
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Au nanobipyramids@mSiO2 core–shell nanoparticles for plasmon-enhanced singlet oxygen photooxygenations in segmented flow microreactors†‡
Carlos
Mendoza
 *a,
Anthony
Désert
b,
Denis
Chateau
b,
Cyrille
Monnereau
b,
Lhoussain
Khrouz
b,
Fréderic
Lerouge
b,
Chantal
Andraud
b,
Jean-Christophe M.
Monbaliu
c,
Stéphane
Parola
b and
Benoît
Heinrichs
a
*a,
Anthony
Désert
b,
Denis
Chateau
b,
Cyrille
Monnereau
b,
Lhoussain
Khrouz
b,
Fréderic
Lerouge
b,
Chantal
Andraud
b,
Jean-Christophe M.
Monbaliu
c,
Stéphane
Parola
b and
Benoît
Heinrichs
a
aNanomaterials, Catalysis & Electrochemistry (NCE), Department of Chemical Engineering, University of Liège, B-4000 Liège, Belgium. E-mail: cmendoza@uliege.be
bUniversité de Lyon, Ecole Normale Supérieure de Lyon, CNRS, Université Lyon 1, UMR 5182, Laboratoire de Chimie, 46 Allée d'Italie, Lyon, F69364, France
cCenter for Integrated Technology and Organic Synthesis (CiTOS), Research Unit MolSys, University of Liège, B-4000 Liège, Belgium
First published on 11th September 2020
Abstract
The plasmonic features of gold nanomaterials provide intriguing optical effects which can find potential applications in various fields. These effects depend strongly on the size and shape of the metal nanostructures. For instance, Au bipyramids (AuBPs) exhibit intense and well-defined plasmon resonance, easily tunable by controlling their aspect ratio, which can act synergistically with chromophores for enhancing their photophysical properties. In Rose Bengal-nanoparticle systems it is now well established that the control of the dye-to-nanoparticle distance ranging from 10 to 20 nm as well as spectral overlaps is crucial to achieve appropriate coupling between the plasmon resonance and the dye, thus affecting its ability to generate singlet oxygen (1O2). We have developed AuBPs@mSiO2 core–shell nanostructures that provide control over the distance between the metal surface and the photosensitizers for improving the production of 1O2 (metal-enhanced 1O2 production – ME1O2). A drastic enhancement of 1O2 generation is evidenced for the resulting AuBPs and AuBPs@mSiO2 in the presence of Rose Bengal, using a combination of three indirect methods of 1O2 detection, namely in operando Electron Paramagnetic Resonance (EPR) with 2,2,6,6-tetramethylpiperidine (TEMP) as a chemical trap, photooxygenation of the fluorescence probe anthracene-9,10-dipropionic acid (ADPA), and photooxygenation of methionine to methionine sulfoxide in a segmented flow microreactor.
1. Introduction
Plasmonic noble metal-based nanostructures have received intensive attention during the last decade owing to the integration of plasmonic functionality with other physicochemical properties, such as catalytic activity,1,2 optical imaging,3,4 and photodynamic/photothermal cancer therapies.5,6 Gold nanoparticles (AuNPs) possess Localized Surface Plasmon Resonance (LSPR), which arises from the collective oscillation of conduction electrons in response to incident light under resonance conditions. The size and geometry of Au NPs can be controlled during their colloidal synthesis by varying the experimental parameters in order to obtain various shapes such as spheres,7 triangles,8,9 rods,10–14 stars15 or bipyramids.16–18 These anisotropic gold bipyramids (AuBPs) possess a Transverse LSPR (T-LSPR) and an intense Longitudinal LSPR (L-LSPR) depending on the nanostructure dimensions and aspect ratio. We have already shown the controlled synthesis of AuBPs with L-LSPR from 600 nm to the NIR region16 and surface modification for preparing hybrid materials.19 We have also demonstrated the potential of such AuBPs for plasmon enhancement effects in various systems: increase of the fluorescence emission and photoresistance of coupled dyes,18 improvement of nonlinear absorption of hybrid materials,20 or enhancement of photocatalytic activity for TiO2/SiO2 thin films.21 In the present work, AuBPs have been investigated for photocatalysis via1O2 production.The transfer from the batch mode to continuous-flow mode in order to improve light–matter interactions on smaller length scales has proved that 1O2 photogeneration benefits from “going to flow”.22 Flow photochemistry has shown numerous potential benefits for this kind of photochemical reaction, such as high light penetration, improved O2 transfer to the liquid phase, ease of processing multiphase reaction mixtures, and potential to include inline analytical technologies.22–28 The most common method to generate 1O2 involves activation of ground state triplet oxygen (3O2), through energy transfer from a light activated photosensitizer (PS). Rose Bengal (RB) shows absorption bands between 480 and 550 nm, and is renowned for its high quantum yield (ϕΔ = 0.76) for the generation of 1O2.28,29 The photophysical properties of RB in close proximity to metal NPs are strongly affected by the plasmon coupling near the metallic surfaces, which may lead to an increase in fluorescence emission.30–32 In addition to such Metal Enhanced Fluorescence (MEF), Metal Enhanced Phosphorescence (MEP) and ME1O2 have also been previously described by Geddes et al.33,34 In RB–AuNP systems, the control of the dye-to-nanoparticle distance is crucial to achieve cooperative coupling between the plasmon resonance and the dye. More precisely, RB reportedly needs to be located at a distance of about 10 nm to achieve maximal enhancement for both fluorescence35 and 1O2 generation efficiencies.36
Typically, 1O2 can be revealed directly by measuring 1O2 phosphorescence at 1270 nm or indirectly, by using chemical probes that react with 1O2 producing compounds that are the detected species. In previous studies, we have studied homogeneous and heterogeneous 1O2 generation by indirect methods.28 In this work, the obtained anisotropic AuBPs@mSiO2 nanostructures with SiO2 thicknesses of 6 and 12 nm were completely characterized in terms of morphology, and spectroscopic and photophysical features. AuBPs@mSiO2 with 12 nm showed higher stability and they were chosen for 1O2 generation tests. Their impact on the 1O2 production by RB in solution was evaluated simultaneously by three complementary protocols for indirect 1O2 detection: in operando Electron Paramagnetic Resonance with 2,2,6,6-tetramethylpiperidine (EPR/TEMP), photooxygenation of anthracene-9,10-dipropionic acid (ADPA), and photooxygenation of methionine (Met) into methionine sulfoxide (MetO) using a segmented flow microreactor.
2. Results and discussion
Characterization of AuBPs and AuBPs@mSiO2 core–shell NPs
Highly truncated AuBPs, synthesized in previous studies,16,20,21 were employed as substrates for the growth of mesoporous SiO2 shells with two different thicknesses to obtain a stable physical separation between the metallic surface and the PS. Surface modifications of Au nanostructures may affect their optical properties. It is well known that the presence of SiO2 shells produces a red-shift in the LSPR of the metal nanoparticle in such core–shell nanostructures. AuBPs@mSiO2 NPs prepared with 50 and 200 μL of TEOS were characterized by UV-vis spectroscopy. The absorbance of the AuBPs and AuBPs@mSiO2 NPs dispersed in H2O and EtOH was measured between 400 and 800 nm (Fig. 1). The UV-vis spectrum of AuBPs suspended in water presented a narrow peak at 643 nm corresponding to the plasmon resonance. AuBPs@mSiO2 prepared with 50 μL of TEOS presented a plasmon resonance shifted to 648 nm, while it was shifted to 652 nm for the core–shell NPs prepared with 200 μL of TEOS. Additionally, the positive surface charge resulting from adsorbed CTA+ cations is observed on the AuBPs and AuBPs@mSiO2 NPs by zeta potential measurements (Table 1). The initial AuBPs and the obtained AuBPs@mSiO2 core–shell NPs were then dispersed in EtOH to verify the effect of SiO2. AuBPs showed a broad peak shifted from 643 to >775 nm, which is characteristic of the aggregation of AuNPs, while all the synthesized AuBPs@mSiO2 nanostructures did not show a change the position of the plasmon resonance when they were dispersed in EtOH. This slight red shift in both solvents indicates the presence of a SiO2 layer around the AuBPs and the possibility of dispersion of the core–shell NPs in EtOH demonstrated the protection of the initial AuBPs from aggregation.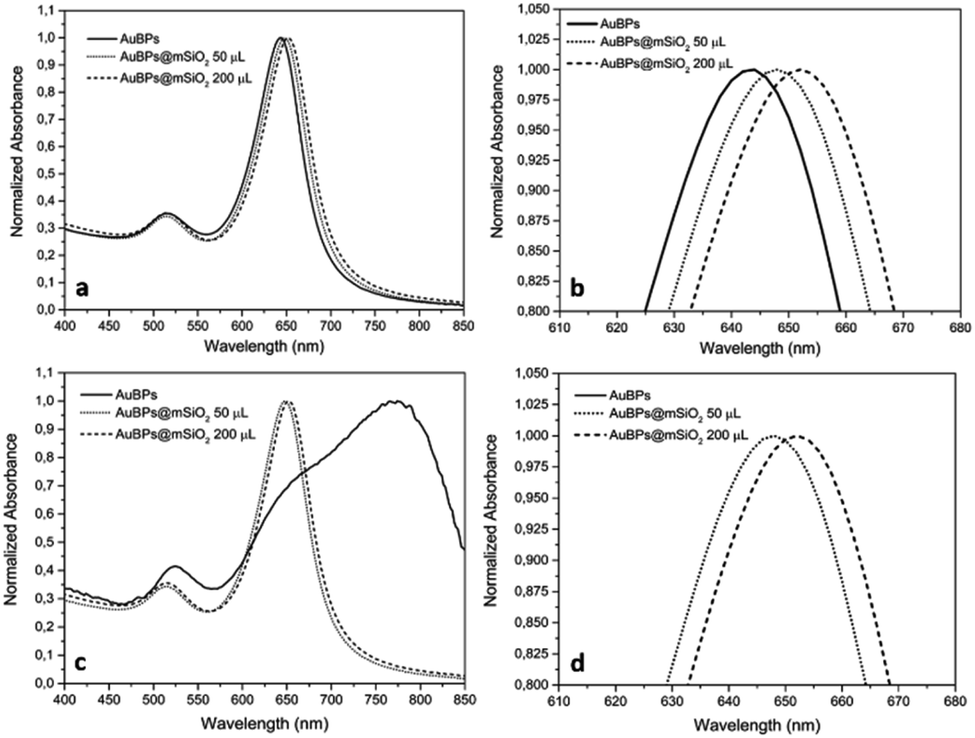 | ||
| Fig. 1 UV-vis spectra of AuBPs and AuBPs@mSiO2 core–shell NPs prepared with 50 and 200 μL of TEOS and then dispersed in (a) H2O, (b) H2O zoomed, (c) EtOH, and (d) EtOH zoomed. | ||
| T-LSPR (nm) | L-LSPR (nm) | T-LSPR (nm) | L-LSPR (nm) | Size DLS (nm) | Zeta potential (mV) | |
|---|---|---|---|---|---|---|
| Solvent | H2O | EtOH | H2O | |||
| AuBPs | 514 | 643 | 524 | Unstable | 30 | 24.0 |
| AuBPs@mSiO2 50TEOS | 514 | 648 | 514 | 648 | 45 | 40.2 |
| AuBPs@mSiO2 200TEOS | 514 | 652 | 515 | 652 | 75 | 44.8 |
Dynamic Light Scattering (DLS) is a simple and accessible technology commonly used to measure the size of nanomaterials with spherical shapes. However, this tool needs to be carefully used when measuring anisotropic nanoparticles. In our case, DLS allowed appropriate measurements of the anisotropic sample. DLS size measurements exhibited one population (monomodal) of stable NPs with hydrodynamic diameters of 30, 45, and 75 nm for AuBPs, AuBPs@mSiO2 obtained with 50 μL of TEOS and AuBPs@mSiO2 with 200 μL, respectively (see the ESI‡). Transmission electron microscopy (TEM) was employed to confirm the size of the AuBPs@mSiO2 core–shells and evaluate the thickness of mSiO2 shells (Fig. 2).
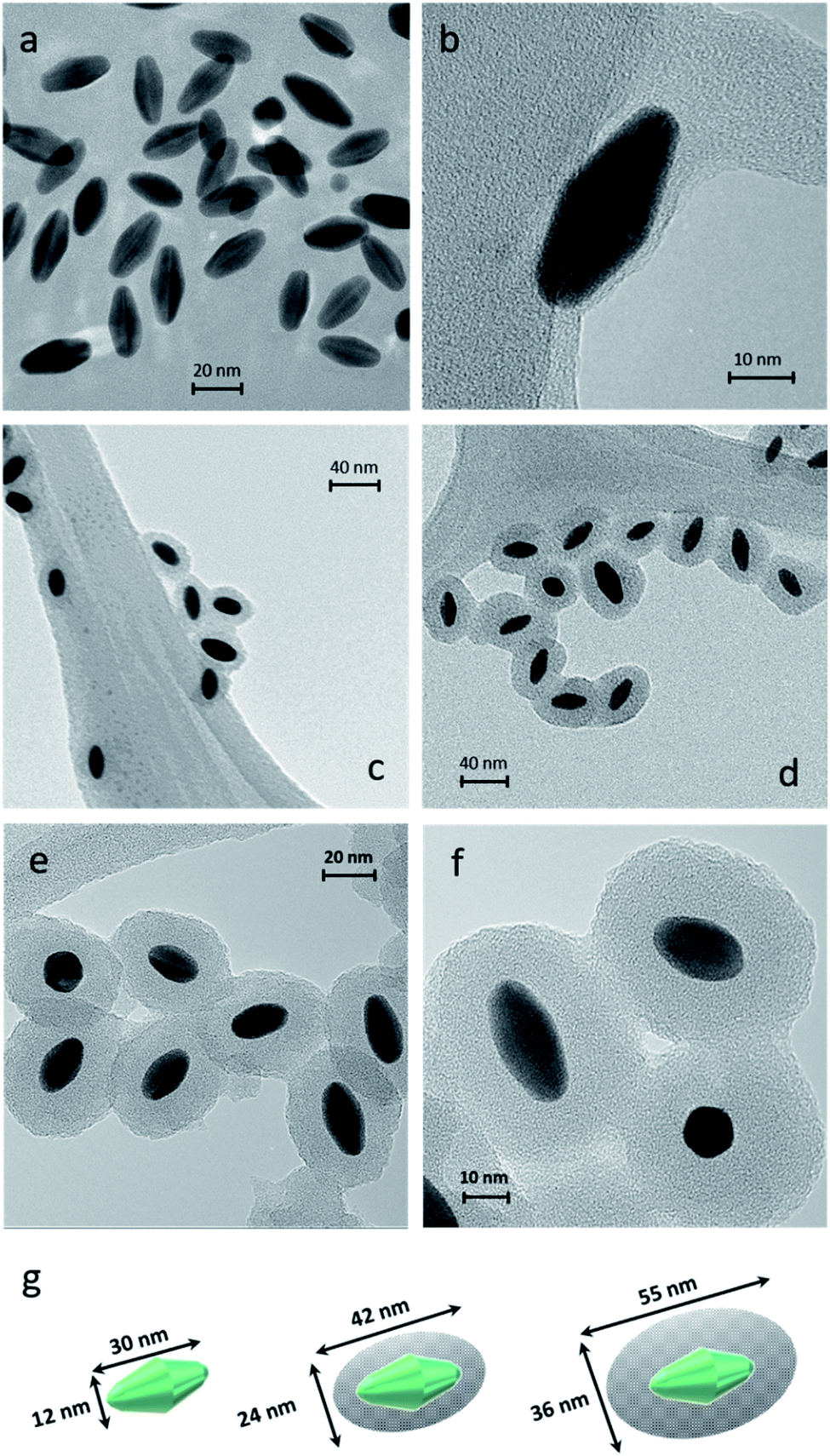 | ||
| Fig. 2 TEM images of AuBPs at a magnification of (a) 280k× and (b) 700k×, AuBPs@mSiO2 (c) 50TEOS and (d) 200TEOS at a magnification of 150k×, AuBPs@mSiO2 200TEOS with a SiO2 thickness of 12 nm at magnification of (e) 280k× and (f) 490k× and (g) size calculations of AuBPs, AuBPs@mSiO2 50TEOS and AuBPs@mSiO2 200TEOS from images (±2.1 nm from 200 particles/20 TEM images). | ||
The effect of the increasing amount of TEOS is shown in Fig. 2b–d that corresponds to the samples AuBPs, AuBPs@mSiO2 50TEOS and AuBPs@mSiO2 200TEOS. More specifically, AuBPs@mSiO2 200TEOS exhibited homogeneous mSiO2 shells with a thickness of 12 nm (±2.1 nm) (Fig. 2e and f). Fig. 2g shows the dimensions of AuBPs and AuBPs@mSiO2 core–shells calculated from TEM images (average values obtained from 200 particles/20 TEM images). The length increased from 30 to 55 nm and the width also increased from 12 to 36 nm with a final surface ratio Au/mSiO2 of (18/82) particle size distribution charts are shown in the ESI.‡ Furthermore, the quality of the encapsulation process was confirmed by UV-vis absorption. The spectra of AuBPs@mSiO2 NPs were slightly redshifted and no peak broadening was observed. These NPs were stable even in ethanol (see LSPR values in Table 1 and Fig. 6). Concordant with those results, DLS size measurements exhibited one population of stable NPs with a hydrodynamic diameter of 75 nm (see the ESI‡). Finally, the positive surface charge resulting from the adsorbed CTA+ cations is observed on the AuBPs and AuBPs@mSiO2 nanoparticles by zeta potential measurements. This is assumed to promote proximity interactions with the negatively charged RB by simple electrostatic attraction. Table 1 summarizes the characterization results of AuBPs and AuBPs@mSiO2 core–shell nanostructures. AuBPs@mSiO2 200TEOS core–shell NPs were chosen to carry out 1O2 generation tests in combination with RB solutions due to their high stability.
1O2 generation by in operando EPR/TEMP
Singlet oxygen reacts with TEMP to produce 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO) which can be observed by EPR spectroscopy.37–391O2 generation was evaluated under white light irradiation (400–800 nm) on the investigated RB/AuBPs@mSiO2 mixtures as well as on different reference samples in order to evaluate (i) the effect of the plasmon resonance and (ii) distance-tuning between RB and AuBPs. First, EPR was carried out with the photosensitizer RB in an aqueous solution (0.045 mM) (Fig. S4a‡). AuBPs and AuBPs@mSiO2 in water were also measured individually as a second and third reference (Fig. S4b and c‡). Finally, AuBPs and AuBPs@mSiO2 in the presence of RB (0.045 mM) were measured in order to estimate the effect of the mSiO2 shell (Fig. S4d and e‡).In all cases, a characteristic signal of a TEMPO radical was present in the sample even in the absence of irradiation, characteristic of a spontaneous oxidation of TEMP by addition of oxygen to generate a TEMPO derivative. This signal is not characteristic of a specific PS mediated photoinduced generation of singlet oxygen, but rather to a contamination of the TEMP sample with TEMPO. However, while no significant evolution of the system was monitored in the absence of a PS or with AuNPs alone, the RB containing sample showed a marked increase of the paramagnetic TEMPO related signal with time unambiguously indicating photosensitized 1O2 production. More significantly, a higher turn-over frequency of 1O2 was observed when RB was mixed with AuBPs in good agreement with a metal enhanced singlet oxygen generation mechanism. Finally, RB mixed with AuBPs@mSiO2 (Fig. S4e‡) showed higher EPR intensities than the sample which contained RB mixed with AuBPs (Fig. S4d‡). This increase in EPR intensities indicated that the presence of a 12 nm SiO2 shell enhances the 1O2 production of the photosensitizer containing free RB. Additionally, relative intensities (I/I0) were calculated from the EPR spectra and kinetics of TEMPO generation were also increased when we introduced the SiO2 shell, as shown in Fig. 3. An effect of saturation with the irradiation time was observed in all cases.
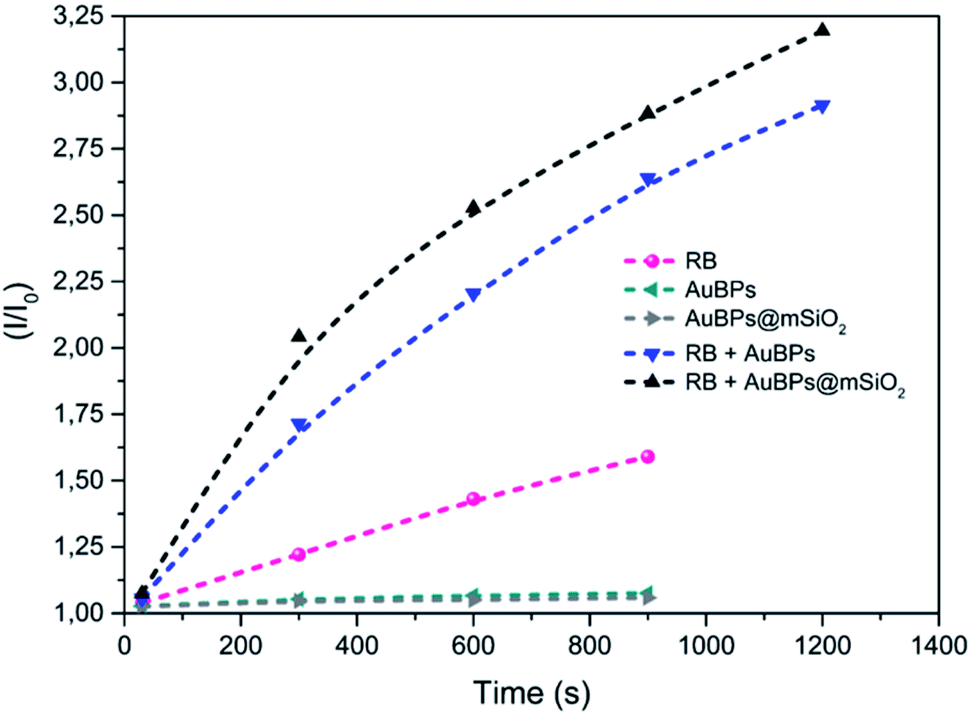 | ||
| Fig. 3 EPR relative intensities of photoinduced generation of TEMPO under visible irradiation for the different systems. | ||
1O2 production by oxidation of ADPA
In the next step, we studied the kinetics of singlet oxygen generation using spectrofluorometry. More precisely ADPA was used as a fluorescent singlet oxygen scavenger. In the presence of singlet oxygen, monitored fluorescence is expected to undergo a first order decrease (see the ESI‡), with a kinetic constant that is proportional to the singlet oxygen generation efficiency of the studied PS. Here the PS systems consisted of RB, AuBPs in the presence of RB or AuBPs@mSiO2 in the presence of RB. In all cases the same concentration of RB and NPs was used. AuBPs and AuBPs@mSiO2 in the absence of RB were also measured as references. However, in good agreement with aforementioned EPR results, these samples did not provide a significant variation of the intensity with time. As shown in Fig. 4, ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (I0/I) (where I stands for the measured luminescence of ADPA after a given irradiation time t, and I0 corresponds to luminescence intensity at irradiation time t = 0) was plotted vs. the irradiation time. ADPA oxidation kinetics were then obtained from the linear fit of the experimental data (see the ESI‡ for details about the kinetic model considered).
(I0/I) (where I stands for the measured luminescence of ADPA after a given irradiation time t, and I0 corresponds to luminescence intensity at irradiation time t = 0) was plotted vs. the irradiation time. ADPA oxidation kinetics were then obtained from the linear fit of the experimental data (see the ESI‡ for details about the kinetic model considered).
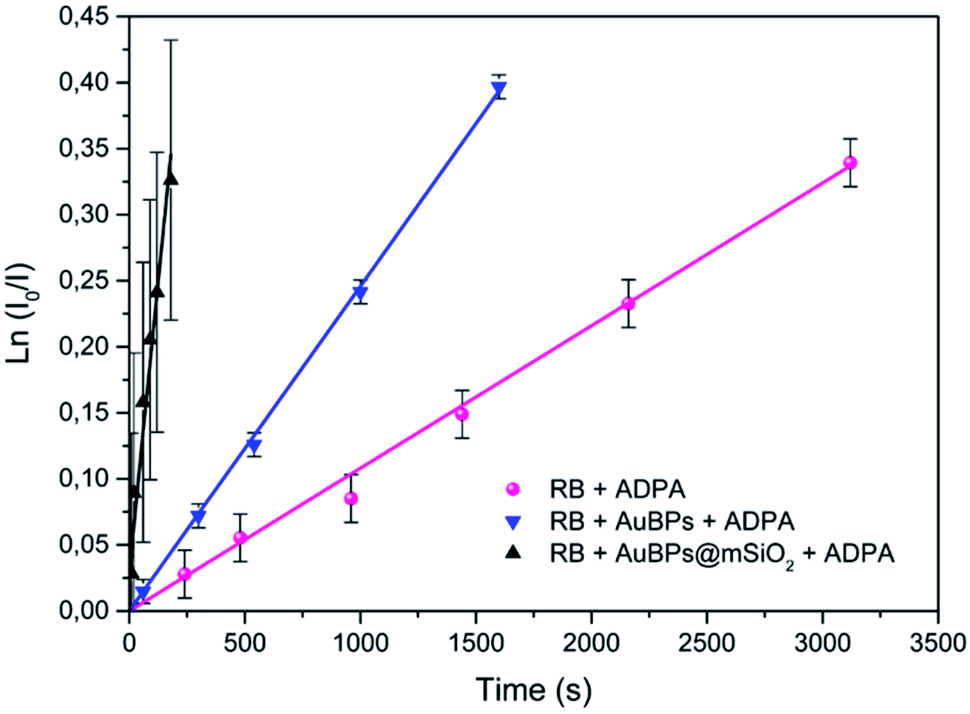 | ||
| Fig. 4 First-order kinetics linearization of the oxidation of ADPA for RB, RB in the presence of AuBPs and RB in the presence of AuBPs@mSiO2. | ||
The initial rate of RB, RB/AuBPs and RB/AuBPs@mSiO2 was 1.08 × 10−4 s−1, 2.46 × 10−4 s−1 and 16.2 × 10−4 s−1, respectively, with a correlation coefficient higher than 0.99 in all cases. It indicates an enhancement in the initial rate of 1O2 production of RB in the presence of AuBPs, which is even greater with the introduction of the 12 nm SiO2 shell for AuBPs@SiO2, maximizing plasmon induced photophysical effects by positioning the PS at an optimal distance from the gold core.
1O2 photooxygenations in a microfluidic reactor
Building upon these promising results, the photooxygenation of Met using AuBPs@mSiO2 core–shell NPs in the presence of RB was attempted under segmented flow conditions in a microfluidic setup constructed from PFA capillaries and using green LEDs as a light source to benefit the various advantages offered by the microsystem technologies.40 The performance of the reaction with AuBPs@mSiO2 NPs was compared with that under the pure RB homogeneous conditions using unsupported RB and the conversion of Met was monitored by 1H NMR. Fig. 5 shows the segmented flow with the proximity of the green LEDs and the conversion obtained with the pure RB system and RB + AuBPs@mSiO2 system for different residence times.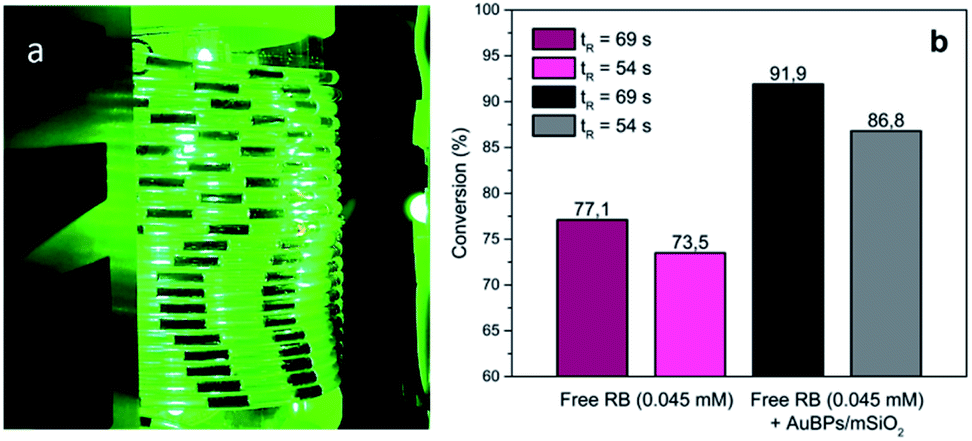 | ||
| Fig. 5 (a) G/L segmented flow, (b) conversion of Met into MetO with RB (0.045 mM) and AuBPs@mSiO2 in the presence of RB (0.045 mM) at a liquid flow rate of 0.5 (tR = 69 s) and 1 mL min−1 (tR = 54 s). | ||
Regarding the conversion of Met to MetO, 77.1 and 73.5% were obtained using free RB for residence times of 69 and 54 s, respectively. With AuBPs@mSiO2 core–shell NPs in the presence of a similar concentration of RB (0.045 mM), a higher efficiency of substrate conversion was observed under the same microfluidic conditions, which we correlate with the previously monitored metal-enhanced generation of 1O2. Namely, conversion was increased to 91.9 and 86.8% for residence times of 69 and 54 s, respectively. Additionally, UV-vis absorption spectra showed a decrease in the RB photodegradation when the PS is combined with AuBPs@mSiO2 NPs; while at short tR (54 s), the difference in the RB photodegradation is marginal, RB degradation can be decreased from 48.2 to 33.5% in the presence of AuBPs@mSiO2 NPs for a longer tR (69 s) (see the ESI‡). This revealed that the AuBPs@mSiO2 can protect the PS against oxidation and photobleaching presumably through more efficient and thus faster energy transfer from RB to surrounding molecular oxygen.
The results obtained through the three quantitative methods clearly demonstrate an enhancement of 1O2 photogeneration via RB in the presence of AuBPs, which we attribute to the coupling effect between the T-LSPR of AuBPs (λT-LSPR ≈ 515 nm) and RB dipole (λabs ≈ 540–550 nm). This increases the excitation and photostability of RB molecules under green irradiation (λlight ≈ 540–560 nm) (because of the spectral overlap between RB absorption and AuBP plasmon resonance, see Fig. 6). It should be noted that the L-LSPR could also be excited, which is the case for EPR experiments with the 400–800 nm irradiation via a halogen lamp. Finally, the significant improvements obtained with AuBPs@mSiO2 NPs, compared to AuBPs, demonstrated the importance of tuning the distance between the metal surface and the molecules for optimizing plasmon enhancement mechanisms, but also the importance of controlling the size and shape of AuNPs to combine them with the spectrum of a specific photosensitizer.6,12,41,42
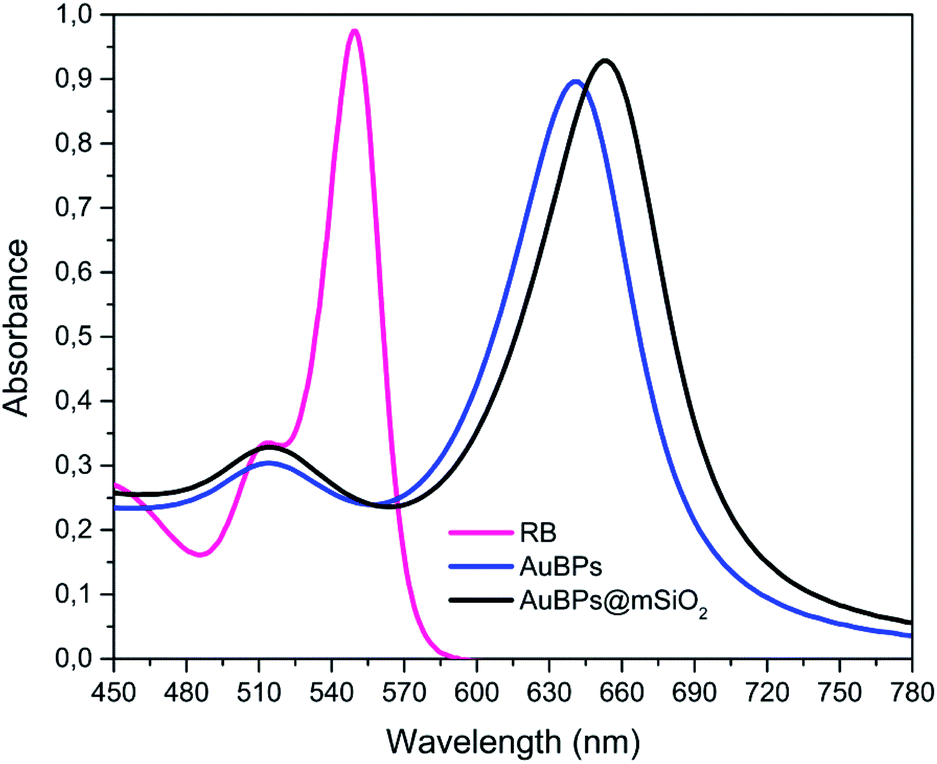 | ||
| Fig. 6 Spectral overlap of AuBPs, AuBPs@mSiO2 and RB. | ||
EPR is a spectroscopic measurement technique, and thus, direct (even using a scavenger) to detect singlet oxygen. It gives us a characteristic fingerprint of its presence. The measurements of photooxygenation of substrates such as ADPA or Met are evaluation of the oxidizing nature of the photosensitized medium, without specific information on the nature of the oxidant species. The advantage of testing ADPA disappearance is the ability to follow 1O2 kinetics in a simple in operando manner at short times and low concentrations. This technique can be carried out with controlled irradiations. Finally, photooxygenation of Met with a synthetic purpose demonstrated the goal of the catalysts though their application. The purpose of previous tests was to detect the presence of 1O2 and its behaviour during the irradiation time, while the objective of the last test was to validate the interest of the method.
3. Experimental
Materials and methods
HCl (37%), ethanol (99.8%), hexadecyltrimethylammonium bromide (CTAB, 98%), tetraethyl orthosilicate (TEOS, 99%), NaOH (98%), Rose Bengal (RB, 95%), TEMP (95%) and TEMPO (98%) were purchased from Sigma Aldrich and used as received. HAuCl4·3H2O (99.9%) and silver nitrate were purchased from Alfa Aesar, and ADPA (>98%) was obtained from Chemodex and L-methionine (Met, >99%) from TCI.Synthesis of AuBPs@mSiO2 core–shell NPs
Preparation of AuBPs using a seed mediated process has been fully described previously.16 In this work, a method adapted from Chateau et al.16 was utilized to prepare a concentrated ([Au] = 3.125 mM) suspension of truncated bipyramids in water with a longitudinal L-LSPR localized at 634 nm and a transverse T-LSPR at 512 nm.43 The concentration of CTAB was previously optimized (2.5 mM) to obtain clearly defined core–shell NPs, avoiding both the formation of AuBP clusters inside of mSiO2 or the homogeneous nucleation of individual mesoporous SiO2 NPs. 20 μL of 0.1 M CTAB solution and 10 μL of 0.1 M NaOH solution were added to 1 mL of a diluted suspension (1/5) of stock AuBPs ([Au] = 0.625 mM). After that, different amounts of TEOS were added to control the thickness of SiO2. 50 μL of TEOS/ethanol (1.75 wt%) solution was injected dropwise under stirring to form the first layer of the mesoporous silica shell (AuBPs@mSiO2 50TEOS) and then 150 μL to obtain a second layer with a 3 h interval between additions (AuBPs@mSiO2 200TEOS). The higher concentrations of TEOS did not lead to the formation of SiO2 shells (individual SiO2 spheres were observed). The reaction was conducted for 24 h, and the resultant colloidal suspension was centrifuged at 10000 rpm for 30 min and washed 3 times with 10 μL of HCl (wt 37%) to remove the maximum amount of unreacted TEOS and the maximum amount of CTAB from the pores.
Characterization
The dispersion and stability of AuBPs and AuBPs@mSiO2 were evaluated as a function of zeta potential by laser doppler velocimetry and their size was measured by DLS using a Malvern Zetasizer Nano for both measurements. The absorption spectra were recorded with a PerkinElmer UV-vis-NIR Lambda 750 spectrometer. TEM images were acquired using an FEI Tecnai G2 20 TWIN instrument operating at 200 kV, which enabled more specific size and thickness calculations of AuBPs and AuBPs@mSiO2.Singlet oxygen generation
The ability of generation of singlet oxygen was evaluated with three different methods: EPR/TEMP, oxidation of ADPA and photooxygenation of Met in a microreactor (Fig. 7).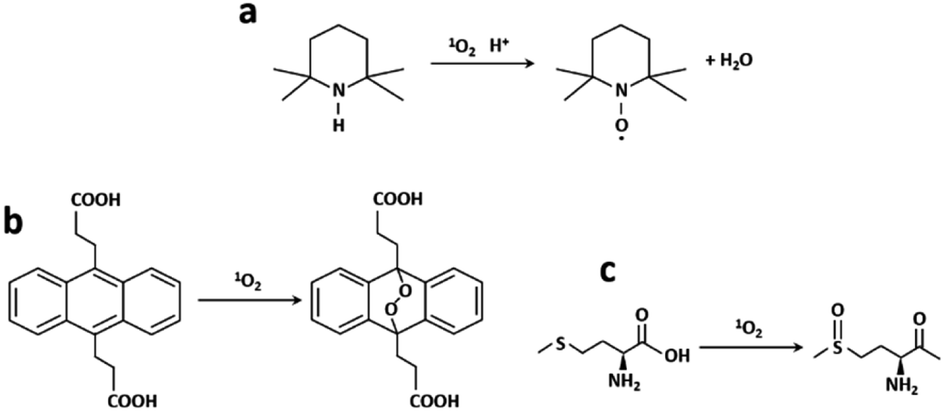 | ||
| Fig. 7 (a) TEMP to paramagnetic TEMPO, (b) ADPA to ADPA–O2 reaction in the presence of 1O2 and (c) 1O2 photooxygenation of Met into MetO. | ||
EPR/TEMP
Photogenerated 1O2 reacts with TEMP leading to the paramagnetic radical TEMPO which has a characteristic three-line EPR spectrum (Fig. 7a). RB, RB/AuBPs and RB/AuBPs@mSiO2 solutions ([RB] = 0.045 mM and [Au] = 0.065 mM) were irradiated with a halogen lamp using a filter which allowed obtaining a wavelength range between 400 and 800 nm. The samples were measured under normal conditions, with 100 kHz magnetic field modulation, 22 mW microwave power and 1.0 G modulation amplitude in a Bruker EMX Plus spectrometer, at room temperature. 1O2 generation was evaluated through double integration of the spectra obtaining I at time t.ADPA method
Polycyclic aromatic hydrocarbons such as anthracenes or rubrene are commonly used as 1O2 traps.44 Among them, ADPA, which can be monitored by both absorption and/or fluorescence spectroscopies,45–48 is highly reactive against 1O2 (reactive rate constant, kr = 8 × 107 M−1 s−1 in heavy water).49 ADPA shows structured absorption and fluorescence spectra with maximums around 380 nm and 430 nm, respectively. Upon reaction with 1O2, its characteristic absorption/fluorescence is bleached concomitant with the formation of an endoperoxide adduct (Fig. 7b).49 In this paper, the changes in the fluorescence intensity of ADPA were used to probe the kinetics of endoperoxide formation and thus to assess 1O2 generation efficiency, using a modified recently reported protocol.50In all cases, freshly prepared solutions of ADPA and of the studied photosensitizer (Rose Bengal) were placed in a 3 mL quartz cuvette (1 × 1 × 3 cm), and stirred at 60 rpm for the whole irradiation period. The absorbance of the photosensitizer was set constant in all experiments (optical density of 0.3 at 378 nm) corresponding to a concentration of Rose Bengal of 0.5 μM. The initial absorbance of ADPA was of 0.1 and the Au concentration used was 0.065 mM. All samples were then irradiated using a spectrofluorometer xenon arc lamp as an irradiation source. The irradiation wavelength was centred at 540 nm, and entrance slits were set at 7 nm opening. RB luminescence was monitored at 600 nm over the whole irradiation time, to ensure that photobleaching of the latter could be considered negligible during the experiment. This setup ensured a constant irradiation power throughout all the irradiation experiments.
Conversion of the ADPA scavenger upon reaction with the photogenerated oxygen was followed by regular measurements of the luminescence signal of the latter (considering it to be linearly proportional to its concentration, which is valid in the investigated range of absorbance) consecutive to an excitation performed at 348 nm.
Segmented flow photooxygenation of methionine
The segmented flow setup for the 1O2 photooxygenation of Met into MetO (Fig. 7c) was constructed from high purity PFA capillaries (800 μm internal diameter) wrapped in coils around a glass cylinder. The PFA coils were surrounded by 4 adjustable heat-sink integrated pillars each supporting 3 high power green LEDs (540 nm, LZ1-00G102, Led Engin) (Fig. S6‡). A Chemyx Nexus 6000 syringe pump was used to handle the suspensions loaded into a 20 mL 316 stainless steel Chemyx syringe. Two different aqueous suspensions were prepared. A first feed solution contained [Met] = 0.1 M and [RB] = 0.045 mM. The second suspension was prepared with [Met] = 0.1 M, [RB] = 0.045 mM and 1 mL of the AuBPs@mSiO2 NP suspension. Oxygen (Air Liquide, Alphagaz 1) was delivered at 10 mL min−1 with a Bronkhorst F210CTM mass flow controller. Liquid flow rates were set at 0.5 and 1 mL min−1, while the flow rate of gas was kept constant (12 mL min−1) during all the experiments. Liquid and gas inlets were then connected by a static mixer (T-Mixer, IDEX-Upchurch, natural PEEK 1/4-28 thread for 1/16′′ o.d. tubing, 0.02′′ through hole) to obtain a segmented flow. A Zaiput Flow Technologies dome-type back-pressure regulator (BPR) was inserted downstream and connected to a cylinder of compressed argon (set point: 8.5 bar). The reaction was monitored by off-line 1H NMR carried out on a Bruker Avance III 400 MHz spectrometer (see the ESI for details‡).4. Conclusions
This paper reports the synthesis of AuBPs@mSiO2 core–shell NPs from previously reported AuBPs, and the ability of the photosensitizer, RB, to enhance 1O2 generation in the presence of AuBPs and AuBPs@mSiO2 core–shell NPs. The SiO2 thickness was previously adjusted with different concentrations of TEOS in order to obtain mSiO2 shells of around 12 nm. 1O2 production of a RB PS was measured with three different methods (EPR, the ADPA method and segmented flow photooxygenation of Met) that were consistent in showing that the generation was significantly enhanced in all cases in the presence of NPs, especially when the distance between AuBPs and RB was tuned through introduction of a 12 nm mesoporous SiO2 shell. Although this distance could be slightly lower due to the porous structure of SiO2, the exact mechanism of metal-enhanced 1O2 generation is under investigation and all these results seem to point out that 1O2 generation enhancement results from plasmon mediated promotion of PS absorption, intersystem crossing and triplet state generation. These results demonstrated the possibility of drastically improving the performance of the photosensitizer Rose Bengal by combining it with AuBPs@mSiO2 core–shell NPs, which may find relevant interesting applications in several fields such as fluorescence imaging (O2 sensing), PDT or 1O2 mediated photoreactions. Segmented flow photooxygenation in microfluidic reactors introduced herein as an applicative example opens the route toward the elaboration of a new generation of devices based on plasmonic and anisotropic core–shell NPs in combination with photosensitizers for improved synthetic oxidative photochemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was funded through the ARC grant for Concerted Research Actions (icFlow - Integrated Continuous Flow Photoreactors), financed by the French Community of Belgium. C. Mendoza acknowledges the French Community of Belgium for the financial support during his scientific stay at the ENS Lyon, France. The federation RENARD, the Département de Chimie Moleculaire, Université Grenoble Alpes and Noémie Emmanuel are acknowledged for their experimental support.References
- X. Zhu, H. Jia, X. M. Zhu, S. Cheng, X. Zhuo, F. Qin, Z. Yang and J. Wang, Adv. Funct. Mater., 2017, 27, 1–15 Search PubMed.
- T. Mallat and A. Baiker, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 11–28 CrossRef CAS.
- T. Zhao, H. Wu, S. Q. Yao, Q. H. Xu and G. Q. Xu, Langmuir, 2010, 26, 14937–14942 CrossRef CAS.
- J. R. G. Navarro, F. Lerouge, C. Cepraga, G. Micouin, A. Favier, D. Chateau, M. T. Charreyre, P. H. Lanoë, C. Monnereau, F. Chaput, S. Marotte, Y. Leverrier, J. Marvel, K. Kamada, C. Andraud, P. L. Baldeck and S. Parola, Biomaterials, 2013, 34, 8344–8351 CrossRef CAS.
- G. Terentyuk, E. Panfilova, V. Khanadeev, D. Chumakov, E. Genina, A. Bashkatov, V. Tuchin, A. Bucharskaya, G. Maslyakova, N. Khlebtsov and B. Khlebtsov, Nano Res., 2014, 7, 325–337 CrossRef CAS.
- N. T. Chen, K. C. Tang, M. F. Chung, S. H. Cheng, C. M. Huang, C. H. Chu, P. T. Chou, J. S. Souris, C. T. Chen, C. Y. Mou and L. W. Lo, Theranostics, 2014, 4, 798–807 CrossRef.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939–13948 CrossRef CAS.
- L. Scarabelli, M. Coronado-Puchau, J. J. Giner-Casares, J. Langer and L. M. Liz-Marzán, ACS Nano, 2014, 8, 5833–5842 CrossRef CAS.
- J. Lv, Y. Yi, G. Q. Wu and W. Liu, Mater. Lett., 2017, 187, 148–150 CrossRef CAS.
- J. Pérez-Juste, L. M. Liz-Marzán, S. Carnie, D. Y. C. Chan and P. Mulvaney, Adv. Funct. Mater., 2004, 14, 571–579 CrossRef.
- K. Park, L. F. Drummy, R. C. Wadams, H. Koerner, D. Nepal, L. Fabris and R. A. Vaia, Chem. Mater., 2013, 25, 555–563 CrossRef CAS.
- X. Ke, D. Wang, C. Chen, A. Yang, Y. Han, L. Ren, D. Li and H. Wang, Nanoscale Res. Lett., 2014, 9, 1–8 CrossRef CAS.
- Q. Sun, Q. You, X. Pang, X. Tan, J. Wang, L. Liu, F. Guo, F. Tan and N. Li, Biomaterials, 2017, 122, 188–200 CrossRef CAS.
- P. Senthil Kumar, I. Pastoriza-Santos, B. Rodríguez-González, F. Javier García De Abajo and L. M. Liz-Marzán, Nanotechnology, 2008, 19, 015606 CrossRef.
- J. R. G. Navarro, A. Liotta, A. Faure, F. Lerouge, F. Chaput, G. Micouin, P. L. Baldeck and S. Parola, Langmuir, 2013, 29, 10915–10921 CrossRef CAS.
- D. Chateau, A. Liotta, F. Vadcard, J. R. G. Navarro, F. Chaput, J. Lermé, F. Lerouge and S. Parola, Nanoscale, 2015, 7, 1934–1943 RSC.
- Z. C. Canbek, R. Cortes-Huerto, F. Testard, O. Spalla, S. Moldovan, O. Ersen, A. Wisnet, G. Wang, J. Goniakowski, C. Noguera and N. Menguy, Cryst. Growth Des., 2015, 15, 3637–3644 CrossRef CAS.
- J. R. G. Navarro, F. Lerouge, G. Micouin, C. Cepraga, A. Favier, M. T. Charreyre, N. P. Blanchard, J. Lermé, F. Chaput, M. Focsan, K. Kamada, P. L. Baldeck and S. Parola, Nanoscale, 2014, 6, 5138–5145 RSC.
- D. Chateau, A. Liotta, D. Gregori and F. Lerouge, J. Sol-Gel Sci. Technol., 2017, 81, 147–153 CrossRef CAS.
- D. Chateau, A. Liotta, H. Lundén, F. Lerouge, F. Chaput, D. Krein, T. Cooper, C. Lopes, A. A. G. El-amay and M. Lindgren, Adv. Funct. Mater., 2016, 26, 6005–6014 CrossRef CAS.
- I. Levchuk, M. Sillanpää, C. Guillard, D. Gregori, D. Chateau, F. Chaput, F. Lerouge and S. Parola, J. Catal., 2016, 342, 117–124 CrossRef CAS.
- C. Mendoza, N. Emmanuel, C. A. Páez, L. Dreesen, J.-C. M. Monbaliu and B. Heinrichs, J. Photochem. Photobiol., A, 2018, 356, 193–200 CrossRef CAS.
- H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noël, Chem. Soc. Rev., 2016, 45, 83–117 RSC.
- B. K. Jähnisch and U. Dingerdissen, Chem. Eng. Technol., 2005, 426–427 CrossRef.
- T. Carofiglio, P. Donnola, M. Maggini, M. Rossetto and E. Rossi, Adv. Synth. Catal., 2008, 350, 2815–2822 CrossRef CAS.
- F. Lévesque and P. H. Seeberger, Org. Lett., 2011, 13, 5008–5011 CrossRef.
- T. H. Rehm, S. Gros, P. Löb and A. Renken, React. Chem. Eng., 2016, 1, 636–648 RSC.
- N. Emmanuel, C. Mendoza, M. Winter, C. R. Horn, A. Vizza, L. Dreesen, B. Heinrichs and J.-C. M. Monbaliu, Org. Process Res. Dev., 2017, 21, 1435–1438 CrossRef CAS.
- E. Gianotti, B. Martins Estevão, F. Cucinotta, N. Hioka, M. Rizzi, F. Renò and L. Marchese, Chem.–Eur. J., 2014, 20, 10921–10925 CrossRef CAS.
- B. Hu, X. Cao, K. Nahan, J. Caruso, H. Tang and P. Zhang, J. Mater. Chem. B, 2014, 2, 7073–7081 RSC.
- S. M. Mooi and B. Heyne, Photochem. Photobiol., 2014, 90, 85–91 CrossRef CAS.
- P. García Calavia, M. J. Marín, I. Chambrier, M. J. Cook and D. A. Russell, Photochem. Photobiol. Sci., 2018, 17, 281–289 RSC.
- Y. Zhang, K. Aslan, M. J. R. Previte and C. D. Geddes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1798–1802 CrossRef CAS.
- Y. Zhang, K. Aslan, M. J. R. Previte and C. D. Geddes, J. Fluoresc., 2007, 17, 345–349 CrossRef CAS.
- H. H. Lin and I. C. Chen, J. Phys. Chem. C, 2015, 119, 26663–26671 CrossRef CAS.
- O. Planas, N. Macia, M. Agut, S. Nonell and B. Heyne, J. Am. Chem. Soc., 2016, 138, 2762–2768 CrossRef CAS.
- G. Nardi, I. Manet, S. Monti, M. A. Miranda and V. Lhiaubet-Vallet, Free Radical Biol. Med., 2014, 77, 64–70 CrossRef CAS.
- J. Al-Nu'airat, B. Z. Dlugogorski, X. Gao, N. Zeinali, J. Skut, P. R. Westmoreland, I. Oluwoye and M. Altarawneh, Phys. Chem. Chem. Phys., 2019, 21, 171–183 RSC.
- C. Mendoza, A. Désert, L. Khrouz, C. A. Páez, S. Parola and B. Heinrichs, Environ. Sci. Pollut. Res., 2019 Search PubMed , in press.
- F. Jamal, G. Jean-Sébastien, P. Maël, P. Edmond and R. Christian, Microsyst. Technol., 2012, 151–158 CrossRef CAS.
- S. Clement, M. Sobhan, W. Deng, E. Camilleri and E. M. Goldys, J. Photochem. Photobiol., A, 2017, 332, 66–71 CrossRef CAS.
- N. Zhou, H. Zhu, S. Li, J. Yang, T. Zhao, Y. Li and Q. H. Xu, J. Phys. Chem. C, 2018, 122, 7824–7830 CrossRef CAS.
- D. Chateau, A. Desert, F. Lerouge, G. Landaburu, S. Santucci and S. Parola, ACS Appl. Mater. Interfaces, 2019, 11, 39068–39076 CrossRef CAS.
- C. S. Foote, Acc. Chem. Res., 1968, 1, 104–110 CrossRef CAS.
- E. Gandin, Y. Lion and A. Van de Vorst, Photochem. Photobiol., 1983, 37, 271–278 CrossRef CAS.
- D. Gao, R. R. Agayan, H. Xu, M. A. Philbert and R. Kopelman, Nano Lett., 2006, 6, 2383–2386 CrossRef CAS.
- X. Shu, V. Lev-Ram, T. J. Deerinck, Y. Qi, E. B. Ramko, M. W. Davidson, Y. Jin, M. H. Ellisman and R. Y. Tsien, PLoS Biol., 2011, 9, e1001041 CrossRef CAS.
- H. K. Yoon, X. Lou, Y. C. Chen, Y. E. Koo Lee, E. Yoon and R. Kopelman, Chem. Mater., 2014, 26, 1592–1600 CrossRef CAS.
- B. A. Lindig, M. A. J. Rodgers and A. P. Schaap, J. Am. Chem. Soc., 1980, 102, 5590–5593 CrossRef CAS.
- A. G. Margaux, T. Le Bahers, A. Banyasz, A. Duperray, A. Grichine, R. Tripier, Y. Guyot, M. Maynadier, C. Nguyen, C. Andraud and C. Monnereau, Chem.–Eur. J., 2019, 25, 9026–9034 CrossRef.
Footnotes |
| † This manuscript is dedicated to the memory of Dr. Carlos A. Páez. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00533a |
| This journal is © The Royal Society of Chemistry 2020 |