
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMass spectrometry imaging and monitoring of in vivo glutathione-triggered cisplatin release from nanoparticles in the kidneys†
Arthur C. K.
Chung
a,
Xuan
Li
b,
Wai-Chung
Li
a,
Tao
Wang
a,
Hin-Kiu
Lee
a,
Lijian
Jin
 b,
Zongwei
Cai
*a and
Ken Cham-Fai
Leung
*ab
b,
Zongwei
Cai
*a and
Ken Cham-Fai
Leung
*ab
aDepartment of Chemistry, State Key Laboratory of Environmental and Biological Analysis, The Hong Kong Baptist University, Kowloon Tong, Kowloon, Hong Kong SAR, P. R. China. E-mail: cfleung@hkbu.edu.hk; zwcai@hkbu.edu.hk
bFaculty of Dentistry, The University of Hong Kong, Sai Ying Pun, Hong Kong SAR, P. R. China
First published on 27th October 2020
Abstract
An increasing number of studies have reported the use of various nanoparticles to encapsulate cisplatin, a frontline chemotherapeutic drug against a broad-spectrum of cancers, for overcoming its inherent drawbacks in clinical applications. Nevertheless, few analytical methods or instruments could provide the precise distribution information on this platinum drug in biological tissues. Herein, we provide the first evidence of applying matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) to assess the spatial distribution of cisplatin released from the cell-penetrating poly(disulfide) (CPD)-modified hollow iron oxide nanoparticles (hFe3O4-MPS-CPD) at the kidneys via an in situ glutathione (GSH) responsive mode. The cisplatin released from the nanoparticles triggered by GSH was successfully examined as [Pt(DDTC)2]+ (m/z 491.01) and [Pt(DDTC)3]+ (m/z 639.04) by MALDI-MS after derivatization using diethyldithiocarbamate. The in situ spatial distribution of [Pt(DDTC)2]+ and [Pt(DDTC)3]+ in the kidneys was then mapped using MALDI-MSI. This study presents an optimized analytical approach to evaluate and map the metallodrug in biological tissue samples in an efficient and convenient manner, offering great assistance in investigating the biodistribution of cisplatin delivered by nanoparticles, and sheds light on facilitating the studies of the pharmacokinetics of cisplatin in biomedical research.
Introduction
Cisplatin (cis-diamminedichloroplatinum(II), CDDP), a frontline anti-neoplastic platinum drug, has served as a panacea against a broad-spectrum of cancers including head, neck, bladder, ovarian and melanoma over decades.1–3 However, its efficacy is strongly limited due to its poor water solubility, nonselective tissue distribution and the glutathione (GSH)-induced drug resistance.1–3 One of the approaches applied to tackle these limitations is to use various nanomaterials with capping systems for enhancing the delivery efficiency and accumulation at tumor sites.4–10 Among the numerous nanomaterials, magnetite nanoparticles (Fe3O4 NPs), a type of superparamagnetic iron oxide nanoparticle (SPION), have received tremendous attention because of their drug delivery and magnetic resonance imaging abilities.11–14 In particular, hollow Fe3O4 nanoparticles (hFe3O4) have emerged as the most promising candidate for theranostic purposes owing to their facile preparation, cargo protection and convertible surface,15,16 and thereby can be developed into a controlled cisplatin delivery system.10Currently, most of the analytical methods used in cisplatin studies are X-ray based techniques,17,18 inductively coupled plasma mass spectrometry (ICP-MS),19 atomic absorption spectroscopy (AAS)20 and high-performance liquid chromatography–mass spectrometry (HPLC-MS);21 however these techniques fail to provide detailed information on the precise localization and distribution of the target drug in biological tissues. Therefore, MSI has come to light and been used as an alternative and innovative technique for spatial mapping of intact drug compounds.
Matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) is one of the powerful label-free imaging tools which are capable of visualizing the distribution of small molecule drugs directly on the target organ surface.22,23 Targeted MALDI-MSI for assessment of pharmaceutical compounds is a useful approach supporting preclinical efficacy, toxicity testing and cancer therapy, as it allows the simultaneous visualization of different metabolites of drugs.24,25 In addition, authentic proof of drug distribution for small molecule drugs and metabolites in target tissue can be convincingly demonstrated with high speed and label-free using MALDI-MSI.26,27 Therefore, the application of MALDI-MSI for different drug substances has gained gradual attraction from the pharmaceutical industry. However, using MALDI-MSI in metallodrug analyses is still limited to animal studies, especially for Pt-based drugs. A few research studies conducted previously using MALDI-MSI were mainly targeted for oxaliplatin,28,29 while imaging studies for cisplatin are still underexplored.
Herein, we constructed an intracellular GSH-responsive controlled release system using cell-penetrating poly(disulfide) (CPD) coated hollow magnetite iron oxide nanoparticles (hFe3O4-MPS-CPD), and then applied it for cisplatin loading. The obtained cisplatin loaded nanoparticles (cisplatin-hFe3O4-MPS-CPD), as a controlled release nanoplatform of the Pt drug, were further tested and their GSH-triggered in situ release of cisplatin on the kidney tissue sections was mapped using MALDI-MSI. This optimized analytical approach could provide great assistance in the identification of cisplatin in the biomedical field, and shed light on investigating nanoparticle-facilitated Pt drug release and distribution in biological samples.
Results and discussion
Characterization of the as-synthesized nanoparticles
As shown in Fig. 1A, the synthesized hFe3O4 with abundant hydroxy groups on the particle surface underwent a two-step modification before cisplatin loading. The as-synthesized particles presented a uniform size distribution and spherical morphology in the TEM image (Fig. 1B–D(i)), and the average diameter of hFe3O4 was 281.1 ± 21.3 nm (Fig. 1F). After the MPS modification, the sulfur signal appeared in the EDX spectrum of hFe3O4-MPS, indicating the successful thiol modification (Fig. 1C(ii)). Then, subsequent in situ polymerization was performed for 30 min on the surface of the nanoparticles. It results in a polymer layer with a thickness around 2–3 nm (Fig. S4†), which may cause insignificant changes to the nanoparticle size as shown by TEM. Moreover, the modified CPD is an organic layer, and has low contrast in TEM compared with the Fe3O4 nanocore. Thus, it may not be easily observed in TEM with the magnification in Fig. 1B–D(i), while the CPD layer can be detected at a higher magnification as shown in Fig. S4.† Meanwhile, the EDX spectrum of hFe3O4-MPS-CPD showed an increased sulfur intensity compared with that of hFe3O4-MPS (Fig. 1D(ii)).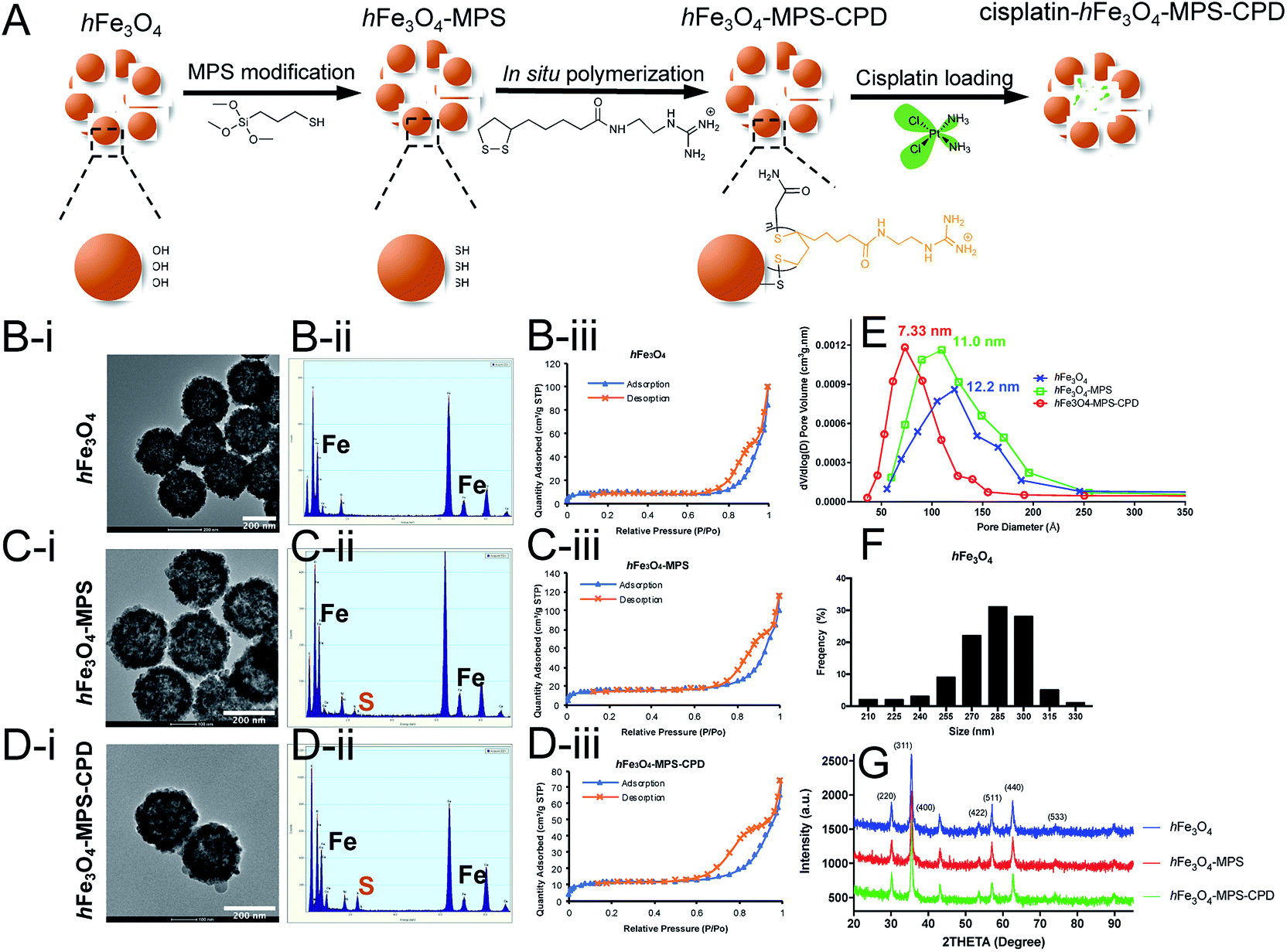 | ||
| Fig. 1 (A) Schematic illustration of synthesis, modification, in situ polymerization and cisplatin loading in hFe3O4 nanoparticles. (i) TEM images, (ii) EDX spectra and (iii) Brunauer–Emmett–Teller (BET) nitrogen adsorption–desorption isotherms of (B) hFe3O4, (C) hFe3O4-MPS and (D) hFe3O4-MPS-CPD. (E) Barrett, Joyner and Halenda (BJH) pore size distribution plots of the as-synthesized nanoparticles. (F) Size distribution of hFe3O4 analyzed. (G) XRD spectra of hFe3O4, hFe3O4-MPS and hFe3O4-MPS-CPD. | ||
Moreover, the porosity of the nanoparticles was assessed using the nitrogen adsorption–desorption isotherms and is presented in Fig. 1B–D(iii). The pore diameter distributions were calculated from the Barrett–Joyner–Halenda (BJH) model shown in Fig. 1E. The narrowing trend of the pore diameters of the iron oxide particles gives evidence for the successful coating of CPDs. The hysteresis loop ranged at 0.8 P/Po, indicating that the dominant pore size is reduced to 5–11 nm and representing the void space between adjacent primary grains of the hollow iron oxide.
The FT-IR spectra in Fig. 2A gave more details of the surface functional groups of the as-prepared nanoparticles with/without CPD polymerization. The obvious peak at around 2900 cm−1 representing the C–H stretching vibration implies the successful in situ polymerization. After treatment with GSH (10 mM) for 30 min, this peak was smaller and shorter compared with that of hFe3O4-MPS-CPD, indicating a GSH-triggered CPD degradation. The organic compositions of the nanoparticles after polymerization and its subsequnt degradation by GSH were verified and analyzed using TGA. The decomposition curves clearly elucidate the increased weight loss (32.0% vs. 10.8%) after polymerization, whereas the weight loss decreased to 7.9% after the GSH treatment (Fig. 2B). This also indicated the GSH-responsive degradation of the CPD layer. The XPS spectra (O1s, N1s and S2p) of hFe3O4, hFe3O4-MPS and hFe3O4-MPS-CPD in Fig. 2C showed the elemental changes after modification and polymerization. For O 1s, the intensified peak at 532.6 eV (pink) confirms the presence of incorporated MPS, while the intensified peak at 531.9 eV (green) representing the enrichment of O–C and N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O contents stemmed from the thioctic component during polymerization. Notably, there is no observable peak for hFe3O4 and hFe3O4-MPS in the N 1s region. After the polymerization, there are a significant peak at 400 eV, corresponding to the presence of an amidine, and an amide peak due to the presence of the CPD layer. A similar result was obtained in the S 2p region. Compared with that of hFe3O4, a new peak appeared in the S 2p of hFe3O4-MPS, suggesting the successful modification of MPS. After the polymerization, the enhanced region at 164.4 eV represented the disulfide bond in the CPD polymer, which supported the in situ polymerization on the nanoparticle surface.
O contents stemmed from the thioctic component during polymerization. Notably, there is no observable peak for hFe3O4 and hFe3O4-MPS in the N 1s region. After the polymerization, there are a significant peak at 400 eV, corresponding to the presence of an amidine, and an amide peak due to the presence of the CPD layer. A similar result was obtained in the S 2p region. Compared with that of hFe3O4, a new peak appeared in the S 2p of hFe3O4-MPS, suggesting the successful modification of MPS. After the polymerization, the enhanced region at 164.4 eV represented the disulfide bond in the CPD polymer, which supported the in situ polymerization on the nanoparticle surface.
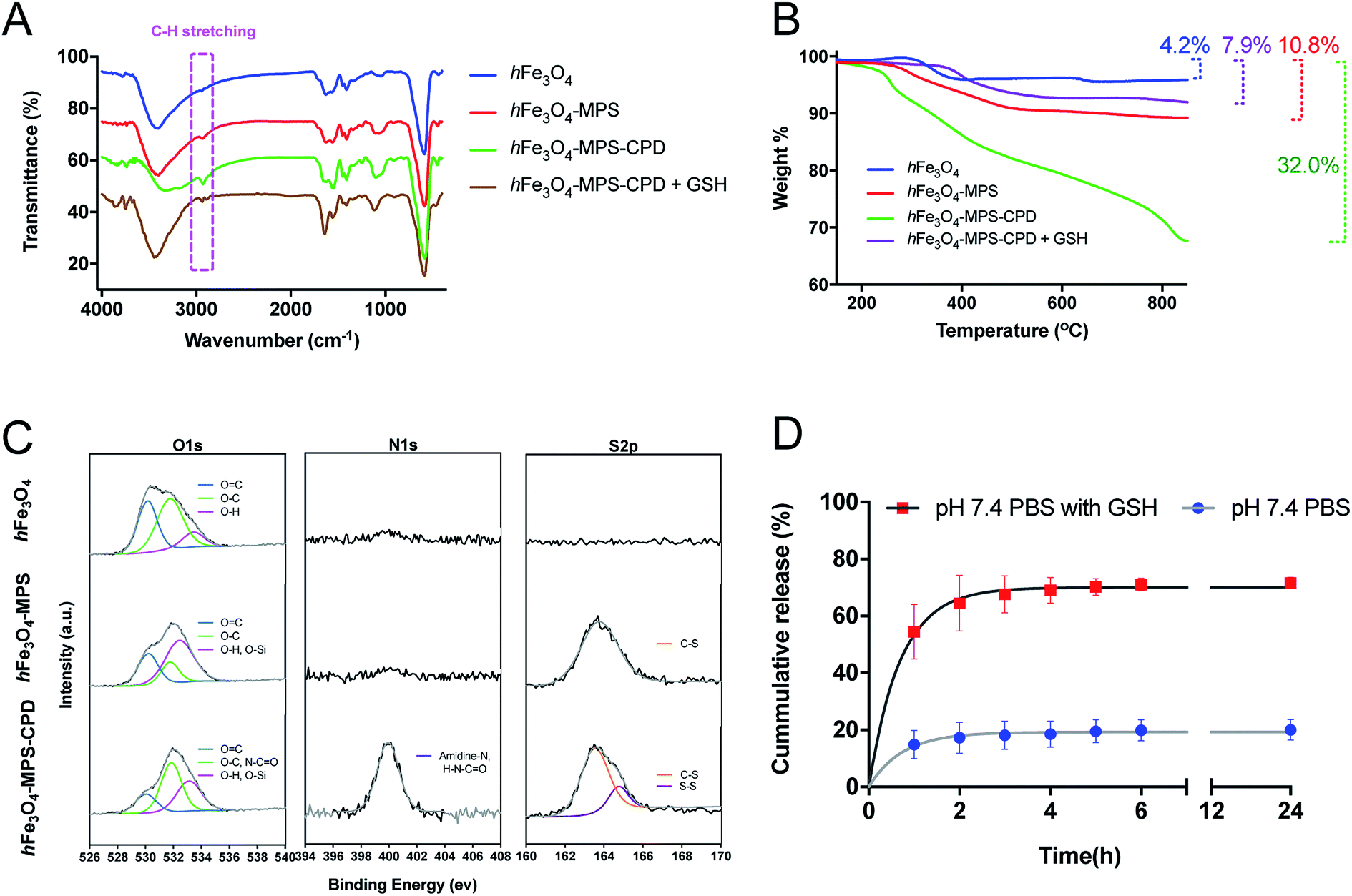 | ||
| Fig. 2 (A) FT-IR spectra and (B) TGA decomposition curves for hFe3O4, hFe3O4-MPS and hFe3O4-MPS-CPD before/after GSH treatment. (C) XPS spectra for O 1s, N 1s and S 2p of hFe3O4, hFe3O4-MPS and hFe3O4-MPS-CPD. (D) Release profiles of cisplatin-hFe3O4-MPS-CPD in pH 7.4 PBS buffer with and without 10 mM GSH at predetermined time intervals over 24 h. | ||
In this study, the influence of different solvents on the cisplatin loading in the as-synthesized hFe3O4-MPS-CPD was also investigated. The resultant loading efficiency was calculated from the Pt/Fe determined using ICP-MS and is summarized in Table S1.† It was found that the cisplatin loading performed in acetone presented the highest Pt/Fe ratio (23.4%), indicating the most optimal loading efficiency. The obtained cisplatin-encapsulated hFe3O4-MPS-CPD (cisplatin-hFe3O4-MPS-CPD) was then dispersed in PBS (pH 7.4) with or without GSH to assess the release profile of cisplatin. The GSH-triggered released Pt drug was determined using ICP-MS and the profiles are presented in Fig. 2D. It was found that only around 20% of cisplatin released from cisplatin-hFe3O4-MPS-CPD in 24 h, while this release was greatly increased to around 70% at 24 h in PBS with 10 mM GSH. The robust release was observed in the first two hours as a consequence of CPD degradation, indicating the successful construction of the GSH-responsive cisplatin release system.
Derivatization of cisplatin for the analysis using MALDI MS
In order to greatly enhance the detection efficiency and sensitivity for Pt-species in this study, an alternative approach was introduced to derivatize cisplatin using a derivatization agent DDTC, as this derivatization agent has been reported to greatly improve the ionization of Pt species.32 DDTC as a reactive nucleophile can react with cisplatin via consecutive substitution reactions with a fast rate, and the resultant hydrophobic chelate complexes can be rapidly ionized and detected by MALDI MS.20,32,33 In this study, we first examined the detection sensitivity of cisplatin without derivatization using MALDI MS. No signal was detected for the cisplatin clusters in Fig. 3B(i), compared with the theoretical cluster spectra of cisplatin (Fig. 3B(ii–v)). After derivatization, cisplatin was coordinated with DDTC and a dimer or trimer of Pt-complexes could be formed (Fig. 3A).28,34,35 The [Pt(DDTC)2]+ and [Pt(DDTC)3]+ were successfully characterized by MALDI MS and the isotope distributions were also detected (Fig. 3C(i–iii)). Our results were consistent with all previous studies based on the characterization of derivatized cisplatin in mice with MALDI MS.19,28,36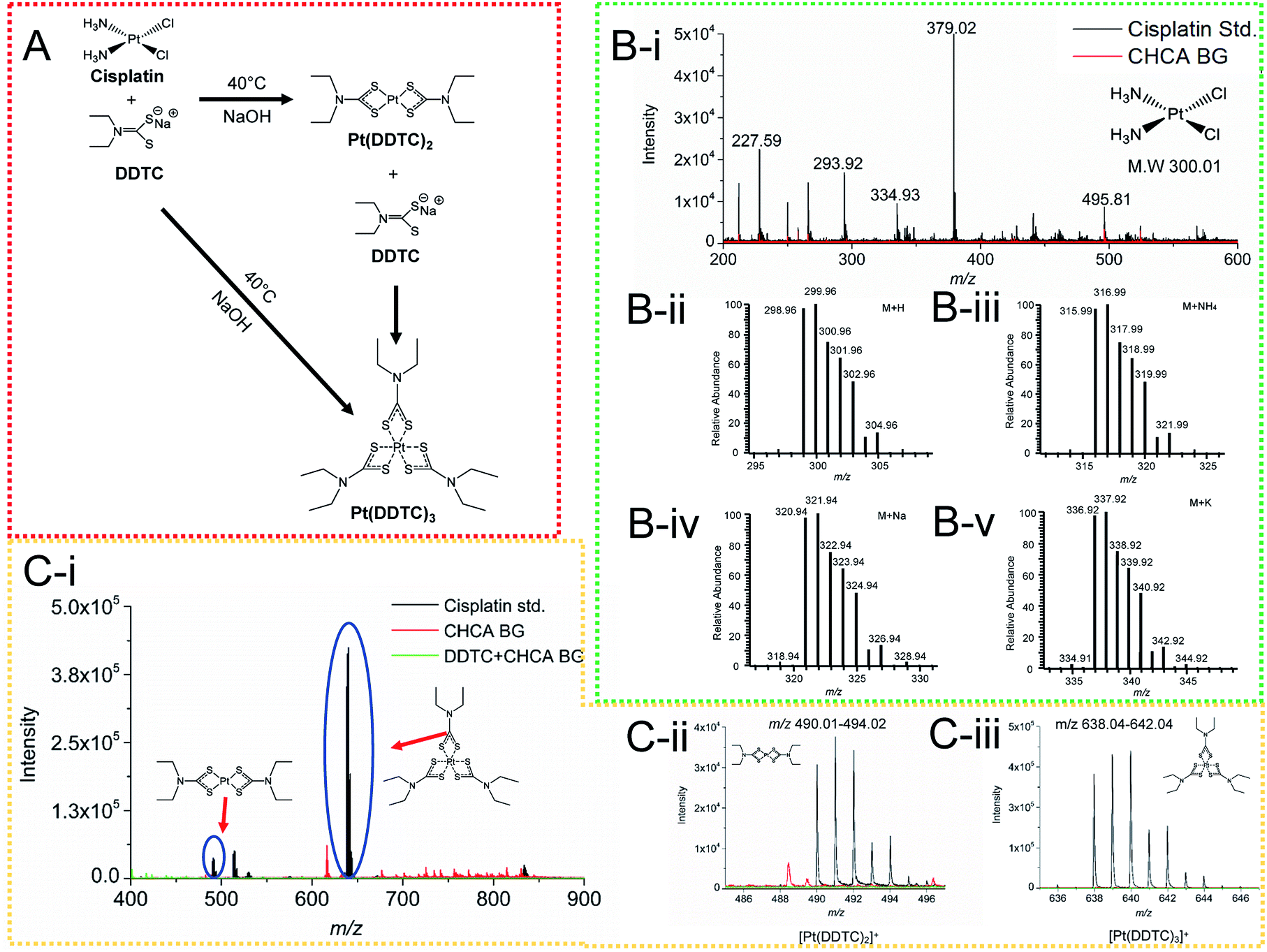 | ||
| Fig. 3 (A) Schematic illustration of derivatization reactions of cisplatin with derivatization agent diethyldithiocarbamate (DDTC). DDTC could directly extract Pt under basic conditions at 40 °C to form both [Pt(DDTC)2]+ (m/z 491.01) and [Pt(DDTC)3]+ (m/z 639.04), and [Pt(DDTC)2]+ could further react with DDTC to form [Pt(DDTC)3]+. (B(i)) MALDI-MS spectrum of cisplatin (0.5 mg mL−1) with matrix CHCA. No signature peaks can be identified for cisplatin from the theoretical calculation for (B(ii)) M + H, (B(iii)) M + NH4, (B(iv)) M + Na and (B(v)) M + K clusters. (C(i)) Overlapping mass spectra of derivatized cisplatin with the blank. The isotope clusters at (C(ii)) m/z 490.01–494.02 and (C(iii)) m/z 638.04–642.04 were assigned to [Pt(DDTC)2]+ and [Pt(DDTC)3]+, respectively. | ||
Imaging of cisplatin spotted on kidney sections
Before mapping derivatized cisplatin in the tissue sections using mass spectrometry imaging (MSI), the interference of background signals including for DDTC and CHCA was first investigated on three tissue sections. As shown in Fig. 4A, the cisplatin + DDTC + CHCA treated section spots showed strong intensity for [Pt(DDTC)2]+m/z 491.01 and [Pt(DDTC)3]+m/z 639.04, while there were no signals observed with DDTC + CHCA or CHCA alone. This suggests that no background noise arising from the tissue could interfere with the target clusters, i.e., [Pt(DDTC)2]+ and [Pt(DDTC)3]+.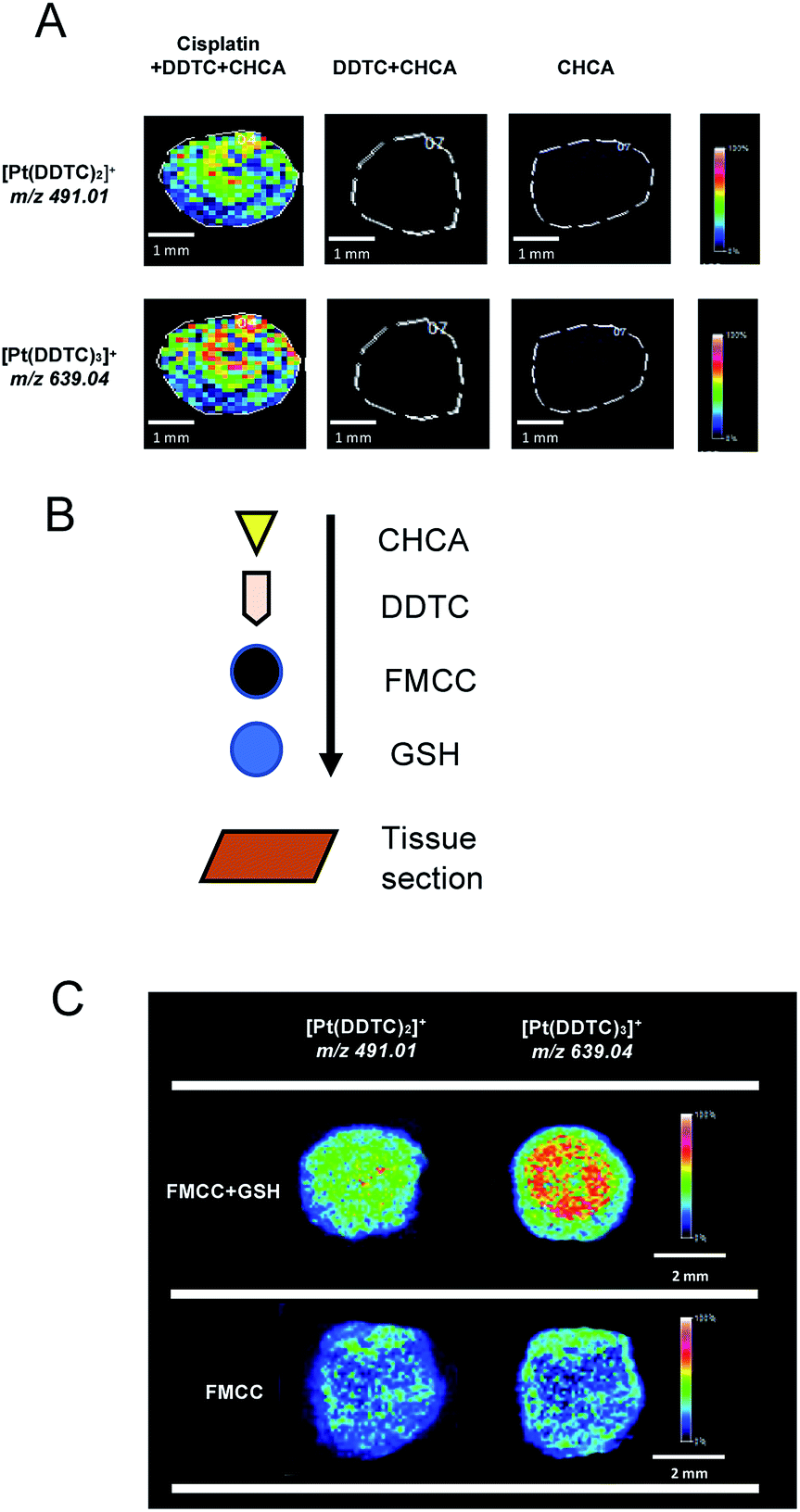 | ||
| Fig. 4 (A) The [Pt(DDTC)2]+ and [Pt(DDTC)3]+ signals were obtained for the tissue sections by MALDI-MSI which were treated with cisplatin (1 mg mL−1) + DDTC + CHCA, DDTC + CHCA, and CHCA, respectively. (B) Schematic illustration showing the addition order of GSH, cisplatin-hFe3O4-MPS-CPD (FMCC), derivatization agent and matrix on the tissue sections. (C) MALDI-MSI of [Pt(DDTC)2]+ and [Pt(DDTC)3]+ in the tissue sections added with cisplatin-hFe3O4-MPS-CPD with or without GSH. | ||
Except for the derivatized cisplatin species, we also examined the detection sensitivity of cisplatin released from cisplatin-hFe3O4-MPS-CPD in the GSH-responsive mode on tissue sections using MALDI-MSI. The GSH, cisplatin-containing nanoparticles, together with the derivatization agent, and matrix were sequentially added on the tissue sections (Fig. 4B), and the signals of [Pt(DDTC)2]+ and [Pt(DDTC)3]+ were compared with those for the tissue section treated with the same agents in the absence of GSH. It was found that the signals of both [Pt(DDTC)2]+ and [Pt(DDTC)3]+ for GSH treated tissue were significantly stronger than those for the tissue only treated with cisplatin-hFe3O4-MPS-CPD (Fig. 4C). This result demonstrated that the GSH-responsive cisplatin releasing from the nanoparticles on the tissue sections could be examined by the current derivatization method and mapped using MALDI-MSI. These findings show that to map the spatial distribution in various biological samples by MALDI-MSI is a viable method.
Mapping the spatial distribution of GSH-responsive derivatized cisplatin by MALDI-MSI
In the animal experiment, one rat was perfused with a cisplatin-hFe3O4-MPS-CPD and GSH solution, while the other one was only perfused with a cisplatin-hFe3O4-MPS-CPD suspension (Fig. 5A). After the treatment, the rats were sacrificed, and their kidneys were removed followed by the frozen section, derivatization and deposition with the matrix for MALDI-MSI analysis (Fig. 5B). The MS spectra showed that both target ions [Pt(DDTC)2]+ and [Pt(DDTC)3]+ were detected. It was noted that the signal intensity of [Pt(DDTC)3]+ was about two fold higher than that of [Pt(DDTC)2]+ (Fig. 5C). This might be due to the continuous derivatization reaction of cisplatin with DDTC. Once [Pt(DDTC)2]+ is formed, it could further react with DDTC to produce [Pt(DDTC)3]+. Thus, the overall intensities of [Pt(DDTC)2]+ were lower than those of [Pt(DDTC)3]+. In addition, the signals of [Pt(DDTC)2]+ and [Pt(DDTC)3]+ in the FMCC + GSH group showed obviously stronger intensities than those of the ones in the FMCC group without GSH, revealing that more cisplatin was released from nanoparticles in the GSH-rich tissue sections. Additionally, a small amount of cisplatin could be released from other reported controlled-released nanosystems without an external trigger in previous studies.37,38 Similar behaviour was also observed in our study where there were traces of cisplatin signals from the FMCC-treated group without applying GSH (Fig. 4C and 5C(i and ii)), revealing that the nanoparticles themselves underwent a restricted drug release.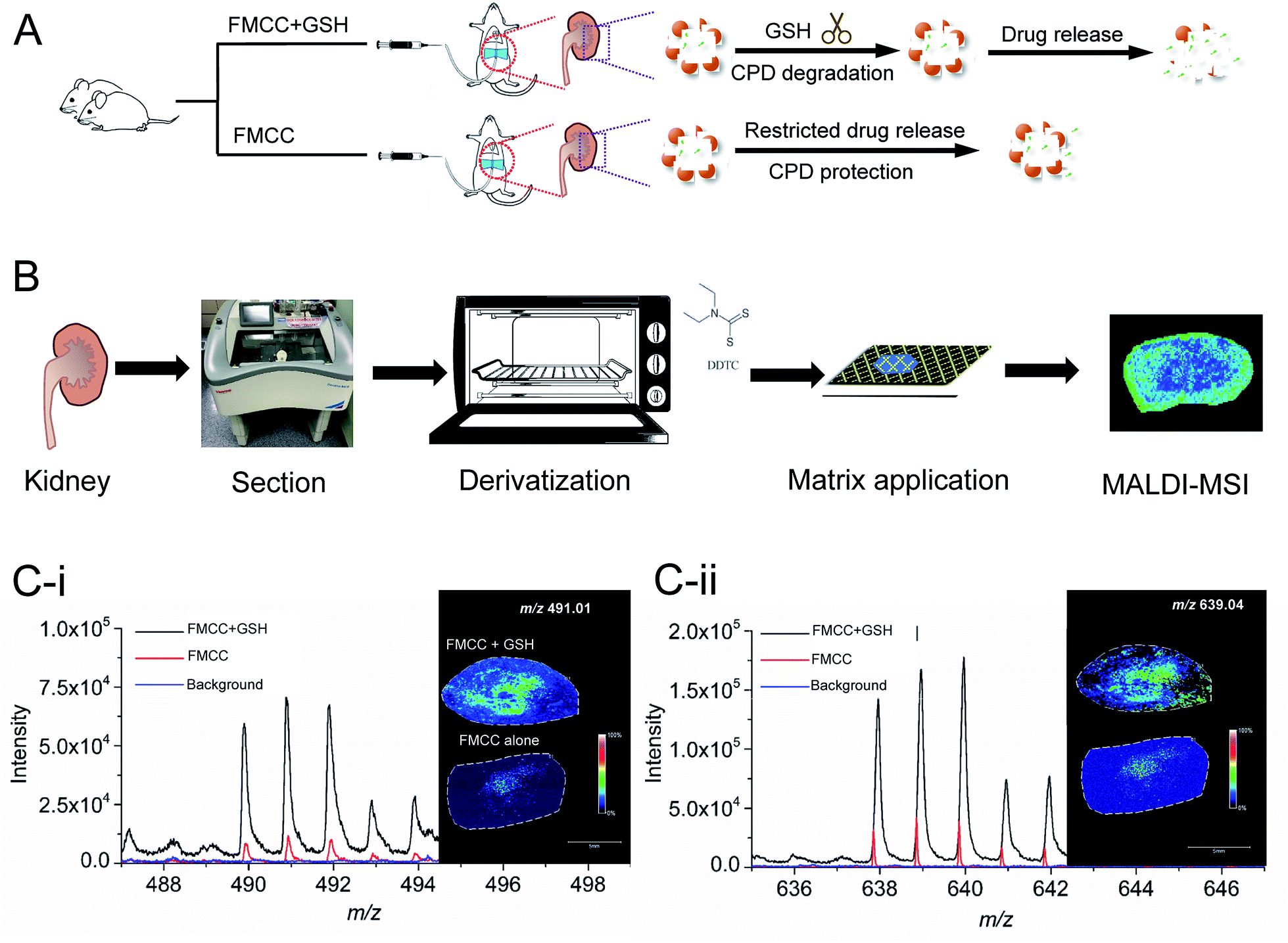 | ||
| Fig. 5 Schematic illustration of the (A) animal experiment of perfusing the kidneys of Sprague Dawley rats with cisplatin-hFe3O4-MPS-CPD (FMCC) with or without GSH, and (B) tissue preparation procedures for MALDI-MSI. The spatial distribution of [Pt(DDTC)2]+ (C(i)) and [Pt(DDTC)3]+ (C(ii)) from cisplatin-hFe3O4-MPS-CPD (FMCC) with or without GSH by MSI. | ||
Conclusions
In summary, cisplatin loaded in CPD-modified nanoparticles (hFe3O4-MPS-CPD) can be easily released in a GSH-responsive mode, and then determined by MALDI MS after derivatization. This optimized approach was further applied for assessing and mapping the derivatized cisplatin in kidney samples by MALDI-MSI. These results demonstrate the potential application of MALDI-MSI in mapping the spatial distribution of derivatized cisplatin in various biological samples.39 The present study on a refined approach to detect the derivatized cisplatin provides the first evidence on assessing the GSH-responsive release of the drug from an in situ smart nanoparticle system by utilizing MALDI-MSI, and this novel method could be extended to determine the pharmacokinetics of cisplatin in biomedical research.Materials and methods
Chemicals
Polyacrylamide (PAM) with a molecular weight of 5–6 megadaltons (MDa) was purchased from International Laboratory USA (South San Francisco, USA). Iron(III) chloride hexahydrate (FeCl3·6H2O) was obtained from Acros (New Jersey, USA). Trisodium citrate dehydrate, urea, ethylenediamine, (3-mercaptopropyl)trimethoxysilane (MPS), urea, sodium diethyldithiocarbamate (DDTC, ≥99%) and matrix α-cyano-4-hydroxycinnamic acid (CHCA) were ordered from Sigma Aldrich (St. Louis, USA). Cisplatin (≥98%), glutathione (GSH), thioctic acid, carbonyldiimidazole, 1H-pyrazole-1-carboxamidine hydrochloride, tris(2-carboxyethyl)phosphine hydrochloride and triethanolamine were bought from Meryer (Shanghai, China).For solvents, ethanol (EtOH), acetonitrile (MeCN), methanol (MeOH), and trifluoroacetic acid (TFA, 99%) were provided by VWR International Company (Radnor, Pennsylvania), while acetone (AC), dichloromethane (DCM), dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) were from RCI Labscan Ltd. (Bangkok, Thailand). All chemicals were of high performance liquid chromatography (HPLC) or analytical grade and utilized with no further purification, and distilled water (18.2 MΩ cm) was purified by using a Milli-Q ICW3000 system (Millipore, Billerica, MA) and degassed with nitrogen prior to usage.
Synthesis of hFe3O4
Monodisperse hollow iron oxide nanoparticles (hFe3O4) were prepared in accordance with a reported hydrothermal method with minor modifications.30 Briefly, 0.562 g of polyacrylamide was first dissolved in 25 mL of water under vigorous stirring for 1 h to obtain a transparent solution. Next, iron(III) chloride hexahydrate (1.01 g), trisodium citrate trihydrate (2.20 g) and urea (0.675 g) were added subsequently to the as-prepared solution. The reaction mixture was stirred at 25 °C for 5 min before transferring to a 100 mL Teflon-lined autoclave and heated at 200 °C for 12 h. After cooling to room temperature, hFe3O4 was acquired by magnetic separation and washed with water and ethanol three times. Finally, 310 mg of black hFe3O4 powder were obtained after drying in an oven at 60 °C for 8 h.Synthesis of hFe3O4-MPS
300 mg of hFe3O4 were dispersed in 100 mL of toluene containing 10 mM MPS and sonicated for 15 min. The solution was then refluxed under vigorous stirring for 18 h with constant nitrogen protection. After cooling to room temperature, the MPS-modified nanoparticles (hFe3O4-MPS) were collected by using a magnet and washed with toluene, ethanol and acetone three times. The residue was then dried under high vacuum for 18 h to obtain 272 mg of the hFe3O4-MPS solid and stored at 4 °C.In situ polymerization of hFe3O4-CPD
Before the polymerization, tris(2-carboxyethyl)phosphine hydrochloride (4.5 mg in 0.5 mL H2O) was added to a suspension of hFe3O4-MPS (50 mg in 2 mL DMF) and sonicated for 30 min. Triethanolamine buffer (2 mL, 0.1 M at pH 9) containing home-synthesized monomers (2 mL, 2 M in MeOH) was then added to the suspension. After 30 min of reaction under sonication, the reaction was terminated using iodoacetamide (90 mg in 1 mL H2O). The resultant hFe3O4-MPS with surface-polymerized CPD (hFe3O4-MPS-CPD) was isolated by using a magnet, washed with 20 mL of DMF, H2O and MeOH three times, and collected after drying under vacuum for 18 h.Characterization of the as-synthesized nanoparticles
The morphology of the particles was observed under a Tecnai G2 20 S-TWIN transmission electron microscope (TEM; FEI, Hillsboro, USA), and the elements were assessed using an equipped energy dispersive X-ray spectrometer (EDX). The size distribution of hFe3O4 was determined from 103 particles randomly selected from the TEM images with ImageJ (Version 1.51s, National Institutes of Health, USA). The pore size of the as-synthesized nanoparticles was assessed from the nitrogen (N2) adsorption–desorption isotherms at around 77 K by using an ASAP 2020M system (Micromeritics, Norcross, GA, USA). The X-ray diffraction (XRD) patterns of the nanoparticles were measured with a Bruker D8 Advance X-ray diffractometer, and the data were retrieved from an X'Pert-Philips X-ray diffractometer having monochromatized CuKα radiation (1.5406 Å, 55 kV and 40 mA). The surface functional groups of the modified or GSH-treated nanoparticles were analyzed by using a Nicolet Magna 550 Series II Fourier transform infrared spectrometer (FT-IR, Nicolet Instrument, Madison, US), while the surface modification was examined by using a TGA-6 thermogravimetric analyzer (TGA; PerkinElmer, Waltham, USA). Elemental analyses (O, N, and S) were performed using X-ray photoelectron spectroscopy (XPS) by using an SKL-12 spectrometer modified with a VG CLAM 4 multichannel hemispherical analyzer.The loading and release of cisplatin-encapsulated hFe3O4-MPS-CPD
Different solvent systems were developed and tested in the loading process, including H2O, DMF, DMF/H2O (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and acetone. In brief, hFe3O4-MPS-CPD (10 mg mL−1) was dispersed in the solvent containing pre-weighed cisplatin, and the suspension was shaken at 300 rpm for 18 h. The cisplatin-encapsulated hFe3O4-MPS-CPD (cisplatin-hFe3O4-MPS-CPD, FMCC) was collected by using a magnet, then washed with acetone/ethanol (v/v, 3:2) three times and dried under high vacuum for 18 h. For the assessment of cisplatin loading efficiency, 5 mg of cisplatin-hFe3O4-MPS-CPD were digested using aqua regia in an autoclave for 6 h at 120 °C. The concentration of the resulting solution was diluted with an acid matrix (2% nitric acid and 2% hydrochloric acid in DI water) to the ppb level and analyzed by using an ELAN DRC II ICP-MS spectrometer (PerkinElmer, Waltman, USA). 56Fe and 195Pt were selected as the analytes of interest during ICP-MS analysis to determine the iron and platinum contents present in the sample solution.
:1) and acetone. In brief, hFe3O4-MPS-CPD (10 mg mL−1) was dispersed in the solvent containing pre-weighed cisplatin, and the suspension was shaken at 300 rpm for 18 h. The cisplatin-encapsulated hFe3O4-MPS-CPD (cisplatin-hFe3O4-MPS-CPD, FMCC) was collected by using a magnet, then washed with acetone/ethanol (v/v, 3:2) three times and dried under high vacuum for 18 h. For the assessment of cisplatin loading efficiency, 5 mg of cisplatin-hFe3O4-MPS-CPD were digested using aqua regia in an autoclave for 6 h at 120 °C. The concentration of the resulting solution was diluted with an acid matrix (2% nitric acid and 2% hydrochloric acid in DI water) to the ppb level and analyzed by using an ELAN DRC II ICP-MS spectrometer (PerkinElmer, Waltman, USA). 56Fe and 195Pt were selected as the analytes of interest during ICP-MS analysis to determine the iron and platinum contents present in the sample solution.
Regarding the drug release, cisplatin-hFe3O4-MPS-CPD was first dispersed in phosphate-buffered saline (PBS) buffer with or without 10 mM GSH at a concentration of 1 mg mL−1 under sonication for 1 min. At each time point, an aliquot was collected and filtered via a syringe membrane (0.2 μm), and then digested in 69% nitric acid at 60 °C for 1 h. The digested aliquots were diluted with an acid matrix (2% nitric acid and 2% hydrochloric acid in double distilled water) for ICP-MS analysis. 195Pt was selected as the monitoring analyte during ICP-MS analysis to determine the platinum content present in the aliquot.
Animal experiments
Six-week-old male Sprague Dawley rats and C57BL/6J mice were obtained from the Chinese University of Hong Kong (CUHK), and they were housed under controlled conditions (22 ± 2 °C, 12 h light/dark cycle daily) with access to food and water. The mice were then sacrificed and the kidneys were removed and frozen immediately in liquid nitrogen followed by storage at −80 °C. These mice tissues were mainly used for establishing the methodology of Pt determination in tissue slices using matrix assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS).In situ derivatization and examination
A stock solution of cisplatin was first prepared in MeOH to achieve a final concentration of 5 mg mL−1. Then, its working calibration solutions were serially diluted with MeOH/H2O (1:1, v/v) from 5.00 to 0.01 mg mL−1. For the kidney sections obtained from C57BL/6J mice, 1 μL of cisplatin solution with different concentrations (5.00, 1.00, 0.50, 0.25, 0.10 and 0.05 mg mL−1) was dropped on the tissue sections respectively, and then dried under vacuum. The in situ derivatization was performed on the well-dried tissue sections. In brief, 5% DDTC prepared in 0.1% NaOH was applied to the tissue slides using an in-house airbrush. Then, the ITO slides were placed in a 50 mL Falcon tube containing moist Kim-wipe tissue paper, and the tube was incubated at 40 °C for 60 min. The slide was then taken out of the tube and dried in an oven for 15 min. At the same time, CHCA (7 mg mL−1) as the matrix solution was prepared in 50% MeCN (MeCN:H2O, 1:1, v/v) containing 0.2% TFA, and then applied using an automatic matrix sprayer (ImagePrep, Bruker Daltonics, Billerica, MA). After the derivatization, the ITO slides were further analyzed with MALDI-TOF MS.
Determination of cisplatin released from cisplatin-hFe3O4-MPS-CPD on tissue slices
In brief, 1 μL GSH was first added on the tissue section and dried immediately under vacuum, and then the same volume of cisplatin-hFe3O4-MPS-CPD (10 mg mL−1 in MeOH/saline with a volume ratio of 1:1) was micro-pipetted onto above the GSH spot. After dry vacuuming, the slide was treated by the derivatization process and matrix application as described above.
In vivo perfusion with cisplatin-hFe3O4-MPS-CPD and assessment of GSH-triggered cisplatin release
For the perfusion experiment, two Sprague Dawley rats were used for the test. Before the perfusion, the rats were anaesthetized with 10% chloral hydrate (0.03 mL/10 g). The kidneys from one rat were perfused with 3 mL of cisplatin-hFe3O4-MPS-CPD (10 mg mL−1 in MeOH/saline with volume ratio of 1:1) and 3 mL of freshly prepared GSH solution (10 mM, in saline) via the abdominal aorta31 at a constant rate (7 mL min−1 g−1) by using a syringe pump, while the other rat was perfused with only 3 mL of cisplatin-hFe3O4-MPS-CPD at the same concentration, rate and conditions. After perfusion, the rats were sacrificed, and the kidneys were immediately frozen in liquid nitrogen, and stored at −80 °C. The collected samples were then sectioned using a CryoStar Nx70 cryostat (Thermal Fisher Scientific, Walldorf, Germany) at −20 °C. The tissue slices with a thickness of 12 μm were mounted on indium-tin oxide (ITO) coated conductive glass slides, and then air-dried and stored at −80 °C for further MALDI-TOF MS analysis. For the tissue sections obtained from the perfusion experiment, the same sample preparation was performed on ITO slides as described above.
MALDI imaging
The MALDI analysis was performed using a rapifleX MALDI Tissuetyper (Bruker Daltonics, Germany) with around 60% of the full laser intensity of the excitation source. The positive ion mode was acquired with a 20 kV source voltage, frequency of 10000 Hz, and 170 ns delayed extraction time. Spectra were acquired with 1000 averaged laser shots with a 100 × 100 μm raster and the mass range of m/z 100–1000. The instrument was calibrated with a mixture of external standards (sinapinic acid, CHCA, 2,5-dihydroxybenzoic acid, and bradykinin (1–7) and angiotensin II) prior to each imaging measurement in positive ionization mode (m/z: 120–1000). All parameters were optimized before MSI analysis to maximize the signals. MALDI-TOF spectrometry was controlled using FlexControl 3.0, and all images were processed using FlexImaging 3.0 (Bruker Daltonics, Germany), while FlexAnalysis 3.0 (Bruker Daltonics, Germany) was applied for all spectral analysis and export.
Abbreviations
| AAS | Atomic absorption spectroscopy |
| MDa | Megadaltons |
| HPLC | High performance liquid chromatography |
| BET | Brunauer–Emmett–Teller |
| BJH | Barrett–Joyner–Halenda |
| CDDP | Cisplatin (cis-diamminedichloroplatinum(II)) |
| CHCA | Matrix α-cyano-4-hydroxycinnamic acid |
| CPDs | Cell-penetrating poly(disulfide)s |
| DCM | Dichloromethane |
| DDTC | Sodium diethyldithiocarbamate |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| EDX | Energy dispersive X-ray spectrometer |
| EtOH | Ethanol |
| FMCC | Cisplatin-hFe3O4-MPS-CPD |
| FT-IR | Fourier transform infrared spectroscopy |
| GSH | Glutathione |
| hFe3O4 | Hollow iron oxide nanoparticles |
| HPLC-MS | High-performance liquid chromatography–mass spectrometry |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| ITO | Indium-tin oxide |
| LC-MS | Liquid chromatography–mass spectrometry |
| MALDI-TOF MS | Matrix assisted laser desorption ionization-time of flight mass spectrometry |
| MALDI-MSI | Matrix-assisted laser desorption/ionization mass spectrometry imaging |
| MeCN | Acetonitrile |
| MeOH | Methanol |
| MPS | (3-Mercaptopropyl)trimethoxysilane |
| MSI | Mass spectrometry imaging |
| PAM | Polyacrylamide |
| PBS | Phosphate-buffered saline |
| SPIONs | Superparamagnetic iron oxide nanoparticles |
| TEM | Transmission electron microscope |
| TFA | Trifluoroacetic acid |
| TGA | Thermogravimetric analyzer |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
Live subject statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of The Hong Kong Baptist University and approved by the Animal Ethics Committee of The Hong Kong Baptist University.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was supported by the State Key Laboratory of Environmental and Biological Analysis (SKLP-1920-P05) and The President's Award for Outstanding Performance in Research Supervision to K. C.-F. L. from HKBU. We are grateful to Ms Winnie Wu and Mr Benson Leung from Surface Analysis & Material Characterization Laboratory, HKBU for their professional assistance. We thank Professor Shouhu Xuan (Department of Modern Mechanics, USTC) for BET and XRD analyses.References
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS.
- B. Köberle, M. T. Tomicic, S. Usanova and B. Kaina, Biochim. Biophys. Acta, Rev. Cancer, 2010, 1806, 172–182 CrossRef.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS.
- J. Peng, T. Qi, J. Liao, B. Chu, Q. Yang, W. Li, Y. Qu, F. Luo and Z. Qian, Biomaterials, 2013, 34, 8726–8740 CrossRef CAS.
- P. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang, Z. Gu and J. Lin, Nano Lett., 2017, 17, 928–937 CrossRef CAS.
- F. Li, T. Li, W. Cao, L. Wang and H. Xu, Biomaterials, 2017, 133, 208–218 CrossRef CAS.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS.
- Y. H. Jeong, H. W. Shin, J. Y. Kwon and S. M. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 23617–23629 CrossRef CAS.
- X. Duan, C. He, S. J. Kron and W. Lin, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 776–791 CAS.
- K. Cheng, S. Peng, C. Xu and S. Sun, J. Am. Chem. Soc., 2009, 131, 10637–10644 CrossRef CAS.
- Y. Pan, X. Du, F. Zhao and B. Xu, Chem. Soc. Rev., 2012, 41, 2912–2942 RSC.
- K. C.-F. Leung, S. Xuan, X. Zhu, D. Wang, C.-P. Chak, S.-F. Lee, W. K.-W. Ho and B. C.-T. Chung, Chem. Soc. Rev., 2012, 41, 1911–1928 RSC.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J. G. Park, T. Y. Ahn, Y. W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS.
- M. Colombo, S. Carregal-Romero, M. F. Casula, L. Gutiérrez, M. P. Morales, I. B. Böhm, J. T. Heverhagen, D. Prosperi and W. J. Parak, Chem. Soc. Rev., 2012, 41, 4306–4334 RSC.
- X.-M. Zhu, J. Yuan, K. C.-F. Leung, S.-F. Lee, K. W. Y. Sham, C. H. K. Cheng, D. W. T. Au, G.-J. Teng, A. T. Ahuja and Y.-X. J. Wang, Nanoscale, 2012, 4, 5744–5754 RSC.
- Y.-X. J. Wang, X.-M. Zhu, Q. Liang, C. H. K. Cheng, W. Wang and K. C.-F. Leung, Angew. Chem., Int. Ed., 2014, 53, 4812–4815 CrossRef CAS.
- J. Z. Zhang, N. S. Bryce, A. Lanzirotti, C. K. Chen, D. Paterson, M. D. de Jonge, D. L. Howard and T. W. Hambley, Metallomics, 2012, 4, 1209–1217 RSC.
- B. Laforce, C. Carlier, B. Vekemans, J. Villanova, R. Tucoulou, W. Ceelen and L. Vincze, Sci. Rep., 2016, 6, 29999 CrossRef CAS.
- J. Bianga, A. Bouslimani, N. Bec, F. Quenet, S. Mounicou, J. Szpunar, B. Bouyssiere, R. Lobinski and C. Larroque, Metallomics, 2014, 6, 1382–1386 RSC.
- M. Chappuy, E. Caudron, A. Bellanger and D. Pradeau, J. Hazard. Mater., 2010, 176, 207–212 CrossRef CAS.
- A. N. Shaik, D. A. Altomare, L. J. Lesko and M. N. Trame, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1046, 243–249 CrossRef CAS.
- D. Gode and D. A. Volmer, Analyst, 2013, 138, 1289–1315 RSC.
- K. Chughtai and R. M. A. Heeren, Chem. Rev., 2010, 110, 3237–3277 CrossRef CAS.
- M. R. Groseclose and S. Castellino, Anal. Chem., 2013, 85, 10099–10106 CrossRef CAS.
- S. Castellino, M. R. Groseclose, J. Sigafoos, D. Wagner, M. de Serres, J. W. Polli, E. Romach, J. Myer and B. Hamilton, Chem. Res. Toxicol., 2012, 26, 241–251 Search PubMed.
- C. W. Chumbley, M. L. Reyzer, J. L. Allen, G. A. Marriner, L. E. Via, C. E. Barry and R. M. Caprioli, Anal. Chem., 2016, 88, 2392–2398 CrossRef CAS.
- I. Rzagalinski and D. A. Volmer, Biochim. Biophys. Acta, Proteins Proteomics, 2017, 1865, 726–739 CrossRef CAS.
- X. Liu, Sci. Rep., 2016, 6, 38507 CrossRef CAS.
- A. Bouslimani, N. Bec, M. Glueckmann, C. Hirtz and C. Larroque, Rapid Commun. Mass Spectrom., 2010, 24, 415–421 CrossRef CAS.
- B. Shen, Y. Ma, S. Yu and C. Ji, ACS Appl. Mater. Interfaces, 2016, 8, 24502–24508 CrossRef CAS.
- X. Liu, Q. Fan, G. Yang, N. Liu, D. Chen, Y. Jiang and L. Wang, Exp. Ther. Med., 2013, 5, 1322–1326 CrossRef.
- D. Theile, J. C. Detering, C. Herold-Mende, G. Dyckhoff, W. E. Haefeli, J. Weiss and J. Burhenne, J. Pharmacol. Exp. Ther., 2012, 341, 51–58 CrossRef CAS.
- S. J. Bannister, L. A. Sternson and A. Repta, J. Chromatogr. A, 1979, 173, 333–342 CrossRef CAS.
- X. Wang and Z. Guo, Anti-Cancer Agents Med. Chem., 2007, 7, 19–34 CrossRef.
- M. Knipp, A. V. Karotki, S. Chesnov, G. Natile, P. J. Sadler, V. Brabec and M. Vašák, J. Med. Chem., 2007, 50, 4075–4086 CrossRef CAS.
- C. Tang, C. Li, C. Tang, W. Zhan, H. Zheng and X. Peng, Anal. Methods, 2013, 5, 7117–7126 RSC.
- L. Liao, J. Liu, E. C. Dreaden, S. W. Morton, K. E. Shopsowitz, P. T. Hammond and J. A. Johnson, J. Am. Chem. Soc., 2014, 136, 5896–5899 CrossRef CAS.
- K. C.-F. Leung, Arch. Chem. Res., 2017, 1, 10 Search PubMed.
- T. Wang, Z. Cai, Y. Chen, W. K. Lee, C.-S. Kwan, M. Li, A. S. C. Chan, Z.-F. Chen, A. K. L. Cheung and K. C.-F. Leung, J. Am. Soc. Mass Spectrom. DOI:10.1021/jasms.0c00198.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, including synthesis, NMR, MS and TEM experiments. See DOI: 10.1039/d0na00708k |
| This journal is © The Royal Society of Chemistry 2020 |