
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHollow N-doped carbon nanofibers provide superior potassium-storage performance†
Ya Ru
Pei
,
Ming
Zhao
,
Hong Yu
Zhou
,
Chun Cheng
Yang
 * and
Qing
Jiang
* and
Qing
Jiang
Key Laboratory of Automobile Materials (Jilin University), Ministry of Education, School of Materials Science and Engineering, Jilin University, Changchun 130022, China. E-mail: ccyang@jlu.edu.cn; Fax: +86-431-85095876; Tel: +86-431-85095371
First published on 20th July 2020
Abstract
Potassium-ion batteries (PIBs) are attractive as an alternative to lithium-ion batteries in emerging energy storage devices. However, a big challenge is to design advanced anode materials with fast charge/discharge and extended lifespan. Herein, a series of hollow N-doped carbon nanofibers (HNCNFs) were derived from polyaniline. As an anode for PIBs, HNCNFs exhibit an ultra-high rate capability of 139.7 mA h g−1 at 30 A g−1 and an ultra-long cycling life of 188.4 mA h g−1 at 1 A g−1 after 4000 cycles. These prominent performances can be ascribed to: (i) the enlarged interlayer spacing, which accommodates more K+ and larger (de)potassiation strain without fracture; (ii) the interconnected hollow nanofibers, which shorten ion diffusion distance and provide enough space to buffer volume change and sufficient electrolyte diffusion paths to ensure enhanced reaction efficiency of active materials; and (iii) high-content pyridinic/pyrrolic N-doping, which improves electrical conductivity, creates more active sites and enhances surface pseudocapacitive behavior, benefiting rapid K+ diffusion. This study provides a facile and cost-effective strategy to fabricate high-performance PIB anode materials on a large scale.
Introduction
With the ever-growing consumption of traditional fossil fuels, searching and developing clean, effective and reproducible energy sources are highly desirable.1,2 Lithium-ion batteries (LIBs) have been widely used in electronic devices, electric vehicles and smart grids by virtue of their high specific energy; however, the scarcity and uneven distribution of lithium sources cause concern about the reliance on LIBs.3,4 This motivates the pursuit of alternative batteries based on earth-abundant elements, such as sodium,5,6 magnesium,7 aluminium8 and potassium ion batteries (PIBs).9 Particularly, PIBs came into the spotlight recently due to the similar intercalation process and closer redox potential of K/K+ (−2.93 V vs. standard hydrogen electrode) to that of Li/Li+ (−3.04 V), implying their high voltage plateau and energy density.10,11 However, the larger ionic radius of K+ (1.38 Å), in contrast to that of Li+ (0.76 Å), usually leads to low reversible capacity and inferior cycling stability as well as insufficient rate capability of the electrodes, severely hindering the development of PIBs.12,13 Thus, it is imperative to explore suitable host materials for achieving convenient and rapid K+ insertion/extraction.Carbonaceous materials appear to be one of the most common anode materials for PIBs owing to their low cost, high natural abundance, tunable interlayer spacing and high electronic conductivity.14 For example, graphite anodes have been demonstrated to possess a high reversible capacity of ∼279 mA h g−1 in PIBs. Even so, the rate capacity and capacity retention of graphite in PIBs are far lower than those in LIBs, which is caused by large interlayer spacing variations, huge volume changes and collapse of the carbon skeleton during repeated K+ insertion/extraction. Amorphous carbon is a good choice to address these issues due to its enlarged interlayer distance, which can facilitate the insertion of K+ and accommodate the volume expansion/shrinkage.15 In this regard, N-doping is an effective approach to tune the interlayer distances and to improve electrical conductivity. Particularly, the types of doped N configurations (pyridinic-N, pyrrolic-N and graphitic-N) play an important role in determining its reactivity. Pyridinic-N and pyrrolic-N, as highly chemically active sites, can supply abundant defects with higher affinity for attracting and storing more alkali metal ions.16 Besides, a hollow architecture can not only provide enough space to buffer volume change, but also afford highly accessible surface areas for sufficient electrode/electrolyte contact to ensure enhanced reaction kinetics of active materials, leading to improved electrochemical performance.17 Motivated by this, Yang et al.18 designed a necklace-like N-doped hollow carbon material with a rate capacity of 204.8 mA h g−1 at 2 A g−1 and cycling performance of 161.3 mA h g−1 at 1 A g−1 after 1600 cycles. Xu et al.19 reported that N-doped carbon nanofibers showed a rate capacity of 101 mA h g−1 at 20 A g−1 and cycling stability of 146 mA h g−1 at 2 A g−1 after 4000 cycles. These previous studies have established some empirical foundation for constructing high-performance carbon anodes for PIBs; however, it is still a great challenge to meet the demand of high-power electric vehicles and large-scale energy storage systems.
Polyaniline (PANI), one of the most potential conducting polymers, has been considered as an important material for application in supercapacitor electrodes.20 Significantly, PANI can be produced with various shapes by controlling the synthesis conditions, such as hollow spheres, nanowires, nanofibers and nanotubes.21 The corresponding carbon materials with specific morphologies can be prepared by maintaining the original shapes of PANI via the annealing treatment in an inert gas atmosphere. More importantly, PANI is one of the N-enriched precursors for carbon materials and its carbonization leads to high-content pyridinic/pyrrolic N, which is promising for energy storage applications.22
Inspired by this, unique hollow N-doped carbon nanofibers (HNCNFs) were fabricated by carbonizing PANI nanofibers synthesized via a facile self-assembly approach. Such an architecture can accommodate larger (de)potassiation strain without fracture, shorten ion diffusion distance, buffer volume change, provide sufficient electrolyte diffusion paths, improve electrical conductivity, create numerous active sites and enhance surface pseudocapacitive behavior. Benefiting from these synergistic effects, the HNCNF electrode presents an extremely high rate capacity (139.7 mA h g−1 at 30 A g−1) and superior long-term cycling stability (188.4 mA h g−1 after 4000 cycles at 1 A g−1). Furthermore, when coupled with a potassium Prussian blue nanoparticle (KPBNP) cathode, the HNCNF anode also shows excellent electrochemical properties in a potassium-ion full cell, demonstrating its great potential for future practical applications.
Experimental section
Materials synthesis
Materials characterization
The surface morphology and microstructure of the specimens were characterized by field-emission scanning electron microscopy (FESEM, JSM-6700F, JEOL, 15 keV) and transmission electron microscopy (TEM, JEM-2100F, JEOL, 200 keV). X-ray diffraction (XRD) measurements were carried out on a D/max2500PC diffractometer using Cu-Kα radiation (λ = 0.15406 nm). Raman spectra were collected using a micro-Raman spectrometer (Renishaw) with a laser of 532 nm excitation wavelength. X-ray photoelectron spectroscopy (XPS) analysis was performed on a Thermo ESCALAB-250Xi spectrometer. The specific area and pore size distribution were determined by nitrogen adsorption and desorption using a Micromeritics ASAP 2020 analyzer. Fourier transform infrared spectroscopy (FT-IR) measurements were performed on an E55 + FRA106 Fourier infrared spectrometer.Electrochemical measurements
The electrochemical measurements for PIBs were conducted on coin-type cells (CR2025), which were assembled in an Ar-filled glove box ([O2] < 1 ppm, [H2O] < 1 ppm). The metallic K foil was used as both counter and reference electrodes. The separator was a Whatman glass fiber (GF/D) and the electrolyte was 0.8 M KPF6 in ethylene carbonate and dimethyl carbonate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in volume). The working electrodes were fabricated by mixing the active materials (HNCNFs), conductive material (Super P) and binder of sodium carboxymethylcellulose (Na–CMC) with a weight ratio of 7:2:1 using deionized water as the solvent and were pasted uniformly on Cu foil and dried in a vacuum at 70 °C for 12 h. Then the electrodes were cut into discoidal pieces with a diameter of 12 mm and the mass loading of active materials is 0.4–0.6 mg cm−2. Galvanostatic charge/discharge cycling tests were performed using a LAND CT2001A battery testing system in a voltage range of 0.01–3 V (vs. K+/K). Cyclic voltammetry (CV) measurements were carried out on an Ivium-n-Stat electrochemical workstation (Ivium Technologies) with a potential scan rate of 0.1 mV s−1 between 0.01 and 3.0 V (vs. K+/K). Electrochemical impedance spectroscopy (EIS) tests were performed with an amplitude of 10 mV in the frequency range of 100 kHz to 10 mHz. In order to investigate the effect of mass loadings of active materials on electrochemical performances of the HNCNFs-700 electrodes, higher mass loadings in a range of 0.6–1.1 mg cm−2 were also used. Moreover, to measure the contribution of Super P to the total capacity, the bare Super P electrode was fabricated by mixing Super P and the binder of Na–CMC with a weight ratio of 9:1 using deionized water as the solvent. Note that the Super P electrode was assembled in the same way as the HNCNF electrodes. For elucidating the reaction mechanism of HNCNFs-700 during the charge/discharge process, the electrodes (after electrochemical tests) were disassembled from the cells in an Ar-filled glove box ([O2] < 1 ppm, [H2O] < 1 ppm) and then rinsed with dimethyl carbonate and ethanol to remove the residual electrolyte before ex situ characterization. For the KPBNP half cell, the KPBNP cathode was fabricated by mixing the active material (KPBNPs), conductive material (Super P) and binder of polyvinylidene fluoride (PVDF) with a weight ratio of 7:2:1 using N-methyl-2-pyrrolidone (NMP) as the solvent and was pasted on Al foil and dried in a vacuum at 100 °C for 12 h. The electrochemical performance of the electrodes was tested in CR2025-type coin cells, which were assembled in an Ar-filled glove box ([O2] < 1 ppm, [H2O] < 1 ppm). Galvanostatic charge/discharge tests were performed with a LAND CT2001A battery testing system in a voltage range of 2.0–4.0 V (vs. K+/K). CV measurements were carried out on an Ivium-n-Stat electrochemical workstation (Ivium Technologies) with a potential scan rate of 0.1 mV s−1 between 2.0 and 4.0 V (vs. K+/K). For assembling the potassium-ion full cells, to ensure the maximized material utilization and a suitable cathode-to-anode capacity ratio, the KPBNP cathode and the HNCNFs-700 anode were paired up in a mass ratio of 4:1.19,24 Note that prior to the full-cell assembly, both the cathode and anode were pre-activated by galvanostatically cycling in a half-cell configuration. Galvanostatic charge/discharge tests of the full cell were performed in a voltage range of 1.0–3.8 V (based on the active material mass of the anode).
:1 in volume). The working electrodes were fabricated by mixing the active materials (HNCNFs), conductive material (Super P) and binder of sodium carboxymethylcellulose (Na–CMC) with a weight ratio of 7:2:1 using deionized water as the solvent and were pasted uniformly on Cu foil and dried in a vacuum at 70 °C for 12 h. Then the electrodes were cut into discoidal pieces with a diameter of 12 mm and the mass loading of active materials is 0.4–0.6 mg cm−2. Galvanostatic charge/discharge cycling tests were performed using a LAND CT2001A battery testing system in a voltage range of 0.01–3 V (vs. K+/K). Cyclic voltammetry (CV) measurements were carried out on an Ivium-n-Stat electrochemical workstation (Ivium Technologies) with a potential scan rate of 0.1 mV s−1 between 0.01 and 3.0 V (vs. K+/K). Electrochemical impedance spectroscopy (EIS) tests were performed with an amplitude of 10 mV in the frequency range of 100 kHz to 10 mHz. In order to investigate the effect of mass loadings of active materials on electrochemical performances of the HNCNFs-700 electrodes, higher mass loadings in a range of 0.6–1.1 mg cm−2 were also used. Moreover, to measure the contribution of Super P to the total capacity, the bare Super P electrode was fabricated by mixing Super P and the binder of Na–CMC with a weight ratio of 9:1 using deionized water as the solvent. Note that the Super P electrode was assembled in the same way as the HNCNF electrodes. For elucidating the reaction mechanism of HNCNFs-700 during the charge/discharge process, the electrodes (after electrochemical tests) were disassembled from the cells in an Ar-filled glove box ([O2] < 1 ppm, [H2O] < 1 ppm) and then rinsed with dimethyl carbonate and ethanol to remove the residual electrolyte before ex situ characterization. For the KPBNP half cell, the KPBNP cathode was fabricated by mixing the active material (KPBNPs), conductive material (Super P) and binder of polyvinylidene fluoride (PVDF) with a weight ratio of 7:2:1 using N-methyl-2-pyrrolidone (NMP) as the solvent and was pasted on Al foil and dried in a vacuum at 100 °C for 12 h. The electrochemical performance of the electrodes was tested in CR2025-type coin cells, which were assembled in an Ar-filled glove box ([O2] < 1 ppm, [H2O] < 1 ppm). Galvanostatic charge/discharge tests were performed with a LAND CT2001A battery testing system in a voltage range of 2.0–4.0 V (vs. K+/K). CV measurements were carried out on an Ivium-n-Stat electrochemical workstation (Ivium Technologies) with a potential scan rate of 0.1 mV s−1 between 2.0 and 4.0 V (vs. K+/K). For assembling the potassium-ion full cells, to ensure the maximized material utilization and a suitable cathode-to-anode capacity ratio, the KPBNP cathode and the HNCNFs-700 anode were paired up in a mass ratio of 4:1.19,24 Note that prior to the full-cell assembly, both the cathode and anode were pre-activated by galvanostatically cycling in a half-cell configuration. Galvanostatic charge/discharge tests of the full cell were performed in a voltage range of 1.0–3.8 V (based on the active material mass of the anode).
Calculation details
In this work, all calculations were performed based on spin-polarized density functional theory (DFT) as implemented in the DMol3 code. The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional was employed to describe the exchange-correlation effects. The van der Waals interactions were determined using the Grimme scheme, and the density functional semi-core pseudopotential (DSPP) with the basis set of double numerical plus polarization (DNP) was used to describe the relativistic effects. The force and energy convergence criteria were set to 2.0 × 10−3 Ha Å−1 and 1.0 × 10−5 Ha, respectively, during the structure relaxation. Supercells consisting of 6 × 6 × 1 graphene unit cells were used and the Brillouin zones were sampled with 4 × 4 × 1 Monkhorst–Pack meshes. Moreover, a 15 Å vacuum space in the z direction was used to prevent possible interactions between the repeated layers.Results and discussion
Material synthesis and characterization
The preparation process of HNCNFs is schematically illustrated in Fig. 1. PANI nanofibers were synthesized via a self-assembly process assisted by the presence of excess oxidant (NH4)2S2O8. During this process, aniline is oxidized to reactive aniline cation-radicals by (NH4)2S2O8, simultaneously producing H2SO4 as the reduzate. Then the initially formed two aniline cation-radicals combine into a dimer and the dimer is further oxidized by (NH4)2S2O8 to a dimer cation-radical, which could act as a surfactant to template the formation of interconnected network-like hollow PANI nanofibers.25,26 The total reaction can be illustrated as follows: | (1) |
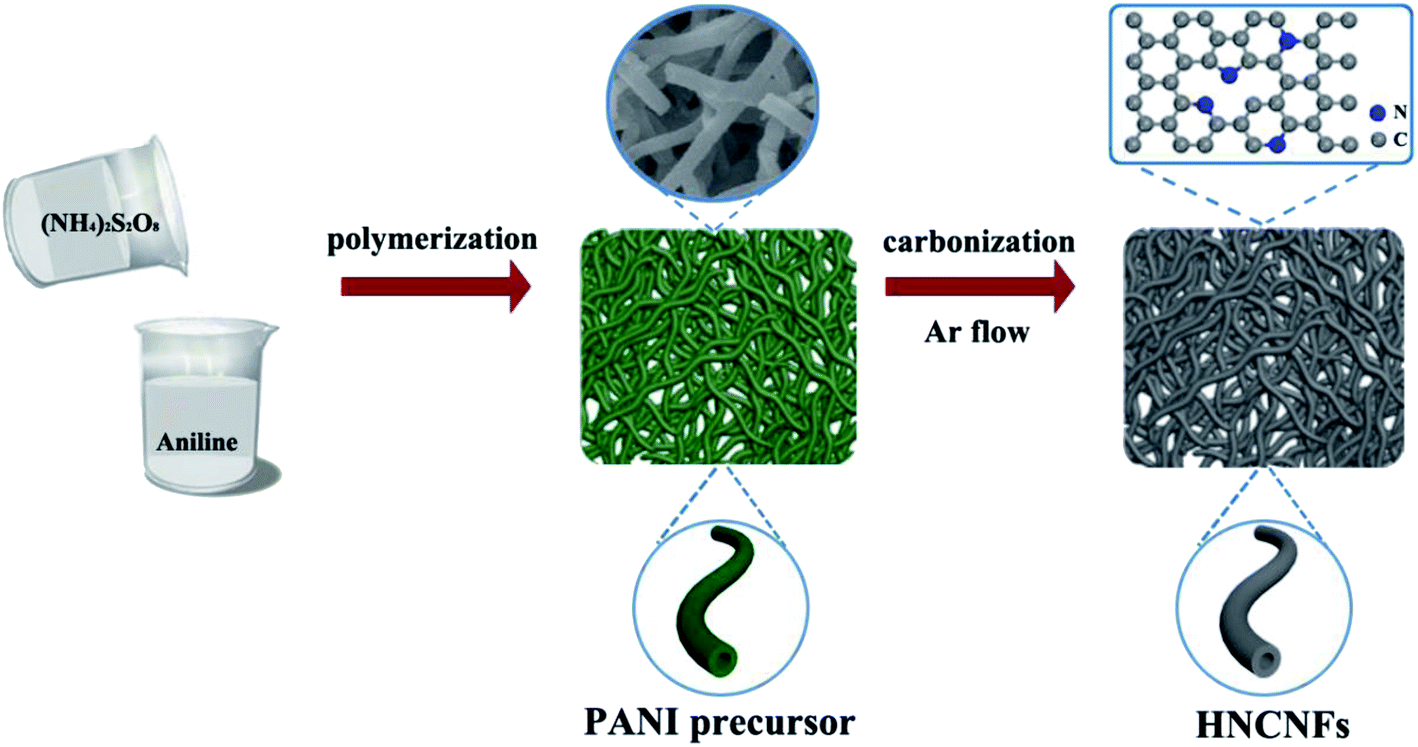 | ||
| Fig. 1 Schematic illustration of the fabrication process of HNCNFs. | ||
As shown in the FT-IR spectrum (see Fig. S1†), the obtained precursor exhibits well-defined peaks with characteristics of PANI.27,28 The HNCNFs were fabricated by subsequently annealing the PANI precursor in an Ar atmosphere.
The results of structure and morphology characterization of HNCNFs are shown in Fig. 2. Fig. 2a shows the comparison of XRD patterns of HNCNFs-600, HNCNFs-700 and HNCNFs-800. Each XRD pattern has two broad diffraction peaks near 22° and 43°, which correspond to (002) and (100) planes, respectively, of carbonaceous materials, indicating the partially graphitic nature of HNCNFs. Furthermore, with increasing temperature, the (002) peak shifts to a higher angle, implying smaller graphene interlayer spaces. According to the Bragg equation, the corresponding interspaces (d002) calculated are 0.416, 0.409 and 0.401 nm for HNCNFs-600, HNCNFs-700 and HNCNFs-800, respectively (summarized in Table S1†), which are all larger than that of graphite (0.335 nm). This is beneficial for K+ insertion.29Fig. 2b shows the Raman spectra of HNCNFs-600, HNCNFs-700 and HNCNFs-800, where two broad peaks at about 1350 and 1588 cm−1 are assigned to the D and G bands of carbon, respectively. The corresponding ID/IG values are 1.67, 1.59 and 1.42, respectively, revealing abundant topological defects and disorders in HNCNFs, which are beneficial for K storage.30 The ratios also suggest an increased graphitization degree with increasing temperature, which is consistent with XRD results. Fig. 2c shows the N2 adsorption/desorption isotherm and pore size distribution curve of HNCNFs-700. The isotherm displays a typical type-IV behavior with a distinct hysteresis loop in a relative pressure range of 0.45–0.95. This indicates the presence of micropores/mesopores in HNCNFs-700, which exhibits a specific surface area of 34.3 m2 g−1, relatively larger than those of HNCNFs-600 and HNCNFs-800 (29.6 and 26.1 m2 g−1, respectively, see Fig. S2†). With increasing the annealing temperature, the polymer precursor was pyrolyzed, forming a large amount of micropores and thus increasing the specific surface area of HNCNFs-700 compared with those of HNCNFs-600. However, further increasing the carbonized temperature to 800 °C, there will be serious decomposition of the polymer precursor and also the transition from micropores to mesopores, decreasing the specific surface area.31–33 Therefore, HNCNFs-700 exhibits the largest surface area in the three samples. From the inset of Fig. 2c, it is seen that the Barrett–Joyer–Halenda pore size of HNCNFs-700 is in a range of 2–50 nm. Pores smaller than 4 nm are generated from mass loss during carbonization, and those larger than 20 nm are ascribed to the hollow interior of the fibers. The micro/mesopores offer short diffusion distance and facilitate rapid diffusion of the electrolyte, contributing to fast charge/discharge properties of HNCNFs-700.19,34–36Fig. 2d exhibits an FESEM image of HNCNFs-700, and those of HNCNFs-600 and HNCNFs-800 are shown in Fig. S3.† Both HNCNFs-600 and HNCNFs-700 well inherit the interconnected morphology of the PANI precursor (see Fig. S3a†). For HNCNFs-800, some carbon nanofibers were destroyed and broken into short fibers with increasing temperature. Fig. 2e presents a TEM image of HNCNFs-700, showing a hollow tubular structure with a diameter of 150–160 nm, providing an inner diameter of 20–50 nm. The TEM images of HNCNFs-600 and HNCNFs-800 are given in Fig. S4.† The high-resolution TEM (HRTEM) image (see Fig. 2f) indicates that HNCNFs-700 contains disarrayed lattice fringes due to a low graphitization degree. The scanning TEM (see Fig. 2g) and elemental mapping images (see Fig. 2h–j) reveal the uniform distribution of C, N and O elements in HNCNFs-700.
 | ||
| Fig. 2 Structure and morphology characterization. (a) XRD patterns of HNCNFs. (b) Raman spectra of HNCNFs. (c) N2 adsorption/desorption isotherms and pore size distribution (the inset) of HNCNFs-700. (d) FESEM image of HNCNFs-700. (e) TEM and (f) HRTEM images of HNCNFs-700. (g) Scanning TEM image of HNCNFs-700. The corresponding elemental mapping images for (h) C, (i) N and (j) O elements. | ||
XPS analysis was performed to quantify the surface elemental composition and chemical characteristics of HNCNFs (see Fig. 3, S5 and S6†). All the HNCNFs are mainly composed of C, N and O elements and the corresponding elemental compositions are listed in Table S1.† The N content decreases with increasing temperature from 600 to 800 °C. The high-resolution C 1s peaks of HNCNFs-700 (see Fig. 3b) located at 284.4, 285.9 and 288.5 eV are assigned to C–C, C–N/C–O and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, respectively.37 The high-resolution O 1s peaks of HNCNFs-700 (see Fig. 3c) can be divided into 531.9 eV (C–O) and 533.1 eV (CO), which can enhance the wettability of carbon materials and induce the capacitance behavior during the electrochemical reaction, enhancing the rate performance and discharge capacity.6,11,18 The high-resolution N 1s peaks of HNCNFs-700 (see Fig. 3d) can be fitted with pyridinic-N (≈398.3 eV), pyrrolic-N (≈400.4 eV) and graphitic-N (≈402.3 eV). The relative atom ratios are 38.1%, 45.7%, and 16.2%, respectively (see the inset table of Fig. 3d). N-doping provides numerous defects in carbon materials, which can capture more K+ and enhance surface pseudocapacitive behavior, facilitating the rate performance and cycling stability of the carbon anode effectively.38–42 From Table S1,† all the HNCNFs possess much higher amounts of pyridinic-N and pyrrolic-N species than that of graphitic-N. Meanwhile, with increasing temperature, the contents of pyridinic-N and pyrrolic-N decrease while that of graphitic-N gradually increases.
O groups, respectively.37 The high-resolution O 1s peaks of HNCNFs-700 (see Fig. 3c) can be divided into 531.9 eV (C–O) and 533.1 eV (CO), which can enhance the wettability of carbon materials and induce the capacitance behavior during the electrochemical reaction, enhancing the rate performance and discharge capacity.6,11,18 The high-resolution N 1s peaks of HNCNFs-700 (see Fig. 3d) can be fitted with pyridinic-N (≈398.3 eV), pyrrolic-N (≈400.4 eV) and graphitic-N (≈402.3 eV). The relative atom ratios are 38.1%, 45.7%, and 16.2%, respectively (see the inset table of Fig. 3d). N-doping provides numerous defects in carbon materials, which can capture more K+ and enhance surface pseudocapacitive behavior, facilitating the rate performance and cycling stability of the carbon anode effectively.38–42 From Table S1,† all the HNCNFs possess much higher amounts of pyridinic-N and pyrrolic-N species than that of graphitic-N. Meanwhile, with increasing temperature, the contents of pyridinic-N and pyrrolic-N decrease while that of graphitic-N gradually increases.
 | ||
| Fig. 3 XPS analysis. (a) XPS survey spectrum of HNCNFs-700. (b), (c) and (d) are high-resolution XPS spectra of C 1s, O 1s and N 1s, respectively. The inset of (d) shows the schematic configurations of different N types and the inset table presents their atomic percentages. | ||
Electrochemical performances
The electrochemical K-storage performances of HNCNFs were investigated using coin-type half-cell configurations vs. K metal as the counter electrode. The CV curves of HNCNFs were measured at a scan rate of 0.1 mV s−1 within a voltage window of 0.01–3.0 V (vs. K+/K) (see Fig. 4a and S7†). For the first cathodic scan, a broad reduction peak at approximately 0.5 V, which disappears at the subsequent scans, can be ascribed to the formation of solid-electrolyte interface (SEI) films. The sharp reversible cathodic peak close to 0.01 V as well as the wide anodic peak are related to the intercalation and deintercalation of K+ into/from the carbon matrix, respectively.43 Two broad peaks around 1.5 V and 0.8 V are attributed to the interaction of K+ with species of N atoms.19 Moreover, from the second cycle to the fifth cycle, the CV curves are well overlapped, demonstrating excellent reversibility of the electrodes. Fig. 4b shows typical charge/discharge voltage profiles for the 1st, 2nd, 50th, 100th and 200th cycles of the HNCNFs-700 electrode at 0.1 A g−1. The initial discharge and charge capacities are 781.9 and 410.6 mA h g−1, respectively, corresponding to an initial coulombic efficiency (CE) of 52.5%. The irreversible capacity loss in the first cycle is caused by the formation of SEI films. With increasing the cycle number, the discharge capacities are 497.8, 297, 277.4 and 274.5 mA h g−1 for the 2nd, 50th, 100th and 200th cycles, respectively. Compared with the HNCNFs-700 electrode, the HNCNFs-600 and HNCNFs-800 electrodes possess lower initial discharge and charge capacities of 624.7 and 312.1 mA h g−1, 715.6 and 374.2 mA h g−1, providing an initial CE of 49.9% and 52.3%, respectively (see Fig. S8†). The above electrochemical properties of HNCNFs are summarized in Table S2.† Besides, the discharge curves of all HNCNF electrodes exhibit sloping characteristics, similar to those of other carbonaceous anode materials as reported.29,30Fig. 4c shows their cycling performance at 0.1 A g−1. The HNCNFs-700 electrode delivers a reversible capacity of 274.5 mA h g−1 after 200 cycles, higher than those of HNCNFs-600 and HNCNFs-800 electrodes (223.2 and 206.4 mA h g−1, respectively). Moreover, the CE gradually reaches close to 100% with the increase of the cycle number, implying its excellent reversibility of K+ insertion/extraction.44 Note that the electrode experiences a gradual capacity decay in the first 30 cycles, which results from the formation and stabilization of SEI films and side effects caused by the irreversible reaction between K+ and residual functional groups from PANI-derived carbon.6 Additionally, the CE of the electrode increases slowly and remains lower than 90% until the 20th cycle. The low initial CEs are commonly found in carbonaceous materials for potassium storage.4,18,19,29,30,45–47 The possible reasons lie in the reductive decomposition of the electrolyte and the formation and stabilization of SEI films.45–47 Nevertheless, the initial CE of HNCNFs-700 is higher than that of most of carbon-based materials reported in the open literature (see Table S3†). Besides, the initial CE can potentially be improved by pre-potassiation treatment, additives in the electrolyte, active materials with an appropriate specific surface area, etc.45,48–50 Typically, to pre-potassiate the electrode material is an effective method, which can reduce the irreversible capacity during this process. Yang et al. demonstrated that the initial CE of N/O dual-doped hierarchical porous hard carbon increases from 25% to 62.9% thanks to the pre-potassiation treatment.29 The improvement of electrolyte with specific additives is beneficial for rapid stabilization of the reversible capacity. Xu and co-workers studied the effects of various electrolyte systems on the CE and capacity in PIBs.51 It is found that more diethyl carbonate is beneficial to form stable SEI films, thus enhancing the initial CE. Furthermore, to design active materials with a relatively low surface area can contribute to higher initial CE since less time is required for the formation of SEI films.52,53Fig. 4d shows the comparison of the rate capabilities of the electrodes at various current densities. It is evident that the HNCNFs-700 electrode shows higher discharge capacity than HNCNFs-600 and HNCNFs-800 electrodes at each current density. The charge/discharge profiles of the HNCNFs-700 electrode at various current densities are shown in Fig. S9.† The average specific capacities are 437.7, 310.6, 268.9, 248.4, 239.6, 211.0, 190.2, 161.7, 150.2 and 139.7 mA h g−1 at 0.1, 0.2, 0.5, 1, 2, 5, 10, 20, 25 and 30 A g−1, respectively, showing much better rate performance than other state-of-the-art carbon-based anode materials for PIBs (summarized in Table S3†). When the current density switches back to 0.1 A g−1, the HNCNFs-700 electrode still recovers a specific capacity of 306.1 mA h g−1 rapidly and then remains stable for further cycling. Recent reports indicate that Super P as a conductive material could contribute a portion of the capacity.5,54,55 We also tested the rate capabilities of Super P at various current densities (see Fig. S10†). Super P possesses a capacity of 33.5 mA h g−1 at 30 A g−1, which is about 1/4 of the total capacity of the electrode (139.7 mA h g−1). Considering the low content (20 wt%) of Super P, its contribution to the overall capacity is only ∼4.8% (=33.5 × 20%/139.7). Further efforts are needed to focus on this issue. The superior rate performance of HNCNFs-700 is attributed to: (i) HNCNFs exhibit a unique interconnected hollow architecture with an enlarged interlayer spacing and thus can accommodate larger (de)potassiation strain without fracture, shorten ion diffusion distance and provide sufficient electrolyte pathways and (ii) the high-content pyridinic/pyrrolic N-doping can provide numerous active sites for K+ insertion/extraction and enhance surface pseudocapacitive behavior, thus promoting the rate performance of the HNCNFs-700 electrode. The ultra-high rate capacity of HNCNFs-700 is also confirmed by their lower charge transfer resistance (Rct) (1320 Ω) than those of HNCNFs-600 (1850 Ω) and HNCNFs-800 (2050 Ω), as shown in Fig. S11.† HNCNFs-600 shows poor electrical conductivity due to its low graphitization degree. For HNCNFs-800, the fibrous structure was destroyed at a high annealing temperature (see Fig. S3c†), resulting in the increased internal resistance. HNCNFs-700 possesses a high degree of graphitization and good interconnection between carbon fibers, in favor of rapid charge transfer and thus the lowest charge transfer resistance.56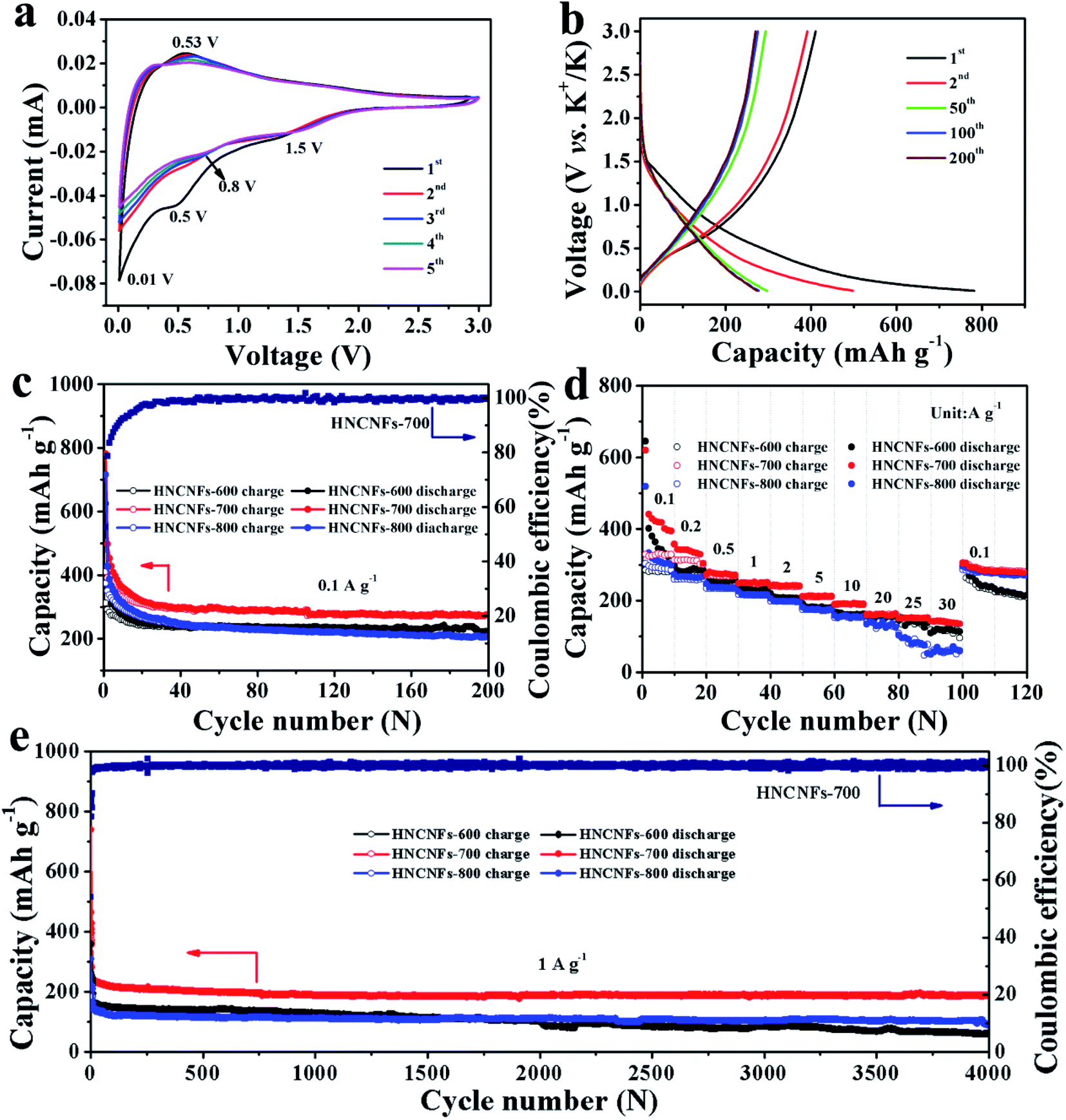 | ||
| Fig. 4 Electrochemical performances of the HNCNF electrodes in PIBs. (a) CV curves of the HNCNFs-700 electrode at a scan rate of 0.1 mV s−1. (b) Galvanostatic charge/discharge curves of the HNCNFs-700 electrode for the 1st, 2nd, 50th, 100th and 200th cycles at a current density of 0.1 A g−1. (c) Cycling performance of HNCNFs-600, HNCNFs-700 and HNCNFs-800 electrodes as well as the coulombic efficiency of the HNCNFs-700 electrode at 0.1 A g−1. (d) Rate performance of HNCNFs-600, HNCNFs-700 and HNCNFs-800 electrodes at various current densities. (e) Long cycling performance of HNCNFs-600, HNCNFs-700 and HNCNFs-800 electrodes as well as the coulombic efficiency of the HNCNFs-700 electrode at 1 A g−1. | ||
The long cycling stability of the HNCNF electrodes was tested at 1 A g−1 for 4000 cycles (see Fig. 4e), where the first five cycles were measured at 0.1 A g−1. The initial discharge capacity at 1 A g−1 (i.e. the discharge capacity of the 6th cycle) is 241.3 mA h g−1. The HNCNFs-700 electrode maintains a capacity of 188.4 mA h g−1 after 4000 cycles (77.2% of the initial discharge capacity at 1 A g−1), exhibiting much better cycling stability than HNCNFs-600 and HNCNFs-800 electrodes (60.5 mA h g−1 and 89.5 mA h g−1 after 4000 cycles, respectively). Note that the capacity decrease in the first 100 cycles may result from the formation of SEI films and the slightly structural destruction caused by the intercalation of large K+.29 Nevertheless, the HNCNFs-700 electrode shows high capacity retention in the subsequent cycles, demonstrating a high reversibility during the potassiation/depotassiation process. In addition, the HNCNFs-700 electrode retains a capacity of 141.7 mA h g−1 at a high current density of 2 A g−1 after 4000 cycles and 132.5 mA h g−1 at 4 A g−1 after 2000 cycles, respectively (see Fig. S12†). The HNCHFs-700 electrode possesses a superior lifespan compared with those in previous studies (except for ref. 30 of the text) summarized in Table S3.† Table S4† summarizes the mass loadings of some carbon-based anode materials in PIBs reported in the recent open literature. It is clear that the mass loading of this work is at a medium level. Moreover, we also tested electrochemical properties of the HNCNFs-700 electrode with higher mass loadings, as shown in Fig. S13.† It is found that the discharged capacities after 4000 cycles at a current density of 1 A g−1 decrease with increasing mass loadings as 188.4 mA h g−1 (0.6 mg cm−2), 161.2 mA h g−1 (0.8 mg cm−2) and 130.4 (1.1 mg cm−2). Nevertheless, the cycling performance of the HNCNFs-700 electrode with a mass loading of 1.1 mg cm−2 is still comparable to most carbon-based anodes (see Table S4†). Moreover, the HNCNFs-700 material also possesses excellent electrochemical Na-storage performances (see Fig. S14 and S15†).
Compared with HNCNFs-600 and HNCNFs-800, the superior K+ storage performance of HNCNFs-700 can be attributed to the following aspects. Firstly, the unique interconnected hollow architecture with an enlarged interlayer spacing is capable of facilitating K+ insertion/extraction with good reversibility. Secondly, HNCNFs-700 possesses the largest specific surface area, which provides efficient electrolyte diffusion paths. Additionally, HNCNFs-700 exhibits the lowest charge transfer resistance, which ensures rapid K+ diffusion. Most importantly, its optimal N-doping content offers numerous electrochemical active sites for efficient K+ absorption. Among different N configurations, pyridinic-N and pyrrolic-N can enhance the K+ rapid diffusion and improve the surface-induced pseudocapacitive processes, while graphitic-N is conducive to the electronic conductivity of the carbon matrix.39,40 With increasing temperature, the contents of pyridinic-N and pyrrolic-N decrease while that of graphitic-N gradually increases. The balance of pyridinic/pyrrolic-N (to induce surface capacitive effects) and graphitic-N (to enhance electronic conductivity) makes HNCNFs-700 more efficient for K+ absorption. Taken together, the hollow architecture, disordered carbon nature, the largest specific surface area and optimal N configurations endow HNCNFs-700 with overwhelmingly superior potassium storage properties among these three samples.
To describe the kinetics and electrochemical reaction mechanism of the HNCNF anode, CV curves at different scan rates from 0.2 to 1 mV s−1 were tested (see Fig. 5a, S16 and S17†). The measured current (i) and sweep rates (v) obey the following relationship:57
| i = avb | (2) |
log(v) + log(a), where a and b are fitting parameters, i is peak current, and v is scan rate. The b value can be confirmed by plotting log(i) − log(v) curves. The b value of 0.5 demonstrates an ideal diffusion-controlled process, while 1.0 represents a capacitive-controlled process.54 In the case of HNCNFs-700, the calculated b values for cathodic and anodic peaks are 0.84 and 0.92, respectively (see Fig. 5b), indicating that the kinetics of HNCNFs-700 are mainly capacitive-controlled. The relative contributions from the capacitive effect (k1v) and the diffusion behavior (k2v1/2) at a fixed rate scan can be obtained according to the following equations:57| i(V) = k1v + k2v1/2 | (3) |
 | ||
| Fig. 5 Electrochemical kinetics analysis. (a) CV curves of the HNCNFs-700 electrode at different scan rates from 0.2 to 1 mV s−1. (b) Measurement of b value with the relationship between log(i) and log(v). (c) Capacitive and diffusion-controlled contributions to the charge storage at 0.8 mV s−1. (d) Normalized contribution ratios of capacitive and diffusion-controlled capacities of the HNCNFs-700 electrode at different scan rates. | ||
To investigate the potassium intercalation/deintercalation process, ex situ Raman analysis, ex situ HRTEM and ex situ XPS were performed for the HNCNFs-700 electrode in the pristine, potassiation and depotassiation states. First, ex situ Raman analysis of the HNCNFs-700 electrode was performed under different states (see Fig. 6a). During the potassiation process, the value of ID/IG decreases from 1.59 to 0.98, indicating a higher degree of graphitization. In the subsequent depotassiation process, the value of ID/IG increases to 1.08, demonstrating a decreased graphitization degree. Fig. 6b–d show HRTEM images of HNCNFs-700 in different stages. In the pristine stage, the interlayer spacing is estimated to be 0.407 nm on average (see Fig. 6b). After full potassiation (see Fig. 6c), the interlayer spacing expands to 0.421 nm. No significant changes in the interlayer spacing are observed during the potassiation process, demonstrating that HNCNFs-700 can effectively adapt to the interlayer spacing change during cycling. The interlayer spacing recovers its original value after full depotassiation (see Fig. 6d), indicating a high reversibility of HNCNFs-700 during the potassiation/depotassiation process. Additionally, elemental mapping images of the HNCNFs-700 electrode after full discharge (see Fig. 6e–i) reveal that C, N, O and K are uniformly distributed, which demonstrates the successful intercalation of K+ into HNCNFs-700. After full charge (see Fig. 6j–n), a relatively weak energy dispersive spectrum intensity from K can be detected, which is attributed to the residual K in SEI films. The homogeneous K elemental mapping also implies that the generated SEI films are uniform.58,59 Moreover, Fig. S18† shows the ex situ XPS analysis for the HNCNFs-700 electrode under the pristine, potassiation and depotassiation states. The peaks of K can be observed after being discharged to 0.01 V (see Fig. S18a†). The high-resolution XPS spectra of K 2p (see Fig. S18b†) can be divided into metallic K (293.2 and 295.6 eV), KC8 (293.7 eV) and –C–O–K/–COOK/NK (292.6 eV), corresponding to the insertion of K+, the formation of KC8 and SEI films, respectively.60 When the electrode was charged to 3.0 V, the peak of KC8 (293.7 eV) disappears. Furthermore, the weak intensity of K is associated with the residual K in SEI films.
 | ||
| Fig. 6 K-storage mechanism of the HNCNFs-700 electrode. (a) Ex situ Raman spectra of the HNCNFs-700 electrode under different potassiation states. Ex situ HRTEM images and the corresponding interlayer spacing of HNCNFs-700 under (b) pristine, (c) full potassiation, and (d) full depotassiation states. (e) Scanning TEM image of the HNCNFs-700 electrode after full discharge. The corresponding elemental mapping images for (f) C, (g) N, (h) O and (i) K elements. (j) Scanning TEM image of the HNCNFs-700 electrode after full charge. The corresponding elemental mapping images for (k) C, (l) N, (m) O and (n) K elements. Top and side views of K atom absorbed in the (o) pristine, (p) pyridinic-N, (q) pyrrolic-N and (r) graphitic-N doped carbon structure. Electron density differences of K absorbed in the (s) pristine, (t) pyridinic-N, (u) pyrrolic-N and (v) graphitic-N structures. Blue and yellow areas represent increased and decreased electron density, respectively. | ||
The K-adsorption abilities at different N-doping sites were also studied, and the relative absorption energy (ΔEa) was calculated based on the DFT simulation. For comparison, the ΔEa value of the K atom on an ideal graphene layer was calculated to be −1.65 eV (see Fig. 6o). Then different N-doping sites (pyridinic-N, pyrrolic-N and graphitic-N) were introduced and the corresponding ΔEa values calculated were −3.18 eV, −3.07 eV and −1.71 eV (see Fig. 6p–r), respectively. This indicates that the pyridinic-N and pyrrolic-N sites have stronger tendencies for K-atom attraction than graphitic-N. The relatively lower ΔEa of the graphitic-N site should be attributed to the electron-rich structure of the graphene layer, which has a negative effect on K adsorption.61 From the side view, the simulation results reveal a shorter distance between the K atom and pyrindic-N (0.271 nm)/pyrrolic-N sites (0.272 nm) than that graphitic-N (0.303 nm), further demonstrating an enhanced affinity of pyrindic-N and pyrrolic-N to K atoms. Moreover, the electron density difference was also calculated by subtracting the charge densities of K atoms and C atoms from those of the combined compounds (see Fig. 6s–v). A net charge gain can be observed in all structures, indicating a charge transfer from the adsorbed K atom to its surrounding C atoms. The charge density tends to accumulate more around the N-doping sites in the doped structures.62 Besides, the tendency is more significant in pyridinic-N and pyrrolic-N than in graphitic-N, also suggesting a stronger K-adsorption in pyridinic and pyrrolic N-doped structures. Owing to the high-content pyridinic and pyrrolic N-doping, the HNCNFs-700 electrode exhibits an enhanced rate capability and cyclability.
To further verify the practical application of HNCNFs-700, potassium-ion full cells were assembled using HNCNFs-700 as the anode and KPBNPs as the cathode, respectively (see Fig. 7a). The structural characterization and electrochemical performance of the KPBNP cathode are provided in Fig. S19.† The XRD pattern (see Fig. S19a†) is in good agreement with previous reports.19,24 Fig. S19b† shows the CV curves of the KPBNP cathode at a scan rate of 0.1 mV s−1 with a voltage window of 2.0–4.0 V (vs. K/K+). The anodic and cathodic peaks can be assigned to the C–FeIII/FeII couple through a reversible K+ intercalation/deintercalation.24 The initial charge and discharge capacities of the cathode at 0.1 A g−1 are 161.8 and 74.0 mA h g−1, respectively (see Fig. S19c†). The electrode retains a discharge capacity of 57.9 mA h g−1 after 100 cycles at 0.1 A g−1 (see Fig. S19d†). Fig. 7b shows the charge/discharge voltage profiles of the HNCNFs-700//KPBNP potassium-ion full cell at 0.1 A g−1. The initial charge and discharge capacities are 414.8 and 138.0 mA h g−1 (based on the active material mass of the anode), respectively, corresponding to an initial CE of 33.3%. The cycling performance of the full cell at 0.1 A g−1 is presented in Fig. 7c. After 100 cycles, the discharge capacity remains 88.9 mA h g−1. Moreover, the energy density (E) of the full cell was calculated by:63
 | (4) |
 | ||
| Fig. 7 Electrochemical performances of the HNCNFs-700//KPBNP potassium-ion full cell. (a) The schematic diagram of the full cell. (b) Galvanostatic charge/discharge curves at 0.1 A g−1. (c) Cycling performance and the coulombic efficiency at 0.1 A g−1. (d) Rate properties at different current densities. (e) Optical photograph of a red LED lit by the full cell. | ||
Conclusions
In summary, HNCNFs were fabricated by a facile pyrolysis process with PANI as the precursor. The as-fabricated HNCNFs-700 possesses a unique hollow interconnected network of carbon nanofibers with an enlarged interlayer spacing, structural defects induced by high-content pyridinic/pyrrolic N-doping and also a disordered carbon nature. Due to the synergistic effects of these structural features, as an anode for PIBs, the HNCNFs-700 electrode exhibits impressive electrochemical properties with ultra-high rate capability and ultra-long cycling life. The superior performances make HNCNFs-700 promising for practical application in energy storage devices and may broaden the pathways of PIBs towards commercialization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is financially supported by the National Natural Science Foundation of China (No. 51671092 and 51631004), the Project of Talent Development in Jilin Province, the Natural Science Foundation of Jilin Province (No. 20200201073JC), the Program for JLU Science and Technology Innovative Research Team (No. 2017TD-09), and the Fundamental Research Funds for the Central Universities.Notes and references
- Y. P. Li, W. T. Zhong, C. H. Yang, F. H. Zheng, Q. C. Pan, Y. Z. Liu, G. Wang, X. H. Xiong and M. L. Liu, Chem. Eng. J., 2019, 358, 1147–1154 CrossRef CAS.
- C. C. Yang, W. T. Jing, C. Li and Q. Jiang, J. Mater. Chem. A, 2018, 6, 3877–3883 RSC.
- C. J. Chen, Z. G. Wang, B. Zhang, L. Miao, J. Cai, L. F. Peng, Y. Y. Huang, J. J. Jiang, Y. H. Huang, L. N. Zhang and J. Xie, Energy Storage Materials, 2017, 8, 161–168 CrossRef.
- W. Wang, J. H. Zhou, Z. P. Wang, L. Y. Zhao, P. H. Li, Y. Yang, C. Yang, H. X. Huang and S. J. Guo, Adv. Energy Mater., 2018, 8, 1701648 CrossRef.
- C. C. Yang, D. M. Zhang, L. Du and Q. Jiang, J. Mater. Chem. A, 2018, 6, 12663–12671 RSC.
- W. T. Jing, Y. Zhang, Y. Gu, Y. F. Zhu, C. C. Yang and Q. Jiang, Matter, 2019, 1, 720–733 CrossRef.
- X. L. Xue, R. P. Chen, C. Z. Yan, P. Y. Zhao, Y. Hu, W. H. Kong, H. N. Lin, L. Wang and Z. Jin, Adv. Energy Mater., 2019, 9, 1900145 CrossRef.
- S. K. Das, Angew. Chem., Int. Ed., 2018, 57, 16606–16617 CrossRef CAS PubMed.
- Q. Zhang, J. F. Mao, W. K. Pang, T. Zheng, V. Sencadas, Y. Z. Chen, Y. J. Liu and Z. P. Guo, Adv. Energy Mater., 2018, 8, 1703288 CrossRef.
- W. Luo, J. Y. Wan, B. Ozdemir, W. Z. Bao, Y. N. Chen, J. Q. Dai, H. Lin, Y. Xu, F. Gu, V. Barone and L. B. Hu, Nano Lett., 2015, 15, 7671–7677 CrossRef CAS PubMed.
- G. Wang, X. H. Xiong, D. Xie, Z. H. Lin, J. Zheng, F. H. Zheng, Y. P. Li, Y. Z. Liu, C. H. Yang and M. L. Liu, J. Mater. Chem. A, 2018, 6, 24317–24323 RSC.
- P. Z. Wang, X. S. Zhu, Q. Q. Wang, X. Xu, X. S. Zhou and J. C. Bao, J. Mater. Chem. A, 2017, 5, 5761–5769 RSC.
- X. X. Zhao, P. X. Xiong, J. F. Meng, Y. Q. Liang, J. W. Wang and Y. H. Xu, J. Mater. Chem. A, 2017, 5, 19237–19244 RSC.
- Z. L. Jian, W. Luo and X. L. Ji, J. Am. Chem. Soc., 2015, 137, 11566–11569 CrossRef CAS PubMed.
- Y. P. Li, C. H. Yang, F. H. Zheng, X. Ou, Q. C. Pan, Y. Z. Liu and G. Wang, J. Mater. Chem. A, 2018, 6, 17959–17966 RSC.
- X. Wang, Q. H. Weng, X. Z. Liu, X. B. Wang, D.-M. Tang, W. Tian, C. Zhang, W. Yi, D. Q. Liu, Y. Bando and D. Golberg, Nano Lett., 2014, 14, 1164–1171 CrossRef CAS PubMed.
- D. Zhang, W. P. Sun, Y. Zhang, Y. H. Dou, Y. Z. Jiang and S. X. Dou, Adv. Funct. Mater., 2016, 26, 7479–7485 CrossRef CAS.
- W. X. Yang, J. H. Zhou, S. Wang, W. Y. Zhang, Z. C. Wang, F. Lv, K. Wang, Q. Sun and S. J. Guo, Energy Environ. Sci., 2019, 12, 1605–1612 RSC.
- Y. Xu, C. L. Zhang, M. Zhou, Q. Fu, C. X. Zhao, M. H. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef PubMed.
- Z. C. Zhao, Y. B. Xie and L. Lu, Electrochim. Acta, 2018, 283, 1618–1631 CrossRef CAS.
- M. M. Yang, B. Cheng, H. H. Song and X. H. Chen, Electrochim. Acta, 2010, 55, 7021–7027 CrossRef CAS.
- J. Quílez-Bermejo, C. González-Gaitán, E. Morallón and D. Cazorla-Amorós, Carbon, 2017, 119, 62–71 CrossRef.
- L. F. Xiao, Y. L. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. M. Nie, G. J. Exarhos and J. Liu, Adv. Mater., 2012, 24, 1176–1181 CrossRef CAS PubMed.
- C. L. Zhang, Y. Xu, M. Zhou, L. Y. Liang, H. S. Dong, M. H. Wu, Y. Yang and Y. Lei, Adv. Funct. Mater., 2017, 27, 1604307 CrossRef.
- N. R. Chiou, L. J. Lee and A. J. Epstein, Chem. Mater., 2007, 19, 3589–3591 CrossRef CAS.
- E. C. Venancio, P.-C. Wang and A. G. MacDiarmid, Synth. Met., 2006, 156, 357–369 CrossRef CAS.
- X. L. Zhang, Q. L. Lin, X. Q. Zhang and K. P. Peng, J. Power Sources, 2018, 401, 278–286 CrossRef CAS.
- H.-P. Cong, X.-C. Ren, P. Wang and S.-H. Yu, Energy Environ. Sci., 2013, 6, 1185–1191 RSC.
- J. L. Yang, Z. C. Ju, Y. Jiang, Z. Xing, B. J. Xi, J. K. Feng and S. L. Xiong, Adv. Mater., 2017, 30, 1700104 CrossRef PubMed.
- D. P. Li, X. H. Ren, Q. Ai, Q. Sun, L. Zhu, Y. Liu, Z. Liang, R. Q. Peng, P. C. Si, J. Lou, J. K. Feng and L. J. Ci, Adv. Energy Mater., 2018, 8, 1802386 CrossRef.
- W. J. Liu, Q. X. Ru, S. X. Zuo, S. Yang, J. Han and C. Yao, Appl. Surf. Sci., 2019, 469, 269–275 CrossRef CAS.
- Z. C. Zhao, Y. B. Xie and L. Lu, Electrochim. Acta, 2018, 283, 1618–1631 CrossRef CAS.
- F. J. Miao, C. L. Shao, X. H. Li, K. X. Wang and Y. C. Liu, J. Mater. Chem. A, 2016, 4, 4180–4187 RSC.
- L. X. Zeng, Y. X. Fang, L. H. Xu, C. Zheng, M.-Q. Yang, J. F. He, H. Xue, Q. R. Qian, M. D. Wei and Q. H. Chen, Nanoscale, 2019, 11, 6766–6775 RSC.
- L. X. Zeng, F. Q. Luo, X. C. Chen, L. H. Xu, P. X. Xiong, X. S. Feng, Y. J. Luo, Q. H. Chen, M. D. Wei and Q. R. Qian, Dalton Trans., 2019, 48, 4149–4156 RSC.
- Y. Wu, S. Hu, R. Xu, J. Wang, Z. Peng, Q. Zhang and Y. Yu, Nano Lett., 2019, 19, 1351–1358 CrossRef PubMed.
- R. Hao, Y. Yang, H. Wang, B. B. Jia, G. S. Ma, D. D. Yu, L. Guo and S. H. Yang, Nano Energy, 2018, 45, 220–228 CrossRef CAS.
- X. Wang, Q. H. Weng, X. Z. Liu, X. B. Wang, D.-M. Tang, W. Tian, C. Zhang, W. Yi, D. Q. Liu, Y. Bando and D. Golberg, Nano Lett., 2014, 14, 1164–1171 CrossRef CAS PubMed.
- J. F. Ruan, Y. H. Zhao, S. N. Luo, T. Yuan, J. H. Yang, D. L. Sun and S. Y. Zheng, Energy Storage Materials, 2019, 23, 46–54 CrossRef.
- D. Nan, Z.-H. Huang, R. T. Lv, L. Yang, J.-G. Wang, W. C. Shen, Y. X. Lin, X. L. Yu, L. Ye, H. Y. Sun and F. Y. Kang, J. Mater. Chem. A, 2014, 2, 19678–19684 RSC.
- R. Hao, H. Lan, C. W. Kuang, H. Wang and L. Guo, Carbon, 2018, 128, 224–230 CrossRef CAS.
- K. Share, A. P. Cohn, R. Carter, B. Rogers and C. L. Pint, ACS Nano, 2016, 10, 9738–9744 CrossRef CAS PubMed.
- Y. L. An, H. F. Fei, G. F. Zeng, L. J. Ci, B. J. Xi, S. L. Xiong and J. K. Feng, J. Power Sources, 2018, 378, 66–72 CrossRef CAS.
- X. Wu, W. Zhao, H. Wang, X. J. Qi, Z. Xing, Q. C. Zhuang and Z. C. Ju, J. Power Sources, 2018, 378, 460–467 CrossRef CAS.
- Y. Liu, H. Dai, L. Wu, W. Zhou, L. He, W. Wang, W. Yan, Q. Huang, L. Fu and Y. Wu, Adv. Energy Mater., 2019, 9, 1901379 CrossRef.
- L. H. Xu, P. X. Xiong, L. X. Zeng, Y. X. Fang, R. P. Liu, J. B. Liu, F. Q. Luo, Q. H. Chen, M. D. Wei and Q. R. Qian, Nanoscale, 2019, 11, 16308–16316 RSC.
- D. M. Zhang, Z. W. Chen, J. Bai, C. C. Yang and Q. Jiang, Batteries Supercaps, 2020, 3, 185–193 CrossRef CAS.
- B. Cao, Q. Zhang, H. Liu, B. Xu, S. Zhang, T. Zhou, J. Mao, W. K. Pang, Z. Guo, A. Li, J. Zhou, X. Chen and H. Song, Adv. Energy Mater., 2018, 8, 1801149 CrossRef.
- Y. Jin, S. Li, A. Kushima, X. Zheng, Y. Sun, J. Xie, J. Sun, W. Xue, G. Zhou, J. Wu, F. Shi, R. Zhang, Z. Zhu, K. So, Y. Cui and J. Li, Energy Environ. Sci., 2017, 10, 580 RSC.
- N. Liu, L. Hu, M. T. McDowell, A. Jackson and Y. Cui, ACS Nano, 2011, 5, 6487 CrossRef CAS PubMed.
- J. Zhao, X. Zou, Y. Zhu, Y. Xu and C. Wang, Adv. Funct. Mater., 2016, 26, 8103 CrossRef CAS.
- Z. Li, L. Ma, T. W. Surta, C. Bommier, Z. Jian, Z. Xing, W. F. Stickle, M. Dolgos, K. Amine, J. Lu, T. Wu and X. Ji, ACS Energy Lett., 2016, 1, 395–401 CrossRef CAS.
- C. Bommier, W. Luo, W.-Y. Gao, A. Greaney, S. Ma and X. Ji, Carbon, 2014, 76, 165 CrossRef CAS.
- C.-M. Wu, P.-I. Pan, Y.-W. Cheng, C.-P. Liu, C.-C. Chang, M. Avdeev and S. Lin, J. Power Sources, 2017, 340, 14–21 CrossRef CAS.
- N.-W. Lee, J.-W. Jung, J.-S. Lee, H.-Y. Jang, I.-D. Kim and W.-H. Ryu, Electrochim. Acta, 2018, 263, 417–425 CrossRef CAS.
- R. Yan, E. Josef, H. Huang, K. Leus, M. Niederberger, J. P. Hofmann, R. Walczak, M. Antonietti and M. Oschatz, Adv. Funct. Mater., 2019, 29, 1902858 CrossRef.
- G. Z. Fang, Z. X. Wu, J. Zhou, C. Y. Zhu, X. X. Cao, T. Q. Lin, Y. M. Chen, C. Wang, A. Q. Pan and S. Q. Liang, Adv. Energy Mater., 2018, 8, 1703155 CrossRef.
- Y. Kim, Y. Kim, Y. Park, Y. N. Jo, Y.-J. Kim, N.-S. Choi and K. T. Lee, Chem. Commun., 2015, 51, 50–53 RSC.
- X. S. Zhou, Z. H. Dai, J. C. Bao and Y.-G. Guo, J. Mater. Chem. A, 2013, 1, 13727–13731 RSC.
- R. Z. Zhao, H. X. Di, X. B. Hui, D. Y. Zhao, R. T. Wang, C. X. Wang and L. W. Yin, Energy Environ. Sci., 2020, 13, 246–257 RSC.
- C. C. Ma, X. H. Shao and D. P. Cao, J. Mater. Chem., 2012, 22, 8911–8915 RSC.
- L.-J. Zhou, Z. F. Hou and L.-M. Wu, J. Phys. Chem. C, 2012, 116, 21780–21787 CrossRef CAS.
- K. Moyer, J. Donohue, N. Ramanna, A. P. Cohn, N. Muralidharan, J. Eaves and C. L. Pint, Nanoscale, 2018, 10, 13335–13342 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00585a |
| This journal is © The Royal Society of Chemistry 2020 |