
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the binding behaviors of a protein YAP65WW domain on graphenic nano-sheets with boron or nitrogen atom doping†
Xiao
Jia
a,
Yanmei
Yang
 *b,
Yang
Liu
a,
Weihua
Niu
b,
Yong-Qiang
Li
a,
Mingwen
Zhao
a,
Yuguang
Mu
*c and
Weifeng
Li
*a
*b,
Yang
Liu
a,
Weihua
Niu
b,
Yong-Qiang
Li
a,
Mingwen
Zhao
a,
Yuguang
Mu
*c and
Weifeng
Li
*a
aSchool of Physics, State Key Laboratory of Crystal Materials, Shandong University, Jinan, Shandong 250100, China. E-mail: lwf@sdu.edu.cn
bCollege of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Shandong Normal University, Jinan, 250014, China. E-mail: yym@sdnu.edu.cn
cSchool of Biological Sciences, Nanyang Technological University, 637551, Singapore. E-mail: ygmu@ntu.edu.sg
First published on 26th August 2020
Abstract
In recent years, nanomaterials have attracted considerable research attention for biological and medical related applications due to their well-recognized physical and chemical properties. However, the deep understanding of the binding process at the protein–nanomaterial interface is essential to solve the concern of nano-toxicity. Here, we study the interactions between the recently reported graphenic nano-sheets, BC3 and C3N, and a prototypical protein (YAP65WW domain) via atomistic molecular dynamics simulations. Our simulations reveal that elemental doping is an effective way to tune the binding characteristics of YAP65WW with two nanomaterials. While YAP65WW can be attracted by two nanomaterials, the BC3 sheet is less able to disrupt the protein structure than C3N. From the energy point of view, this is because protein residues demonstrate a binding preference with the trend from electron rich nitrogen to electron deficient boron. Structural analyses of the bio-nano interface revealed the formation of an ordered water shell on the BC3 surface, which was compatible to the crystal pattern of BC3. When a protein binds with BC3, these interfacial water molecules protect the protein from being disrupted. We suggest that elemental doping is efficient to produce fruitful biological-effects of graphenic nanomaterials, which make it a prospective solution for the future design and fabrication of advanced nanomaterials with desired function.
1. Introduction
In recent years, graphene has attracted considerable research interest in numerous fields such as electronic1,2 and optical materials,3,4 catalytic agents, sensors and biomedical materials5 due to its well-recognized physical and chemical properties. Particularly, in biological and medical applications, graphene has been widely studied in tumor therapy,6 biosensors7 and gene sequencing platforms.8,9 However, the major concern of the potential nano-toxicity has been attracting enormous attention of researchers worldwide. Numerous studies have shown that graphene can cause severe damage to biomacromolecules, such as protein unfolding, DNA unwinding, and destructive extraction of the cell membrane.10–14 For instance, our previous theoretical simulations demonstrate that when a graphene monolayer has defects, the protein unfolding speed is significantly accelerated by specific interactions between defects and charged residues.15,16 To reduce the toxicity, surface morphology engineering17–19 and functionalization with polyethylene glycol or serum proteins have been demonstrated to be a success.20 Meanwhile, our previous studies have demonstrated that a nitrogenized graphene (abbreviated as C2N) monolayer is biocompatible to both protein and DNA systems.21,22 Thus, modifying the graphenic structures by elemental doping is expected to be an efficient way to tune the biological functions of the nanomaterials.Recently, the successful experimental syntheses of boron-doped graphene, BC3 (ref. 23) and nitrogen-doped graphene, C3N24 have been achieved. BC3 and C3N monolayers have excellent material chemistry properties and are used in batteries25–27 and high sensitive gas sensors.28–32 Structurally, the B and N atoms are arranged in a patterned manner in graphenic nanosheets. Each B or N atoms is covalently connected to three C atoms (Fig. 1a). Due to the electronegativity difference between the three constitute elements, B, C and N, there is an intrinsic electron transfer within the planar structure of BC3 and C3N, resulting in periodically distributed dipoles. A recent theoretical study on the interactions of BC3 and water clearly showed that BC3 induced a significant re-arrangement of water dipoles at the interface, making BC3 a promising candidate for the transport of drug species.33 Thus, element doping to graphene is expected to bring out interesting behaviors at the nano-bio interface compared to the non-polar graphene.
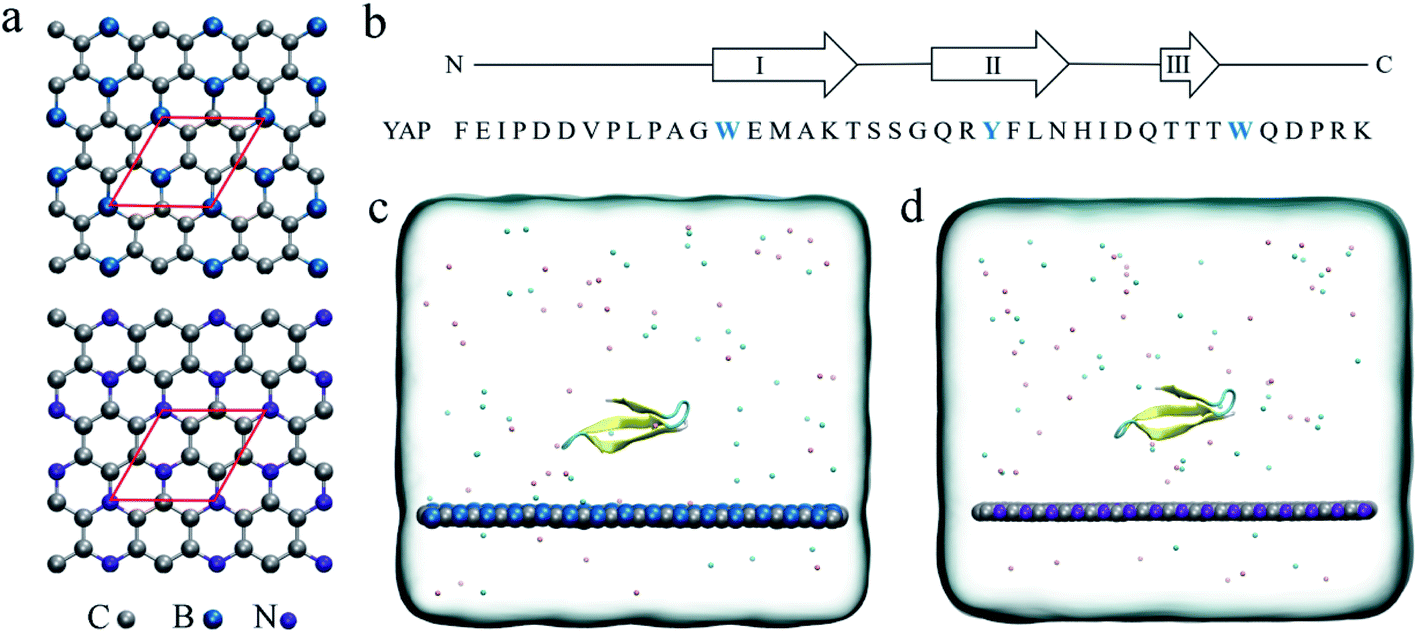 | ||
| Fig. 1 (a) Crystal structures of the BC3 and C3N monolayer. The red rhombus indicates the primitive cell of BC3/C3N. (b) The sequence of the YAP65WW domain. The positions of strands I–III, connected by hairpins, are indicated above. The initial configurations of the YAP65WW domain on the surface of BC3 (c) and C3N (d) in the simulation box. Carbon, boron and nitrogen atoms are colored in silver, blue and violet, respectively. Na+ and Cl− ions are shown as pink and cyan spheres. Water molecules are not shown for clarity. | ||
In this study, all-atom molecular dynamics (MD) simulations have been conducted to explore the dynamic and energetic characteristics of a prototypical protein, YAP65WW domain, binding with BC3 and C3N nanosheets. The adsorption processes and structure evolutions of the protein were systematically analyzed and compared. The simulation results revealed that both BC3 and C3N were capable of attacking YAP65WW. It is interesting to find that C3N is more capable of inducing the structural unfolding of YAP65WW. This mechanism is because of the binding preference of the protein with the trend from electron-rich nitrogen to electron-deficient boron. We believe that the perspective insights obtained from our study would be beneficial to guide and potentiate the practical applications of graphenic materials and bring about a flourishing new branch in the design of functional bio-nano materials.
2. Computational methods
The initial coordinates of YAP65WW were obtained from the crystal structure (PDB code 1JMQ34 and truncated to include residues 15–40 (ref. 16)). We selected YAP65WW as a model protein because it has been widely used in the study of protein folding kinetics and the interactions with nanomaterials.35–42 As shown in Fig. 1b, YAP65WW is a signaling and regulatory protein, which exists as a three-stranded, antiparallel β-sheet topology.43–46 The BC3 nano-sheet has dimensions of 7.76 × 8.06 nm2 and is composed of 1620 carbon and 540 boron atoms. Similarly, C3N contains 1620 carbon and 540 nitrogen atoms with dimensions of 7.29 × 7.59 nm2. The nano-sheet was placed in the xy plane and the cross-section of the simulation box was equal to the size of the nano-sheet. Following our previous study,21 the positions of C3N and BC3 nano-sheets were restrained using a harmonic potential with a force constant of 1000 kJ mol−1 nm−2 in all the simulations to mimic a local surface in the whole sample. As depicted in Fig. 1c and d, YAP65WW was then placed above the nano-sheet with a separation of 2 nm (measured from the center of mass). Then, the complex was solvated in a water box with a height of 7 nm. Periodic boundary conditions were applied on the three directions. For water, the SPC/E water model47,48 was used. Then, 40 Na+ and 42 Cl− ions were added to neutralize the net charge of YAP65WW and mimic the physiological condition.The MD simulations were performed using the GROMACS package.49 The YAP65WW was described by the AMBER99sb force field.50 For the two nano-sheets, the force field parameters were adopted from our previous work.18 The long-range electrostatic interactions were treated using the particle mesh Ewald (PME) method.51,52 The van der Waals (vdW) interactions were calculated with a cutoff distance of 1.2 nm. The covalent bonds involving hydrogen atoms were constrained by the LINCS algorithm.53 After energy minimization, both systems were equilibrated for 1 ns in the NVT ensemble using the v-rescale thermostat at 300 K,54 and simulated for another 1 ns in the NPT ensemble with a pressure coupling of 1 bar at the z-direction of the simulation box and temperature of 300 K using the Parrinello–Rahman coupling method. During these two stages, position restrains were applied to the heavy atoms of the protein. Then, 1000 ns production simulations were conducted in the NVT ensemble at 300 K. For each nano-sheet, three parallel simulations with different protein orientations and initial velocities were generated for data analysis. In addition to the BC3 and C3N simulations, three control simulations of YAP65WW in water solvent have also been conducted with similar setups to verify the intrinsic stability of protein itself.
3. Results and discussions
3.1 Quick loading of YAP65WW onto BC3 and C3N
First, we accessed the binding behavior of YAP65WW with two substrates. Initially, YAP65WW was placed above each substrate with a distance of 2 nm. As summarized in Fig. 2, the separation dropped to around 1.2 nm during the first tens of nanoseconds in the simulations (stage one), indicating the quick loading of the protein to the substrates. Then, the separations uniformly decreased to around 0.55 to 0.60 nm in three trajectories after hundred nanoseconds of simulations (stage two), although the specific time differed in different trajectories. A similar tendency was also observed when monitoring the number of contacts (Nc) between the protein and the substrates. Along with the decrease in distance, YAP65WW established firm contacts with the two substrates. A pair of atoms from the protein and BC3/C3N, respectively, separated within 0.6 nm was defined as a contacting pair. The Nc profiles first quickly increased to a plateau of around 80 within 100 ns in two simulations, except that it took a slightly longer time (∼150 ns) to reach this plateau in the third trajectory for C3N, corresponding to the first stage in the distance analyses. Then, in the second stage, YAP65WW adjusted its conformation until the end of the simulations. It is worth noticing that YAP65WW demonstrated different adsorption behaviors on BC3 and C3N at the end of the simulations. In detail, there are 124.38 ± 2.64 and 157.90 ± 4.61 atomic contacting pairs (each value is averaged over three trajectories) for binding of YAP65WW to BC3 and C3N, respectively, which accounted for 57.58 ± 1.22% and 73.10 ± 2.13% of heavy atoms of the protein. From this phenomenon, it is expected that C3N is energetically more attractive to YAP65WW because more intimate bindings are established.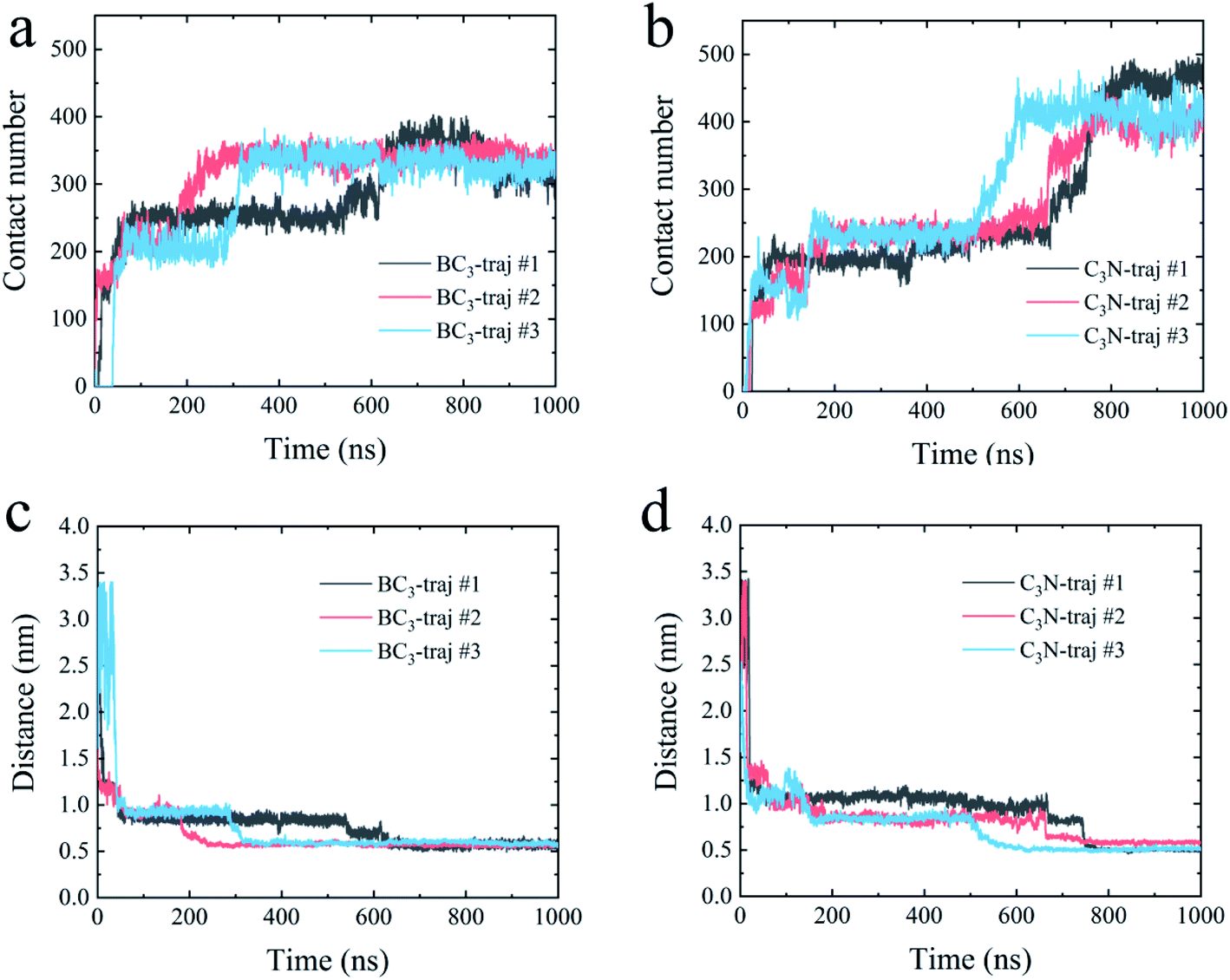 | ||
| Fig. 2 The contact number between the YAP65WW and BC3 (a) and C3N (b); the separation distance between the center of mass of YAP65WW and BC3 (c) and C3N (d) along the normal directions of the nanosheets. | ||
3.2 Structure disruption of YAP65WW and key residues at the interface
Accompanied by the binding, obvious structure changes were observed for YAP65WW on two nano-sheets. Fig. 3 depicts eight representative snapshots for YAP65WW binding with BC3 and C3N. At the early binding stage, YAP65WW approaches to the BC3 atoms at ∼7 ns (Fig. 3a) through five residues, S10, G11, Q12, R13 and Q26 (the contacting time is determined by monitoring the Nc of each residue with BC3/C3N, as depicted in Fig. S1 in the ESI†). Then, the protein laid down with its β-sheets stacking parallel to the BC3 surface (Fig. 3b). Three aromatic key residues, W3, F15 and W25, were found to form intimate π–π stacking55 with BC3, which caused a quick flipping over for the strand-3 and more residues (S9, N17 and I19) adsorbed on the BC3 surface (Fig. 3c). Then, the protein was found to undergo a severe unfolding process (Fig. 3d).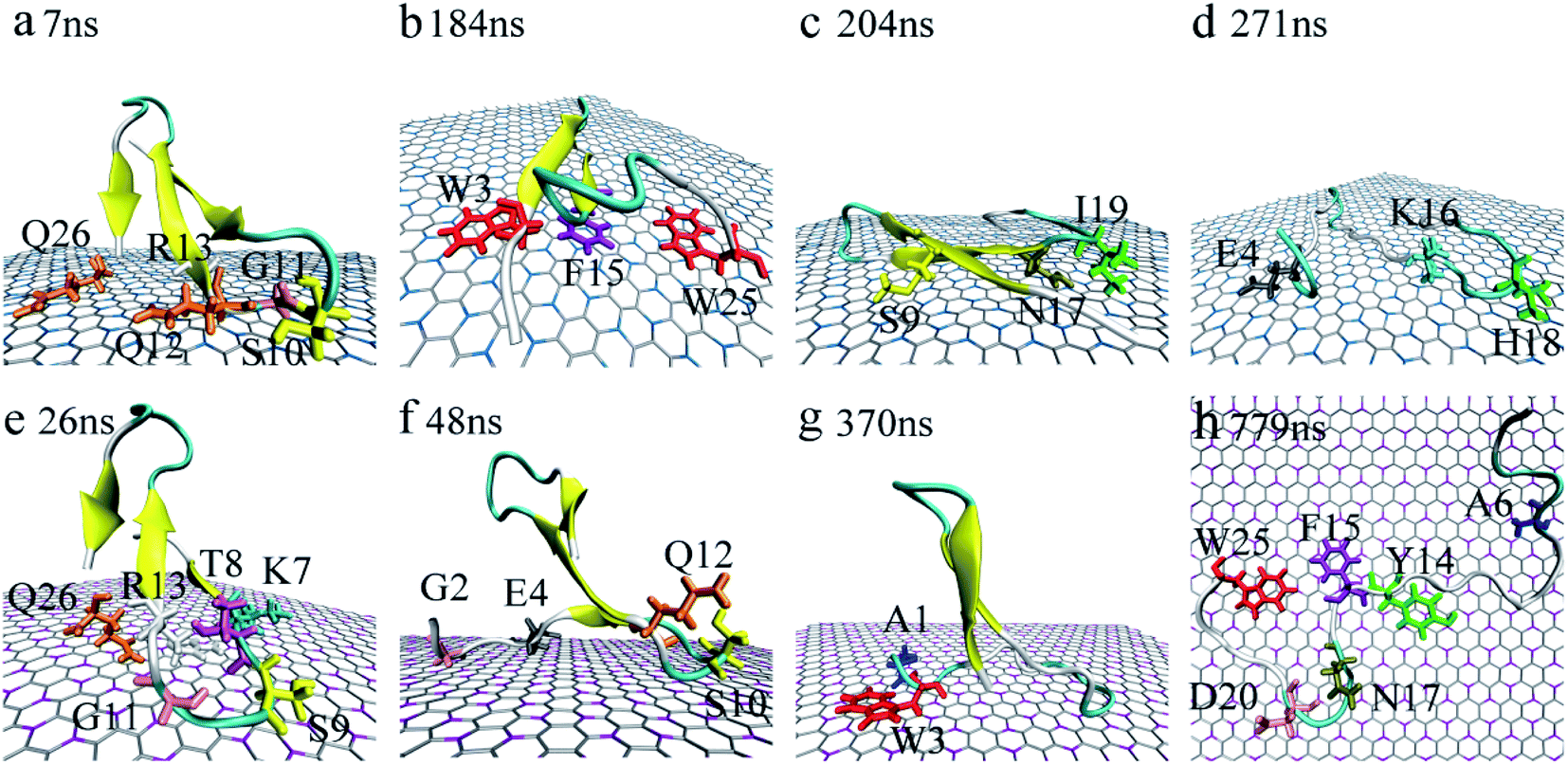 | ||
| Fig. 3 Structure evolution of the YAP65WW domain during the adsorption process on the surface of BC3 (a–d) and C3N (e–h). Residues that play key roles at the bio-nano interface are highlighted. | ||
While for C3N, YAP65WW contacted to C3N at 26 ns by the K7, T8, S9, G11, R13 at the β-hairpin region and Q26 (Fig. 3e). At about 48 ns, the strand-1 of YAP65WW formed firm contacts with C3N through G2, E4, S10 and Q12 (Fig. 3f). The unfolding of the β-sheets happened at ∼370 ns at strand-1 (Fig. 3g). At around 749 ns, YAP65WW completely unfolded (Fig. 3h) with most residues (A6, Y14, F15, N17, D20 and W25) adsorbed on the C3N surface. Despite the different binding processes, it is notable that the π–π stacking interactions between the aromatic residues and the substrate appear in both systems. As shown in Fig. S2,† the interfacial water molecules were squeezed once the aromatic residues approached, resulting in nano-scale dewetting. From previous studies, the π–π stacking is commonly found in the interactions of graphenic nanomaterials with aromatic groups of biomolecules, resulting in severe structural distorsions.15,16,55–58
We further quantitatively accessed the structural evolution of the protein by calculating the root mean squared deviation (RMSD) of YAP65WW with respect to the crystal structure (only heavy atoms were used in the calculations). As summarized in Fig. 4, the RMSD profiles first increased to around 0.5 nm, particularly for YAP65WW with C3N, revealing a slight conformational change from the native structure at the early binding stage. For YAP65WW binding to BC3, the totally unfolded state has an RMSD value of around 0.75 nm (Fig. 4a). In contrast, for the case of YAP65WW on C3N, RMSD reached 0.8, 1.0 and 1.75 in three trajectories, indicating that the protein structure change is more severe (Fig. 4b). In the control simulations of an isolate YAP65WW in water, the protein maintained a highly ordered native structure with no obvious unfolding being observed (Fig. S3†). This confirms that the unfolding of YAP65WW in the BC3 and C3N simulations is mainly caused by the nano-surface.
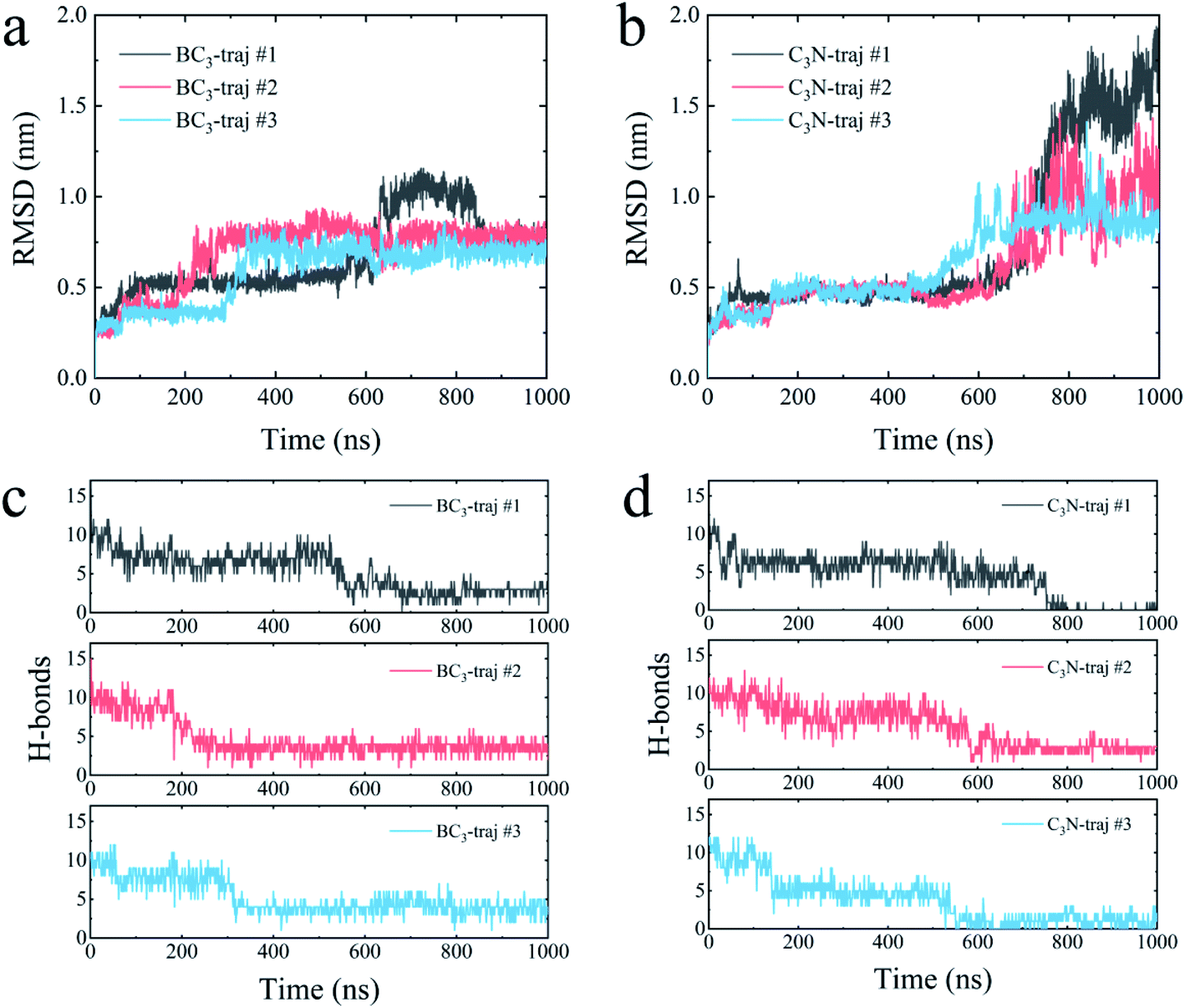 | ||
| Fig. 4 Time evolutions of the root mean square deviation (RMSD) of the YAP65WW heavy atom relative to the crystal structure upon binding to BC3 (a) and C3N (b). Time evolution of number of intra-protein H-bonds upon binding to BC3 (c) and C3N (d). | ||
It is well known that the spatial structure of protein is mainly maintained by the intra-protein hydrogen bonds (H-bonds). During the unfolding process, a clear decrease in the number of H-bonds was observed. As shown in Fig. 4c, the H-bonds decreased from 17 for native YAP65WW to 2.89 ± 0.56, 3.57 ± 0.78 and 3.77 ± 0.99 after 1000 ns simulations for YAP65WW binding to BC3. However, for C3N (Fig. 4d), relatively less H-bonds were maintained and the corresponding values were 0.10 ± 0.30, 2.71 ± 0.56 and 0.95 ± 0.83. This phenomenon confirms that C3N causes more severe YAP65WW disruptions than BC3. It is also worth mentioning that, although BC3 induced a smaller ratio of structure loss in YAP65WW, the beginning of YAP65WW unfolding on the BC3 surface was earlier than that on C3N.
3.3 Secondary structure evolution of YAP65WW
The biological functions of proteins mainly depend on the secondary structure. Considering this, the in-depth secondary structure analyses of YAP65WW were further conducted using the define secondary structure of protein (DSSP)59 program, as summarized in Fig. 5. At the native conformation, YAP65WW contains three antiparallel β-sheet segments: β-sheet 1 (residues 2–8), β-sheet 2 (residues 12–18), and β-sheet 3 (residues 21–25). These three segments were initially preserved during the first 200–300 nanosecond simulations, after which the β-sheets almost disappeared except that small segments showed a dynamic manner of unfolding and folding in the first trajectory of the YAP65WW–BC3 system and the second trajectory of the YAP65WW–C3N system. At the end of all the trajectories, no obvious segments with ordered secondary structures were maintained.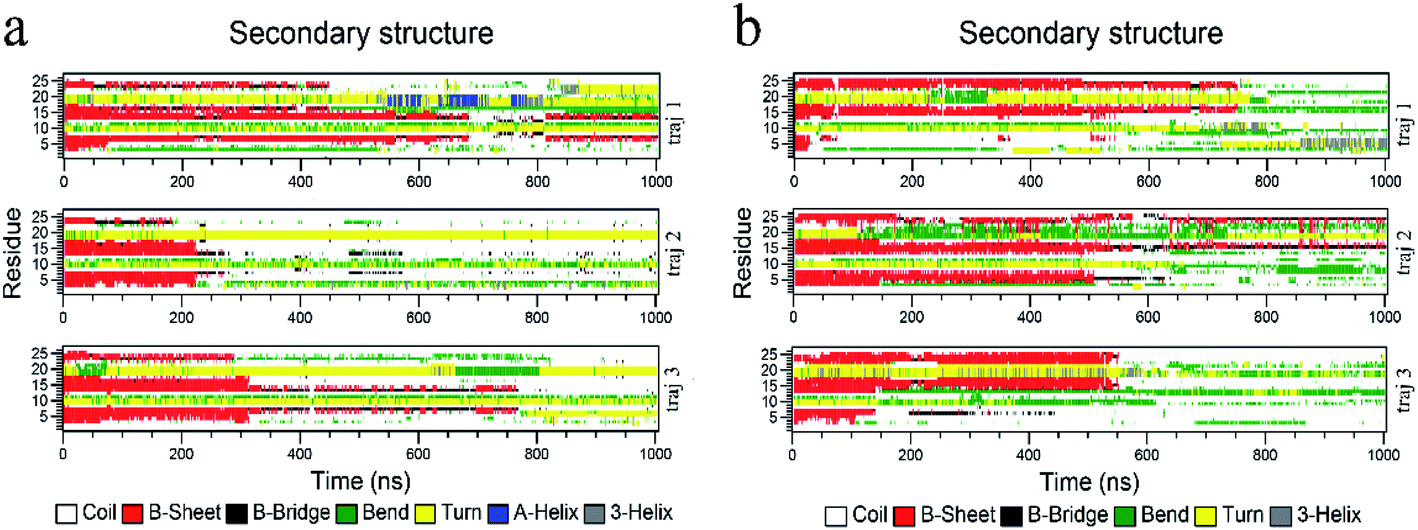 | ||
| Fig. 5 Time evolution of the secondary structure of YAP65WW upon binding to BC3 (a) and C3N (b). The calculations of the secondary structures were conducted by the do_dssp module implemented in the GROMACS package. | ||
3.4 Transverse diffusion of YAP65WW on BC3 and C3N
From the above analyses, both BC3 and C3N are found to be rather attractive to protein because the YAP65WW binding was found to be constantly preserved throughout the simulations. However, differences were also observed between the two substrates. In Fig. 6, we plotted the transverse diffusion of YAP65WW on the two surfaces, which revealed distinct behaviors. For BC3, the diffusion of protein is relatively slow, as shown in Fig. 6a–c. During the 1000 ns simulations, the protein only travelled to certain regions of the BC3 surface. In contrast, during the 1000 ns simulations, the trajectories of YAP65WW almost covered the C3N surface, as shown in Fig. 6d–f. This reveals that the transverse diffusion of protein on the BC3 surface is, to a certain extent, restricted. To further understand the reasons for the different diffusivities, we quantitatively analyzed the bio-nano interface. In Fig. S4 in the ESI,† we plotted the distributions of surface water on BC3 and C3N, respectively. It was found that water molecules on BC3 form a highly ordered structure, which is compatible with the crystal pattern of BC3. On the top of hexagonal carbon rings, obvious water clusters are found with high densities. When protein binds with BC3, these localized water clusters will definitely hinder the protein diffusion and protect protein from being denatured.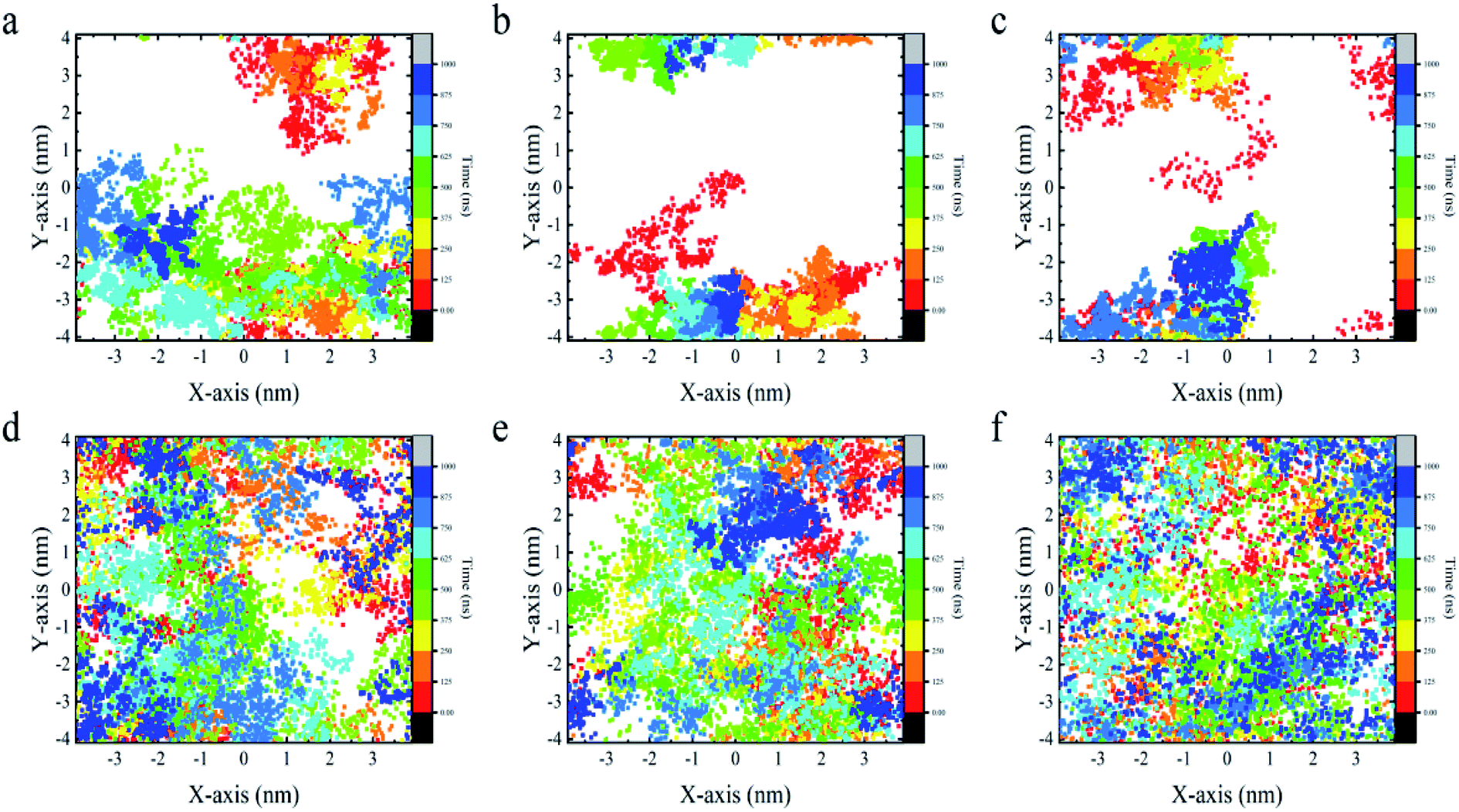 | ||
| Fig. 6 Transverse diffusion of YAP65WW on the surface of BC3 (a–c) and C3N (d–f) from three parallel trajectories for each substrate. Each dot in the figure depicts the position of the center of mass of protein at certain time. | ||
3.5 Energetic characteristics of YAP65WW binding to BC3 and C3N
To dissect the origins of the driving force for the YAP65WW and BC3/C3N binding, we have analyzed the interaction energy components from: (i) YAP65WW interacts with substrate and (ii) YAP65WW interacts with water. As shown in Fig. 7a, the adsorptions of YAP65WW to BC3 and C3N are both exergonic. The YAP65WW–C3N binding releases more energy (averagely 752.20 kJ mol−1) than the YAP65WW–BC3 (averagely 589.15 kJ mol−1), indicating more intimate binding of the former. Particularly, for YAP65WW–C3N from 500–800 ns, sharp decreases can be observed for the energy profiles that are consistent with the Nc analyses in Fig. 2a.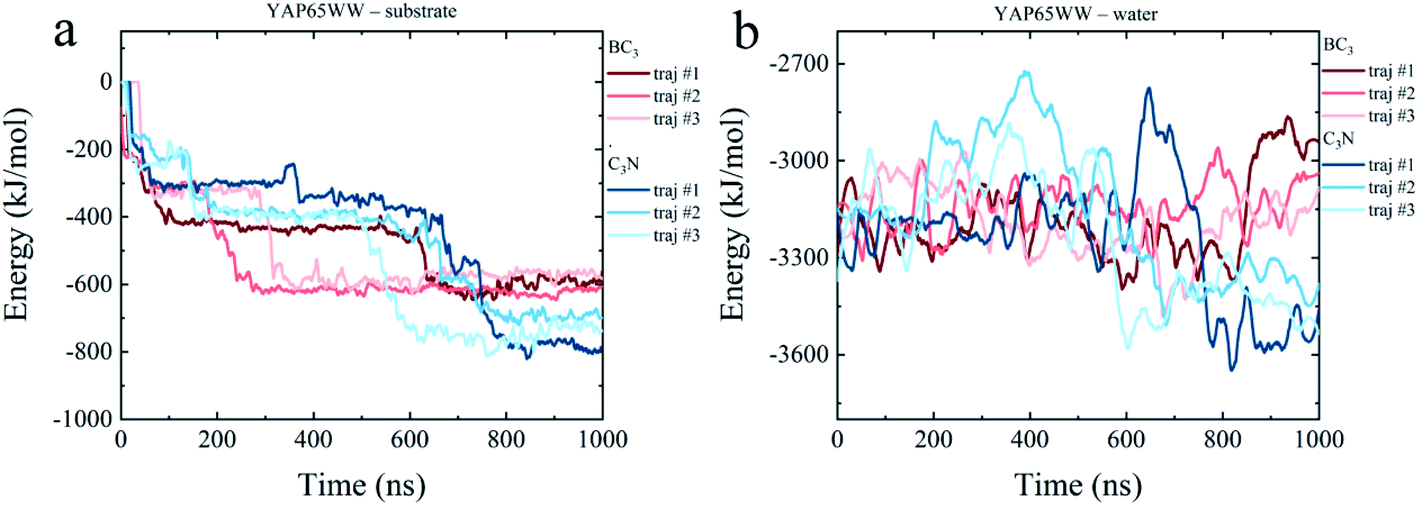 | ||
| Fig. 7 Time evolutions of interaction energy terms of (a) YAP65WW – substrate and (b) YAP65WW – water. | ||
The energy term representing the direct interactions of YAP65WW and substrate drives the binding process. In general, the energy released from binding partially compensates the increase in intra-protein potential energy that maintains the folded state. In addition, the interaction energy of the protein with water molecules (abbreviated as Ehydra) also changes because of the conformational unfolding upon adsorption. As summarized in Fig. 7b, for the two systems, the time evolutions of two Ehydra are found to be quite different. Particularly, it is interesting to find a clear energy decrease for YAP65WW – water in the C3N system at 400–700 ns. We attribute this phenomenon to the more severe unfolding of YAP65WW on C3N than that on BC3, as revealed by the RMSD in Fig. 4a and b. This resulted in more residues exposed to water, which lower the interaction energy to water solvent. This is further supported by monitoring the contacting water around YAP65WW.
To provide a deep understanding of the atomic contribution to the overall binding energy, we plotted the dependence of interaction energy between YAP65WW and substrate with respect to number of contacts (Nc) between them (only heavy atoms are considered). As summarized in Fig. 8, a clear linear dependence of the binding strength on Nc was observed. Second, the larger slopes of the C3N–protein represent a stronger binding strength than BC3–protein. This reflects that C3N is more attractive towards protein, which is consistent to Fig. 7a. Further decomposition of total interaction energy to the elemental level (Fig. S5†) indicates a binding preference with the trend from nitrogen boron. Based on these observations, it is believed that elemental doping can efficiently regulate the interactions of graphenic materials with biomacromolecules, which deserve further experimental efforts.
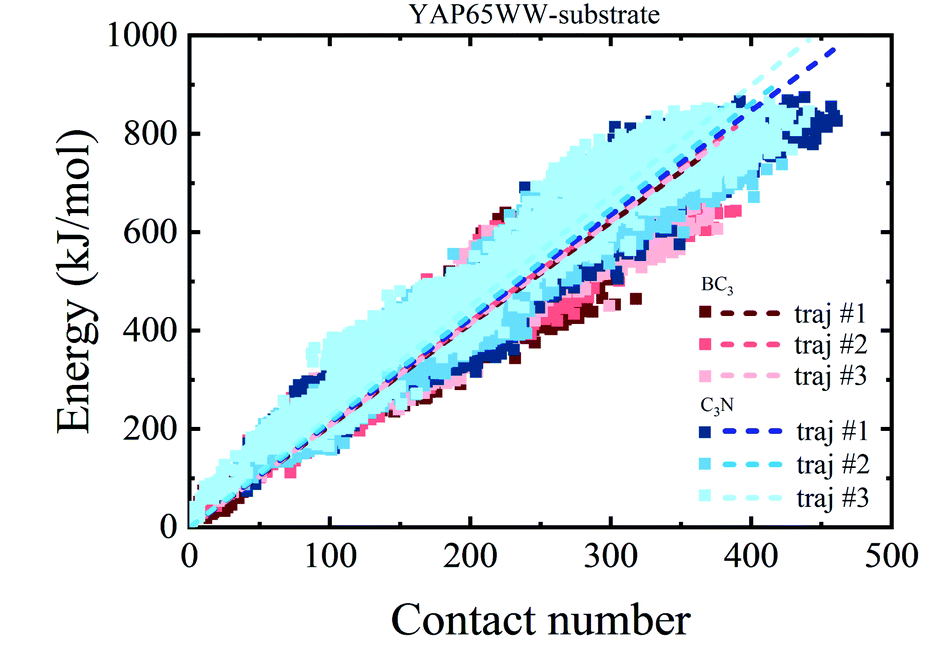 | ||
| Fig. 8 Relationship between the interaction energy and number of contacts between YAP65WW and substrate (the dash lines indicate linear fit of all data from whole trajectories). | ||
4. Conclusion
Using MD simulations, the characteristics of the binding process and structural evolution of protein with graphenic layered materials, B3C and C3N, were systematically studied. The effects of the elemental doping of boron and nitrogen atoms were compared. Our data clearly revealed that both BC3 and C3N are rather attractive to protein. The binding further induced distortions in protein secondary structures. More importantly, C3N demonstrated a stronger capability to destroy the protein structure than BC3. In terms of interaction energy, the binding preference of protein follows the trend from electron-rich nitrogen to electron-deficient boron. Localized high density water clusters are formed on the B3C surface, which can effectively hinder the transverse diffusion of the protein on it. These findings could be useful to guide the design and fabrication of novel layered nanomaterials with fruitful and tunable biological effects through the elemental doping technique.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant no. 11874238), the Basic Research Project of Natural Science Foundation of Shandong Province (Grant no. ZR2018MA034) and Fundamental Research Funds of Shandong University.References
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- F. Schwierz, Nat. Nanotechnol., 2010, 5, 487–496 CrossRef CAS.
- U. Kürüm, O. Ö. Ekiz, H. G. Yaglioglu, A. Elmali, M. Ürel, H. Güner, A. K. Mızrak, B. Ortaç and A. Dâna, Appl. Phys. Lett., 2011, 98, 141103 CrossRef.
- K. V. Sreekanth, S. Zeng, J. Shang, K. T. Yong and T. Yu, Sci. Rep., 2012, 2, 737–743 CrossRef.
- V. C. Sanchez, A. Jachak, R. H. Hurt and A. B. Kane, Chem. Res. Toxicol., 2012, 25, 15–34 Search PubMed.
- K. Yang, S. Zhang, G. Zhang, X. Sun, S. T. Lee and Z. Liu, Nano Lett., 2010, 10, 3318–3323 CrossRef CAS.
- C. Li, J. Adamcik and R. Mezzenga, Nat. Nanotechnol., 2012, 7, 421–427 CrossRef CAS.
- J. Wilson, L. Sloman, Z. He and A. Aksimentiev, Adv. Funct. Mater., 2016, 26, 4830–4838 CrossRef CAS.
- S. M. Avdoshenko, D. Nozaki, C. Gomes da Rocha, J. W. Gonzalez, M. H. Lee, R. Gutierrez and G. Cuniberti, Nano Lett., 2013, 13, 1969–1976 CrossRef CAS.
- J. Mao, R. Guo and L. T. Yan, Biomaterials, 2014, 35, 6069–6077 CrossRef CAS.
- R. Guo, J. Mao and L. T. Yan, Biomaterials, 2013, 34, 4296–4301 CrossRef CAS.
- G. Zuo, S. G. Kang, P. Xiu, Y. Zhao and R. Zhou, Small, 2013, 9, 1546–1556 CrossRef CAS.
- Y. Chong, C. Ge, Z. Yang, J. A. Garate, Z. Gu, J. K. Weber, J. Liu and R. Zhou, ACS Nano, 2015, 9, 5713–5724 CrossRef CAS.
- B. Luan, T. Huynh, L. Zhao and R. Zhou, ACS Nano, 2015, 9, 663–669 CrossRef CAS.
- Z. Gu, W. Song, S. H. Chen, B. Li, W. Li and R. Zhou, Nanoscale, 2019, 11, 19362–19369 RSC.
- B. Li, D. R. Bell, Z. Gu, W. Li and R. Zhou, Carbon, 2019, 146, 257–264 CrossRef CAS.
- W. Zhang, T. Huynh, P. Xiu, B. Zhou, C. Ye, B. Luan and R. Zhou, Carbon, 2015, 94, 895–902 CrossRef CAS.
- Y. Deng, F. Wang, Y. Liu, Y. Yang, Y. Qu, M. Zhao, Y. Mu and W. Li, Nanoscale, 2020, 12, 5217–5226 RSC.
- Y. Liu, X. Song, Y. Yang, Y.-Q. Li, M. Zhao, Y. Mu and W. Li, Nanoscale, 2020, 12, 5209–5216 RSC.
- G. Duan, S. G. Kang, X. Tian, J. A. Garate, L. Zhao, C. Ge and R. Zhou, Nanoscale, 2015, 7, 15214–15224 RSC.
- B. Li, W. Li, J. M. Perez-Aguilar and R. Zhou, Small, 2017, 13, 1603685 CrossRef.
- Z. Gu, L. Zhao, S. Liu, G. Duan, J. M. Perez-Aguilar, J. Luo, W. Li and R. Zhou, ACS Nano, 2017, 11, 3198–3206 CrossRef CAS.
- T. C. King, P. D. Matthews, H. Glass, J. A. Cormack, J. P. Holgado, M. Leskes, J. M. Griffin, O. A. Scherman, P. D. Barker, C. P. Grey, S. E. Dutton, R. M. Lambert, G. Tustin, A. Alavi and D. S. Wright, Angew. Chem., Int. Ed. Engl., 2015, 54, 5919–5923 CrossRef CAS.
- J. Mahmood, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6 CrossRef.
- Y. Liu, V. I. Artyukhov, M. Liu, A. R. Harutyunyan and B. I. Yakobson, J. Phys. Chem. Lett., 2013, 4, 1737–1742 CrossRef CAS.
- Y. Qie, J. Liu, S. Wang, S. Gong and Q. Sun, Carbon, 2018, 129, 38–44 CrossRef CAS.
- J. Xu, J. Mahmood, Y. Dou, S. Dou, F. Li, L. Dai and J. B. Baek, Adv. Mater., 2017, 29, 1702007 CrossRef.
- M. S. Mahabal, M. D. Deshpande, T. Hussain and R. Ahuja, ChemPhysChem, 2015, 16, 3511–3517 CrossRef CAS.
- S. Mehdi Aghaei, M. M. Monshi, I. Torres, S. M. J. Zeidi and I. Calizo, Appl. Surf. Sci., 2018, 427, 326–333 CrossRef CAS.
- Y. Tang, X. Cui, W. Chen, D. Zhu, H. Chai and X. Dai, Appl. Phys. A: Mater. Sci. Process., 2018, 124, 1–13 CAS.
- H. Cui, K. Zheng, Y. Zhang, H. Ye and X. Chen, IEEE Electron Device Lett., 2018, 39, 284–287 CAS.
- N. Prasetyo and T. S. Hofer, J. Chem. Theory Comput., 2018, 14, 6472–6483 CrossRef CAS.
- E. Chigo-Anota, M. A. Alejandro, A. B. Hernández, J. J. S. Torres and M. Castro, RSC Adv., 2016, 6, 20409–20421 RSC.
- M. J. Macias, M. Hyvonen, E. Baraldi, J. Schultz, M. Sudol, M. Saraste and H. Oschkinat, Nature, 1996, 382, 646–649 CrossRef CAS.
- Z. Gu, P. De Luna, Z. Yang and R. Zhou, Phys. Chem. Chem. Phys., 2017, 19, 3039–3045 RSC.
- G. T. Ibragimova and R. C. Wade, Biophys. J., 1999, 77, 2191–2198 CrossRef CAS.
- M. Jager, H. Nguyen, M. Dendle, M. Gruebele and J. W. Kelly, Protein Sci., 2007, 16, 1495–1501 CrossRef CAS.
- H. Hu, J. Columbus, Y. Zhang, D. Wu, L. Lian, S. Yang, J. Goodwin, C. Luczak, M. Carter, L. Chen, M. James, R. Davis, M. Sudol, J. Rodwell and J. J. Herrero, Proteomics, 2004, 4, 643–655 CrossRef CAS.
- F. Cecconi, C. Guardiani and R. Livi, Biophys. J., 2006, 91, 694–704 CrossRef CAS.
- M. Petrovich, A. L. Jonsson, N. Ferguson, V. Daggett and A. R. Fersht, J. Mol. Biol., 2006, 360, 865–881 CrossRef CAS.
- W. P. Russ, D. M. Lowery, P. Mishra, M. B. Yaffe and R. Ranganathan, Nature, 2005, 437, 579–583 CrossRef CAS.
- J. Ricardo Pires, F. Taha-Nejad, F. Toepert, T. Ast, U. Hoffmüller, J. Schneider-Mergener, R. Kühne, M. J. Macias and H. Oschkinat, J. Mol. Biol., 2001, 314, 1147–11156 CrossRef.
- B. Andre and J. Y. Springael, Biochem. Biophys. Res. Commun., 1994, 205, 1201–1205 CrossRef CAS.
- P. Bork and M. Sudol, Trends Biochem. Sci., 1994, 19, 531–533 CrossRef CAS.
- K. Hofmann and P. Bucher, FEBS Lett., 1995, 358, 153–157 CrossRef CAS.
- J. R. Pires, F. Taha-Nejad, F. Toepert, T. Ast, U. Hoffmuller, J. Schneider-Mergener, R. Kuhne, M. J. Macias and H. Oschkinat, J. Mol. Biol., 2001, 314, 1147–1156 CrossRef CAS.
- J. Zielkiewicz, J. Chem. Phys., 2005, 123, 104501 CrossRef.
- B. Hess and N. F. van der Vegt, J. Phys. Chem. B, 2006, 110, 17616–17626 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef.
- Z. Yang, Z. Wang, X. Tian, P. Xiu and R. Zhou, J. Chem. Phys., 2012, 136, 025103 CrossRef.
- G. Zuo, X. Zhou, Q. Huang, H. Fang and R. Zhou, J. Phys. Chem. C, 2011, 115, 23323–23328 CrossRef CAS.
- S. M. Tomásio and T. R. Walsh, J. Phys. Chem. C, 2009, 113, 8778–8785 CrossRef.
- Y. Chong, C. Ge, Z. Yang, J. A. Garate, Z. Gu, J. K. Weber, J. Liu and R. Zhou, ACS Nano, 2015, 6, 5713–5724 CrossRef.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00365d |
| This journal is © The Royal Society of Chemistry 2020 |