compressive strength up to 0.8 strain, irrespective of charges (Fig. 7a). The dry compression stress–strain curve has three distinct regions, i.e., initial linear elastic region at low strain (<0.05), plastic deformation plateau region at up to ca. 0.4 strain, and final densification stage up to 0.8 strain. Impressively, the Young's modulus reached 518 kPa and ultimate stress 84.7 kPa for the PC-CNF-8 h aerogel (Fig. 7a). The fully hydrated aerogel could withstand increasing cyclic compression from 0.4 to 0.8 strain, showing no yield behavior and one order of magnitude lower ultimate wet compression stress than in the dry state (Fig. 7b). In fact, the ultimate wet stress at 0.4 strain decreased significantly with increasing charges (Fig. 7c), in contrast to dry compression stress. Clearly, the lowered wet compressive behavior is associated with increasing aldehyde–carboxylate conversion, indicative of increasing water accessibility to PC-CNF interfaces with increasing surface charges. Impressively, the dry PC-CNF-8 h aerogel (Fig. S9a†) did not buckle under 3000× its weight (Fig. S9b†), but wet gel saturated for 15 min would fracture upon pressing with a spatula (Fig. S9c†). Most intriguingly, PC-CNF-8 h aerogels remained intact and stable in chloroform and decane for at least 60 days (Fig. S9d and e†). The excellent resiliency of the PC-CNF aerogel in nonpolar, low dielectric constant organic liquids further supports the idea that interfacial recrystallization along the most homogeneous 200 crystallographic planes is impenetrable to non-polar liquids.
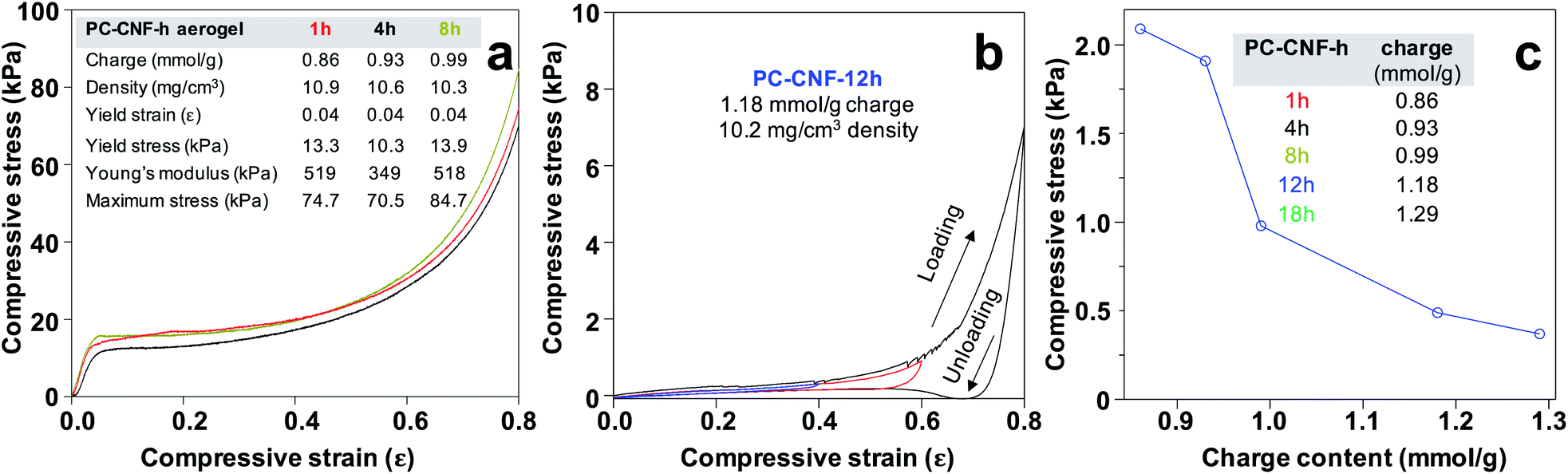 | ||
| Fig. 7 Uniaxial compression of PC-CNF aerogels (1.0%): (a) dry stress–strain curves up to 0.8 strain; (b) consecutive wet compression of PC-CNF-12 h (1.18 mmol g−1) at increasing strains of 0.4, 0.6, and 0.8; (c) ultimate wet stress at 0.4 strain of aerogels from PC-CNFs of increasing charge. | ||
Normalized by density (Fig. 7a), the PC-CNF-8 h aerogel's dry compressive 50.2 kPa mg−1 cm−3 specific modulus and 8.2 kPa mg−1 cm−3 ultimate stress were 7.5 and 2.5 times those of the TEMPO-CNF aerogels (3.1 and 6.7 kPa mg−1 cm−3), respectively,32 and even 2.8 and 1.4 times those of TEMPO-CNF aerogels cross-linked with diisocyanate (18.2 and 5.7 kPa mg−1 cm−3),33 all from rice straw cellulose. The most impressive higher dry strength of the PC-CNF aerogel than TEMPO-CNF aerogels is therefore attributed to the larger lateral CNF dimensions and their ability to associate more extensively through non-polar interactions and, in particular, interfacial recrystallization along the 200 planes. Compared to the aerogel reported from periodate–chlorite oxidation of wood pulp (ρ = 18 mg cm−3, ϕ = 98.8%, 4.5 kPa mg−1 cm−3 specific modulus, 5.6 kPa mg−1 cm−3 ultimate stress),20 the 75% lighter and more porous PC-CNF-8 h aerogel is 11 times stronger, where wet compressive strength was not reported. Furthermore, the PC-CNF-8 h aerogel is tougher and 5 times more water absorbing than the 8 times denser (ρ = 82 mg cm−3, 1.5 kPa mg−1 cm−3 yield strength, 13 kPa mg−1 cm−3 specific modulus, 13 mL water absorption) aerogel from periodate–hemiacetal cross-linked and ambient dried acetone gel.34 Both were fabricated from organogels,20,34 and neither reported amphiphilicity.
Intriguingly, dry PC-CNF-8 h aerogel could fully recover from 0.2 strain even with very low elastic strain (<0.05). When compressed by an extreme 14 kg or 800000 times the aerogel mass to 0.8 strain, dry axial recovery was ca. 40% of its original 1 cm height (Video S1†). Most impressively, a PC-CNF-8 h aerogel dry compressed to 0.8 strain could fully expand and recover to its original dimensions in water (Video S2†), exhibiting excellent wet recovery. In the context of C2,C3 carboxylated CNFs, these sequential periodate–chlorite oxidations represent the most systematically and significantly optimized approach to tunable surface aldehyde and carboxyl chemistries. Aerogels from these diverse PC-CNFs not only offer similar controlled densities, porosities, and amphiphilicities, but also unmatched dry compressive toughness and unique wet recovery behavior, reported for the first time to our knowledge.
Conclusions
New insight into sequential periodate–chlorite oxidation of cellulose has been ilucidated by delineating the significance of stoichiometrically optimized primary oxidation, the charge effect from the secondary oxidation, and deprotonation in the disintegration into regioselective 2,3-dialdehyde/dicarboxylate-nanocelluloses with tunable geometries and surface chemistries. While the primary periodate oxidant quantity dictated the nanocellulose length, i.e., CNFs at slightly higher NaIO4 to non-crystalline AGU stoichiometry and CNCs with two times excess oxidant, the secondary chlorite oxidation time controlled the aldehyde to carboxyl conversion, and thus surface charges. PC-CNFs from optimal sequential 0.5:1 NaIO4/AGU and 1:1 NaClO2/AGU oxidation coupled with minimal blending were on average 3.83 nm wide, 1.26 nm thick, and ca. 1 μm long, with tunable surface aldehyde (0.71 to 0.0 mmol g−1) and carboxylate (0.64–1.35 mmol g−1) contents and excellent yields (94–98%). The distinctive C2–C3 glucosidic ring opening and oxidation along the 110 or 10 lattice planes led to more heterogeneous surface chemistries and flexible nanofibrils, so the aqueous PC-CNFs remained fluidic up to 0.9% and gelled at 1.0%, far less viscous than TEMPO-CNFs, also from rice straw. Freezing induced self-assembling of PC-CNFs was clearly evident via both polar and nonpolar interfaces, specifically interfacial recrystallization along the 200 crystallographic planes of the highly charged PC-CNF-72 h (1.35 mmol g−1) at −196 °C and varying charges and concentrations of PC-CNFs at −20 °C into fibrous and amphiphilic aerogel structures, respectively. In particular, the ultra-low density (1.7–10.9 mg cm−3) and ultra-high porosity (99.9–99.3%) aerogels are amphiphilic super-absorbents, absorbing water (up to 103 mL g−1) as well as chloroform (up to 72 mL g−1) and decane (up to 129 mL g−1). The excellent dry compressive specific modulus (50.2 kPa mg−1 cm−3) and ultimate stress at 0.8 strain (8.2 kPa mg−1 cm−3) of these PC-CNF aerogels were significantly higher than those of TEMPO-CNF aerogels, i.e., 7.5 and 2.8 times, respectively. Most impressively, the dry PC-CNF-8 h aerogel exhibited full recovery from 0.2 strain, 40% recovery from under 800000 times mass load or 0.8 strain, and uniquely, 100% wet recovery from 0.8 strain dry compression. Not only is this sequential periodate–chlorite oxidation robust and environmentally friendly, its versatility to synthesize a variety of nanocelluloses is easily tuned for greater potential applications, and also leads to far stronger aerogels without further reactions or use of additional organic solvent or chemicals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this research from the California Rice Research Board (Project RU-9) and the Jastro Shields Research Award, University of California, Davis, and exploratory work conducted by Dr F. Jiang are greatly appreciated.References
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- S. Iwamoto, W. Kai, A. Isogai and T. Iwata, Biomacromolecules, 2009, 10, 2571–2576 CrossRef CAS.
- T. Nishino, I. Matsuda and K. Hirao, Macromolecules, 2004, 37, 7683–7687 CrossRef CAS.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS.
- T. Saito, Y. Nishiyama, J.-L. Putaux, M. Vignon and A. Isogai, Biomacromolecules, 2006, 7, 1687–1691 CrossRef CAS.
- R. K. Johnson, A. Zink-Sharp, S. H. Renneckar and W. G. Glasser, Cellulose, 2009, 16, 227–238 CrossRef CAS.
- R. Baati, A. Magnin and S. Boufi, ACS Sustainable Chem. Eng., 2017, 5, 2350–2359 CrossRef CAS.
- Food and Agriculture Organization of the United Nations, http://www.fao.org/, accessed May 15, 2020.
- P. Lu and Y.-L. Hsieh, Carbohydr. Polym., 2012, 87, 564–573 CrossRef CAS.
- F. Jiang, S. Han and Y.-L. Hsieh, RSC Adv., 2013, 3, 12366–12375 RSC.
- P. Lu and Y.-L. Hsieh, Powder Technol., 2012, 225, 149–155 CrossRef CAS.
- S. Hu and Y.-L. Hsieh, ACS Sustainable Chem. Eng., 2014, 2, 726–734 CrossRef CAS.
- B. T. Hofreiter, I. A. Wolff and C. L. Mehltretter, J. Am. Chem. Soc., 1957, 79, 6457–6460 CrossRef CAS.
- E. Maekawa and T. Koshijima, J. Appl. Polym. Sci., 1984, 29, 2289–2297 CrossRef CAS.
- L. Münster, M. Fojtů, Z. Capáková, T. Vaculovič, M. Tvrdoňová, I. Kuřitka, M. Masařík and J. Vícha, Biomacromolecules, 2019, 20, 1623–1634 CrossRef.
- A. Tejado, M. N. Alam, M. Antal, H. Yang and T. G. M. van de Ven, Cellulose, 2012, 19, 831–842 CrossRef CAS.
- H. Liimatainen, M. Visanko, J. A. Sirviö, O. E. O. Hormi and J. Niinimaki, Biomacromolecules, 2012, 13, 1592–1597 CrossRef CAS.
- S. D. Hujaya, G. S. Lorite, S. J. Vainio and H. Liimatainen, Acta Biomater., 2018, 75, 346–357 CrossRef CAS.
- S. F. Plappert, F. W. Liebner, J. Konnerth and J.-M. Nedelec, Carbohydr. Polym., 2019, 226, 115306 CrossRef CAS.
- S. F. Plappert, J.-M. Nedelec, H. Rennhofer, H. C. Lichtenegger and F. W. Liebner, Chem. Mater., 2017, 29, 6630–6641 CrossRef CAS.
- M. W. Lister and P. Rosenblum, Can. J. Chem., 1961, 39, 1645–1651 CrossRef CAS.
- H. Liimatainen, J. Sirviö, H. Pajari, O. Hormi and J. Niinimäki, J. Wood Chem. Technol., 2013, 33, 258–266 CrossRef CAS.
- S. Koprivica, M. Siller, T. Hosoya, W. Roggenstein, T. Rosenau and A. Potthast, ChemSusChem, 2016, 9, 825–833 CrossRef CAS.
- W. A. MacCrehan, M. Bedner and G. R. Helz, Chemosphere, 2005, 60, 381–388 CrossRef CAS.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS.
- J. Ganster and H.-P. Fink, in Polymer Handbook, ed. E. H. Immergut and E. A. Grulke, Wiley & Sons, New York, 4th edn., 1999, Sect. V, pp. 135–157 Search PubMed.
- U. J. Kim and S. Kuga, J. Chromatogr. A, 2001, 919, 29–37 CrossRef CAS.
- F. Jiang and Y.-L. Hsieh, ACS Sustainable Chem. Eng., 2016, 4, 1041–1049 CrossRef CAS.
- F. Jiang and Y.-L. Hsieh, Carbohydr. Polym., 2013, 95, 32–40 CrossRef CAS.
- F. Jiang, T. Kondo and Y.-L. Hsieh, ACS Sustainable Chem. Eng., 2016, 4, 1697–1706 CrossRef CAS.
- F. Jiang and Y.-L. Hsieh, J. Mater. Chem. A, 2014, 2, 350–359 RSC.
- F. Jiang and Y.-L. Hsieh, J. Mater. Chem. A, 2014, 2, 6337–6342 RSC.
- F. Jiang and Y.-L. Hsieh, ACS Appl. Mater. Interfaces, 2017, 9, 2825–2834 CrossRef CAS.
- V. López Durán, J. Erlandsson, L. Wågberg and P. A. Larsson, ACS Sustainable Chem. Eng., 2018, 6, 9951–9957 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Reaction comparisons from cited work, aqueous dispersible PC-cell and aqueous non-dispersible P-cell, kinks and splitting of PC-CNFs, assembled PC-CNF fibril dimensions and aqueous re-dispersibility, SEM of the aerogel cellular wall, aerogel interaction in liquids, and dry and water-activated shape recovery. See DOI: 10.1039/d0na00771d |
| This journal is © The Royal Society of Chemistry 2020 |