
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermally induced intra-molecular transformation and metalation of free-base porphyrin on Au(111) surface steered by surface confinement and ad-atoms†
Dipayan
Sen
 a,
Piotr
Błoński
a,
Bruno de la
Torre
ab,
Pavel
Jelínek
ab and
Michal
Otyepka
*a
a,
Piotr
Błoński
a,
Bruno de la
Torre
ab,
Pavel
Jelínek
ab and
Michal
Otyepka
*a
aRegional Centre of Advanced Technologies and Materials, Faculty of Science, Palacký University, Olomouc, Czech Republic. E-mail: michal.otyepka@upol.cz
bInstitute of Physics, Academy of Sciences of the Czech Republic, Prague, Czech Republic
First published on 27th May 2020
Abstract
We investigated chemical transformations of a fluorinated free-base porphyrin, 5,10,15,20-tetrakis(4-fluorophenyl)-21,23H-porphyrin (2H-4FTPP) under a Au(111) surface confinement and including gold adatoms by using an experiment and density functional theory based first-principles calculations. Annealing of 2H-4FTPP led to cyclodehydrogenation of the molecule to a π-extended fused aromatic planar compound, 2H-4FPP, and metallation of the porphyrin ring by Au atoms to Au-4FPP complex. Noticeable lowering of bond-dissociation energies of the pyrrole's C–H bonds of the Au(111) supported molecule with respect to their values in the gas phase explained the observed on-surface planarization. Our findings also indicate that Au adatoms may catalyze cleavage of C–H/F bonds in temperature-initiated processes on Au surfaces. BDEs and explicit inclusion of Au adatoms helps to rationalize thermally induced chemical reactions on the respective surface.
Introduction
The recent developments in on-surface synthesis enable the construction of tailored covalent nanostructures under ultrahigh vacuum (UHV) conditions on metal surfaces which are unattainable in solution.1–3 Moreover, the capability of acquiring submolecular imaging using scanning probe microscopy (SPM) with functionalized probes4,5 provided the unique method to determine intermediates and final products of on-surface reactions.6 The on-surface synthesis inherently taking place on a metal surface poses many fundamental questions, e.g., to what extent the surface confinement i.e., either a physical or chemical adsorption, may affect studied chemical processes? Furthermore, the on-surface chemical processes examined by UHV-SPM are usually thermally induced, which opens another question: since it is well known that metal atoms may detach from edges of terraces and move on the surface as adatoms7 and such adatoms do not have saturated valences, may they represent spots enabling catalysis of the surface chemical reactions?Despite the unprecedented chemical resolution provided by the high-resolution SPM technique, we still lack detailed understanding of reaction mechanisms due to limited temporal resolution. The reaction mechanism is hence deduced from the identified intermediates and reaction products.8 Theoretical calculations help to search for an optimal reaction pathway.9–13 In order to follow the real processes happening during on-surface chemistry, the involvement of the catalytic adatoms has to be explicitly considered.
Porphyrins belong to a class of molecular systems that already demonstrated key utility in diverse fields ranging from catalysis, spintronics, molecular sensing and photosynthesis to biological applications such as metabolism.14,15 Such high potential of porphyrin molecules thus enthused researchers to pursue investigations on porphyrin derivatives and their self-assembly. Controlling chemical reactions of surface restricted porphyrin molecules by rational designing could open up a plethora of prospective research avenues including modifications of the physicochemical properties of surface-confined porphyrin derivatives in situ by macrocycle metalation.16–18 However, to realize its potential to the fullest, an in-depth understanding of the various conformations of the concerning species at the atomic level is necessary, which is unfortunately still largely lacking. In the current work we addressed (i) the chemical evolution of the fluorinated free-base porphyrin under surface confinement on Au substrate and (ii) the catalytic role of gold adatoms on Au(111) in the 2H-4FTPP → 2H-4FPP → Au-4FPP transformation by combining UHV-SPM experiments and density functional theory (DFT) computations. Our results indicate that the gold adatom significantly affected the reaction barriers of various processes thus modulating the reaction mechanism. Generally, a role of catalytic adatoms has to be carefully analysed in order to obtain plausible reaction mechanism of on-surface catalysed processes.
Methods
Computational details
DFT based first principles computations were carried out by using Vienna Ab initio Simulation Package (VASP)19,20 which incorporates projector-augmented-wave (PAW) method21,22 along with plane wave basis sets with an energy cut-off of 500 eV. Electronic exchange and correlation effects were treated by using Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional within the generalized gradient approximation (GGA).23 All computations were performed in spin unrestricted manner. Zero damping DFT-D3 method of Grimme24 was utilized in all computations to account for the dispersion corrections. Brillouin zone samplings were kept restricted to Γ point only due to the supercell dimensions being sufficiently large.The Au(111) surface was modelled by using 7 × 7 and 8 × 8 supercells consisting of 4 Au layers of which the bottom two layers were kept constrained in all structural relaxations. Vacuum layer of length ∼12 Å was deployed along the off-planar direction to ward off spurious interactions with the periodic images. Bare Au(111) substrate (cell parameters and the internal atomic positions), the Au(111) slab with pre-adsorbed Au adatom (the internal atomic positions), freestanding fluorinated free-base porphyrin, 2H-4FTPP, molecular model (the internal atomic positions) and the 2H-4FPP/Au(111) heterostructures (the internal atomic positions) were relaxed until all forces were reduced to less than 0.01 eV Å−1. Their respective total energies and electronic properties were re-computed using PBE0 hybrid functional25 (i.e., by mixing the PBE exchange energy and Hartree–Fock (HF) exchange energy in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, along with the full PBE correlation energy). Partial charges of various atomic species were computed by Bader analysis.26 The VASP data (for selected configurations) were also compared against non-periodic DFT results performed by using Gaussian code27 and B3P86 hybrid functional28 (which uses the P86 correlation functional instead of LYP but retains the three parameters derived for B3LYP29).
:1 ratio, along with the full PBE correlation energy). Partial charges of various atomic species were computed by Bader analysis.26 The VASP data (for selected configurations) were also compared against non-periodic DFT results performed by using Gaussian code27 and B3P86 hybrid functional28 (which uses the P86 correlation functional instead of LYP but retains the three parameters derived for B3LYP29).
Relative stabilities of various surface-adsorbed species (i.e., Au adatom on an Au(111) slab and the 2H-4FPP (bis-(2,10-difluoro-5H-diindeno[2,1-b:1′,2′-d]pyrrole)-5,7-bis(1H-pyrrol-2-yl)) molecule on a bare Au(111) slab and the Au(111) slab with pre-adsorbed Au adatom) were obtained from their adsorption energies, Eads, calculated using the following expression:
| Eads = Etot − Eslab − Eadsorb, | (1) |
The H and F bond dissociation energies of the 2H-4FTPP molecule were computed by using the following formula:
| BDE = Edis + EH/F − E2H-4FTPP | (2) |
Experimental details
The experiments were performed with an ultra-high vacuum combined noncontact-AFM/STM system (Createc) at 5 K using a qPlus sensor equipped with a Pt/Ir tip, where the base pressure was below 5 × 10−10 mbar. The Au(111) substrate was prepared by standard cycles of Ar+ sputtering and subsequent annealing. 2H-4FTPP species are commercial from Porphyrin Systems and were sublimated from a tantalum pocket onto a clean Au(111) at room temperature. Next, samples were transferred to the SPM stage held at 5 K. In nc-AFM imaging, a metal tip mounted onto a qplus sensor (resonant frequency ≈ 30 kHz; stiffness ≈ 1800 N m−1) was oscillated with a constant amplitude of 50 pm. The tip apex was functionalized by a CO-molecule picked up from Au(111) substrate. All images were taken at a constant height mode.Results
Experimental findings
Fig. 1a shows the free-base porphyrin 2H-4FTPP used in this study. Deposition of small amount of molecules on the Au(111) substrate at room temperature led single 2H-4FTPP featuring a two-fold symmetric characteristics attributed to the nonplanar macrocycle deformation of the porphyrin upon surface stabilization as depicted in Fig. 1b. High-resolution AFM image clearly shows the saddle shape deformations attributed to the twisted fluorine-aryl moieties whereas the inner depression is assigned to the free-base macrocycle (Fig. 1b). After annealing to 500 K and 575 K molecular chains stabilized by intermolecular C–H⋯F–C interactions confined to fcc regions of the Au(111) herringbone reconstruction are observed. Fig. 1c shows that thermal annealing of the sample to 575 K gave rise to the formation of one-dimensional supramolecular arrays, stabilized by weak intermolecular interactions.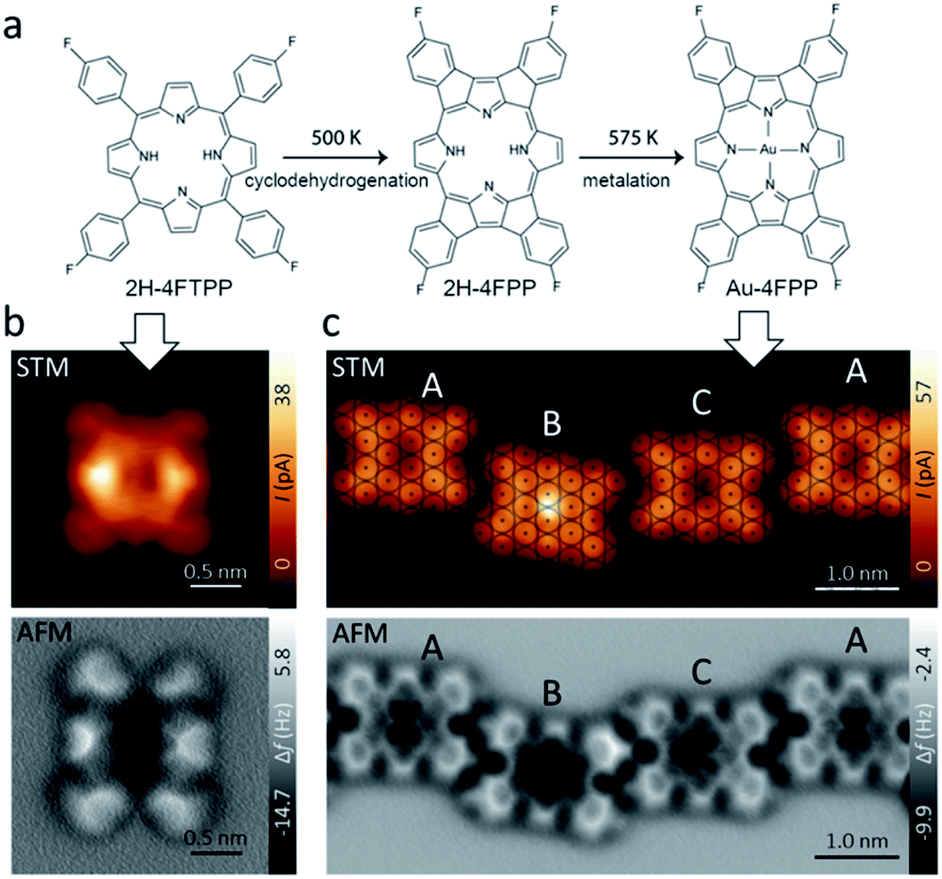 | ||
| Fig. 1 (a) Chemical sketch of thermal-assisted reaction pathway of 4F-TPP on Au(111) affording: planarization of the molecular backbone via cyclodehydrogenation and electrocyclic ring closure and self-metallation with an Au atom from Au(111) surface. (b) Constant height STM (top) and AFM (below) images with functionalized CO-tip of a single 2H-4FTPP molecule after adsorption on Au(111) at RT. (c) Constant height STM (top) with superimposed atomic register of Au(111) substrate and AFM (below) images of A (non metallated), B (adatom-metallated) and C (surface-metallated) types of 2H-4FPP molecules after annealing to 575 K. | ||
High-resolution AFM images (Fig. 1b and c) unambiguously show that the 2H-4FTPP molecules underwent planarization through dehydrogenation and electrocyclic ring closure of their aryl termini and macrocyclic pyrroles, i.e., 2H-4FTPP → 2H-4FPP. Note, that the first annealing step at 500 K planarized the fluorinated porphyrin precursor but preserved the integrity of the macrocycle and its C–F bonds. Regarding of featuring the macrocycle, both constant height high-resolution STM and AFM images display up to three different planarized species, to be termed A, B and C which are assigned to non-metallated, Au-adatom metallated (AuB-4FPP) and Au-surface metallated (AuC-4FPP) molecules, respectively. The second annealing step at 575 K for 30 minutes induced self-metalation in approximately 20% of the planarized molecules. Statistically we found approximately 65% of A molecules, 20% of B and 15% of C type. Molecule A shows a void at the core in both STM and AFM images as expected for empty macrocycles. On the other hand, on molecules B and C, a cross-shaped feature is visible in the AFM images, a similar contrast as has also been observed on other metal phthalocyanines and porphyrins.30,31 Interestingly, significant differences are clear in STM images of molecules B and C. The former one displays a fain protrusion at the core whereas a void is featured at the latter one. This observation points to a two different metallation mechanism in B and C molecules. One shall note, that previous works on similar molecules have also reported intermediate metallated-states.17,18 In fact, when we have a detailed look on the adsorption sites of the three species on Au(111) substrate with atomic resolution, we found significant differences (Fig. 1c top). The centre of molecule C was found to be close to top site, which allowed to develop coordination of the macrocycle with Au-surface atom, whereas, the centre of molecule A was found to be close to a bridge site so any Au atoms from the surface were too far to develop any coordination and localization of Au-adatoms is energetically unfavourable. In a different manner, the centre of B molecule was found close to a hollow site which is far from any surface atoms but energetically favourable for Au-adatom thus facilitating its coordination with the macrocycle.
Theoretical results
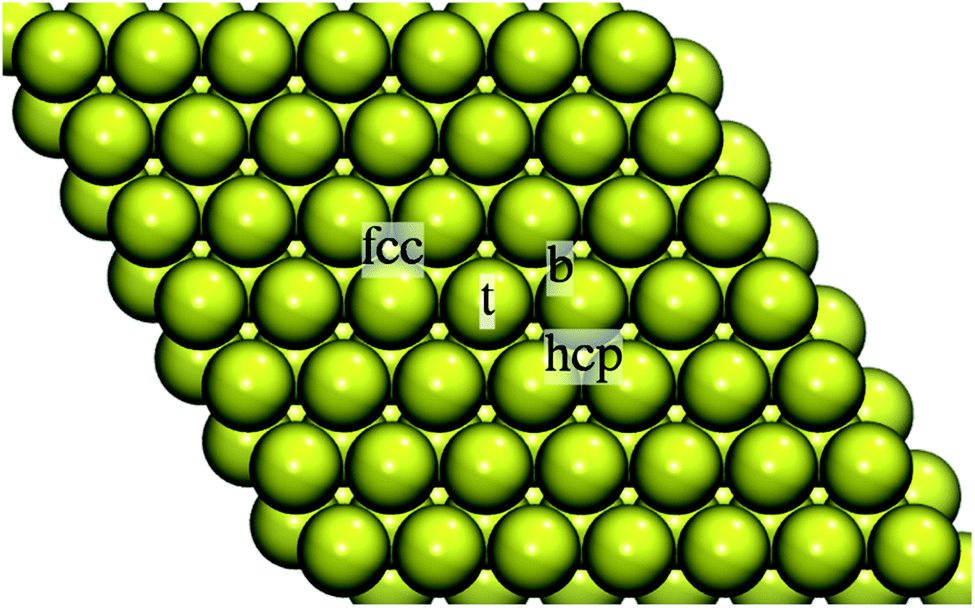 | ||
| Fig. 2 Top view of the 7 × 7 4-layer slab representing the Au(111) surface with the on-surface adsorption sites indicated: on-top (t), bridge (b), hollow fcc, and hollow hcp. | ||
The stability of Au adatom in the hollow hcp site was marginally lower (Eads = −67.91 kcal mol−1). This was also reflected in the adatom-surface distance: 2.678 Å vs. 2.688 Å for Au in fcc and hcp hollow, respectively. The on-top and bridge positions were unstable, the Au adatom moved to the nearest hollow fcc site. Our results are in line with the combined experimental and theoretical work reporting the gold adatom located at a three-fold hollow site.32 Test calculations by using a smaller 4 × 4 Au supercell and 4 × 4 × 1 k-point grid yielded Eads = −70.17 kcal mol−1 at the adatom-surface distance of 2.684 Å for Au in the fcc hollow and Eads = −68.87 kcal mol−1 at the adatom-surface distance of 2.695 Å for the adsorption in the hollow hcp.
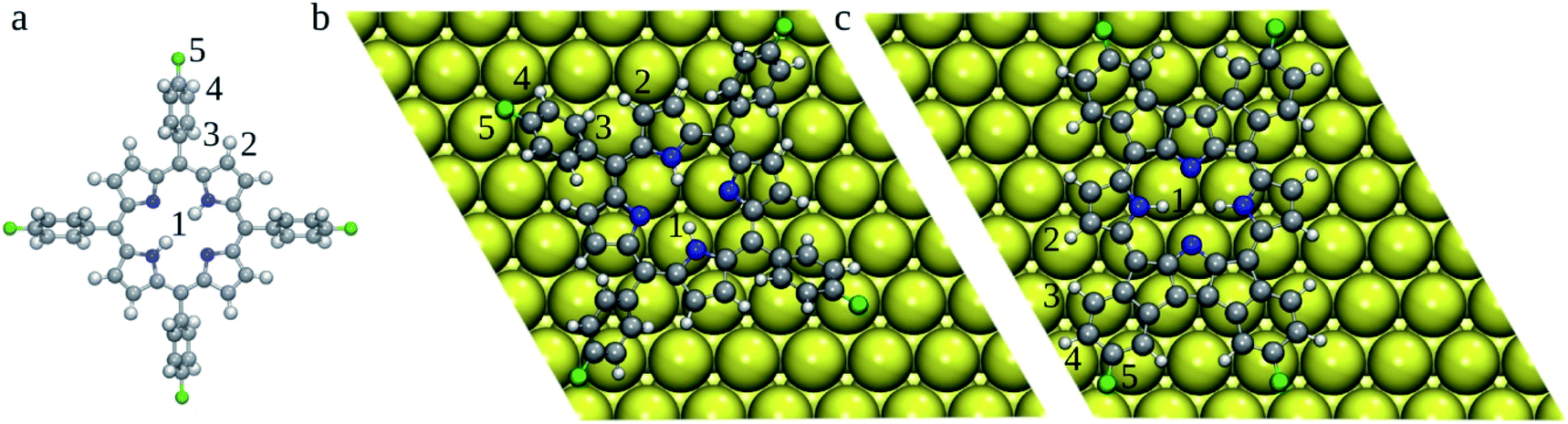 | ||
| Fig. 3 Optimized 2H-4FTPP in the gas-phase (a) and 2H-4TFPP deposited on the Au(111) surface (top-view) (b). 2H-4FPP on Au(111) (c). Au atoms are shown in yellow, C in grey, N in blue, F in green, and H in white. BDEs of bonds labelled by numbers are gathered in Table 1. | ||
To rationalize the heat induced sequential intra-molecular transformations, (i) planarization of the molecular backbone via cyclodehydrogenation and electrocyclic ring closure (2H-4FTPP → 2H-4FPP), (ii) metallation with an surface Au atom (2H-4FPP → AuC-4FPP), we calculated BDEs of the relevant H (1–4) and F (5) atoms (Fig. 3) both of freestanding 2H-4FTPP and 2H-4FPP deposited on Au(111) (Table 1). It was shown that B3P86 provides good estimates of BDEs,33,34 however, this functional was not implemented for periodic DFT calculations, therefore we tested performance of PBE0 + D3 against B3P86 functional. Both B3P86 and PBE0 + D3 provide the same orders of calculated BDEs and differ each other less than 3%, thus periodic DFT computations with PBE0 + D3 functional could reliably be deployed to study energetics and structural transformation of 2H-4FTPP → 2H-4FPP → Au-4FPP under surface confinement, a scope which falls outside of the purview of the non-periodic DFT implementations that are standard for stand-alone molecular models. C–F bond exhibited highest BDE, which implies, during chemical transformation induced by surface confinement or external stimuli, integrity of the fluorine functionalization is least likely to be perturbed. This observation directly corroborates the experimental observations.35 The adsorption of 2H-4FTPP on the Au(111) surface led also to significant lowering of the H/F BDEs with respect to the gas phase (Table 1). Particularly the C–H2 bond can be easily cleaved, which can lead to ring fusion at this particular C atom in agreement with the experimental observations.35 The liberated H atoms will either bind to the Au surface or Au adatom (vide infra), and if two H atoms meet, they may form H2, which would desorb to the gas-phase. However, atomic hydrogen can also passivate the defluorinated carbon atoms that do not participate in oligomers formation.35 It should be noted that the surface confinement changed significantly order of BDEs, i.e., susceptibility of individual bonds for reaction. The susceptibility of C–H2 bond with respect to the vacuum can originate from the clash of H2 and H3 atoms caused by the rotation of phenyl ring due to molecule surface planarization. The barriers against the rotation of phenyl rings could be calculated by using Nudged Elastic Band method,9 which however, will not be feasible for current systems. Therefore, here we have focused on rather simple indices (BDEs), which can be readily calculated even for large systems.
| Bond | Freestanding | Freestanding | Au (111) adsorbed 2H-4FTPP | Au (111) adsorbed 2H-4FPP |
|---|---|---|---|---|
| B3P86 | PBE0 + D3 | PBE0 + D3 | PBE0 + D3 | |
| N–H1 | 111.8 (0.0) | 111.8 (0.0) | 143.3 (70.3) | 95.8 (3.8) |
| C–H2 | 124.5 (12.7) | 121.8 (10.0) | 120.1 (47.1) | 92.0 (0.0) |
| C–H3 | 119.3 (7.5) | 116.2 (4.4) | 74.0 (1.0) | 97.2 (5.2) |
| C–H4 | 121.5 (9.7) | 117.7 (5.9) | 73.0 (0.0) | 95.9 (3.9) |
| C–F | 129.9 (18.1) | 127.8 (16.0) | 127.6 (54.6) | 105.5 (13.5) |
|
|
|||
|---|---|---|---|
| Position | E ads | Position | E ads |
| 1fcc | −133.4 (0.0) | 4 | −125.8 (−7.6) |
| 1hcp | −131.4 (−2.0) | 34 | −127.8 (−5.6) |
| C b | −111.4 (−22.0) | 5 | −122.9 (−10.5) |
| 2 | −126.6 (−6.8) | 45 | −124.5 (−8.9) |
| 3 | −125.1 (−8.3) | 56 | −104.3 (−29.1) |
| 23 | −121.7 (−11.7) | 6 | −97.1 (−36.3) |
| Au(111) | −124.9 (−8.5) | ||
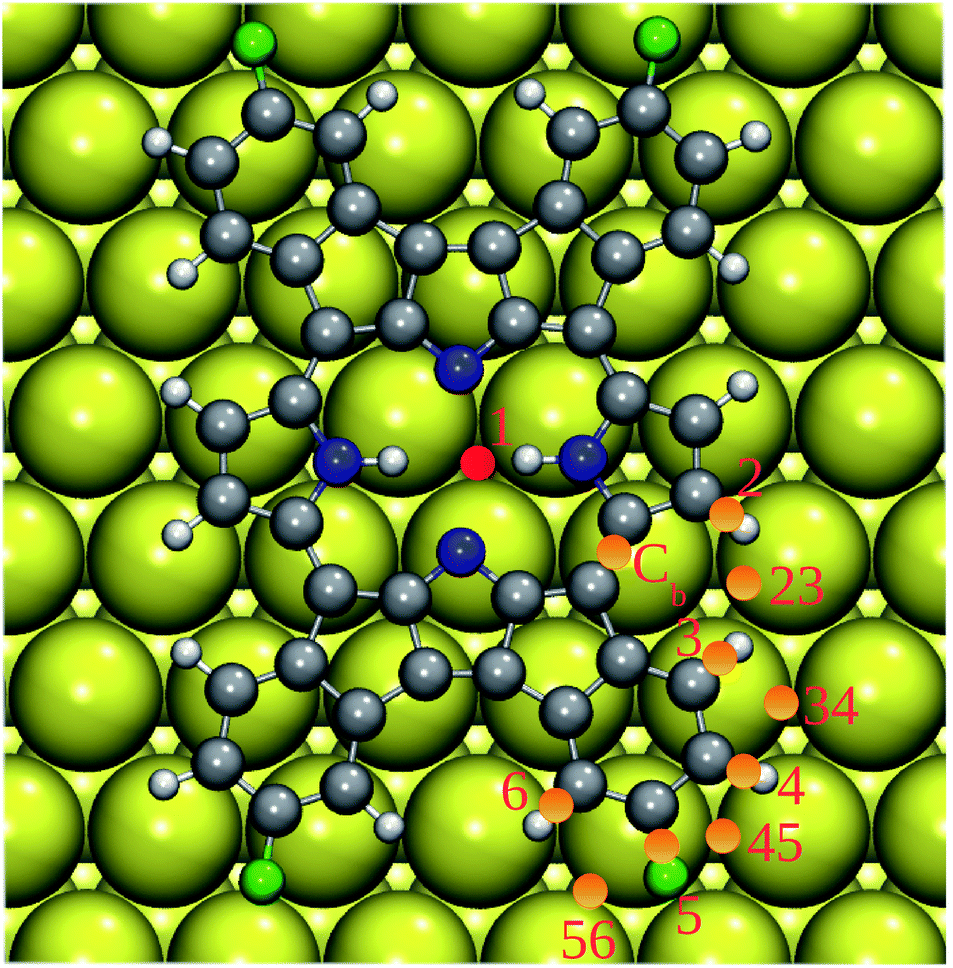
To further explore this point, we computed the BDEs of the relevant C(N)–H and C–F bonds of 2H-4FPP deposited on Au(111) with pre-adsorbed Au adatom. We used following notation: 1@Au1 for BDE of the N–H bond (1) of the central ring and Au adatom in the GS position (Au1), 2@Au23 for BDE of the C–H bond (2) and Au adatom in the rim position between two terminal H atoms (2 and 3), etc. The computed BDEs are listed in Table 3. The important point to note is that the Au adatom on the Au(111) surface changed BDEs of 2H-4FPP considerably. In the close proximity to the Au adatom, the drop in BDEs of the concerned bonds was most prominent. Conversely, if the Au adatom was located far away from the concerned bonds, their dissociation energies approached to that of them on the pristine Au(111) surface. Among all configurations considered, the central H atom(s) exhibited highest susceptibility to cleavage due to the Au adatom. This further supports the experimentally observed metallation of 2H-4FPP to Au-4FPP.35
| Configuration | BDE | Configuration | BDE |
|---|---|---|---|
| 1@Au1 | 84.4 | 5@Au5 | 90.8 |
| 2@Au1 | 92.7 | 1@Au34 | 101.3 |
| 3@Au1 | 99.7 | 2@Au34 | 96.5 |
| 4@Au1 | 96.7 | 3@Au34 | 88.2 |
| 5@Au1 | 104.5 | 4@Au34 | 81.8 |
| 2@Au2 | 85.9 | 5@Au34 | 128.0 |
| 3@Au3 | 83.0 | 5@Au56 | 116.5 |
| 4@Au4 | 83.1 | 6@Au56 | 105.4 |
Bader charge analysis revealed an accumulation of −0.046e on the Au adatom on the Au(111) surface. Conversely, for all configurations in which the Au adatom was at the nearest most stable position of the respective C(N)–H and C–F bonds of 2H-4FPP, it showed a small partial charge. Hence, a small electron transfer between the Au adatom and the molecule could be expected. Indeed, Fig. S3† displaying the charge-density-difference isosurfaces for these systems, confirmed the Au adatom donated electrons in all cases to 2H-4FPP. The charge transfer between Au and the molecule was significantly increased in the presence of adatom. The increased charge transfer between the Au adatom and the molecule and the Au–N bond-lengths (Fig. S4†) indicate on the formation of the respective chemical bonds.
Conclusions
In the current study we investigated the chemical evolution of the fluorinated free-base porphyrin (2H-4FTPP) under surface confinement on Au substrate and the catalytic role of gold adatoms on Au(111) in the 2H-4FTPP → 2H-4FPP → Au-4FPP transformation from both experimental and theoretical perspectives.To rationalize the experimental findings, we investigated the energetics of 2H-4FTPP, 2H-4FPP, and metallated AuB-4FPP configurations through DFT based first principle calculations. The obtained results readily indicated the H/F BDEs of the 2H-4FPP on the Au(111) surface are considerably lower in comparison to the same in gas phase. The pyrrole's C–H bond was found to be particularly vulnerable for cleavage which could explain the dehydrogenation and the subsequent planarization observed during experimental measures.
Various configurations of 2H-4FPP adsorbed on Au (111) with pre-adsorbed Au adatom were also investigated in detail. Among them, in direct agreement with experimental observations, a configuration in which the centre of 2H-4FPP rests on the Au adatom was found to be energetically most stable. Importantly, the Au adatom decreased BDEs of 2H-4FPP quite a considerably. The effect was most prominent for the bonds in direct vicinity to the Au adatom, thus conforming its catalytic role. Also, Bader charge analysis confirmed small amount of electron transfer between the Au adatom and the 2H-4FPP in these cases. These findings provided an atom-level insight into the mechanism of on-surface synthesis and transformation of porphyrin-derivative, and thus may stimulate the development of this emerging field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support of the Operational Programme for Research, Development and Education of the European Regional Development Fund (Project No. CZ.02.1.01/0.0/0.0/16_019/0000754). MO acknowledges the ERC grant (683024) from the EU Horizon 2020 Research and Innovation Programme.Notes and references
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687 CrossRef CAS PubMed.
- On-surface synthesis, Proceedings of the International Workshop On-Surface Synthesis, ed A. Gourdon, Springer, 2014 Search PubMed.
- S. Clair and D. G. de Oteyza, Chem. Rev., 2019, 119, 4717 CrossRef CAS PubMed.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110 CrossRef CAS PubMed.
- P. Jelínek, J. Phys.: Condens. Matter, 2017, 29, 343002 CrossRef PubMed.
- D. G. de Oteyza, P. Gorman, Y.-Ch. Chen, S. Wickenburg, A. Riss, D. J. Mowbray, G. Etkin, Z. Pedramrazi, H.-Z. Tsai, A. Rubio, M. F. Crommie and F. R. Fischer, Science, 2013, 340, 1434 CrossRef CAS PubMed.
- G. Antczak and G. Ehrlich, Surface Diffusion: Metals, Metal Atoms, and Clusters. Cambridge University Press, 2010 Search PubMed.
- A. Riss, A. Pérez Paz, S. Wickenburg, H.-Z. Tsai, D. G. De Oteyza, A. J. Bradley, M. M. Ugeda, P. Gorman, H. S. Jung, M. F. Crommie, A. Rubio and F. R. Fischer, Nat. Chem., 2016, 8, 678 CrossRef CAS PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9901 ( J. Chem. Phys. , 2000 , 113 , 9978 ) CrossRef CAS.
- J. Björk, J. Phys.: Condens. Matter, 2016, 28, 083002 CrossRef PubMed.
- M. Bieri, M.-T. Nguyen, O. Gröning, J. Cai, M. Treier, K. Aït-Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2010, 132, 16669 CrossRef CAS PubMed.
- A. Lunghi, M. Iannuzzi, R. Sessoli and F. Totti, J. Mater. Chem. C, 2015, 3, 7294 RSC.
- M. Telychko, J. Su, A. Gallardo, Y. Gu, J. I. Mendieta-Moreno, D. Qi, A. Tadich, S. Song, P. Lyu, Z. Qiu, H. Fang, M. J. Koh, J. Wu, P. Jelínek and J. Lu, Angew. Chem., Int. Ed., 2019, 58, 18591 CrossRef CAS PubMed.
- W. Auwärter, D. Ecija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105 CrossRef PubMed.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259 CrossRef CAS.
- H. Marbach, Acc. Chem. Res., 2015, 48, 2649 CrossRef CAS PubMed.
- T. E. Shubina, H. Marbach, K. Flechtner, A. Kretschmann, N. Jux, F. Buchner, H.-P. Steinrück, T. Clark and J. M. Gottfried, J. Am. Chem. Soc., 2007, 129, 9476 CrossRef CAS PubMed.
- G. Mette, D. Sutter, Y. Gurdal, S. Schnidrig, B. Probst, M. Iannuzzi, J. Hutter, R. Alberto and J. Osterwalder, Nanoscale, 2016, 8, 7958 RSC.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 ( Phys. Rev. Lett. , 1997 , 78 , 1396 ) CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. P. Perdew and M. Ernzerhof, J. Chem. Phys., 1996, 105, 9982 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Wallingford CT, 2009 Search PubMed.
- B3P86 functional is defined by its implementation in the Gaussian program.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98(45), 11623 CrossRef CAS.
- B. de la Torre, M. Švec, P. Hapala, J. Redondo, O. Krejčí, R. Lo, D. Manna, A. Sarmah, D. Nachtigallová, J. Tuček, P. Błoński, M. Otyepka, R. Zbořil, P. Hobza and P. Jelínek, Nat. Commun., 2018, 9, 2831 CrossRef PubMed.
- K. Kaiser, L. Gross and F. Schulz, ACS Nano, 2019, 13, 6947 CrossRef CAS PubMed.
- J. Kestell, J. Walker, Y. Bai, J. A. Boscoboinik, M. Garvey and W. T. Tysoe, J. Phys. Chem. C, 2016, 120(17), 9270 CrossRef CAS.
- Y. Feng, L. Liu, J.-T. Wang, H. Huang and Q.-X. Guo, J. Chem. Inf. Comput. Sci., 2003, 43, 2005 CrossRef CAS PubMed.
- P. Trouillas, P. Marsal, D. Siri, R. Lazzaroni and J.-L. Duroux, Food Chem., 2006, 97, 679 CrossRef CAS.
- B. Cirera, B. de la Torre, D. Moreno, M. Ondráček, R. Zbořil, R. Miranda, P. Jelínek and D. Écija, Chem. Mater., 2019, 31, 3248 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00401d |
| This journal is © The Royal Society of Chemistry 2020 |