
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVapour confinement as a strategy to fabricate metal and bimetallic nanostructures†
Haritha
V. S.
ab,
Maya
Balan
b,
J. Th. M. De
Hosson
c and
Gopi
Krishnan
 *d
*d
aDepartment of Physics, University of Kerala, Kariayavattom, Thiruvananthapuram, 695581, India
bAmrita Centre for Nanosciences and Molecular Medicine, Amrita Vishwa Vidyapeetham, Kochi, Kerala 682041, India
cDepartment of Applied Physics, Zernike Institute for Advanced Materials, Faculty of Science and Engineering, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
dRenjord AS, Asbjørn Øverås veg 12 B, 7036 Trondheim, Norway. E-mail: gopi.k.krish@gmail.com
First published on 6th August 2020
Abstract
Metal nanostructures have attracted much attention in biomedical, plasmonic, hydrogen storage, and high-energy battery applications. However, the synthesis of various nanostructures of highly reactive elements (e.g. Mg) is still a difficult task and no single-approach has been reported for synthesizing such nanostructures. In this work, we produced magnesium nanoparticles (NPs), nanowires (NWs) and nanoneedles (NNs) in a single-approach, based on thermal evaporation without any carrier gas. Importantly, we employed rapid heating and cooling via a rapid thermal processing (RTP) furnace to control the nucleation and growth of nanostructures. The testing of Zn and Mg–Zn nanostructures was done to validate our approach and design for other metals and bimetallics. Interestingly, Cu and Ag nanoparticles were produced from metal salts (metal acetates and nitrates) with a reasonable control. The tuning of various nanostructures was possible by interplaying (i) with the curvature/outer diameter of the quartz bottle used for evaporation and (ii) by varying the position of the substrates. More specifically, the curvature of the quartz bottle increased the vapour collisions and effectively reduced the thermal energy of the vapour. Altogether, this favoured the control and confinement of vapour onto substrates and achieved supersaturation. Simultaneously, it led to the formation of various nanostructures without any carrier gas. The presented experimental set up is a versatile, simple, single-step and cost-effective solution for producing high-quality nanostructures.
Introduction
Metallic nanostructures are extremely interesting from both a fundamental and application point of view, as tailoring of the size, shape and aspect ratios of nanostructures can significantly enhance a wide variety of mechanical, plasmonic, electronic and catalytic properties.1–5Noble metals like Au, Ag, Pt, Pd and other transition metals such as Fe, Co, and Ni are mainly explored for diverse applications such as plasmonic sensing, bioimaging and magnetic recording.6,7 These nanostructures are mostly synthesized using wet chemical methods with an appropriate solvent, reducing agent and surfactant. Importantly, the selection of the reagents is exceptionally crucial for their controlled size distribution and design. However, the primary concern with such synthesis methods is the (i) use of organic solvents, (ii) reaction by-products, (iii) multistep reaction processes, and (iv) high concentration of surfactants required to stabilize the nanostructures.8 Although the noble and transition metal nanoparticles are well studied, there are only a few reports available on the synthesis of nanostructures of highly abundant and reactive alkaline earth metals like magnesium.9–12 Mg is a light and abundant element in the earth's crust and is recognized as an exciting candidate in the field of hydrogen storage, batteries, aerospace and biomedical applications.13–17 Recently, Mg nanostructures have shown an increasing influence of surface energy, grain boundary, and surface roughness for improved performances as compared to the bulk Mg.18
As a result of their excellent plasmonic properties at higher frequencies, Mg nanostructures like nanodisks, nanoparticles and nanohelices have been used for chiral sensing and hydrogen sensing in the UV and visible ranges.19–21 Further, electrodes in Mg/air batteries built from Mg nanostructures have shown several advantages including high energy density and theoretical voltage.22 Irrespective of their applicability in many fields, Mg/Mg-based nanoparticles are heavily investigated in the field of solid-state hydrogen storage;23–26 Nevertheless, Mg nanostructures have been least explored till date as it is exceptionally challenging to synthesize them. At the same time, any development in the form of simple synthesis techniques to produce distinct Mg nanostructures will pave the way for different applications in the near future.
Generally, in addition to the wet chemical approaches, CVD and PVD are also used for the fabrication of different nanostructures, most commonly for metal-oxide nanostructures.27–29 However, the synthesis of reactive nanostructures using CVD based-approaches is limited due to the involvement of trace impurities, toxic precursors and prolonged reaction and waiting time. In contrast, Inert Gas Condensation (IGC) based on PVD can be used to synthesize high-purity metallic/bimetallic nanoparticles.30,31 However, the production cost for such ultra-high vacuum-based deposition systems is exceptionally high. Moreover, IGC, and other synthesis methods like hydrogen plasma metal reaction,32 can provide solutions for the synthesis of metallic/bimetallic nanoparticles. Conversely, nanostructures other than nanoparticles are still challenging to produce by these methods.
In this work, a simple design is proposed to serve as an alternative for the fabrication of metallic/bimetallic nanostructures. A low-cost and vacuum-based thermal evaporation set up is adopted for the production of various nanostructures of Mg, Zn and further tested for bimetallic Mg–Zn and noble elements like Ag and Cu. Notably, we have strategically introduced a curved quartz bottle, for controlling and confining the metal vapours to the substrate without using a carrier gas, thus allowing the fine-tuning of local supersaturation ratios on the substrates to produce different nanostructures.
Results and discussion
To synthesize nanostructures via the thermal evaporation method in a tube furnace, it is necessary to transport the vapours from the evaporation zone to the substrate; simultaneously, control over the supersaturation on the substrate is necessary. However, for attaining supersaturation condition, it is necessary to confine and control the vapour and its pressure over the substrate. More frequently, both requirements are satisfied using a carrier or inert gas, e.g., Ar or N2. Trace impurities (e.g. O2 and H2O) present in the carrier gas influence the processes of nucleation and growth and often lead to the oxidation of reactive elements, e.g., Mg.31 Moreover, the use of inert/carrier gas in an ultra-high vacuum set up would also require an efficient pumping system to produce Mg nanostructures, which increases the production and set up costs. As such, to solve this issue and showcase an alternative approach without using a carrier gas, we have strategically designed and incorporated a quartz bottle inside the tubular furnace to produce different nanostructures. The designed bottle consists of a neck portion (a curvature) at one end to control and confine the vapour on the substrates.Fig. 1(a) shows Mg nanoparticles produced with sizes of 140–400 nm at a sublimation temperature of 600 °C with a holding duration of 1 min. The Mg vapour was directly deposited on the carbon-coated TEM grids using a quartz bottle of outer diameter (OD) 5 cm and neck diameter (ND) of 3 cm. The nanoparticles reflect a hexagonal prismatic shape that is commonly observed for Mg nanoparticles with a MgO shell of thickness ∼3–5 nm. The SAED pattern shown in Fig. 1(b) shows the indexed (100), (002), (101) planes that match the characteristic reflections of the Mg and (200) plane of MgO. Once the neck diameter of the quartz bottle decreased to 2 cm, the average diameter of Mg nanoparticles was reduced from 240 ± 3 nm to 60 ± 3 nm. Fig. 1(c) confirms the reduction of Mg nanoparticle size as it varies from ∼30–100 nm. As the size is reduced, the Kirkendall void due to oxidation is observed, as reported previously for Mg nanoparticles.30 The inset in Fig. 1(d) shows a void formation in Mg nanoparticles. Fig. 1(d) shows the HRTEM image of Mg nanoparticles resolving the (101) plane of Mg and (200) plane of MgO.
 | ||
| Fig. 1 (a) Bright-field TEM image of Mg nanoparticles produced at a sublimation temperature of 600 °C in the 3 cm OD quartz bottle; (b) the corresponding SAED pattern. (c) Bright-field TEM image of Mg nanoparticles produced at 600 °C in the 2 cm OD quartz bottle and (d) the corresponding HRTEM image of the particle; the inset shows a higher magnification image of a single particle, and the arrow indicates the void formation. | ||
In comparison, as the neck diameter (ND) of the quartz bottle is reduced, the size of the particle decreases, which can be explained with the help of the schematic representation shown in Fig. 2. Fig. 2(d) and (e) represent the influence of quartz bottles having ND of 4 and 2 cm in controlling the Mg vapour deposition on to the substrate. The observation from the schematic indicates that the deposition on the substrate with the 2 cm ND quartz bottles is less as compared to that with 4 cm. Moreover, this indicates that the Mg vapour concentration is reduced on the substrate. Thus, the rate of Mg deposition was also reduced after the neck region. This schematic is based on our experimental evidence for a constant holding duration. Importantly, Mg vapour can be controlled and confined by the curvature before the substrate. Thus, it reduces the thermal energy of Mg vapour as the mean free path of Mg atoms is reduced due to collision. In principle, a pressure gradient is created before/after the curvature that favours the control of the Mg deposition. Nevertheless, the deposition rate and yield can be increased by various other means such as (i) increasing the initial quantity of the source powder, (ii) or by increasing the holding duration and (iii) by decreasing the heating rate. Please see Fig. S1 and S2 in the ESI,† which show a higher deposition yield on stainless steel and other metal substrates.
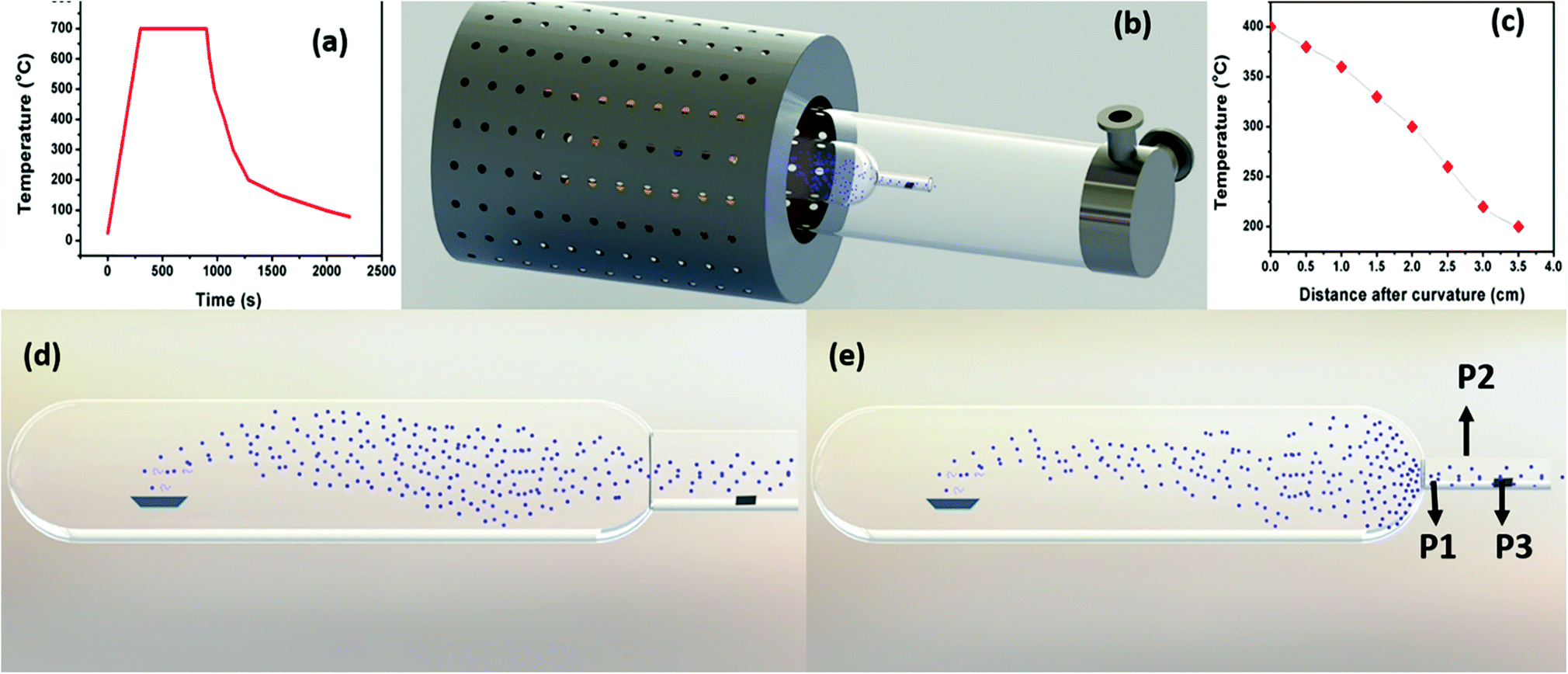 | ||
| Fig. 2 (a) Temperature profile of the RTP furnace at a source temperature of 700 °C for a holding duration of 15 minutes. (b) Schematic representation of the overall set up. (c) Temperature gradient profile concerning the distance from the curvature at a source temperature of 700 °C. (d) Schematic representation of Mg sublimation and deposition in the 4 cm OD quartz bottle and (e) the 2 cm OD quartz bottle. | ||
Conversely, once the ND increases to 3 cm, the neck region is not efficient in confining the Mg vapour. Thus, this subsequently allows more high energy vapour to pass through the neck region and deposit on the colder region. This observation was confirmed by our experimental evidence, which indicates that by the introduction of the quartz bottle, the rate of Mg vapour deposited on the substrates can be controlled, and also depending on the ND (3 cm or 2 cm), the total area of the deposition region behind the curvature can also be varied. For a curvature OD of 3 cm, the deposition area was greater as compared to the OD of 2 cm. However, in contrast, the control over the vapour confinement becomes impossible once the curvature/neck region is not present, or the diameter of the bottle is increased.
To further validate the necessity of the curvature, we also performed Mg sublimation in a test tube having an OD of 2 cm with the same length as that of the quartz bottle mentioned in the previous section. This tube has a uniform length without any neck/curvature. Fig. S3 in the ESI† shows bright-field TEM images of Mg structures of size between 4–7 micrometres deposited on the carbon-coated TEM grid. The replacement of the quartz bottle with a test tube tremendously increased the deposition rate over the substrate. However, this change resulted only in the formation of bulk Mg structures, thus justifying that the reduction in the OD from 3 cm to 2 cm is not critical in the formation of nanostructures, but rather the increased curvature of the bottle is a crucial aspect of our observation. Moreover, this difference has allowed the formation of smaller Mg nanoparticles on the substrate even without a carrier gas. A detailed analysis of the effect of the curvature using theoretical calculation and simulation will be necessary to uncover the complete understanding of the process.
In contrast, using a quartz tube of OD 8 cm, without the quartz bottle and the test tube, we could only observe the formation of Mg or MgO thin film; this was predicted and observed in a normal thermal evaporation process. The formation of the MgO film is due to the oxidation of the Mg film during its exposure to the outside atmosphere. The bright-field TEM image of MgO film and the corresponding MgO SAED pattern are shown in Fig. S4 of the ESI.† This suggests that in a normal thermal evaporation set-up, the vapour collected on the substrate generally possesses high energy and thus collisions on the substrate are transformed into a thin film. However, once the mean free path of the vapour is reduced, it favours the formation of nanoparticles. Nevertheless, such a condition in a standard set-up is possible only when the pressure of a carrier gas/sputter gas is interplayed and increased. Alternatively, without such a possibility, we can alter the mean free path, the energy of Mg atoms and vapour concentration by introducing a quartz bottle. It is essential to point out that without this modification, it is impossible to deposit Mg or have nanoparticle formation on substrates via thermal evaporation in a tube furnace (Fig. S2 in the ESI† shows aggregated Mg nanoparticles on copper, nickel and stainless-steel substrates). The problem associated with Mg deposition has already been reported during Mg nanowire formation.33 Nonetheless, without the use of carrier gas, we can control the supersaturation over the substrate. The significant temperature gradient created by the rapid heating furnace is one of the prerequisites observed for nanostructure formation. Moreover, by eliminating the carrier gas, we have forcefully stopped any impurities, especially (O2) playing a role in the oxidation of Mg.
To understand the effects of the temperature and position of a substrate, the 2 cm ND quartz bottle was placed as constant, and Mg sublimation was performed at 700 °C with a holding duration of 15 minutes to synthesize various Mg nanostructures on different substrates (e.g., TEM grid and stainless steel). The substrates were kept at three different positions, P1 being closer to the neck region, P2 is an intermediate position and P3 in the colder region, which is farther from the neck as shown in Fig. 2(e). Fig. 3(a) shows the bright-field TEM image of hollow Mg microparticles that are formed at position P1 (∼350–400 °C). These particles appeared to be faceted having a size of around 1 μm with a hollow inner core and an outer MgO shell. This suggests that the process of sublimation follows the formation of Mg particles. In general, the sublimation temperature of Mg decreases as the vapour pressure of Mg increases in a vacuum (10−6 mbar).34 Moreover, as the particle size decreases, the vapour pressure of Mg further increases, leading to the sublimation of nanoparticles at 350–400 °C30 and hollow MgO particles are formed. A high-magnification bright-field TEM image shown in Fig. 3(b) clearly shows hollow MgO microparticles and the presence of small crystals on the surface of the particles. These smaller particles are formed due to Mg condensation after the sublimation process. The formation of hollow MgO particles was confirmed from the SAED pattern shown in Fig. 3(c), which shows the presence of the (111), (200), (220) and (311) reflections of MgO.
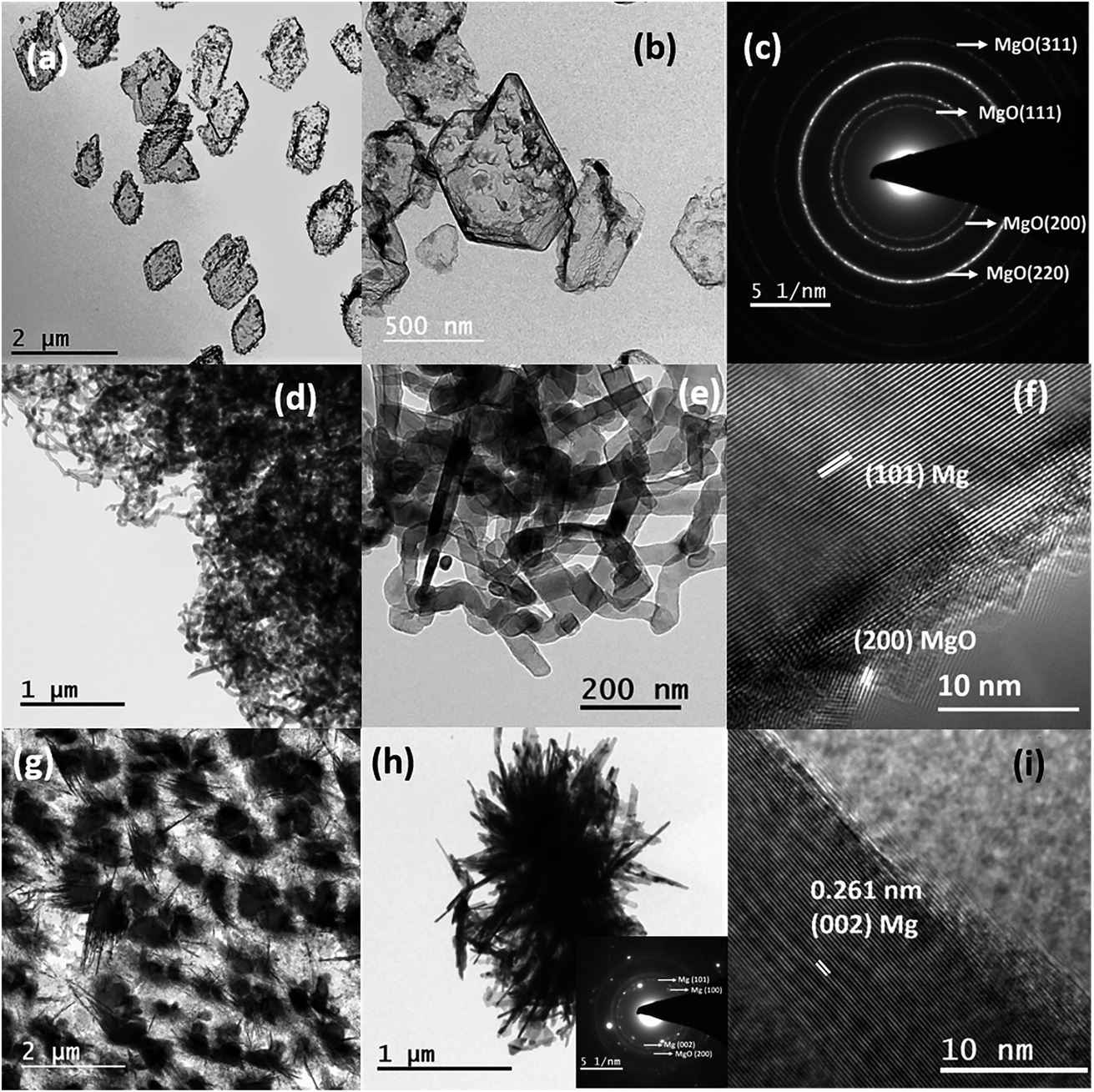 | ||
| Fig. 3 (a and b) The bright-field TEM images of hollow MgO microstructures at a sublimation temperature of 700 °C and a holding duration of 15 minutes at position P1; (c) the corresponding SAED pattern. (d and e) The serpentine-like Mg nanowires at 700 °C, position P2. (f) The HRTEM image of a single nanowire. (g and h) Bright-field TEM images of Mg needles; the inset represents the corresponding SAED pattern. (i) HRTEM image, deposited at position P3. | ||
Once the position of the substrate shifted to an intermediate position P2 and a temperature regime between 300–350 °C, instead of hollow particles, the formation of the Mg nanowire was observed as shown in Fig. 3(d) and (e). The Mg nanowires had a size in the range of 30–60 nm with a MgO shell of thickness ∼3–5 nm. The HRTEM image of the nanowire shown in Fig. 3(f) confirmed an interplanar distance (d) of 0.245 nm that corresponds to the (101) lattice planes of Mg. The serpentine-like nanowires tend to show different growth directions for different nanowires, e.g. [101], [100], [002], etc. This could be due to the random orientation of nanowires at higher growth temperatures as well as increased vapour concentrations that affect the kinetics of nanowire growth. The serpentine-like Mg nanowires were formed from the melted Mg microparticles using the vapour-solid growth mechanism; the growth process for these Mg nanowires is discussed in detail elsewhere.33 Subsequently, once the position of the substrate was moved to a colder region P3 (∼250 °C), the formation of the needle-like structure of diameter less than ∼20 nm was observed as shown in Fig. 3(g) and (h). Moreover, the HRTEM image shown in Fig. 3(i) again confirms the formation of Mg by indicating the (002) lattice plane.
As compared to Mg NWs, the NNs are shorter in length and the diameter decreases from the base to the tip. Once the NNs start to grow from the microparticles as mentioned earlier for nanowires, the Mg atoms need to diffuse from the microparticles (base) to the tip, since the source for Mg growth was found to be from the microparticles and not from the direct vapour. Moreover, during Mg vapour deposition at position P3, the temperature and vapour concentration on the substrate were less in comparison to the other positions, so the number of atoms diffusing from the base to tip decreased.35 This resulted in the gradual reduction in the diameter from the base to the tip and led to the formation of Mg nanoneedles. Since our experiments were conducted in a rapid thermal processing (RTP) tubular, single-zone furnace, the sublimation/source temperature significantly affected the temperature gradient between the source and the substrate. The overall temperature profile for the RTP furnace at a sublimation temperature of 700 °C and a holding duration of 15 minutes is represented as Fig. 2(a), and the temperature profile (in the deposition region) with respect to the distance from the curvature at a constant source temperature of 700 °C is also represented in Fig. 2(c).
Fig. 2(c) further indicates a gradual reduction in the temperature after the curvature, which is directly dependent on the source temperature. To demonstrate this, Mg deposition was carried out at a lower sublimation temperature of 600 °C at various positions, P1, P2 and P3, identical to those used at 700 °C. Once the temperature was reduced from 700 °C to 600 °C, the effect of temperature on each substrate decreased in the corresponding identical positions (P1, P2 and P3). Fig. (S5) (ESI†) represents the bright-field TEM images of Mg nanostructures that were deposited when the substrate was placed at position P1 (∼250–300 °C). Although at the same substrate temperature (∼250–300 °C), at P2 for 700 °C, the deposition resulted in nanowire formation; at 600 °C when the substrates were placed closer to the heating zone at P1, the partially melted Mg microstructures were obtained. This distinctly infers that in addition to the temperature change with respect to the position, the Mg vapour concentration/vapour pressure also varies (will be higher at P1, closer to the curvature), which affects the growth of different structures. From this, it can be understood that the temperature at position P1 was not sufficient to completely sublimate the particles. As a result, partially melted microparticles were observed at this position. At position P2 (∼200–250), shorter and straighter Mg nanowires were found to be growing from Mg microparticles as shown in Fig. S6(a) and (b) in the ESI,† and these nanowires had diameters (10–15 nm) that were less than that of the serpentine-like Mg nanowires that are formed at a sublimation temperature of 700 °C. The SAED pattern shown in Fig. S6(c)† confirms the presence of Mg. The growth process for the straight Mg nanowires is different from that of serpentine wires, and the former shows a nanoparticle attached growth mechanism from the microparticles, which was explained in detail elsewhere.33
The most important observation to be noted from the obtained morphologies is the absence of defects (e.g. screw dislocations, stacking faults or twin boundaries), which mostly favours the growth of one-dimensional structures.35 In our case, the growth of one-dimensional nanostructures is entirely dependent on the supersaturation conditions and the surface energy of the growing planes. As the substrates were further moved apart at P3 (150–200 °C), instead of needle-like structures originating from the microparticles, smaller spherical Mg nanoparticles with size between 20–40 nm were observed from Fig. S6(d) and (e).† Compared to the Mg nanoparticles shown in Fig. 1, these particles were formed from the melting of the microparticles during solidification. They were not transformed by the processes of adsorption, nucleation, coalescence and growth of particles on the substrate. Moreover, the difference in the morphologies observed at 700 °C and 600 °C at identical positions clearly indicates that the growth of the nanostructures is sensitive to both source temperature and position. The formation and growth of different nanostructures on the substrates with respect to the position and the sublimation temperatures directly pinpoints the role of supersaturation. The degree of supersaturation strongly depends upon the experimental parameters like temperature and pressure. Under our experimental conditions, the temperature of the substrates plays a crucial role in determining the supersaturation conditions, which can be altered with respect to the sublimation temperature, holding duration and the position of the substrates. It is well known that the supersaturation ratio is given as follows:
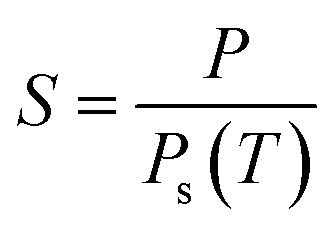
With this general concept of local supersaturation ratios and the formation of different nanostructures on the substrates, if we try to interpret the formation of different Mg nanostructures with respect to the substrate position and temperature, the substrate, which is kept closer to the heating zone (position P1), will have a higher substrate temperature and the corresponding local supersaturation ratio will be much less (S = 0). These conditions will favour the formation of Mg microstructures only. However, at the same time, sublimation temperatures of 700 °C and 600 °C resulted in the formation of hollow MgO microstructures and melted Mg microstructures with the variations in the substrate temperatures. The formation of microstructures clearly pinpoints that low supersaturation conditions only favour the formation of bulk structures. Alternatively, once the substrate position is changed to a lower temperature, instead of Mg microstructures, anisotropy is favoured on the substrate, leading to a supersaturation ratio of S > 1. This has resulted in the formation of one-dimensional structures like NWs and NNs. Generally, the anisotropic growth of nanostructures depends on the fundamental crystal symmetry and structure of the material.39 Mg possesses a hexagonal closed packed crystal structure and has low crystal symmetry as compared to face centred cubic (FCC) metals like Cu, Al, etc., which results in higher anisotropic surface diffusions of atoms. This will favour the formation and growth of one-dimensional nanostructures.9,40,41 Consequently, two types of Mg nanowires, namely serpentine-like and straight nanowires, and nanoneedles were formed at different source temperatures and substrate positions.
A decrease in temperature with respect to the sublimation zone will further increase the degree of supersaturation (S ≫ 1) and simultaneously favour the spontaneous nucleation and isotropic growth of Mg nanoparticles. It is important to note that the Mg nanoparticles do not show any void formation in comparison to the Mg nanoparticles that were grown on the substrate, as shown in Fig. 1. This indicates that the nanoparticles grown from the bulk or melt particles have less chance of defect formation.
Fig. 4(a) shows a schematic representation of the overall Mg nanostructures with respect to the substrate position and sublimation temperatures. Fig. 4(b) also represents a qualitative supersaturation curve concerning the temperature gradient in the substrates. The supersaturation ratio was found to increase as the temperatures of the substrate decreased, and the curve of our set up represents a similar trend as observed in the normal thermal evaporation in tube furnaces with a carrier gas flow.37 Finally, with the favour of the curvature and the temperature gradient induced by rapid thermal heating, the supersaturation can be controlled and varied to produce different nanostructures.
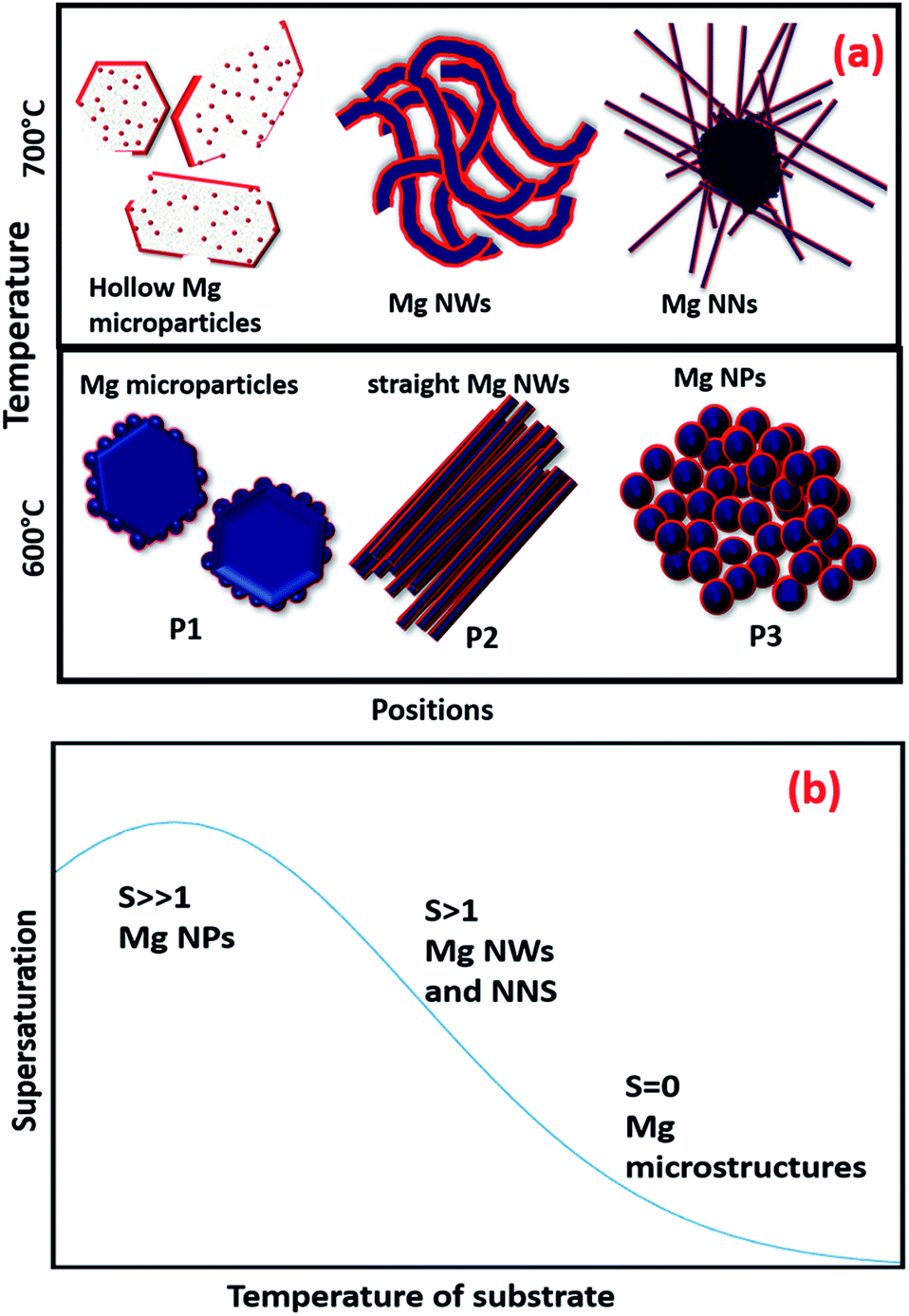 | ||
| Fig. 4 (a) Schematic representation of different Mg nanostructures and (b) the supersaturation curve with respect to temperature. | ||
After the complete understanding of the formation of Mg nanostructures, to further highlight the potential and versatility of our experimental set up, we adopted the same methodology for the fabrication of nanostructures of other lower melting point and higher vapour pressure metals like Zn. The same 2 cm ND quartz bottle that was adopted for the production of Mg nanostructures was utilized for Zn as well. The bulk Zn powder was taken as the source material and sublimation was carried out at 600 °C. Fig. 5(a) and (c) show the bright-field TEM images of Zn nanostructures that were deposited on the carbon-coated TEM grids at different substrate positions P2 (∼200–250 °C) and P3 (∼150–200 °C). Similar to the Mg nanoparticles, Fig. 5(a) shows a uniform distribution of Zn nanoparticles/nanodisks with hexagonal shapes. The SAED pattern of a single Zn nanoparticle, shown in Fig. 5(b), confirmed the formation of Zn nanoparticles with a ZnO shell by indicating the (101) and (100) characteristic reflections of Zn and ZnO. Zn nanowires were densely distributed over the substrates in comparison to Mg nanowires. They also showed a zigzag or serpentine-like morphology, which is generally reported for higher vapour pressure/low melting metals. The diameter of the wires ranged from ∼50–250 nm with the length of a few micrometres.42 The HRTEM image of a single Zn nanowire in Fig. 5(d) shows a thin oxide layer having a thickness of ∼2.5 nm. This confirmed the formation of the ZnO shell on the Zn nanowire by showing the (100) and (002) lattice orientations. The growth of Zn nanostructures can be understood similar to Mg nanostructures, which starts from the sublimation of Zn powder to Zn vapour. The vapour condenses on the substrate to form Zn nanoclusters, followed by the growth/aggregation of nanoclusters that result in the formation of nanoparticles/nanodisks. As the temperature of the substrates increases (at a low supersaturation ratio of S > 1), the nanodisks start to attach themselves through the orientation attachment process, to reduce the surface energy. This acts as the root for the formation and growth of serpentine-like Zn nanowires, similar to the previous reports.36,39 Moreover, the Zn nanodisk acts as a nucleation point for the incoming vapours, and the anisotropy of the HCP crystal structure favours the formation of Zn nanowires. The as-produced Zn wires show a vapour-solid growth mechanism with a growth direction of [100].
 | ||
| Fig. 5 (a) Bright-field TEM image of Zn nanoparticles and (b) the corresponding SAED pattern. (c) Bright-field TEM images of Zn nanowires and (d) the corresponding HRTEM image. | ||
Since we are using a vacuum-based thermal evaporation set up with the added advantage of rapid heating and cooling rates, there is a natural possibility for the atomic-scale mixing of different metal vapours. To understand and check the possibilities, we chose both Mg and Zn as the source materials, having lower melting points and high vapour pressures. Fig. 6(a) shows the bright-field TEM image of Mg–Zn nanoparticles deposited on a carbon-coated TEM grid, which were kept at the P3 position at a sublimation temperature of 550 °C. Unlike the Mg and Zn nanoparticles, which have hexagonal prismatic shapes, the Mg–Zn nanoparticles tend to show hexagonal, triangular, platelet like structures and cube and rod-like projections with a reduced average particle size of 45 nm. The random shape distribution may be a consequence of an alteration in the surface energy of different facets due to the presence of Mg. This affects the growth rate, and the Mg composition varies from ∼7–15 at% in the overall distribution of nanoparticles produced. Triangular platelet-shaped particles showed the minimum concentration of Mg and hexagonal platelet-shaped particles are showed the highest composition of Mg in the nanoparticles. The EDX spectrum of a single particle (hexagonal platelet-shaped), shown in Fig. 6(c), confirms the formation of Zn-rich Mg–Zn alloy nanoparticles by indicating an ∼80 at% of Zn and ∼14 at% of Mg and ∼5 at% of O. Moreover, the HRTEM image of a single Mg–Zn particle shown in Fig. 6(b) reflects an increased lattice spacing for the (101) orientation of Zn. The corresponding lattice parameters for the Mg–Zn nanoparticles, calculated using the SAED pattern (a = 2.725 Å and c = 5.031 Å), were larger than the standard lattice parameters of Zn (a = 2.6650 Å and c = 4.9470 Å). This supports the formation of the Zn-rich Mg–Zn solid solution. Since Mg has a larger atomic diameter (Mg: 160 pm and Zn: 134 pm) than Zn, the partial substitution of Zn by Mg will lead to an increased d-spacing as well as lattice constants. The source temperature of 400 °C is sufficient for the sublimation of both Mg and Zn powders in a vacuum as both Mg and Zn have a high vapour pressure. The simultaneous sublimation followed by the vapour confinement by the curvature will lead to an increase in the number of collisions between Mg and Zn vapours. This will further lead to the deposition of the vapour on the substrate. Moreover, since Zn has a higher vapour pressure than Mg at all temperatures, the rate of Zn diffusion is faster as compared to Mg, and this leads to the formation of Zn-rich nanostructures.43 Nonetheless, the final composition and morphology can be varied by tuning the vapour pressure, initial powder composition and supersaturation. Moreover, other Mg–Zn nanostructures can also be fabricated similar to Mg and Zn, as mentioned earlier. Correspondingly, we were able to produce Mg–Zn nanowires with intermetallic structures such as MgZn2 and MgZn. A detailed understanding of the control of the Mg–Zn composition is still underway, and more studies have to be performed to further understand the morphology-composition relationship for the Mg–Zn nanostructures. Nevertheless, this indicates that by controlling and confining the vapour, the alloy nanoparticles can be produced and deposited on various substrates as indicated earlier.
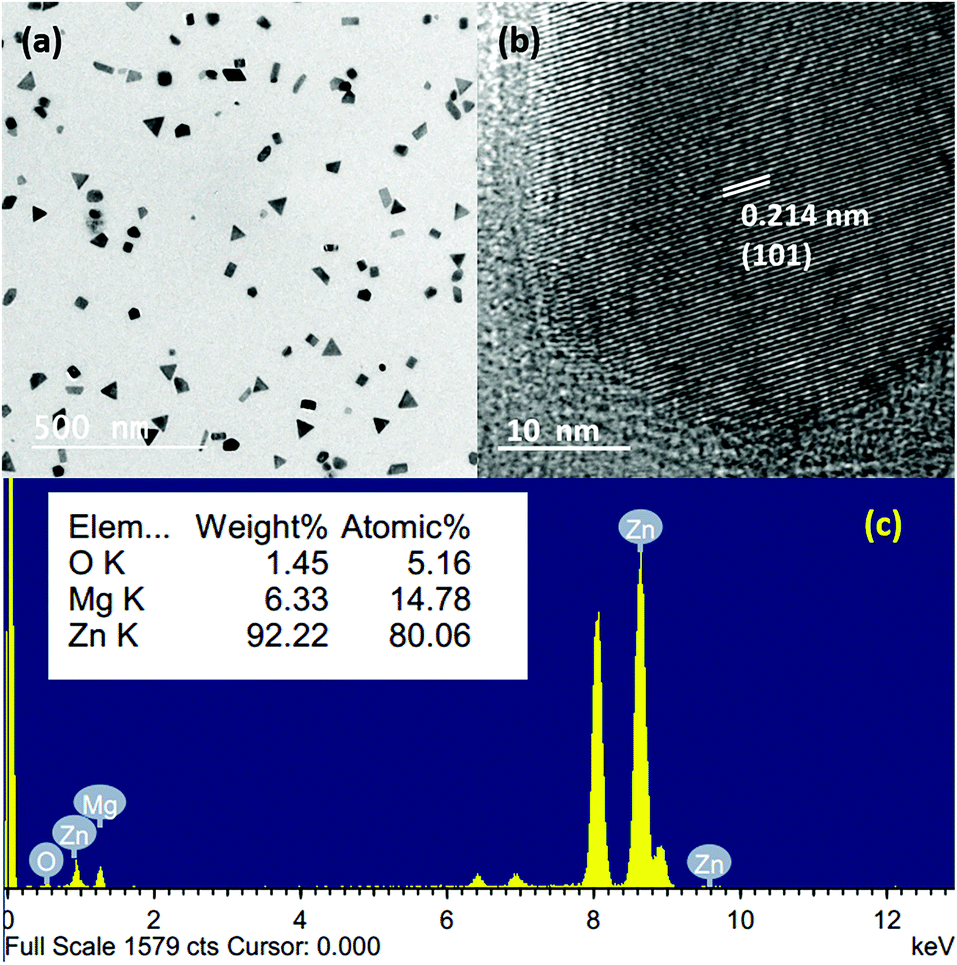 | ||
| Fig. 6 (a and b) Bright-field TEM images of Mg–Zn nanoparticles; (b) HRTEM image of a single Mg–Zn nanoparticle; (c) EDX spectrum from a single Mg–Zn nanoparticle. | ||
To further expand this to various other metal nanostructures, we have chosen other high melting point/lower vapour pressure metals, such as noble and transition metal as they have a higher demand for various applications. Nevertheless, when we consider their synthesis in a standard tubular furnace, the most critical limitation is the extremely high evaporation/sublimation temperature needed to produce metal vapours. In our RTP furnace, the maximum reachable temperature is ∼1000 °C, which is not sufficient for the evaporation of other metals. In order to overcome this challenge, we incorporated metal precursors as a source, so that they can decompose at lower temperatures to give metal vapours. Although such approaches already exist, the decomposition of metal salt in a high vacuum without a carrier gas to produce metal nanostructures has not been reported until now. For the preparation of silver and copper nanoparticles, we chose silver nitrate and copper acetate as the metal precursors, which can yield pure Ag and Cu through their decomposition. Fig. 7(a) and (b) depict the bright-field TEM images of uniformly dispersed silver nanoparticles deposited on a TEM grid. It has an average size of ∼9 nm at a source temperature of 700 °C. Fig. 7(c) and (d) show the corresponding SAED pattern and the HRTEM image, which confirm the formation of Ag by displaying its characteristic reflections. Fig. 8(a) and (b) represent the bright-field TEM images of copper nanoparticles with bimodal size distributions having smaller nanoparticles in the range of ∼3–4 nm and bigger particles of size ∼30 nm obtained at an evaporation temperature of 800 °C. The as-deposited copper nanoparticles, in contrast to silver nanoparticles, tend to show a random size distribution due to the higher substrate temperatures (>500 °C) and lower heating rates (20 °C min−1). Moreover, Cu nanoparticles also tend to show an amorphous shell; EDX analysis confirmed that this is due to the Cu2O formation following its exposure to the outside atmosphere. The HRTEM image and SAED pattern shown in Fig. 7(c) and (d) confirmed that the core of the nanoparticles was pure Cu.
 | ||
| Fig. 7 (a and b) Bright-field TEM images of Ag nanoparticles, (c) the corresponding SAED pattern and (d) HRTEM image. | ||
 | ||
| Fig. 8 (a and b) Bright-field TEM images of Cu nanoparticles, (c) SAED pattern and (d) HRTEM image. | ||
On considering the thermal decomposition of silver nitrate, a temperature between 250–500 °C was found to be sufficient for its decomposition to form Ag, NO2 and O2 species. However, we observed the fragmentation of silver nitrate at even lower temperatures, similar to the decomposition of gold acetate.44 These fragmented molecules were effectively transported to the substrates with the aid of pumping and curvature to the deposition region. Further, the higher substrate temperatures (>300 °C) led to the decomposition of nitrates over the substrates to form silver nanoparticles. For copper acetate, a similar kind of decomposition to volatile products like acetone, acetic acid, acetaldehyde, CO2 and solid products like copper and copper oxide was observed.45 Alternatively, in our case, the sublimation of copper acetate and its deposition on the substrates was observed; a photograph of copper acetate deposition on the quartz bottle is shown in Fig. S7 in the ESI.† Conversely, for copper acetate, the complete removal of the volatile species was achieved only at lower heating rates, so the decomposition of copper acetate was conducted at a lower heating rate of 20 °C min−1. This lower heating rate decreased the temperature gradient and further increased the substrate temperatures (>500 °C). This increase in the substrate temperature allowed the formation of an amorphous copper film over the substrates by the decomposition of acetate species. Subsequently, crystalline copper nanoparticles nucleated and grew from the amorphous copper film; this will be discussed in detail elsewhere. This shows that metal powders, as well as other metal precursors, can be used in our design/set up. Moreover, the thermal decomposition of acetates and nitrates in air/inert condition will decompose into metal/metal oxides at the source position45 itself; the remaining substances will be in powder form only. However, with the aid of a quartz bottle in high vacuum, we can transport the evolved volatile species during heating, or the sublimed nitrate/acetate species from the source to the appropriate substrate's position, where the final deposition is carried out. Although this cannot be applicable for all acetates and nitrates, if a metal precursor can be sublimated in vacuum conditions, then our approach will work. It will be interesting to try other acetates and nitrates as well.
Finally, all the nanostructures produced showed a similar trend of nucleation and growth on the substrates, irrespective of the choice of metal sources (metal powder/metal salts), high/low vapour pressure or low/high melting points. Also, the produced metal/bimetallic nanostructures showed monodisperse size distributions, except for the copper nanoparticles. This highlights the importance of faster heating and cooling rates. A faster heating rate will lead to a rapid sublimation of metal sources, followed by the condensation on the substrate to form metal clusters. These metallic clusters will diffuse and aggregate on the substrates to form different nanostructures. Generally, a higher heating rate allows a better-defined nucleation step. It results in a low dispersity of morphology and size. At the same time, lower heating rates show a higher dispersity.46 For all our depositions, carbon-coated copper grids were used as the substrates; it is crucial to pinpoint the importance of the substrate in the formation and growth of nanostructures.
The relation between the surface energy of the substrate, the material and interface, given by Young's equation, will systematically determine the growth models for the thin film formation. If the substrate surface energy (γB) is less than the sum of the surface energies of the film (γA) and the interface energy (between the film and substrate) (γ*) (γB < γA + γ*), island growth is favoured,47 where the metallic interactions will be higher, and the substrate will be weak. It is an essential criterion to obtain particles in the first stage of thin-film formation and to attract more particles in the later stages. Moreover, in addition to the surface energy, the adsorption energy, as well as the diffusion mechanisms of adatoms on the substrates, will also determine the growth (3D island growth or 2D layer growth).48 Since our TEM grid (substrate) is coated by an amorphous carbon layer, its surface energy will be comparatively lower than the depositing metal species. Therefore, the interactions between the substrate and the metal atoms (adsorption energy) will be less. Furthermore, as adsorption energy and diffusion barriers are directly correlated, a lower adsorption energy will favour a lower diffusion barrier. It will always lead to higher metallic interactions, resulting in island/3D growth,48 which favours the formation of various metal nanostructures. Alternatively, we were also able to tune different metal nanostructures on other substrates (SS foil, Pd, Cu, SiN membranes, etc.) as the substrate-metal interactions became negligible at higher substrate temperatures.49 The SEM images of Mg nanoparticles and Zn nanowires formed on the SS foils are shown in Fig. S8 in the ESI.† In short, this simple and cost-effective thermal evaporation set up can be used as a straight-forward approach to producing various metal nanostructures by the effective use of the vapour confinement, rapid heating and cooling rates.
Conclusion
We have demonstrated a simple, cost-effective and robust design based on thermal evaporation, for fabricating metallic/bimetallic nanostructures of various elements. By adopting this set-up, different nanostructures of lower melting point elements like Mg (NPs, NWs, NNs), Zn (NPs and NWs) and Mg–Zn alloy nanoparticles were successfully fabricated through the sublimation of metal powders. Copper and silver nanoparticles of less than 30 nm were also produced by the decomposition of metal salts and confining the deposition on the substrates. The tuning of various nanostructures was possible using a strategy of vapour confinement by incorporating a quartz bottle along with rapid thermal heating to confine the vapour and control the local supersaturation conditions over the substrates. The confinement of vapour using the curvature/neck diameter of a quartz bottle plays a significant role in altering the thermal energy and the mean free path of the metal atoms. This controls the rate and vapour pressure over the substrate without a carrier gas flow. Finally, the presented strategy can be very well applied to the formation of other nanostructures using a simple thermal evaporation technique.Experimental procedure
Fig. 2(b) shows the schematic diagram of the vacuum-based thermal evaporation set-up. Two separate quartz bottles with outer body diameters of 5 cm and curvature/neck OD of 3 and 2 cm were used for the thermal evaporation process. Commercially available metal powders were used as the source materials for Mg and Zn sublimation. In contrast, for Cu and Ag, anhydrous copper acetate and silver nitrate were used because the maximum reachable temperature in the furnace was only 1000 °C. Next, 50 mg of metal powder/metal salt was loaded on a Mo boat, which was placed inside the quartz bottle. The quartz bottle, along with the Mo boat, was placed in a tubular RTP furnace. The whole set up was evacuated to a base pressure of 10−5–10−6 mbar. Metal powders/salts were sublimated/evaporated at temperatures ranging from 600–800 °C at different holding durations ranging from 1 to 15 minutes. Metal vapours were collected from the colder regions beyond the curvature/neck at different substrate positions of P1, P2 and P3, which were at distances of 16, 18 and 20 cm from the source. Carbon-coated TEM grids, stainless steel foils, Cu foils, SiN grids, Pd grids, etc., were used as the substrates, and prepared samples were further examined by transmission electron microscopy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the support of this work from the Department of Science and Technology (DST) India through DST INSPIRE FACULTY Program, grant no DST/INSPIRE/04/2013/000470. We also would like to thank, Mr Vimal M, for his contribution to the schematic drawing.References
- U. Y. Qazi and R. Javaid, Adv. Nanoparticles, 2016, 5, 27–43 CrossRef CAS.
- M. Tian, J. Wang, J. Kurtz, T. E. Mallouk and M. H. W. Chan, Nano Lett., 2003, 3, 919–923 CrossRef CAS PubMed.
- M. Hu, J. Chen, Z. Li, L. Au, G. V Hartland, X. Li and M. Hu, Chem. Soc. Rev., 2006, 35, 1084 RSC.
- B. Wiley and Y. Sun, Acc. Chem. Res., 2007, 40, 1067–1076 CrossRef CAS PubMed.
- Z. Liu, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- P. K. Jain, X. Huang, I. H. El-sayed and M. A. El-sayed, Acc. Chem. Res., 2008, 41, 7–9 CrossRef PubMed.
- J. A. Scott, D. Totonjian, A. A. Martin, T. T. Tran, J. Fang, M. Toth, A. M. McDonagh, I. Aharonovich and C. J. Lobo, Nanoscale, 2016, 8, 2804–2810 RSC.
- T. Sakai and P. Alexandridis, langmuir, 2004, 20, 8426–8430 CrossRef CAS PubMed.
- F. Tang, T. Parker, H. Li, G. Wang and T. Lu, J. Nanosci. Nanotechnol., 2007, 7, 3239–3244 CrossRef CAS PubMed.
- H. Wang, X. Song, L. You and B. Zhang, J. Cryst. Growth, 2015, 432, 78–82 CrossRef CAS.
- S. U. Bayca, M. F. Cansizoglu, A. S. Biris and F. Watanabe, Int. J. Hydrogen Energy, 2011, 36, 5998–6004 CrossRef CAS.
- K. Zhang, C. Rossi, C. Tenailleau, P. Alphonse, K. Zhang, C. Rossi, C. Tenailleau and P. Alphonse, Appl. Phys. Lett., 2009, 063123 Search PubMed.
- B. Peng, J. Liang, Z. Tao and J. Chen, J. Mater. Chem., 2009, 19, 2877–2883 RSC.
- M. F. Ryan, Ann. Clin. Biochem., 1991, 28, 19–26 CrossRef PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- M. Villeta, B. De Agustina, J. M. S. De Pipaón and E. M. Rubio, Int. J. Adv. Manuf. Technol., 2012, 60, 1237–1246 CrossRef.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283–1315 CrossRef CAS PubMed.
- L. Weng and T. J. Webster, Int. J. Nanomed., 2013, 8, 1773–1781 Search PubMed.
- A. Manuscript, Chem. Commun., 2016, 52, 12179–12182 RSC.
- F. Sterl, N. Strohfeldt, R. Walter, R. Griessen, A. Tittl and H. Giessen, Nano Lett., 2015, 15, 7949–7955 CrossRef CAS PubMed.
- X. Duan, S. Kamin and N. Liu, Nat. Commun., 2017, 8, 1–9 CrossRef PubMed.
- W. Li, C. Li, C. Zhou and H. Ma, Angew. Chem. Int. Ed., 2006, 45, 6009–6012 CrossRef CAS PubMed.
- M. Calizzi, F. Venturi, M. Ponthieu, F. Cuevas, V. Morandi, T. Perkisas, S. Bals and L. Pasquini, Phys. Chem. Chem. Phys., 2015, 18, 7–11 Search PubMed.
- N. S. Norberg, T. S. Arthur, S. J. Fredrick and A. L. Prieto, J. Am. Chem. Soc., 2011, 133, 10679–10681 CrossRef CAS PubMed.
- K.-F. A.-Z. insou Liu, Int. J. Hydrogen Energy, 2015, 61, 1–13 Search PubMed.
- L. Pasquini, E. Callini, E. Piscopiello, A. Montone, M. V. Antisari and E. Bonetti, Appl. Phys. Lett., 2009, 94, 10–13 CrossRef.
- W. Lee, M. Jeong and J. Myoung, Acta Mater., 2004, 52, 3949–3957 CrossRef CAS.
- J. Benson, S. Boukhalfa, A. Magasinski, A. Kvit and G. Yushin, ACS Nano, 2012, 6, 118–125 CrossRef CAS PubMed.
- J. Huotari, J. Lappalainen, J. Puustinen, T. Baur, C. Alépée, T. Haapalainen, S. Komulainen, J. Pylvänäinen and A. Lloyd, Procedia Eng., 2015, 120, 1158–1161 CrossRef CAS.
- G. Krishnan, B. J. Kooi, G. Palasantzas, Y. Pivak and B. Dam, J. Appl. Phys., 2010, 107, 053504 CrossRef.
- G. Krishnan, S. D. Graaf, G. H. ten Brink, P. O. Å. Persson, B. J. Kooi and G. P. Palasantzas, Nanoscale, 2017, 9, 8149–8156 RSC.
- T. Liu, H. Shao and X. Li, J. Phys.: Condens. Matter, 2003, 15, 2507 CrossRef CAS.
- H. V. Sheela, V. Madhusudhanan and G. Krishnan, Nanoscale Adv., 2019, 1, 1754–1762 RSC.
- Q. Yu, M. M. Mao, Q. J. Li, X. Q. Fu, H. Tian, J. X. Li, S. X. Mao and Z. Zhang, Nano Lett., 2016, 16, 1156–1160 CrossRef CAS PubMed.
- D. Yuvaraj and K. N. Rao, Mater. Sci. Eng., B, 2009, 164, 195–199 CrossRef CAS.
- R. López, E. Vigueras-Santiago, P. E. Acuña-Avila, S. Hernández-López, G. López-Téllez, E. A. Zaragoza-Contreras, C. A. Hernández-Escobar, W. Antünez and N. Torres-Gómez, Bull. Mater. Sci., 2015, 38, 1777–1781 CrossRef.
- C. Ye, X. Fang, Y. Hao, X. Teng and L. Zhang, J. Phys. Chem. B, 2005, 109, 19758–19765 CrossRef CAS PubMed.
- P. Mohanty, T. Kang, B. Kim and J. Park, J. Phys. Chem. B, 2006, 110, 791–795 CrossRef CAS PubMed.
- Q. Wang, G. Chen and N. Zhou, Nanotechnology, 2009, 20, 085602 CrossRef PubMed.
- Y. He and Y. Zhao, Cryst. Growth Des., 2010, 10, 440–448 CrossRef CAS.
- F. Tang, G. Wang, T. Lu, F. Tang, G. Wang and T. Lu, J. Appl. Phys., 2008, 102, 014306 CrossRef.
- S. Kar, T. Ghoshal and S. Chaudhuri, Chem. Phys. Lett., 2006, 419, 174–178 CrossRef CAS.
- B. B. Argent, Trans. Faraday Soc., 1964, 61, 655–664 Search PubMed.
- S. D. Bakrania, G. K. Rathore and M. S. Wooldridge, J. Therm. Anal. Calorim., 2009, 95, 117–122 CrossRef CAS.
- Z. Lin, D. Han and S. Li, J. Therm. Anal. Calorim., 2012, 107, 471–475 CrossRef CAS.
- P. Guardia, J. Pe and L. M. Liz-marza, Chem. Commun., 2010, 46, 6108–6110 RSC.
- N. Kaiser, Appl. Opt., 2002, 41, 3053–3060 CrossRef CAS PubMed.
- F. Ruffino and F. Giannazzo, Crystals, 2017, 7, 24–30 CrossRef.
- C. Langlois, Z. L. Li, J. Yuan, D. Alloyeau, J. Nelayah and D. Bochicchio, Nanoscale, 2012, 4, 3381–3388 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, including TEM images Mg/MgO films, Mg microstructures are available. See DOI: 10.1039/d0na00467g |
| This journal is © The Royal Society of Chemistry 2020 |