DOI:
10.1039/D0NA00535E
(Review Article)
Nanoscale Adv., 2020,
2, 4986-4995
The role of oxygen defects in metal oxides for CO2 reduction
Received
28th June 2020
, Accepted 22nd August 2020
First published on 25th August 2020
Abstract
The abuse of fossil fuels release large amount of CO2, causing intense global warming. Using photoreduction and electroreduction to convert CO2 into highly valuable fuels such as CO and CH4 can effectively solve this problem. However, due to the limited activity and selectivity, pristine catalyst materials cannot meet the requirements of practical applications, which means that some modifications to these catalysts are necessary. In this review, a series of research reports on oxygen defect engineering have been introduced. First, the methods of preparing oxygen defects by heat treatment, doping, and photoinduction combined with influencing factors in the preparation are introduced. Subsequently, common characterization methods of oxygen defects including EPR, Raman, XPS, EXAFS, and HRTEM are summarized. Finally, the mechanisms of introducing oxygen defects to improve CO2 reduction are discussed, and include enhancing light absorption, improving CO2 adsorption and activation, as well as promoting stability of the reaction intermediates. The summary of research on oxygen defects provides guidance for researchers who focus on CO2 reduction and accelerate the realization of its industrial applications in the future.
 Zesheng Deng | Zesheng Deng is a PhD student at East China University of Science and Technology (ECUST, Shanghai, China). He obtained his bachelor's degree from ECUST, then joined Prof. Jinlong Zhang's group in 2018. His research interest focuses on CO2 photoreduction. |
 Jiahui Ji | Jiahui Ji is PhD student at East China University of Science and Technology (ECUST, Shanghai, China). He obtained his bachelor's degree from Nanjing Tech University, then joined Prof. Mingyang Xing's group in 2017. His research interest focuses on Advanced Oxidation Technologies (AOTs) such as photocatalysis and Fenton reactions. |
 Mingyang Xing | Dr Mingyang Xing is a Professor and supervisor of postgraduate students at the School of Chemistry and Molecular Engineering, East China University of Science and Technology (ECUST). He obtained his doctoral degree in 2012 from ECUST and then worked at University of California, Riverside, as a visiting scholar for one year. His research focuses on the design and preparation of functional nanomaterials and applications to the environment and energy fields. He has published more than 90 papers in SCI journals in these areas, which have been cited more than 6800 times (H-index: 46). |
 Jinlong Zhang | Prof. Dr Jinlong Zhang obtained his bachelor's degree in 1985 and his PhD in 1993 from East China University of Science and Technology, Shanghai, China. Professor Zhang joined the East China University of Science and Technology in 1993. He worked at Osaka Prefecture University as a Postdoctoral candidate (JSPS) while collaborating with Prof. Anpo from 1996 to 2000. He became a full professor in 2000. He has published over 350 original papers, which were cited more than 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times (H-index: 76). He is currently on the editorial boards of “Applied Catalysis B: Environmental” and “Scientific Reports”. He is also the associate editor of “Res. Chem. Intermed.”. His research interests include photocatalysis, mesoporous materials, and materials science. He has been selected as one of the “Most Cited Chinese Researchers” in China by “Elsevier” in 2014, 2015, 2016, 2017, and 2018. 000 times (H-index: 76). He is currently on the editorial boards of “Applied Catalysis B: Environmental” and “Scientific Reports”. He is also the associate editor of “Res. Chem. Intermed.”. His research interests include photocatalysis, mesoporous materials, and materials science. He has been selected as one of the “Most Cited Chinese Researchers” in China by “Elsevier” in 2014, 2015, 2016, 2017, and 2018. |
1. Introduction
1.1 Background
For decades, the consumption of fossil fuels has caused a sharp rise in the level of CO2 emission, and has resulted in some serious problems related to global warming and climate change.1,2 Therefore, it is necessary to take some measures to limit and reduce CO2 emission. The capture and conversion of CO2 into reusable value-added chemicals is considered an effective method to remove excess CO2 from air.3,4 Therefore, the photoreduction and electroreduction of CO2 are considered to be environment-friendly methods for converting CO2 into combustible chemicals (such as CO and CH4), followed by the development of a large number of photocatalysts and electrocatalysts.5–8 The defects contained in the catalysts play an important role in capturing CO2 and lowering the activation energy of adsorbed CO2.9–11 In this review, various materials with oxygen defects have been discussed to gain more insight into the correlation between oxygen defects and the activity and selectivity of CO2 photoreduction and electroreduction. Finally, the role of oxygen defect-rich catalysts in the conversion of CO2 to reusable fuel is summarized and their future applications are investigated.
1.2 Mechanisms of CO2 photoreduction and electroreduction
Photoreduction and electroreduction processes have been proved to be effective methods for the removal of excessive CO2 in air.12–14 Moreover, there are some differences in the mechanisms between CO2 electroreduction and photoreduction.
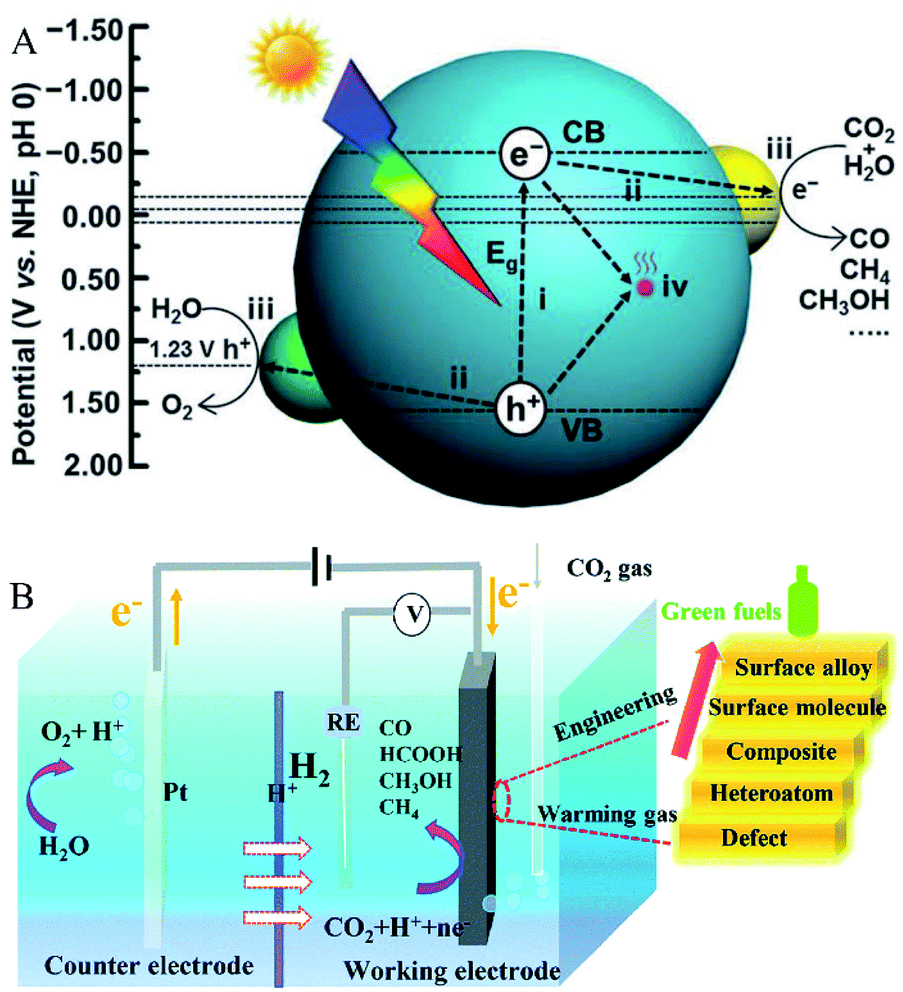
Since the pioneering work on photoelectrochemical water splitting that was reported by Fujishima and Honda in 1972,15 CO2 photoreduction has gradually developed and studied extensively. As shown in Fig. 1A, the generally accepted process of CO2 photoreduction is as follows: first, the photoexcited electrons are excited from the valence band to the conduction band, leaving holes in the valence band. Then, the electrons and holes are separated, which migrate to the surface of the catalysts to reduce CO2 to combustible chemicals (such as CO and CH4).16 The valence state of carbon in CO2 is the highest oxidation state; thus, there are various reduction products of CO2, ranging from CO and CH4 to higher hydrocarbons and oxygenates such as CH3OH and HCOOH. On the other hand, the actual process of CO2 photoreduction is slightly complicated. Consider the case of CO as the only photocatalytic product. As eqn (1)–(5) show, first, the photogenerated electrons combine with CO2 and H+ to form the intermediate *COOH. Then, *COOH combines with the photogenerated electrons and H+ to release H2O and to obtain CO absorbed on the catalyst's surface. Finally, CO is desorbed from the catalyst surface. In the meantime, photogenerated holes combine with water to produce oxygen and H+. Due to the complex process of CO2 reduction, there are many factors that influence the activity and selectivity of CO2 photoreduction. Solar light harvesting, charge separation and transportation, as well as surface reactions, are the three main factors, which is recognized as crucial for improving the selectivity and activity of CO2 photoreduction. In fact, it is difficult for the pristine catalyst to meet all requirements. While, introducing oxygen defects is an effective method to solve these problems.
| | | CO2 + e− + * + H+ → *COOH | (1) |
| | | *COOH + e− + H+ → *CO + H2O | (2) |
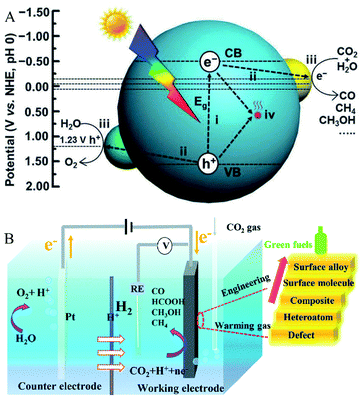 |
| | Fig. 1 (A) Schematic illustration of CO2 photoreduction. Reproduced with permission: Copyright The Royal Society of Chemistry 2016;16 (B) schematic illustration of CO2 electroreduction. Reproduced with permission: Copyright The Royal Society of Chemistry 2019.17 | |
As for the electric reduction process of CO2, it mainly relies on the electrons generated in the electrode to reduce the CO2 adsorbed on the electrocatalyst electrode. As shown in Fig. 1B, electrocatalytic CO2 reduction usually occurs in a three-electrode system, including a working electrode loaded with the electrocatalyst for CO2 reduction, a reference electrode for determining the potential, and a counter electrode that balances the charge and the current. In the process of CO2 electroreduction, electrons transfer to the working electrode and reduce the adsorbed CO2 to valuable fuels such as HCOOH and CO.17 There are many factors that affect CO2 electroreduction, such as the applied potential, the nature of the electrolyte solution, and the electrocatalyst. In this review, the roles of oxygen-defect electrocatalysts applied in CO2 electroreduction are mainly discussed. As published in the literature, the process for converting CO2 to CO involves the steps shown in eqn (6)–(8) and the process of hydrogen evolution involves the steps shown in eqn (9) and (10).18–20 At the beginning of the reaction, CO2 is adsorbed on the electrocatalyst first and then reduced to the intermediate *COOH, which is further transformed to H2O and CO, and then desorbed from the electrocatalysts. When the electrocatalyst has the best CO binding energy, it can promote further reaction with other reduction products, such as CH4 and CH3OH. As reported in the literature, the two main limitations of CO2 electroreduction activity are *COOH formation and hydrogen evolution.
| | | CO2(g) + e− + * +H+ → *COOH | (6) |
| | | *COOH + e− + * +H+ → *CO + H2O(l) | (7) |
| | | *H + H+ + e− → * + H2 | (10) |
1.3 The role of oxygen defects
Among the strategies for improving catalytic CO2 reduction, introducing oxygen defects into the catalysts has become one of the most likely ways to achieve this goal. The introduction of oxygen defects can enhance the conductivity of the catalysts, and generate a large number of activation sites and adsorption sites on these catalysts, which help to improve their ability for CO2 reduction. A large number of studies have reported that oxygen defect-rich catalysts play a very effective role in CO2 reduction.11,21–24 The formation of oxygen defects can modify the optical absorption by introducing a defective level in order to reduce the bandgap, which further enhances the activity of the catalysts in CO2 reduction. Moreover, the formation of the oxygen defects is usually accompanied by the exposure of the metal cation of the metal oxide catalysts and surface atomic structure modification of the catalysts, which may expose more active sites on the catalysts, thus leading to the enhancement in the adsorption and activation of CO2 in CO2 reduction.25 According to the studies, the oxygen defects exposed on the surface of the catalysts can offer chemical adsorption sites to reduce the CO2 adsorption energy, activate CO2 by bending or stretching the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, and lower the energy barrier by stabilizing the intermediates. It can be anticipated that the introduction of oxygen defects can really improve the CO2 reduction efficiency.
O bond, and lower the energy barrier by stabilizing the intermediates. It can be anticipated that the introduction of oxygen defects can really improve the CO2 reduction efficiency.
Therefore, in order to further intuitively understand the role of oxygen defects in CO2 reduction and the corresponding mechanism, we have summarized the different preparation methods of oxygen defect-rich catalysts, the characterization of oxygen defects, and the mechanism involving promotion of CO2 reduction by the introduction of oxygen defects.
2. The introduction of oxygen defects
Oxygen defect engineering has been widely utilized to adjust the electronic structure of the target catalysts and to improve their performance in CO2 reduction. According to the published reports, oxygen defects are usually introduced by methods such as heat treatment, irradiation, or doping modification.
2.1 Introducing oxygen defects by heat treatment
Oxygen defects are created by removing oxygen atoms from the catalyst. The common methods for removing oxygen atoms include using a strong reducing agent to reduce the catalyst, reducing the ambient oxygen partial pressure (such as anaerobic condition), and introducing interfacial lattice strain to reduce the Gibbs free energy of oxygen defect formation, so that the reaction balance can move toward the direction of oxygen defects' generation.
There are many methods of introducing oxygen defects through thermal induction operated in vacuum or inert gas environment.26–28 Wang et al.29 reported a strategy to control the oxygen defect concentration by annealing ZnO nanoparticles in different atmospheres with different oxygen partial pressures at 550 °C for 2 h. By comparing the oxygen defect concentration of the ZnO samples annealed in oxygen, air, and nitrogen atmospheres, it was found that the order of oxygen defect concentration on the surface of the ZnO samples was nitrogen > air > oxygen annealing environments. This result was consistent with the assumption that a lower oxygen defect concentration was obtained in a higher oxygen partial pressure during the heat treatment, demonstrating that the oxygen partial pressure during the heat treatment did have a great effect on the oxygen defect concentration.
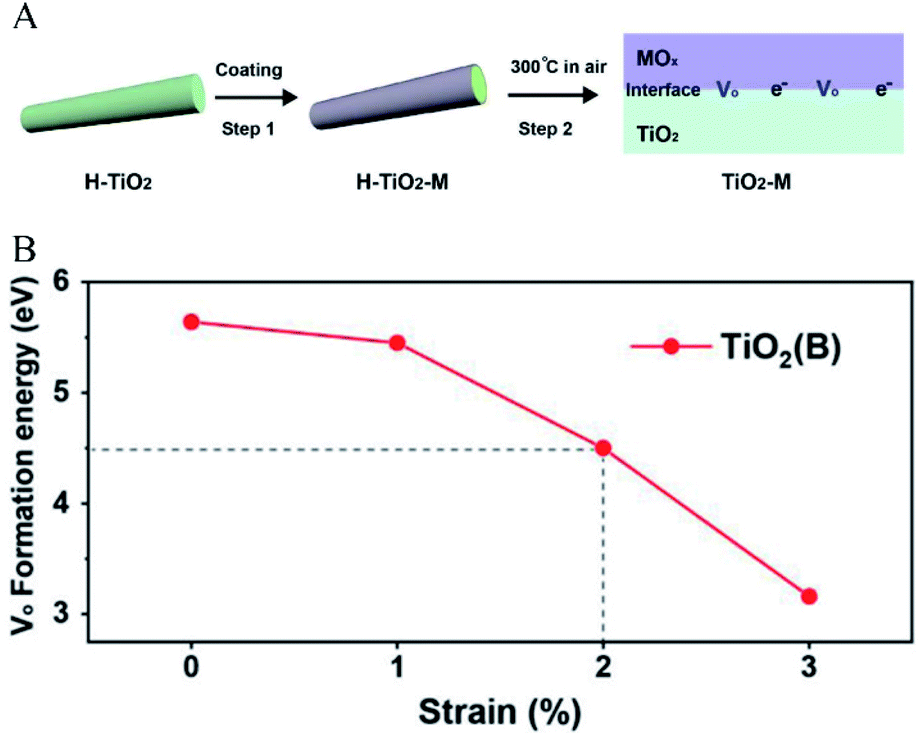
In addition to decreasing the partial pressure of oxygen, studies have shown that interfacial lattice strain is another important factor that controls the generation of oxygen defects. Wang et al.30 reported a method for introducing oxygen defects under mild heating conditions by introducing interfacial lattice strain. In this case, as shown in Fig. 2A, interfacial lattice strain was introduced in TiO2(B) by coating transition metal oxides. The results showed that TiO2(B) with coated transition metal oxides had a much higher ratio of oxygen defects than that with uncoated transition metal oxides. As shown in Fig. 2B, it could be calculated that the potential barrier gradually decreased as the interface lattice strain increased. The Gibbs free energy (ΔG) of oxygen defect formation could be defined as a function of epitaxial strain (η) and temperature (T), according to the following relationship, which confirmed the mechanism that the interfacial strain affected the energy barrier of oxygen defect generation (eqn (11)):
| | | ΔG = ΔE(η) + ΔF(η, T) + 0.5 × μ0(T) | (11) |
where
μ0 is the oxygen chemical potential, Δ
E represents the free energy at zero temperature, and Δ
F represents the thermal contribution to free energy. According to the equilibrium, the increase in the interfacial lattice strain led to the decrease in Δ
G. Therefore, it could be concluded that the interfacial lattice strain introduced by coating made the oxygen defect generation thermodynamically favorable.
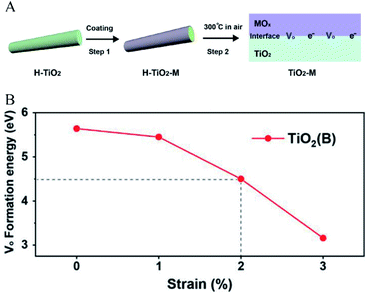 |
| | Fig. 2 (A) Illustration of the introduction of interfacial lattice strain by coating transition metal oxides, (B) the relationship between interfacial strain and oxygen defect formation energy. Reproduced with permission: Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.30 | |
The temperature is another significant influencing factor of oxygen defects. According to eqn (11), temperature has a great influence on the Gibbs free energy of oxygen defect formation. The temperature determines whether the oxygen defects can be generated or not. Liu et al.31 reported a method of controlling the concentration of oxygen vacancies (OVs) by annealing TiO2 powder in hydrogen gas at different temperatures ranging from 120 °C to 700 °C. In this case, the result of the thermal treatment demonstrated that the content of oxygen defect increased at first and then decreased with the continuous increase in the annealing temperature. The whole process could be divided into three steps: firstly, hydrogen was physically adsorbed on the surface of TiO2 below 300 °C; secondly, when the temperature was higher than 300 °C, the electrons were transferred from the hydrogen atoms to the oxygen atoms, and then the oxygen atoms were removed by the hydrogen atoms in the form of water, leaving oxygen defects on TiO2. As a result, the oxygen defect content increased with the temperature increasing from 300 °C to 450 °C. Thirdly, when the temperature rose up to 450 °C, the electrons were transferred from the oxygen defects to Ti4+, and Ti3+ was formed at the same time, leading to a decrease in the oxygen defects content as the temperature increased over 450 °C. The result of temperature control on the oxygen defect content showed that the OVs formation was determined by thermodynamic control. This means that the oxygen defect content increase for thermodynamically favorable processes and decrease for thermodynamically unfavorable processes.
In addition to introducing oxygen defects under the gas–solid condition by means of heating at high temperature, using a strong reducing agent in the liquid–solid condition can also effectively introduce oxygen defects at a mild temperature. Kim et al.32 reported a strategy for the preparation of BaSnO3−x with abundant oxygen defects under the solid–liquid condition at room temperature with NaBH4 as the reducing agent. In this case, they could effectively adjust the concentration of oxygen defects by controlling the amount of NaBH4. Compared with the other methods for introducing oxygen defects at high temperature, this mild liquid–solid method could produce oxygen defects without high-temperature processing, which prevented the high temperature from destroying the material structure of the catalysts.
2.2 Introducing oxygen defects by doping
The traditional method of generating oxygen defects is heat treatment at a high temperature, which has some problems in keeping the structure and morphology unchanged. The method of introducing oxygen defects by doping heteroatoms is feasible for triggering oxygen defects in order to maintain the stability even under oxygen-rich circumstances. Liu et al.33 reported a facile strategy for introducing oxygen defects by annealing commercial MO3 at 300 °C in a NH3 atmosphere. The N-doped MO3 obtained from the heat treatment had advantages in attaining stable oxygen defects by decreasing the formation energy of the oxygen defects. Zhang et al.34 reported another method of doping boron atom into transition metal oxides to form stable oxygen defects by the pyrolysis approach. The result indicated that boron doping reduced the generation energy of oxygen defect formation in transition metal oxides, which made the generation of oxygen defects thermodynamically favorable. Moreover, substituting the transition metal with the doping cation is also an effective way to introduce oxygen defects. Zhao et al.35 reported a method for precisely controlling the concentration of the oxygen defects by Cu doping under the hydrothermal condition. Cu/TiO2 was synthesized with different amounts of Cu ions, which determined the concentration of the oxygen defects. It was proved that the Ti–O coordination number was decreased as the amount of the doping Cu ions increased, which made it easy for to release oxygen and thereby increase the oxygen defect content. These evidences demonstrate that the concentration of oxygen defects could be controlled by cation substitution. Tong et al.25 also reported an Fe-doping method for the introduction of oxygen defects in WO3−x. Fe-doped WO3−x was prepared by the hydrothermal method in ethanol solvent, with FeCl3 and WCl6 used as the precursors. In this case, with the increase in the amount of doped iron, the oxygen defect concentration increased gradually. The low valence state cation substituted the high valence state cation, which would reduce M–O (M is the metal cation) bond and release oxygen; thus, with the increase in cation-doping, the oxygen defect concentration increased gradually. Usually, the activity of the catalysts has an optimal point; thus, that the cation-doping method could control the oxygen defect concentration for obtaining the optimal activity.
2.3 Introducing oxygen defects by illumination
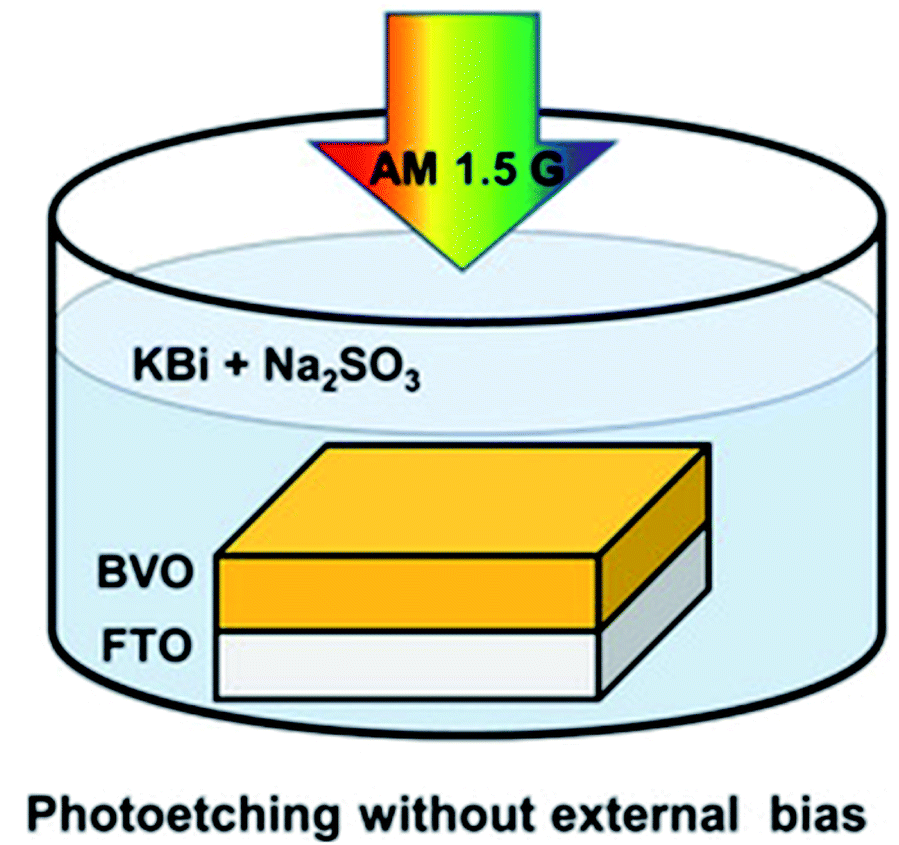
The creation of oxygen defects by photoinduction is a facile method. As for the metal oxide semiconductors, the photoelectrons can be excited by illumination and transferred from oxygen to metal cation, then the oxygen atoms are released and simultaneously form the oxygen defects. Feng et al.36 reported a method for introducing oxygen defects by illumination. The whole process of generating oxygen defects was operated at room temperature by the photoetching method. As shown in Fig. 3, BiVO4 with abundant oxygen defects was obtained by immersing the BiVO4 photoanode into the electrolyte containing alkali solution and sodium sulfate with illumination. This photoetching method increased the donor density without introducing a heteroatom and maintained the structure of the BiVO4 photoanode. Therefore, the method offered a facile modification strategy to attain oxygen defects. Ye and co-workers37 have reported a photoinduced oxygen defect engineering method to fabricate black In2O3−x nanosheets. They adopted a 300 W Xe lamp as the irradiation source and introduced oxygen defects in an H2/CO atmosphere. During the process of introducing oxygen defects, h+ was reduced by CO or H2 and the photogenerated electron reduced In2O3 to In2O3−x so as to introduce oxygen defects. Zhang et al.38 have reported a method of introducing oxygen defects in BiOCl by illumination. BiOCl was irradiated under a 500 W Xe lamp for 5 h, then the white BiOCl turned to gray defective BiOCl. The introduction of oxygen defects can be effectively controlled by the illumination method. These obtained oxygen defects can effectively widen the photoabsorption range of the catalysts, and improve the CO2 adsorption and the catalytic activity. The photoinduction method is an ideal strategy to regenerate oxygen defects in catalysts without introducing a heteroatom and breaking the structure of the catalysts.
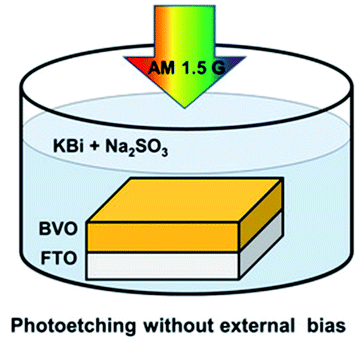 |
| | Fig. 3 Illustration of the introduction of oxygen defects by photoetching. Reproduced with permission: Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.36 | |
3. Characterization of the oxygen defects
In the process of studying the role of defective catalysts in CO2 reduction, it is usually required to characterize the type and quantity of defects introduced into the catalysts, including the oxygen defects. Common defects characterization methods include high resolution transmission electron microscopy (HRTEM), X-ray photoelectron spectroscopy (XPS), Raman spectroscopy, electron paramagnetic resonance (EPR), X-ray absorption fine structure (XAFS), and photoluminescence (PL); it is usually necessary to combine some of these methods for defect characterization.
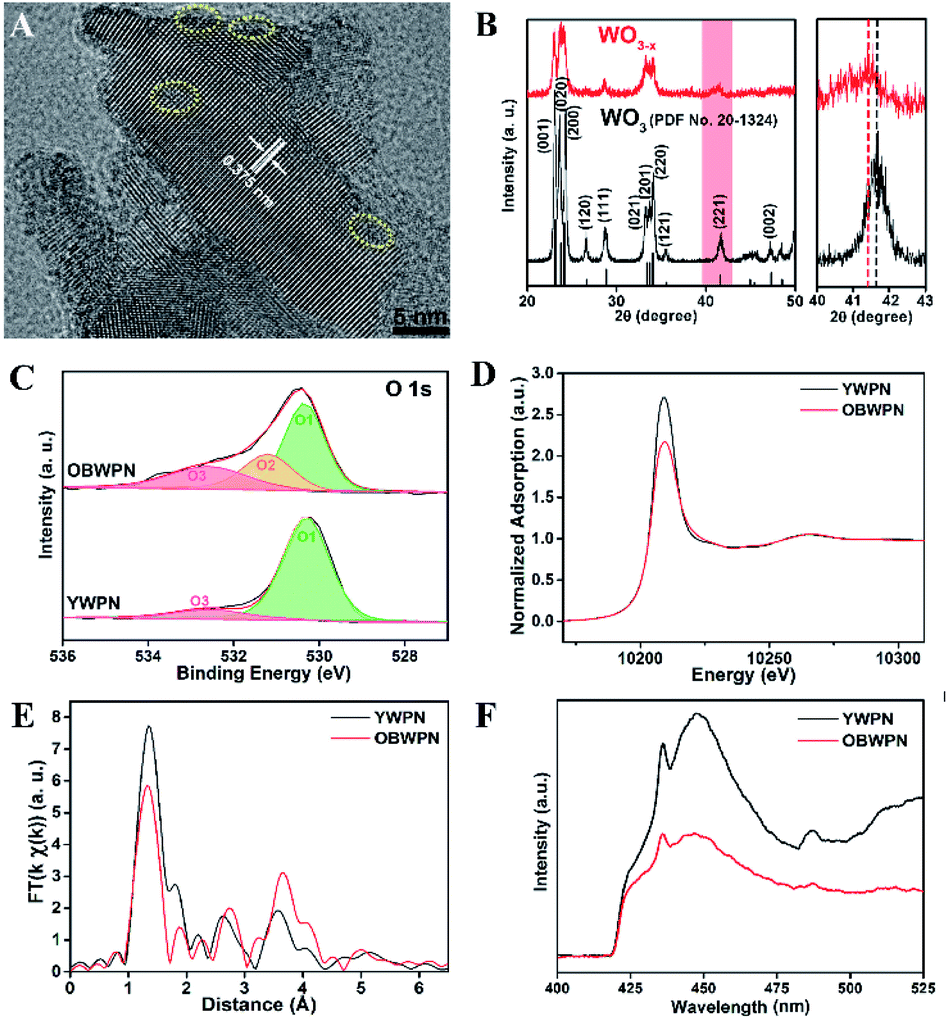
Liu et al.39 synthesized OVs-enriched blue WO3−x porous nanorods (OBWPN) without N-doping by a simple method. Fig. 4A is the HRTEM image of OBWPN, in which the lattice fringe of 0.375 nm is clearly observed, indexed to the (020) plane of WO3. The amorphous areas of OBWPN marked with a yellow circle proved that its crystallinity had decreased. Comparing the XRD spectra of yellow WO3 porous nanorods (YWPN) and OBWPN (Fig. 4B), the two XRD patterns are similar but the peak intensity of OBWPN is relatively weak, further proving that its crystallinity was reduced, which was attributed to the formation of OVs. XPS was employed to confirm the formation of OVs as well. As shown in the O 1s XPS spectra (Fig. 4C), both YWPN and OBWPN contain two typical peaks at 530.3 and 532.6 eV, which represent the W–O bonds and the hydroxyl species of water molecules adsorbed on the surface, respectively. Moreover, the unique peak of OBWPN at 531.2 eV indicates the existence of OVs, proving that the ammonia-assisted reduction strategy introduced OVs in OBWPN. In order to search for more evidences for the existence of OVs, XAFS at W L3-edge and PL spectroscopy were carried out. Fig. 4D is the X-ray absorption near-edge structure (XANES) of YWPN and OBWPN. Compared with YWPN, the peak of OBWPN decreased significantly, indicating that OVs caused the decrease in the W valence of OBWPN. Fig. 4E exhibits the fourier transform (FT) of k-weighted W EXAFS spectra for YWPN and OBWPN. The peak at ∼1.33 Å represents the W–O bond. The W–O peak magnitude of OBWPN was lower than that of YWPN, indicating that the formation of OVs reduced the W–O coordination number in OBWPN. Comparing the fluorescence peaks at 440 nm in the PL spectra of YWPN and OBWPN (Fig. 4F), it can be observed that the peak of OBWPN is weaker, which means that its recombination rate of photogenerated electrons and holes is lower, caused by OVs.
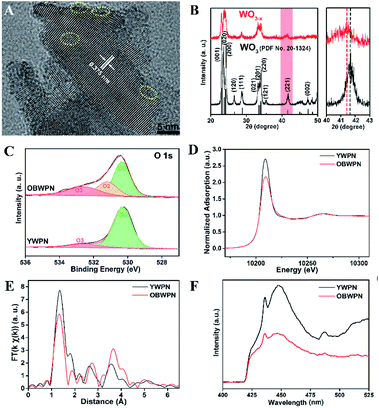 |
| | Fig. 4 (A) HRTEM image of OBWPN, (B) XRD patterns of YWPN (black) and OBWPN (red), (C) O 1s XPS spectra of YWPN and OBWPN, (D) normalized W L3-edge XANES spectra of YWPN and OBWPN, (E) fourier transform (FT) of k-weighted W EXAFS spectra of YWPN and OBWPN, (F) photoluminescence spectra of YWPN and OBWPN. Reproduced with permission: Copyright 2018 Elsevier Inc.39 | |
Ahmed et al.40 has synthesized TG (TiO2 modified by reduced graphene oxide) to reduce the ratio of the relative concentration of the bulk defects to the surface defects so as to improve CO2 photoreduction. PALS (positron annihilation lifetime spectroscopy) was employed to quantify the relative concentrations of these defects. As observed from Table 1, three positron lifetime components τ1, τ2, τ3 and their relative intensities I1, I2, I3 were recorded. The positron lifetimes τ1, τ2, and τ3 are attributed to the bulk defects, surface defects, and micropores as well as the annihilation of orthopositronium atoms, respectively. In addition, I1, I2, I3 are the relative intensity of τ1, τ2, τ3, and the ratio of I1/I2 is assigned to the relative defect concentration of the bulk to the surface. In this case, the TG had a lower ratio of I1/I2, which indicated that the coupling of graphene reduced the relative concentration of the bulk defects to the surface defects. Considering that TG has a much higher CO2 reduction activity, this result effectively proved that the surface oxygen defects can improve the photocatalytic activity more effectively.
Table 1 Positron lifetime and relative intensities of TiO2 and TG.40 Reproduced with permission: Copyright 2019, Springer Nature
| Sample |
τ
1 (ns) |
τ
2 (ns) |
τ
3 (ns) |
I
1 (%) |
I
2 (%) |
I
3 (%) |
I
1/I2 |
| TiO2 |
0.1985 |
0.3937 |
2.2900 |
54.4 |
43.9 |
1.76 |
1.23 |
| TG |
0.2069 |
0.3864 |
2.3130 |
50.1 |
48.3 |
0.67 |
1.03 |
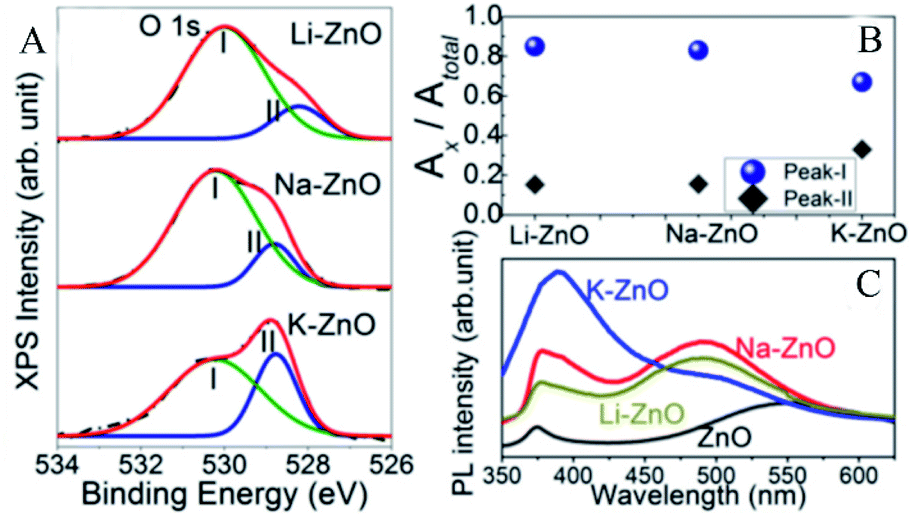
Karmakar et al.41 explored the role of OVs in enhancing the photoelectrochemical properties of alkali metal (Li, Na, and K) doped ZnO nanorods. The doped alkali metal ions replaced the zinc ions, leaving extra holes, which required electrons to neutralize, thus forming the OVs simultaneously. Through XPS analysis, it could be found that the O 1s spectra of the alkali metal doped ZnO NRs had broad peaks and could be divided into two peaks located at 531.6 eV and 529.9 eV, which represented OVs and O2− ions, respectively. The peak intensity of the OVs was related to their concentration. As shown in Fig. 5A, the peak intensities of OVs of Li and Na–ZnO NRs were higher than that of K–ZnO NRs. In this order, the corresponding peak area ratios of their OVs were 0.85, 0.83, and 0.67, respectively, which demonstrated that Li and Na–ZnO NRs contained almost equal OVs concentrations, more than that of the K–ZnO NRs (Fig. 5B). The room temperature PL emission spectra display that the doped ZnO NRs showed significant sub-band gap defect emission near 490 nm, as shown in Fig. 5C, and this deep level emission (DLE) is usually attributed to oxygen-vacancy defects.
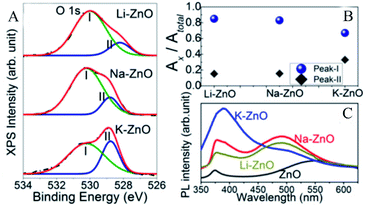 |
| | Fig. 5 High-resolution XPS spectra of (A) O 1s for the doped ZnO NRs, (B) the peak area ratios of peak-I and peak-II to the total area, (C) room-temperature PL emission spectra of the alkali metal doped ZnO NRs. Reproduced with permission: Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.41 | |
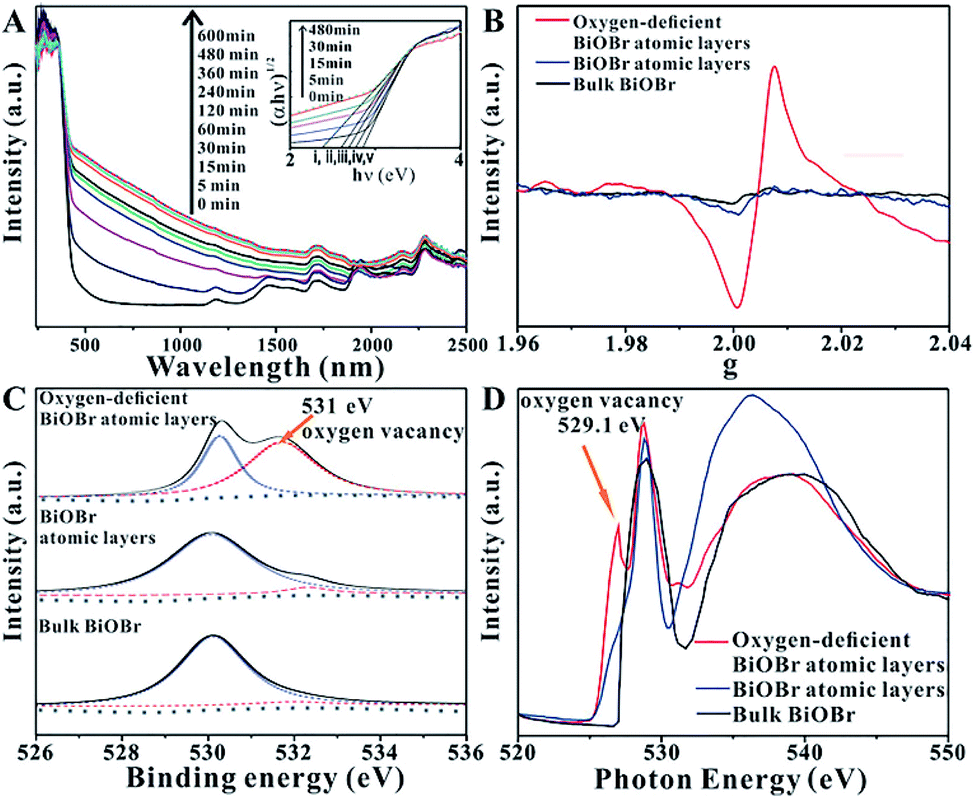
In order to enhance the efficiency of photocatalytic CO2 reduction, Wu et al.42 used high-energy ultraviolet irradiation to produce a large number of OVs on the (001) surface of BiOBr, and characterized the existence of OVs by means of XPS, XAFS, EPR, etc. As shown in Fig. 6A, UV-vis diffuse reflectance spectroscopy was used to confirm the existence of OVs in the BiOBr atomic layer. With the increase in the ultraviolet radiation to the BiOBr atomic layer, a gradually increasing new absorption peak appeared in the visible region, which was due to the formation of OVs. The absorption of visible light by the BiOBr atomic layer reached its maximum value when irradiated with ultraviolet light for 480 minutes, indicating that the oxygen defect concentration reached its maximum value at this time. In response to this situation, EPR technology was used to further prove the existence of the OVs. Fig. 6B shows that the bulk BiOBr and BiOBr atomic layers have no obvious EPR signal, indicating that there are almost no OVs. In contrast, the BiOBr atomic layer, after ultraviolet irradiation for 480 min, showed the strongest EPR signal at g = 2.004, indicating that ultraviolet irradiation did indeed cause OVs in the BiOBr atomic layer. As observed from Fig. 6C, the high-resolution O 1s XPS spectrum shows that after 480 min of ultraviolet irradiation, a new peak at 531.4 eV appears, which is a peak formed by the oxygen atoms near the OVs. In addition, as shown in Fig. 6D, the same conclusion can be obtained from the O K-edge XANES spectra. The extra peak centered at 529.1 eV can be attributed to the OVs caused by ultraviolet light irradiation.
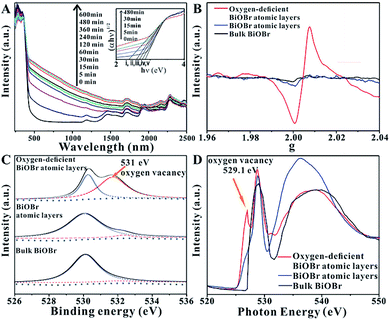 |
| | Fig. 6 (A) UV-vis diffuse reflectance spectra for the BiOBr atomic layers after UV irradiation for different periods of time, (B) EPR spectra, (C) high-resolution O 1s XPS spectra, (D) O K-edge XANES spectra for oxygen-deficient BiOBr atomic layers, BiOBr atomic layers, and bulk BiOBr (oxygen-deficient BiOBr atomic layers represent BiOBr atomic layers after UV irradiation for 480 min). Reproduced with permission: Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.42 | |
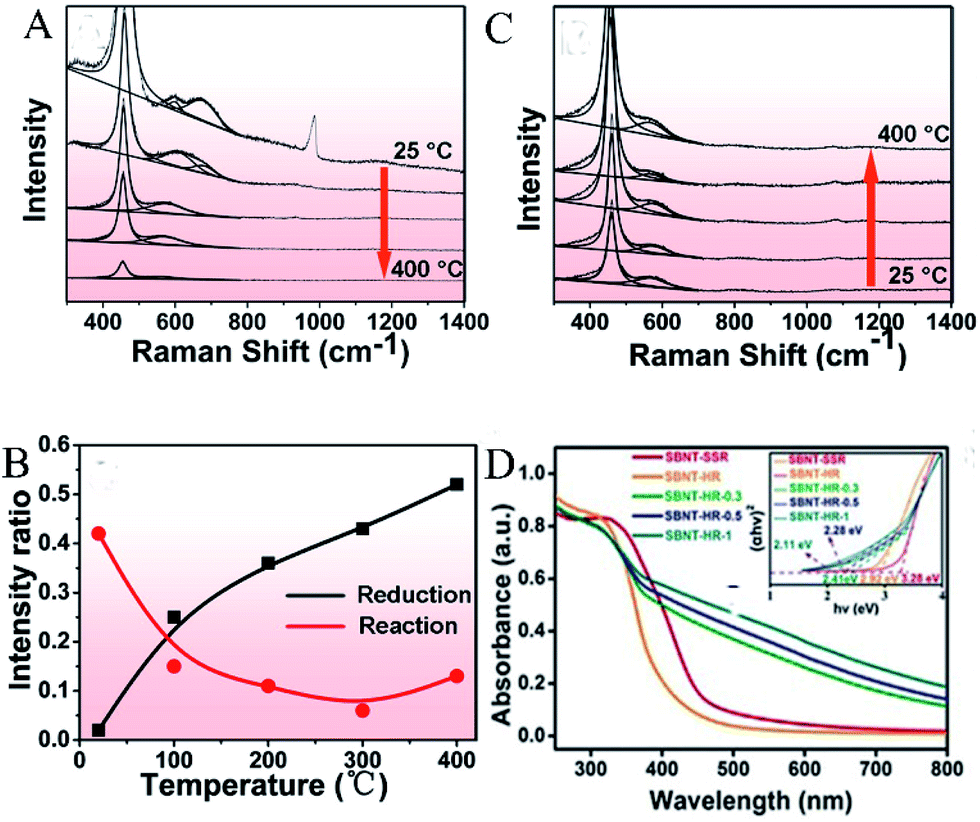
In order to understand the active site-dependent reaction mechanism, Wang et al.43 used operando spectroscopy methodology to study the Ru/CeO2 catalytic CO2 methanation reaction. OVs usually play an important role in the dissociation of oxidative bonds. Therefore, operando Raman was used to detect the changes in the OVs concentration in CeO2 during the reaction. As shown in Fig. 7A and B, the relative peak intensity ratio between the defect-induced (D) mode peak (∼570 cm−1) and the first-order F2g peak (∼460 cm−1) is related to the OV concentration in CeO2, and Fig. 7C records the value of ID/IF2g. During the reduction, as the temperature increased from 25 °C to 400 °C, the value of ID/IF2g increased from 0.02 to 0.52, proving that OVs were gradually generated during the catalyst reduction process. However, when the reaction gases (CO2 and H2) participated, the intensity ratio dropped sharply from 0.43 (at 25 °C) to 0.15 (at 100 °C), and remained low in the temperature range of 200–400 °C.
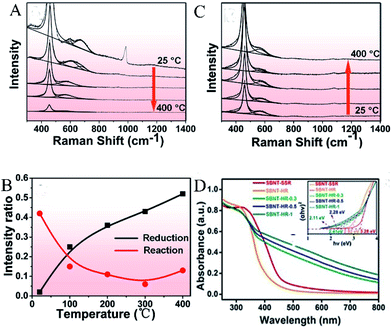 |
| | Fig. 7 Operando visible Raman spectra of the Ru/CeO2 catalyst in (A) the reduction process, (B) the reaction process. The temperature points selected in each process: 25, 100, 200, 300, and 400 °C. (C) The corresponding ID/IF2g value as a function of temperature in the reduction and reaction process. Reproduced with permission: Copyright 2020 American Chemical Society.43 (D) UV-vis diffuse reflectance spectra (DRS), band gap (inset). Reproduced with permission: Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.44 | |
4. Mechanism of oxygen-defects improving the CO2 reduction
Due to poor light absorption, slow charge separation, and inefficient surface-active sites, developing effective catalysts for achieving the conversion of CO2 into useful fuels faces enormous challenges. In recent years, the introduction of oxygen defects has become one of the most effective ways to improve the catalytic efficiency of catalysts.
4.1 Increasing the optical absorption
The light absorption capacity of a photocatalyst determines its efficiency of using solar energy. Yu et al.44 reported a simple and controllable in situ reduction method for the introduction of oxygen defects on the surface of Aurivillius-phase Sr2Bi2Nb2TiO12 (SBNT-HR). By the in situ treatment of SBNT-HR with 0.3, 0.5, and 1 mL glyoxal, various OVs-rich Sr2Bi2Nb2TiO12 (SBNT-HR-X, X = 0.3, 0.5, 1) were obtained. As shown in Fig. 7D, the light absorption range of SBNT-HR-X extended to 800 nm and as the amount of glyoxal used increased, its light absorption capacity also gradually increased, which might be caused by the increase in the oxygen defect concentration. SBNT-HR (absorption edge of 420 nm) and bulk WO3 obtained by the high-temperature solid–state reaction (SBNT-SSR, absorption edge of 460 nm) are relatively weak in the visible region. The inset in Fig. 7D shows that the band gaps of SBNT-SSR, SBNT-HR, and SBNT-HR-X (X = 0.3, 0.5, 1) are 3.28, 2.92, 2.41, 2.28, and 2.11 eV, respectively. This is because the introduced OVs brings a new defect level to the band gap, thus extending the excitation light range to the entire visible light region.
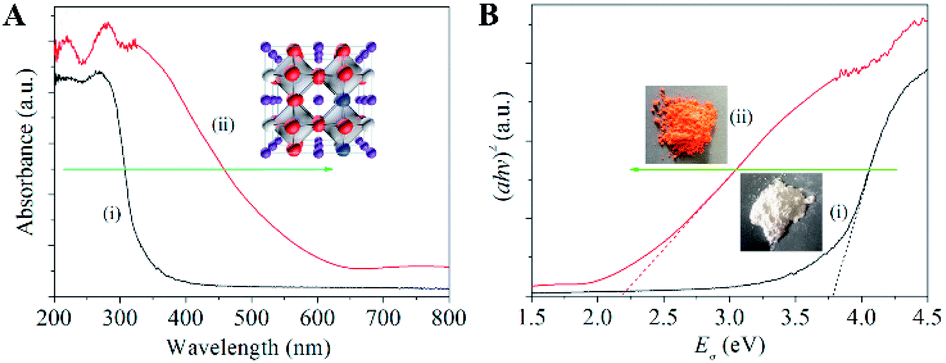
Similarly, in order to enhance the absorption of visible light, Hou et al.45 prepared sodium tantalate nanocubes with nitrogen and oxygen-vacancy confined (OVs-NaTaON) for efficient photocatalytic CO2 reduction. Fig. 8A shows the optical absorption of NaTaO3 and OVs-NaTaON crystals through UV-vis diffuse reflection spectroscopy. The absorption edge of original NaTaO3 is about 315 nm and its band gap is 3.78 eV (Fig. 8B), while the visible light absorption wavelength range of the OVs-NaTaON nanocrystals is up to 600 nm, corresponding to the band gap of 2.18 eV, as shown in the plots of the transformed Kubelka–Munk function vs. the light energy. The greatly reduced band gap proved that the introduction of OVs significantly improved the ability of the catalyst to absorb visible light. The illustration in Fig. 8B shows that the macro color of the catalyst changed from white to orange, which more clearly showed its enhanced visible light absorption capacity.
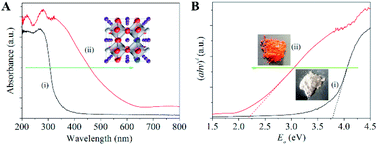 |
| | Fig. 8 (A) UV-vis diffuse reflectance spectra of (i) NaTaO3 and (ii) Vo-NaTaON crystals. (B) Band-gap energy of (i) NaTaO3 and (ii) Vo-NaTaON crystals. Reproduced with permission: Copyright 2016 Elsevier Ltd. All rights reserved.45 | |
4.2 Increasing the active sites and enhancing CO2 adsorption
The CO2 reduction processes includes four steps: adsorption, activation, reaction, and desorption. The introduction of oxygen defects can not only contribute to the enrichment of CO2 on the catalyst surface but can also serve as an electron capture center to reduce the recombination of electrons and holes as well as to provide more CO2 activation sites.
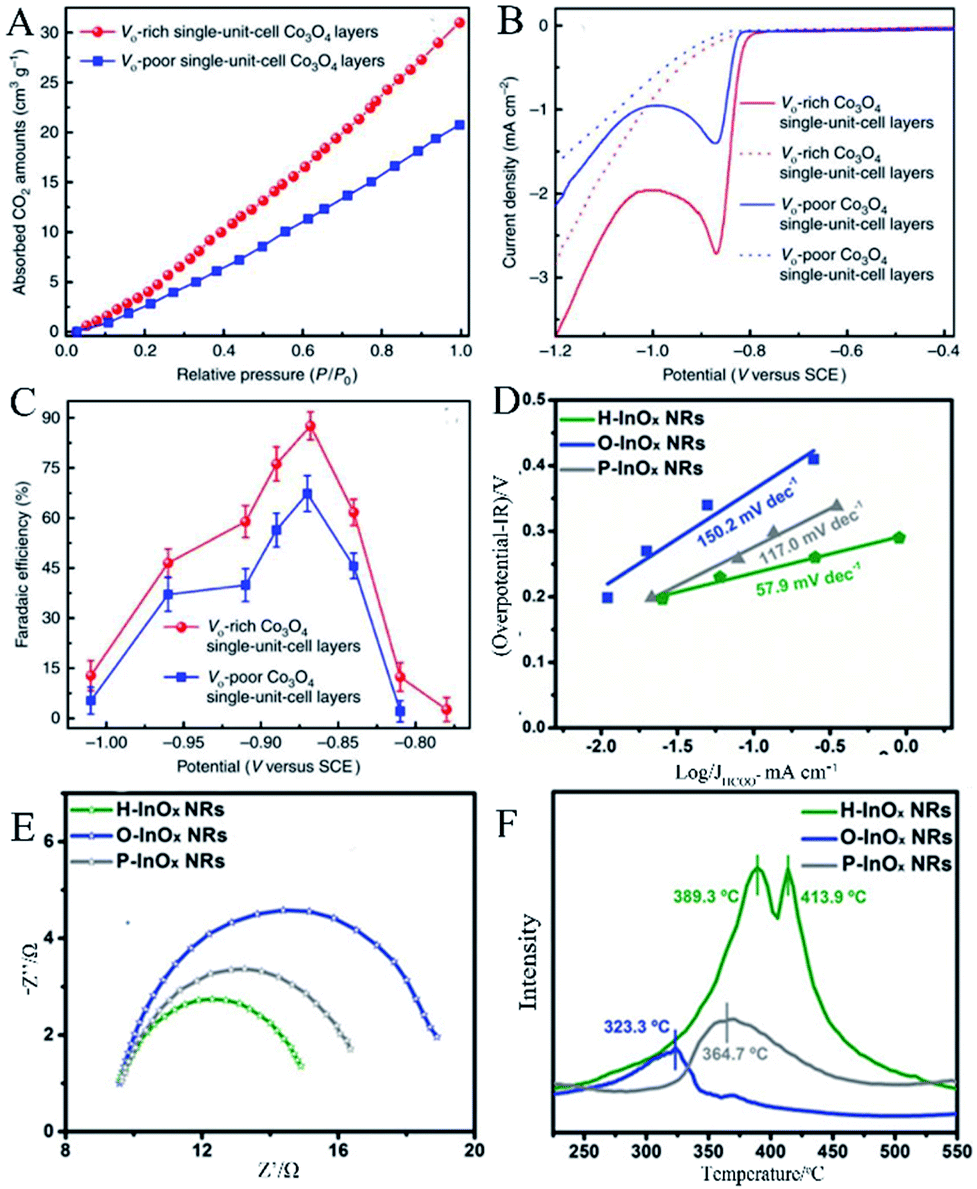
Gao et al.46 used the synthesized single-unit-cell layers of anoxic cobalt oxide as an example to construct a model for confining OVs in the atomic layer. As shown in the results of linear sweep voltammetry (LSV) in Fig. 9A, the large cathodic peaks at ca. −0.87 V versus saturated calomel electrode (SCE) could be ascribed to CO2 reduction, where the current density of the OVs-rich Co3O4 single-unit-cell layers was approximately twice that of the OVs-poor Co3O4 single-unit-cell layers, indicating that the introduction of OVs was able to improve the ability of catalytic CO2 reduction. Fig. 9B shows the result that the maximum faradaic efficiency of the OVs-rich and OVs-poor Co3O4 single-unit-cell layers was 87.6% and 67.3% for the production of formate at the potential of −0.87 V versus SCE, respectively, further proving the superior selectivity of the former in formate production. The improvement in CO2 reduction activity and selectivity could be attributed to the involvement of OVs. The increased OVs could provide more active sites for stable reduction intermediates, thereby reducing the activation energy barrier. In the measurement of CO2 adsorption, the result in Fig. 9C exhibits that the OVs-rich Co3O4 single-unit-cell layers had a higher CO2 adsorption capacity than that of the OVs-poor Co3O4 single-unit-cell layers, indicating that a higher OVs concentration contributed to an increase in CO2 adsorption.
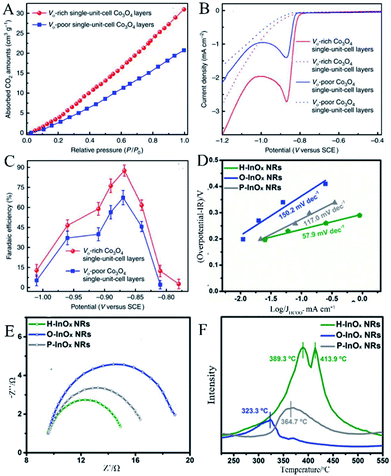 |
| | Fig. 9 (A) Linear sweep voltammetric curves in a CO2-saturated (solid line) and N2-saturated (dashed line) 0.1 M KHCO3 aqueous solution, (B) faradaic efficiencies of formate at different applied potentials, (C) CO2 adsorption isotherms. Reproduced with permission: Copyright 2020 Springer Nature Limited.46 (D) Tafel plots of HCOO−, (E) Nyquist plots, (F) CO2-TPD profiles of the P-InOx NRs, O-InOx NRs, and H-InOx NRs.Reproduced with permission: Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.47 | |
Zhang et al.47 prepared a series of OV-engineered InOx nanoribbons to enhance the absorption and activation of CO2, thereby improving the CO2 electrical reduction ability. Fig. 9D shows the current density of HCOO− under various overpotentials and the corresponding fitted Tafel slopes. The Tafel slopes of O-InOx NR, P-InOx NR, and H-InOx NR are 150.2, 117.0, and 57.9 mV dec−1, respectively, in which the Tafel slope of H-InOx NR was the lowest, demonstrating that the adsorption capacity was enhanced due to the presence of OVs. Nyquist plots were used to evaluate the interface charge transfer resistance, as shown in Fig. 9E. Obviously, the charge transfer resistance of the H-InOx NRs was the smallest, which means fastest CO2 adsorption, charge transfer, and intermediate formation processes. This is because the introduction of OVs enhanced the selectivity of CO2 reduction. To further investigate the effect of the presence of OVs on the adsorption capacity of CO2 molecules, temperature programmed CO2 desorption (CO2-TPD) was carried out and is shown in Fig. 9F. The CO chemical desorption temperature is arranged in the order O-InOx NRs (323.3 °C) < P-InOx NRs (364.7 °C) < H-InOx NRs (389.3 °C and 413.9 °C), indicating the most capable adsorption of CO2 by H-InOx NRs. The enhanced peaks of the H-InOx NRs at higher temperature further showed that the introduction of the OVs could significantly promote the chemisorption capacity of CO2, leading to high activity for CO2 reduction.
5. Conclusions
Climate change caused by excessive CO2 emission has received global attention. Using photoreduction and electric reduction methods, CO2 can be converted into valuable fuel substances such as CO and CH4. However, limited by the lack of activity and selectivity of the original catalytic material, CO2 reduction still cannot be put into practical use. Therefore, a variety of methods for introducing oxygen defects are listed, including efficient techniques such as heat treatment, doping, and illumination. Various characterization methods of oxygen defects are also summarized. The key point is to perform a qualitative analysis of the oxygen defects contained in the material by combining these characterization methods and to compare the content of oxygen defects generated. Oxygen defects mainly play a role in increasing the light absorption of the catalyst, providing the adsorption and activation sites, effectively improving the stability of the reaction intermediate during the CO2 reduction process, and maintaining great influence on the catalyst activity and selectivity. A deeper understanding of the structural modification produced by these materials themselves will further promote the industrialization of CO2 emission reduction, enabling it to be put into practical use on a large scale, and effectively improving actual energy problems and excessive CO2 emissions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the State Key Research Development Program of China (no. 2016YFA0204200). Project supported by Shanghai Municipal Science and Technology Major Project (grant no. 2018SHZDZX03) and the Program of Introducing Talents of Discipline to Universities (B16017). National Natural Science Foundation of China (no. 21822603, 21811540394, 21677048, 21773062 and 21577036), and the Fundamental Research Funds for the Central Universities.
References
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS.
- K. Caldeira and M. E. Wickett, Nature, 2003, 425, 365 CrossRef CAS.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS.
- D. W. Keith, G. Holmes, D. S. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- Y. Pu, Y. Luo, X. Wei, J. Sun, L. Li, W. Zou and L. Dong, Appl. Catal., B, 2019, 254, 580–586 CrossRef CAS.
- H.-P. Liang, A. Acharjya, D. A. Anito, S. Vogl, T.-X. Wang, A. Thomas and B.-H. Han, ACS Catal., 2019, 9, 3959–3968 CrossRef CAS.
- S. Gao, B. Gu, X. Jiao, Y. Sun, X. Zu, F. Yang, W. Zhu, C. Wang, Z. Feng and B. J. Ye, J. Am. Chem. Soc., 2017, 139, 3438–3445 CrossRef CAS.
- X. Jiao, Z. Chen, X. Li, Y. Sun, S. Gao, W. Yan, C. Wang, Q. Zhang, Y. Lin and Y. J. Luo, J. Am. Chem. Soc., 2017, 139, 7586–7594 CrossRef CAS.
- Y. Ji and Y. Luo, J. Am. Chem. Soc., 2016, 138, 15896–15902 CrossRef CAS.
- N. Guo, H. Xue, A. Bao, Z. Wang, J. Sun, T. Song, X. Ge, W. Zhang, K. Huang and F. He, Angew. Chem., Int. Ed., 2020, 59, 13778–13784 CrossRef CAS.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J. L. Wang, D. Q. Yuan and Y. Q. Lan, Angew. Chem., Int. Ed., 2019, 131, 12522–12527 CrossRef.
- P. Wu, Y. Li, J.-J. Zheng, N. Hosono, K.-I. Otake, J. Wang, Y. Liu, L. Xia, M. Jiang and S. Sakaki, Nat. Commun., 2019, 10, 1–8 CrossRef.
- T. Fogeron, P. Retailleau, L. M. Chamoreau, Y. Li and M. Fontecave, Angew. Chem., Int. Ed., 2018, 57, 17033–17037 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- X. Sun, L. Lu, Q. Zhu, C. Wu, D. Yang, C. Chen and B. Han, Angew. Chem., Int. Ed., 2018, 57, 2427–2431 CrossRef CAS.
- W. Zhu, L. Zhang, S. Liu, A. Li, X. Yuan, C. Hu, G. Zhang, W. Deng, K. Zang and J. Luo, Angew. Chem., Int. Ed., 2020, 59, 12664–12668 CrossRef CAS.
- F. Y. Gao, S. J. Hu, X. L. Zhang, Y. R. Zheng, H. J. Wang, Z. Z. Niu, P. P. Yang, R. C. Bao, T. Ma and Z. Dang, Angew. Chem., Int. Ed., 2020, 132, 8784–8790 CrossRef.
- J. Di, C. Zhu, M. Ji, M. Duan, R. Long, C. Yan, K. Gu, J. Xiong, Y. She and J. Xia, Angew. Chem., Int. Ed., 2018, 57, 14847–14851 CrossRef CAS.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS.
- X. Jin, C. Lv, X. Zhou, L. Ye, H. Xie, Y. Liu, H. Su, B. Zhang and G. Chen, ChemSusChem, 2019, 12, 2740–2747 CrossRef CAS.
- L. Lu, B. Wang, S. Wang, Z. Shi, S. Yan and Z. Zou, Adv. Funct. Mater., 2017, 27, 1702447 CrossRef.
- Y. Tong, H. Guo, D. Liu, X. Yan, P. Su, J. Liang, S. Zhou, J. Liu, G. Q. Lu and S. X. Dou, Angew. Chem., Int. Ed., 2020, 59, 7356–7361 CrossRef CAS.
- M. Vasilopoulou, A. M. Douvas, D. G. Georgiadou, L. C. Palilis, S. Kennou, L. Sygellou, A. Soultati, I. Kostis, G. Papadimitropoulos and D. Davazoglou, J. Am. Chem. Soc., 2012, 134, 16178–16187 CrossRef CAS.
- F. C. Calaza, Y. Xu, D. R. Mullins and S. H. Overbury, J. Am. Chem. Soc., 2012, 134, 18034–18045 CrossRef CAS.
- T. W. Kim, Y. Ping, G. A. Galli and K.-S. Choi, Nat. Commun., 2015, 6, 1–10 Search PubMed.
- Y. Wang, B. Wang, Y. Xu, M. Fang, Z. Wu, W. Zhu, J. Hong and C. Li, J. Chin. Chem. Soc., 2017, 64, 188–194 CrossRef CAS.
- W. Zhang, L. Cai, S. Cao, L. Qiao, Y. Zeng, Z. Zhu, Z. Lv, H. Xia, L. Zhong and H. Zhang, Adv. Mater., 2019, 31, 1906156 CrossRef CAS.
- H. Liu, H. Ma, X. Li, W. Li, M. Wu and X. Bao, Chemosphere, 2003, 50, 39–46 CrossRef CAS.
- M. Kim, B. Lee, H. Ju, J. Y. Kim, J. Kim and S. W. Lee, Adv. Mater., 2019, 31, 1903316 CrossRef.
- K. Liu, W. Zhang, F. Lei, L. Liang, B. Gu, Y. Sun, B. Ye, W. Ni and Y. Xie, Nano Energy, 2016, 30, 810–817 CrossRef CAS.
- K. Zhang, G. Zhang, J. Qu and H. Liu, Small, 2018, 14, 1802760 CrossRef.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef.
- S. Feng, T. Wang, B. Liu, C. Hu, L. Li, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 2044–2048 CrossRef CAS.
- Y. Qi, L. Song, S. Ouyang, X. Liang, S. Ning, Q. Zhang and J. Ye, Adv. Mater., 2020, 32, 1903915 CrossRef CAS.
- L. Zhang, W. Wang, D. Jiang, E. Gao and S. Sun, Nano Res., 2015, 8, 821–831 CrossRef CAS.
- D. Liu, C. Wang, Y. Yu, B.-H. Zhao, W. Wang, Y. Du and B. Zhang, Chem, 2019, 5, 376–389 CAS.
- G. Ahmed, F. Raziq, M. Hanif, J. Khan, K. S. Munawar, M. Wu, X. Cao and Z. Liu, Sci. Rep., 2019, 9, 1–8 CrossRef.
- K. Karmakar, A. Sarkar, K. Mandal and G. G. Khan, ChemElectroChem, 2018, 5, 1147–1152 CrossRef CAS.
- J. Wu, X. Li, W. Shi, P. Ling, Y. Sun, X. Jiao, S. Gao, L. Liang, J. Xu and W. Yan, Angew. Chem., Int. Ed., 2018, 130, 8855–8859 CrossRef.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS.
- H. Yu, J. Li, Y. Zhang, S. Yang, K. Han, F. Dong, T. Ma and H. Huang, Angew. Chem., Int. Ed., 2019, 58, 3880–3884 CrossRef CAS.
- J. Hou, S. Cao, Y. Wu, F. Liang, L. Ye, Z. Lin and L. Sun, Nano Energy, 2016, 30, 59–68 CrossRef CAS.
- S. Gao, Z. Sun, W. Liu, X. Jiao, X. Zu, Q. Hu, Y. Sun, T. Yao, W. Zhang and S. Wei, Nat. Commun., 2017, 8, 1–9 CrossRef.
- J. Zhang, R. Yin, Q. Shao, T. Zhu and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 5609–5613 CrossRef CAS.
Footnote |
| † Zesheng Deng and Jiahui Ji contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Jinlong
Zhang
* and
Jinlong
Zhang