
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilization of reduced copper on ceria aerogels for CO oxidation†
Catherine L.
Pitman
 a,
Ashley M.
Pennington
a,
Todd H.
Brintlinger
b,
Daniel E.
Barlow
c,
Liam F.
Esparraguera
c,
Rhonda M.
Stroud
b,
Jeremy J.
Pietron‡
d,
Paul A.
DeSario
*c and
Debra R.
Rolison
*c
a,
Ashley M.
Pennington
a,
Todd H.
Brintlinger
b,
Daniel E.
Barlow
c,
Liam F.
Esparraguera
c,
Rhonda M.
Stroud
b,
Jeremy J.
Pietron‡
d,
Paul A.
DeSario
*c and
Debra R.
Rolison
*c
aNRL/NRC Postdoctoral Associate, U.S. Naval Research Laboratory, Washington, D.C. 20375, USA
bMaterials Science and Technology Division (Code 6300), U.S. Naval Research Laboratory, Washington, D.C. 20375, USA
cChemistry Division (Code 6100), U.S. Naval Research Laboratory, Washington, D.C. 20375, USA. E-mail: paul.desario@nrl.navy.mil; rolison@nrl.navy.mil
dFormer Employee, Surface Chemistry Branch (Code 6170), U.S. Naval Research Laboratory, Washington, D.C. 20375, USA
First published on 3rd September 2020
Abstract
Photodeposition of Cu nanoparticles on ceria (CeO2) aerogels generates a high surface area composite material with sufficient metallic Cu to exhibit an air-stable surface plasmon resonance. We show that balancing the surface area of the aerogel support with the Cu weight loading is a critical factor in retaining stable Cu0. At higher Cu weight loadings or with a lower support surface area, Cu aggregation is observed by scanning and transmission electron microscopy. Analysis of Cu/CeO2 using X-ray photoelectron spectroscopy and Fourier-transform infrared spectroscopy finds a mixture of Cu2+, Cu+, and Cu0, with Cu+ at the surface. At 5 wt% Cu, Cu/CeO2 aerogels exhibit high activity for heterogeneous CO oxidation catalysis at low temperatures (94% conversion of CO at 150 °C), substantially out-performing Cu/TiO2 aerogel catalysts featuring the same weight loading of Cu on TiO2 (20% conversion of CO at 150 °C). The present study demonstrates an extension of our previous concept of stabilizing catalytic Cu nanoparticles in low oxidation states on reducing, high surface area aerogel supports. Changing the reducing power of the support modulates the catalytic activity of mixed-valent Cu nanoparticles and metal oxide support.
1. Introduction
Stabilizing reduced copper nanoparticles (Cu NPs) under ambient conditions is desirable for catalytic applications where low-valent Cu is more active than CuO and for plasmonic applications as a nonprecious alternative to Au and Ag. One way to achieve low-valent Cu is through interaction with reducing oxide supports. Well-engineered materials favor Cu2O over the more prevalent but less active CuO.1,2 Extending this concept, Vovchok et al. reported reduction of Cu2+ to Cu0 on their microporous cerium oxide (CeO2) support at elevated temperature under operando conditions, but did not discuss whether sufficient Cu0 was stabilized to produce surface plasmon resonance (SPR).3 We are aware of only two reports of nanoscale Cu supported on CeO2 that demonstrate SPR.4,5We recently reported stable metallic Cu0 and Cu+ content in Cu NPs photodeposited from Cu2+ salts on titania aerogels (Cu/TiO2(aero)).6 We attribute the stability of low-valent Cu to a close interaction between the oxide support and the deposited Cu NPs, made possible by the high surface area and small particle size of the oxide support. Donation of electron density from the reducing oxide to the supported metal NP stabilizes sufficient metallic Cu for Cu/TiO2(aero) to exhibit a Cu SPR.6 In contrast, Cu photodeposition on larger diameter, low surface-area particulate TiO2 (Cu/TiO2(partic)) does not produce materials that exhibit a SPR. The importance of the morphology of TiO2 in supporting a Cu SPR calls into question whether (1) other reducible metal oxides will display the same stabilization of low-valent Cu and (2) if similar morphology constraints are required when employing a more easily reducible metal oxide.
Copper NPs on CeO2 supports are extensively studied catalysts for CO oxidation,7–10 the water–gas shift reaction,11–13 and volatile organic compound oxidation.14,15 A complicating factor is that CeO2 is active on its own due to facile Ce4+/3+ redox interchange allowing the material to act as an oxygen buffer.16 This reducibility is a key feature of CeO2 catalytic cycles.
The reducibility of the metal oxide is further increased by nanostructuring the support. The formation energy of a corner oxygen vacancy in small CeO2 clusters is calculated to be as low as 0.8 eV in comparison to the 2.25 eV calculated on the extended (111) surface, the most thermodynamically stable facet of fluorite CeO2.17 This effect is attributed to the presence of under-coordinated surface sites in nanostructured supports as well as the flexibility of nanoparticles to relax around oxygen vacancies and thereby stabilize them.18 Several groups have focused on developing nanostructured CeO2 supports to produce highly defective materials, including by controlling the shape of the nanoparticles,7,8 template syntheses,19 and using specific calcination protocols.9 The addition of supported metal nanoparticles can increase the reducibility of the support by providing an electron sink to stabilize the charge produced by oxygen-vacancy generation.18 In addition to altering the reducibility of the oxide, deposition also introduces Cu‖CeO2 interfaces that are implicated in reactivity.20 The aerogel expression of the reducing oxide offers critical attributes: increased reducibility of the support through nanostructuring and more oxide NP‖Cu NP junctions.
We now show that another reducible metal oxide aerogel stabilizes metallic Cu. Using CeO2, a reducible metal oxide amenable to aerogel synthesis, we report herein that Cu/CeO2 aerogel stabilizes a significant fraction of reduced Cu species (Cu0 and Cu+) in the Cu nanoparticles. Furthermore, copper nanoparticle–modified ceria aerogel exhibits an SPR and demonstrates high activity for the low-temperature heterogeneous catalytic oxidation of carbon monoxide (CO).
2. Results and discussion
2.1 Synthesis of Cu/CeO2 materials
The ceria aerogel (CeO2(aero)) is prepared following literature procedures for sol–gel synthesis and then supercritically drying the wet gel.21 The gel is formed by epoxide-driven hydrolysis of CeCl3 in methanol. After forming dispersed CeO2 particles (the “sol”), the hydrolysis continues to covalently crosslink the oxide nanoparticles to form a gel. The aged gel is rinsed with isopropanol and acetone, and then the acetone-filled gel is dried with supercritical CO2 to produce an amorphous CeO2(aero). The aerogel is calcined in static air at 500 °C for 2 h (5 °C min−1 heating and cooling ramp) to crystallize the networked CeO2 domains. Post-calcination, CeO2(aero) has an average of 82(11) m2 g−1 surface area, 0.35(7) cm3 g−1 pore volume, and 17(3) nm average pore diameter (Table S1, Fig. S1 and S2†). As is typical for sol–gel syntheses, slight variation occurs between nominally identical batches of the aerogel support; we report values that are an average of 7 replicate batches. The fluorite crystal structure expected for CeO2 after calcination at 500 °C is confirmed using powder X-ray diffraction (XRD). Scherrer analysis estimates 6–9 nm diameter crystallites from the (111) reflection at 2θ = 28.55° (Fig. S3†), with the range reflecting batch-to-batch variation, in good agreement with the highly textured morphology seen by scanning electron microscopy (SEM, Fig. 1). | ||
| Fig. 1 Scanning electron micrographs of (left) CeO2(aero) and (right) CeO2(partic). | ||
In order to determine the effects of the aerogel morphology on the properties of the CeO2 support, we examined a commercially obtained particulate nanopowder ceria (CeO2(partic)). The commercial CeO2 has lower surface area (26 m2 g−1vs. 82(11) m2 g−1) and pore volume (0.09 cm3 g−1vs. 0.35(7) cm3 g−1) than the aerogel and lacks the covalent bonding between CeO2 nanoparticles present in the aerogel network. Crystalline regions averaged 21(1) nm diameter by XRD, suggesting particulate CeO2 is 2–3 times larger than the bonded CeO2 nanoparticles in the aerogel network. By SEM, particles as large as 75 nm across are observed (Fig. 1), which may comprise several crystalline domains or reflect the size range of crystallites in CeO2(partic).
Ceria has a direct band gap with an energy (Eg) that depends on crystallite size;22 the energies for CeO2(aero) and CeO2(partic) are calculated from Tauc plots of the diffuse-reflectance UV-Vis spectra (Fig. S4†). The values are 2.95 eV (420 nm) and 3.08 eV (403 nm) for CeO2(aero) and CeO2(partic), respectively. Although the CeO2(aero) crystallite size is less than the CeO2 Bohr radius (7–9 nm) and quantum confinement has been observed for dispersed CeO2 nanoparticles,23 the covalent attachment of the CeO2 crystallites in the aerogel framework likely prevents confinement. Indeed, the opposite effect is instead observed, with CeO2(aero) showing a decrease in bandgap energy compared to bulk CeO2 (3.19 eV, 389 nm). This red-shift was previously observed and attributed to the presence of surface Ce ions (in either the 4+ or 3+ oxidation states) and the presence of oxygen vacancies.24 The red-shift of the band-gap energy for CeO2(aero) relative to CeO2(partic) is consistent with this interpretation, given the smaller size of the crystallites and higher surface area of the aerogel.
Copper nanoparticles are photodeposited using a modification of the procedure described by Wu et al.25 A slurry of ethanol, water, ceria, and copper(II) nitrate at pH 9–10 is stirred under inert atmosphere while irradiating with broadband light, adjusting the ratio of Ce![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu to achieve a nominal 2.5–10 wt% copper loading. Excitation of the CeO2 band gap leads to mobile electrons that can reduce Cu2+ adsorbed on the surface. Charge compensation is provided by oxidation of a sacrificial reagent (ethanol). To maintain the large surface area of the CeO2 support, gentle drying techniques are needed following photodeposition. When samples dried at ambient conditions are heated under vacuum, pore collapse is observed. To minimize this deleterious loss of pore volume, after collecting the solids from the photodeposition on a membrane filter, the material is first dried at ambient conditions overnight, and then dried at 70 °C for 24 h. Porosimetry of materials dried in this manner do not reveal pore collapse, although a small reduction in pore diameter occurs, consistent with partial filling of pores by Cu NPs (Fig. 2A).
:Cu to achieve a nominal 2.5–10 wt% copper loading. Excitation of the CeO2 band gap leads to mobile electrons that can reduce Cu2+ adsorbed on the surface. Charge compensation is provided by oxidation of a sacrificial reagent (ethanol). To maintain the large surface area of the CeO2 support, gentle drying techniques are needed following photodeposition. When samples dried at ambient conditions are heated under vacuum, pore collapse is observed. To minimize this deleterious loss of pore volume, after collecting the solids from the photodeposition on a membrane filter, the material is first dried at ambient conditions overnight, and then dried at 70 °C for 24 h. Porosimetry of materials dried in this manner do not reveal pore collapse, although a small reduction in pore diameter occurs, consistent with partial filling of pores by Cu NPs (Fig. 2A).
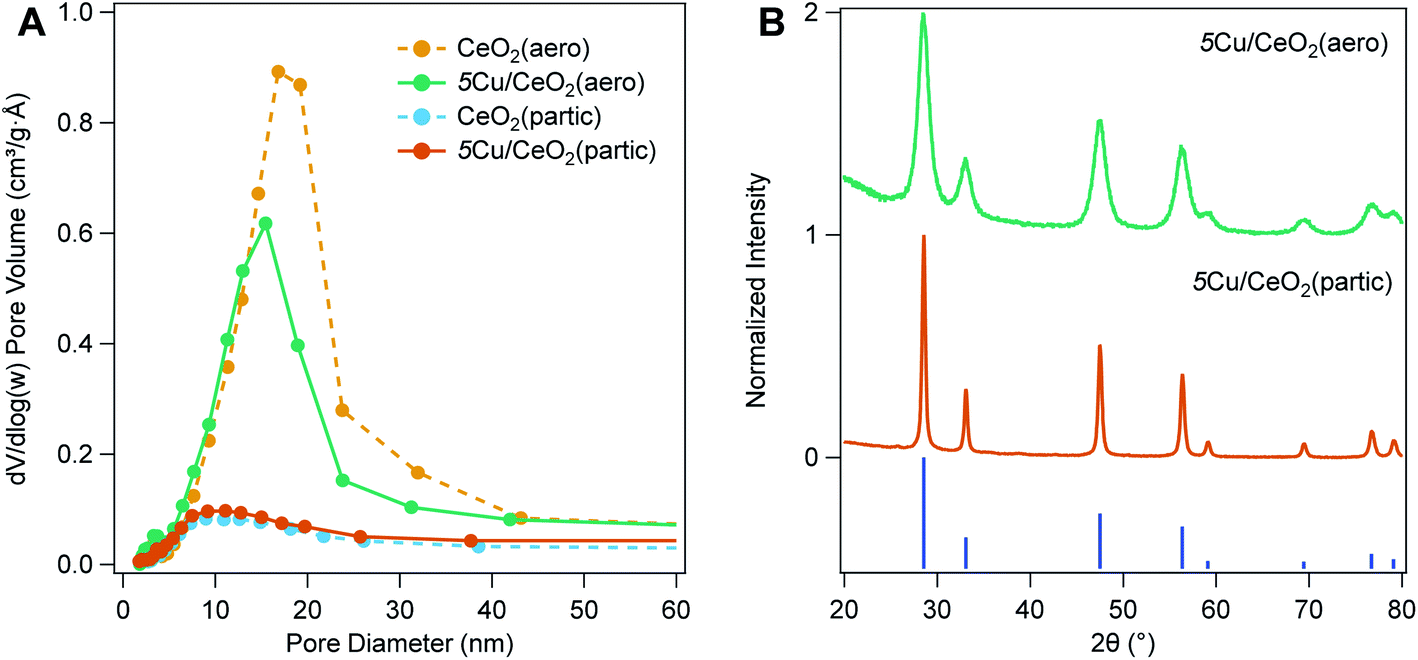 | ||
| Fig. 2 (A) Differential pore volume as a function of pore width for CeO2(aero) and CeO2(partic) with and without photodeposited copper. (B) Representative X-ray diffraction patterns of 5Cu/CeO2(aero) and 5Cu/CeO2(partic) materials compared to CeO2 (JCPDS# 01-089-8436). Scherrer analysis of the CeO2 (111) reflection at 2θ = 28.55° with FWHM of 1.28(1)° and 0.38(1)° gives 6.3(1) nm and 21.3(6) nm CeO2 particles for 5Cu/CeO2(aero) and 5Cu/CeO2(partic), respectively. | ||
Copper weight loading is assessed by X-ray fluorescence spectroscopy, comparing Cu:Ce fluorescence intensity to a standard curve of physically mixed CuO and CeO2 (both averaging <50 nm diameter particles). Copper weight loadings closely approximate the nominal weight loading (Table 1). Nominally 5 wt% Cu/CeO2 aerogel (5Cu/CeO2(aero)) has between 5.4–5.8 wt% Cu, while nominally 5 wt% Cu/CeO2 particulate (5Cu/CeO2(partic)) has 4.8–4.9 wt% Cu. The nominally 2.5 wt% and 10 wt% Cu/CeO2 aerogels ((2.5Cu/CeO2(aero) and 10Cu/CeO2(aero)), respectively) are 2.8 wt% and 10.5 wt% Cu, respectively. Following deposition, CeO2 maintains its fluorite structure, but no Cu reflections are observed at either 2.5 wt% or 5 wt% Cu loading (Fig. 2B). For 10Cu/CeO2(aero), reflections for CuO, but not Cu2O or Cu(0), are observed by XRD (Fig. S5†).
| Sample | CeO2 diametera (nm) | % Cub | % Cu0,+c | Surface aread (m2 g−1) | Pore volumed (cm3 g−1) | Pore widthd (nm) |
|---|---|---|---|---|---|---|
| a Determined via XRD, calculated across all aerogel or particulate samples. b Determined via XRF. c Determined via XPS. d Determined via desorption arm of nitrogen physisorption. | ||||||
| CeO2(aero) | 8.8(14) | — | — | 79 | 0.35 | 15.7 |
| 2.5Cu/CeO2(aero) | 8.8(14) | 2.8 | 44(10) | 82 | 0.28 | 11.2 |
| 5Cu/CeO2(aero) | 8.8(14) | 5.6 | 50(19) | 83 | 0.30 | 13.0 |
| 10Cu/CeO2(aero) | 8.8(14) | 10.5 | 42(10) | 65 | 0.27 | 11.8 |
| CeO2(partic) | 21.3(6) | — | — | 26 | 0.08 | 12.0 |
| 5Cu/CeO2(partic) | 21.3(6) | 4.8 | 30(10) | 31 | 0.11 | 12.6 |
For microstructural characterization, both scanning transmission electron microscopy with high-angle annular dark-field (STEM-HAADF) imaging as well as conventional high-resolution transmission electron microscopy (HR-TEM) imaging were performed, as seen in Fig. 3A–C. These analyses show the ceria support to be extended aerogel structures with crystallites ∼5–15 nm in size and a CeO2(111) lattice spacing of 0.31 nm, comparable to the values found by XRD. The high atomic number (Z) of Ce compared to Cu precludes an intensity-based identification and imaging of Cu NPs.
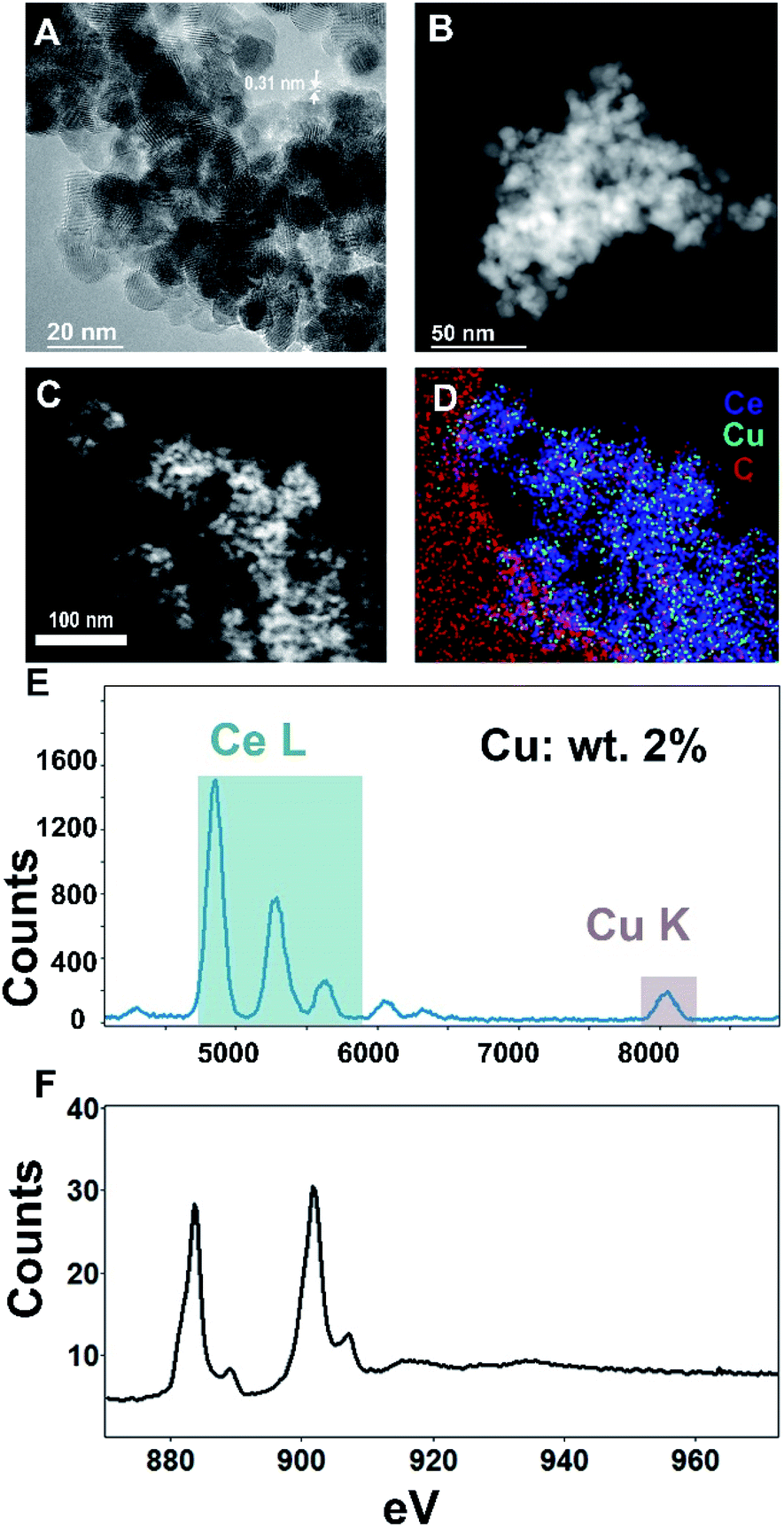 | ||
| Fig. 3 Transmission electron microscopy (TEM) of copper nanoparticles supported on 5Cu/CeO2(aero). (A) High-resolution (HR-TEM) of 5Cu/CeO2(aero). (B) High-angle annular dark-field image in scanning mode (STEM-HAADF). (C) STEM-HAADF image corresponding with (D) energy-dispersive spectroscopy in scanning mode (STEM-EDS). (E) EDS plot of broad-beam irradiated 5Cu/CeO2(aero) showing Ce and Cu present. (F) Electron energy-loss-spectroscopy of broad-beam irradiated 5Cu/CeO2(aero). | ||
For compositional analysis, we relied on energy-dispersive spectroscopy (EDS) and electron energy-loss spectroscopy (EELS) (Fig. 3D–F). The EDS-measured composition (2% copper by weight) was similar to that obtained by XRF. Acquiring an unambiguous image of the Cu nanoparticle morphology was elusive—neither EDS nor EELS show significant enough signal above the Ce background that is localized to ‘nanoparticle’-type morphologies. Additional transmission electron micrographs are found in the ESI.†
2.2 Influence of support surface area and mesostructure on electronic state of Cu
The diffuse reflectance UV-visible spectra of 5Cu/CeO2(aero) and 2.5Cu/CeO2(aero) (Fig. 4A and B), prepared with 48 h of irradiation, both exhibit a broad feature at 740 nm consistent with Cu SPR.6 Irradiation of 5Cu/CeO2(partic) did not produce an SPR, despite using the same photodeposition conditions, and instead displayed features consistent with CuO and Cu2O.6 At a higher weight loading, even 96 h of photoreduction does not produce a visible SPR for 10Cu/CeO2(aero). In contrast, 10Cu/TiO2(aero) exhibits a Cu SPR feature after only 24 h of photoreduction. Theoretical work predicts that the presence of a Cu SPR suggests that the Cu NPs are at least 2 nm in diameter.26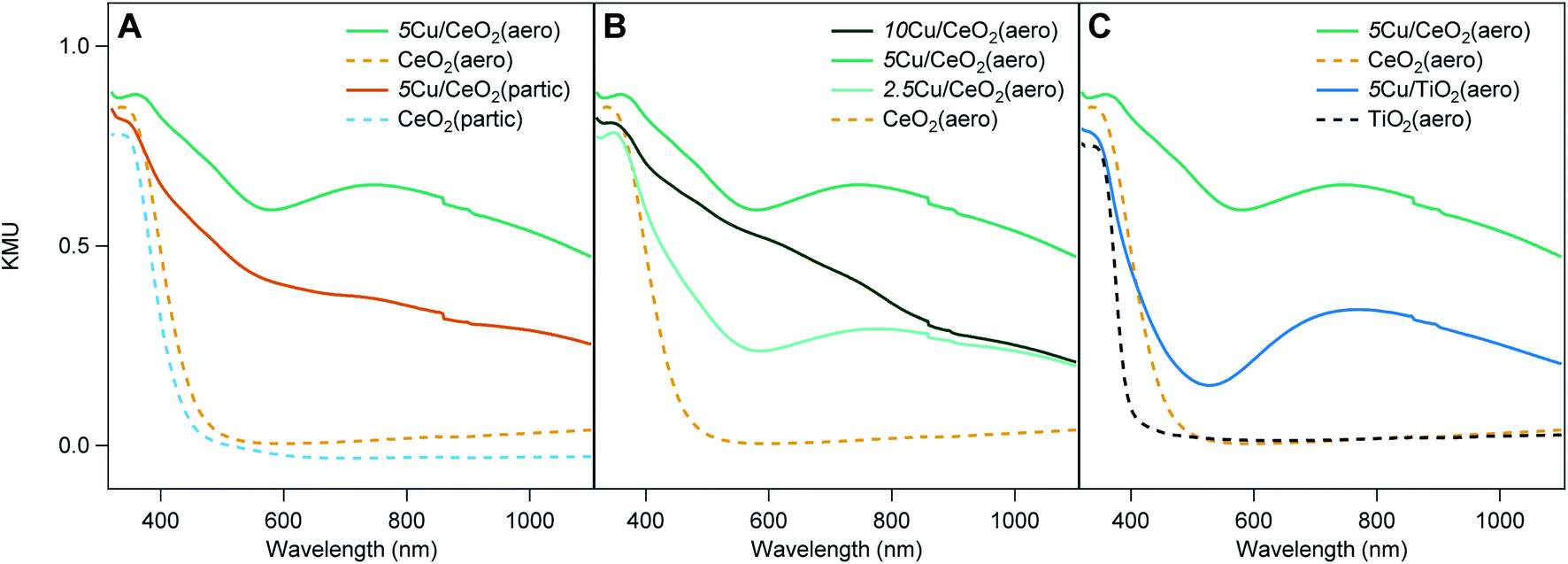 | ||
| Fig. 4 Diffuse reflectance UV-Vis spectra in Kubelka–Munk units (KMU) for (A) 5Cu/CeO2(aero) (green), CeO2(aero) (dashed yellow), 5Cu/CeO2(partic) (amber), and CeO2(partic) (dashed blue); (B) 10Cu/CeO2(aero) (dark green), 5Cu/CeO2(aero) (green), 2.5Cu/CeO2(aero) (light green), and CeO2(aero) (dashed yellow); (C) 5Cu/CeO2(aero) (green), 5Cu/TiO2(aero) (blue), CeO2(aero) (dashed yellow), and TiO2(aero) (dashed black). | ||
When comparing Cu SPRs produced at 5 wt% Cu supported on the two metal oxide aerogels, the intensity of the SPR is brighter on TiO2 than on CeO2 (Fig. 4C), suggesting more metallic Cu is stabilized on TiO2(aero) than CeO2(aero). Notably, there is a higher absorbance between 400 nm and 600 nm on 5Cu/CeO2(aero) relative to 5Cu/TiO2(aero), which may be an indication of more Cu+ on CeO2 due to the band-gap adsorption of Cu2O at ∼580 nm.27
Ceria appears less able than TiO2 to support metallic copper given that (1) longer irradiation times are required to produce SPRs, (2) the SPR response is not as bright on CeO2 relative to TiO2, and (3) an SPR is not observed on CeO2 aerogels at ≥5 wt% Cu. As the ratio of Cu to CeO2 surface area increases, either by increasing the copper loading or by decreasing the surface area of the support, SPRs are not observed. Rather, we observe large Cu features in the SEM suggesting that the available sites for Cu nucleation have been exhausted, and that further Cu deposition results in particle growth (Fig. S11–S13†). These large copper particulates interact less extensively with the CeO2 support (have a lower ratio of interface to bulk volume), which is consistent with their oxidized state.
On a per mass basis, CeO2 has less available surface area (∼80 m2 g−1) than TiO2 (∼150 m2 g−1) to support a given weight loading of Cu NPs. On a per mole basis, however, the surface area of the two metal oxide aerogels is comparable (14000 m2 (mol CeO2)−1 to 12000 m2 (mol TiO2)−1). The disparity in atomic weight is highlighted by the fact that 10Cu/TiO2(aero) and 5Cu/CeO2(aero) both support 12 mol% Cu while 10Cu/CeO2(aero) supports 23 mol% Cu. Surface area and available nucleation sites are clearly important considerations as to whether or not the photodeposited material can support a Cu SPR. Further illustrating this surface-area dependence, when nanoparticulate supports rather than aerogel supports of either oxide are utilized, the SPR feature is absent.
In addition to surface-area effects, we use X-ray photoelectron spectroscopy (XPS) to assess if differences in the Ce4+:Ce3+ ratio in the aerogel and nanoparticulate supports contribute to the differences in speciation of supported Cu NPs. In the Ce 3d region (Fig. 5A, 875 to 925 eV), the Ce4+ signature consists of 6 peaks between 882.6 eV and 916.7 eV while Ce3+ gives rise to 4 peaks between 880.6 eV and 904.1 eV.28 Recently published results have shown that with these 10 peaks in the Ce 3d region, small modifications to the peak-fitting procedure can vary the estimated Ce3+ content from 2.3% to 38.9%.29 Given the similarity of the Ce peaks in our particulate and aerogel samples, we have elected to not fit this region and qualitatively assert that, at least under vacuum conditions, both supports are dominated by Ce4+ and have similar Ce3+ content. The similarity in Ce3+ content suggests that the differences in Cu speciation that we observe are due more to the surface area and morphology of the support than to their degree of reducibility.
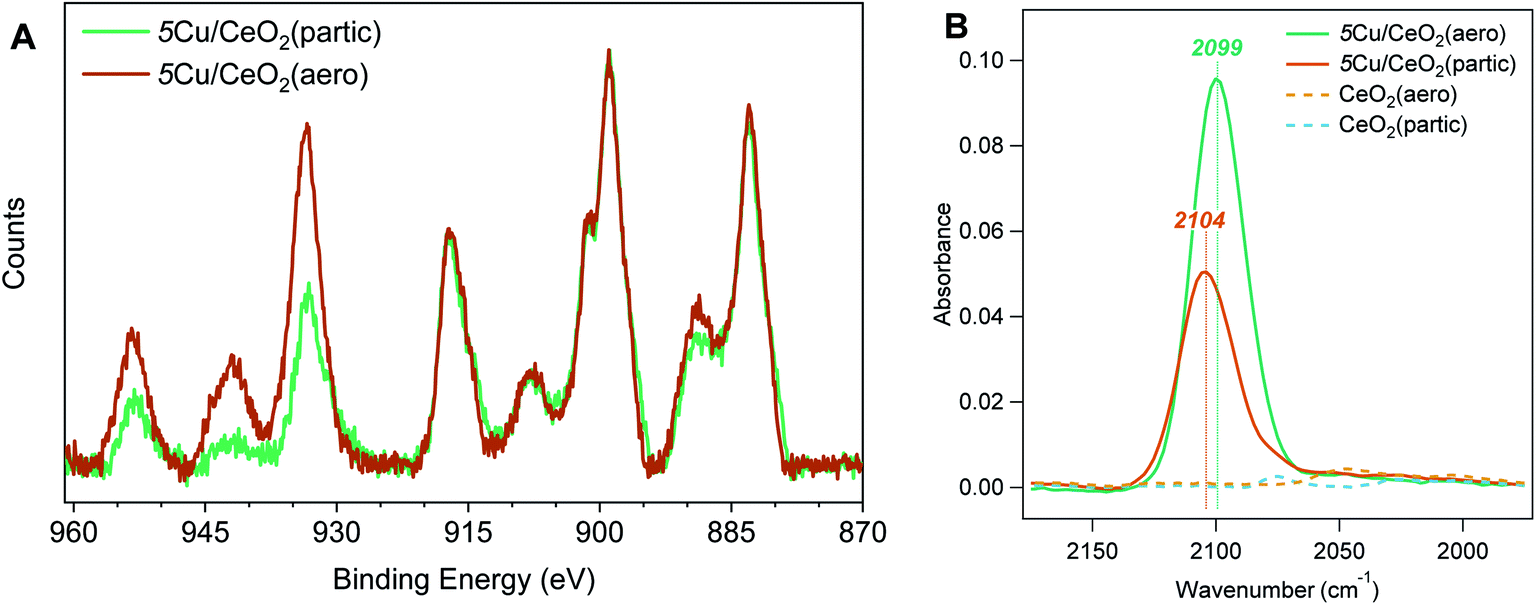 | ||
| Fig. 5 (A) Background-subtracted X-ray photoelectron spectra of 5Cu/CeO2(partic) (amber) and 5Cu/CeO2(aero) (green) in the Ce 3d (875 to 925 eV) and Cu 2p (925 to 965 eV) regions. (B) ATR-FTIR spectra for 5Cu/CeO2(aero) (green), 5Cu/CeO2(partic) (amber), CeO2(aero) (dashed yellow), and CeO2(partic) (dashed blue), showing CO binding at 2099 cm−1 and 2104 cm−1 for 5Cu/CeO2(aero) and 5Cu/CeO2(partic), respectively. | ||
The differences in Cu speciation between 5Cu/CeO2(aero) and 5Cu/CeO2(partic) are apparent in the Cu 2p region of the XPS spectra (Fig. 5A, 925 to 965 eV). The Cu 2p3/2 region consists of a peak at 932 eV for Cu0 and Cu+, low-valent states indistinguishable in these spectra, and a peak at 934 eV for Cu2+. Notably, Cu2+ gives rise to a shake-up feature around 941 eV, which is more prominent for 5Cu/CeO2(partic) than 5Cu/CeO2(aero). We attempted to resolve Cu0 and Cu+ by fitting the Auger CuLMM line; however, given the mixture of oxidation states, peak broadening inherent to insulating materials, and low Cu content of our catalysts, satisfactory fits were not obtained (Fig. S14†).
To quantify the proportion of reduced Cu in these materials (Cu0 and Cu+), we use the method described by Biesinger to compare the area under the 2p3/2 and shake-up features.30 The advantage of this method is that it is insensitive to peak broadening caused by surface charging and peak shifts expected for Cu particles of different sizes on supports.31 We find a higher percentage of reduced Cu for 5Cu/CeO2(aero) (50 ± 19%) than for 5Cu/CeO2(partic) (30 ± 10%), which is consistent with the presence of an SPR on the aerogel and lack of an SPR on the particulate. For 5Cu/TiO2(aero) this method gives 30% reduced Cu, which given the prominence of the SPR, suggests that the reduced Cu is predominantly metallic. In contrast, given its lack of an SPR, the 30% reduced Cu content in 5Cu/CeO2(partic) is likely Cu+. The signal intensity of Cu (relative to Ce) for 5Cu/CeO2(partic) is also higher than for 5Cu/CeO2(aero), despite XRF confirming similar Cu loadings. This difference is expected for a support with lower surface area. XPS is a surface-sensitive technique, and a larger fraction of the surface of the low surface-area particulate is covered with Cu at the same mass loading as on the high surface-area aerogel. We observed a similar overestimation by XPS of Cu loading on TiO2(partic) supports, which have a surface area of 10 m2 g−1.32
We utilize CO as a probe molecule in attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy to interrogate the morphology and electronic structure of the photodeposited Cu. Binding modes are observed at 2099 cm−1 and 2104 cm−1 for 5Cu/CeO2(aero) and 5Cu/CeO2(partic), respectively, on 4 mg films cast on ZnSe ATR crystals under CO flow (Fig. 5B). These positions are consistent with the reported binding energy for CO on Cu+ sites. Stacchiola et al. reported a peak at 2097 cm−1, which they attributed to small islands of Cu2O on metallic Cu surfaces, and saw this mode blue-shift to 2114 cm−1 as CeO2 coverage of their Cu substrate increased.33 Both peaks are assigned to formally Cu+ species, where increasing contact with CeO2 withdraws electron density from Cu+, which reduces back-donation to bound carbonyl, and thus results in a stronger C–O bond. The weaker carbonyl stretch observed on 5Cu/CeO2(aero) (2099 cm−1) is consistent with subsurface metallic Cu underlying Cu+, as is suggested by the presence of a Cu SPR. The stronger carbonyl stretch observed on 5Cu/CeO2(partic) (2104 cm−1) shows that this Cu+ is in a less reducing environment, as is expected for a material that shows no Cu SPR and a lower fraction of reduced Cu by XPS. The smaller peak intensity on the particulate support derives from less accessible Cu surface area and larger Cu particles, as substantiated by the large Cu features observed in SEM and TEM.
At ambient temperature, no features associated with Ce4+–CO (2168 cm−1), Ce3+–CO (2161 cm−1), or Cu0–CO (2073 cm−1)33,34 are observed on any of the materials studied. Carbonyl association on these sites is expected to be weak, and typically, low temperatures are required for their observation.34
2.3 Thermal CO oxidation
We next examine the effect of reducible CeO2(aero) mesoporous metal oxide support on the catalytic activity of Cu NPs for CO oxidation. Catalyst beds are pre-treated at 150 °C in 64 mL min−1 of an anoxic flow (0.8 mL min−1 CO, 63 mL min−1 He). After activation, O2 is added into the feed stream (16 mL min−1), and CO consumption and CO2 production are monitored by an in-line GC. We choose the modest activation temperature to compare to our earlier work on TiO2 and to minimize Cu particle ripening.32,35–37After activation, catalyst beds of CeO2(aero) show no activity for CO oxidation below 150 °C (Fig. S15†). The commercial CeO2(partic) shows slightly more activity than CeO2(aero), achieving a maximum of 15% CO conversion at 150 °C. Duplicate beds packed using catalyst from the same batch show minimal difference in activity indicating that preparation of the catalyst bed has negligible impact. Thermogravimetric analysis does not show substantial weight loss indicating that surface-adsorbed species also have a negligible impact on catalysis (Fig. S16†). Activity is consistent over multiple regeneration cycles of the bare CeO2(aero) while a small loss is observed after the first regeneration of the CeO2(partic) material (Fig. S17†). The bare supports are used as diluent in the catalyst bed of Cu/CeO2 samples, and this lack of reactivity confirms that the diluent in the catalyst bed is a minor contributor to CO oxidation.
With the addition of 5Cu/CeO2(aero) into the catalyst bed (10 mg of catalyst to 40 mg of Cu-free CeO2(aero)), at 150 °C, 94% conversion of CO is observed (3.5 molCO min−1 molCu−1). Conversion falls as the temperature is decreased, reaching 50% conversion (T50) at 91 °C (Table 2) and falling to 0.5% conversion at the lowest temperature for which we recorded data: 50 °C. The catalyst is regenerated using the same method as the activation. Upon regeneration, minimal difference in activity from the fresh bed (Fig. 6B) indicates that the catalyst is stable and not likely undergoing significant, irreversible changes during the course of the reaction.32 The catalyst is stable over four regenerations (5 cycles), lasting a total of 27.5 h on stream. At the same weight loading, the CO oxidation activity of 5Cu/CeO2(aero) far exceeds that of 5Cu/CeO2(partic) (10 mg of catalyst, 40 mg of CeO2(partic)), presumably due to the higher surface area of the aerogel and the more reduced Cu it supports.
| Sample | molCO min−1 molCu−1 at 150 °C | X CO at 150 °C | T 50 (°C) |
|---|---|---|---|
| CeO2(aero) | — | 0.03 | — |
| CeO2(partic) | — | 0.15 | — |
| 5Cu/CeO2(partic) | 2.4 | 0.55 | 144 |
| 2.5Cu/CeO2(aero) | 4.7 | 0.64 | 119 |
| 5Cu/CeO2(aero) | 3.5 | 0.94 | 91 |
| 10Cu/CeO2(aero) | 1.7 | 0.88 | 91 |
| 5Cu/TiO2(aero) | 0.8 | 0.20 | — |
| 10Cu/TiO2(aero) | 1.9 | 0.90 | 120 |
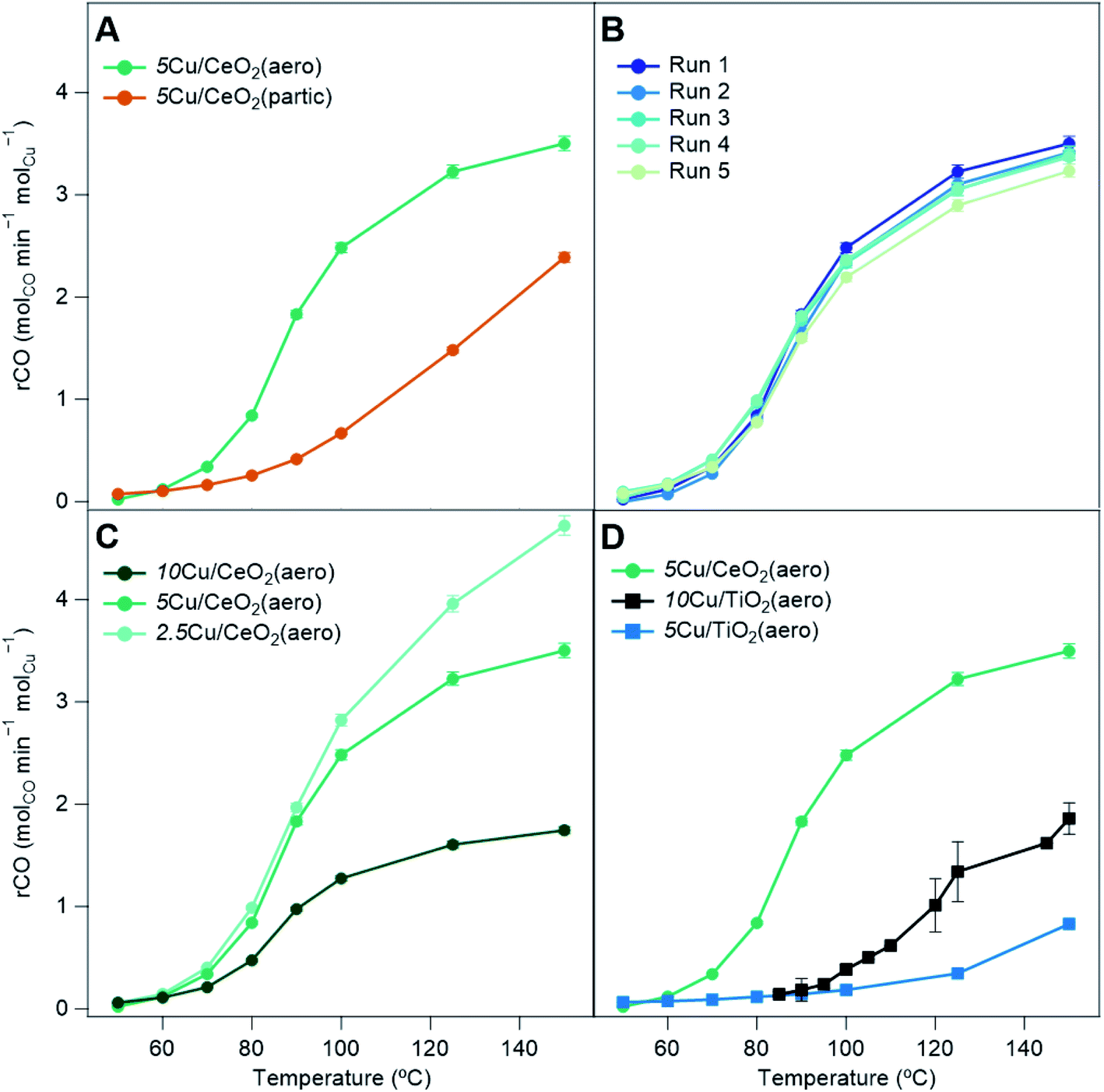 | ||
| Fig. 6 CO conversion as a fraction of mol CO converted per min per mol Cu in catalyst bed versus temperature for (A) catalysts with different CeO2 supports: 5Cu/CeO2(aero) (green) and 5Cu/CeO2(partic) (amber); (B) 5 runs of 5Cu/CeO2(aero) separated by regeneration of the catalyst bed; (C) different weight loading of Cu on CeO2(aero): 10Cu/CeO2(aero) (dark green), 5Cu/CeO2(aero) (green), 2.5Cu/CeO2(aero) (light green); and (D) catalysts with CeO2vs. TiO2 supports: 5Cu/CeO2(aero) (green), 5Cu/TiO2(aero) (blue), 10Cu/TiO2(aero) (black). | ||
We compare the effect of Cu weight loadings on CeO2(aero) for the extent of CO conversion (Fig. 6C). Although 5Cu/CeO2(aero) shows higher CO conversion than 2.5Cu/CeO2(aero), on a Cu-normalized basis, the lower weight loading material is more active (4.7 molCO min−1 molCu−1). Increasing the weight loading to 10 wt% did not impact CO conversion, so the Cu-normalized conversion falls (1.7 molCO min−1 molCu−1). The Cu deposited for 10Cu/CeO2(aero) beyond that deposited for 5 wt% does not contribute to CO oxidation, likely because the additional Cu is mostly oxidized and contained in the large “bicone” structures observed by SEM (Fig. S11†). The large, oxidized Cu regions in 10Cu/CeO2(aero) are evidenced by the lack of an SPR in its absorption spectrum (Fig. 4B) and by the prominent CuO reflections in its XRD pattern (Fig. S5†).
We also contrast the activity of Cu/CeO2 catalysts to both 5Cu/TiO2(aero) and 10Cu/TiO2(aero) catalysts (Fig. 6D). For TiO2-based samples, the catalyst bed comprises 10 mg of active material diluted with 40 mg of TiO2(aero) (and similarly the diluent is a negligible contributor to CO oxidation activity). The pretreatment and data collection procedures are identical to those of the Cu/CeO2 materials. We find little activity for CO conversion with our 5Cu/TiO2(aero): 20% at 150 °C (0.8 molCO min−1 molCu−1) and negligible conversion below 100 °C. The 10Cu/TiO2(aero), the subject of our previous study,32 had a conversion of about 90% at 150 °C (1.9 molCO min−1 molCu−1), but conversion decreased rapidly, falling to below 50% conversion at 120 °C.
The reduction in activity upon halving the Cu weight loading on TiO2 was much more severe than the modest reduction in CO conversion observed for Cu/CeO2(aero). This reduction in activity on TiO2 may suggest that Cu0 and Cu+, which are expected to be the primary drivers of CO oxidation, may be inaccessible or that Cu particles do not contain the appropriate synergistic mixture of Cu0 and Cu+ to aid in turnover.
Despite having lower surface area and lower Cu weight loading, 5Cu/CeO2(aero) shows higher CO conversion than 10Cu/TiO2(aero). When photodepositing Cu, sufficient surface area is required to prevent the formation of Cu aggregates and to ensure the extended interaction between the metal oxide support and the Cu NPs. But with this surface area requirement met in both 5Cu/CeO2(aero) and 10Cu/TiO2(aero), the superior CO oxidation activity shown on the CeO2(aero) supports at a lower Cu weight loading is likely due to the increased reducibility of CeO2. The increased reducibility of CeO2 facilitates generation of oxygen vacancies that activate molecular O2.16–18 Activated oxygen species react with CO bound to Cu+ sites located near the oxide interface20 to facilitate CO2 generation.
3. Conclusions
We show that reduced Cu NPs displaying SPRs are supported on high surface-area CeO2 aerogel, similar to our prior report for Cu supported on TiO2 aerogel. The high surface area of the support is a critical factor in creating the close contact between the Cu NPs and the reducing oxide required to stabilize Cu0. When the surface area of the support relative to Cu content is lower, either by using a lower surface area support or by increasing Cu loading, the fraction of oxidized Cu is higher, and the Cu SPR is not observed. The consequence of the ability of the CeO2 morphology to tune activity is seen in the low activity of Cu/CeO2(partic) for low-temperature CO oxidation. The Cu/CeO2(aero) catalysts far outperform their Cu/TiO2(aero) counterparts for low-temperature CO oxidation even at lower Cu weight loadings or equal mole fractions, showing that the transition to the more reducible CeO2 produces more catalytically active materials. Even with the high surface area CeO2(aero), in the absence of Cu, low-temperature CO oxidation is not observed. The generalization of the Cu/MOx(aero) motif for stabilizing reduced states of Cu offers a design principle for reactions that benefit from these low-valent states.4. Experimental
4.1 Materials
Copper(II) nitrate hemipentahydrate (Cu(NO3)2·2.5H2O, Sigma-Aldrich, 98%), cerium chloride heptahydrate (CeCl3·7H2O, Sigma-Aldrich, 99.9%), (±)-propylene oxide (Sigma-Aldrich, ≥99%), ceria nanopowder (Sigma-Aldrich, <50 nm particles, 99.95%, Lot# MKCD8228), ethanol (absolute ethanol, Warner-Graham), and nitric acid (HNO3, Fisher 69.9%) were used as received. Methanol (Fisher, 99.9%) for CeO2 aerogel synthesis was stored over 3 Å sieves and filtered before use. Ultrapure 18 MΩ cm water (Barnstead Micropure) was used in all syntheses requiring water.4.2 Synthesis of cerium oxide (CeO2) aerogels
Cerium oxide aerogels were prepared by a modification of a literature procedure.21 In a plastic beaker, 2.39 g of CeCl3·7H2O (6.4 mmol) was dissolved in 10 g of anhydrous MeOH, followed by adding 6 g of propylene oxide while stirring. The solution was allowed to stir until a gel formed and then covered and aged overnight. The aged gel was transferred to a jar, washed several times with isopropanol and then acetone. The wet gels were loaded into a supercritical dryer under acetone, and the acetone was then exchanged for liquid CO2 at 10 °C over the course of several rinses. The temperature of the dryer was raised to 42 °C, forming a supercritical CO2 phase and vented slowly to atmospheric pressure. The aerogels were calcined in air at 500 °C (5 °C min−1 ramp, 2 h dwell, 5 °C min−1 cooling ramp) to yield nanocrystalline fluorite CeO2 aerogel.4.3 Deposition of Cu nanoparticles at CeO2 supports
Copper deposition followed a modification of the method used to photodeposit Cu nanoparticles on TiO2 aerogels.6 As an exemplar for 5 wt% Cu loading, a slurry of 100 mg of CeO2 aerogel or commercial CeO2 (0.27 mmol) and 19 mg of Cu(NO3)2·2.5H2O (0.08 mmol, 5.2 mg Cu) in 68 mL 9:1 H2O:EtOH in a Pyrex round bottom flask was brought to pH 9.5 ± 0.5 with aqueous NaOH and HNO3. The solution was sparged with Ar for 30 min prior to irradiating the sample with a 500 W Xe arc lamp (Newport-Oriel) for 48 h. The solids were collected by filtration (0.1 μm polyvinylidene fluoride membrane filter), washed with several aliquots of water, air-dried overnight, and then dried at 70 °C for 16 h.
4.4 Structural, physical and chemical characterization
The Brunauer–Emmett–Teller (BET) surface area and Barrett–Joyner–Halenda (BJH) pore size distributions were derived from the desorption arm of the N2 physisorption isotherms (Micromeritics ASAP2020). The isotherm data were fit with Micromeritics DFTplus software using the Harkins and Jura thickness curve employing the Faas correction. Prior to N2 physisorption, samples were degassed at 150 °C for 10 h. The crystalline phases were characterized using X-ray diffraction (Rigaku SmartLab, 40 kV and 44 mA, 4° min−1 scan rate). The average crystallite diameter was determined via the Scherrer equation using the full width half maximum (FWHM) of the CeO2 (111) diffraction peak at 2θ = 28.55°. Diffuse-reflectance UV-visible spectra (PerkinElmer 750, 60 mm integrating sphere) were converted to absorbance using the Kubelka–Munk transformation. Optical band gaps were determined from Tauc plots by extrapolating the linear portion of the plot near the absorption edge to the energy axis. The X-ray photoelectron spectra (Thermo Scientific K-Alpha, Al-Kα radiation) were recorded using a flood gun to minimize charging on the insulating samples. High-resolution spectra were recorded in the Cu 2p, Ce 3d, and O 1s regions. Peak integration was performed with a Smart background on Avantage software. All peak positions were referenced to the Ce4+ 3d3/2 shake-up peak at 916.7 eV.28 The scanning electron micrographs were recorded using a LEO Supra 55 field-emission microscope operating at 10 keV in secondary electron mode. Transmission electron microscopy was performed in a JEOL JEM2200FS operating at 200 kV for HR-TEM, preliminary STEM-HAADF, and STEM-EDS while an aberration-corrected STEM Nion UltraSTEM200X operating at 200 kV was used for additional STEM-HAADF, STEM-EELS, and STEM-EDS. Samples of Cu/CeO2 were prepared via sonication in isopropyl alcohol with dropcasting onto lacey-carbon supports on gold TEM grids.4.5 Attenuated total reflectance FTIR (ATR)
We prepared suspensions of CeO2 and Cu/CeO2 by sonicating 20 mg of sample in 1.0 mL DI water for 15 min and drop cast 2 μL of each sample onto a ZnSe ATR prism for a loading of 4 mg. To increase wettability of the ZnSe ATR prisms prior to drop casting, we plasma oxidized the clean prism under 310 mTorr and 250 mL min−1 O2 for 5 min (March Fluorine Reactive Ion Etcher). The ATR spectra were monitored with a Nicolet 6700 (ThermoFisher) equipped with a liquid nitrogen-cooled MCT/B detector. Spectra were taken during the fill as well as purging segments with a resolution of 4 cm−1 in the range of 650–4000 cm−1 as a product of a 32-scan average with no attenuation and an aperture setting to 74%.The binding of CO on CeO2 and Cu/CeO2 was investigated at room temperature under anaerobic conditions (10 mL min−1 CO, 25 mL min−1 He). The sample was dried overnight under He flow prior to CO exposure. The feed stream flowed over the sample for 10 min to saturate the catalyst surface, then CO(g) was purged from the system with 25 mL min−1 He for 5 h.
4.6 Carbon monoxide oxidation
Carbon monoxide (CO) oxidation was performed in a glass tube (3/8′′ OD, 0.22′′ ID) continuous-flow packed bed reactor (PBR) housed in a programmable ceramic tube oven. Reactions were conducted under dark conditions. The temperature controller (Digi-Sense 1-Zone, Cole Parmer) was interfaced to a K-type thermocouple affixed to the outer wall of the reactor. The temperature differential between the center of the catalyst bed and the outer wall of the reactor is a possible source of error; however, the thin, 0.15′′, glass-tube reactor wall should not be sufficient to generate a significant temperature gradient. Reaction conditions were chosen such that heat and mass transport effects were inconsequential.32,37,38 The 50 mg catalyst bed was sandwiched between glass wool. We diluted 10 mg of Cu/CeO2 catalyst with 40 mg of native (i.e., Cu-free) CeO2 in order to increase space time and limit conversion.Prior to reaction, the catalyst bed was activated at 150 °C (ramp rate 10 °C min−1, dwell time 30 min) in an anoxic gas stream of 1.25% CO in He (64 mL min−1). These conditions were chosen to minimize ripening, to highlight the ability of the catalyst to function without high-temperature oxidation and reduction treatments, and to facilitate comparison to earlier work.32,35,39 During reaction, the reactor was kept at atmospheric pressure while feeding 80 mL min−1 of 1% CO (Airgas, 10000 ppm), 20% O2 (Keen, 4.4 grade, 200000 ppm), and 79% He (Praxair, 5.0 UHP), giving a GHSV of 39000 h−1. The temperature was decreased stepwise at a ramp rate of 2 °C min−1. Reactor effluent was fed into an in-line GC (GC-2014, Shimadzu) equipped with a Pulsed Discharge Detector for product analysis. Reaction products were analyzed at steady state by allowing the reactor to dwell at each temperature (150 °C, 125 °C, 100 °C, 90 °C, 80 °C, 70 °C, 60 °C, and 50 °C) for 35 min. The durability and aging of the catalyst was tested via four regenerations for a total of 5 cycles.
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors acknowledge funding provided by the Office of Naval Research and the Defense Threat Reduction Agency (DTRA, HDTRA1825787). C. L. P. and A. M. P. gratefully acknowledge the National Research Council for support through a Naval Research Laboratory/National Research Council Postdoctoral Associateship.References
- A. E. Baber, X. Yang, H. Y. Kim, K. Mudiyanselage, M. Soldemo, J. Weissenrieder, S. D. Senanayake, A. Al-Mahboob, J. T. Sadowski, J. Evans, J. A. Rodriguez, P. Liu, F. M. Hoffmann, J. G. Chen and D. J. Stacchiola, Angew. Chem., Int. Ed., 2014, 53, 5336–5340, DOI:10.1002/anie.201402435.
- S. D. Senanayake, N. A. Pappoe, T.-D. Nguyen-Phan, S. Luo, Y. Li, W. Xu, Z. Liu, K. Mudiyanselage, A. C. Johnston-Peck, A. I. Frenkel, I. Heckler, D. Stacchiola and J. A. Rodriguez, Surf. Sci., 2016, 652, 206–212, DOI:10.1016/j.susc.2016.02.014.
- D. Vovchok, C. J. Guild, J. Llorca, W. Xu, T. Jafari, P. Toloueinia, D. Kriz, I. Waluyo, R. M. Palomino, J. A. Rodriguez, S. L. Suib and S. D. Senanayake, Phys. Chem. Chem. Phys., 2017, 19, 17708–17717, 10.1039/c7cp02378b.
- Y. Bu, S. Er, J. W. Niemantsverdriet and H. O. A. Frederiksson, J. Catal., 2018, 357, 176–187, DOI:10.1016/j.jcat.2017.11.014.
- Y. Zhang, L. Liang, Z. Chen, J. Wen, W. Zhong, S. Zou, M. Fu, L. Chen and D. Ye, Appl. Surf. Sci., 2020, 516, 146035, DOI:10.1016/j.apsusc.2020.146035.
- P. A. DeSario, J. J. Pietron, T. H. Brintlinger, M. McEntee, J. F. Parker, O. Baturina, R. M. Stroud and D. R. Rolison, Nanoscale, 2017, 9, 11720–11729, 10.1039/c7nr04805j.
- D. Gamarra, A. López Cámara, M. Monte, S. B. Rasmussen, L. E. Chinchilla, A. B. Hungría, G. Munuera, N. Gyorffy, Z. Schay, V. Cortés Corberán, J. C. Conesa and A. Martínez-Arias, Appl. Catal., B, 2013, 130–131, 224–238, DOI:10.1016/j.apcatb.2012.11.008.
- M. Lykaki, E. Pachatouridou, S. A. C. Carabineiro, E. Iliopoulou, C. Andriopoulou, N. Kallithrakas-Kontos, S. Boghosian and M. Konsolakis, Appl. Catal., B, 2018, 230, 18–28, DOI:10.1016/j.apcatb.2018.02.035.
- C. Wang, Q. Cheng, X. Wang, K. Ma, X. Bai, S. Tan, Y. Tian, T. Ding, L. Zheng, J. Zhang and X. Li, Appl. Surf. Sci., 2017, 422, 932–943, DOI:10.1016/j.apsusc.2017.06.017.
- L. Du, W. Wang, H. Yan, X. Wang, Z. Jin, Q. Song, R. Si and C. Jia, J. Rare Earths, 2017, 35, 1186–1196, DOI:10.1016/j.jre.2017.04.005.
- R. Si, J. Raitano, N. Yi, L. Zhang, S.-W. Chan and M. Flytzani-Stephanopoulos, Catal. Today, 2012, 180, 68–80, DOI:10.1016/j.cattod.2011.09.008.
- X. Wang, J. A. Rodriguez, J. C. Hanson, D. Gamarra, A. Martínez-Arias and M. Fernández-García, J. Phys. Chem. B, 2006, 110, 428–434, DOI:10.1021/jp055467g.
- K. Mudiyanselage, S. D. Senanayake, L. Feria, S. Kundu, A. E. Baber, J. Graciani, A. B. Vidal, S. Agnoli, J. Evans, R. Chang, S. Axnanda, Z. Liu, J. F. Sanz, P. Liu, J. A. Rodriguez and D. J. Stacchiola, Angew. Chem., Int. Ed., 2013, 52, 5101–5105, DOI:10.1002/anie.201210077.
- D. Delimaris and T. Ioannides, Appl. Catal., B, 2009, 89, 295–302, DOI:10.1016/j.apcatb.2009.02.003.
- C. Hu, Q. Zhu, Z. Jiang, Y. Zhang and Y. Wang, Microporous Mesoporous Mater., 2008, 113, 427–434, DOI:10.1016/j.micromeso.2007.11.043.
- M. Konsolakis, Appl. Catal., B, 2016, 198, 49–66, DOI:10.1016/j.apcatb.2016.05.037.
- A. Migani, G. N. Vayssilov, S. T. Bromley, F. Illas and K. M. Neyman, Chem. Commun., 2010, 46, 5936–5938, 10.1039/c0cc01091j.
- A. R. Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, ACS Catal., 2017, 7, 6493–6513, DOI:10.1021/acscatal.7b01913.
- Y. Su, Z. Tang, W. Han, Y. Song and G. Lu, Catal. Surv. Asia, 2015, 19, 68–77, DOI:10.1007/s10563-015-9185-3.
- A. Chen, X. Yu, Y. Zhou, S. Miao, Y. Li, S. Kuld, J. Sehested, J. Liu, T. Aoki, S. Hong, M. F. Camellone, S. Fabris, J. Ning, C. Jin, C. Yang, A. Nefedov, C. Wöll, Y. Wang and W. Shen, Nat. Catal., 2019, 2, 334–341, DOI:10.1038/s41929-019-0226-6.
- C. Laberty-Robert, J. W. Long, E. M. Lucas, K. A. Pettigrew, R. M. Stroud, M. S. Doescher and D. R. Rolison, Chem. Mater., 2006, 18, 50–58, DOI:10.1021/cm051385t.
- V. Ramasamy and G. Vijayalakshmi, Mater. Sci. Semicond. Process., 2016, 42, 334–343, DOI:10.1016/j.mssp.2015.10.026.
- N. S. Arul, D. Mangalaraj, P. C. Chen, N. Ponpandian and C. Viswanathan, Mater. Lett., 2011, 65, 2635–2638, DOI:10.1016/j.matlet.2011.05.022.
- A. Corma, P. Atienzar, H. García and J.-Y. Chane-Ching, Nat. Mater., 2004, 3, 394–397, DOI:10.1038/nmat1129.
- G. J. Wu, N. J. Guan and L. D. Li, Catal. Sci. Technol., 2011, 1, 601–608, 10.1039/c1cy00036e.
- E. Cottancin, G. Celep, J. Lermé, M. Pellarin, J. R. Huntzinger, J. L. Vialle and M. Broyer, Theor. Chem. Acc., 2006, 116, 514–523, DOI:10.1007/s00214-006-0089-1.
- D. Tahir and S. Tougaard, J. Phys.: Condens. Matter, 2012, 24, 175002, DOI:10.1088/0953-8984/24/17/175002.
- M. Romeo, K. Bak, J. El Fallah, F. Le Normand and L. Hilaire, Surf. Interface Anal., 1993, 20, 508–512, DOI:10.1002/sia.740200604.
- C. M. Sims, R. A. Maier, A. C. Johnston-Peck, J. M. Gorham, V. A. Hackley and B. C. Nelson, Nanotechnology, 2019, 30, 085703, DOI:10.1088/1361-6528/aae364.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334, DOI:10.1002/sia.6239.
- J. P. Espinós, J. Morales, A. Barranco, A. Caballero, J. P. Holgado and A. R. González-Elipe, J. Phys. Chem. B, 2002, 106, 6921–6929, DOI:10.1021/jp014618m.
- P. A. DeSario, C. L. Pitman, D. J. Delia, D. M. Driscoll, A. J. Maynes, J. R. Morris, A. M. Pennington, T. H. Brintlinger, D. R. Rolison and J. J. Pietron, Appl. Catal., B, 2019, 252, 205–213, DOI:10.1016/j.apcatb.2019.03.073.
- K. Mudiyanselage, H. Y. Kim, S. D. Senanyake, A. E. Baber, P. Liu and D. Stacchiola, Phys. Chem. Chem. Phys., 2013, 15, 15856–15862, 10.1039/c3cp52295d.
- C. Binet, M. Daturi and J.-C. Lavalley, Catal. Today, 1999, 50, 207–225, DOI:10.1016/s0920-5861(98)00504-5.
- C.-S. Chen, J.-H. You, J.-H. Lin and Y.-Y. Chen, Catal. Commun., 2008, 9, 2381–2385, DOI:10.1016/j.catcom.2008.06.003.
- F. Boccuzzi, A. Chiorino, G. Martra, M. Gargano, N. Ravasio and B. Carrozzini, J. Catal., 1997, 165, 129–139, DOI:10.1006/jcat.1997.1475.
- C. S. Chen, T. C. Chen, C. C. Chen, Y. T. Lai, J. H. You, T. M. Chou, C. H. Chen and J.-F. Lee, Langmuir, 2012, 28, 9996–10006, DOI:10.1021/la301684h.
- B. Schumacher, Y. Denkwitz, V. Plzak, M. Kinne and R. J. Behm, J. Catal., 2004, 224, 449–462, DOI:10.1016/j.jcat.2004.02.036.
- B. Schumacher, V. Plzak, M. Kinne and R. J. Behm, Catal. Lett., 2003, 89, 109–114, DOI:10.1023/a:1024731812974.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional porosimetry details, XRD and UV-Vis characterization, SEM and TEM images, catalytic runs. See DOI: 10.1039/d0na00594k |
| ‡ Lawrence Livermore National Lab, 7000 East Avenue, Livermore, CA, 94550. |
| This journal is © The Royal Society of Chemistry 2020 |